

Abstract
Introduction
Traditional cancer therapy has many disadvantages such as low selectivity and high toxicity of chemotherapy, as well as insufficient efficacy of targeted therapy. To enhance the cytotoxic effect and targeting ability, while reducing the toxicity of antitumor drugs, an antibody drug conjugate (ADC) was developed to deliver small molecular cytotoxic payloads directly to tumor cells by binding to specific antibodies via linkers.
Method
By reviewing published literature and the current progress of ADCs, we aimed to summarize the basic characteristics, clinical progress, and challenges of ADCs to provide a reference for clinical practice and further research.
Results
ADC is a conjugate composed of three fundamental components, including monoclonal antibodies, cytotoxic payloads, and stable linkers. The mechanisms of ADC including the classical internalization pathway, antitumor activity of antibodies, bystander effect, and non‐internalizing mechanism. With the development of new drugs and advances in technology, various ADCs have achieved clinical efficacy. To date, nine ADCs have received US Food and Drug Administration (FDA) approval in the field of hematologic tumors and solid tumors, which have become routine clinical treatments.
Conclusion
ADC has changed traditional treatment patterns for cancer patients, which enable the same treatment for pancreatic cancer patients and promote individualized precision treatment. Further exploration of indications could focus on early‐stage cancer patients and combined therapy settings. Besides, the mechanisms of drug resistance, manufacturing techniques, optimized treatment regimens, and appropriate patient selection remain the major topics.
Keywords: antibody drug conjugate, cancer, targeting ability, therapy
Antibody drug conjugate (ADC) has been under rapid development in recent years, the application of ADC has changed traditional treatment patterns for pan‐cancer patients and has become a great breakthrough for individualized precision treatment. We conducted this review to summarize basic characteristics, clinical progress, and challenges of ADCs to provide a reference for clinical practice and further researches.
1. OVERVIEW
The efficacy of traditional antitumor therapies, which include nonspecific chemotherapy and molecular targeted therapy, is unsatisfactory, owing to the high toxicity of the former and insufficient cytotoxicity and labeling ability of target genes in the latter. 1 Thus, aiming to combine the strong cytotoxicity of chemotherapy with the high specificity of targeted therapy, antibody drug conjugates (ADCs) are designed to selectively deliver cytotoxic payloads directly to target cancer cells. 2 Antibody drug conjugates can overcome several traditional problems, including the narrow therapeutic window, low selectivity, and rapid plasma clearance of chemotherapy, as well as the unsatisfactory antitumor efficacy of targeted therapy. 3 Since 2000, nine ADCs have been approved by the US Food and Drug Administration (FDA) for various treatment settings in both hematologic and solid tumors, and hundreds of studies and clinical trials are currently being explored. However, there are still many challenges in the development of ADC; thus, this review aimed to investigate the current progress and development of ADCs in various types of cancers to provide a reference for clinical applications and further exploration.
2. BASIC CHARACTERISTICS OF ADCS
Antibody drug conjugate is a conjugate composed of three fundamental components, namely, monoclonal antibodies that target‐specific tumor antigens, high‐potency small molecular cytotoxic payloads, and stable linkers. 4 The basic characteristics of each component are shown in Figure 1.
FIGURE 1.
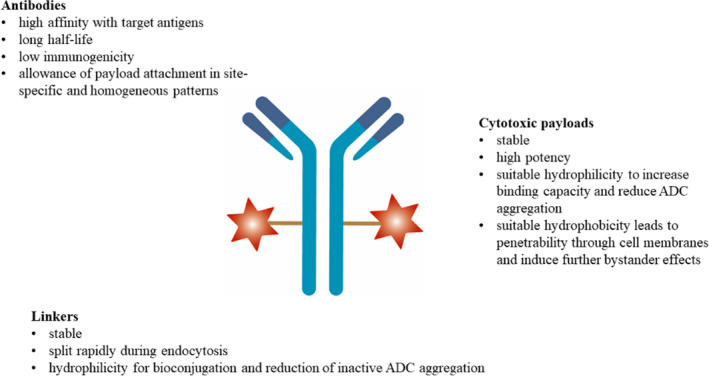
Basic characteristics of ADC. ADC, antibody drug conjugate
Antibody drug conjugate antibodies contain two antigen‐binding fragments (Fabs) that mediate antigen recognition and a constant fragment (Fc) that mediates immune interaction by binding to receptors (FcR) on effector cells. 5 Appropriate antibodies should have high affinity for target antigens, long half‐life, allowance of site‐specific and homogeneous attachment of payloads, and low immunogenicity to avoid immunoreactions. 6 , 7 , 8 Antibodies are mainly immunoglobulin G (IgG) molecules with high affinity and a long half‐life in the blood circulation system. 9 The IgG1 isotype is easily produced, with relatively strong antibody‐dependent cytotoxicity (ADCC) and complement‐dependent cytotoxicity (CDC), and is the most commonly used antibody subtype. 10 , 11
Cytotoxic payloads are effective components at sub‐nanomolar concentrations with linker‐conjugated functional groups and should be stable in blood. Suitable hydrophilicity is required to increase binding capacity and reduce ADC aggregation, and proper hydrophobicity leads to penetrability through the cell membranes, mediating the bystander effect. 12 , 13 The two major categories are microtubule‐targeting agents and DNA‐damaging agents. 14 Microtubule‐targeting agents can induce cell cycle arrest (G2/M phase) by inhibiting mitotic spindles during chromosome segregation and apoptosis. 15 , 16 , 17 Deoxyribonucleic acid‐damaging agents can induce cell cycle arrest and apoptosis through alkylation, scission, cross‐linking, or intercalation after binding to double helix minor grooves, which have higher cytotoxic efficacies at various cell cycle phases compared with microtubule‐targeting agents. 18 , 19 , 20 Other alternative payloads under development include RNA polymerase inhibitors and spliceosome inhibitors. 13
Linkers are connections between antibodies and cytotoxic payloads with specific cleavage mechanisms at the target site, which are significant influential factors of pharmacokinetics, pharmacodynamics, and therapeutic windows. 21 , 22 Linkers should be stable in plasma to prevent off‐target toxicity due to pyrolysis and should split rapidly during endocytosis to efficiently release payloads. 12 , 23 In addition, hydrophilicity is needed for the bioconjugation and reduction of inactive ADC aggregation. 3 , 8 Linkers can be categorized as cleavable and non‐cleavable linkers. 24 Cleavable linkers are the most common type that can exploit the difference in physiological conditions between circulatory and target‐cell conditions. 25 Non‐cleavable linkers are non‐reducible bonds to amino acid residues on antibodies, and the release of payloads relies on the proteolytic degradation of antibodies in lysosomes; thus, effective internalization and transfer to lysosomes are required. 26 , 27 In addition, cleavable linkers can induce bystander effects after the release of cytotoxic payloads, whereas non‐cleavable linkers generally lack a bystander effect because the charged amino acid residues cannot cross cell membranes effectively. 3 , 12
3. MECHANISMS OF ADCS
The classical mechanism of ADC is as follows: ADCs can specifically bind to target antigens on the cell surface after intravenous injection, and then ADC‐antigen complexes could be internalized via antigen‐dependent endocytosis or antigen‐independent pinocytosis, and endocytosis mediated by clathrin is the major mode. 17 , 28 After intracellular trafficking and processing through endosomal and/or lysosomal pathways that rely on organelle acidification, 29 payloads can be released into the cytoplasm through linker cleavage in the chemical and enzymatic environment or lysosomal proteolytic antibody degradation for non‐cleavable linkers. 30 , 31 Payloads play cytotoxic roles by damaging DNA or inhibiting microtubule assembly. 32 , 33
Antibody drug conjugate can also exert antitumor effects through other mechanisms. First, antibodies could retain the antitumor activity, including the ability to interfere with the function of targets mediated by the Fab region and induce ADCC, CDC, and antibody‐dependent cellular phagocytosis mediated by the Fc region. 34 , 35 Second, after ADC molecules are internalized into antigen‐positive tumor cells, cytotoxic payloads with suitable hydrophobicity could permeate cell membranes or be released after the apoptosis of target cells and, subsequently, induce the bystander effect, which can not only kill adjacent tumor cells with negative antigen expression, but also destroy the environment of tumor growth, such as tumor stromal cells and tumor blood vessels. 3 , 36 The bystander effect could facilitate the homogenous distribution of payloads and lead to indications for solid tumors with heterogeneously expressed antigens. 37 Finally, the non‐internalizing mechanism is also under exploration, that is, linker cleavage and payload release could occur extracellularly in the redox and acidic tumor microenvironment with extracellular proteases. 38 , 39 A detailed schematic of the mechanism of ADC is shown in Figure 2.
FIGURE 2.
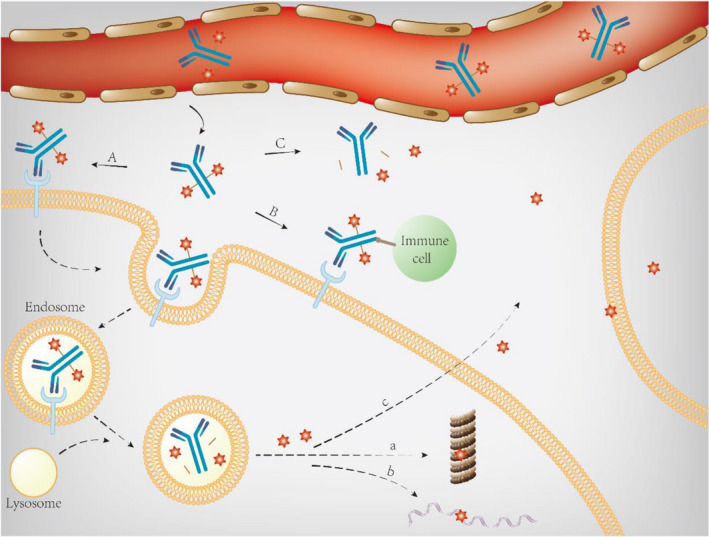
Schematic diagram of ADC mechanism. (A) classical internalizing pathway; (B) retained antitumor activity of antibodies; (C) non‐internalizing pathway; (a) inhibiting microtubule; (b) damaging DNA; (c) bystander effect. ADC, antibody drug conjugate
4. CLINICAL APPLICATION OF ADCS
Since 2000, five ADCs have received FDA approval for hematologic tumors: gemtuzumab ozogamicin (GO, CD33‐targeting) for acute myeloid leukemia (AML) patients, 40 , 41 brentuximab vedotin (BV, CD30‐targeting) for Hodgkin's lymphoma (HL) and non‐Hodgkin's lymphoma (NHL) patients, 42 , 43 inotuzumab ozogamicin (INO, CD22‐targeting) for acute lymphoblastic leukemia (ALL) patients, 44 polatuzumab vedotin‐piiq (PV, CD79b‐targeting) for diffuse large B‐cell lymphoma (DLBCL) patients, 45 and belantamab mafodotin (BM, B‐cell maturation antigen [BCMA]‐targeting) for multiple myeloma (MM) patients. 46
In addition, considering that the complex microenvironment of solid tumors hinders the penetration and accessibility of ADCs, the development of ADCs in solid tumors is later than that in hematologic malignancies. Currently, four ADCs have been approved by the FDA in 2013, including three ADCs, ado‐trastuzumab emtansine (T‐DM1, human epidermal growth factor receptor 2 [HER2]‐targeting), 47 trastuzumab deruxtecan (T‐DXd, HER2‐targeting), 48 , 49 and sacituzumab govitecan (SG, trophoblast cell surface antigen 2 [Trop‐2]‐targeting) for breast cancer patients 50 and enfortumab vedotin (EV, Nectin‐4‐targeting) for urothelial cancer patients. 51
Time sequences and indications for FDA approval are shown in Figure 3. The characteristics and pivotal clinical trials of different ADCs are summarized in Tables 1 and 2, respectively. In the following section, we summarize the application of ADCs in different clinical settings.
FIGURE 3.

Time sequences and indications of FDA approval. FDA, US Food and Drug Administration
TABLE 1.
Summary of FDA approved ADCs
| ADC | Abbreviation/trade name | Time | Antigen | Antibody | Linker | Cytotoxic payload | Mechanism | Approved disease |
|---|---|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin | GO; Mylotarg; CMA‐676 | 2000/2017 | CD33 | Humanized IgG4 | Cleavable acid‐labile linker | N‐acetyl gamma calicheamicin | DNA‐damaging agents | AML |
| Brentuximab vedotin | BV; Adcetris; SGN‐35 | 2011 | CD30 | Chimeric IgG1 | Cleavable protease linker | MMAE | Microtubule‐targeting agents | HL, NHL |
| Inotuzumab ozogamicin | INO; Besponsa; CMC‐544 | 2017 | CD22 | Humanized IgG4 | Cleavable acid linker | N‐acetyl gamma calicheamicin | DNA‐damaging agents | ALL |
| Polatuzumab vedotin‐piiq | PV; Polivy | 2019 | CD79b | Humanized IgG1 | Cleavable protease linker | MMAE | Microtubule‐targeting agents | DLBCL |
| Belantamab mafodotin | Blenrep; belantamab mafodotin‐blmf; GSK2857916 | 2020 | BCMA | Humanized IgG1 | Non‐cleavable protease‐resistant maleimidocaproyl linker | MMAF | Microtubule‐targeting agents | MM |
| Ado‐trastuzumab emtansine | T‐DM1; Kadcyla | 2013 | HER2 | Humanized IgG1 | Non‐cleavable thioether linker | DM1 | Microtubule‐targeting agents | Breast cancer |
| Trastuzumab deruxtecan | T‐DXd; DS‐8201a; [fam‐]trastuzumab deruxtecan‐nxki; Enhertu | 2019 | HER2 | Humanized IgG1 | Cleavable tetrapeptide‐based linker | Deruxtecan | DNA‐damaging agents | Breast cancer |
| Sacituzumab govitecan | SG; Sacituzumab govitecan‐hziy; IMMU‐132; Trodelvy | 2020 | Trop‐2 | Humanized IgG1 | Cleavable pH‐sensitive linker | SN‐38 | DNA‐damaging agents | Breast cancer |
| Enfortumab vedotin | EV; enfortumab vedotin‐ejfv; Padcev | 2019 | Nectin‐4 | Fully human IgG1 | Cleavable protease linker | MMAE | Microtubule‐targeting agents | Urothelial cancer |
Abbreviations: ADC, antibody drug conjugate; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; CD, cluster of differentiation; CTCL, cutaneous T‐cell lymphoma; DLBCL, diffuse large B‐cell lymphoma; DM1, maytansinoids; HL, Hodgkin lymphoma; IgG, immunoglobulin G; MM, multiple myeloma; MMAE, monomethyl auristatin E; MMAF, monomethyl auristatin F; NHL, non‐Hodgkin lymphoma; PTCL, peripheral T‐cell lymphoma; SN‐38, camptothecin analogs.
TABLE 2.
Pivotal clinical trials of FDA approved ADCs
| ADC | Clinical trials | Phase | Line | Regimen | Disease | Drug | ADC dosage | Citation |
|---|---|---|---|---|---|---|---|---|
| GO | Sievers, et al | 2 | First relapse | Monotherapy | AML | GO | 9 mg/m2, every 14 days | 195 |
| SWOG S0106 | 3 | ND | Combined therapy | AML | Induction: DA+GO versus DA; post‐consolidation: GO versus observation | Induction: 6 mg/m2, day 4; post‐consolidation: 5 mg/m2, every 28 days | 184 | |
| ALFA‐0701 | 3 | ND | Combined therapy | AML | DA+GO versus DA | Induction: 3 mg/m2, days 1, 4, 7; consolidation: 3 mg/m2, day 1, 2 cycles | 100, 101 | |
| EORTC‐GIMEMA AML‐19 | 3 | ND | Monotherapy | AML | GO versus best supportive care | Induction: 6 mg/m2 day 1, 3mg/m2 day 8; consolidation: 2 mg/m2, monthly | 99 | |
| MyloFrance‐1 | 2 | First relapse | Monotherapy | AML | GO | 3 mg/m2, days 1, 4, 7 | 54 | |
| NCT00909168 | 2 | ND | Combined therapy | AML | GO+FLAI | 3 mg/m2, day 6 | 103 | |
| EORTC‐GIMEM AML‐17 | 3 | ND | Combined therapy | AML | Induction: GO followed by MICE versus MICE; consolidation: GO+ICE versus ICE | Induction: 6 mg/m2, days 1 and 15; consolidation: 3 mg/m2, day 0 | 104 | |
| SWOG0535 | 2 | ND | Combined therapy | APL | GO+ATRA+ATO | 9 mg/m2, day 1 | 105 | |
| NCT00143975 | 2 | R/R | Combined therapy | AML | GO+cytarabine+mitoxantrone+ATRA | 3 mg/m², day 1 | 57 | |
| NCT00895934 | 1/2 | R/R | Combined therapy | AML | GO+azacytidine+vorinostat | 3 mg/m2, days 4, 8 | 59 | |
| NCT00766116 | 1/2 | R/R | Combined therapy | AML | GO+azacytidine | 6 mg/m2, days 7, 21 | 60 | |
| NCT00882102 | 2 | R/R, ND | Combined therapy | AML, MDS | GO+decitabine | 3 mg/m2, day 5 | 58 | |
| BV | NCT00848926 | 2 | R/R | Monotherapy | HL | BV | 1.8 mg/kg, every 21 days, 16 cycles | 65, 69 |
| AETHERA | 3 | Consolidation therapy after ASCT | Monotherapy | HL | BV | 1.8 mg/kg, every 21 days | 67, 68 | |
| NCT01393717 | 2 | R/R | Monotherapy | HL | BV | 1.8 mg/kg, every 21 days | 70 | |
| NCT02243436 | 1/2 | R/R | Combined therapy | HL | BV+ESHAP | 1.8 mg/kg, day 1, every 21 days | 71 | |
| NCT02227199 | 1/2 | R/R | Combined therapy | HL | BV+ICE | 1.5 mg/kg, days 1, 8, every 21 days | 72 | |
| NCT01874054 | 1/2 | R/R | Combined therapy | HL | BV+bendamustine | 1.8 mg/kg, day 1, every 21 days | 73 | |
| NCT02280993 | 2 | R/R | Combined therapy | HL | BV+DHAP | 1.8 mg/kg, day 1, every 21 days | 74 | |
| NCT02572167 | 1/2 | R/R | Combined therapy | HL | BV+nivolumab | 1.8 mg/kg, day 1, every 21 days, 4 cycles | 75 | |
| ECHELON‐1 | 3 | ND | Combined therapy | HL | BV+AVD versus ABVD | 1.2 mg/kg, days 1, 15, every 28 days, 6 cycles | 106 | |
| NCT00866047 | 2 | R/R | Monotherapy | sALCL | BV | 1.8 mg/kg, every 21 days, 16 cycles | 76, 77 | |
| NCT01421667 | 2 | R/R | Monotherapy | NHL | BV | 1.8 mg/kg, every 21 days | 78 | |
| ALCANZA | 3 | R/R | Monotherapy | CTCL | BV | 1.8 mg/kg, every 21 days, 16 cycles | 79 | |
| ECHELON‐2 | 3 | ND | Combined therapy | PTCL | BV+CHP versus CHOP | 1.8 mg/kg, every 21 days, 6–8 cycles | 108 | |
| CheckMate 436 | 1/2 | R/R | Combined therapy | PMBL | BV+nivolumab | 1.8 mg/kg, every 21 days | 81 | |
| NCT01925612 | 2 | ND | Combined therapy | DLBCL | BV+R‐CHP | 1.8 mg/kg, every 21 days, 6 cycles | 111 | |
| NCT01994850 | 1/2 | ND | Combined therapy | B‐cell lymphoma | BV+R‐CHP | 1.8 mg/kg, every 21 days, 6 cycles | 112 | |
| INO | INOVATE | 3 | R/R | Monotherapy | ALL | INO | Total 1.8 mg/m² per cycle: 0.8 mg/m² on day 1; 0.5 mg/m² on day 8, day 15. Cycle 1, 21 days; subsequent cycles, 28 days. For patients achieving CR: 0.5 mg/m², days 1, 8, and 15 | 83, 84, 85 |
| NCT01371630 | 2 | ND /salvage | Combined therapy | ALL | INO+mini‐hyper‐CVD | 1.3–1.8 mg/m² in cycle 1, 1.0–1.3 mg/m² in cycle 2–4, every 4 weeks | 86, 87, 113 | |
| NCT00299494 | 1/2 | R/R | Combined therapy | NHL | INO+rituximab | 1.8 mg/m², every 28 days, 8 cycles | 88 | |
| NCT01232556 | 3 | R/R | Combined therapy | NHL | INO+rituximab versus chemotherapy (bendamustine or gemcitabine)+rituximab | 1.8 mg/m², every 28 days, 3–6 cycles | 91 | |
| PV | GO29365 | 1/2 | R/R | Combined therapy | DLBCL | PV+bendamustine+rituximab | 1.8 mg/kg, every 21 days | 93 |
| NCT01992653 | 1/2 | ND | Combined therapy | DLBCL | PV+rituximab or obinutuzumab+CHP | 1.8 mg/kg, every 21 days | 115 | |
| BM | DREAMM‐2 | 2 | PT | Monotherapy | MM | BM | 2.5 or 3.4 mg/kg, every 3 weeks | 95 |
| T‐DM1 | EMILIA | 3 | PT | Monotherapy | Breast cancer | T‐DM1 | 3.6 mg/kg, every 21 days | 121, 122, 123 |
| TH3RESA | 3 | PT | Monotherapy | Breast cancer | T‐DM1 | 3.6 mg/kg, every 21 days | 126, 127 | |
| NCT02236000 | 1b | PT | Combined therapy | Breast cancer | T‐DM1+Neratinib | 3.6 mg/kg, every 21 days | 128 | |
| MARIANNE | 3 | ND | Monotherapy / Combined therapy | Breast cancer | T‐DM1/T‐DM1+pertuzumab | 3.6 mg/kg, every 21 days | 146 | |
| KRISTINE | 3 | Neoadjuvant | Combined therapy | Breast cancer | T‐DM1 plus pertuzumab | 3.6 mg/kg, every 21 days | 151, 152 | |
| KATHERINE | 3 | Adjuvant | Monotherapy | Breast cancer | T‐DM1 | 3.6 mg/kg, every 21 days | 149 | |
| KAMILLA trial | 3 | PT | Monotherapy | Breast cancer | T‐DM1 | 3.6 mg/kg, every 21 days | 125 | |
| T‐DXd | DESTINY‐Breast01 | 2 | PT | Monotherapy | Breast cancer | T‐DXd | 5.4 mg/kg, every 21 days | 134 |
| DESTINY‐Gastric01/NCT03329690 | 2 | PT | Monotherapy | Gastric cancer | T‐DXd | 6.4 mg/kg, every 21 days | 135 | |
| NCT02564900 | 1 | PT | Monotherapy | Breast cancer | T‐DXd | 6.4 mg/kg, every 21 days | 136 | |
| SG | NCT01631552 | 1/2 | PT | Monotherapy | Breast cancer | IMMU‐132 | 8 or 10 mg/kg, days 1 and 8, every 21 days | 138, 139, 140, 141, 142, 143 |
| EV | EV‐101 | 1 | PT | Monotherapy | Urothelial cancer | EV | 1.25 mg/kg, days 1, 8, 15, every 29 days | 144 |
| EV‐201 | 2 | PT | Monotherapy | Urothelial cancer | EV | 1.25 mg/kg, days 1, 8, 15, every 28 days | 145 |
Abbreviations: ABVDH, doxorubicin+bleomycin+vinblastine+dacarbazine; ADC, antibody drug conjugate; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; ATO, arsenic trioxide; ATRA, all‐trans retinoic acid; AVD, doxorubicin+vinblastine+dacarbazine; BM, belantamab mafodotin; BV, brentuximab vedotin; CHOP, cyclophosphamide+doxorubicin+vincristine+prednisone; CHP, cyclophosphamide+doxorubicin+prednisone; CR, complete remission; CTCL, cutaneous T‐cell lymphoma; DA, daunorubicin+cytarabine; DHAP, dexamethasone+cisplatin+cytarabine; DLBCL, diffuse large B‐cell lymphoma; ESHAP, etoposide+solumedrol+high‐dose AraC+cisplatin; EV, enfortumab vedotin; FLAI, fludarabine+cytarabine+idarubicin; GO, gemtuzumab ozogamicin; HL, Hodgkin lymphoma; ICE, ifosfamide+carboplatin+etoposide; INO, inotuzumab ozogamicin; MDS, myelodysplastic syndrome; MICE, mitoxantrone+etoposide+cytarabine; mini‐hyper‐CVD, cyclophosphamide+vincristine+methotrexate+cytarabine; MM, multiple myeloma; ND, newly diagnosed; NHL, non‐Hodgkin lymphoma; PMBL, primary mediastinal B‐cell lymphoma; PT, previously treated; PTCL, peripheral T‐cell lymphoma; PV, polatuzumab vedotin‐piiq; R‐CHP, rituximab+cyclophosphamide+doxorubicin+prednisone; R/R, relapsed/refractory; sALCL, systemic anaplastic large‐cell lymphoma; SG, sacituzumab govitecan; T‐DM1, ado‐trastuzumab emtansine; T‐DXd, trastuzumab deruxtecan.
4.1. Hematologic malignancies
4.1.1. Relapsed or refractory cancer
First, anti‐CD33 GO was explored mostly in AML patients. Acute myeloid leukemia is a common type of aggressive hematologic malignancy characterized by abnormal proliferation and differentiation of immature myeloid cells, which accounts for 20% of hematologic malignancy‐related deaths. 52 , 53 The fractionated dosage of GO was observed in the phase 2 MyloFrance‐1 trial for the first Relapsed or refractory (R/R) CD33‐positive AML patients; 26% of patients achieved complete remission (CR) with a median relapse‐free survival (RFS) of 11 months and manageable toxicities, which led to FDA approval. 54 Several studies explored the efficacy of GO in combination with chemotherapy as salvage therapy, including the combination of GO and DA therapy (daunorubicin plus cytarabine) (overall response rate [ORR]: 38.8%; CR rate: 22.2%; 2‐year RFS rate: 18.5%; 2‐year overall survival [OS] rate: 26%), 55 the combination of GO and MYLODAM schema (cytarabine and mitoxantrone) (ORR: 67%; 2‐year RFS rate: 36%; 2‐year OS rate: 54%), 56 the combination of GO and high‐dose cytarabine, mitoxantrone, and all‐trans retinoic acid (ATRA) (CR/CR with incomplete hematologic recovery [CRi] rate: 51%; ORR: 61.5%; 4‐year OS rate: 32%), 57 as well as the combination of GO and decitabine (CR/CRi rate: 18%; median OS: 3.5 months). 58 Moreover, two phase 1/2 trials showed the enhanced efficacy of hypomethylating agent therapy in addition to GO through epigenetic effects in R/R AML patients (NCT00766116 trial, GO, plus azacytidine; CR/CRi rate: 24%; NCT00895934 trial, GO, azacytidine, plus vorinostat; CR/CRi rate: 41.9%; median OS: 224.5 days). 59 , 60 Furthermore, a retrospective study also showed the efficacy of GO combined with intermediate‐dose cytarabine for relapsed patients after stem‐cell transplantation (ORR: 60%; median OS: 103 days; median EFS: 76 days). 61 Consequently, GO‐based regimens might be considered as a salvage and bridge therapy to transplant for R/R AML patients and may also be a potential therapy for patients after transplantation.
Second, anti‐CD30 BV was detected in both HL and NHL patients. Hodgkin's lymphoma is a relatively rare B‐cell malignancy that contributes to 10% of lymphomas, while NHL is common and contains a series of heterogeneous malignancies with various pathological and clinical characteristics. 62 , 63 , 64 The phase 2 NCT00848926 trial showed the efficacy of monotherapy BV for R/R CD30‐positive HL patients after an autologous stem‐cell transplantation (ASCT) or at least two lines of multiagent chemotherapies (ORR: 75%; CR rate: 34%; adverse event [AE] over grade 3: 55%; 5‐year progression‐free survival [PFS] rate: 22%; 5‐year OS rate: 41%). 65 , 66 The phase 3 AETHERA trial also demonstrated the efficacy of BV compared with placebo as consolidation therapy for HL patients at high risk of R/R after ASCT (median PFS: 42.9% vs. 24.1%; p = 0.0013; 5‐year PFS rate: 59% vs. 41%; hazard ratio [HR]: 0.521; 95% confidence interval [CI]: 0.379–0.717). 67 , 68 Based on these two trials, BV received FDA approval for patients with R/R CD30‐positive HL and patients with a high risk of R/R after ASCT. 42 , 69 For R/R CD30‐positive HL patients prior to ASCT, the phase 2 NCT01393717 study demonstrated that an ORR of 68%, besides, 49% of patients received ASCT without salvage chemotherapy. 70 In addition, in terms of combination therapy, the combination of BV and chemotherapy was explored widely for R/R HL patients before ASCT, with an approximate ORR of 90% and a CR rate in the range of 70–80%, including BV plus etoposide, solumedrol, high‐dose AraC, and cisplatin (ESHAP, NCT02243436 trial) 71 ; BV plus ifosfamide, carboplatin, and etoposide (ICE, NCT02227199 trial) 72 ; BV plus bendamustine (NCT01874054 trial) 73 ; and BV plus dexamethasone, cisplatin, and cytarabine (DHAP, NCT02280993 trial). 74 Moreover, a phase 1/2 NCT02572167 trial investigated the combination of BV and immunotherapy as initial salvage therapy for patients with R/R HL; 82% of patients receiving BV plus nivolumab achieved ORR and 61% achieved CR. Although 98% of patients had AEs, the majority were in grades 1–2. 75 Thus, the combination therapy of BV showed activity with manageable AEs for R/R HL patients prior to ASCT and should be further confirmed in phase 3 trials.
The phase 2 NCT00866047 trial showed the efficacy of BV monotherapy for previously treated CD30‐positive systemic anaplastic large cell lymphoma (sALCL) patients (CR rate: 66%; median PFS: 20.0 months; 5‐year PFS: 39%; 5‐year OS rate: 60%). 76 , 77 In the phase 2 NCT01421667 study, patients with R/R CD30‐positive peripheral T‐cell lymphoma (PTCL) receiving BV achieved an ORR of 41%; for patients with angioimmunoblastic T‐cell lymphoma, the ORR was 54%, and the median PFS was 6.7 months. 78 In addition, the phase 3 ALCANZA trial explored the benefit of BV on R/R CD30‐positive cutaneous T‐cell lymphomas patients; patients receiving BV had a superior clinical benefit compared with the control group (methotrexate or bexarotene), including ORR (67% vs. 20%; p < 0.0001), complete response rate (16% vs. 2%; p = 0.0046), and median PFS (17.2% vs. 3.5%; p < 0·0001). The most frequently reported AE is peripheral sensory neuropathy. 79 Subsequently, BV therapy has been approved by the FDA for NHL. 42 , 43 For B‐cell NHL, the subset of phase 2 NCT01421667 study showed the efficacy of BV for patients with R/R CD30‐positive B‐cell NHL, mainly DLBCL, with an ORR of 44% and a CR rate of 17%. 80 The combination therapy was detected in the phase 1/2 CheckMate436 trial; the combination of BV and nivolumab for R/R CD30‐positive primary mediastinal B‐cell lymphoma (PMBL) patients with an ORR of 73%, a CR rate of 37%, besides, 53% of patients developed grade 3–4 AEs 81 AEs.
Third, CD22‐directed INO was applied in patient with ALL, a heterogeneous neoplasm of lymphoid progenitors, which consists of 85% B‐cell lineage and 15% T‐cell lineage. Patients with ALL have a poor prognosis, especially in adults. 82 Inotuzumab ozogamicin was explored as a monotherapy for adult R/R CD22‐positive B‐cell precursor ALL patients in the phase 3 INOVATE trial. Results demonstrated that INO group achieved a better CR rate (80.7% vs. 29.4%; p < 0.001), median PFS (5.0 vs. 1.8 months; p < 0.001), and median OS (7.7 vs. 6.7 months; p = 0.04) compared with the chemotherapy group. 83 A long‐term survival report showed sustained benefit (CR/CRi rate: 73.8% vs. 30.9%, p < 0.0001; median OS: 7.7 vs. 6.2 months, p = 0.0105), despite a higher incidence of hepatotoxicities (51% vs. 34%). 84 , 85 Thus, INO received FDA approval for adult patients with R/R B‐cell precursor ALL. 44 In addition, the application of INO in combination therapy was explored in both patients with ALL and B‐cell NHL. The phase 2 NCT01371630 trial found the efficacy of INO plus mini‐hyper‐CVD chemotherapy regimen (cyclophosphamide, vincristine, methotrexate, an cytarabine) for Philadelphia chromosome‐negative R/R ALL patients (ORR: 78%; CR rate: 59%; median RFS: 8 months; median OS: 11 months), 86 while for patients in the first relapse, blinatumomab was also an additional option for combination therapy (ORR: 92%; CR rate: 73%; median RFS: 11 months; median OS: 25 months). 87 For B‐cell NHL, the phase 1/2 NCT00299494 trial investigated the efficacy of INO plus rituximab for R/R CD20/CD22‐positive B‐cell NHL patients; the ORR was 87% and 74%, and the 2‐year PFS rates were 68% and 42% for follicular lymphoma (FL) and DLBCL, respectively. 88 Efficacy was also found in the combination therapy of INO with both rituximab, gemcitabine, dexamethasone, and cisplatin (R‐GDP) as well as rituximab, cyclophosphamide, vincristine, and prednisone (R‐CVP) in R/R CD22‐positive B‐cell NHL patients in a phase 1 trial (NCT01055496). 89 , 90 However, phase 3 NCT01232556 trial compared INO plus rituximab with chemotherapy plus rituximab for R/R aggressive B‐cell NHL, and no significant benefit was shown, while two patients suffered from grade 3 veno‐occlusive disease (VOD)/sinusoidal obstruction syndrome (SOS). 91
Fourth, CD79b‐targeted PV was explored in patients with DLBCL. Diffuse large B‐cell lymphoma contributes to approximately 25% of NHL cases, and despite its curability, 40% of patients suffer from R/R disease. 92 The combination therapy of PV plus bendamustine and rituximab (pola‐BR) was evaluated in comparison with bendamustine and rituximab (BR) in a randomized phase 1b/2 GO29365 trial for patients with transplantation‐ineligible R/R DLBCL. Results showed that the pola‐BR group achieved superior CR rate (40.0% vs. 17.5%, p = 0.026), PFS (median PFS: 9.5 vs. 3.7 months, p < 0.001), and OS (median OS: 12.4 vs. 4.7 months; p = 0.002), while higher incidences were found in grades 3–4 neutropenia, anemia, and thrombocytopenia. 93 The results led to the accelerated FDA approval of PV combined with bendamustine plus rituximab for R/R adult patients with DLBCL after two previous treatments. 45
Finally, BCMA‐targeted BM was investigated in bone marrow cancer and MM patients. Although the 5‐year survival rate of MM patients is nearly 70%, more than 10% of patients still have a poor prognosis. 94 The phase 2 DREAMM‐2 trial showed benefits for R/R MM patients after at least four previous therapies, including a proteasome inhibitor, an anti‐CD38 monoclonal antibody, and an immunomodulatory drug (BM at 2.5 mg/kg every 3 weeks; ORR: 31%; median PFS: 2.9 months; BM at 3.4 mg/kg every 3 weeks; ORR: 34%; median PFS: 4.9 months) with a most common AE of keratopathy (27%; 21%). 95 Thus, BM has received FDA approval for R/R MM patients. 46 In addition, efficacies were also found in combined therapies, the phase 1/2 DREAMM‐4 trial (BM+pembrolizumab) showed an ORR of 67% (2.5 mg/kg cohort) and 43% (3.4 mg/kg cohort) for R/R MM patients receiving ≥3 previous therapy; the phase 1/2 DREAMM‐6 study (BM [2.5 mg/kg]+bortezomib/dexamethasone) showed an ORR of 78% for R/R MM patients receiving ≥1 previous therapy. 96 , 97 Several phase 3 trials are ongoing to verify the benefits of both monotherapy and combined therapies (DREAMM‐3, DREAMM‐7, DREAMM‐8, and DREAMM‐9). 98
4.1.2. Newly diagnosed cancer
First, the phase 3 EORTC‐GIMEMA AML‐19 trial explored the front‐line monotherapy of GO compared with best supportive care for older AML patients who were ineligible for intensive chemotherapy. The results showed that the administration of GO achieved a CR/CRi rate of 27% and a superior OS benefit (median OS: 4.9 vs. 3.6 months, p = 0.005) compared with that of the control group with similar serious AE rates. 99 The combination therapy of GO with chemotherapy for older patients with newly diagnosed AML was explored in the phase 3 ALFA‐0701 trial. Comparing with the standard DA induction therapy, the combination of GO with DA showed a significant event‐free survival (EFS) benefit (median EFS: 17.3 vs. 9.5 months, p = 0.0002), but there was no significant OS benefit. Manageable AEs were observed with a fractionated dosage of GO. 100 , 101 A meta‐analysis further confirmed the efficacy of GO plus standard induction chemotherapy for AML patients, which showed a significantly superior OS (5‐year OS rate: 34.6% vs. 30.7%, p = 0.01), especially for patients with favorable cytogenetics or intermediate risk. 102 Thus, the FDA approved both monotherapy and combined therapy of GO for adult patients with CD33‐positive untreated AML in September 2017. 40 , 41
In addition, other combination regimens included the combination of GO and FLAI regimen (fludarabine, cytarabine, and idarubicin) (CR rate: 82%; 5‐year OS rate: 52%), 103 the combination of GO and decitabine (CR/CRi rate: 45%; median OS: 7 months), 58 and the combination of GO and MICE regimen (mitoxantrone, etoposide, and cytarabine). No survival benefits were observed, but higher mortality rates compared with that of chemotherapy were recorded. 104 Furthermore, a phase 2 study (SWOG0535 trial) showed the benefits and tolerances of GO, ATRA, and arsenic trioxide in newly diagnosed high‐risk acute promyelocytic leukemia patients (CR rate: 86%; 3‐year EFS rate: 78%; 3‐year OS rate: 86%; 6‐week mortality rate: 11%). 105 Thus, GO with various regimens may be an option for newly diagnosed AML patients.
Second, the first‐line setting of BV was explored in both HL and NHL. The randomized phase 3 ECHELON‐1 trial explored the efficacy of the combination of BV and chemotherapy for previously untreated stage III/IV classic HL patients and led to an FDA approval. Patients receiving BV, doxorubicin, vinblastine, and dacarbazine (A+AVD) were compared with those receiving doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD); the results showed a significant benefit for PFS (2‐year PFS rate: 82.1% vs. 77.2%; p = 0.04), despite a higher rate of AEs over grade 3 (83% vs. 66%). 106 , 107 For NHL patients, the phase 3 ECHELON‐2 trial showed that the combination group (BV, cyclophosphamide, doxorubicin, and prednisone [A+CHP]) also achieved clinical benefit in terms of PFS (median PFS: 48.2 vs. 20.8 months; p = 0.0110) and OS (HR: 0.66; 95% CI: 0.46–0.95; p = 0.0244) compared with chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) for patients with treatment‐naive CD30‐positive PTCL, 108 which led to an FDA approval. 109 , 110 In addition, the phase 2 NCT01925612 trial explored the first‐line application of BV in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone (R‐CHP) for high‐intermediate/high‐risk DLBCL patients, and the results showed an ORR of 91%, while for CD30+ patients, the 18‐month PFS rate was 79% and the OS rate was 92%. 111 Another phase 1/2 NCT01994850 trial showed an ORR of 100% when combining BV with R‐CHP as first‐line therapy for CD30‐positive B‐cell lymphoma patients, with a CR rate of 86% and a 2‐year PFS and OS of 85% and 100%, respectively. 112
Third, INO was explored in the first‐line setting in the phase 2 NCT01371630 trial for Philadelphia chromosome‐negative ALL patients aged over 60 years, and the application of INO plus mini‐hyper‐CVD chemotherapy regimen showed a 2‐year PFS rate of 59%, with manageable toxicities. The most common grade 3–4 AEs were prolonged thrombocytopenia, and VOD occurred in 8% of patients. 113 Veno‐occlusive disease should be considered for patients with abnormal liver function during the administration of INO, while the application of blinatumomab in consolidation therapy could prolong the duration between INO and ASCT, which might decrease the VOD risk. 114
Fourth, in terms of PV, the phase 1b–2 trial (NCT01992653) investigated the combination therapy of PV in addition to rituximab or obinutuzumab plus CHP in treatment‐naïve DLBCL patients, with a complete response rate of 77% and an ORR of 89%. 115 The treatment regimen was validated in the Phase 3 POLARIX trial.
4.2. Solid tumors
The development of ADC in solid tumors has mainly focused on breast and urothelial cancers.
Breast cancer is the second most common cancer, with various subtypes according to histopathology and the expression of both hormone receptors and growth factors. 116 , 117 Urothelial carcinoma is the major type of bladder cancer, and it can occur in the upper urinary tract and proximal urethra. 118 , 119
4.2.1. Previously treated advanced cancer
The HER2‐targeted ADC, T‐DM1, was the first FDA‐approved ADC in solid tumors. The FDA approval of T‐DM1 for previously treated (trastuzumab and a taxane) patients with HER2‐positive metastatic breast cancer was based on the phase 3 EMILIA trial. 47 , 120 Results showed patients receiving T‐DM1 had a significant prolonged PFS (median PFS: 9.6 months vs. 6.4 months; p < 0.001) and OS (median OS: 30.9 months vs. 25.1 months; p < 0.001) compared with lapatinib plus capecitabine group, as well as less AEs of grade 3 or above (41% vs. 57%). 121 The final descriptive analysis showed sustained benefit (29.9 months vs. 25.9 months; HR: 0.75; 95% CI: 0.64–0.88). 122 In addition, subgroup analysis showed that T‐DM1 could penetrate the blood–brain barrier and a significantly superior OS benefit was observed for patients with brain metastases (median OS: 26.8 months vs. 12.9 months, p = 0.008) for brain metastases patients. 123 , 124 The efficacy for brain metastases was further confirmed in the phase 3 KAMILLA trial (ORR: 21.4%; clinical benefit rate [CBR]: 42.9%; median PFS: 5.5 months; median OS: 18.9 months). 125 Additionally, in the phase 3 TH3RESA study, T‐DM1 was compared with the treatment of the physician's choice in advanced HER2‐positive breast cancer patients who had previously received at least two HER2‐directed agents. Significant superior survival was shown in patients receiving T‐DM1 (median PFS: 6.2 months vs. 3.3 months, p < 0.0001; median OS: 22.7 months vs. 15.8 months, p = 0.0007), with a lower rate of AEs over grade 3 (40% vs. 47%) but a higher rate of serious AEs (25% vs. 22%). 126 , 127 The survival benefits and acceptable toxicity suggested that T‐DM1 could be used as a posterior line therapy for advanced HER2‐positive breast cancer. Moreover, new combination therapies are still being explored, including T‐DM1 combined with neratinib, which showed efficacy (ORR: 63%) in a phase 1b trial (NCT02236000) and needed further investigation. 128
In addition, T‐DM1 was also investigated in other HER2‐positive solid tumors, including gastric cancer (GATSBY trial, T‐DM1 vs. taxane, without superior benefit) 129 , 130 and lung cancer (NCT02675829 trial, PR rate: 44%, median PFS: 5 months; NCT02289833 trial, ORR: 20%; CBR: 30%). 131 , 132 Further studies have focused on other solid cancers, including bladder cancers, urinary tract cancers, pancreatic cancer, and colorectal cancers, and combined therapies of T‐DM1 and immunotherapy have also been explored.
Second, for HER2‐targeted T‐DXd, the payload of T‐DXd (deruxtecan) could be released and permeate cell membranes, inducing effective cytotoxicity on neighboring tumor cells despite the expression levels of HER2, which expanded its indications for tumors with heterogeneous HER2 expression. 133 In phase 2, the DESTINY‐Breast01 trial explored the efficacy of T‐DXd for advanced HER2‐positive breast cancer patients who were heavily pre‐treated and already received T‐DM1; the median PFS was 16.4 months, with 60.9% of patients responding; interstitial lung disease needed extra attention and was observed in 13.6% of the patients. 134 Based on these results, T‐DXd received accelerated FDA approval for advanced HER2‐positive breast cancer patients who received at least two lines of anti‐HER2‐based regimens in December 2019. 48 , 49
The efficacy of T‐DXd has also been observed in other types of tumors. The phase 2 DESTINY‐Gastric01 trial showed the benefit of T‐DXd compared with chemotherapy for previously treated patients with HER2‐positive advanced gastric cancer (ORR: 51% vs. 14%, p < 0.001; median OS: 12.5 months vs. 8.4 months, p = 0.01). 135 In addition, T‐DXd was also evaluated in pretreated, HER2‐expressing, or HER2‐mutant advanced solid tumors (including non‐small cell lung cancer [NSCLC], colorectal cancer, and other solid cancers) in the phase 1 NCT02564900 trial. For the entire population, an ORR of 28.3% and a median PFS of 7.2 months were found, while HER2‐mutant NSCLC patients showed an ORR of 72.7% and a median PFS of 11.3 months. 136 In addition, several phase 3 trials are ongoing, including the confirmation of T‐DXd for breast cancer patients as well as the exploration of T‐DXd for patients with HER‐low expression and combination therapies. 48
Third, for the Trop‐2‐targeted SG, the novel pH‐sensitive hydrolyzable CL2A linker enabled SG to release SN‐38 in both intracellular tumors and the tumor microenvironment, which could induce bystander effects. 137 The phase 1/2 NCT01631552 trial in small cell lung cancer (SCLC) patients with advanced epithelial cancers 138 and metastatic urothelial carcinoma patients (ORR: 33.3%; CBR: 45.4%; median PFS: 5.5 months; median OS: 13.0 months) 139 were hormone receptor‐positive, while metastatic urothelial carcinoma patients (ORR: 31%; CBR: 44.4%; median PFS: 5.5 months; median OS: 12 months) 140 ; metastatic urothelial carcinoma patients (ORR: 31%; CBR: 47%; median PFS: 7.3 months; median OS: 18.9 months) 141 ; metastatic NSCLC patients (ORR: 19%; CBR: 43%; median PFS: 5.2 months; median OS: 9.5 months) 142 ; and metastatic SCLC patients (ORR: 14%; CBR: 34%; median PFS: 3.7 months; median OS: 7.5 months) were HER2‐negative. 143 Thus, with durable efficacy and tolerable AEs, SG received accelerated FDA approval for metastatic triple‐negative breast cancer patients after at least two lines of therapies in 2020, and fast‐track designations were also granted for patients with metastatic urothelial carcinoma, NSCLC, and SCLC. 50 Further confirmatory and exploratory trials are ongoing, including the phase 3 ASCENT trial for breast cancer patients and phase 2 studies for urothelial cancer as well as other endometrial cancer patients. In the meantime, combination therapies of SG are also being explored.
Furthermore, the Nectin‐4‐directed EV was first evaluated in the phase 1 study EV‐101 trial for heavily pretreated advanced urothelial carcinoma patients, with an ORR of 43% and a median OS of 12.3 months with tolerable AEs. 144 The FDA approval was based on the results of the phase 2 EV‐201 trial for advanced urothelial cancer patients after treatment with platinum‐containing chemotherapy and anti‐PD‐1/L1 therapy. Efficacy was found with an ORR of 44%, a median PFS of 5.8 months, and a median OS of 11.7 months, wherein the most common AEs were fatigue and peripheral neuropathy. 51 , 145 A confirmation phase 3 EV‐301 trial exploring EVs compared with chemotherapy is ongoing.
4.2.2. Newly diagnosed advanced cancer
First‐line application of TDM‐1 was evaluated for advanced HER2‐positive breast cancer patients, wherein the phase 3 MARIANNE trial compared T‐DM1 plus pertuzumab and T‐DM1 monotherapy with trastuzumab plus taxane (control group). Results showed that T‐DM1 with or without pertuzumab had similar efficacy with the control group (median PFS: 15.2 months vs. 14.1 months vs. 13.7 months; response rate: 64.2% vs. 59.7% vs. 67.9%; median response duration: 21.2 months vs. 20.7 months vs. 12.5 months), while there was a lower rate of AEs over grade 3 (46.2% vs. 45.4% vs. 54.1%). 146 Considering the longer duration of response and safety of T‐DM1, the National Comprehensive Cancer Network recommended first‐line T‐DM1 for HER2‐positive breast cancer patients who were not candidates for preferred standard treatment. 147
Besides, first‐line applications of other ADCs were under investigation, the ongoing phase 1 EV‐103 trial explored the combination therapy of EV and chemotherapy or immunotherapy as first‐line therapy for advanced urothelial cancer patients, with a 71% ORR for patients receiving EV and pembrolizumab. 148
4.2.3. Early stage cancer
For early stage cancer, TDM‐1 was explored in both adjuvant and neoadjuvant settings. In phase 3, the KATHERINE trial explored the application of adjuvant T‐DM1 compared with trastuzumab for HER2‐positive early breast cancer patients with residual invasive disease after the administration of neoadjuvant chemotherapy and HER2‐targeted therapy. Patients receiving T‐DM1 achieved better invasive disease‐free survival (iDFS) than patients receiving trastuzumab (3‐year iDFS rate: 88.3% vs. 77.0%). The application of adjuvant T‐DM1 reduced the risk of recurrence or death by 50% (p < 0.001), but a higher rate of AEs of grade 3 (25.7% vs. 15.4%). 149 Thus, the FDA approved T‐DM1 as adjuvant treatment for HER2‐positive early stage breast cancer patients with residual invasive disease, which further expanded the indications for T‐DM1. 150 In terms of neoadjuvant therapy for HER2‐positive operable breast cancer patients, the phase 3 KRISTINE trial explored T‐DM1 plus pertuzumab (T‐DM1+P, also as adjuvant therapy) versus docetaxel, carboplatin, and trastuzumab plus pertuzumab (TCH+P, followed by adjuvant trastuzumab plus pertuzumab), and patients in the T‐DM1+P group had a lower pathologic complete response rate (44.4% vs. 55.7%; p = 0.016) and fewer grade ≥3 AEs (13% vs. 64%). 151 In the long‐term follow‐up, patients receiving TDM‐1+P had a higher risk of EFS (HR: 2.61; 95% CI: 1.36–4.98) due to more locoregional preoperative progression (6.7% vs. 0%), while the risk of iDFS after surgery was similar (HR: 1.11; 95% CI: 0.52–2.40); patients in the T‐DM1+P group had fewer grade ≥3 AEs (31.8% vs. 67.7%), but a higher rate of subsequent AE‐caused treatment discontinuation (18.4% vs. 3.8%) during adjuvant setting. 152
5. CHALLENGES AND PROSPECTS
The greatest advantage of ADCs is that they eliminate tumor cells, avoid healthy cells, and expand the therapeutic index. 153 Three oncology drugs were selected as blockbuster drugs in the “2020 Cortellis Drugs to Watch,” two of which were ADCs for solid tumors, including SG for triple‐negative breast cancer patients and T‐DXd for HER2‐positive breast cancer patients. 154 Many new ADCs are under development, such as the MMAE‐trastuzumab ADC for HER2‐positive breast cancer and rituximab‐vcMMAE ADC for CD20‐positive B‐cell lymphoma. 155 , 156 However, the ADC still has a huge potential for improvement.
5.1. Optimization of ADC structure
First, restrictively selecting extracellular antigens with highly homogeneous expression on tumors but limited expression in healthy tissues could improve the targeted selectivity of ADC and reduce drug toxicity. 12 , 36 However, antigens on solid tumors are highly heterogeneous and dynamic. Considering the bystander effect and non‐internalizing mechanism, the range of antigen selection is expanded because tumor cells with negative antigen expression or antigens without induction of sufficient internalization are also candidates for ADC targets, and the impact of heterogeneous antigen expression is reduced. 25 , 157 Potential risks include lower cellular selectivity and off‐target toxicity. 3 Additionally, oncogenic mutant targets are potential antigens for ADCs. Mutant antigens with high and homogeneous expression have higher possibilities of ubiquitylation and internalization. 131 , 158 Oncogenic antigens might also avoid the downregulation of expression to elicit resistance and exert additional antitumor effects through antibody‐mediated inhibition of downstream signaling pathways. 159 , 160
In addition, rapid technological advancements have emerged given that the fundamental components and conjugation strategies of ADCs are significant factors that need to be improved. 161 , 162 Promotions of fundamental components include producing more optimized antibodies, controllable linkers, and efficient payloads. For antibody engineering, despite the advantages of IgG antibodies, partial unconjugated antibodies induce additional toxicities via ADCC and CDC, while adjustments to the ADC structure could reduce Fc gamma receptor affinity and reduce intrinsic immunological effects. 163 , 164 , 165 , 166 In addition, high molecular weight and retention in the perivascular space limit the diffusion of ADC into the tumor tissues in solid tumors. 2 , 167 Thus, improved antibody structures, including antibody fragments, alternative skeletons, and natural ligands, are being studied further. 168 , 169 Attaching payloads to small molecule fragments could improve the penetration into tumor tissues, especially tumors with poor blood supply and central nervous system tumors; however, rapid clearance remains a major problem. 170 , 171 In addition, the development of bispecific antibodies could enhance tumor specificity and rapid internalization, which might reduce target binding in non‐tumor tissues. 172 As for linkers, novel metal‐mediated cleavage linkers based on simple caging moieties are under development with a well‐controlled drug release by biorthogonal bond‐cleavage reaction and a reduction in toxicity because substoichiometric amounts of metals could achieve the intended efficacy, considering the catalytic activity. Developed metal‐mediated linkers include palladium‐mediated, ruthenium‐mediated, copper‐mediated, and platinum‐mediated cleavages. 25 , 173 Furthermore, because of the limited number of ADCs reaching tumor cells, payloads with high efficacies are of significance, and the potential innovation of payload is not limited to cytotoxic drugs. Other agents including enzymes, protein toxins, targeted drugs, radionuclides, and immunotherapeutic drugs are also under investigation. 13 , 174
The conjugation strategy affects the homogeneity of the drug‐to‐antibody ratio (DAR, the average number of cytotoxic molecules attached to each antibody), the release time of cytotoxic payloads, and off‐target toxicity. 4 Compared with traditional nonspecific conjugation, site‐specific conjugations could increase the homogeneity, stability, pharmacokinetics, and decrease toxicity. 175 , 176 , 177 However, considering the inefficient chemistry and immunogenicity, new technologies are still under development for a better controlled DAR and homogeneity of ADC. For example, utilizing the dolaflexin platform, the new ADC, XMT‐1536, which targets sodium‐phosphate cotransporter protein type II (NaPi2b), is connected by a water‐soluble polymer, “Fleximer,” to improve the water‐soluble, pharmacokinetic, and immunogenicity with a DAR of 10–12. Efficacy was found with an ORR of 34% and tolerability for heavily pretreated ovarian cancer patients, which led to an FDA fast‐track designation. 178 , 179 Furthermore, studies have also reported computational approaches for self‐assembled synthesizing ADCs by molecular docking and dynamics simulations to overcome the instability and heterogeneity of ADCs. 180
5.2. Remained challenges
Nevertheless, the development of ADCs still faces great challenges, including drug resistance and toxicity. Drug resistance remains challenging without explicit mechanisms. Current hypothetic mechanisms include decreased penetration caused by tumor microenvironment changes, downregulation of antigens, deficiencies in pathways of internalization, and resistance to payloads. 4 , 26 Some payloads might be transported by ATP‐binding cassette transporter proteins such as multidrug resistance protein 1 (MDR1), which induces active efflux of the payload and leads to drug resistance. 181 Studies on resistance mechanisms could lead to promising directions for further optimization of ADCs. 147
Toxicity is a significant factor that limits the clinical application of ADCs. Toxicity mainly depends on the positive rate and physiological function of antigens in non‐tumor tissues, the stability of linkers, the quantity and characteristics of payloads, and the bystander effect. Adverse effects of various ADCs are specific; several ADCs received black box warning, including VOD/SOS for patients receiving INO and ocular toxicity for patients receiving BM. 46 , 182 Adverse effects should be closely monitored, actively prevented, and timely treated with appropriate adjustments to regimens in clinical applications. Both the optimization of the ADC structure and adjustment of the administration regimen are potential solutions for reducing toxicities. The structure of ADC has been developed throughout three generations. First‐generation ADCs such as GO are mostly a combination of murine monoclonal antibodies and nondegradable linkers, 167 which can hardly target tumor tissues accurately and fail to achieve a therapeutic effect with high toxicity. 4 , 13 Thus, further improvement was developed in second‐generation ADCs with improved target selectivity, reduced immunogenic humanized antibodies, more effective payloads, and stable linkers, which showed increased clinical efficacy and safety. Most of the currently approved ADCs, including BV and T‐DM1, belong to the second generation. However, disadvantages still exist, such as the presence of unbound antibodies and high DAR of ADC, which lead to off‐target toxicity, ADC aggregation, increased drug metabolic rate, and rapid clearance. Furthermore, third‐generation ADCs further optimized the previous deficiencies, including SG. Optimized site‐specific conjugation techniques, DAR of 2–4, and reduction of unbound antibodies could reduce the off‐target rate and improve the efficacy of ADCs. In addition, the fractionated dosing regimen is an approach to expand the therapeutic index, which could reduce toxicity caused by the peak concentration of ADCs in blood, extend the exposure time of ADCs in tumors, while maintaining or increasing dose intensity to ensure antitumor efficacy. 183 For example, despite the withdrawal of GO by the FDA due to the high incidence of fatal toxicities, 184 fractionated doses and alternative administration strategies led to FDA re‐approval with therapeutic efficacy and manageable toxicity. 40 , 185
5.3. Further exploration on clinical application
Although ADC has been widely applied in various tumor settings, most of the indications were applied to patients with treatment‐refractory cancer due to the hypoxic and immunosuppressive tumor microenvironment, hindrance of drug penetration, and the high heterogeneity of cancer. 186 To date, all five approved ADCs for hematologic malignancies have been indicated for R/R patients, of which only BV and GO received FDA approval for first‐line treatment. In solid tumors, all four ADCs were approved for advanced previously treated patients, while only T‐DM1 was approved in the adjuvant setting. The efficacy of ADCs after resistance to traditional therapies suggests that different pathways of cytotoxic drugs and different payloads of ADCs might provide more possibilities for various sequential therapies. 29 Other treatment settings, including first‐line therapy, are under further investigation.
Besides, despite initial exploration being mainly based on monotherapy, several ADCs showed therapeutic efficacy in combination with other drugs, including chemotherapy, targeted therapy, and immunotherapy. Cytotoxic agents with non‐overlapping mechanisms might be an option for combined therapy of ADC with chemotherapy. 187 , 188 The combination with targeted therapy aims to promote the overexpression or degradation of target antigens and enhance the susceptibility to ADCs, while the combination with antiangiogenic drugs might affect the efficiency of drug delivery by altering the vascular supply of the tumor. 158 , 189 , 190 As for the combination of ADC and immunotherapy, ADC might increase tumor infiltrating lymphocytes (TILs) and affect the tumor microenvironment, which might improve the responses to immunotherapy. 147 , 191 However, the mechanisms of combined therapy, drug interactions, additive toxicities, subsequent treatments, optimal selection of patients, and further validation in clinical data are still needed. 192 , 193
The treatment efficacies of ADCs varied among patients, emphasizing the significance of predictive biomarkers and selection of patients. Common biomarkers are the expression and density of specific target antigens on tumor cells, which are associated with the internalization and metabolization of ADC. 194 However, the target antigen was not sufficient to predict the efficacy of ADCs, and the detection methods and cutoff values still need to be determined. Besides, further developments of ideal biomarkers are significant, which should be able to distinguish the sensitivity of ADCs, guide treatment selections, reflect signals for early response, and monitor the therapeutic process. 2 , 183
6. CONCLUSIONS
The application of ADC has changed traditional treatment patterns for cancer patients, especially the posterior line treatment for patients with refractory tumors. Antibody drug conjugates enable the same treatment for pancreatic cancer patients and have become a great breakthrough for individualized precision treatment. Currently, with the development of ADCs, the therapeutic window is expanded and the limitation of heterogeneously expressed antigens is overcome through the bystander effect and non‐internalizing mechanism. Further exploration of indications includes patients with early stage cancer and combined therapy settings, which is of great potential. The mechanisms of drug resistance, manufacturing techniques, optimized treatment regimens, and appropriate patient selection remain as the major topics.
CONFLICT OF INTEREST
None.
Li W‐Q, Guo H‐F, Li L‐Y, Zhang Y‐F, Cui J‐W. The promising role of antibody drug conjugate in cancer therapy: Combining targeting ability with cytotoxicity effectively. Cancer Med. 2021;10:4677–4696. 10.1002/cam4.4052
Funding information
This work was supported by Jilin Scientific and Technological Development Program (CN) (20190303146SF) and General Program of National Natural Science Foundation of China (81874052).
Contributor Information
Wen‐Qian Li, Email: wqli20@mails.jlu.edu.cn.
Jiu‐Wei Cui, Email: cuijw@jlu.edu.cn.
DATA AVAILABILITY STATEMENT
Clinical data of the current study were extracted from published clinical trials, which were listed in the reference. No other date was available.
REFERENCES
- 1. Miao R, Chen H‐H, Dang QI, et al. Beyond the limitation of targeted therapy: improve the application of targeted drugs combining genomic data with machine learning. Pharmacol Res. 2020;159: 104932. [DOI] [PubMed] [Google Scholar]
- 2. Birrer MJ, Moore KN, Betella I, Bates RC. Antibody‐drug conjugate‐based therapeutics: state of the science. J Natl Cancer Inst. 2019;111(6):538‐549. [DOI] [PubMed] [Google Scholar]
- 3. Bargh JD, Isidro‐Llobet A, Parker JS, Spring DR. Cleavable linkers in antibody‐drug conjugates. Chem Soc Rev. 2019;48(16):4361‐4374. [DOI] [PubMed] [Google Scholar]
- 4. Xie H, Adjei AA. Antibody‐drug conjugates for the therapy of thoracic malignancies. J Thorac Oncol. 2019;14(3):358‐376. [DOI] [PubMed] [Google Scholar]
- 5. Melgarejo‐Rubio G, Pérez‐Tapia SM, Medina‐Rivero E, Velasco‐Velázquez MA. Antibody‐drug conjugates: the new generation of biotechnological therapies against cancer. Gac Med Mex. 2020;156(3):228‐235. [DOI] [PubMed] [Google Scholar]
- 6. Hedrich WD, Fandy TE, Ashour HM, Wang H, Hassan HE. Antibody‐drug conjugates: pharmacokinetic/pharmacodynamic modeling, preclinical characterization, clinical studies, and lessons learned. Clin Pharmacokinet. 2018;57(6):687‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McDonagh CF, Turcott E, Westendorf L, et al. Engineered antibody‐drug conjugates with defined sites and stoichiometries of drug attachment. Protein Eng Des Sel. 2006;19(7):299‐307. [DOI] [PubMed] [Google Scholar]
- 8. Nicolaou KC, Rigol S. The role of organic synthesis in the emergence and development of antibody‐drug conjugates as targeted cancer therapies. Angew Chem Int Ed Engl. 2019;58(33):11206‐11241. [DOI] [PubMed] [Google Scholar]
- 9. Zhang A, Fang J, Chou RY, Bondarenko PV, Zhang Z. Conformational difference in human IgG2 disulfide isoforms revealed by hydrogen/deuterium exchange mass spectrometry. Biochemistry. 2015;54(10):1956‐1962. [DOI] [PubMed] [Google Scholar]
- 10. Schlothauer T, Herter S, Koller CF, et al. Novel human IgG1 and IgG4 Fc‐engineered antibodies with completely abolished immune effector functions. Protein Eng Des Sel. 2016;29(10):457‐466. [DOI] [PubMed] [Google Scholar]
- 11. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chari RV, Miller ML, Widdison WC. Antibody‐drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53(15):3796‐3827. [DOI] [PubMed] [Google Scholar]
- 13. Yaghoubi S, Karimi MH, Lotfinia M, et al. Potential drugs used in the antibody‐drug conjugate (ADC) architecture for cancer therapy. J Cell Physiol. 2020;235(1):31‐64. [DOI] [PubMed] [Google Scholar]
- 14. Diamantis N, Banerji U. Antibody‐drug conjugates—an emerging class of cancer treatment. Br J Cancer. 2016;114(4):362‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dumontet C, Jordan MA. Microtubule‐binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abdollahpour‐Alitappeh M, Lotfinia M, Gharibi T, et al. Antibody‐drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol. 2019;234(5):5628‐5642. [DOI] [PubMed] [Google Scholar]
- 17. Kovtun YV, Goldmacher VS. Cell killing by antibody‐drug conjugates. Cancer Lett. 2007;255(2):232‐240. [DOI] [PubMed] [Google Scholar]
- 18. Gébleux R, Casi G. Antibody‐drug conjugates: current status and future perspectives. Pharmacol Ther. 2016;167:48‐59. [DOI] [PubMed] [Google Scholar]
- 19. Fu Y, Ho M. DNA damaging agent‐based antibody‐drug conjugates for cancer therapy. Antib Ther. 2018;1(2):33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maugeri‐Saccà M, Bartucci M, De Maria R. DNA damage repair pathways in cancer stem cells. Mol Cancer Ther. 2012;11(8):1627‐1636. [DOI] [PubMed] [Google Scholar]
- 21. Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC linker chemistry. Pharm Res. 2015;32(11):3526‐3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCombs JR, Owen SC. Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. Aaps j. 2015;17(2):339‐351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody‐drug conjugates. MAbs. 2016;8(4):659‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tsuchikama K, An Z. Antibody‐drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018;9(1):33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Oliveira BL, Stenton BJ, Unnikrishnan VB, et al. Platinum‐triggered bond‐cleavage of pentynoyl amide and N‐propargyl handles for drug‐activation. J Am Chem Soc. 2020;142(24):10869‐10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu B, Liu D. Antibody‐drug conjugates in clinical trials for lymphoid malignancies and multiple myeloma. J Hematol Oncol. 2019;12(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frigerio M, Kyle AF. The chemical design and synthesis of linkers used in antibody drug conjugates. Curr Top Med Chem. 2017;17(32):3393‐3424. [DOI] [PubMed] [Google Scholar]
- 28. Jedema I, Barge RMY, van der Velden VHJ, et al. Internalization and cell cycle‐dependent killing of leukemic cells by Gemtuzumab Ozogamicin: rationale for efficacy in CD33‐negative malignancies with endocytic capacity. Leukemia. 2004;18(2):316‐325. [DOI] [PubMed] [Google Scholar]
- 29. Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody‐drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsui CK, Barfield RM, Fischer CR, et al. CRISPR‐Cas9 screens identify regulators of antibody‐drug conjugate toxicity. Nat Chem Biol. 2019;15(10):949‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambert JM, Berkenblit A. Antibody‐drug conjugates for cancer treatment. Annu Rev Med. 2018;69:191‐207. [DOI] [PubMed] [Google Scholar]
- 32. Chau CH, Steeg PS, Figg WD. Antibody‐drug conjugates for cancer. Lancet. 2019;394(10200):793‐804. [DOI] [PubMed] [Google Scholar]
- 33. Nicolaou KC, Rigol S. Total synthesis in search of potent antibody‐drug Conjugate payloads. From the fundamentals to the translational. Acc Chem Res. 2019;52(1):127‐139. [DOI] [PubMed] [Google Scholar]
- 34. Redman JM, Hill EM, AlDeghaither D, Weiner LM. Mechanisms of action of therapeutic antibodies for cancer. Mol Immunol. 2015;67(2):28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tai Y‐T, Mayes PA, Acharya C, et al. Novel anti‐B‐cell maturation antigen antibody‐drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128‐3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kovtun YV, Audette CA, Ye Y, et al. Antibody‐drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66(6):3214‐3221. [DOI] [PubMed] [Google Scholar]
- 37. Singh AP, Seigel GM, Guo L, et al. Evolution of the systems pharmacokinetics‐pharmacodynamics model for antibody‐drug conjugates to characterize tumor heterogeneity and in vivo bystander effect. J Pharmacol Exp Ther. 2020;374(1):184‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen‐dependent internalisation required? Br J Cancer. 2017;117(12):1736‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szot C, Saha S, Zhang XM, et al. Tumor stroma‐targeted antibody‐drug conjugate triggers localized anticancer drug release. J Clin Invest. 2018;128(7):2927‐2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jen EY, Ko C‐W, Lee JE, et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33‐positive acute myeloid leukemia. Clin Cancer Res. 2018;24(14):3242‐3246. [DOI] [PubMed] [Google Scholar]
- 41. Norsworthy KJ, Ko C‐W, Lee JE, et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33‐positive acute myeloid leukemia. Oncologist. 2018;23(9):1103‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nikolaenko L, Nademanee A. Brentuximab vedotin and its use in the treatment of advanced Hodgkin's lymphoma. Future Oncol. 2020;16(29):2273‐2282. [DOI] [PubMed] [Google Scholar]
- 43. Shea L, Mehta‐Shah N. Brentuximab vedotin in the treatment of peripheral T cell lymphoma and cutaneous T cell lymphoma. Curr Hematol Malig Rep. 2020;15(1):9‐19. [DOI] [PubMed] [Google Scholar]
- 44. Samra B, Jabbour E, Ravandi F, Kantarjian H, Short NJ. Evolving therapy of adult acute lymphoblastic leukemia: state‐of‐the‐art treatment and future directions. J Hematol Oncol. 2020;13(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deeks ED. Polatuzumab vedotin: first global approval. Drugs. 2019;79(13):1467‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Markham A. Belantamab mafodotin: first approval. Drugs. 2020;80(15):1607‐1613. [DOI] [PubMed] [Google Scholar]
- 47. Amiri‐Kordestani L, Blumenthal GM, Xu QC, et al. FDA approval: ado‐trastuzumab emtansine for the treatment of patients with HER2‐positive metastatic breast cancer. Clin Cancer Res. 2014;20(17):4436‐4441. [DOI] [PubMed] [Google Scholar]
- 48. Cesca MG, Vian L, Cristóvão‐Ferreira S, Pondé N, de Azambuja E . HER2‐positive advanced breast cancer treatment in 2020. Cancer Treat Rev. 2020;88:102033. [DOI] [PubMed] [Google Scholar]
- 49. Keam SJ. Trastuzumab deruxtecan: first approval. Drugs. 2020;80(5):501‐508. [DOI] [PubMed] [Google Scholar]
- 50. Syed YY. Sacituzumab govitecan: first approval. Drugs. 2020;80(10):1019‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hanna KS. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs. 2020;80(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 52. Burd A, Levine RL, Ruppert AS, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26(12):1852‐1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. He C, Wang X, Luo J, Ma Y, Yang Z. Long noncoding RNA maternally expressed gene 3 is downregulated, and its insufficiency correlates with poor‐risk stratification, worse treatment response, as well as unfavorable survival data in patients with acute myeloid leukemia. Technol Cancer Res Treat. 2020;19:1533033820945815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taksin A‐L, Legrand O, Raffoux E, et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia. 2007;21(1):66‐71. [DOI] [PubMed] [Google Scholar]
- 55. Chantepie SP, Reboursiere E, Mear J‐B, et al. Gemtuzumab ozogamicin in combination with intensive chemotherapy in relapsed or refractory acute myeloid leukemia. Leuk Lymphoma. 2015;56(8):2326‐2330. [DOI] [PubMed] [Google Scholar]
- 56. Debureaux P‐E, Labopin M, Mamez A‐C, et al. Fractionated gemtuzumab ozogamicin in association with high dose chemotherapy: a bridge to allogeneic stem cell transplantation in refractory and relapsed acute myeloid leukemia. Bone Marrow Transplant. 2020;55(2):452‐460. [DOI] [PubMed] [Google Scholar]
- 57. Hutter‐Kronke M‐L, Benner A, Dohner K, et al. Salvage therapy with high‐dose cytarabine and mitoxantrone in combination with all‐trans retinoic acid and gemtuzumab ozogamicin in acute myeloid leukemia refractory to first induction therapy. Haematologica. 2016;101(7):839‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Daver N, Kantarjian H, Ravandi F, et al. A phase II study of decitabine and gemtuzumab ozogamicin in newly diagnosed and relapsed acute myeloid leukemia and high‐risk myelodysplastic syndrome. Leukemia. 2016;30(2):268‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walter RB, Medeiros BC, Gardner KM, et al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a phase I/II study. Haematologica. 2014;99(1):54‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Medeiros BC, Tanaka TN, Balaian L, et al. A phase I/II trial of the combination of azacitidine and gemtuzumab ozogamicin for treatment of relapsed acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2018;18(5):346‐352.e5. [DOI] [PubMed] [Google Scholar]
- 61. Koren‐Michowitz M, Maayan H, Apel A, et al. Salvage therapy with ARA‐C and gemtuzumab ozogamicin in AML patients relapsing after stem cell transplantation. Ann Hematol. 2015;94(3):375‐378. [DOI] [PubMed] [Google Scholar]
- 62. Maruyama D, Terui Y, Yamamoto K, et al. Final results of a phase II study of nivolumab in Japanese patients with relapsed or refractory classical Hodgkin lymphoma. Jpn J Clin Oncol. 2020;50(11):1265‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Voorhees TJ, Beaven AW. Therapeutic updates for relapsed and refractory classical hodgkin lymphoma. Cancers (Basel). 2020;12(10):2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abuamsha H, Kadri AN, Hernandez AV. Cardiovascular mortality among patients with non‐Hodgkin lymphoma: differences according to lymphoma subtype. Hematol Oncol. 2019;37(3):261‐269. [DOI] [PubMed] [Google Scholar]
- 65. Younes A, Gopal AK, Smith SE, et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol. 2012;30(18):2183‐2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen R, Gopal AK, Smith SE, et al. Five‐year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2016;128(12):1562‐1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moskowitz CH, Nademanee A, Masszi T, et al. Brentuximab vedotin as consolidation therapy after autologous stem‐cell transplantation in patients with Hodgkin's lymphoma at risk of relapse or progression (AETHERA): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2015;385(9980):1853‐1862. [DOI] [PubMed] [Google Scholar]
- 68. Moskowitz CH, Walewski J, Nademanee A, et al. Five‐year PFS from the AETHERA trial of brentuximab vedotin for Hodgkin lymphoma at high risk of progression or relapse. Blood. 2018;132(25):2639‐2642. [DOI] [PubMed] [Google Scholar]
- 69. de Claro RA, McGinn K, Kwitkowski V, et al. Food and Drug Administration approval summary: brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large‐cell lymphoma. Clin Cancer Res. 2012;18(21):5845‐5849. [DOI] [PubMed] [Google Scholar]
- 70. Chen R, Palmer JM, Martin P, et al. Results of a multicenter phase II trial of brentuximab vedotin as second‐line therapy before autologous transplantation in relapsed/refractory hodgkin lymphoma. Biol Blood Marrow Transplant. 2015;21(12):2136‐2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Garcia‐Sanz R, Sureda A, de la Cruz F, et al. Brentuximab vedotin and ESHAP is highly effective as second‐line therapy for Hodgkin lymphoma patients (long‐term results of a trial by the Spanish GELTAMO Group). Ann Oncol. 2019;30(4):612‐620. [DOI] [PubMed] [Google Scholar]
- 72. Cassaday RD, Fromm J, Cowan AJ, et al. Safety and activity of Brentuximab Vedotin (BV) plus Ifosfamide, Carboplatin, and Etoposide (ICE) for Relapsed/Refractory (Rel/Ref) classical Hodgkin Lymphoma (cHL): initial results of a phase I/II trial. Blood. 2016;128(22):1834.27465916 [Google Scholar]
- 73. LaCasce AS, Bociek RG, Sawas A, et al. Brentuximab vedotin plus bendamustine: a highly active first salvage regimen for relapsed or refractory Hodgkin lymphoma. Blood. 2018;132(1):40‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kersten MJ, Driessen J, Zijlstra JM, et al. Combining brentuximab vedotin with dexamethasone, high‐dose cytarabine and cisplatin as salvage treatment in relapsed or refractory Hodgkin lymphoma: the phase II HOVON/LLPC Transplant BRaVE study. Haematologica. 2021;106(4):1129‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Herrera AF, Moskowitz AJ, Bartlett NL, et al. Interim results of brentuximab vedotin in combination with nivolumab in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2018;131(11):1183‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN‐35) in patients with relapsed or refractory systemic anaplastic large‐cell lymphoma: results of a phase II study. J Clin Oncol. 2012;30(18):2190‐2196. [DOI] [PubMed] [Google Scholar]
- 77. Pro B, Advani R, Brice P, et al. Five‐year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood. 2017;130(25):2709‐2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T‐cell lymphomas with single‐agent brentuximab vedotin. Blood. 2014;123(20):3095‐3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Prince HM, Kim YH, Horwitz SM, et al. Brentuximab vedotin or physician's choice in CD30‐positive cutaneous T‐cell lymphoma (ALCANZA): an international, open‐label, randomised, phase 3, multicentre trial. Lancet. 2017;390(10094):555‐566. [DOI] [PubMed] [Google Scholar]
- 80. Jacobsen ED, Sharman JP, Oki Y, et al. Brentuximab vedotin demonstrates objective responses in a phase 2 study of relapsed/refractory DLBCL with variable CD30 expression. Blood. 2015;125(9):1394‐1402. [DOI] [PubMed] [Google Scholar]
- 81. Zinzani PL, Santoro A, Gritti G, et al. Nivolumab combined with brentuximab vedotin for relapsed/refractory primary mediastinal large B‐cell lymphoma: efficacy and safety from the phase II CheckMate 436 study. J Clin Oncol. 2019;37(33):3081‐3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhao Y, Huang H, Wei G. Novel agents and biomarkers for acute lymphoid leukemia. J Hematol Oncol. 2013;6:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: final report and long‐term survival follow‐up from the randomized, phase 3 INO‐VATE study. Cancer. 2019;125(14):2474‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kantarjian HM, DeAngelo DJ, Advani AS, et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: results from the open‐label, randomised, phase 3 INO‐VATE study. Lancet Haematol. 2017;4(8):e387‐e398. [DOI] [PubMed] [Google Scholar]
- 86. Jabbour E, Ravandi F, Kebriaei P, et al. Salvage chemoimmunotherapy with inotuzumab ozogamicin combined with mini‐hyper‐CVD for patients with relapsed or refractory Philadelphia chromosome‐negative acute lymphoblastic leukemia: a phase 2 clinical trial. JAMA Oncol. 2018;4(2):230‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jabbour E, Sasaki K, Ravandi F, et al. Chemoimmunotherapy with inotuzumab ozogamicin combined with mini‐hyper‐CVD, with or without blinatumomab, is highly effective in patients with Philadelphia chromosome‐negative acute lymphoblastic leukemia in first salvage. Cancer. 2018;124(20):4044‐4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fayad L, Offner F, Smith MR, et al. Safety and clinical activity of a combination therapy comprising two antibody‐based targeting agents for the treatment of non‐Hodgkin lymphoma: results of a phase I/II study evaluating the immunoconjugate inotuzumab ozogamicin with rituximab. J Clin Oncol. 2013;31(5):573‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ogura M, Tobinai K, Hatake K, et al. Phase I study of inotuzumab ozogamicin combined with R‐CVP for relapsed/refractory CD22+ B‐cell non‐Hodgkin lymphoma. Clin Cancer Res. 2016;22(19):4807‐4816. [DOI] [PubMed] [Google Scholar]
- 90. Sangha R, Davies A, Dang NH, et al. Phase 1 study of inotuzumab ozogamicin combined with R‐GDP for the treatment of patients with relapsed/refractory CD22+ B‐cell non‐Hodgkin lymphoma. J Drug Assess. 2017;6(1):10‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dang NH, Ogura M, Castaigne S, et al. Randomized, phase 3 trial of inotuzumab ozogamicin plus rituximab versus chemotherapy plus rituximab for relapsed/refractory aggressive B‐cell non‐Hodgkin lymphoma. Br J Haematol. 2018;182(4):583‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lugtenburg PJ, de Nully Brown P, van der Holt B, et al. Rituximab‐CHOP with early rituximab intensification for diffuse large B‐cell lymphoma: a randomized phase III trial of the HOVON and the Nordic Lymphoma Group (HOVON‐84). J Clin Oncol. 2020;38(29):3377‐3387. [DOI] [PubMed] [Google Scholar]
- 93. Sehn LH, Herrera AF, Flowers CR, et al. Polatuzumab vedotin in relapsed or refractory diffuse large B‐cell lymphoma. J Clin Oncol. 2020;38(2):155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Danziger SA, McConnell M, Gockley J, et al. Bone marrow microenvironments that contribute to patient outcomes in newly diagnosed multiple myeloma: a cohort study of patients in the Total Therapy clinical trials. PLoS Med. 2020;17(11):e1003323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM‐2): a two‐arm, randomised, open‐label, phase 2 study. Lancet Oncol. 2020;21(2):207‐221. [DOI] [PubMed] [Google Scholar]
- 96. Nooka AKMM, Bahlis N, Weisel K, et al. DREAMM‐4: evaluating safety and clinical activity of belantamab mafodotin in combination with pembrolizumab in patients with relapsed/refractory multiple myeloma (RRMM). European Haematology Association (EHA) Congress. 2020;EP955. [Google Scholar]
- 97. Nooka AKGK, Quach H, Forbes A, et al. DREAMM‐6: safety and tolerability of belantamab mafodotin in combination with bortezomib/dexamethasone in relapsed/refractory multiple myeloma. Blood. 2020;136(Supplement 1):19‐20. [Google Scholar]
- 98. Yu B, Jiang T, Liu D. BCMA‐targeted immunotherapy for multiple myeloma. J Hematol Oncol. 2020;13(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Amadori S, Suciu S, Selleslag D, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized phase III EORTC‐GIMEMA AML‐19 trial. J Clin Oncol. 2016;34(9):972‐979. [DOI] [PubMed] [Google Scholar]
- 100. Lambert J, Pautas C, Terré C, et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open‐label, phase III ALFA‐0701 trial. Haematologica. 2019;104(1):113‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Castaigne S, Pautas C, Terré C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de‐novo acute myeloid leukaemia (ALFA‐0701): a randomised, open‐label, phase 3 study. Lancet. 2012;379(9825):1508‐1516. [DOI] [PubMed] [Google Scholar]
- 102. Hills RK, Castaigne S, Appelbaum FR, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta‐analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15(9):986‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Candoni A, Papayannidis C, Martinelli G, et al. Flai (fludarabine, cytarabine, idarubicin) plus low‐dose Gemtuzumab Ozogamicin as induction therapy in CD33‐positive AML: final results and long term outcome of a phase II multicenter clinical trial. Am J Hematol. 2018;93(5):655‐663. [DOI] [PubMed] [Google Scholar]
- 104. Amadori S, Suciu S, Stasi R, et al. Sequential combination of gemtuzumab ozogamicin and standard chemotherapy in older patients with newly diagnosed acute myeloid leukemia: results of a randomized phase III trial by the EORTC and GIMEMA consortium (AML‐17). J Clin Oncol. 2013;31(35):4424‐4430. [DOI] [PubMed] [Google Scholar]
- 105. Lancet JE, Moseley AB, Coutre SE, et al. A phase 2 study of ATRA, arsenic trioxide, and gemtuzumab ozogamicin in patients with high‐risk APL (SWOG 0535). Blood Adv. 2020;4(8):1683‐1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Connors JM, Jurczak W, Straus DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin's lymphoma. N Engl J Med. 2018;378(4):331‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Straus DJ, Długosz‐Danecka M, Alekseev S, et al. Brentuximab vedotin with chemotherapy for stage III/IV classical Hodgkin lymphoma: 3‐year update of the ECHELON‐1 study. Blood. 2020;135(10):735‐742. [DOI] [PubMed] [Google Scholar]
- 108. Horwitz S, O'Connor OA, Pro B, et al. Brentuximab vedotin with chemotherapy for CD30‐positive peripheral T‐cell lymphoma (ECHELON‐2): a global, double‐blind, randomised, phase 3 trial. Lancet. 2019;393(10168):229‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Richardson NC, Kasamon YL, Chen H, et al. FDA approval summary: brentuximab vedotin in first‐line treatment of peripheral T‐cell lymphoma. Oncologist. 2019;24(5):e180‐e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Barta SK, Gong JZ, Porcu P. Brentuximab vedotin in the treatment of CD30+ PTCL. Blood. 2019;134(26):2339‐2345. [DOI] [PubMed] [Google Scholar]
- 111. Budde LE, Halwani A, Yasenchak CA, et al. Results of an ongoing phase 2 study of brentuximab vedotin with Rchp as frontline therapy in patients with high‐intermediate/high‐risk Diffuse Large B Cell Lymphoma (DLBCL). Blood. 2016;128(22):104.27207787 [Google Scholar]
- 112. Svoboda J, Bair SM, Landsburg DJ, et al. Brentuximab vedotin in combination with rituximab, cyclophosphamide, doxorubicin, and prednisone as frontline treatment for patients with CD30‐positive B‐cell lymphomas. Haematologica. 2021;106(6):1705–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kantarjian H, Ravandi F, Short NJ, et al. Inotuzumab ozogamicin in combination with low‐intensity chemotherapy for older patients with Philadelphia chromosome‐negative acute lymphoblastic leukaemia: a single‐arm, phase 2 study. Lancet Oncol. 2018;19(2):240‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Aujla A, Aujla R, Liu D. Inotuzumab ozogamicin in clinical development for acute lymphoblastic leukemia and non‐Hodgkin lymphoma. Biomark Res. 2019;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tilly H, Morschhauser F, Bartlett NL, et al. Polatuzumab vedotin in combination with immunochemotherapy in patients with previously untreated diffuse large B‐cell lymphoma: an open‐label, non‐randomised, phase 1b–2 study. Lancet Oncol. 2019;20(7):998‐1010. [DOI] [PubMed] [Google Scholar]
- 116. Britt KL, Cuzick J, Phillips KA. Key steps for effective breast cancer prevention. Nat Rev Cancer. 2020;20(8):417‐436. [DOI] [PubMed] [Google Scholar]
- 117. Pashayan N, Antoniou AC, Ivanus U, et al. Personalized early detection and prevention of breast cancer: ENVISION consensus statement. Nat Rev Clin Oncol. 2020;17(11):687‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Grivas PD, Melas M, Papavassiliou AG. The biological complexity of urothelial carcinoma: insights into carcinogenesis, targets and biomarkers of response to therapeutic approaches. Semin Cancer Biol. 2015;35:125‐132. [DOI] [PubMed] [Google Scholar]
- 119. Felsenstein KM, Theodorescu D. Precision medicine for urothelial bladder cancer: update on tumour genomics and immunotherapy. Nat Rev Urol. 2018;15(2):92‐111. [DOI] [PubMed] [Google Scholar]
- 120. Lambert JM, Chari RV. Ado‐trastuzumab Emtansine (T‐DM1): an antibody‐drug conjugate (ADC) for HER2‐positive breast cancer. J Med Chem. 2014;57(16):6949‐6964. [DOI] [PubMed] [Google Scholar]
- 121. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med. 2012;367(19):1783‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Diéras V, Miles D, Verma S, et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2‐positive advanced breast cancer (EMILIA): a descriptive analysis of final overall survival results from a randomised, open‐label, phase 3 trial. Lancet Oncol. 2017;18(6):732‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Krop IE, Lin NU, Blackwell K, et al. Trastuzumab emtansine (T‐DM1) versus lapatinib plus capecitabine in patients with HER2‐positive metastatic breast cancer and central nervous system metastases: a retrospective, exploratory analysis in EMILIA. Ann Oncol. 2015;26(1):113‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Dong R, Ji J, Liu H, He X. The evolving role of trastuzumab emtansine (T‐DM1) in HER2‐positive breast cancer with brain metastases. Crit Rev Oncol Hematol. 2019;143:20‐26. [DOI] [PubMed] [Google Scholar]
- 125. Montemurro F, Delaloge S, Barrios CH, et al. Trastuzumab emtansine (T‐DM1) in patients with HER2‐positive metastatic breast cancer and brain metastases: exploratory final analysis of cohort 1 from KAMILLA, a single‐arm phase IIIb clinical trial(☆). Ann Oncol. 2020;31(10):1350‐1358. [DOI] [PubMed] [Google Scholar]
- 126. Krop IE, Kim S‐B, González‐Martín A, et al. Trastuzumab emtansine versus treatment of physician's choice for pretreated HER2‐positive advanced breast cancer (TH3RESA): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2014;15(7):689‐699. [DOI] [PubMed] [Google Scholar]
- 127. Krop IE, Kim S‐B, Martin AG, et al. Trastuzumab emtansine versus treatment of physician's choice in patients with previously treated HER2‐positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open‐label phase 3 trial. Lancet Oncol. 2017;18(6):743‐754. [DOI] [PubMed] [Google Scholar]
- 128. Abraham J, Montero AJ, Jankowitz RC, et al. Safety and efficacy of T‐DM1 plus neratinib in patients with metastatic HER2‐positive breast cancer: NSABP Foundation trial FB‐10. J Clin Oncol. 2019;37(29):2601‐2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Thuss‐Patience PC, Shah MA, Ohtsu A, et al. Trastuzumab emtansine versus taxane use for previously treated HER2‐positive locally advanced or metastatic gastric or gastro‐oesophageal junction adenocarcinoma (GATSBY): an international randomised, open‐label, adaptive, phase 2/3 study. Lancet Oncol. 2017;18(5):640‐653. [DOI] [PubMed] [Google Scholar]
- 130. Shah MA, Kang Y‐K, Thuss‐Patience PC, et al. Biomarker analysis of the GATSBY study of trastuzumab emtansine versus a taxane in previously treated HER2‐positive advanced gastric/gastroesophageal junction cancer. Gastric Cancer. 2019;22(4):803‐816. [DOI] [PubMed] [Google Scholar]
- 131. Li BT, Shen R, Buonocore D, et al. Ado‐trastuzumab emtansine for patients with HER2‐mutant lung cancers: results from a phase II basket trial. J Clin Oncol. 2018;36(24):2532‐2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Peters S, Stahel R, Bubendorf L, et al. Trastuzumab Emtansine (T‐DM1) in patients with previously treated HER2‐overexpressing metastatic non‐small cell lung cancer: efficacy, safety, and biomarkers. Clin Cancer Res. 2019;25(1):64‐72. [DOI] [PubMed] [Google Scholar]
- 133. Bartsch R. Trastuzumab‐deruxtecan: an investigational agent for the treatment of HER2‐positive breast cancer. Expert Opin Investig Drugs. 2020;29(9):901‐910. [DOI] [PubMed] [Google Scholar]
- 134. Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2‐positive breast cancer. N Engl J Med. 2020;382(7):610‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Shitara K, Bang Y‐J, Iwasa S, et al. Trastuzumab deruxtecan in previously treated HER2‐positive gastric cancer. N Engl J Med. 2020;382(25):2419‐2430. [DOI] [PubMed] [Google Scholar]
- 136. Tsurutani J, Iwata H, Krop I, et al. Targeting HER2 with trastuzumab deruxtecan: a dose‐expansion, phase I study in multiple advanced solid tumors. Cancer Discov. 2020;10(5):688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Perrone E, Lopez S, Zeybek B, et al. Preclinical activity of sacituzumab govitecan, an antibody‐drug conjugate targeting Trophoblast Cell‐Surface Antigen 2 (Trop‐2) linked to the active metabolite of irinotecan (SN‐38), in ovarian cancer. Front Oncol. 2020;10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Starodub AN, Ocean AJ, Shah MA, et al. First‐in‐human trial of a novel Anti‐Trop‐2 antibody‐SN‐38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. 2015;21(17):3870‐3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bardia A, Mayer IA, Vahdat LT, et al. Sacituzumab govitecan‐hziy in refractory metastatic triple‐negative breast cancer. N Engl J Med. 2019;380(8):741‐751. [DOI] [PubMed] [Google Scholar]
- 140. Kalinsky K, Diamond JR, Vahdat LT, et al. Sacituzumab govitecan in previously treated hormone receptor‐positive/HER2‐negative metastatic breast cancer: final results from a phase I/II, single‐arm, basket trial. Ann Oncol. 2020;31(12):1709‐1718. [DOI] [PubMed] [Google Scholar]
- 141. Tagawa ST, Faltas BM, Lam ET, et al. Sacituzumab govitecan (IMMU‐132) in patients with previously treated metastatic urothelial cancer (mUC): results from a phase I/II study. J Clin Oncol. 2019;37(7_suppl):354. [Google Scholar]
- 142. Heist RS, Guarino MJ, Masters G, et al. Therapy of advanced non‐small‐cell lung cancer with an SN‐38‐Anti‐Trop‐2 drug conjugate, sacituzumab govitecan. J Clin Oncol. 2017;35(24):2790‐2797. [DOI] [PubMed] [Google Scholar]
- 143. Gray JE, Heist RS, Starodub AN, et al. Therapy of Small Cell Lung Cancer (SCLC) with a topoisomerase‐I‐inhibiting Antibody‐Drug Conjugate (ADC) targeting Trop‐2, sacituzumab govitecan. Clin Cancer Res. 2017;23(19):5711‐5719. [DOI] [PubMed] [Google Scholar]
- 144. Rosenberg J, Sridhar SS, Zhang J, et al. EV‐101: a phase I study of single‐agent enfortumab vedotin in patients with nectin‐4‐positive solid tumors, including metastatic urothelial carcinoma. J Clin Oncol. 2020;38(10):1041‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rosenberg JE, O'Donnell PH, Balar AV, et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti‐programmed death 1/programmed death ligand 1 therapy. J Clin Oncol. 2019;37(29):2592‐2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Perez EA, Barrios C, Eiermann W, et al. Trastuzumab emtansine with or without pertuzumab versus trastuzumab plus taxane for human epidermal growth factor receptor 2‐positive, advanced breast cancer: primary results from the phase III MARIANNE study. J Clin Oncol. 2017;35(2):141‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. García‐Alonso S, Ocaña A, Pandiella A. Trastuzumab emtansine: mechanisms of action and resistance, clinical progress, and beyond. Trends Cancer. 2020;6(2):130‐146. [DOI] [PubMed] [Google Scholar]
- 148. Hoimes CJ, Rosenberg JE, Srinivas S, et al. EV‐103: initial results of enfortumab vedotin plus pembrolizumab for locally advanced or metastatic urothelial carcinoma. Ann Oncol. 2019;30(Supplement_5). [Google Scholar]
- 149. von Minckwitz G , Huang C‐S, Mano MS, et al. Trastuzumab emtansine for residual invasive HER2‐positive breast cancer. N Engl J Med. 2019;380(7):617‐628. [DOI] [PubMed] [Google Scholar]
- 150. Wedam S, Fashoyin‐Aje L, Gao X, et al. FDA approval summary: ado‐trastuzumab emtansine for the adjuvant treatment of HER2‐positive early breast cancer. Clin Cancer Res. 2020;26(16):4180‐4185. [DOI] [PubMed] [Google Scholar]
- 151. Hurvitz SA, Martin M, Symmans WF, et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2‐positive breast cancer (KRISTINE): a randomised, open‐label, multicentre, phase 3 trial. Lancet Oncol. 2018;19(1):115‐126. [DOI] [PubMed] [Google Scholar]
- 152. Hurvitz SA, Martin M, Jung KH, et al. Neoadjuvant trastuzumab emtansine and pertuzumab in human epidermal growth factor receptor 2‐positive breast cancer: three‐year outcomes from the phase III KRISTINE study. J Clin Oncol. 2019;37(25):2206‐2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Makawita S, Meric‐Bernstam F. Antibody‐drug conjugates: patient and treatment selection. Am Soc Clin Oncol Educ Book. 2020;40:1‐10. [DOI] [PubMed] [Google Scholar]
- 154. The 2020 Cortellis Drugs to Watch. https://www.bioworld.com/articles/432945‐the‐2020‐cortellis‐drugs‐to‐watch
- 155. Abdollahpour‐Alitappeh M, Hashemi Karouei SM, Lotfinia M, Amanzadeh A, Habibi‐Anbouhi M. A developed antibody‐drug conjugate rituximab‐vcMMAE shows a potent cytotoxic activity against CD20‐positive cell line. Artif Cells Nanomed Biotechnol. 2018;46(sup2):1‐8. [DOI] [PubMed] [Google Scholar]
- 156. Yaghoubi S, Gharibi T, Karimi MH, et al. Development and biological assessment of MMAE‐trastuzumab antibody‐drug conjugates (ADCs). Breast Cancer. 2021;28(1):216‐225. [DOI] [PubMed] [Google Scholar]
- 157. Siena S, Marsoni S, Sartore‐Bianchi A. Breaking barriers in HER2+ cancers. Cancer Cell. 2020;38(3):317‐319. [DOI] [PubMed] [Google Scholar]
- 158. Li BT, Michelini F, Misale S, et al. HER2‐mediated internalization of cytotoxic agents in ERBB2 amplified or mutant lung cancers. Cancer Discov. 2020;10(5):674‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Damelin M, Zhong W, Myers J, Sapra P. Evolving strategies for target selection for antibody‐drug conjugates. Pharm Res. 2015;32(11):3494‐3507. [DOI] [PubMed] [Google Scholar]
- 160. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26(45):6469‐6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Dan N, Setua S, Kashyap VK, et al. Antibody‐drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals (Basel). 2018;11(2):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Mehrling T, Soltis D. Challenges in optimising the successful construction of antibody drug conjugates in cancer therapy. Antibodies (Basel). 2018;7(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab‐DM1 (T‐DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347‐356. [DOI] [PubMed] [Google Scholar]
- 164. McDonagh CF, Kim KM, Turcott E, et al. Engineered anti‐CD70 antibody‐drug conjugate with increased therapeutic index. Mol Cancer Ther. 2008;7(9):2913‐2923. [DOI] [PubMed] [Google Scholar]
- 165. Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody‐drug conjugates. Nat Rev Drug Discov. 2017;16(5):315‐337. [DOI] [PubMed] [Google Scholar]
- 166. Peters C, Brown S. Antibody‐drug conjugates as novel anti‐cancer chemotherapeutics. Biosci Rep. 2015;35(4):e00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Teicher BA, Chari RV. Antibody conjugate therapeutics: challenges and potential. Clin Cancer Res. 2011;17(20):6389‐6397. [DOI] [PubMed] [Google Scholar]
- 168. Goulet DR, Atkins WM. Considerations for the design of antibody‐based therapeutics. J Pharm Sci. 2020;109(1):74‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Richards DA. Exploring alternative antibody scaffolds: antibody fragments and antibody mimics for targeted drug delivery. Drug Discov Today Technol. 2018;30:35‐46. [DOI] [PubMed] [Google Scholar]
- 170. Zhuang C, Guan X, Ma H, Cong H, Zhang W, Miao Z. Small molecule‐drug conjugates: a novel strategy for cancer‐targeted treatment. Eur J Med Chem. 2019;163:883‐895. [DOI] [PubMed] [Google Scholar]
- 171. Casi G, Neri D. Antibody‐drug conjugates and small molecule‐drug conjugates: opportunities and challenges for the development of selective anticancer cytotoxic agents. J Med Chem. 2015;58(22):8751‐8761. [DOI] [PubMed] [Google Scholar]
- 172. Li JY, Perry SR, Muniz‐Medina V, et al. A biparatopic HER2‐targeting antibody‐drug conjugate induces tumor regression in primary models refractory to or ineligible for HER2‐targeted therapy. Cancer Cell. 2019;35(6):948‐949. [DOI] [PubMed] [Google Scholar]
- 173. Wang X, Liu Y, Fan X, et al. Copper‐triggered bioorthogonal cleavage reactions for reversible protein and cell surface modifications. J Am Chem Soc. 2019;141(43):17133‐17141. [DOI] [PubMed] [Google Scholar]
- 174. Mehta A, Forero‐Torres A. Development and integration of antibody‐drug conjugate in non‐Hodgkin lymphoma. Curr Oncol Rep. 2015;17(9):41. [DOI] [PubMed] [Google Scholar]
- 175. Dai Z, Zhang X‐N, Nasertorabi F, et al. Synthesis of site‐specific antibody‐drug conjugates by ADP‐ribosyl cyclases. Sci Adv. 2020;6(23):eaba6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Junutula JR, Raab H, Clark S, et al. Site‐specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925‐932. [DOI] [PubMed] [Google Scholar]
- 177. Nejadmoghaddam MR, Minai‐Tehrani A, Ghahremanzadeh R, Mahmoudi M, Dinarvand R, Zarnani AH. Antibody‐drug conjugates: possibilities and challenges. Avicenna J Med Biotechnol. 2019;11(1):3‐23. [PMC free article] [PubMed] [Google Scholar]
- 178. Hamilton EBM, Tolcher A, Buscema J, et al. Safety and efficacy of XMT‐1536 in ovarian cancer: A subgroup analysis from the phase I expansion study of XMT‐1536, a NaPi2b antibody‐drug conjugate. Annals of Oncology. 2020;31:S627–S628. [Google Scholar]
- 179. Mersana Therapeutics. XMT‐1536. https://www.mersana.com/pipeline/xmt‐1536/
- 180. Gupta N, Ansari A, Dhoke GV, et al. Computationally designed antibody‐drug conjugates self‐assembled via affinity ligands. Nat Biomed Eng. 2019;3(11):917‐929. [DOI] [PubMed] [Google Scholar]
- 181. Jackson D, Stover D. Using the lessons learned from the clinic to improve the preclinical development of antibody drug conjugates. Pharm Res. 2015;32(11):3458‐3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Yurkiewicz IR, Muffly L, Liedtke M. Inotuzumab ozogamicin: a CD22 mAb‐drug conjugate for adult relapsed or refractory B‐cell precursor acute lymphoblastic leukemia. Drug Des Devel Ther. 2018;12:2293‐2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Coats S, Williams M, Kebble B, et al. Antibody‐drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin Cancer Res. 2019;25(18):5441‐5448. [DOI] [PubMed] [Google Scholar]
- 184. Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854‐4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Burnett A, Cavenagh J, Russell N, et al. Defining the dose of gemtuzumab ozogamicin in combination with induction chemotherapy in acute myeloid leukemia: a comparison of 3 mg/m2 with 6 mg/m2 in the NCRI AML17 Trial. Haematologica. 2016;101(6):724‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299‐309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Zhong H, Chen C, Tammali R, et al. Improved therapeutic window in BRCA‐mutant tumors with antibody‐linked pyrrolobenzodiazepine dimers with and without PARP inhibition. Mol Cancer Ther. 2019;18(1):89‐99. [DOI] [PubMed] [Google Scholar]
- 188. Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. Synthetic lethality exploitation by an Anti‐Trop‐2‐SN‐38 antibody‐drug conjugate, IMMU‐132, plus PARP inhibitors in BRCA1/2‐wild‐type triple‐negative breast cancer. Clin Cancer Res. 2017;23(13):3405‐3415. [DOI] [PubMed] [Google Scholar]
- 189. O'Malley DM, Matulonis UA, Birrer MJ, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)‐targeting antibody‐drug conjugate (ADC), in combination with bevacizumab in patients with platinum‐resistant ovarian cancer. Gynecol Oncol. 2020;157(2):379‐385. [DOI] [PubMed] [Google Scholar]
- 190. Moore KN, Birrer MJ, Marsters J, et al. Phase 1b study of anti‐NaPi2b antibody‐drug conjugate lifastuzumab vedotin (DNIB0600A) in patients with platinum‐sensitive recurrent ovarian cancer. Gynecol Oncol. 2020;158(3):631‐639. [DOI] [PubMed] [Google Scholar]
- 191. Gerber HP, Sapra P, Loganzo F, May C. Combining antibody‐drug conjugates and immune‐mediated cancer therapy: what to expect? Biochem Pharmacol. 2016;102:1‐6. [DOI] [PubMed] [Google Scholar]
- 192. Boswell CA, Mundo EE, Zhang C, et al. Impact of drug conjugation on pharmacokinetics and tissue distribution of anti‐STEAP1 antibody‐drug conjugates in rats. Bioconjug Chem. 2011;22(10):1994‐2004. [DOI] [PubMed] [Google Scholar]
- 193. Shen BQ, Bumbaca D, Saad O, et al. Catabolic fate and pharmacokinetic characterization of trastuzumab emtansine (T‐DM1): an emphasis on preclinical and clinical catabolism. Curr Drug Metab. 2012;13(7):901‐910. [DOI] [PubMed] [Google Scholar]
- 194. Lambert JM, Morris CQ. Antibody‐Drug Conjugates (ADCs) for personalized treatment of solid tumors: a review. Adv Ther. 2017;34(5):1015‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33‐positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19(13):3244‐3254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Clinical data of the current study were extracted from published clinical trials, which were listed in the reference. No other date was available.