

Abstract
Background
Lung adenocarcinoma (LUAD), as the most common subtype of lung cancer, is the leading cause of cancer deaths in the world. The accumulation of driver gene mutations enables cancer cells to gradually acquire growth advantage. Therefore, it is important to understand the functions and interactions of driver gene mutations in cancer progression.
Methods
We obtained gene mutation data and gene expression profile of 506 LUAD tumors from The Cancer Genome Atlas (TCGA). The subtypes of tumors with driver gene mutations were identified by consensus cluster analysis.
Results
We found 21 significantly mutually exclusive pairs consisting of 20 genes among 506 LUAD patients. Because of the increased transcriptomic heterogeneity of mutations, we identified subtypes among tumors with non‐silent mutations in driver genes. There were 494 mutually exclusive pairs found among driver gene mutations within different subtypes. Furthermore, we identified functions of mutually exclusive pairs based on the hypothesis of functional redundancy of mutual exclusivity. These mutually exclusive pairs were significantly enriched in nuclear division and humoral immune response, which played crucial roles in cancer initiation and progression. We also found 79 mutually exclusive triples among subtypes of tumors with driver gene mutations, which were key roles in cell motility and cellular chemical homeostasis. In addition, two mutually exclusive triples and one mutually exclusive triple were associated with the overall survival and disease‐specific survival of LUAD patients, respectively.
Conclusions
We revealed novel mutual exclusivity and generated a comprehensive functional landscape of driver gene mutations, which could offer a new perspective to understand the mechanisms of cancer development and identify potential biomarkers for LUAD therapy.
Keywords: mutations, functional associations, mutual exclusivity, non small cell lung cancer, cancer genetics
We revealed novel mutual exclusivity and generated a comprehensive functional landscape of driver gene mutations, which could offer a new perspective to understand the mechanisms of cancer development and identify biomarkers for LUAD therapy.

1. INTRODUCTION
Lung cancer is a major public health problem all over the world and the second leading cause of death in the world. 1 , 2 Lung adenocarcinoma (LUAD) is the most common histologic type of primary lung cancer, accounting for about 40% of all lung cancers. 3 Its incidence rate is increasing in people who have never smoked, or are aged 20–46 years. 4 LUAD patients can be divided into five groups using the TNM classifications system. 5 , 6 The higher the grade is, the more malignant the tumor is. The standard treatment of LUAD patients is surgery, radiation therapy, chemotherapy, and targeted therapies. 7 , 8 , 9
Some studies have shown that solid tumors contain hundreds or thousands of genetic alterations across multiple cancers, including lung adenocarcinoma. 10 , 11 , 12 , 13 The vast majority of them are point mutations, which are composed of driver mutations and passenger mutations. The former confer selective growth advantages to tumor cells, 12 , 14 , 15 , 16 while the latter occur during a large number of cell divisions. 17 , 18 The driver gene is defined as one whose mutations increase cell growth under the specific conditions in cells. 19 There are some driver gene mutations found in LUAD, for example, driver gene EGFR. 20 , 21 Targeted therapies against several oncogenic drivers, such as EGFR and BRAF mutations, have been approved for the precision treatment of LUAD. 22 , 23 , 24 However, patients with the same driver genetic alterations showed extensive genetic and transcriptomic heterogeneity. 25 , 26
The heterogeneity of tumors could be a major obstacle for anticancer treatment, which can occur at genetic, transcriptomic, and histological levels. Transcriptomic heterogeneity is very important in cancer researches because mRNAs can be treated as a bridge that links genetic variations and physiological traits. 27 , 28 Nowadays, gene expression‐based molecular subtyping has been used in cancers to aid treatment decisions due to the existence of transcriptomic heterogeneity. For example, breast cancer patients can be divided into four intrinsic molecular subtypes (Basal‐like, HER‐2 positive, Luminal A, and Luminal B), which has been proven to be clinically effective. 29 However, the degree of transcriptomic heterogeneity of LUAD tumors with different driver gene mutations is still unclear, which may be the key to understand the functions of driver gene mutations and improve the therapeutic efficacy in cancers.
The accumulating genetic alterations in cancers do not occur at random, but mutually depend on each other. 30 Some co‐occurring events were observed in multiple cancers by high‐throughput sequencing data. 31 For example, driver gene CTNNB1 and PIK3CA cooperatively promote tumor metastasis. 32 Mutual exclusivity refers to the phenomenon that genetic alterations of genes do not tend to occur in the same sample, which has been widely observed in numerous cancer cohorts. 33 , 34 Some well‐known cancer driver genes are mutual exclusivity. The mutually exclusive events, including BRAF and KRAS mutations (two members of MAPK–ERK pathway), are mutual exclusivity and undergo genetic alterations in lung cancer patients. 35 , 36 Similarly, KRAS and EGFR mutations are also mutually exclusive in LUAD. 37 What's more, the co‐occurring or mutually exclusive events have been reported to be clinically relevant. For example, the mutual exclusivity of ATM and TP53 mutations in mantle cell lymphoma patients was associated with significantly reduced overall survival. 38 Thus, it is necessary to comprehensively identify and analyze co‐occurring or mutually exclusive events of mutations to enhance the understanding of tumorigenesis and improve the treatment strategies for precision medicine.
In the present study, we comprehensively characterized the functions of 178 driver gene mutations within specific subtypes across 506 LUAD patients from The Cancer Genome Atlas (TCGA) project. The tumors with driver gene mutations were divided into diverse subtypes based on transcriptomic heterogeneity. Interestingly, we found more co‐occurring and mutually exclusive pairs of subtypes of tumors with driver gene mutations. Furthermore, these mutually exclusive pairs exhibited crucial roles in cancers, including nuclear division, humoral immune response, cell motility, cell differentiation, and blood circulation. Finally, we observed functional and prognostic mutually exclusive triples.
2. MATERIALS AND METHODS
2.1. Data source
In this study, the mutation profile, gene expression profile, and corresponding clinical metadata (including clinicopathological factors, overall survival [OS], disease‐specific survival [DSS]) of LUAD patients were accessed through The Cancer Genome Atlas (TCGA) portal (https://portal.gdc.cancer.gov). Non‐silent mutations are of great significance for the functional analysis of mutated genes, 39 so subsequent analysis focused on non‐silent mutations. For the mutation profile, non‐silent somatic mutations (missense mutation, nonsense mutation, translation start site, in‐frame deletion, in‐frame insert, frame‐shift deletion, frame‐shift insert, splice site, and nonstop mutation) remained according to UCSC Genome Browser (http://genome.ucsc.edu). 40 The data were filtered to exclude patients without mutation data or clinical information. There were 506 LUAD patients having mutation data and gene expression data. A total of 497 patients have available overall survival data and 464 patients have disease‐specific survival data. Besides, the cancer driver genes (n = 846) were obtained from public sources in our research. The Catalogue Of Somatic Mutations In Cancer (COSMIC) gene census manually collected more than 700 cancer genes that were mutated and causally implicated in cancer development from literatures. 41 Matthew H. Bailey et al. used 26 computational tools to identify 299 driver genes in multiple cancers. 42 The Cancer Genome Atlas Research Network identified 18 significant mutated genes among 412 lung adenocarcinomas. 43 And, Joshua D Campbell et al. identified 66 driver genes in lung cancers. 44
2.2. Identifying subtypes of tumors with mutations in each driver gene
For each driver gene, we classified the tumor samples harboring mutations in this driver gene into subtypes in three steps. First, the top 2000 most variant genes were selected according to the value of median absolute deviation (MAD) across tumors. Expression values of the above variable genes were log2 transformed and then median‐centered across samples for each gene. Second, we performed consensus clustering to divide the driver gene‐mutated tumors into subtypes by ConsensusClusterPlus R package. 45 We used Partitioning Around Medoids (PAM) algorithm to implement the unsupervised consensus clustering the Pearson's correlation coefficient as a similarity measure based on the expression data of most variable genes. The optimal number of subtypes was assessed based on 80% sample resampling over 1000 iterations. For driver genes mutations that appeared in more than 20 samples, the optimal number was determined by consensus membership heatmap. For driver gene mutations that mutated in no more than 20 samples, they were divided directly into two subtypes. Third, we calculated the silhouette width of each tumor sample within the same subtypes based on the Pearson distance. Samples with non‐positive silhouette width were regarded as unstable and removed. Subtypes with samples less than five were also removed.
2.3. Identifying the functions of mutually exclusive pairs
We selected 17,887 genes which showed detectable expression (counts >1 in at least 30% of LUAD samples). At first, differential expression analysis was performed based on the gene expression profile of LUAD patients by the “DESeq2” R package. Genes with the cutoff criteria of |log2‐fold change| ≥1 and FDR <0.05 between some subtype of tumors with driver gene mutations and wild‐type (WT) patients were regarded as differentially expressed genes (DEGs). Functional enrichment in Gene Ontology (GO) biological processes of the above DEGs was performed using g: profiler, 46 , 47 setting a threshold of 0.05 for statistical significance. At this point, the functions of driver gene mutations within subtypes were found.
Then, we identified the functions of mutually exclusive pairs of driver gene mutations within subtypes, which were functional intersections between two subtypes of driver gene mutations of some mutually exclusive pair. Furthermore, the functions of mutually exclusive triples were functional intersection in at least two mutually exclusive pairs.
Visualization of GO enrichment was performed using the EnrichmentMap plugin 48 in Cytoscape. 49 Similar GO terms were clustered together based on the similarity between each other using the overlap coefficient. Clusters were manually circled and labeled to highlight the prevalent biological functions among related GO terms.
2.4. Survival analyses
For survival analysis, overall survival and disease‐specific survival were used as the end points. The Kaplan–Meier method was performed for visualization purposes and the differences between survival curves were calculated by log‐rank test. Univariate and multivariate Cox proportional hazards regression models were applied to estimate the prognostic capability of mutually exclusive triples. The p values smaller than 0.05 were considered to be statistically significant. All of the statistical analyses were performed using R software (www.r‐project.org).
3. RESULTS
3.1. The mutually exclusive/co‐occurring events of driver gene mutations across LUAD patients
Driver gene mutations promote tumorigenesis and play major impacts on patient outcome. 50 , 51 To explore the functions and associations among driver gene mutations in LUAD, we obtained 846 driver genes from public sources and mutation profile of patients from TCGA. Among the 506 LUAD patients, we identified a total of 11,519 non‐silent mutations in driver genes. The non‐silent mutations consisted of 9509 missense mutations (83%), 1003 nonsense mutations (9%), 403 splice site mutations (3%), 9 translation start site (0.08%), 2 nonstop mutation (0.02%), 514 frameshift indels (4%), and 79 in‐frame indels (0.7%). There were 783 driver genes with at least one mutation among LUAD patients. And, 178 driver genes were mutated in more than 15 patients (> 3%) among the above genes 51 , 52 , 53 (Figure 1A), which were used for subsequent analysis. Seven of these genes appeared in at least 100 patients, including driver genes TP53 (n = 257), MUC16 (n = 211), CSMD3 (n = 201), LRP1B (n = 171), KRAS (n = 147), SPTA1 (n = 128), and FAT3 (n = 103). These seven driver genes could be verified by the oncoKB database 54 and by other researches. 55 , 56 , 57 , 58
FIGURE 1.
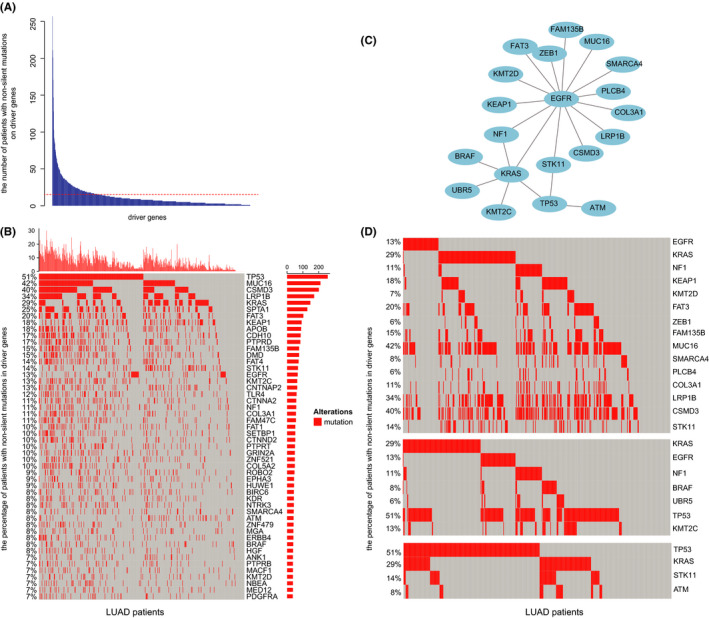
The mutually exclusive events of driver gene mutations across LUAD patients. (A) The numbers of LUAD patients with non‐silent mutations in driver genes. The red line indicated 15 patients. (B) Heatmap of the top 50 frequently mutated driver genes. The red square represented the mutations of driver genes. The X‐axis means LUAD patients and the Y‐axis means the percentage of patients with non‐silent mutations in driver genes. (C) All 21 mutually exclusive events were identified in 178 driver gene mutations, including 20 driver genes. (D) The mutual exclusivity of EGFR mutations, KRAS mutations, or TP53 mutations. Each column represents mutated tumors. The X‐axis means LUAD patients and the Y‐axis means the percentage of patients with non‐silent mutations in driver genes
In the profile of non‐silent mutations in driver genes, which presented in at least 7% of LUAD patients, mutually exclusive or co‐occurring phenomena were observed (Figure 1B). Therefore, 21 mutually exclusive events were significantly identified in LUAD using the DISCOVER method, 59 involving 20 driver gene mutations (q value <0.05, Figure 1C). We observed that EGFR mutations were mutually exclusive with 14 driver gene mutations, such as driver oncogene KRAS and KMT2D mutations (Figure 1D). KRAS mutations and six driver gene mutations were mutually exclusive (Figure 1D). In addition, oncogene TP53 mutations were not only mutually exclusive with KRAS mutations, but also with STK11 and ATM mutations (Figure 1D). However, no significantly co‐occurring events were found in our research. These results pointed to the existence of mutually exclusive driver gene mutations across LUAD patients.
3.2. High transcriptomic heterogeneity of tumors with driver gene mutations
We sought to examine the extent of the transcriptomic heterogeneity of 506 LUAD tumors and 59 adjacent normal samples. Genes exhibited stable high or low expression in normal samples, while the expression of genes was quite messy in tumors through unsupervised cluster analysis on the top 300 variant genes. This phenomenon indicated that transcriptomic heterogeneity increased in tumors (Figure 2A). To quantificationally estimate the transcriptomic heterogeneity for samples, we calculated the median absolute deviation of all genes in tumor samples. The MAD values of tumors were significantly higher than the normal samples (p < 0.001, Wilcoxon rank sum paired test, Figure 2B).
FIGURE 2.
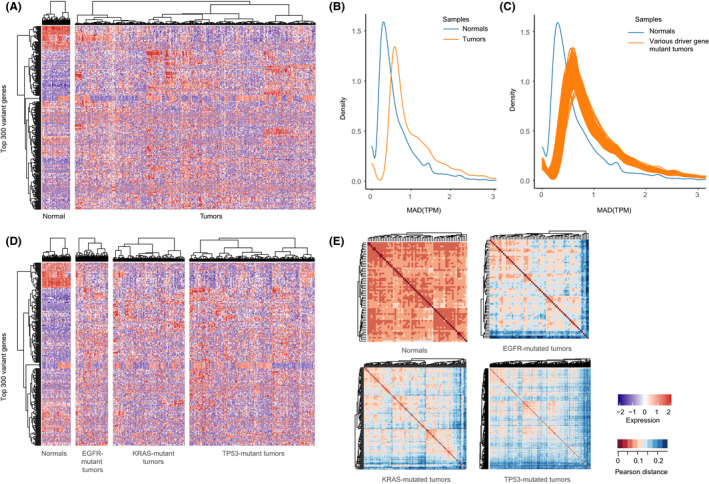
High transcriptomic heterogeneity in tumors with driver gene mutations. (A) A heatmap showed the unsupervised hierarchical clustering of LUAD tumors (n = 506) and normal samples (n = 59) by the most variable genes (n = 300). (B) The MAD distribution of all genes across all tumor and normal samples. The blue and orange lines representing tumor and normal samples, respectively. (C) The MAD distribution of all genes across samples with driver gene mutations. The blue and orange lines representing gene‐mutated tumors and normal samples, respectively. (D) A heatmap was shown by unsupervised hierarchical clustering of the most variable genes (n = 300) among EGFR‐, KRAS‐, and TP53‐mutated tumors and normal samples. (E) Pairwise comparison of transcriptomic profiles in normal samples and EGFR, KRAS, and TP53 mutant tumors. The heatmaps were plotted using unsupervised hierarchical clustering with Pearson distance (1‐Pearson correlation coefficient)
Tumors with various driver gene mutations also consistently had higher transcriptomic heterogeneity compared with the normal samples (p < 0.001, Wilcoxon rank‐sum paired test, Figure 2C). Besides, cluster heatmap showed that genes expressed inconsistently in their respective tumor samples with driver gene mutations, such as EGFR‐, KRAS‐, and TP53‐mutated tumors (Figure 2D). Meanwhile, we compared the transcriptomic similarity within tumors with driver gene mutations and normal samples by calculating the Pearson distance. A tight correlation among normal samples was observed (Figure 2E). In contrast, tumors with various driver gene mutations showed great diversity, for example, EGFR‐mutated tumors, KRAS‐mutated tumors, and TP53‐mutated tumors (Figure 2E). This phenomenon showed that unknown homogeneous clusters were hidden in the driver gene‐mutated tumors. These results implied that tumors with driver gene mutations were needed to be divided into diverse subtypes.
3.3. Identification of subtypes of tumors with driver gene mutations
Next, we identified molecular subtypes of LUAD tumors in three steps (Figure 3A). First, LUAD tumors were assigned to 178 groups by driver gene mutations. Then, the consensus clustering algorithm was applied on the transcriptomic profiles of each group of driver gene‐mutated tumors to determine subtypes. In order to retain representative samples in each subtype, we eventually performed silhouette width analysis to exclude the unstable samples. To ensure sufficient statistical power, there were five subtypes (COL2A1 subtype two, SMAD4 subtype two, IL7R subtype two, MUC6 subtype one, and NUP98 subtype two) with the small sample size (n < 5) excluded in our study. We finally divided the tumors with 178 driver gene mutations into various subtypes and the majority of driver‐mutated tumor groups had two subtypes (143/178, Figure 3B).
FIGURE 3.
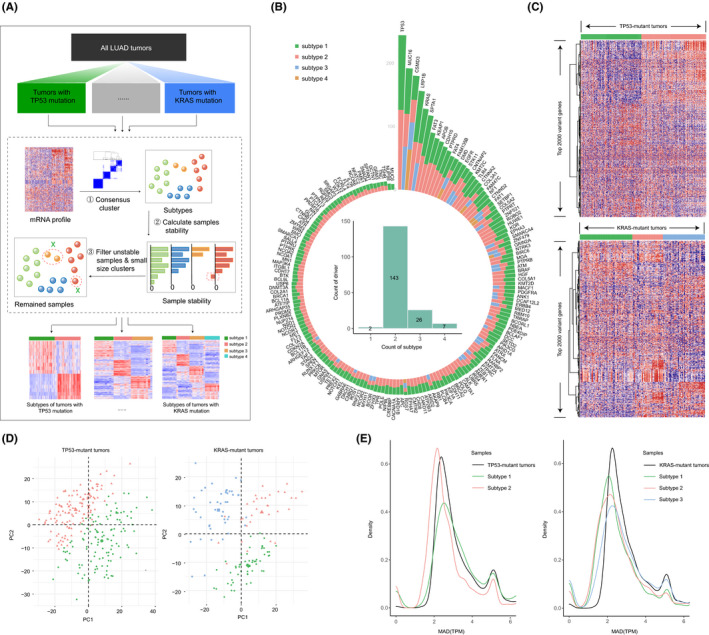
Subtypes of tumors with driver gene mutations. (A) The overview of identification of subtypes of tumors with driver gene mutations. (B) Ring bar plots (outer) showing the count of subtypes and samples of each driver gene. Bar plots (inner) showing the count of driver genes divided into various subtypes. (C) Gene expression heatmap (red =high expression; blue =low expression) of TP53‐ (n = 244) and KRAS (n = 131)‐mutated tumors by the 2000 most variable genes. (D) A principal component analysis (PCA) of TP53‐ and KRAS‐mutated tumors. (E) The MAD distribution of the most variable genes in TP53 (left) and KRAS (right) mutant tumors and corresponding subtypes
Here, we took the critical driver gene TP53 and KRAS as examples. As shown in the cluster heatmap on top 2000 variable genes, a discriminable expression pattern between two subtypes of TP53 mutations was observed (Figure 3C top). KRAS‐mutated tumors, divided into three subtypes, showed a similar phenomenon (Figure 3C bottom). The clustering robustness was assessed with principal component analysis, TP53‐ and KRAS‐mutated tumors formed two and three separate groups, respectively, which supported the classification of subtypes (Figure 3D). In order to observe the changes in transcriptomic heterogeneity between tumors with driver gene mutations and their corresponding subtypes, we compared the MAD of top variable genes. Transcriptomic heterogeneity of TP53 subtype two decreased significantly (p < 0.001, Wilcoxon rank‐sum paired test) compared with all tumors with TP53 mutation, while subtype one changed little (Figure 3E). Besides, all the three subtypes of KRAS‐mutated tumor samples decreased significantly (p < 0.0001, Wilcoxon rank‐sum paired test, Figure 3E). These results indicated that the subtypes of each driver gene‐mutated tumors were composed of stable subtypes with lower transcriptomic heterogeneity.
For further analysis of functions of driver gene mutations in subtypes, we identified DEGs between the subtypes of tumors with driver gene mutations and wild‐type samples and performed functional enrichment analysis based on these DEGs using g: profiler. 46 , 47 We found some key biological functions of driver gene mutations within subtypes. TP53 subtype one was significantly enriched in cell cycle, cell division, and nuclear division (adjusted enrichment p < 0.05, Figure S1A). The cell cycle is associated with a cancer hallmark, self‐sufficiency in growth signals. 60 , 61 , 62 We also identified functions related to other cancer hallmarks. The humoral immune process of TP53 subtype two was related to tumor‐promoting inflammation and evading immune detection (Figure S1A). 60 , 61 , 62 Importantly, KRAS subtype three was specifically enriched in the immune system process, such as lymphocyte proliferation, leukocyte proliferation, and T cell proliferation (Figure S1B). These results suggested that different subtypes of the same driver gene mutations in LUAD patients had diverse functions.
3.4. The co‐occurring or mutually exclusive pairs of driver gene mutations within subtypes
We wanted to explore whether the co‐occurring or mutually exclusive pairs existed in driver gene mutations within subtypes. At first, we identified 21 significantly co‐occurring pairs among 394 subtypes of 178 driver gene mutations using the DISCOVER method, including 24 subtypes and 24 driver genes (q value <0.05, Figure 4A). Among these co‐occurring pairs, we found TP53 subtype one was co‐occurring with FAT3 subtype two (q value =0.025, Figure 4B). There were 22 common samples shared by these two subtypes, encompassing 78 samples of TP53 subtype one and 25 samples of FAT3 subtype two. However, driver gene TP53 mutations were not co‐occurring with FAT3 mutations (q value =0.983). Another example of TP53 subtype one and LRP1B subtype one was observed, which was identified as a co‐occurring pair (q value =0.011, Figure 4C). This co‐occurring pair was commonly detected in 33 samples (54 samples of LRP1B subtype one). A similar phenomenon was also observed between ROBO2 subtype two and NSD1 subtype two (q value =0.036, Figure 4D). These results revealed that more co‐occurring pairs of subtypes of tumors with driver gene mutations were found than tumors with mutations in driver genes.
FIGURE 4.
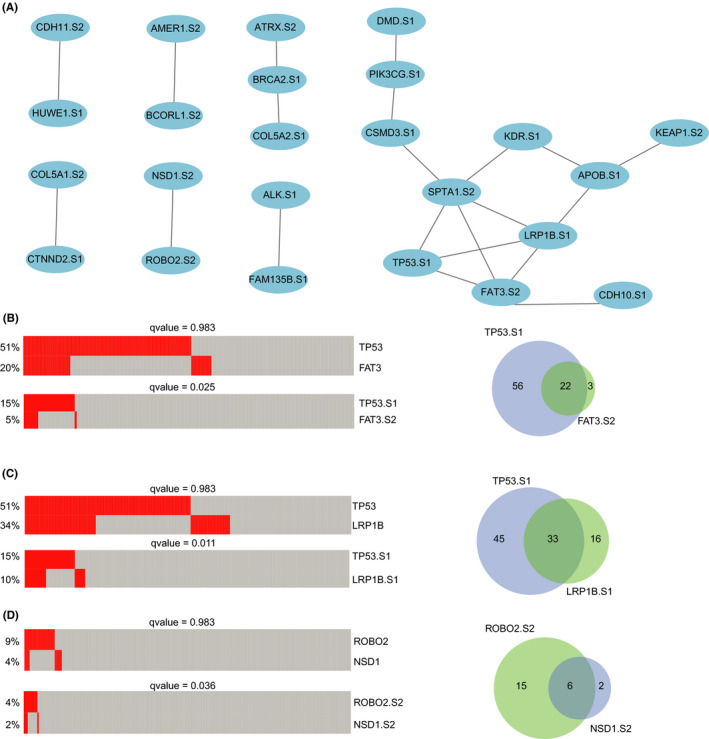
The co‐occurring pairs of driver gene mutations within subtypes. (A) Twenty‐one co‐occurring pairs of 20 driver gene mutations within subtypes. S1, S2, and S3 mean subtype one, subtype two, and subtype three, respectively. (B–D) The co‐occurring pairs of TP53 subtype one–FAT3 subtype two, TP53 subtype one–LRP1B subtype one, ROBO2 subtype two–NSD1 subtype two, respectively. The heatmap on the left showed the mutational status of driver gene mutations within subtypes (red for mutation) for each sample (each column). Venn diagrams on the right showing the overlap of samples with co‐occurring pairs
We also identified 494 mutually exclusive pairs of driver gene mutations within subtypes (Figure 5A), including 179 subtypes of 112 driver gene mutations. There was an interesting phenomenon that up to 168 subtypes were mutually exclusive with two subtypes of TP53. TP53 subtype one and TP53 subtype two were mutually exclusive with 82 subtypes and 91 subtypes of driver gene mutations, respectively. Only five subtypes (SETD2 subtype one, KEAP1 subtype one, KRAS subtype two, ATM subtype three, and KRAS subtype three) were mutually exclusive with both two subtypes of TP53 mutations (Table S1). Most subtypes were mutually exclusive with one of the TP53 mutations’ subtypes. Although PIK3CA subtype two was mutually exclusive with TP53 subtype one, it was not mutually exclusive with TP53 subtype two. The same result was observed between TP53 subtype two and PTPRT subtype two (Figure S2).
FIGURE 5.
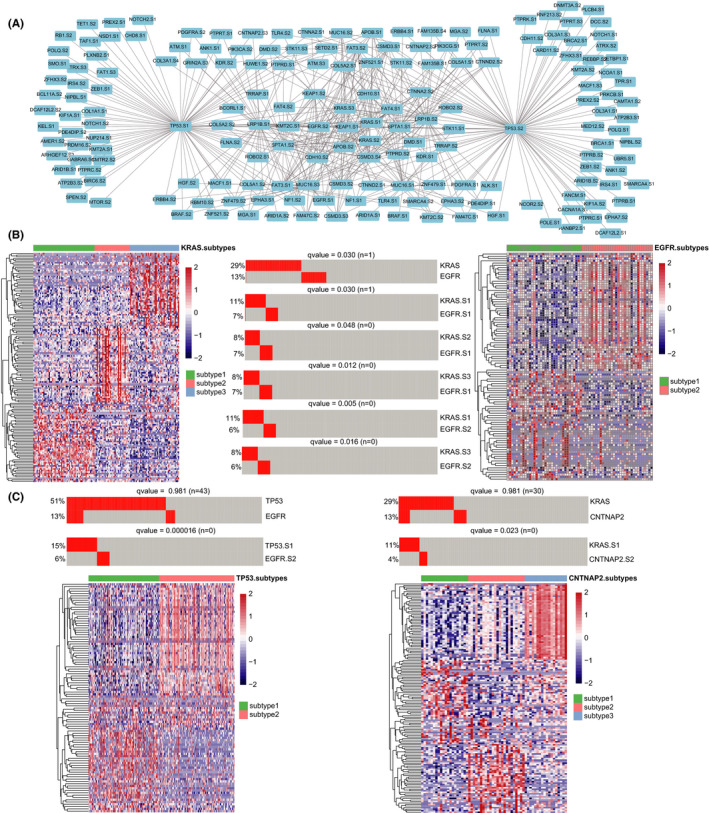
The mutually exclusive pairs of driver gene mutations within subtypes. (A) A total of 494 mutually exclusive pairs of 112 driver gene mutations within subtypes. S1, S2, and S3 mean subtype one, subtype two, and subtype three, respectively. (B) The mutually exclusive pair of subtypes of KRAS mutations and EGFR mutations (middle). The heatmap showed the top 50 differentially expressed genes between subtypes of KRAS‐mutated (left) and TP53‐mutated (right) tumors. (C) The mutually exclusive pairs of TP53 subtype one–EGFR subtype two and KRAS subtype one–CNTNAP2 subtype two
Among 494 mutually exclusive pairs, some pairs were not only identified among subtypes of tumors with driver gene mutations but also among driver gene mutations. Let us take an example of the driver gene KRAS and EGFR. KRAS mutations and EGFR mutations were significantly mutually exclusive events (q value =0.030, Figure 5B). At the subtype level, tumors with KRAS mutations and EGFR mutations were divided into three and two groups, respectively. Except for the pair of KRAS subtype two and EGFR subtype two, the other five pairs of subtypes were all significantly mutually exclusive (q value <0.05). Surprisingly, most of the mutually exclusive pairs were only observed at the subtype level. TP53 subtype one was mutually exclusive with EGFR subtype two (q value =0.000016), where tumors with TP53 mutations were divided into two groups (Figure 5C). However, TP53 mutations were not significantly mutually exclusive with EGFR mutations (q value =0.981). A similar phenomenon was found in the pair of KRAS subtype one and CNTNAP2 subtype two (Figure 5C). These results revealed that more mutually exclusive or co‐occurring pairs were identified among subtypes of tumors with driver gene mutations based on transcriptomic heterogeneity.
3.5. Identification of biological functions of driver gene mutations within subtypes
Subsequently, we identified the functions of mutually exclusive pairs among driver gene mutations within subtypes by functional enrichment analysis (see Methods). As a result, numerous biological processes were significantly enriched for mutually exclusive pairs, such as cell cycle, cell adhesion, and cell differentiation. There were 78 important biological functions found among two subtypes of EGFR mutations and their mutually exclusive events, including 25 subtypes of 13 driver gene mutations (Figure 6A). The mutually exclusive pair (TP53 subtype one and EGFR subtype two) was specifically enriched in nuclear chromosome segregation and nuclear division (Figure 6B). Besides, the humoral immune response was found in the mutually exclusive events (CSMD3 subtype two and EGFR subtype two) (Figure 6C), which was useful for autoimmunogenic human tumor antigens and cancers. 63 , 64 The similar results were observed in other subtypes of driver gene mutations. Take KRAS mutations as an example, the pair of KRAS subtype three and TP53 subtype one was associated with cell motility, cell differentiation, and blood circulation (Figure S3). These functions played key roles in cancer progression. 65 , 66 , 67 In addition, TP53 mutations and RB1 mutations were synthetic lethality in the SynLethDB 68 database. The common presence of the two driver genes could kill cells. All findings suggested that we could identify crucial biological processes of driver gene mutations according to the analysis of mutual exclusion of subtypes of driver gene‐mutated tumors.
FIGURE 6.
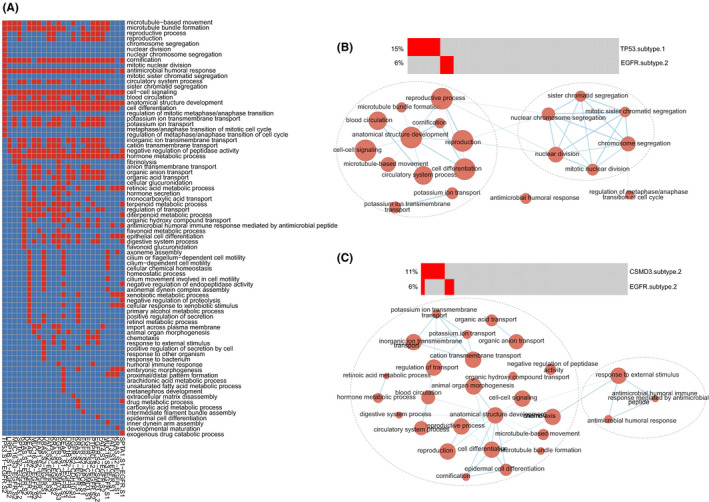
The functions of mutually exclusive pairs of EGFR mutations within subtypes. (A) The heatmap showed the functions of mutually exclusive pairs of EGFR mutations within subtypes. Red represented some mutually exclusive pair was significantly enriched in one function. (B–C) The functions of mutually exclusive pairs of TP53 subtype one–EGFR subtype two and CSMD3 subtype two–EGFR subtype two. Functional enrichment results were visualized using EnrichmentMap plugin in Cytoscape. Node size was proportional to the size of the functional gene set. Clusters were manually circled
3.6. The functions of mutually exclusive triples
Furthermore, we identified 79 mutually exclusive triples among driver gene mutations within subtypes (Table S2). In the mutually exclusive triple, every two subtypes were mutually exclusive. There were a number of important biological processes found related to these mutually exclusive triples. Among these mutually exclusive triples, the triple (TP53 subtype two–KEAP1 subtype one–TLR4 subtype one) harbored the most functions (n = 70), including cell differentiation, homeostatic process, vascular circulatory system, cell–cell signaling, and cilium‐dependent cell motility (Figure 7A). We found 38 mutually exclusive triples were significantly enriched in cell motility, which were associated with cancer invasion and metastasis. 69 , 70 For example, the functions of a mutually exclusive triple (KEAP1 subtype two–FAT3 subtype one–EGFR subtype one) were cilium‐dependent cell motility, cell differentiation, microtubule bundle formation, and cellular chemical homeostasis (Figure 7B). The triple, FAT3 subtype two–KRAS subtype one–KEAP1 subtype one, was significantly enriched in cell–cell adhesion, circulatory system process, and immune‐mediated antimicrobial (Figure 7C).
FIGURE 7.
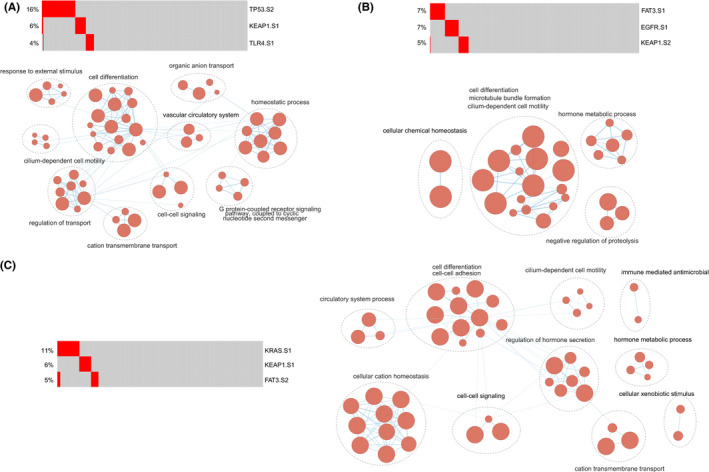
The functions of mutually exclusive triples. (A–C) The functions of mutually exclusive triples of TP53 subtype two–KEAP1 subtype one–TLR4 subtype one, KEAP1 subtype two–FAT3 subtype one–EGFR subtype one, and FAT3 subtype two–KRAS subtype one–KEAP1 subtype one. Functional enrichment results were visualized using EnrichmentMap plugin in Cytoscape. Node size was proportional to the size of the functional gene set. Clusters were manually circled and labeled
3.7. The prognostic value of mutually exclusive triples
Finally, we explored the prognostic value of 78 mutually exclusive triples. There were six mutually exclusive triples significantly predicting the overall survival of LUAD patients using the log‐rank test (p < 0.05, Figure 8). The samples with these mutually exclusive triples had significantly shorter OS compared with WT sequences (p < 0.05). To assess whether the survival prediction ability of the prognostic triples was independent of other clinicopathologic factors in LUAD, univariate and multivariable Cox regression analyses were performed. The covariables included age, gender, AJCC stage, T stage, N stage, and these six triples. The patients harboring “TP53 subtype one–KRAS subtype 3–FAM47C subtype 2” (HR =1.405, 95% CI 1.014–1.945, p = 0.041, Table 1), “KEAP1 subtype two – FAT3 subtype one–EGFR subtype one” (HR =1.440, 95% CI 1.023–2.027, p = 0.036, Table 2) independently predicted poor OS of the patients with LUAD. The other clinical factors, N stage, was also independently associated with shorter OS of patients (p = 0.019 and p = 0.013, respectively).
FIGURE 8.
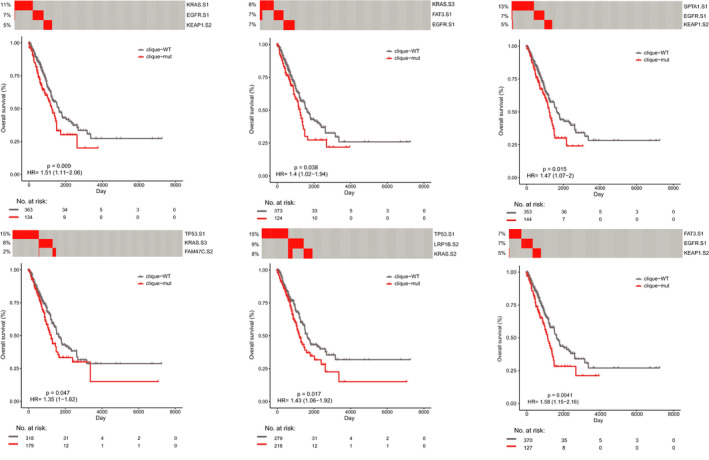
The associations of mutually exclusive triples with clinical outcome. Comparison of overall survival among patients carrying mutually exclusive triples (red line) and cases harboring unmutated genes (gray line) by Kaplan–Meier analysis (with log‐rank p values) in the cohort of LUAD patients from TCGA
TABLE 1.
Multivariate analysis for a mutually exclusive triple of overall survival in the cohort (TP53 subtype one–KRAS subtype three–FAM47C subtype two)
| Variables | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Age | 1.009 | 0.994–1.025 | 0.249 | 1.012 | 0.996–1.029 | 0.140 |
| AJCC stage | ||||||
| Stage II versus I | 2.345 | 1.624–3.388 | <0.001* | 1.046 | 0.556–1.968 | 0.890 |
| Stage III versus I | 3.495 | 2.379–5.133 | <0.001* | 1.59 | 0.626–4.034 | 0.329 |
| Stage IV versus I | 3.341 | 1.834–6.088 | <0.001* | 2.123 | 0.995–4.531 | 0.052 |
| T stage | ||||||
| T2 versus T1 | 1.409 | 0.984–2.018 | 0.061 | 1.232 | 0.845–1.795 | 0.278 |
| T3 versus T1 | 3.027 | 1.798–5.097 | <0.001* | 2.534 | 1.341–4.788 | 0.004 |
| T4 versus T1 | 3.117 | 1.641–5.920 | <0.001* | 1.667 | 0.778–3.571 | 0.189 |
| N stage | ||||||
| N1 versus N0 | 2.363 | 1.669–3.346 | <0.001* | 2.025 | 1.124–3.648 | 0.019* |
| N2 versus N0 | 3.115 | 2.123–4.569 | <0.001* | 1.630 | 0.698–3.085 | 0.258 |
| N3 versus N0 | 3.842 | 0‐Inf | 0.994 | 1.751 | 0‐Inf | 0.994 |
| Gender | ||||||
| Male versus Female | 1.050 | 0.779–1.416 | 0.747 | 0.882 | 0.641–1.214 | 0.441 |
| TP53 subtype one–KRAS subtype three–FAM47C subtype two | ||||||
| Mutations versus wild type | 1.345 | 0.993–1.820 | 0.055 | 1.405 | 1.014–1.945 | 0.041* |
Significant p values are labeled with *(p < 0.05).
TABLE 2.
Multivariate analysis for a mutually exclusive triple of overall survival in the cohort (KEAP1 subtype two–FAT3 subtype one–EGFR subtype one)
| Variables | Univariate | Multivariate | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p value | HR | 95% CI | p value | |
| Age | 1.009 | 0.994–1.025 | 0.249 | 1.010 | 0.994–1.026 | 0.237 |
| AJCC stage | ||||||
| Stage II versus I | 2.345 | 1.624–3.388 | <0.001* | 0.998 | 0.527–1.890 | 0.995 |
| Stage III versus I | 3.495 | 2.379–5.133 | <0.001* | 1.110 | 0.429–2.877 | 0.829 |
| Stage IV versus I | 3.341 | 1.834–6.088 | <0.001* | 1.641 | 0.746–3.607 | 0.218 |
| T stage | ||||||
| T2 versus T1 | 1.409 | 0.984–2.018 | 0.061 | 1.264 | 0.866–1.843 | 0.224 |
| T3 versus T1 | 3.027 | 1.798–5.097 | <0.001* | 2.446 | 1.299–4.607 | 0.006* |
| T4 versus T1 | 3.117 | 1.641–5.920 | <0.001* | 2.062 | 0.948–4.484 | 0.068 |
| N stage | ||||||
| N1 versus N0 | 2.363 | 1.669–3.346 | <0.001* | 2.127 | 1.176–3.846 | 0.013* |
| N2 versus N0 | 3.115 | 2.123–4.569 | <0.001* | 2.274 | 0.975–5.302 | 0.057 |
| N3 versus N0 | 3.842 | 0–Inf | 0.994 | 2.570 | 0–Inf | 0.994 |
| Gender | ||||||
| Male versus Female | 1.05 | 0.779–1.416 | 0.747 | 0.951 | 0.695–1.302 | 0.755 |
| KEAP1 subtype two–FAT3 subtype one–EGFR subtype one | ||||||
| Mutations versus wild type | 1.573 | 1.144–2.163 | 0.005* | 1.440 | 1.023–2.027 | 0.036* |
Significant p values are labeled with *(p < 0.05).
In addition, we investigated the associations of the mutually exclusive triples and disease‐specific survival of LUAD patients. We found that eight mutually exclusive triples significantly predicted shorter DSS of patients by log‐rank test (p < 0.05, Figure S4). Furthermore, one mutually exclusive triple (TP53 subtype one–MUC16 subtype one–KRAS subtype three) significantly independently predicted the survival of patients by univariate and multivariable Cox regression analyses (HR = 1.475, 95% CI 0.986–2.207, p = 0.048, Table S3), adjusting for other clinicopathologic factors. These results indicated that the predictive ability of two mutually exclusive triples and one mutually exclusive triple was independent of clinicopathological factors for OS and DSS in LUAD, respectively.
4. DISCUSSION
In this study, we comprehensively characterized the functions of 178 driver gene mutations within specific subtypes across 506 LUAD patients from TCGA. We identified 21 significantly mutually exclusive events of driver gene mutations. Because of increasing transcriptomic heterogeneity of tumors with driver gene mutations, they were divided into different subtypes. Notably, using mutual exclusivity, more co‐occurring (n = 21) and mutually exclusive pairs (n = 494) were found among driver gene mutations within subtypes. The observation motivated us to explore the functions of mutually exclusive pairs. These mutually exclusive pairs of mutations within subtypes played crucial roles in cancers, including nuclear chromosome segregation, nuclear division, humoral immune response, cell motility, cell differentiation, and blood circulation. These results indicated that we could identify more crucial and refined functions of driver gene mutations according to the analysis of mutual exclusion of subtypes of driver gene‐mutated tumors. At last, 79 mutually exclusive triples were significantly enriched in cell differentiation, cell motility, cilium‐dependent cell motility, and cellular chemical homeostasis. We also observed three prognostic mutually exclusive triples, which independently predicted the survival of LUAD patients. In summary, we found novel mutual exclusivity and functional associations of driver gene mutations based on transcriptomic heterogeneity, which could offer a new perspective to understand the mechanisms of cancer development.
In recent studies, accumulating evidence supported that the heterogeneity of LUAD tumors plays an important role in tumor progression, and LUAD is composed of subtypes distinguishable only at the molecular level. 71 , 72 , 73 Toshiyuki Takeuchi et al. established a basis for expression profile‐defined classification, which can classify adenocarcinomas into two major types. 74 It should be noted that even the tumors with the same driver gene mutations can cause different progression. 75 In this study, we have observed high transcriptomic heterogeneity in LUAD tumor samples with various driver gene mutations. And, most tumor samples with driver gene mutation can be divided into at least two subtypes. Interestingly, subtypes of the same driver gene mutations were enriched in different functions related to cancer progression. This suggested that transcriptomic heterogeneity, as a confounding factor, may hide the various function of driver genes, which can be resolved with subtypes.
In this research, we identified many biological functions of mutually exclusive pairs of driver gene mutations within subtypes, which played crucial roles in tumorigenesis. Some of these functions were associated with cancer hallmarks, including self‐sufficiency in growth signals, tumor‐promoting inflammation, and evading immune detection. 60 , 61 , 62 For example, the mutually exclusive pair, TP53 subtype one–EGFR subtype two, was not only significantly enriched in known functions of driver genes, but also in more refined functions, including sister chromatid segregation, mitotic sister chromatid segregation, and microtubule bundle formation. The famous driver gene TP53, a tumor suppressor gene, is the most frequently mutated gene (>50%) in cancer, which plays a crucial role in preventing cancer formation, such as cell cycle and cell differentiation. 76 , 77 , 78 , 79 EGFR mutations are related to a number of important functions in cancers, including LUAD and glioblastoma, 80 , 81 which regulate cell–cell adhesion division and cell differentiation. 82 , 83 A similar phenomenon was observed in mutually exclusive triples, such as TP53 subtype two–KEAP1 subtype one–TLR4 subtype one. These findings revealed that the functions of driver gene mutations within subtypes were more refined and were associated with cancer hallmarks, which demonstrated the functions we identified were reliable.
In this research, there were three mutually exclusive triples that independently predicted shorter survival of LUAD patients than the wild‐type samples, namely “TP53 subtype one–KRAS subtype three–FAM47C subtype two,” “KEAP1 subtype two–FAT3 subtype one ‐ EGFR subtype one,” and “TP53 subtype one–MUC16 subtype one–KRAS subtype three.” Although, TP53 mutations, 84 , 85 KRAS mutations, 86 , 87 and KEAP1 mutations 88 were predictive of survival of lung cancer patients. This result revealed that mutually exclusive triples could serve as biomarkers of LUAD patients or therapeutic targets. However, these two prognostic triples were needed more data to evaluate their effectiveness in cancers.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
Supporting information
Fig S1‐S4‐Table S1‐S3
Lan Y, Liu W, Zhang W, et al. Transcriptomic heterogeneity of driver gene mutations reveals novel mutual exclusivity and improves exploration of functional associations. Cancer Med. 2021;10:4977–4993. 10.1002/cam4.4039
Yujia Lan and Wei Liu contributed equally to this work as first authors.
Yun Xiao and Yanyan Ping contributed equally to this work as corresponding authors.
Funding information
This work was supported by the National Natural Science Foundation of China (Grant No. 31871336), National Science Foundation of Heilongjiang Province (Grant No. YQ2019C012), the Heilongjiang Postdoctoral Foundation (Grant No. LBH‐Q18099), the Heilongjiang Postdoctoral Foundation (LBH‐Z19082), and Special Funds for the Construction of Higher Education in Heilongjiang Province (grant UNPYSCT‐2018068).
Contributor Information
Yanyan Ping, Email: pingyanyan@hrbmu.edu.cn.
Yun Xiao, Email: xiaoyun@ems.hrbmu.edu.cn.
DATA AVAILABILITY STATEMENT
The data of this study were freely available from public resources. Because this study did not include humans or animals, the ethical approval was not applicable.
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7‐30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 2. DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438‐451. 10.3322/caac.21583. [DOI] [PubMed] [Google Scholar]
- 3. Zappa C, Mousa SA. Non‐small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288‐300. 10.21037/tlcr.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Groot PM, Wu CC, Carter BW, Munden RF. The epidemiology of lung cancer. Transl Lung Cancer Res. 2018;7(3):220‐233. 10.21037/tlcr.2018.05.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amin MB, Greene FL, Edge SB, et al. The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population‐based to a more "personalized" approach to cancer staging. CA Cancer J Clin. 2017;67(2):93‐99. 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 6. Chansky K, Detterbeck FC, Nicholson AG, et al. The IASLC lung cancer staging project: external validation of the revision of the TNM stage groupings in the eighth edition of the TNM classification of lung cancer. J Thorac Oncol. 2017;12(7):1109‐1121. 10.1016/j.jtho.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 7. Yuan M, Huang LL, Chen JH, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non‐small‐cell lung cancer. Signal Transduct Target Ther. 2019;4(1):61. 10.1038/s41392-019-0099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meador CB, Hata AN. Acquired resistance to targeted therapies in NSCLC: updates and evolving insights. Pharmacol Ther. 2020;210: 10.1016/j.pharmthera.2020.107522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nokin MJ, Ambrogio C, Nadal E, Santamaria D. Targeting infrequent driver alterations in non‐small cell lung cancer. Trends Cancer. 2020. 10.1016/j.trecan.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 10. Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan‐Cancer analysis project. Nat Genet. 2013;45(10):1113‐1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333‐339. 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546‐1558. 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463(7278):191‐196. 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17‐37. 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 15. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719‐724. 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alderton GK. Genomics: driving cancer biology. Nat Rev Cancer. 2011;11(2):79. [PubMed] [Google Scholar]
- 17. Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self‐renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA. 2013;110(6):1999‐2004. 10.1073/pnas.1221068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pon JR, Marra MA. Driver and passenger mutations in cancer. Annu Rev Pathol. 2015;10:25‐50. 10.1146/annurev-pathol-012414-040312. [DOI] [PubMed] [Google Scholar]
- 19. Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci USA. 2016;113(50):14330‐14335. 10.1073/pnas.1616440113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang S, Shi J, Ye Z, et al. Predicting EGFR mutation status in lung adenocarcinoma on computed tomography image using deep learning. Eur Respir J. 2019;53(3). 10.1183/13993003.00986-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seow WJ, Matsuo K, Hsiung CA, et al. Association between GWAS‐identified lung adenocarcinoma susceptibility loci and EGFR mutations in never‐smoking Asian women, and comparison with findings from Western populations. Hum Mol Genet. 2017;26(2):454‐465. 10.1093/hmg/ddw414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299‐311. 10.1016/S0140-6736(16)30958-8. [DOI] [PubMed] [Google Scholar]
- 23. Liu N, Yu M, Yin T, et al. Progression of malignant pleural effusion during the early stage of gefitinib treatment in advanced EGFR‐mutant lung adenocarcinoma involving complex driver gene mutations. Signal Transduct Target Ther. 2020;5(1):63. 10.1038/s41392-020-0161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mayekar MK, Bivona TG. Current landscape of targeted therapy in lung cancer. Clin Pharmacol Ther. 2017;102(5):757‐764. 10.1002/cpt.810. [DOI] [PubMed] [Google Scholar]
- 25. Ansah GA, Segura JC, Buckland RB. Semen production, sperm quality, and their heritabilities as influenced by selection for fertility of frozen‐thawed semen in the chicken. Poult Sci. 1985;64(9):1801‐1803. 10.3382/ps.0641801. [DOI] [PubMed] [Google Scholar]
- 26. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature. 2013;499(7457):214‐218. 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chesler EJ, Lu LU, Shou S, et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat Genet. 2005;37(3):233‐242. 10.1038/ng1518. [DOI] [PubMed] [Google Scholar]
- 28. Hubner N, Wallace CA, Zimdahl H, et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet. 2005;37(3):243‐253. 10.1038/ng1522. [DOI] [PubMed] [Google Scholar]
- 29. Parker JS, Mullins M, Cheang MCU, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160‐1167. 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gatenby RA, Cunningham JJ, Brown JS. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nat Commun. 2014;5:5499. 10.1038/ncomms6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wilkinson SM, Uhl JR, Kline BC, Cockerill FR 3rd. Assessment of invasion frequencies of cultured HEp‐2 cells by clinical isolates of Helicobacter pylori using an acridine orange assay. J Clin Pathol. 1998;51(2):127‐133. 10.1136/jcp.51.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blakely CM, Watkins TBK, Wu W, et al. Evolution and clinical impact of co‐occurring genetic alterations in advanced‐stage EGFR‐mutant lung cancers. Nat Genet. 2017;49(12):1693‐1704. 10.1038/ng.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22(2):398‐406. 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cancer Genome Atlas Research N . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061‐1068. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Licar A, Cerkovnik P, Novakovic S. Distribution of some activating KRAS and BRAF mutations in slovene patients with colorectal cancer. Med Oncol. 2011;28(4):1048‐1053. 10.1007/s12032-010-9631-z. [DOI] [PubMed] [Google Scholar]
- 36. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature. 2002;418(6901):934. 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 37. Unni AM, Lockwood WW, Zejnullahu K, Lee‐Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife. 2015;4:e06907. 10.7554/eLife.06907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mareckova A, Malcikova J, Tom N, et al. ATM and TP53 mutations show mutual exclusivity but distinct clinical impact in mantle cell lymphoma patients. Leuk Lymphoma. 2019;60(6):1420‐1428. 10.1080/10428194.2018.1542144. [DOI] [PubMed] [Google Scholar]
- 39. Zhou Q, Li Z, Song L, et al. Whole‐exome mutational landscape of metastasis in patient‐derived hepatocellular carcinoma cells. Genes Dis. 2020;7(3):380‐391. 10.1016/j.gendis.2020.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldman M, Craft B, Swatloski T, et al. The UCSC cancer genomics browser: update 2015. Nucleic Acids Res. 2015;43(Database issue):D812‐D817. 10.1093/nar/gku1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941‐D947. 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173(2):371‐385 e18. 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cancer Genome Atlas Research N . Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543‐550. 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607‐616. 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572‐1573. 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raudvere U, Kolberg L, Kuzmin I, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191‐W198. 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Reimand J, Arak T, Adler P, et al. g:Profiler‐a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016;44(W1):W83‐W89. 10.1093/nar/gkw199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network‐based method for gene‐set enrichment visualization and interpretation. PLoS One. 2010;5(11):e13984. 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498‐2504. 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cousin S, Khalifa E, Crombe A, et al. Targeting ERBB2 mutations in solid tumors: biological and clinical implications. J Hematol Oncol. 2018;11(1):86. 10.1186/s13045-018-0630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Metzeler KH, Herold T, Rothenberg‐Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686‐698. 10.1182/blood-2016-01-693879. [DOI] [PubMed] [Google Scholar]
- 52. Zhang H, Deng Y, Zhang Y, et al. Cooperative genomic alteration network reveals molecular classification across 12 major cancer types. Nucleic Acids Res. 2017;45(2):567‐582. 10.1093/nar/gkw1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao C, Zhuang J, Zhou C, et al. SNP mutation‐related genes in breast cancer for monitoring and prognosis of patients: a study based on the TCGA database. Cancer Med. 2019;8(5):2303‐2312. 10.1002/cam4.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017. 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Turajlic S, Xu H, Litchfield K, et al. Deterministic evolutionary trajectories influence primary tumor growth: TRACERx renal. Cell. 2018;173(3):595‐610 e11. 10.1016/j.cell.2018.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang A, Zhou B, Sun W. Integrated genomic characterization of cancer genes in glioma. Cancer Cell Int. 2017;17:90. 10.1186/s12935-017-0458-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Masoodi T, Siraj S, Siraj AK, et al. Genetic heterogeneity and evolutionary history of high‐grade ovarian carcinoma and matched distant metastases. Br J Cancer. 2020;122(8):1219‐1230. 10.1038/s41416-020-0763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang K, Yuen ST, Xu J, et al. Whole‐genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46(6):573‐582. 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 59. Canisius S, Martens JW, Wessels LF. A novel independence test for somatic alterations in cancer shows that biology drives mutual exclusivity but chance explains most co‐occurrence. Genome Biol. 2016;17(1):261. 10.1186/s13059-016-1114-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 61. Plaisier CL, Pan M, Baliga NS. A miRNA‐regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22(11):2302‐2314. 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lin K, Song LJ, Ma J, Zhang TS, You DY, He YW. Identification of cancer hallmark‐associated gene and lncRNA cooperative regulation pairs and dictate lncRNA roles in oral squamous cell carcinoma. J Cell Mol Med. 2020;24(9):5213‐5223. 10.1111/jcmm.15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Murphy MA, O'Leary JJ, Cahill DJ. Assessment of the humoral immune response to cancer. J Proteomics. 2012;75(15):4573‐4579. 10.1016/j.jprot.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 64. Stockert E, Jäger E, Chen Y‐T, et al. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187(8):1349‐1354. 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang J, Li HF, Fan W, et al. Regularity of drugs compatibility of anti‐hepatoma traditional Chinese medicine ancient prescriptions and risk evaluation of anti‐hepatoma new drug research and development. Zhongguo Zhong Yao Za Zhi. 2014;39(19):3870‐3875. [PubMed] [Google Scholar]
- 66. Melo SA, Luecke LB, Kahlert C, et al. Glypican‐1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177‐182. 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755‐1766. [PubMed] [Google Scholar]
- 68. Guo J, Liu H, Zheng J. SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 2016;44(D1):D1011‐D1017. 10.1093/nar/gkv1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stuelten CH, Parent CA, Montell DJ. Cell motility in cancer invasion and metastasis: insights from simple model organisms. Nat Rev Cancer. 2018;18(5):296‐312. 10.1038/nrc.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kassis J, Lauffenburger DA, Turner T, Wells A. Tumor invasion as dysregulated cell motility. Semin Cancer Biol. 2001;11(2):105‐117. 10.1006/scbi.2000.0362. [DOI] [PubMed] [Google Scholar]
- 71. Hua X, Zhao W, Pesatori AC, et al. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat Commun. 2020;11(1):2459. 10.1038/s41467-020-16295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Daemen A, Cooper J, Myrta S, et al. Transcriptional subtypes resolve tumor heterogeneity and identify vulnerabilities to MEK inhibition in lung adenocarcinoma. Clin Cancer Res. 2020. 10.1158/1078-0432.CCR-20-1835. [DOI] [PubMed] [Google Scholar]
- 73. Best SA, Ding S, Kersbergen A, et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS‐mutant lung adenocarcinoma. Nat Commun. 2019;10(1):4190. 10.1038/s41467-019-12164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Takeuchi T, Tomida S, Yatabe Y, et al. Expression profile‐defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J Clin Oncol. 2006;24(11):1679‐1688. 10.1200/JCO.2005.03.8224. [DOI] [PubMed] [Google Scholar]
- 75. Dong X, Huang D, Yi X, et al. Diversity spectrum analysis identifies mutation‐specific effects of cancer driver genes. Commun Biol. 2020;3(1):6. 10.1038/s42003-019-0736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57‐68. 10.2147/OTT.S53876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Westphal CH, Schmaltz C, Rowan S, Elson A, Fisher DE, Leder P. Genetic interactions between atm and p53 influence cellular proliferation and irradiation‐induced cell cycle checkpoints. Cancer Res. 1997;57(9):1664‐1667. [PubMed] [Google Scholar]
- 78. Öner MG, Rokavec M, Kaller M, et al. Combined inactivation of TP53 and MIR34A promotes colorectal cancer development and progression in mice via increasing levels of IL6R and PAI1. Gastroenterology. 2018;155(6):1868‐1882. 10.1053/j.gastro.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 79. Rucker FG, Dolnik A, Blatte TJ, et al. Chromothripsis is linked to TP53 alteration, cell cycle impairment, and dismal outcome in acute myeloid leukemia with complex karyotype. Haematologica. 2018;103(1):e17‐e20. 10.3324/haematol.2017.180497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Walker F, Abramowitz L, Benabderrahmane D, et al. Growth factor receptor expression in anal squamous lesions: modifications associated with oncogenic human papillomavirus and human immunodeficiency virus. Hum Pathol. 2009;40(11):1517‐1527. 10.1016/j.humpath.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 81. da Cunha SG, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6:49‐69. 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 82. Kim S, Schein AJ, Nadel JA. E‐cadherin promotes EGFR‐mediated cell differentiation and MUC5AC mucin expression in cultured human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289(6):L1049‐L1060. 10.1152/ajplung.00388.2004. [DOI] [PubMed] [Google Scholar]
- 83. Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. May‐Jun. 1999;82(2–3):241‐250. 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 84. Freudenstein D, Litchfield C, Caramia F, et al. TP53 Status, patient sex, and the immune response as determinants of lung cancer patient survival. Cancers (Basel). 2020;12(6). 10.3390/cancers12061535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Aisner DL, Sholl LM, Berry LD, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations—The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038‐1047. 10.1158/1078-0432.CCR-17-2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Uras IZ, Moll HP, Casanova E. Targeting KRAS mutant non‐small‐cell lung cancer: past, present and future. Int J Mol Sci. 2020;21(12). 10.3390/ijms21124325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tomasini P, Walia P, Labbe C, Jao K, Leighl NB. Targeting the KRAS pathway in non‐small cell lung cancer. Oncologist. 2016;21(12):1450‐1460. 10.1634/theoncologist.2015-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kadara H, Choi M, Zhang J, et al. Whole‐exome sequencing and immune profiling of early‐stage lung adenocarcinoma with fully annotated clinical follow‐up. Ann Oncol. 2017;28(1):75‐82. 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4‐Table S1‐S3
Data Availability Statement
The data of this study were freely available from public resources. Because this study did not include humans or animals, the ethical approval was not applicable.