

Abstract
Pathogenic germline variants in DMD gene, which encodes the well-known cytoskeletal protein named dystrophin, are associated with a wide range of dystrophinopathies disorders, such as Duchenne muscular dystrophy (DMD, severe form), Becker muscular dystrophy (BMD, mild form) and intermediate muscular dystrophy (IMD). Muscle biopsy, immunohistochemistry, molecular (multiplex ligation-dependent probe amplification (MLPA)/next-generation sequencing (NGS) and Sanger methods) and in silico analyses were performed in order to identify alterations in DMD gene and protein in a patient with a clinical manifestation and with high creatine kinase levels. Herein, we described a previously unreported intronic variant in DMD and reduced dystrophin staining in the muscle biopsy. This novel DMD variant allele, c.9649+4A>T that was located in a splice donor site within intron 66. Sanger sequencing analysis from maternal DNA showed the presence of both variant c.9649+4A>T and wild-type (WT) DMD alleles. Different computational tools suggested that this nucleotide change might affect splicing through a WT donor site disruption, occurring in an evolutionarily conserved region. Indeed, we observed that this novel variant, could explain the reduced dystrophin protein levels and discontinuous sarcolemmal staining in muscle biopsy, which suggests that c.9649+4A>T allele may be re-classified as pathogenic in the future. Our data show that the c.9649+4A>T intronic sequence variant in the DMD gene may be associated with an IMD phenotype and our findings reinforce the importance of a more precise diagnosis combining muscle biopsy, molecular techniques and comprehensive in silico approaches in the clinical cases with negative results for conventional genetic analysis.
Key words: DMD gene, muscular dystrophy, dystrophinopathies, intronic sequence variant
Introduction
Dystrophinopathies are X-linked recessive disorders associated with pathogenic variants in the DMD gene (OMIM # 300377) which result in abnormal synthesis of dystrophin protein, a cytoskeletal protein with a major structural role in muscle 1. These disorders lead to muscle weakness and progressive degeneration of muscle function 1,2. Although most of the pathogenic variants in DMD gene are large rearrangements, it is estimated that 25-35% of affected patients have small-scale sequence variants affecting the dystrophin structure and/or function 3. These disorders are progressive neuromuscular commonly diagnosed between the ages of 2 and 6 years due to delay in walking, unsteady gait, repeated stumbling, frequent falls, and difficulty at climbing stairs as well as increased levels of serum creatine kinase (CK) 4.
The functional impact of genetic variants is believed to be the main driver of variability in clinical manifestations. Based on that, these disorders can be classified as Duchenne muscular dystrophy (DMD; OMIM #310200), when a patient presents the severe form or Becker muscular dystrophy (BMD; OMIM #300376) and intermediate muscular dystrophy (IMD), both characterized by early-onset, and by milder forms caused by a partially functional dystrophin 5.
Several clinical cases with an IMD phenotype have recently been reported in the literature mainly due to widely spread, availability and cost reduction of genetic analysis such as multiplex ligation-dependent probe amplification (MLPA) and next-generation sequencing (NGS) techniques 6. These molecular methods provide precise and early diagnosis combined with the microscopic study of invasive muscle biopsy 7. Also, advances in NGS technologies have allowed improvements in molecular diagnosis and identification of new sequence variants in gene regions not previously evaluated, such as intronic alterations which constitute less than 0.5% of the currently reported causative variants but their value is presumably underestimated in dystrophinopathies 8.
In this study, we described a novel point variant in intron 66 of the DMD gene in a patient with intermediate manifestation of dystrophinopathy.
Case presentation
A 9-year-old boy patient from Rio Grande do Sul state (Southern region) of Brazil was born by cesarean delivery at 36 weeks, weighing 2495 g, head circumference 34 cm and discharged from hospital 48 hours later. His parents were not consanguineous. He sat independently at 9 months, never crawled, and walked at 22 months. He had no other comorbidities, never suffered any surgical procedure. His parents noticed that since he was three years old, he often fell on the ground, and had difficulties to go up the stairs, stand up or do physical activities.
On physical examination, the patient exhibited head circumference of 53cm, medium and photoreactive pupils, eye movements preserved in all directions, posture with hyperlordosis and global hypotonia. Moreover, he had proximal muscle weakness in upper limbs and deep hyporeflexia, bilateral flexion-cutaneous-plantar reflex, calf pseudohypertrophy, and Gowers sign. Wechsler Intelligence Scale for Children IV (WISC-IV) intelligence tests revealed a low intelligence quotient (IQ) of 59, characterizing mild cognitive deficit. An elevated determination of creatine phosphokinase (CPK; 11.150 U/I) and aldolase (10.9 IU/L) enzymes were also observed. Other laboratorial tests showed: aspartate transaminase (AST) 282 mg/dL; thyroid stimulating hormone (TSH) 4.06 nIU/L; B12 vitamin 494 pg/mL; lactic acid 6.5 mg/dL. Skull magnetic resonance imaging (MRI) and transthoracic Doppler echocardiogram indicated no evidence of significant abnormalities. Considering these clinical features, specially impaired muscle function and high levels of CPK, this patient was referred to a molecular analysis of the DMD gene and muscle biopsy. There was no family history of muscle weakness or cardiac abnormalities. Indeed, the patient’s mother showed normal CPK levels (62 U/I) and normal echocardiogram.
Methods and results
Muscle biopsy was collected at the quadriceps muscle, frozen in isopentane cooled in liquid nitrogen and fresh-frozen cryostat sections were used for histochemistry, enzyme histochemistry and immunohistochemistry (IHC) analysis. Transverse serial frozen sections were stained with hematoxylin-eosin (HE) (Fig. 1A) and modified trichrome gomori (Fig. 1B) reveled great variation in muscle fiber sizes, round hypotrophic fibers and myonuclei internalization. Necrotic muscle fibers surrounded by myophagocytosis, increased endomysial connective tissue (masson trichrome stain) and interstitial adipose tissue was observed in several muscle areas. No intracytoplasmic vacuoles and no rods or ragged red fibers (RRF) were observed. The normal distribution of glycogen in muscle biopsy was evaluated by Periodic Acid Schiff (PAS) staining. Enzyme histochemistry did not show cytochrome c oxidase (COX) -negative/SDH-positive muscle fibers (Fig. 1C). Additionally, no conspicuous intra-vacuolar or perimysial amyloid deposits were revealed by Congo Red staining. Core- and target-defects in succinate dehydrogenase (SDH) (Fig. 1D) and nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) were not observed in this sample.
Figure 1.
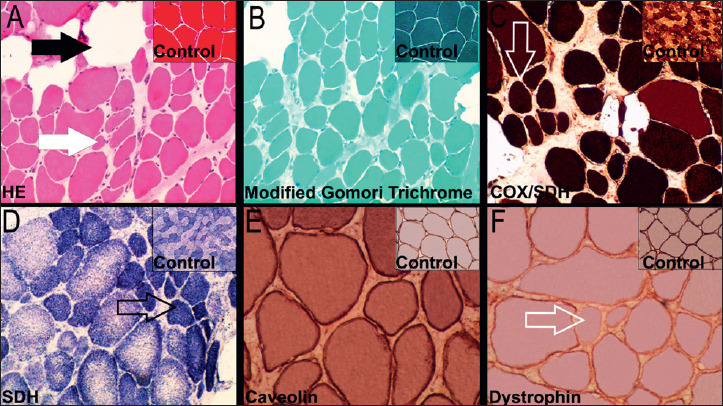
Quadriceps muscle biopsy with features of dystrophy. A) Muscle fibres showing variation in size and atrophic fibres (white arrow) surrounded by endomysial connective tissue/adipose tissue (dark arrow) (Hematoxylin and Eosin, 10x); B) Muscle section with variation in fibre size, internal nuclei, increased endomysial connective tissue and adipose tissue (modified Gomori trichrome, 10x); C) and D) Oxidative enzymes showing variation in fibre size with slight predominance of type 1 fibres (open arrow) [cytochrome c oxidase/succinate dehydrogenase (COX/SDH) and succinate dehydrogenase (SDH), respectively; 10x]. (E) Immunolabelling of caveolin-3 using peroxidase label in the same muscle (20x). (F) Immunolabelling of dystrophin revealing some fibres with weak and uneven (open arrow; 20x) labelling compared with controls.
Muscle fibers exhibited diffuse sarcolemmal immunoreactivity for dysferlin (DYSF), neuronal nitric oxide synthase (nNOS) and laminin alpha 2. Strong diffuse immunoreactivity with alpha-sarcoglycan (data not shown) and caveolin-3 (Fig. 1E) in the membranes of muscle fibers were companied by diffuse emerin staining in myonuclei. Reduced and discontinuous sarcolemmal staining on hypotrophic fibers were evidenced in the immunoreactivity using a human -dystrophin monoclonal antibody that reacts with the N-terminal domain of this protein (Fig. 1F). Sparse satellite and regenerated muscle fibers were highlighted by immunoreaction with CD56 and scattered macrophages (CD68 positivity) and lymphocytes (CD45 positivity) were evidenced in the endomysial area (data not shown). A significant hypotrophy of type 1 fibers was verified in the slow myosin heavy chain (MHC) class I immunoreaction (data not shown). The source, clone and dilution of antibodies used for each staining are as follows: anti-dystrophin (Accurate Chemical & Scientific Corporation; Dy10/12B2;1:20), anti-nNOS (Santa Cruz Biotechnology; R-20; 1:70), anti-caveolin-3 (Abcam; ab2912; 1:100), anti-laminin-2 (Enzo; 4H8-2; 1:100), anti-SGCA (Sigma-Aldrich; polyclonal; 1:50), anti-DYSF (Abnova; Ham1/7B6; 1:20), anti-MHC class I (Abcam; W6/32; 1:130), anti-CD56 (Bio-Rad; Eric-1; 1:50), anti-CD68 (Dako; KP1; RTU) and anti-CD45 (Dako; 2B11 + PD7/26; RTU).
Molecular and in silico analysis
Afterwards, genomic DNA of the proband was isolated from peripheral blood leukocytes and the screening for exon deletions or duplications in the DMD gene was performed by MLPA technique using the SALSA® MLPA® P034 and P035 (DMD/Becker) kits (MRC Holland, Amsterdam, Netherlands), according to the manufacturer’s instructions. A reference DNA (no deletions and/or duplications in the DMD gene) was used as a normal copy number control. MLPA amplified fragments were separated by capillary gel electrophoresis in an ABI 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) and the results were analyzed using the Coffalyser.Net Software (https://coffalyser.wordpress.com/) (MRC® Holland, Amsterdam, Netherlands). MLPA analysis identified no DMD deletions and/or duplications in the proband. In parallel, we performed target sequencing of the DMD gene using a NGS approach. The DMD gene regions were amplified using standard multiplex polymerase chain reaction (PCR) reactions through an Ion AmpliSeq customized panel (Thermo Fisher Scientific), which covers the 79 exons and at least 5 bp of exon-intron boundaries of DMD. PCR products were then sequenced by Ion Torrent Personal Genome machine (Ion Torrent Systems Inc, Gilford, NH), according to manufacturer’s instructions. Human DMD sequence corresponding to the NM_004006.2 was used as a wild-type (WT) reference. A novel hemizygous DMD variant, described as c.9649+4A>T, was detected in intron 66 (mean coverage of this genic region = 2080x).
To confirm the origin of this DMD intronic sequence variant, genomic DNA was obtained from his mother (an obligatory carrier) and, after amplification by PCR using primers that flank the variant region previously described by Lenk and colleagues 9, the purified PCR product was analyzed by Sanger sequencing. The sequencing analysis showed the presence in heterozygosis of the c.9649+4A>T variant (Fig. 2).
Figure 2.
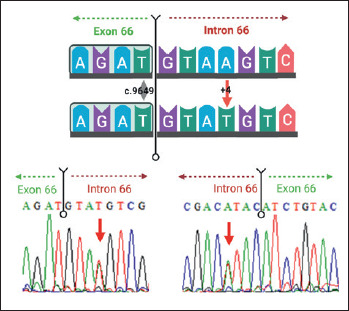
Schematic representation of the DMD gene region encompassing the novel sequence variant c.9649+4A>T (indicated by the red arrow) in the splice donor site within intron 66 (upper panel; Created with BioRender.com), and bidirectional Sanger sequencing analysis from maternal DNA showing the presence of both variant and wild-type alleles (lower panel). Sense and antisense DMD sequences are shown by indicating the orientation of the DNA strands in the panels, as well as a range of 4-8 base pairs are underlined in both directions at the exon-intron junction in order to highlight this boundary. WT, wild-type sequence.
Considering the lack of information regarding DMD c.9649+4A>T variant, an in silico approach was employed. First, the search for this intronic alteration in several population databases, including 1000 genomes Project, Exome Sequencing Project (ESP), Exome Aggregation Consortium database (ExAC), Genome Aggregation Database (gnomAD), and Online Archive of Brazilian Mutations (AbraOM), indicated that c.9649+4A>T was not previously reported in healthy individuals. Additionally, the variant was not described neither in Leiden Open Variation Database (LOVD) 10, ClinVar nor in a specific DMD/BMD mutation database (TREAT-NMD DMD Global database) 11. Based on this, it was considered a novel DMD sequence variant. Next, in silico analysis was performed in order to investigate the biological effect on splicing motifs (including exonic enhancers and silencers). Mutation Taster 12, Human Splicing Finder 13 and Berkeley Drosophila Genome Project (BDGP) 14 algorithms suggested that the c.9649+4A>T intronic variant might affect splicing through a WT donor site disruption. Supplementary Figure 1 depicts the output provided by Human Splicing Finder algorithm, showing the reduction in the splicing prediction raw scores comparing WT DMD sequence vs. variant sequence. Moreover, PhyloP score derived from alignment of 46 vertebrate species genomic sequences 15 indicated that this nucleotide change occurs in an evolutionarily conserved region. The SpliceAI, a deep learning-based tool 16, predicts the identified variant to affect most probably the splicing (Delta score = 0.58). Finally, the c.9649+4A>T was classified according to the guidelines proposed by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) 12 and by Sherloc classification system 17 for the interpretation of sequence variants. After careful analysis of all available evidence about this novel DMD variant, it was classified as a variant of uncertain significance (VUS) by both classification approaches. Briefly, PM2 (pathogenic moderate evidence: absent from controls), PP3 and PP4 (supporting pathogenic evidence: multiple lines of computational evidence support a deleterious effect and patient’s phenotype is highly specific for a disease with a single genetic etiology, respectively) were the applied criteria using the ACMG guidelines. In contrast, EV0135, EV0184 and EV0024 (pathogenic evidence: absent from general population; variant involving a donor +3A/G, +4A or +5G; and weak functional evidence for protein function disrupted, respectively), as well as EV0211 (neutral evidence: first case report with the variant) were the selected criteria based on Sherloc framework.
Supplementary Figure 1.
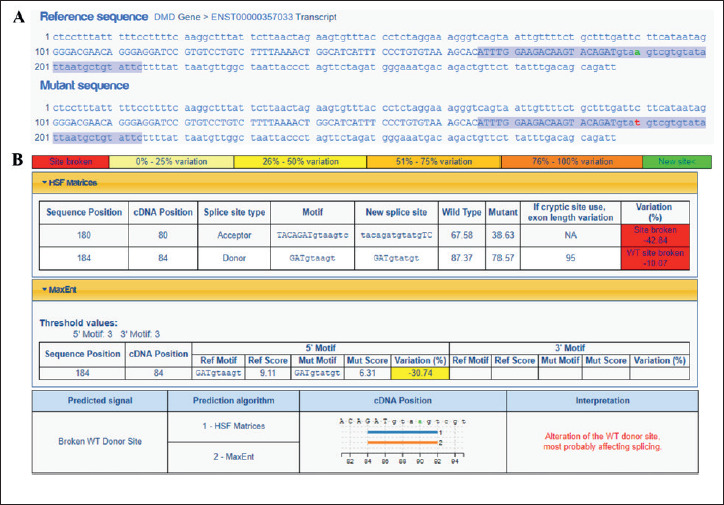
Analysis of the novel intronic DMD mutation identified in this case report (c.9649+4A>T, intron 66) using the Human Splicing Finder (HSF) algorithm. A) Comparison between the reference (wild-type, WT) and mutated DMD sequences, encompassing exon 66 (upper case nucleotides letters, 86 base pairs) and 100 intronic nucleotides at exon ends (lower case letters). The highlighted blue sequences were analyzed by HSF and MaxEntScan predictors. Green and red letters indicate the WT and mutated nucleotide change, respectively. B) Raw and interpreted tables show relevant results related to the mutation position and context. Variations in the tables were depicted in colored boxes, according to the scale showed in the upper panel. c.9649+4A>T was predicted to affect splicing by disrupting a WT donor site (note the reduction in the raw scores when compared WT DMD vs mutant sequence).
Discussion
More than 890 DMD pathogenic variants have been described so far (detailed information in: https://clinvarminer.genetics.utah.edu/variants-by-gene/DMD/condition/Duchenne%20muscular%20dystrophy/pathogenic), covering the different functional domains of dystrophin protein. The majority (~65%) of these causative variants are intragenic deletions/duplications that often lead to frameshift errors. Among the remaining ones, we find intronic alterations that usually create cryptic exons by activating potential splice sites 8,18. Of note, the pre-mRNA of DMD gene is composed by 99% of introns 19 that exhibit a very complex pattern of expression and different alternative splicing 20, contributing to high rates of point variants, insertions and deletions 21,22. This study presents a novel intronic sequence variant in the DMD gene, c.9649+4A>T in a patient at 9 years of age with an intermediate manifestation of muscular dystrophy. Remarkably, the variant was absent from all queried databases (1000 Genomes Project, ExAC, ESP, GnomAD, LOVD).
The novel DMD sequence variant described here is at the 5′ donor splice site of intron 66 which is essential during the post-transcriptional modifications, specifically the pre-mRNA splicing process 23. Disruptions on donor site, defined by the three terminal nucleotides of each exon and the first seven bases of the downstream intron, tended to generate an alternative 5’ splice site, resulting in different protein isoforms 23 that leads to a wide variety of clinical Duchenne phenotypes 24.
Importantly, at the same splice donor site of intron 66, two different germline DMD variants at the nucleotide position +5 were previously reported in the LOVD database 10, namely c.9649+5 G>T 25 and c.9649+5 G>A 26, being classified as pathogenic and likely pathogenic alterations, respectively. In another sequence variant, also in the same position, c.9649+5G>C, was classified as pathogenic in the ClinVar database 11. This finding represents an indirect evidence that nucleotide changes at c.9649+4 position might have functional impact associated with the splicing efficiency alteration in the DMD intron 66. In accordance with it, previous studies showed that the point intronic variants c.9649+1G>A 24, c.9649+2T>C 24 and c.9649+2insT 27 also at the 5′ donor splice site of intron 66, led to severe phenotype (Duchenne dystrophy). Other point variants within or in the vicinity of the intron 66 region also leads to a range of variety clinical Duchene phenotype as variants c.9649+15T>C 9,28, c.9807+5G>A 28 and c.9857+15C>T 29 but its pathogenic effects are unknown.
Indeed, the novel DMD intronic variant is located within in the cysteine-rich domain of the protein, consisting in a region required for a β-dystroglycan interaction with dystrophin complex 30. Therefore, we can speculate that this genetic alteration might destabilize this complex, a functional consequence which could explain the discontinuous sarcolemmal staining observed in our muscle biopsy analysis, leading to the clinical manifestation of muscle weakness observed in the proband. As note, it is well known that germline DMD pathogenic variants in the cysteine-rich domain are among the possible underlying genetic defects associated with DMD phenotype 31,32.
As recently well reviewed 33,34, the global cognition functions are often affected in DMD patients, even in severe or mild phenotypes and, likewise, our patient also shows a mild cognitive deficit. At the same time, our patient has a healthy heart function, suggesting that the intronic variant reported here produces enough amounts of dystrophin protein in the cardiac muscle as observed in normal echocardiogram. Based on the muscle biopsy analysis and in silico results obtained in the current study, we may suggest that, even considering its uncertain clinical significance using both ACMG-AMP and Sherloc criteria, the c.9649+4A>T variant leads to a decrease in the dystrophin protein production levels and hypotrophic fibers as showed in Figure 1. Moreover, the reduced levels of dystrophin on patient’s muscle might explain his high levels of serum CK which can be released from dystrophic fibers 35. Further functional and molecular approaches such as patient-derived induced pluripotent stem (iPS) cells 36 and analyses based on dystrophin reporter minigene 37 are needed in order to characterize the tissue-specific effects of this novel variant in the dystrophin protein in detail. Indeed, segregation of clinical phenotype within the family may clarify the impact of this variant on its classification.
As limitations of this study, the RNA isolation from the biopsied muscle tissue of proband could not be performed due to the small amount of material collected. It would be important in order to evaluate the DMD transcript expression levels and abundance of specific isoforms. Furthermore, the amount of dystrophin protein in the brain or cardiac muscle in our patient was not evaluated. Finally, although this is the first report of the intronic DMD variant c.9649+4A>T and the clinical suspicion of molecular alterations in this gene was strong, our molecular approach was based on the single-gene analysis of one patient, not involving additional whole exome or genome sequencing tests to screen for potential causative variants in other genes.
Conclusions
Our data show that the c.9649+4A>T intronic sequence variant in the DMD gene may be associated with an IDM phenotype and further studies are needed to clarify the complete functional effects of this genetic alteration, specially its functional impact in the mRNA processing. Overall, our findings reinforce that the variant described here initially classified as VUS, may be re-classified in the future as likely pathogenic or pathogenic. Indeed, our study reinforces the importance of a more precise diagnosis combining muscle biopsy, new generation molecular techniques and comprehensive in silico approaches in the clinical cases with negative results for conventional genetic analysis.
Figures and tables
Acknowledgement
We appreciate the cooperation of the patient and his parents for this study. Schematic representations were created with BioRender.com.
Footnotes
Ethical consideration
Written informed consent was obtained from the parents of the index patient/proband for publication of this case report and accompanying laboratory tests results and images.
Funding
APSB was supported by a post doc fellowship from CAPES/PNPD (Programa Nacional de Pós-Doutorado).
Conflict of interest
The authors declare that there is no conflict of interest.
Author contributions
APBS, IAV, GB and RS designed the study, coordinated the project, performed sequence analysis, analyzed the data and wrote the paper; RS, FQ, provided patient samples and patient data. IVA designed and performed bioinformatics analysis. JCB, MLB performed immunohistochemical experiments and assisted in drafting and critical reading. ACBF, GB performed molecular experiments, assisted in drafting and critical reading. All authors read and approved the final manuscript.
References
- 1.Gao QQ, McNally EM. The dystrophin complex: structure, function, and implications for therapy. Comprehensive Physiol 2015;5:1223-1239. https://doi.org/10.1002/cphy.c140048 10.1002/cphy.c140048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen DG, Whitehead NP, Froehner SC. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiological Rev 2016;96:253-305. https://doi.org/10.1152/physrev.00007.2015 10.1152/physrev.00007.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med 2019;70:239-255. https://doi.org/10.1146/annurev-med-081117-010451 10.1146/annurev-med-081117-010451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat 2016;12:1795-1807. https://doi.org/10.2147/NDT.S93873 10.2147/NDT.S93873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimizu-Motohashi Y, Komaki H, Motohashi N, et al. Restoring dystrophin expression in Duchenne muscular dystrophy: current status of therapeutic approaches. J Personalized Med 2019;9:1. https://doi.org/10.3390/jpm9010001 10.3390/jpm9010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elhawary NA, Jiffri EH, Jambi S, et al. Molecular characterization of exonic rearrangements and frame shifts in the dystrophin gene in Duchenne muscular dystrophy patients in a Saudi community. Hum Genomics 2018;12:18. PMID PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Xu M, Li H, et al. Genotypes and phenotypes of DMD small mutations in Chinese patients with dystrophinopathies. Front Genet 2019;10:114. https://doi.org/10.3389/fgene.2019.00114 10.3389/fgene.2019.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 2016;53:145-151. https://doi.org/10.1136/jmedgenet-2015-103387 10.1136/jmedgenet-2015-103387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenk U, Hanke R, Thiele H, et al. Point mutations at the carboxy terminus of the human dystrophin gene: implications for an association with mental retardation in DMD patients. Hum Mol Genet 1993;2:1877-1881. https://doi.org/10.1093/hmg/2.11.1877 10.1093/hmg/2.11.1877 [DOI] [PubMed] [Google Scholar]
- 10.Fokkema IF, Taschner PE, Schaafsma GC, et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011;32:557-563. https://doi.org/10.1002/humu.21438 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- 11.Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat 2015;36:395-402. https://doi.org/10.1002/humu.22758 10.1002/humu.22758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361-362. https://doi.org/10.1038/nmeth.2890 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 13.Desmet FO, Hamroun D, Lalande M, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. https://doi.org/10.1093/nar/gkp215 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reese MG, Eeckman FH, Kulp D, et al. Improved splice site detection in Genie. J Comput Biol 1997;4:311-323. https://doi.org/10.1089/cmb.1997.4.311 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- 15.Pollard KS, Hubisz MJ, Rosenbloom KR, et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110-121. https://doi.org/10.1101/gr.097857.109 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176:535-48 e24. https://doi.org/10.1016/j.cell.2018.12.015 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 17.Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med 2017;19:1105-1117. https://doi.org/10.1038/gim.2017.37 10.1038/gim.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD Mutations in 576 dystrophinopathy families: a step forward in genotype-phenotype correlations. PloS One 2015;10:e0135189. https://doi.org/10.1371/journal.pone.0135189 10.1371/journal.pone.0135189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731-740. https://doi.org/10.1016/s1474-4422(03)00585-4 10.1016/s1474-4422(03)00585-4 [DOI] [PubMed] [Google Scholar]
- 20.Tuffery-Giraud S, Miro J, Koenig M, et al. Normal and altered pre-mRNA processing in the DMD gene. Hum Genet 2017;136:1155-1172. https://doi.org/10.1007/s00439-017-1820-9 10.1007/s00439-017-1820-9 [DOI] [PubMed] [Google Scholar]
- 21.Gualandi F, Rimessi P, Cardazzo B, et al. Genomic definition of a pure intronic dystrophin deletion responsible for an XLDC splicing mutation: in vitro mimicking and antisense modulation of the splicing abnormality. Gene 2003;311:25-33. https://doi.org/10.1016/s0378-1119(03)00527-4 10.1016/s0378-1119(03)00527-4 [DOI] [PubMed] [Google Scholar]
- 22.Bovolenta M, Neri M, Fini S, et al. A novel custom high density-comparative genomic hybridization array detects common rearrangements as well as deep intronic mutations in dystrophinopathies. BMC Genomics 2008;9:572. https://doi.org/10.1186/1471-2164-9-572 10.1186/1471-2164-9-572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaz-Drago R, Custodio N, Carmo-Fonseca M. Deep intronic mutations and human disease. Hum Genet 2017;136:1093-1111. https://doi.org/10.1007/s00439-017-1809-4 10.1007/s00439-017-1809-4 [DOI] [PubMed] [Google Scholar]
- 24.Deburgrave N, Daoud F, Llense S, et al. Protein- and mRNA-based phenotype-genotype correlations in DMD/BMD with point mutations and molecular basis for BMD with nonsense and frameshift mutations in the DMD gene. Hum Mutat 2007;28:183-195. https://doi.org/10.1002/humu.20422 10.1002/humu.20422 [DOI] [PubMed] [Google Scholar]
- 25.Takeshima Y, Yagi M, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 2010;55:379-388. https://doi.org/10.1038/jhg.2010.49 10.1038/jhg.2010.49 [DOI] [PubMed] [Google Scholar]
- 26.Toksoy G, Durmus H, Aghayev A, et al. Mutation spectrum of 260 dystrophinopathy patients from Turkey and important highlights for genetic counseling. Neuromuscul Disord 2019;29:601-613. https://doi.org/10.1016/j.nmd.2019.03.012 10.1016/j.nmd.2019.03.012 [DOI] [PubMed] [Google Scholar]
- 27.Vieitez I, Gallano P, Gonzalez-Quereda L, et al. Mutational spectrum of Duchenne muscular dystrophy in Spain: study of 284 cases. Neurologia 2017;32:377-385. https://doi.org/10.1016/j.nrl.2015.12.009 10.1016/j.nrl.2015.12.009 [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Li Y, Song T, et al. A retrospective analysis of 237 Chinese families with Duchenne muscular dystrophy history and strategies of prenatal diagnosis. J Clin Lab Anal 2018;32:e22445. https://doi.org/10.1002/jcla.22445 10.1002/jcla.22445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett RR, den Dunnen J, O’Brien KF, et al. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet 2001;2:17. https://doi.org/10.1186/1471-2156-2-17 10.1186/1471-2156-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung D, Yang B, Meyer J, et al. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem 1995;270:27305-27310. https://doi.org/10.1074/jbc.270.45.27305 10.1074/jbc.270.45.27305 [DOI] [PubMed] [Google Scholar]
- 31.Bies RD, Caskey CT, Fenwick R. An intact cysteine-rich domain is required for dystrophin function. J Clin Invest 1992;90:666-672. https://doi.org/10.1172/JCI115909 10.1172/JCI115909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rafael JA, Cox GA, Corrado K, et al. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol 1996;134:93-102. https://doi.org/10.1083/jcb.134.1.93 10.1083/jcb.134.1.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Doorenweerd N. Combining genetics, neuropsychology and neuroimaging to improve understanding of brain involvement in Duchenne muscular dystrophy – a narrative review. Neuromuscul Disord 2020;30:437-442. https://doi.org/10.1016/j.nmd.2020.05.001 10.1016/j.nmd.2020.05.001 [DOI] [PubMed] [Google Scholar]
- 34.Naidoo M, Anthony K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol Neurobiol 2020;57:1748-1767. https://doi.org/10.1007/s12035-019-01845-w 10.1007/s12035-019-01845-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sumita DR, Vainzof M, Campiotto S, et al. Absence of correlation between skewed X inactivation in blood and serum creatine-kinase levels in Duchenne/Becker female carriers. Am J Med Genet 1998;80:356-361. PMID: [PubMed] [Google Scholar]
- 36.Kazuki Y, Hiratsuka M, Takiguchi M, et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol Ther 2010;18:386-393. https://doi.org/10.1038/mt.2009.274 10.1038/mt.2009.274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorain S, Peccate C, Le Hir M, et al. Dystrophin rescue by trans-splicing: a strategy for DMD genotypes not eligible for exon skipping approaches. Nucleic Acids Res 2013;41:8391-8402. https://doi.org/10.1093/nar/gkt621 10.1093/nar/gkt621 [DOI] [PMC free article] [PubMed] [Google Scholar]