

Conspectus
Over the last century, malaria deaths have decreased by more than 85%. Nonetheless, there were 405 000 deaths in 2018, mostly resulting from Plasmodium falciparum infection. In the 21st century, much of the advance has arisen from deployment of insecticide-treated bed nets and artemisinin combination therapy. However, over the last decade parasites with a delayed artemisinin clearance phenotype have appeared in Southeast Asia threatening further gains. The effort to find new drugs is thus urgent. A prominent process in blood stage malaria parasites, which we contend remains a viable drug target, is hemozoin formation. This crystalline material consisting of heme can be readily seen when parasites are viewed microscopically. The process of its formation in the parasite, however, is still not fully understood.
In early work, we recognized hemozoin formation as a biomineralization process. We have subsequently investigated the kinetics of synthetic hemozoin (β-hematin) crystallization catalyzed at lipid-aqueous interfaces under biomimetic conditions. This led us to the use of neutral detergent-based high throughput screening (HTS) for inhibitors of β-hematin formation. A good hit rate against malaria parasites was obtained. Simultaneously we developed a pyridine-based assay which proved successful in measuring concentrations of hematin not converted to β-hematin.
The pyridine assay was adapted to determine the effects of chloroquine and other clinical antimalarials on hemozoin formation in the cell. This permitted determination of the dose dependent amounts of exchangeable heme and hemozoin in P. falciparum for the first time. These studies have shown that hemozoin inhibitors cause a dose-dependent increase in exchangeable heme, correlated with decreased parasite survival. Electron spectroscopic imaging (ESI) showed a relocation of heme iron into the parasite cytoplasm, while electron microscopy provided evidence of disruption of hemozoin crystals. This cellular assay was subsequently extended to the top-ranked hits from a wide range of scaffolds found by HTS. Intriguingly, the amounts of exchangeable heme at the parasite growth IC50 values of these scaffolds showed substantial variation. The amount of exchangeable heme was found to be correlated with the amount of inhibitor accumulated in the parasitized red blood cell. This suggests that heme-inhibitor complexes, rather than free heme, lead to parasite killing. This was supported by ESI using a Br-containing compound which showed co-localization of Fe and Br, as well as by confocal Raman microscopy which confirmed the presence of a complex in the parasite. Current evidence indicates that inhibitors block hemozoin formation by surface adsorption. Indeed, we have successfully introduced molecular docking with hemozoin to find new inhibitors. It follows that the resulting increase in free heme leads to formation of the parasiticidal heme-inhibitor complex. We have reported crystal structures of heme-drug complexes for several aryl methanol antimalarials in non-aqueous media. These form coordination complexes, but most other inhibitors interact non-covalently and determination of their structures remains a major challenge.
It is our view that key future developments will include improved assays to measure cellular heme levels, better in silico approaches for predicting β-hematin inhibition, and a concerted effort to determine the structure and properties of heme-inhibitor complexes.
Graphical Abstract
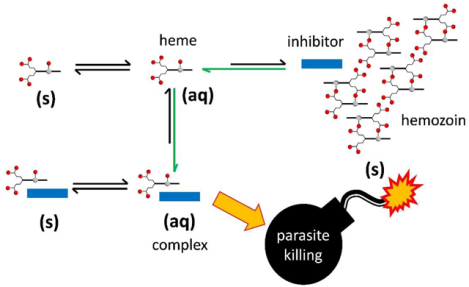
Introduction
In 1900 malaria caused almost three million deaths.5 Mortalities due to this blood borne parasitic disease declined following the introduction of the insecticide DDT and the synthetic 4-amino quinoline drug, chloroquine, during the 1940s.6 However, chloroquine resistance emerged in the late 1950s and 1960s in the Greater Mekong Subregion (GMS) and Latin America, respectively, before spreading to the Pacific Islands (1960 to mid-1970s) and Africa (1980s). Consequently, worldwide deaths increased again, largely due to rampant infections in sub-Saharan Africa,5 which today accounts for 90–95% of global malaria statistics.7 The introduction of pyrethroid treated bed nets and artemisinin combination therapies (ACTs), consisting of an artemisinin-derived drug and a second partner drug (usually a quinoline or related drug) with a longer half-life, has seen a major breakthrough in malaria control from the start of the 21st century.8 ACTs are the first-line malaria treatment today, but treatment failure arising from prolonged clearance times has been detected in the GMS since 2009.9 The World Health Organization reported a total of 405 000 malaria deaths in 2018.7 Thus despite major advances discussed above, malaria remains a serious international health problem. This is exacerbated by the fact that to date, there is no licensed vaccine for the disease, although one candidate (RTS,S) is undergoing clinical trials.7
Malaria is caused by protozoan parasites of the genus Plasmodium, transmitted to mammalian hosts by the female Anopheles mosquito.10 Four species cause human malaria, P. malariae, P. ovale, P. vivax and P. falciparum, with the last being the most virulent. Following sexual reproduction in the mosquito, parasites enter the human host during a blood meal and develop asexually in hepatic cells before invading red blood cells (RBCs), Figure 1. The blood stage is associated with fevers that re-occur every 2–4 days.5 During this stage, the parasite develops from merozoite, through ring and trophozoite to a mature schizont, before erupting from the RBC and invading new RBCs.10 The metabolically-active trophozoite digests large quantities of RBC hemoglobin (Figure 1). A manifold of enzymes is involved in this process, releasing ferroprotoporphyrin IX into the parasite’s digestive vacuole (DV). Subsequently, this must be oxidized to ferriprotoporphyrin IX since the Fe in crystalline malaria pigment (hemozoin) is indisputably in the ferric state based on spectroscopic evidence.11 Autoxidation has been widely assumed but no direct experimental observation of this process in the parasite has been reported. Heme12 contributes to membrane rupture among other things.13 Since parasites lack a heme oxygenase function,14 its disposal is primarily via sequestration as hemozoin,11c comprising cyclic μ-propionato dimers of heme (Figure 1).15 While several antimalarials, including components of ACT, have been shown to inhibit formation of synthetic hemozoin (β-hematin),16 their mechanism(s) of drug action remain poorly understood.
Figure 1.
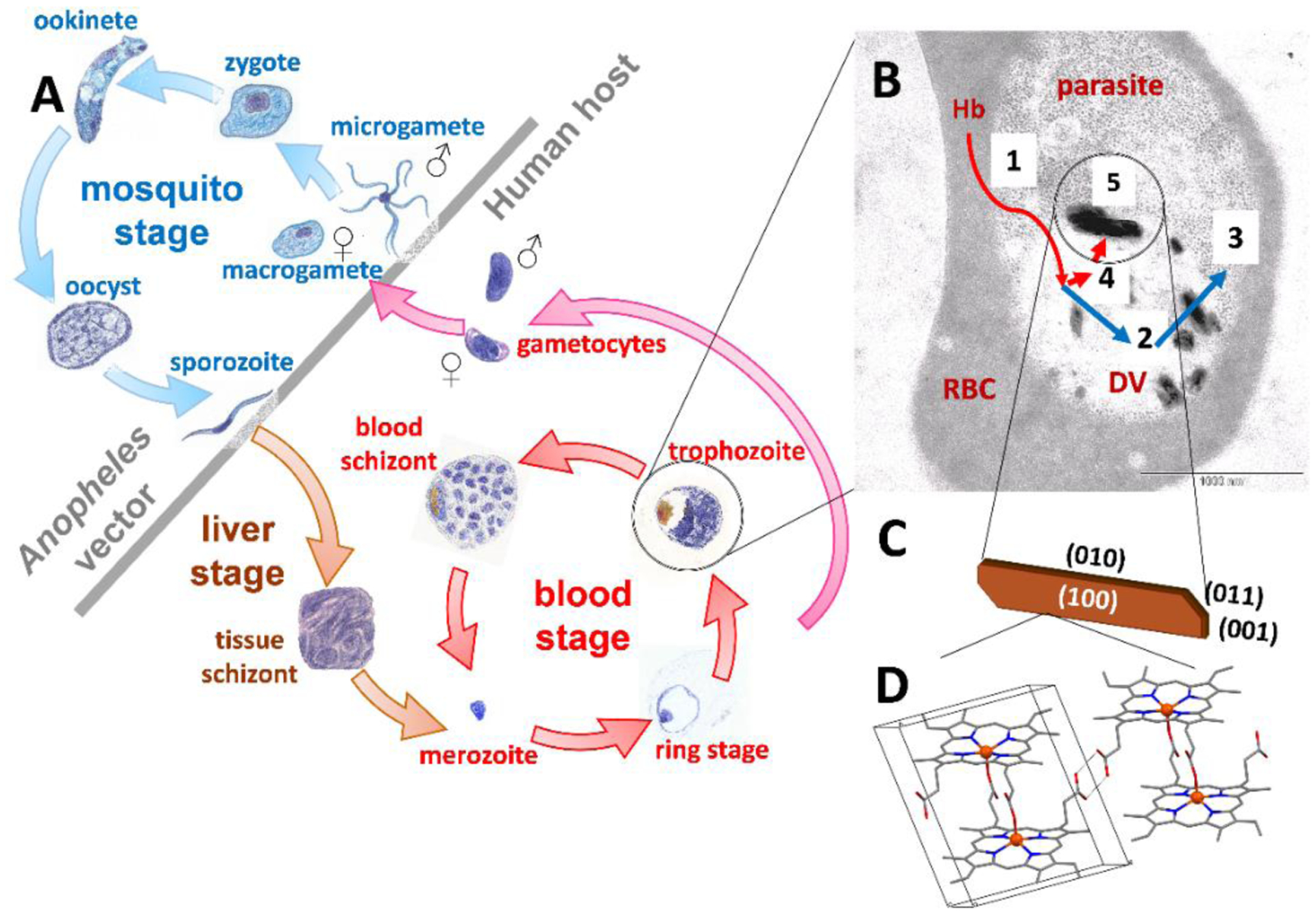
Lifecycle of P. falciparum (A), showing the mosquito, liver, and pathogenic blood stages. The trophozoite (B), exhibits electron dense hemozoin crystals. Host RBC cytoplasm (1), mostly Hb, is ingested into the acidic parasite DV and the globin is digested by aspartic, histoaspartic, cysteine and zinc proteases to peptides (2), and finally by aminopeptidases to amino acids (3). Released heme is oxidized from the Fe(II) to Fe(III) state (4) and the latter is finally crystallized as hemozoin (5). A schematic illustration of hemozoin (C) shows its lath-like shape and oblique ends, with crystal faces labelled. The molecular structure is shown in (D), consisting of a cyclic dimer, in which the heme propionate group of each monomer coordinates to the Fe(III) center of its partner, while the propionic acid groups hydrogen bond to neighboring dimers.
Here, we review our contributions to understanding mechanisms of hemozoin formation and drug action. We maintain that hemozoin remains a unique and viable drug target.
Mechanism of Hemozoin Formation
Mössbauer spectroscopy, chemical analysis and electron spectroscopic imaging have demonstrated that hemozoin accounts for ≥95% of the iron in trophozoites.11c In contrast to the conclusive knowledge of its composition,17 the mechanism of hemozoin formation is still not fully understood. An early study suggested involvement of a so-called heme polymerase,18 and a number of proteins have been considered for this role. Histidine-rich protein (HRP) II and III bind free heme and mediate its conversion to β-hematin under acidic conditions.19 This function has also been attributed to heme detoxification protein (HDP) present across Plasmodium species.20 In 2020, a homolog of lipocalin PV5 was reported to be involved in the control of hemozoin crystallization in the malaria parasite. Disruption of this protein results in changes to the external morphology of hemozoin crystals.21
Lipids have also been proposed to catalyze heme detoxification. Bendrat et al. demonstrated that an acetonitrile extract from hemozoin contained oleic, palmitic and stearic acids, and promoted formation of β-hematin.22 Subsequent studies confirmed the activity of lipids in β-hematin formation23 independent of parasite-derived proteins.24
β-Hematin can be prepared in the absence of proteins or lipids. Indeed, our early work employed carboxylic acids to mediate conversion of amorphous heme to β-hematin.25 Sigmoidal kinetics of β-hematin formation were modeled using the Avrami equation with an Avrami constant of four. The interpretation is that crystal nucleation is an ongoing (sporadic) process, while crystal growth occurs in three dimensions. We likened this to biomineralization which often proceeds via an amorphous phase and suggested that acetate may be a phase-transfer catalyst, a role which lipids could fulfil in vivo. Lipids may, of course, also provide fixed sites of nucleation and templated (epitaxial) growth. We later pioneered a method for forming β-hematin at octanol-, pentanol- and lipid-water interfaces,26 while others demonstrated the effectiveness of solvents27 and detergents.28 Notably, the interface method proceeds under biomimetic conditions (37 °C and 0.05 M buffer), with a physiologically-relevant half-life.26 First-order kinetics were observed, consistent with an Avrami constant of one, indicative of a fixed number of nucleation sites (provided by the solvent interface or lipid molecules) and crystal growth in one dimension along the interface.29 Transmission electron micrograph (TEM) images of hemozoin crystals within neutral lipid droplets provided initial support for this mechanism,30 although later cryogenic soft X-ray tomography data reported by Kapishnikov et al. suggested that hemozoin crystals form at the inner membrane of the digestive vacuole.31 Studies of β-hematin formation at interfaces have provided important insights regarding the mechanism of crystal formation. Grazing incidence X-ray diffraction (GIXD) experiments reported by Leiserowitz and co-workers indicated that β-hematin crystals align with their (100) face parallel to an air-water interface,32 and we later confirmed this observation at neutral lipid (monomyristoylglycerol) interfaces.33 TEM and confocal imaging were used to investigate β-hematin formation in lipid-water emulsions; notably, a blend of neutral lipids (including monoacyl- and diacylglycerides) previously found associated with hemozoin by Pisciotta et al.34 Observation of crystals at lipid droplet surfaces strongly supported the epitaxial growth model where the lipid head group orientates the amphiphilic heme molecules. Diacylglycerides were found to be able to mediate the formation of β-hematin formation faster than monoacylglcerides,35 which we previously attributed to a lower activation energy.34b A density functional theory (DFT) study supported the role of diacylglycerides in promoting heme aggregation, likely an essential step in crystal nucleation.36 Finally, the size of β-hematin crystals was found to correlate with the diameter of the lipid droplets, with average crystal length approximately 60% that of the droplet diameter.34c This size-control may not be evident for native hemozoin, however, if the crystals form at inner membrane surfaces where the curvature is less pronounced than in synthetic droplets.31
Our work on the mechanism of β-hematin formation has demonstrated the efficacy of lipids in the process. The observation of β-hematin crystals aligned with their (100) surfaces parallel to the lipid-water interface is also consistent with their theoretical morphology.37 The (001) and (011) faces have been identified as the fastest-growing, and it has been proposed that sites on these surfaces are available for the adsorption of heme from solution, explaining the predominant crystal growth in the c-direction.29, 38 It has been hypothesized that antimalarial drugs may target these binding sites to impede crystal growth.29
Evidence of Cellular Hemozoin Inhibition and Cellular Mechanism of Action
Despite an extensive literature on β-hematin inhibition by antimalarials and experimental compounds, only in the last seven years has direct evidence of hemozoin inhibition in the malaria parasite been demonstrated. Key to this has been the development of a reliable method for measuring Fe(III) heme in aqueous solution, which we called the pyridine hemichrome inhibition of β-hematin (Phiβ) assay,39 inspired by the well-known pyridine hemochrome assay. Although originally intended for use in high throughput screening (HTS), its employment to quantify free heme has proven to be its most useful application. In 2013 we used cellular fractionation, electron spectroscopic imaging (ESI) based on electron energy loss spectroscopy, and transmission electron microscopy (TEM) to show that the well-known antimalarial chloroquine inhibits hemozoin formation in the parasite, with a corresponding increase in “free” (non-hemozoin, non-hemoglobin) heme, which we hereafter refer to as exchangeable heme in this article.1 Chloroquine has been known to inhibit β-hematin formation since 1992,18 but this was the first direct evidence in P. falciparum. It must be noted that the exact nature of this exchangeable heme has not been elucidated. In untreated parasites it is likely to be precipitated Fe(III)protoporphyin IX, probably in the form of hematin or hemin, or a mixture of the two. It is very unlikely to be freely dissolved in solution, given its extremely low Ksp value at the low pH of the DV.40 In treated parasites, it may also include complexes with the drug or experimental inhibitor.
The cellular fractionation assay mentioned above involves isolation of cultured trophozoites from RBCs by saponin lysis, followed by freeze-thaw lysis of the trophozoites.1 This releases a cytoplasmic fraction with a weak hemoglobin (Hb) spectrum, corresponding to undigested intraparasitic Hb. The insoluble pellet is split into an exchangeable heme fraction soluble upon treatment with 2% sodium dodecyl sulfate (SDS) and 2.5% aqueous pyridine (pH 7.5) and an insoluble hemozoin pellet which is subsequently solubilized with 0.1 M NaOH. All three heme fractions are finally converted into a monomeric low spin heme-pyridine complex, permitting direct comparison. This assay demonstrated that chloroquine causes a dose-dependent decrease in hemozoin from 95% to ≈74% of total heme, while exchangeable heme and Hb increase from ≈2.5% each to ≈15% and ≈11%, respectively. The increase in exchangeable heme closely parallels the decrease in parasite survival, while the increase in undigested Hb only becomes apparent at about 2×IC50 of chloroquine, suggesting that exchangeable heme is responsible for chloroquine activity. ESI showed a redistribution of heme Fe into the parasite cytoplasm at the IC50 of chloroquine, possibly to the endoplasmic reticulum, while TEM demonstrated disruption of hemozoin crystal growth by chloroquine, since multiple crystalline domains or thin crystalline layers were seen.
Later development of a multiwell plate-based assay permitted investigation of a wider range of drugs and experimental compounds. The 4-aminoquinoline antimalarials amodiaquine and piperaquine were both found to inhibit hemozoin formation with increases in exchangeable heme similar to chloroquine (Figure 2).41 The aryl methanols mefloquine and lumefantrine, on the other hand, inhibited hemozoin formation without an increase in exchangeable heme.42 This indicates that these are not true hemozoin inhibitors since their behavior resembles atovaquone, a known inhibitor of the parasite mitochondrial cytochrome bc1 complex which does not even inhibit β-hematin formation. The decrease in hemozoin is likely an indirect effect and shows that the crucial signal for direct hemozoin inhibition in the cell is both a decrease in hemozoin and increase in exchangeable heme. The assay has been extended to a range of benzamides, triarylimidazoles and benzimidazoles that were shown to be direct cellular hemozoin inhibitors.43
Figure 2.
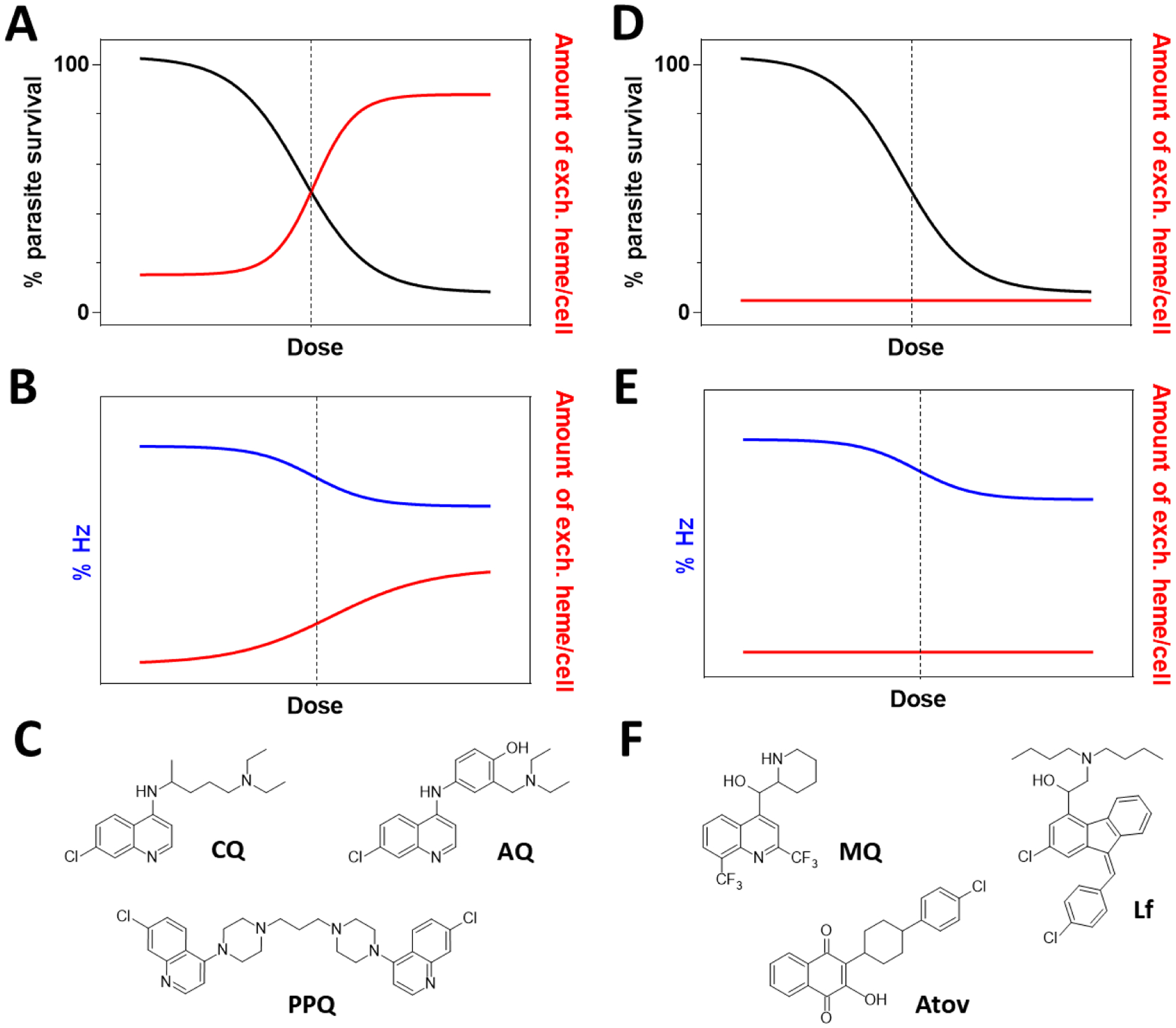
Two modes of action of antimalarial drugs: parasite survival (left axis, black) decreases in (A) and (D), but hemozoin inhibitors induce a corresponding increase in exchangeable (exch.) heme (right axis, red) that coincides with a decrease (left axis, blue) in hemozoin, abbreviated Hz (B); no change in exchangeable heme levels is observed for non-hemozoin inhibitors despite a decrease in hemozoin (E). Dashed vertical lines indicate parasite growth IC50. (C) Examples of hemozoin inhibitors include chloroquine (CQ), amodiaquine (AQ) and piperaquine (PPQ) while (F) mefloquine (MQ), lumefantrine (Lf) and atovaquone (Atov) are examples of non-hemozoin inhibitors. In these schematic graphs which illustrate expected trends, no quantities are shown on the right axes or on the left axes in B and E because the values are strongly dependent on the identity of the inhibitor. Hemozoin levels in untreated parasites are ≈95%.
Most, but not all compounds found to inhibit both parasite growth and β-hematin formation have been found to be hemozoin inhibitors when tested using the cellular heme fractionation assay. This strongly supports the hypothesis that these compounds act by inhibiting hemozoin formation, but also illustrates the importance of measuring inhibition in the parasite as a validation step. Further indirect support for the hemozoin inhibition hypothesis by 4-aminoquinoline antimalarials has come from proteomics studies that have found no protein targets in the parasite.44
High Throughput Screening
HTS for hemozoin inhibitors has utilized different β-hematin inhibition assays, often influenced by the contemporary ideas about mechanisms of hemozoin formation. The first such study was based on incorporation of radiolabeled heme onto preformed β-hematin crystals,45 consistent with an earlier paper suggesting that formation of hemozoin was autocatalytic.46 A later study made use of the Phiβ assay using 4.5 M acetate at 60 °C.47 These studies screened ≈100,000 and ≈16,000 compounds, respectively, yielding new β-hematin inhibitors, but a relatively disappointing fraction were active against malaria parasites (14% and 2.5%, respectively).
Later discovery of neutral lipid catalyzed β-hematin formation prompted development of a detergent mediated assay using Nonidet P40 (NP-40).48 Subsequent combination with aqueous pyridine for measuring unconverted hematin concluded in the screening of a 144,330 compound library.49 Here, 32% of the β-hematin inhibitors with varied scaffolds were active against parasites, with 15% thereof exhibiting nanomolar activity. This provided a trove of data, permitting subsequent development of Bayesian models to predict β-hematin inhibition and β-hematin-inhibiting antimalarial activity.50 When this model was used to rank 1510 US Food and Drug Administration (FDA) approved drugs, 17/32 of the top-ranked compounds had reported activity against malaria parasites and only 5/32 were reported inactive. For the bottom-ranked compounds, 25/32 had been reported inactive, with only one compound reported active against malaria parasites. Established hemozoin-inhibiting antimalarials, amodiaquine and chloroquine were ranked second and twenty-first, respectively. A subsequent investigation of 9/19 other drugs on the top 21 list revealed that eight were β-hematin inhibitors and five inhibited parasite growth. The hit rate for β-hematin inhibition among the eleven compounds for which experiments have been performed is thus 91% (10/11), while that for parasite growth inhibition is 64% (7/11). Three of the top 21 non-quinoline compounds, namely lapatinib, nilotinib and lomitapide, were found to have nanomolar IC50 values and little to no cross-resistance with chloroquine. They inhibited cellular hemozoin formation with the signature increase in cellular exchangeable heme and decrease in hemozoin.51
Benzamide, triarylimidazole and 2-phenylbenzimidazole scaffolds were among the most prominent in the NP-40 based HTS study (Figure 3).49b These were investigated in detail in three separate studies. In the first, symmetric N1,N3-diarylbenzene-1,3-dicarboxamides were found to be potent β-hematin inhibitors when the central benzene ring bore an electron withdrawing group or the two aryl rings were pyridyl rings with the N atom meta or para to the amide group. Activity against parasites increased with increasing lipophilicity, so the most potent compound with an IC50 of 600 nM and no cross-resistance with chloroquine was poorly water soluble.43a In a second study, triarylimidazoles proved intractable to modification, since substantial alterations to the original hit compounds tended to abolish activity. All had IC50 values >1 μM against cultured P. falciparum.43b The most potent compounds in both series were found to inhibit cellular hemozoin formation.
Figure 3.
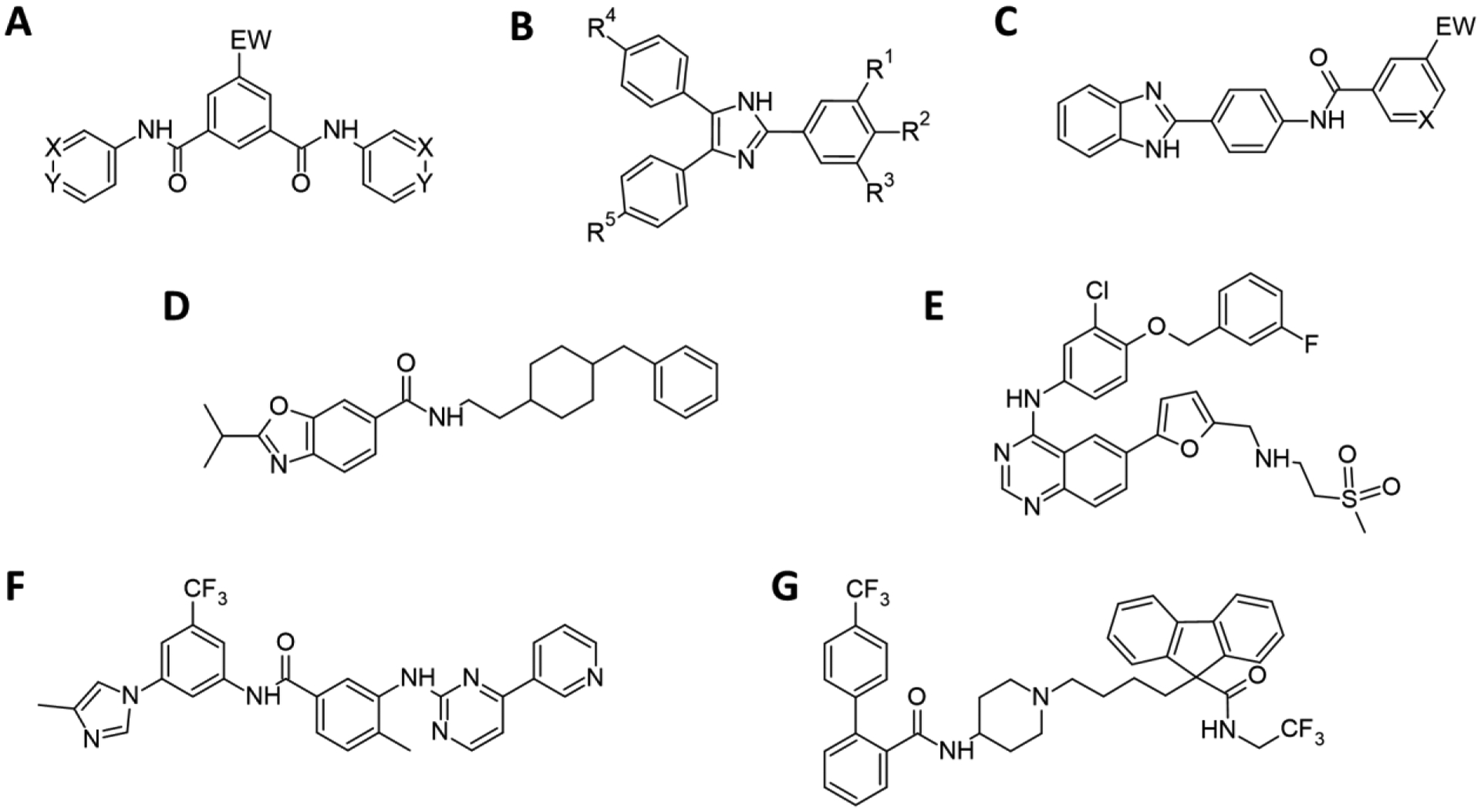
Scaffolds and compounds identified through HTS. (A) The symmetric N1,N3-diarylbenzene-1,3-dicarboxamide series demonstrated potent β-hematin inhibitory activity with either EW being an electron withdrawing group or with X or Y = N. (B) Active triarylimidazoles had R1 and R3 = OMe. R2 = OH favored activity, but if R2 = H, R4/R5 were required to be halogens or OH/OMe. (C) A series of 2-phenylbenzimidazoles identified through enumeration. For β-hematin inhibition EW = an electron withdrawing group was most favorable, as was X = N. The test compound used in subsequent electron energy loss and Raman spectroscopy studies contained EW = Br and X = N. (D) A benzoxazole with both β-hematin inhibition activity and activity against cultured parasites identified by virtual screening. USFDA approved drugs (E) lapatinib, (F) nilotinib, and (G) lomitapide highly ranked using a Bayesian approach, also found by virtual screening. All three were confirmed to be inhibitors of cellular hemozoin formation.
In a third study, benzimidazoles were synthesized from three building blocks, an o-phenylenediamine, a p-aminobenzoic acid and an aromatic carboxylic acid.43c All possible compounds based on 26 commercially available o-phenylenediamines, 24 p-aminobenzoic acids and 522 aromatic carboxylic acids were enumerated. The 325 728 candidate compounds were then filtered using the abovementioned Bayesian model for activity and for water solubility, reducing the list to 35 124 candidate compounds. Eventually, eighteen compounds were selected for further study. Sixteen (89%) had potent β-hematin inhibitory properties and all exhibited activity against cultured parasites, four (25%) with nanomolar activities. The best hits showed little to no cross-resistance with chloroquine and good selectivity against malaria parasites. The most active compound, with IC50=410 nM against the NF54 chloroquine sensitive parasite strain, inhibited cellular hemozoin formation. Unfortunately, this series also suffered from inadequate water solubility.
Mechanism of Inhibition of Hemozoin Formation
Heme was identified as a target of chloroquine and related drugs in 1980.52 Chou et al. attributed activity to complexation with heme, leading to membrane damage.13a Later these drugs were shown to inhibit β-hematin formation in the presence of parasite extracts, which was attributed to inhibition of a putative heme polymerase.18 Soon afterwards, we showed that they inhibit β-hematin formation directly in acetate medium, and proposed that this results from complexation.16a Later we found that the association constants between heme and quinine, an active inhibitor, and heme and inactive 9-epiquinine are almost identical, precluding complexation as the sole determinant of β-hematin inhibition.53
Improved understanding of β-hematin crystallization has advanced our knowledge of the mechanism of inhibition. Shortly after publication of the structure of β-hematin in 2000,15 Leiserowitz and co-workers proposed that inhibitors adsorb onto the corrugated fastest-growing (001) crystal face.37 They later showed that chloroquine and quinine caused tapered ends of the usually lath-like β-hematin crystals which they ascribed to inhibition of crystal growth.32
In 2013, we reported the first single crystal X-ray diffraction (SCD) structure of the β-hematin dimer as a DMSO solvate.29 The sample was grown in the presence of chloroquine, which we proposed acted as a growth-rate inhibitor, affording sufficiently large crystals for SCD analysis, although it is possible that it simply acted as a base, facilitating Fe-carboxylate bond formation. We found a decrease in the observed first-order rate constant for the formation of β-hematin in the presence of quinoline antimalarials. This occurred at low drug concentrations without a decrease in yield, while at higher concentrations there was a substantial reduction in final yield of β-hematin. We proposed reversible adsorption to free binding sites on the crystal surface at low concentration. The corresponding drug adsorption constant, Kads, was directly related to kinetic inhibition of lipid mediated β-hematin formation. At high concentration, we proposed that irreversible formation of a heme-drug complex as an insoluble precipitate accounts for decreased β-hematin yields. We later demonstrated a linear correlation between Kads and β-hematin inhibitory activity for both quinolines and benzamides as well as between Kads and parasite growth IC50 in the D10 chloroquine-sensitive strain.2
More recent atomic force microscopy measurements by Vekilov and co-workers in a citric acid/octanol model system has supported the “adsorption hypothesis”.38, 54 In particular, chloroquine and other antimalarials have been shown to adsorb onto the large, flat (100) crystal face impeding growth of new islands by step-pinning and kink-blocking. Adsorption was shown to be more efficient than heme-drug complex formation, accounting for inhibition of crystal growth at significantly lower concentrations. In 2019, Kapishnikov et al. used X-ray fluorescence microscopy to show that bromoquine, the bromine analogue of chloroquine, adsorbs onto hemozoin crystals in the parasite itself.55 Thus, current evidence strongly supports the hypothesis that hemozoin inhibiting drugs poison crystal growth via adsorption onto the crystal surface.
Heme-inhibitor Complexes
Inhibition of hemozoin formation causes a build-up of free heme, widely held to be responsible for parasite killing. This would imply that the toxicity of heme is independent of the inhibitor. Consequently, all hemozoin inhibitors should effect the same level of exchangeable heme at their respective IC50 values.4 In fact, diverse hemozoin inhibitors showed quite the opposite, with exchangeable heme varying markedly at the inhibitor IC50. Compounds with higher IC50 values often led to higher levels of exchangeable heme. For eleven active inhibitors, the amount of inhibitor accumulated in the parasite as determined by the inoculum effect, was found to correlate directly with exchangeable heme. These observations point to the formation of heme-inhibitor complexes in the parasite as the key to toxicity to the organism.
We recently provided the first direct evidence for the formation of a such a heme-inhibitor complex in an NF54 chloroquine-sensitive strain of P. falciparum.4 A bromo-substituted benzimidazole (Figure 3c) was selected for investigation owing to the high fraction of exchangeable heme it caused (≈25%). The low natural abundance of bromine in parasites was also advantageous for cellular imaging using electron energy loss spectroscopy (EELS). Signals for Fe and Br co-localized in the DV of parasitized RBCs. Given that heme accounts for ≥95% of iron in the parasite,11c the signal for Fe in the DV was attributed to both hemozoin and exchangeable heme. The co-localization of inhibitor with hemozoin is consistent with adsorption discussed above, while speciation of the exchangeable heme was initially less clear. The notion of a heme-inhibitor complex has been proposed by Kapishnikov et al. following the detection of bromoquine in the DV membrane of treated parasites.55 Using confocal Raman microscopy we could definitively identify a heme complex of the bromo-substituted benzimidazole. Principal component analysis showed that Raman peak positions from a region near the hemozoin in the DV were distinct from those of oxy- and deoxyhemoglobin, as well as hematin, hemin, hemozoin and free inhibitor. On the other hand, they were indistinguishable from those of a hemin-inhibitor mixture, corresponding to a heme-inhibitor complex. We propose that such complexes are responsible for parasite killing, either directly, by inhibiting enzyme targets or transporters in the DV or indirectly, by transferring free heme to the cytoplasm and effecting damage through the generation of reactive oxygen species. The importance of such complexation does not negate the fact that an inhibitor must first be able to inhibit hemozoin formation, likely by poisoning crystal growth (Figure 4). Thus, as discussed above, despite its strong association with heme, 9-epiquinine is inactive against β-hematin formation and has only very weak activity against parasite growth. Presumably, only following inhibition of hemozoin formation do exchangeable heme levels exceed basal levels, which then drives complexation.
Figure 4.
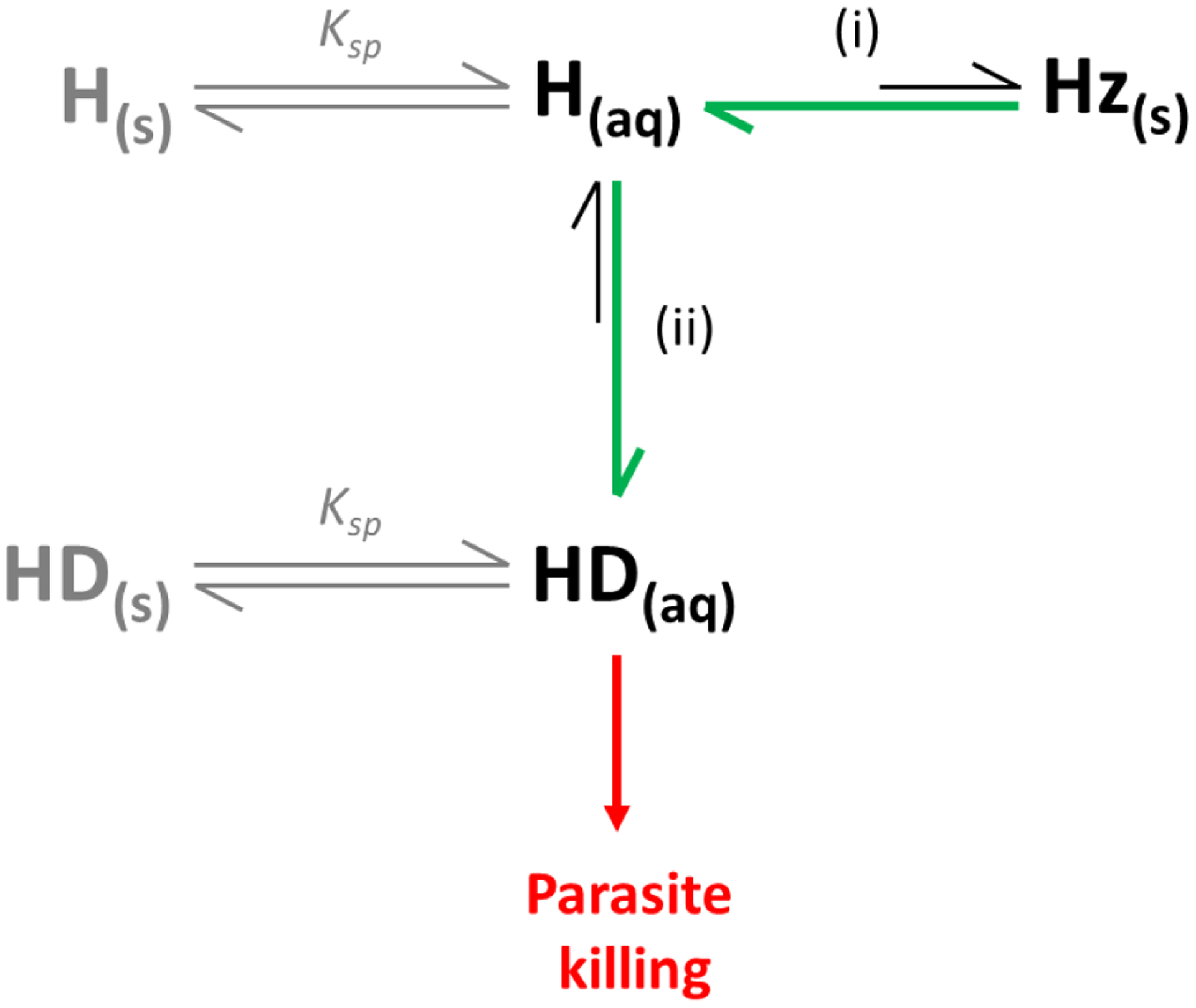
Proposed heme speciation in the presence of hemozoin inhibitors. Drugs targeting the hemozoin pathway (i) cause a decrease in hemozoin (Hz) formation via adsorption, resulting in a build-up of exchangeable heme (H(aq)). These drugs form complexes (HD(aq)) with free heme (ii), that are likely responsible for parasite killing (red arrow). Green arrows indicate the shift in equilibria in the presence of a Hz inhibitor. Precipitation of free heme (H(s)) and heme-drug complexes (HD(s)) represent competing processes (grey) that depend on their solubility products (Ksp).
To date, difficulty in isolating precipitates from the parasite milieu has precluded structure determination in situ. Attempts to obtain definitive solid-state structures of the heme-chloroquine complex have been unsuccessful. Consequently, numerous solution state and in silico studies have been conducted. The avid reader is directed to a review of the early contributions in this regard.56 Briefly, early 1H NMR studies of heme complexes of chloroquine and quinine based on a ring current model concluded that chloroquine intercalates between two heme molecules in a sandwich-type fashion. Based on the supposition that the μ-oxo dimer was the prevalent heme species in aqueous solution, a chloroquine-μ-oxo dimer complex was proposed, consistent with the 1:2 stoichiometry inherent in a sandwich structure.
In 2006, we showed that heme is a π-π dimer in aqueous solution,57 and that alternative species, namely the monomer, μ-oxo dimer, or even higher aggregates, may form in mixed solvents, at high salt concentration or as a function of pH.58 Molecular dynamics (MD) and time-resolved density functional theory (DFT) experiments, together with extended X-ray absorption fine structure (EXAFS) data, provided further insights into the electronics, structure and hydration of monomeric and dimeric heme in solution.59 We later demonstrated that chloroquine induces formation of the heme μ-oxo dimer in solution.60 MD simulations were carried out for two hypothetical structures, in which chloroquine was either stacked on the unligated face of the μ-oxo dimer or docked between the two oxo-bridged porphyrins.61 EXAFS, infrared and magnetic data supported the latter structure. Interestingly, a crystal structure of a Ga(III)protoporphyrin IX complex of chloroquine has been reported.62 In addition to π-stacking of the quinoline ring with the porphyrin, it was shown that the protonated quinoline N atom hydrogen bonds to an axial methoxide ligand on the metalloporphyrin. These spectroscopic and structural studies are indicative of specific stereochemical requirements for strong interactions in these complexes.
We reported the first SCD structure of a heme-halofantrine complex in 2008.63 Prior to recrystallization from a water/pyridine/acetone solution, the sample was collected from an aqueous-lipid emulsion, consistent with the prevailing proposal that lipid environments may serve as a site of drug (or heme-drug complex) action in the parasite.64 The complex was observed to involve coordination of the alkoxide group of halofantrine to the Fe center of heme. This was later also confirmed in structures of heme-quinine, quinidine,65 and mefloquine.66 In the latter study, EXAFS measurements of single crystals and solution samples indicated that this interaction persists in non-aqueous solution for all three aryl methanol drug complexes. The question of course remains whether these coordination complexes occur in parasitized red blood cells. Until answered, this presents a challenge to the rational design of new inhibitors to target heme detoxification.
Structure-activity and Docking Studies
Extensive studies of chloroquine analogues, both as parasite growth and β-hematin formation inhibitors has provided a well-developed structure activity relationship model for 4-aminoquinolines.67 The 4-aminoquinoline nucleus is the minimal substructure for strong heme complexation but does not inhibit β-hematin formation. 4-amino-7-chloroquinoline exhibits the latter activity but is not strongly active against parasites. This requires a basic amine-containing side chain. Electron withdrawing groups, such as CN, can replace the 7-Cl group while antiparasitic activity is related to the pKa of the compound, consistent with pH trapping in the acidic parasite DV.68 While not transferable to other scaffolds,43a this model has proven useful for designing new 4-aminoquinolines, such as the dibemequines. These contain a dibemethine side chain that chemosensitizes chloroquine-resistant strains of P. falciparum to chloroquine, thus serving a dual function.69 They are strongly active in both cultured parasites and mouse malaria and inhibit chloroquine transport by the P. falciparum chloroquine resistance transporter (PfCRT). The related pyridodibemequines are also active, but both series are metabolically unstable in human and mouse liver microsomes, precluding further development.70 Work is continuing on the metabolites that are themselves active.
Recently, we have explored molecular docking to discover new β-hematin inhibitors with activity against malaria parasites. A tranche of the ZINC15 database consisting of 7 070 compounds that were in stock for purchase, unreactive, neutral, with LogP<5 and molecular weight <450 were docked with β-hematin. This yielded 324 compounds with docking energies <−10 kcal/mol. These compounds were filtered to remove toxic liabilities (mutagenicity, tumorigenicity or irritant effects) and to comply with Lipinski’s rule of five. All remaining hits were visually inspected to confirm that they were suitably docked, and fifteen were selected for further investigation. Eleven (73%) inhibited β-hematin formation and nine (60%) inhibited growth of P. falciparum. The two most active compounds were investigated in greater detail. One showed excellent selectivity against malaria parasites over mammalian cells and inhibited hemozoin formation in the parasite. This benzoxazole derivative (Figure 3d) is the first hemozoin inhibiting compound active against malaria parasites to be discovered using molecular docking with the crystal surface.3 This approach was also applied to the USFDA approved drugs, identifying some of the compounds previously found using the Bayesian model, including lapatinib, nilotinib, lomitapide (Figure 3e–g) and chloroquine. The hit rate for both β-hematin inhibition and parasite growth inhibition was lower in this case (26%).51 Molecular docking is thus a promising technique for future identification of new hemozoin-inhibiting scaffolds.
Future Prospects
Hemozoin remains an important research topic in the malaria field despite widespread chloroquine resistance. Hemozoin is still a viable target for new drugs because chloroquine resistance arises from mutations in the pfcrt gene, which encodes a DV membrane transporter. Resistance is compound-specific and not linked to changes in hemozoin formation.71 Furthermore, widespread phenotypic screening for activity against malaria parasites subsequently necessitates target identification. Inevitably, hemozoin inhibitors are among the active compounds discovered. Consequently, there is a need for improved understanding of hemozoin formation and inhibition, including resolving the role of lipids and proteins in these processes.
Substantial progress is still required on several fronts. Firstly, to identify a hemozoin inhibitor, it is not adequate to demonstrate only β-hematin inhibition. Rather, it is necessary to show inhibition of hemozoin formation in the parasite itself. The current method is cumbersome, consumes considerable parasite material and uses toxic pyridine to complex heme.41a There is thus a need for improved assays. Development of liquid chromatography methods with colorimetric or mass spectrometric detection that use much smaller samples, preferably involving 96-well plates, would represent a major advance in this area. This would improve throughput and make the assay more accessible to many medicinal chemistry laboratories. Secondly, predicting β-hematin inhibition would assist in selection of molecules for medicinal chemistry campaigns. Here, molecular docking with hemozoin, which has already proven useful, could be expanded and improved. Additionally, methods representing chemical space in reduced dimensions, for example principal component analysis, may permit enrichment of in silico libraries with compounds active against specific targets, whether hemozoin or enzymes. Machine learning methods are soon also likely to assist in this process as more screening data become available. Thirdly, a better understanding of the overall hemozoin formation pathway and effects of inhibition of specific targets along this pathway could assist in target deconvolution based on changes in cellular hemozoin formation. This is a crucial consideration since there are several points along the pathway that can be inhibited, resulting in changes in hemozoin formation. Not all necessarily involve a direct interaction with heme or hemozoin. Considerable advances in all these areas are likely in the next few years.
Lastly, the recent discovery that hemozoin inhibitors probably act as heme complexes, returns the spotlight to these complexes. Key questions are the strengths of association of heme and inhibitor in aqueous and lipid environments, solubilities, lipophilicities and structures of these complexes. These are all challenging problems because of complicated speciation, low solubilities at DV pH and heme aggregation. Furthermore, the paramagnetic nature of ferriheme reduces the utility of techniques such as NMR. Innovative approaches will therefore be needed to address these challenges.
ACKNOWLEDGMENTS
The work in our laboratories discussed in this article has been supported by generous funding from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health in the form of successive grants in the IRIDA program (R01AI083145, R01AI110329 and R01AI143521). It has also been supported by the National Research Foundation of South Africa through competitive grants for rated researchers (grant numbers 73901, 91516 and 111696) and the Thuthuka Programme (87962) and self-initiated grants from the South African Medical Research Council. We also acknowledge the many students and collaborators who have worked with us and apologize to authors whose work we have not cited due to limitations of space. We acknowledge Dr Tebogo Mabotha, Department of Chemistry, University of Cape Town and Prof. Delia Haynes, Department of Chemistry and Polymer Science, Stellenbosch University for the TEM and β-hematin structure shown in Figure 1B and D, respectively.
Biographies
Katherine de Villiers was born in Bulawayo, Zimbabwe. She received her Ph.D. in chemistry in 2008 from the University of Cape Town, South Africa. She joined Stellenbosch University in 2009, where she is currently a senior lecturer in the Department of Chemistry and Polymer Science. She leads the “HaemTeamSU” research group, where her interests are in antimalarial drug development for both enzyme and non-enzyme (hemozoin) targets.
Timothy J. Egan was born in Johannesburg South Africa. He received his Ph.D. in bioinorganic chemistry in 1988 from the University of the Witwatersrand in Johannesburg, South Africa. He was a postdoctoral research associate at the Albert Einstein College of Medicine in Bronx, New York from 1991 – 1993. He joined the University of Cape Town in 1996, where he is currently Jamison Professor of Inorganic Chemistry and an affiliate member of the Institute of Infectious Disease and Molecular Medicine. He leads the bioinorganic research group, where his interests center on hemozoin inhibiting antimalarials and antimalarial drug mechanisms.
Footnotes
The authors declare no competing financial interests.
Key References
- Combrinck JM; Mabotha TE; Ncokazi KK; Ambele MA; Taylor D; Smith PJ; Hoppe HC; Egan TJ, Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol 2013, 8, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1 This paper describes the effects of chloroquine on intracellular hemozoin formation in the malaria parasite Plasmodium falciparum, demonstrating inhibition of hemozoin formation, an increase in exchangeable heme, a relocation of heme iron into the parasite cytoplasm and disruption of hemozoin crystal growth.
- Fitzroy S-M; Gildenhuys J; Olivier T; Tshililo NO; Kuter D; de Villiers KA, The effects of quinoline and non-quinoline inhibitors on the kinetics of lipid-mediated β-hematin crystallization. Langmuir 2017, 33, 7529–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2 We demonstrated a significant correlation between the strength of adsorption of an inhibitor to β-hematin (Kads) and IC50 in a biomimetic system, which provides strong support for the adsorption hypothesis with respect to hemozoin inhibition.
- de Sousa ACC; Combrinck JM; Maepa K; Egan TJ, Virtual screening as a tool to discover new β-haematin inhibitors with activity against malaria parasites. Sci. Rep 2020, 10, 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]; 3 In this paper, we use molecular docking to discover new hemozoin inhibiting compounds from an in silico library, demonstrate their activity against β-hematin formation, identify a sub-set active against P. falciparum and show that they inhibit intracellular hemozoin formation with the signature increase in exchangeable heme.
- Openshaw R; Maepa K; Benjamin SJ; Wainwright L; Combrinck JM; Hunter R; Egan TJ, A diverse range of hemozoin inhibiting scaffolds act on Plasmodium falciparum as heme complexes. ACS Infect. Dis 2021, 7, 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]; 4 In this paper we conclude that heme-inhibitor complexes are central to parasiticidal activity of antimalarial drugs. We found a 1:1 correlation between amounts of free heme and accumulated inhibitor and confirmed the presence of a heme-inhibitor complex in parasites.
References
- 1.Combrinck JM; Mabotha TE; Ncokazi KK; Ambele MA; Taylor D; Smith PJ; Hoppe HC; Egan TJ, Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem. Biol 2013, 8, 133–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fitzroy S-M; Gildenhuys J; Olivier T; Tshililo NO; Kuter D; de Villiers KA, The effects of quinoline and non-quinoline inhibitors on the kinetics of lipid-mediated β-hematin crystallization. Langmuir 2017, 33, 7529–7537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Sousa ACC; Combrinck JM; Maepa K; Egan TJ, Virtual screening as a tool to discover new β-haematin inhibitors with activity against malaria parasites. Sci. Rep 2020, 10, 3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Openshaw R; Maepa K; Benjamin SJ; Wainwright L; Combrinck JM; Hunter R; Egan TJ, A diverse range of hemozoin inhibiting scaffolds act on Plasmodium falciparum as heme complexes. ACS Infect. Dis 2021, 7, 362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter R; Mendis KN, Evolutionary and historical aspects of the burden of malaria. Clin. Microbiol. Rev 2002, 15, 564–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slater AFG, Chloroquine: mechanism of drug action and resistance in Plasmodium falciparum. Pharmacol. Ther 1993, 57, 203–235. [DOI] [PubMed] [Google Scholar]
- 7.WHO World malaria report; World Health Organisation: 2019; p 142.
- 8.White NJ, Antimalarial drug resistance and combination chemotherapy. Philos. Trans. R. Soc. Lond. B Biol. Sci 1999, 354, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dondorp AM; Nosten F; Yi P; Das D; Phyo AP; Tarning J; Lwin KM; Ariey F; Hanpithakpong W; Lee SJ; Ringwald P; Silamut K; Imwong M; Chotivanich K; Lim P; Herdman T; An SS; Yeung S; Singhasivanon P; Day NP; Lindegardh N; Socheat D; White NJ, Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 2009, 361, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherman IW, A brief history of malaria and discovery of the parasites’s life cycle. In Malaria: Parasite biology, pathogenesis and protection, Sherman IW, Ed. ASM Press: Washington D.C., 1998; pp 3–10. [Google Scholar]
- 11.(a) Wood BR; Langford SJ; Cooke BM; Glenister FK; Lim J; McNaughton D, Raman imaging of hemozoin within the food vacuole of Plasmodium falciparum trophozoites. FEBS Lett. 2003, 554, 247–252; [DOI] [PubMed] [Google Scholar]; (b) Sienkiewicz A; Krzystek J; Vileno B; Chatain G; Kosar AJ; Bohle DS; Forró L, Multi-frequency high-field EPR study of iron centers in malarial pigments. J. Am. Chem. Soc 2006, 128, 4534–4535; [DOI] [PubMed] [Google Scholar]; (c) Egan TJ; Combrinck JM; Egan J; Hearne GR; Marques HM; Ntenteni S; Sewell BT; Smith PJ; Taylor D; van Schalkwyk DA; Walden JC, Fate of haem iron in the malaria parasite Plasmodium falciparum. Biochem. J 2002, 365, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strictly speaking heme refers to ferroprotoporphyrin IX, however since details of oxidation state and speciation in the parasite are largely unknown, we have used the term heme generically to refer to both ferro- and ferriprotoporphyrin IX. There is no doubt that it is in the ferric state in cell-free studies discussed herein, as well as in the case of hemozoin and in observed inhibitor complexes. On the other hand, the speciation of exchangeable heme in the parasite is not definitively known. It should also be noted that some authors refer to β-hematin as hematin anhydride.
- 13.(a) Chou AC; Fitch CD, Hemolysis of mouse erythrocytes by ferriprotoporphyrin IX and chloroquine. Chemotherapeutic implications. J. Clin. Invest 1980, 66, 856–858; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar S; Bandyopadhyay U, Free heme toxicity and its detoxification systems in humans. Toxicol. Lett 2005, 157, 175–188. [DOI] [PubMed] [Google Scholar]
- 14.Sigala PA; Crowley JR; Hseih S; Henderson JP; Goldberg DE, Direct tests of enzymatic heme degradation by the malaria parasite Plasmodium falciparum. J. Biol. Chem 2012, 287, 37793–37807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagola S; Stephens PW; Bohle DS; Kosar AD; Madsen SK, The structure of malaria pigment (β-haematin). Nature 2000, 404, 307–310. [DOI] [PubMed] [Google Scholar]
- 16.(a) Egan TJ; Ross DC; Adams PA, Quinoline anti-malarials inhibit spontaneous formation of ß-haematin (malaria pigment). FEBS Lett. 1994, 352, 54–57; [DOI] [PubMed] [Google Scholar]; (b) Hawley SR; Bray PG; Mungthin M; Atkinson JD; O’Neill PM; Ward SA, Relationship between antimalarial drug activity, accumulation, and inhibition of heme polymerisation in Plasmodium falciparum in vitro. Antimicrob. Agents Chemother 1998, 42, 682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Fitch CD; Kanjananggulpan P, The state of ferriprotoporphyrin IX in malaria pigment. J. Biol. Chem 1987, 262, 15552–15555; [PubMed] [Google Scholar]; (b) Slater AFG; Swiggard WJ; Orton BR; Flitter WD; Goldberg DE; Cerami A; Henderson GB, An iron-carboxylate bond links the heme units of malaria pigment. Proc. Natl. Acad. Sci. USA 1991, 88, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slater AFG; Cerami A, Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature 1992, 355, 167–169. [DOI] [PubMed] [Google Scholar]
- 19.(a) Sullivan DJJ; Gluzman IY; Goldberg DE, Plasmodium haemozoin formation mediated by histidine-rich proteins. Science 1996, 271, 219–222; [DOI] [PubMed] [Google Scholar]; (b) Papalexis V; Siomos M-A; Campanale N; Guo X-G; Kocak G; Foley M; Tilley L, Histidine-rich protein 2 of the malaria parasite, Plasmodium falciparum, is involved in detoxification of the by-products of haemoglobin degradation. Mol. Biochem. Parasitol 2001, 115, 77–86. [DOI] [PubMed] [Google Scholar]
- 20.Jani D; Nagarkatti R; Beatty W; Angel R; Slebodnick C; Andersen J; Kumar S; Rathore D, HDP-A novel haem detoxification protein from the malaria parasite. PLoS Pathog. 2008, 4, e1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matz JM; Drepper B; Blum TB; van Genderen E; Burrel A; Martin P; Stach T; Collinson LM; Abrahams JP; Matuschewski K; Blackman MJ, A lipocalin mediates unidirectional heme biomineralization in malaria parasites. Proc. Natl. Acad. Sci 2020, 117, 16546–16556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendrat K; Berger BJ; Cerami A, Haem polymerisation in malaria. Nature 1995, 378, 138–139. [DOI] [PubMed] [Google Scholar]
- 23.Fitch CD; Cai G.-z.; Chen Y-F; Shoemaker JD, Involvement of lipids in ferriprotoporphyrin IX polymerisation in malaria. Biochim. Biophys. Acta 1999, 1454, 31–37. [DOI] [PubMed] [Google Scholar]
- 24.(a) Dorn A; Stoffel R; Matile H; Bubendorf A; Ridley RG, Malarial haemozoin/ß-haematin supports haem polymerisation in the absence of protein. Nature 1995, 374, 269–271; [DOI] [PubMed] [Google Scholar]; (b) Dorn A; Vippagunta SR; Matile H; Bubendorf A; Vennerstrom JL; Ridley RG, A comparison and analysis of several ways to promote haematin (haem) polymerisation and an assessment of its initiation in vitro. Biochem. Pharmacol 1998, 55, 737–747. [DOI] [PubMed] [Google Scholar]
- 25.(a) Egan TJ; Mavuso WW; Ncokazi KK, The mechanism of ß-haematin formation in acetate solution. Parallels between haemozoin formation and biomineralisation processes. Biochemistry 2001, 40, 204–213; [DOI] [PubMed] [Google Scholar]; (b) Egan TJ; Tshivase MG, Kinetics of ß-haematin formation from suspensions of haematin in aqueous benzoic acid. Dalton Trans. 2006, 5024–5032. [DOI] [PubMed] [Google Scholar]
- 26.Egan TJ; Chen JY-J; de Villiers KA; Mabotha TE; Naidoo KJ; Ncokazi KK; Langford SJ; McNaughton D; Pandiancherri S; Wood BR, Haemozoin (ß-haematin) biomineralisation occurs by self assembly near the lipid/water interface. FEBS Lett. 2006, 580, 5105–5110. [DOI] [PubMed] [Google Scholar]
- 27.Huy NT; Maeda A; Uyen DT; Trang DTX; Sasai M; Shiona T; Oida T; Harada S; Kamei K, Alcohols induce beta-haematin formation via the dissociation of aggregated haem and reduction in interfacial tension of the solution. Acta Trop. 2007, 101, 130–138. [DOI] [PubMed] [Google Scholar]
- 28.Sandlin RD; Fong KY; Stiebler R; Gulka CP; Nesbitt JE; Oliveira MP; Oliveira MF; Wright DW, Detergent-mediated formation of β-hematin: Heme crystallization promoted by detergents implicates nanostructure formation for use as a biological mimic. Cryst. Growth Des 2016, 16, 2542–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gildenhuys J; le Roex T; Egan TJ; de Villiers KA, The single crystal X-ray structure of β-hematin DMSO solvate grown in the presence of chloroquine, a β-hematin growth-rate inhibitor. J. Am. Chem. Soc 2013, 135, 1037–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pisciotta JM; Coppens I; Tripathi AK; Scholl PF; Shuman J; Bajad S; Shulaev V; Sullivan DJ Jr, The role of neutral lipid nanospheres in Plasmodium falciparum heme crystallisation. Biochem. J 2007, 402, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapishnikov S; Weiner A; Shimoni E; Guttmann P; Schneider G; Dahan-Pasternak N; Dzikowski R; Leiserowitz L; Elbaum M, Oriented nucleation of hemozoin at the digestive vacuole membrane in Plasmodium falciparum. Proc. Natl. Acad. Sci 2012, 109, 11188–11193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Solomonov I; Osipova M; Feldman Y; Baehtz C; Kjaer K; Robinson IK; Webster GT; McNaughton D; Wood BR; Weissbuch I; Leiserowitz L, Crystal nucleation, growth, and morphology of the synthetic malaria pigment ß-haematin and the effect thereon by quinoline additives: The malaria pigment as a target of various antimalarial drugs. J. Am. Chem. Soc 2007, 129, 2615–2627. [DOI] [PubMed] [Google Scholar]
- 33.de Villiers KA; Osipova M; Mabotha TE; Solomonov I; Feldman Y; Kjaer K; Weissbuch I; Egan TJ; Leiserowitz L, Oriented nucleation of beta-hematin crystals induced at various interfaces: Relevance to haemozoin formation. Cryst. Growth Des 2009, 9 (1), 626–632. [Google Scholar]
- 34.(a) Hoang AN; Ncokazi KK; de Villiers KA; Wright DW; Egan TJ, Crystallization of synthetic haemozoin (beta-haematin) nucleated at the surface of lipid particles. Dalton Trans. 2010, 39, 1235–1244; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoang AN; Sandlin RD; Omar A; Egan TJ; Wright DW, The neutral lipid composition present in the digestive vacuole of Plasmodium falciparum concentrates heme and mediates β-hematin formation with an unusually low acticvation energy. Biochemistry 2010, 49, 10107–10116; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ambele MA; Sewell BT; Cummings FR; Smith PJ; Egan TJ, Synthetic Hemozoin (β-Hematin) Crystals Nucleate at the Surface of Neutral Lipid Droplets that Control Their Sizes. Cryst. Growth Des 2013, 13, 4442–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambele MA; Egan TJ, Neutral lipids associated with haemozoin mediate efficient and rapid β-haematin formation at physiological pH, temperature and ionic composition. Malar. J 2012, 11 (337). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuter D; Mohunlal R; Fitzroy S-M; Asher C; Smith PJ; Egan TJ; de Villiers KA, Insights into the initial stages of lipid-mediated haemozoin nucleation Cryst. Eng. Comm 2016, 18, 5177–5187. [Google Scholar]
- 37.Buller R; Peterson ML; Almarsson Ö; Leiserowitz L, Quinoline binding site on malaria pigment crystal: A rational pathway for antimalarial drug design. Cryst. Growth Des 2002, 2, 553–562. [Google Scholar]
- 38.Olafson KN; Ketchum MA; Rimer JD; Vekilov PG, Mechanisms of hematin crystallization and inhibition by the antimalarial drug chloroquine. Proc. Natl. Acad. Sci 2015, 112, 4946–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ncokazi KK; Egan TJ, A colorimetric high-throughput β-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal. Biochem 2005, 338, 306–319. [DOI] [PubMed] [Google Scholar]
- 40.Morrison DB; Williams EFJ, The solubility and titration of hemin and ferrihemic acid. J. Biol. Chem 1941, 137, 461–473. [Google Scholar]
- 41.(a) Combrinck JM; Fong KY; Gibhard L; Smith PJ; Wright DW; Egan TJ, Optimization of a multi-well colorimetric assay to determine haem species in Plasmodium falciparum in the presence of anti-malarials. Malaria J. 2015, 14, e253; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dinghra SK; Redhi D; Combrinck JM; Yeo T; Okombo J; Henrich PP; Cowell AN; Gupta P; Stegman ML; Hoke JM; Cooper RA; Winzeler E; Mok S; Egan TJ; Fidock DA, A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. mBio 2017, 8, e00303–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanaerschot M; Lucatoni L; Li T; Combrinck JM; Ruecker A; Tiruppadiripuliyur SK; Rubiano K; Ferreira PE; Siciliano G; Gulati S; Henrich PP; Ng CL; Murithi JM; Corey VC; Duffy S; Lieberman OJ; Sinden RE; Alano P; Delves MJ; Sim KL; Winzeler EA; Egan TJ; Hoffman SL; Avery VM; Fidock DA, Identifying hexahydroquinolines as new antimalarial candidates with potent blood stage and transmission-blocking activity. Nat. Microbiol 2017, 2, 1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.(a) Wicht KJ; Combrinck JM; Smith PJ; Hunter R; Egan TJ, Identification and SAR evaluation of hemozoin-inhibiting benzamides active against Plasmodium falciparum. J. Med. Chem 2016, 59, 6512–6530; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wicht KJ; Combrinck JM; Smith PJ; Hunter R; Egan TJ, Identification and mechanistic evaluation of hemozoin-inhibiting triarylimidazoles active against Plasmodium falciparum. ACS Med. Chem. Lett 2017, 8, 201–205; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) L’abbate FP; Müller R; Openshaw R; Combrinck JM; de Villiers KA; Hunter R; Egan TJ, Hemozoin inhibiting 2-phenylbenzimidazoles active against malaria parasites. Eur. J. Med. Chem 2018, 159, 243–254; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fong KY; Sandlin RD; Wright DW, Identification of β-hematin inhibitors in the MMV Malaria Box. Int. J. Parasitol. Drugs Drug Resist 2015, 5, 84–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.(a) Graves PR; Kwiek JJ; Fadden P; Ray R; Hardeman K; Coley AM; Foley M; Haystead TAJ, Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol. Pharmacol 2002, 62, 1364–1372; [DOI] [PubMed] [Google Scholar]; (b) Woodland J; Hunter R; Smith PJ; Egan TJ, Chemical proteomics and super-resolution imaging reveal that chloroquine interacts with Plasmodium falciparum multidrug resistance-associated protein and lipids. ACS Chem. Biol 2018, 13, 2939–2948. [DOI] [PubMed] [Google Scholar]
- 45.Kurosawa Y; Dorn A; Kitsuji-Shirane M; Shimada H; Satoh T; Matile H; Hofheinz W; Masciadri R; Kansy M; Ridley RG, Hematin polymerization assay as a high-throughput screen for identification of new antimalarial pharmacophores. Antimicr. Agents Chemother 2000, 44, 2638–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorn A; Stoffel R; Matile H; Bubendorf A; Ridley RG, Malarial haemozoin/β-haematin supports haem polymerization in the absence of protein. Nature 1995, 374, 269–271. [DOI] [PubMed] [Google Scholar]
- 47.Rush MA; Baniecki ML; Mazitschek R; Cortese JF; Wiegand R; Clardy J; Wirth DF, Colorimetric high-throughput screen for detection of heme crystallization inhibitors. Antimicr. Agents Chemother 2009, 53, 2564–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carter MD; Phelan VV; Sandlin RD; Bachmann BO; Wright DW, Lipophilic mediated assays for β-hematin inhibitors. Combinatorial Chem. High Througput Scr 2010, 13, 285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(a) Sandlin RD; Carter MD; Lee PJ; Auschwitz JM; Leed SE; Johnson JD; Wright DW, Use of the NP-40 detergent-mediated assay in discovery of inhibitors of β−hematin crystallization. Antimicr. Agents Chemother 2011, 55, 3363–3369; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sandlin RD; Fong KY; Wicht KJ; Carrell HM; Egan TJ; Wright DW, Indentification of β-hematin inhibitors in a high-throughput screening effort reveals scaffolds with in vitro antimalarial activity. Int. J. Parasitol. Drugs Drug Resist 2014, 4, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wicht KJ; Combrinck JM; Smith PJ; Egan TJ, Bayesian models trained with HTS data for predicting β-haematin inhibition and in vitro antimalarial activity. Bioorg. Med. Chem 2015, 23, 5210–5217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Sousa ACC; Maepa K; Combrinck JM; Egan TJ, Lapatinib, nilotinib and lomitapide inhibit haemozoin formation in malaria parasites. Molecules 2020, 25, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chou AC; Chevli R; Fitch CD, Ferriprotoporphyrin IX fulfills the criteria for identification as the chloroquine receptor of malaria parasites. Biochemistry 1980, 19, 1543–1549. [DOI] [PubMed] [Google Scholar]
- 53.Egan TJ; Mavuso WW; Ross DC; Marques HM, Thermodynamic factors controlling the interaction of quinoline antimalarial drugs with ferriprotoporphyrin IX. J. Inorg. Biochem 1997, 68, 137–145. [DOI] [PubMed] [Google Scholar]
- 54.Olafson KN; Nguyen TQ; Rimer JD; Vekilov PG, Antimalarials inhibit hematin crystallization by unique drug–surface site interactions. Proc. Natl. Acad. Sci 2017, 114, 7531–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kapishnikov S; Staalsø T; Yang Y; Lee J; Pérez-Berná AJ; Pereiro E; Yang Y; Werner S; Guttmann P; Leiserowitz L; Als-Nielsen J, Mode of action of quinoline antimalarial drugs in red blood cells infected by Plasmodium falciparum revealed in vivo. Proc. Natl. Acad. Sci 2019, 116, 22946–22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Egan TJ, Interactions of quinoline antimalarials with hematin in solution. J. Inorg. Biochem 2006, 100, 916–926. [DOI] [PubMed] [Google Scholar]
- 57.de Villiers KA; Kaschula CH; Egan TJ; Marques HM, Speciation and structure of ferriprotoporphyrin IX in aqueous solution: spectroscopic and diffusion measurements demonstrate dimerization, but not μ-oxo dimer formation. J. Biol. Inorg. Chem 2007, 12, 101–117. [DOI] [PubMed] [Google Scholar]
- 58.Asher C; De Villiers KA; Egan TJ, Speciation of ferriprotoporphyrin IX in aqueous and mixed aqueous solution is controlled by solvent identity, pH, and salt concentration. Inorg. Chem 2009, 48, 7994–8003. [DOI] [PubMed] [Google Scholar]
- 59.(a) Kuter D; Venter GA; Naidoo KJ; Egan TJ, Experimental and Time-Dependent Density Functional Theory Characterization of the UV−Visible Spectra of Monomeric and μ-Oxo Dimeric Ferriprotoporphyrin IX. Inorg. Chem 2012, 51, 10233–10250; [DOI] [PubMed] [Google Scholar]; (b) Kuter D; Streltsov V; Davydova N; Venter GA; Naidoo KJ; Egan TJ, Molecular structures and solvation of free monomeric and dimeric ferriheme in aqueous solution: Insights from molecular dynamics simulations and extended X-ray absorption fine structure spectroscopy. Inorg. Chem 2014, 53, 10811–10824. [DOI] [PubMed] [Google Scholar]
- 60.Kuter D; Benjamin SJ; Egan TJ, Multiple spectroscopic and magnetic techniques show that chloroquine induces formation of the μ-oxo dimer of ferriprotoporphyrin IX. J Inorg Biochem 2014, 133, 40–49. [DOI] [PubMed] [Google Scholar]
- 61.Kuter D; Streltsov V; Davydova N; Venter GA; Naidoo KJ; Egan TJ, Solution structures of chloroquine–ferriheme complexes modeled using MD simulation and investigated by EXAFS spectroscopy. J. Inorg. Biochem 2016, 154, 114–125. [DOI] [PubMed] [Google Scholar]
- 62.Dodd EL; Bohle DS, Orienting the heterocyclic periphery: a structural model for chloroquine’s antimalarial activity. Chem. Commun 2014, 50, 13765–13768. [DOI] [PubMed] [Google Scholar]
- 63.de Villiers KA; Marques HM; Egan TJ, The crystal structure of halofantrine-ferriprotoporphyrin IX and the mechanism of action of arylmethanol antimalarials. J. Inorg. Biochem 2008, 102, 1660–1667. [DOI] [PubMed] [Google Scholar]
- 64.Pisciotta JM; Coppens I; Tripathi AK; Scholl PF; Shuman J; Bajad S; Shulaev V; Sullivan DJ, The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem. J 2007, 402, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Villiers KA; Gildenhuys J; le Roex T, Iron(III) protoporphyrin IX complexes of the antimalarial Cinchona alkaloids quinine and quinidine. ACS Chem. Biol 2012, 7, 666–671. [DOI] [PubMed] [Google Scholar]
- 66.Gildenhuys J; Sammy CJ; Müller R; Streltsov VA; le Roex T; Kuter D; de Villiers KA, Alkoxide coordination of iron(III) protoporphyrin IX by antimalarial quinoline methanols: a key interaction observed in the solid-state and solution. Dalton Trans. 2015, 44, 16767–16777. [DOI] [PubMed] [Google Scholar]
- 67.(a) Vippagunta SR; Dorn A; Matile H; Bhattacharjee AK; Karle JM; Ellis WY; Ridley RG; Vennerstrom JL, Structural specificity of chloroquine-hematin binding related to inhibition of hematin polymerization and parasite growth. J. Med. Chem 1999, 42, 4630–4639; [DOI] [PubMed] [Google Scholar]; (b) Egan TJ; Hunter R; Kaschula CH; Marques HM; Misplon A; Walden JC, Structure-function relationships in aminoquinolines: effect of amino and chloro groups on quinoline-hematin complex formation, inhibition of β-hematin formation, and antiplasmodial activity. J. Med. Chem 2000, 43, 283–291. [DOI] [PubMed] [Google Scholar]
- 68.Kaschula CH; Egan TJ; Hunter R; Basilico N; Parapini S; Taramelli D; Pasini E; Monti D, Structure-activity relationships in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. J. Med. Chem 2002, 45, 3531–3539. [DOI] [PubMed] [Google Scholar]
- 69.(a) Zishiri VK; Hunter R; Smith PJ; Taylor D; Summers G; Kirk K; Martin RE; Egan TJ, A series of structurally simple chloroquine chemosensitizing dibemethin derivatives that inhibit chloroquine transport by PfCRT. Eur. J. Med. Chem 2011, 46, 1729–1742; [DOI] [PubMed] [Google Scholar]; (b) Zishiri VK; Joshi MC; Hunter R; Chibale K; Smith PJ; Summers RL; Martin RE; Egan TJ, Quinoline antimalarials containing a dibemethin group are active against chloroquinone-resistant Plasmodium falciparum and inhibit chloroquine transport via the P. falciparum chloroquine-resistance transporter (PfCRT). J. Med. Chem 2011, 54, 6956–6968. [DOI] [PubMed] [Google Scholar]
- 70.Joshi MC; Okombo J; Nsumiwa S; Ndove J; Taylor D; Wiesner L; Hunter R; Chibale K; Egan TJ, 4-Aminoquinoline antimalarials containing a benzylmethylpyridylmethylamine group are active against drug resistant Plasmodium falciparum and exhibit oral activity in mice J. Med. Chem 2017, 60, 10245–10256. [DOI] [PubMed] [Google Scholar]
- 71.Summers RL; Nash MN; Martin RE, Know your enemy: understanding the role of PfCRT in drug resistance could lead to new antimalarial tactics. Cell. Mol. Life Sci 2012, 69, 1967–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]