

Abstract
Ovarian cancer selective metastasizes to the omentum contributing to the poor prognosis associated with ovarian cancer. However, the mechanism underlining this propensity and therapeutic approaches to counter this process have not been fully elucidated. Here, we show that MCP-1 produced by omental adipocytes binding to its cognate receptor CCR-2 on ovarian cancer cells facilitates migration and omental metastasis by activating the PI3K/AKT/mTOR pathway and its downstream effectors HIF-1α and VEGF-A in cell lines, xenografts, and transgenic murine models. MCP-1 antibody significantly decreased tumor burden and increased survival of mice in vivo. Interestingly, metformin decreased omental metastasis at least partially by inhibiting MCP-1 secretion from adipocytes independent of direct effects on cancer cells. Together this suggests a novel target of MCP-1/CCR-2 axis that could benefit ovarian cancer patients.
Keywords: Omentum adipocytes, ovarian cancer, metastasis, MCP-1, metformin
Introduction
Epithelial ovarian cancer remains the leading cause of death of gynecological malignancies with a five-year survival rate of less than 40% [1]. The majority of ovarian cancer patients are diagnosed at late stage with widespread metastases that are mostly located in the abdomen [2–4], especially in the omentum [5]. Although most patients have an initial response to therapy, which includes aggressive debulking surgery followed by chemotherapy [6, 7], overall prognosis is limited by a high recurrence rate primarily in the abdomen.
Obesity has been associated with late stage as well as omental metastases of ovarian cancer [8, 9]. Recently, adipocytes have been proposed to contribute to the aggressiveness of ovarian cancer metabolic reprogramming [10], activating insulin signaling [11], altering the tumor microenvironment or inducing systemic inflammation [12, 13]. These mechanisms likely contribute to the role of omental adipocytes in omental metastases. However, whether additional mechanisms are relevant as well as methods to intervene therapeutically in the actions of omental adipocytes remains lacking.
Monocyte Chemotactic Protein-1 (MCP-1), also known as chemokine (C-C motif) ligand 2 (CCL-2), is a member of chemokine branch of the cytokine superfamily [14]. MCP-1 through binding to its receptor CCR-2, which is commonly expressed in monocytes/macrophages, promotes cancer development by attracting circulating monocytes/macrophages and then reprograming the tumor microenvironment into an immune suppressive status [15–17]. However, whether MCP-1 also directly affects tumor cells is less well characterized.
In this study, using cell line models, xenografts, and transgenic mice models, we demonstrate a key role for the MCP-1/CCR-2 axis in facilitating ovarian cancer cell migration and omental metastases. We also show that metformin inhibits MCP-1 secretion from adipocytes, which contribute to interrupts the signaling axis and has the potential to benefit ovarian cancer patients.
Results
Omental adipocytes promote ovarian cancer peritoneal metastasis
Five weeks after intraperitoneal injection of SKOV3-ERFP cells into NOD/SCID mice, we found red fluorescence was specifically enriched in omentum metastasis by fluorescence stereozoom microscopy (Figure 1A), confirming omentum as a favorable site for metastasis of this model. To assess the role of the omentum in tumor aggressiveness, we performed a sham-operation or omentectomy in NOD/SCID mice prior to intraperitoneal injection of SKOV3 cells (Figure 1B). Strikingly, peritoneal metastases were significantly reduced, by both numbers and volume, in mice that had omentectomy, further supporting the pivotal role of the omentum in intra-abdominal metastasis (Figure 1C).
Figure 1. Adipocytes from the omentum facilitate ovarian cancer metastasis and chemotherapy resistance in ovarian cancer.
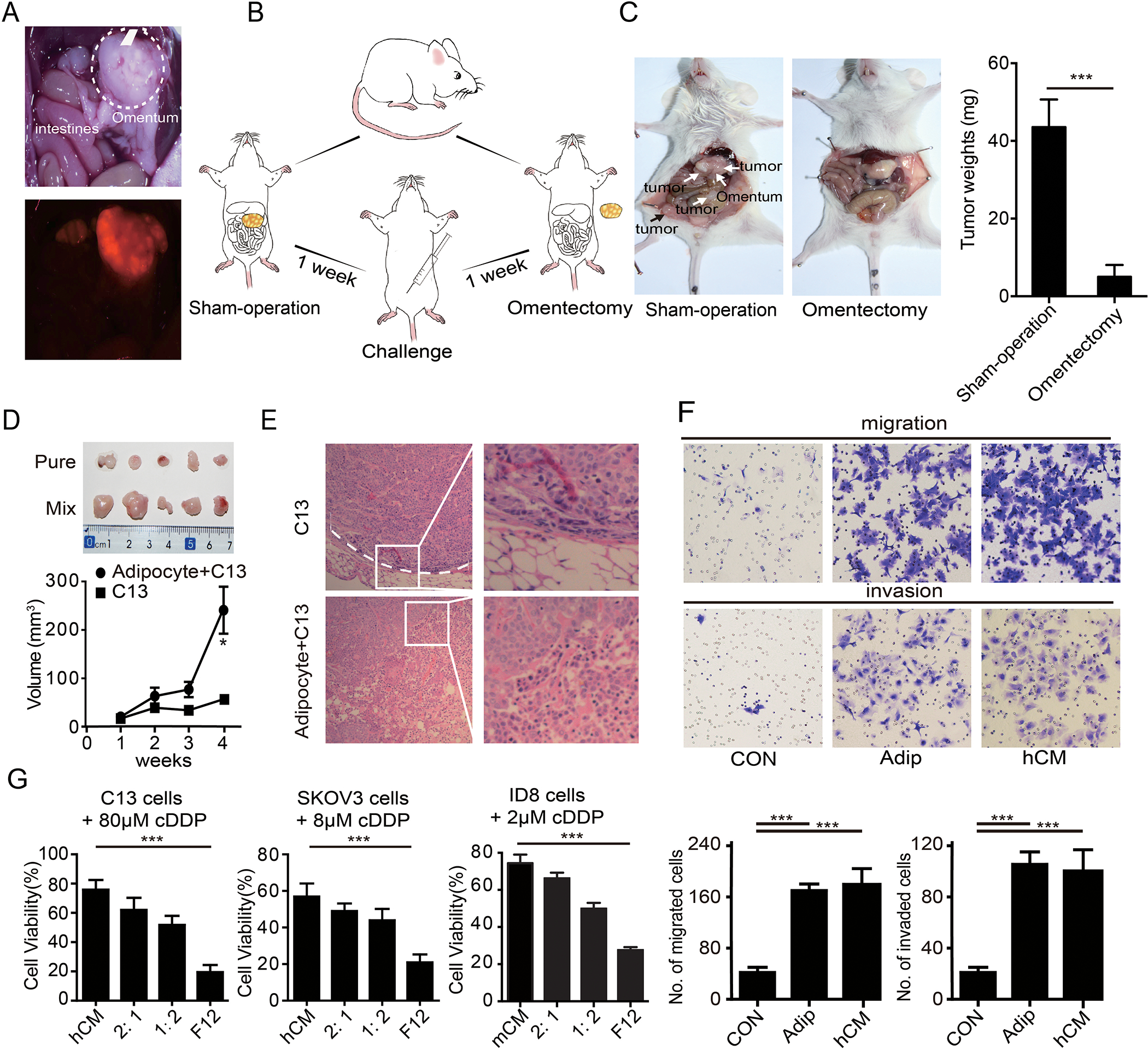
(A) Representative images of omental metastases of ovarian cancer as shown by the red fluorescent in SKOV3-ERFP cells. (B) Diagram of the NOD/SCID mice challenged with the SKOV3 cells after a sham-operation and omentectomy. (C) Tumor burdens of ovarian metastases in NOD/SCID mice with a sham-operation and omentectomy. Tumors are marked by the arrowheads. The quantification bar graph of tumor weights is displayed on the right panel. (D) Gross morphology of the tumors sizes and quantification of tumor weights were shown. (E) Representative H&E staining of the tumor in (D). Tumor border is marked by a white dotted line. Magnified images are displayed at right. (F) Representative images of the migration and invasion of the SKOV3 cells toward complete medium, adipocytes (Adip) and the conditional medium from the primary adipocytes (hCM). Quantifications of migrated cells are displayed on the lower panel. (n=3 technical replicates for three independent experiments) (G) Bar graph of the C13, SKOV3 and ID8 cell viability after treatment with cDDP in F12 medium, conditional medium of primary adipocytes (hCM), or diluted hCM (CM: F12 = 2:1, or = 1:2). (n=3 technical replicates from three independent experiments). Date are presented as the mean ± SEM, Student’s t test. *P<0.05, ***p< 0.001.
Given adipocytes are the dominant components of omentum, we hypothesized that adipocytes contributed to peritoneal metastasis of ovarian cancer. Therefore, we injected a mix of C13 cells and adipocytes derived from the omentum or C13 cells alone into mice subcutaneously. Consistent with the hypothesis, co-injection with adipocytes increased tumor size compared to C13 cells alone (Figure 1D). Further, tumors arising from co-injection of C13 with adipocytes did not have discrete boundaries with normal tissue, consistent with invasion and propensity for metastasis (Figure 1E). We thus assessed the ability of adipocytes to promote cancer cell migration and invasion in vitro. Consistent with our previous findings and others [18], we observed that both adipocytes and their CM (conditional medium of the primary adipocytes) increased migration and invasion of cancer cells (Figure 1F and Supplementary figure 1A), suggesting that adipocytes modulate migration and invasion through secretion and action of adipokines. Moreover, CM not only enhanced migration and invasion, but also induced a notable resistance to cisplatin (cDDP), the main therapeutic approach for ovarian cancer (Figure 1G). Thus, omental adipocytes contribute to tumor aggressiveness by promoting migration, invasion, and chemo-resistance through the action of secreted cytokines.
MCP-1 secreted by omental adipocyte induces an aggressive phenotype of ovarian cancer cells
We next sought to determine the identity of cytokines that mediate the effects of omental adipocytes using cytokine protein arrays. Of the cytokines on the arrays, MCP-1 was present specifically in supernatants of adipocytes but not in supernatants of cancer cells (Figure 2A). We then performed ELISAs to quantify MCP-1 levels in supernatants from a panel of cancer cell lines and primary human omental adipocytes. MCP-1 levels were higher in supernatants from omental adipocytes than ovarian cancer cell lines, which was consistent with the data in GTEx database which shows the MCP-1 is highest in omentum adipocytes (Figure 2B and Supplementary 2A). Furthermore, MCP-1 was markedly higher in paired omental adipocytes and primary ovarian cancer cells from 10 independent ovarian cancer patients (Figure 2C). To determine whether MCP-1 contributed to the activity of omental adipocytes, we verified the effect of MCP-1 on ovarian cancer cells. Besides migration and invasion enhancement (Figure 2D and supplementary figure 2B), MCP-1 enhanced cisplatin resistance (Figure 2E) similar but not fully equal to omental adipocytes (Figure 1F-1G). Neutralization antibodies of MCP-1 completely abolished the enhanced migration and invasion ability of CM in SKOV3 (Figure 2F). Furthermore, as expected, tumor burden and peritoneal metastases were significantly reduced by neutralization antibodies of MCP-1 as omentectomy did (Figure 2G), strongly supporting that MCP-1 is one of the mainstays of omental adipocytes to promote ovarian cancer aggressiveness.
Figure 2: MCP-1 secreted by omental adipocyte induces an aggressive phenotype of ovarian cancer cells.
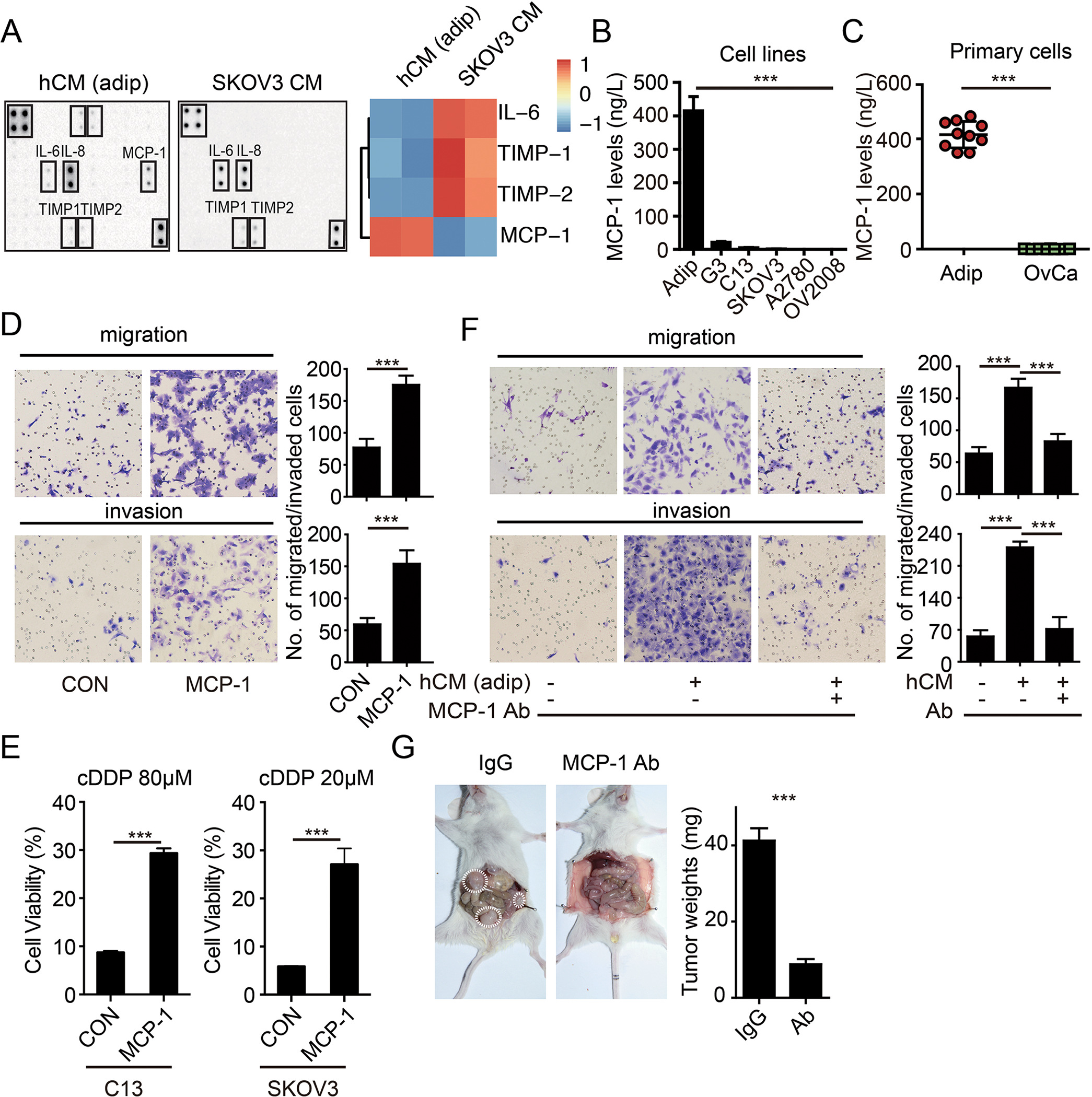
The cytokine protein array analysis of the hCM and SKOV3 cells. Heat map of the expression levels of adipokines is displayed on the right panel. (B) Bar graph of the MCP-1 expression levels in the adipocytes, SKOV3-G3, C13, SKOV3, A2780 and OV2008 cells, as detected by ELISA. (n=3 replicates for three independent experiments) (C) Bar graph of MCP-1 levels in the paired omental adipocytes and ovarian cancer cells from ovarian cancer patients (OvCa) by ELISA. (n=3 technical replicates) (D) Representative images of migration and invasion of SKOV3 cells toward complete medium with or without with MCP-1 (20μg/L). (n=3 technical replicates for three independent experiments) (E) Bar graph of the C13, SKOV3 cell viability after treatment with cDDP as indicated for 48 hours in completed medium with or without MCP-1 (20μg/L). (n=3 technical replicates for three independent experiments) (F) Representative images of migration and invasion of SKOV3 cells toward complete medium, and conditional medium from primary adipocytes (hCM), and conditional medium from primary adipocytes with neutralization antibody of MCP-1 (Ab, 1 mg/L). (n=3 technical replicates for three independent experiments) (G) Tumor burdens of ovarian metastases in NOD/SCID mice with IgG or MCP-1 neutralization antibodies. Tumors are marked by the dotted circles. The quantification bar graph of tumor weights is displayed on the right panel. Date are presented as the mean ± SEM, Student’s t test. ***p< 0.001.
Binding of MCP-1 to its cognate receptor CCR-2 on ovarian cancer cells promotes tumor growth and metastasis
CCR-2, the receptor for MCP-1, is expressed by macrophages and has been proposed to mediate the effects of MCP-1 through altering the tumor microenvironment [19, 20]. Alternatively, CCR-2 expression by cancer cells could mediate the effects of MCP-1 by altering cell survival [21]. To determine whether MCP-1 facilitates tumor growth and metastasis through actions in microenvironment or on cancer cells directly, we took advantage of MCP-1−/− and CCR-2−/− mice. MCP-1−/− mice are defective in MCP-1 secretion but retain CCR-2 expression on microenvironment cells, including peritoneal monocytes, macrophages, and Kupffer cells that have been proposed to mediate the activity of MCP-1. In contrast, CCR-2−/− mice retain MCP-1 secretion but are defective in CCR-2 expression on cells in the microenvironment. Tumor omental metastasis and ascites production by ID8 cells were significantly suppressed in MCP-1−/− mice (which retain CCR-2 expression and do not produce MCP-1) but not in CCR-2−/− mice (which were with CCR-2 defect but injected with CCR-2+ ovarian cancer cells ) compared to WT mice (Figure 3A-C). This was associated with a more prolonged median survival (P<0.05) of ID8 tumor bearing MCP-1−/− mice, but not CCR-2−/− mice (Figure 3D). These results are supportive of the concept that MCP-1 secreted by adipocytes, facilitates ovarian cancer growth, and peritoneal metastasis through binding to CCR-2 on cancer cells, independent of effects in the microenvironment can contribute to tumor growth and metastasis to the omentum.
Figure 3. CCR-2 in ovarian cancer cells, but not in microenvironment cells, interact with the of MCP-1 and then promote tumor growth and metastasis.
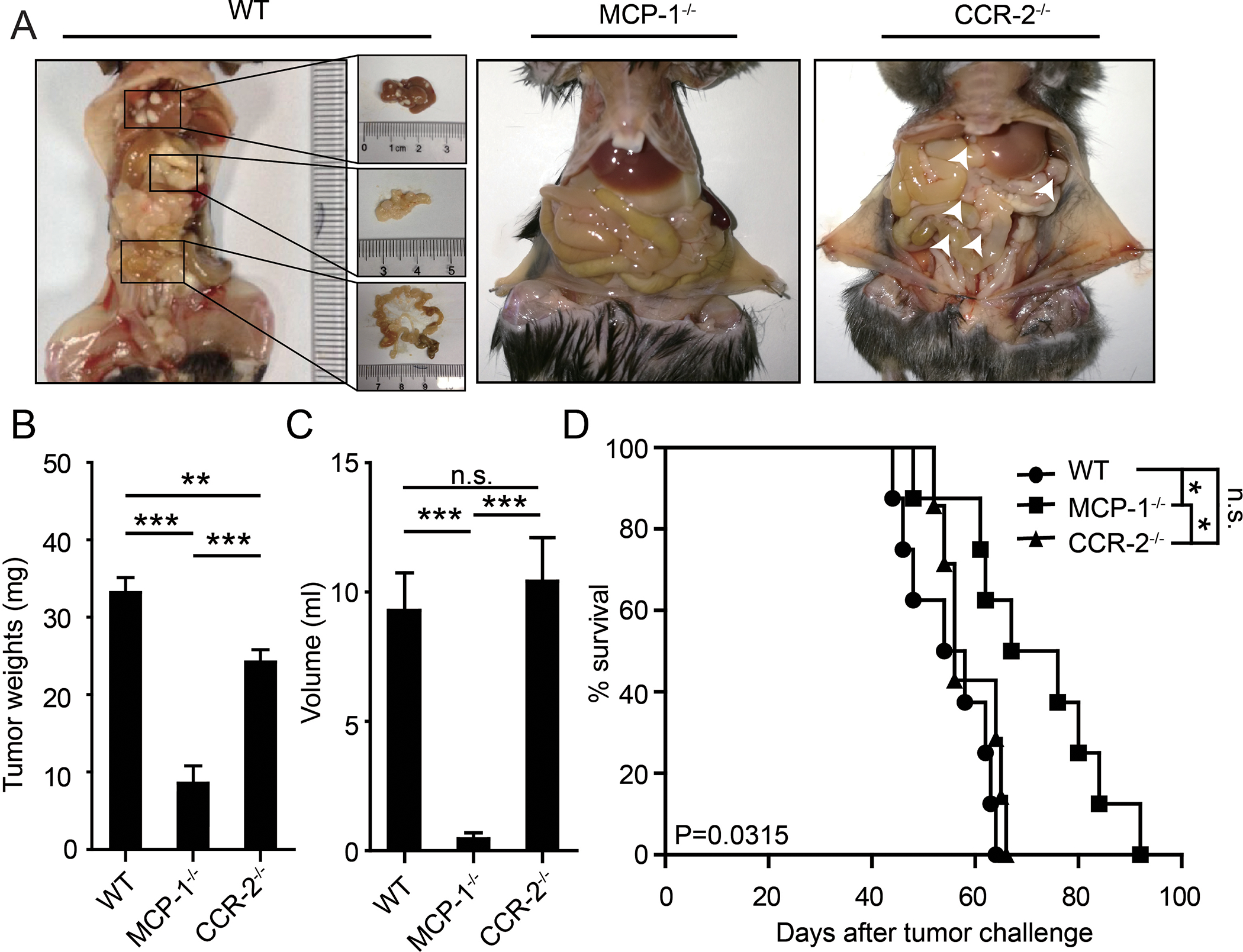
(A) Gross anatomy of tumor burdens in WT, MCP-1−/−, and CCR-2−/− mice. The liver, omentus, and mesentery metastases in WT mice were enlarged. The white arrows indicated micrometastases in CCR-2−/− mice. (B) Bar graph of tumor weights. (C) Bar graph of the ascites volumes in WT, MCP-1−/−, and CCR-2−/− mice. Date are presented as the mean ± SEM, Student’s t test. ***p< 0.001. n.s. no siginificancy. (D) The K-M survival curves of the overall survival of WT, MCP-1−/−, and CCR-2−/− mice. The statistical significance (P value) was test by log-rank test.
CCR-2 expressed in ovarian cancer cells is associated with metastasis
To confirm that CCR-2 is expressed by ovarian cancer cells, we performed Western blots and demonstrated varying degrees of CCR-2 expression in 5 frequently studied ovarian cancer cell lines as well as two cell lines derived from the omentum metastasis of SKOV3 mice models (SKOV3-M1 and SKOV3-M2). ID8, SKOV3, SKOV3-M1 and SKOV3-M2 cells had elevated expression of CCR-2, which was correlated with increased invasion and migration potential compared to the other cell lines’ (Figure 4A, B and Supplementary figure 3A-I). Secondly, we assessed CCR-2 levels in paired specimens of primary ovarian tumors and omental metastases by immunohistochemistry. CCR-2 levels were negative in the most of all normal tissue and borderline tumor, were low in primary cancer cells, but were enriched in omental metastases (Figure 4C-E). Indeed, of the 47 patients assessed, CCR-2 levels were elevated in omental metastases, with the exception of the one tumor that had high levels in the primary site. Consistently, CCR2 is much higher in omental metastases than ovarian cancer cells in primary in GSE2109 database (Figure 4F). This supports the contention that MCP-1-CCR-2 interactions contribute to omental metastases.
Figure 4. CCR-2 expressed in ovarian cancer cells is associated with metastasis.
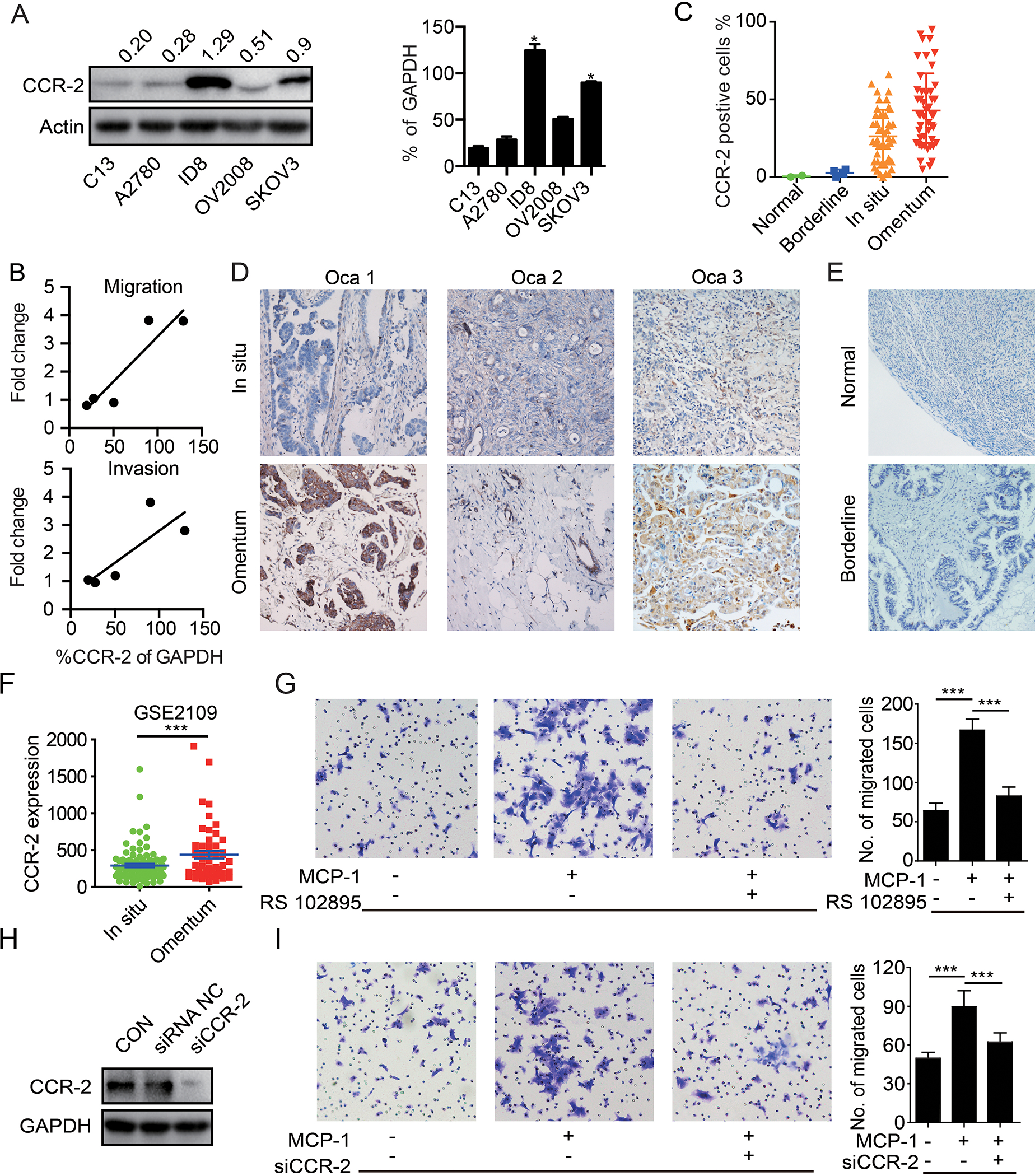
(A) Western blot of CCR-2 in the C13, A2780, ID8, OV2008 and SKOV3 cells.The quantification bar graph of Western blot is displayed on the right panel. (n=3 independent experiments) (B) Correlation between CCR-2 expression level and migration/invasion fold change of C13, A2780, ID8, OV2008 and SKOV3 cells. (C) Quantification of CCR-2 positive cells in normal tissue, borderline tumor tissue, primary ovarian tumors and paired omental metastasis. (D) Representative immunohistochemical staining of CCR-2 in primary ovarian tumor (Ovca 1–3) and paired omental metastasis. (E) Representative immunohistochemical staining of CCR-2 in normal ovary tissue and borderline tumor tissue. (F) Quantification of CCR-2 in primary ovarian cancers and omentum metastases in GSE2019 database. (G) Migration of the SKOV3 cells toward complete medium, complete medium with MCP-1 (20 μg/L), and complete medium with MCP-1 (20 μg/L) plus CCR-2 antagonist (RS 102895, 2 μg/L). The corresponding bar graph is displayed on the right side. (n=3 technical replicates for three independent experiments) (H) The CCR-2 knockdown efficacy with siRNA was verified with Western blot. (n=3 independent experiments) (I) Migration of the parental or CCR-2 knockdown SKOV3 cells toward complete medium, or complete medium with MCP-1 (20 μg/L). The corresponding bar graph is displayed on the right side. (n=3 technical replicates for three independent experiments). Date are presented as the mean ± SEM, Student’s t test. ***p< 0.001.
Effects of MCP-1 on ovarian cancer cells are mediated by CCR-2
To verify that the effects of MCP-1 on migration were mediated by CCR-2 on ovarian cancer cells, we applied a CCR-2 specific antagonist, RS102895 [22], in Transwell assays. Notably, RS102895 abrogated the effects of MCP-1 on migration (Figure 4G and Supplementary Figure 4A). Furthermore, depletion of CCR-2 with siRNA abolished the effects of MCP-1 on migration (Figure 4H and 4I). Interesting, CCR-2 is highest in ovarian cancer cells than other cell lines in NCI60 database (Supplementary Figure 4B). Intriguingly, GO (Gene Ontology) analysis of NCI60 cell lines indicated that higher CCR-2 expression was associated with pathways implicated in tumor metastasis including chemotaxis, cell migration and blood vessel development (Supplementary Figure 4C). Otherwise, CCR-2 antagonist (RS 102895) also partially abolished the DDP resistance of CM in ovarian cancer cell lines (Supplementary figure 4D). Together the data implicate binding of MCP-1 to its cognate receptor CCR-2 on cancer cells metastasis of ovarian cancer cells to the omentum and partially contributes to cisplatin resistance of ovarian cancers.
MCP-1/CCR-2 axis activates the PI3K-AKT-mTOR pathway leading to HIF-1α mediated VEGF-A expression
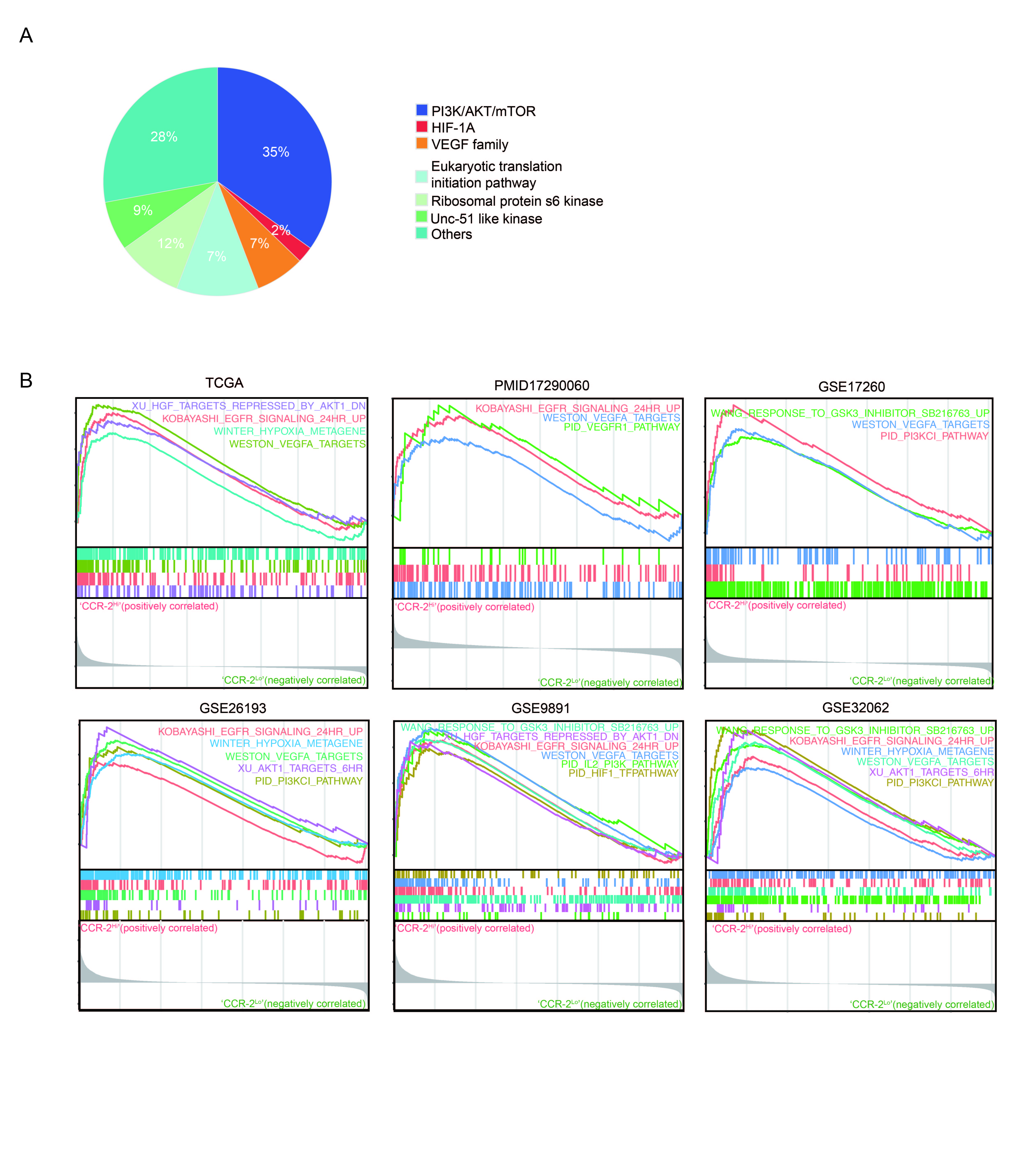
To explore the mechanistic underpinnings of the actions of MCP-1 on ovarian cancer cells, ID8 tumors from MCP-1−/− and WT mice were assessed by microarray analysis. Gene set enrichment analysis (GSEA) revealed that PI3K/AKT/mTORs, VEGF, and HIF-1α pathway were significantly lower in tumors from MCP-1−/− mice compared to WT mice (Figure 5A, Supplementary Figure 5A). Furthermore, tumors with lower CCR-2 concurrently attenuated PI3K/AKT/mTORs, VEGF, and HIF-1α pathway activity in TCGA, PMID17290060, GSE17260, GSE26193, GSE9891, GSE32062 (Supplementary Figure 5B). Furthermore, MCP-1 activated multiple components of the PI3K/AKT/mTOR signaling pathway including p-mTOR and pS6, PI3K p100/p85 complex, AKT, Rictor, Raptor and TSC1 as well as VEGFR and EGFR in human SKOV3 cells as assessed by RPPA. MCP-1 also altered the apoptotic pathway as indicated by decreased p-Bad, Bid, Bax and cleaved Caspase-7 (Figure 5B). Third, with Human phospho-RTK array, we further confirmed that MCP-1 phosphorylated ATK, mTOR, and EGFR both in C13 and SKOV3 cells (Figure 5C).
Figure 5. MCP-1/CCR-2 axis enhances VEGF-A expression via HIF-1α following the activation of the PI3K-AKT-mTOR pathway.
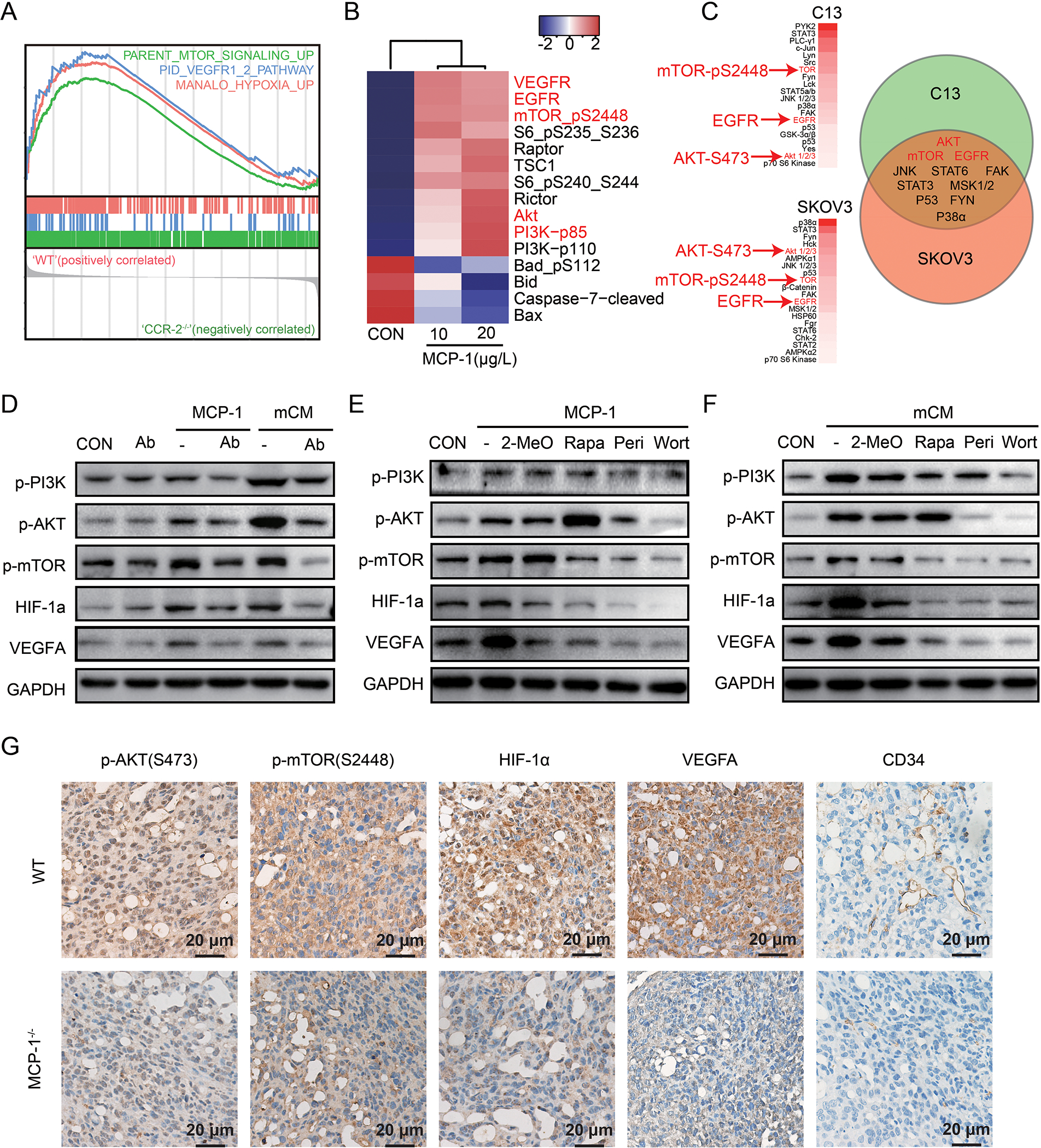
(A) GSEA analysis in tumor from MCP-1−/− mice. (B) Heatmap of differentially expressed proteins cultured with or without MCP-1 for 24 hours in SKOV3 cells detected by RPPA compared to vehicle. (C) Phosphorylated proteins list after treatment with MCP-1 (20μg/L) for 24 hours in C13 and SKOV3 detected by Human phospho-RTK array (more than1.5 folds change were listed) and a Venn diagram of the overlap changing proteins in C13 and SKOV3 were shown on right panel. (D-F) Western blot of phosphorylated PI3K/AKT/mTOR pathway, along with HIF-1α and VEGF-A, treated as indicated in ID8 cells. (n=3 independent experiments). Wort for wortmannin (a PI3K inhibitor, 100nM for 24h); Peri for perifosine (an AKT inhibitor, 10μM for 24h); Rapa for rapamycin (a mTOR inhibitor, 5nM for 24h); 2-Meo for 2-MeOE2 (a HIF-1α inhibitor, 2.5μM for 24h). (G) Representative immunohistochemical staining of p-AKT, p-mTOR, HIF-1α, VEGF-A and CD34 in tumors from the WT and MCP-1−/− mice. The scale bar corresponds to 50 μm.
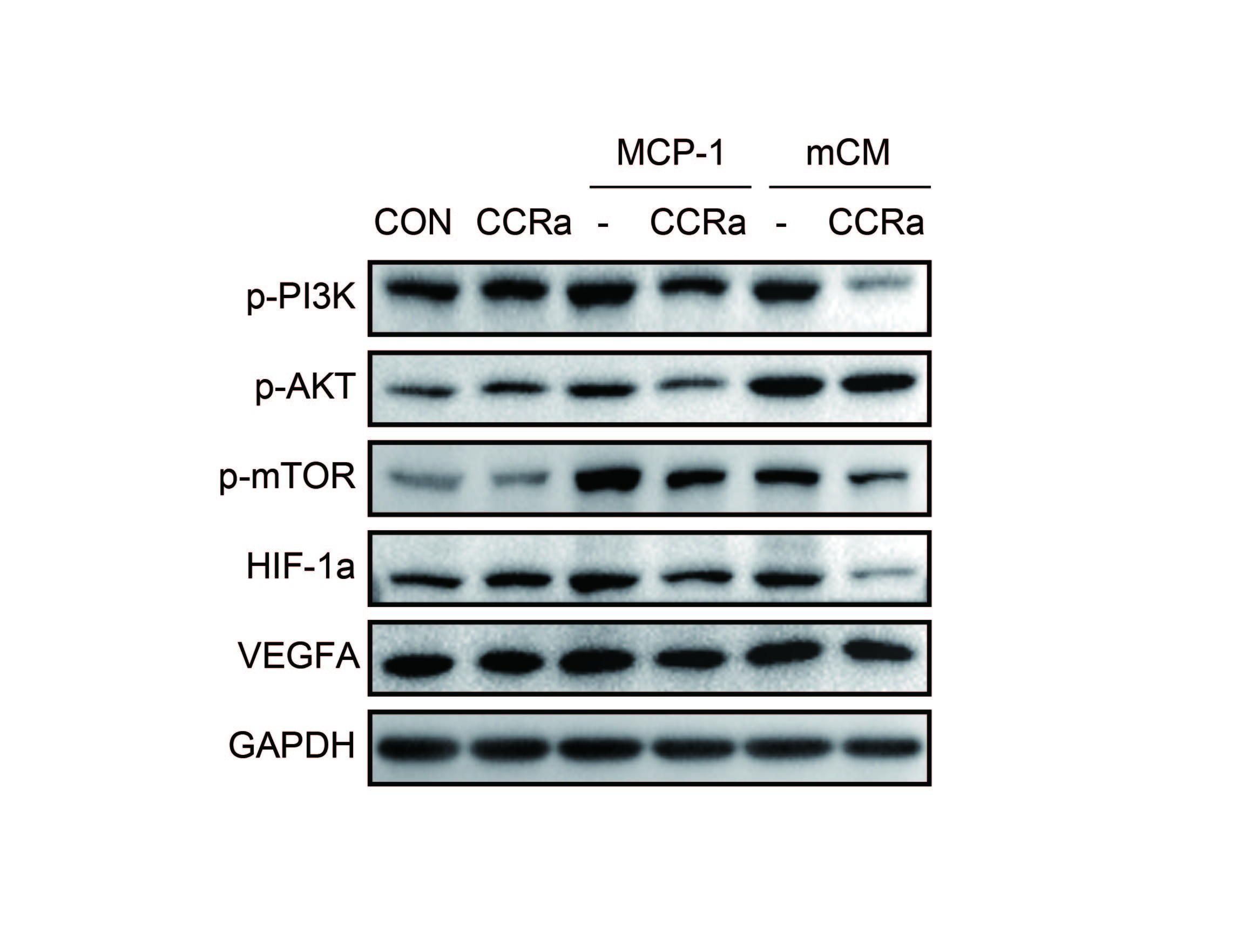
HIF-1α, which is downstream of the PI3K/AKT/mTOR signaling pathway, promotes the expression of VEGF-A that has been implicated in tumor growth and metastasis [23]. Importantly, both conditional medium from the primary murine adipocytes (mCM) and mouse MCP-1 activated the PI3K/AKT/mTOR signaling pathway and increased HIF-1α and VEGF-A in ID8 cells (Figure 5D). MCP-1 neutralization antibody attenuated the effects of MCP-1 or mCM (Figure 5D). Similarly, the CCR-2 antagonist blocked pathway activation by both MCP-1 and mCM (Supplementary Figure 6). Consistent with a role for the PI3K/AKT/mTOR and HIF-1α pathways in production of VEGF-A, wortmannin (a PI3K inhibitor), perifosine (an AKT inhibitor), or rapamycin (a mTOR inhibitor) attenuated activation of the PI3K/AKT/mTOR pathway as well as downstream HIF-1α and VEGF-A expression induced by either MCP-1 or mCM (Figure 5E and F). In addition, 2-MeOE2 (a HIF-1α inhibitor) also decreased VEGF-A without altering PI3K/AKT/mTOR activation (Figure 5E and F). Consistent with the with in vitro data, p-AKT, p-mTOR, HIF-1α, and VEGF-A were lower in tumors from MCP-1−/− than in tumors from WT mice by immunohistochemical analyses (Figure 5G). CD34 indicative of tumor vessel formation was also lower.
Metformin decreases MCP-1 secretion by adipocytes and prevents ovarian cancer metastasis
Metformin has been previously reported to suppress the secretion of several cytokines from adipocytes, including necrosis factor (TNF), MCP-1 and leptin [24]. We verified that metformin, with the dose didn’t affect adipocyte viability (Supplementary figure 7 A and B), decreased MCP-1 secreted by omental adipocytes using cytokine protein array (Figure 6A), and ELISA (Figure 6B) from omental adipocytes isolated from ovarian cancer patients. Further, MCP-1 was suppressed by metformin in time-dependent manner in murine adipocyte-like cells differentiated from 3T3-L1 (Figure 6C). Notably, conditional media from metformin treated adipocytes, CM (MET) (see methods), failed to upregulate p-AKT, HIF-1α, and VEGF-A in ID8 cells (Figure 6D). Although metformin has been reported to inhibit p-AKT and induced apoptosis [25], ID8 cells were resistant to the effects of metformin on both of cell signaling and cell toxicity (Figure 6E and F). Consistent with the effects on cell signaling, CM (MET) did not promote migration of ID8 cells (Figure 6G). In vivo, metformin delayed tumorigenesis and omental metastasis of ID8 cells (Figure 6H), and prolonged survival of mice (Figure 6I). Immunohistochemical analysis confirmed downregulation of MCP-1 in tumor microenvironment of mice treated with metformin. Further, VEGF-A was decreased and fewer CD34 positive endothelial cells were observed in metformin treated mice (Figure 6J). Thus, metformin inhibits ovarian cancer metastasis at least partially owing to its suppresses effects on secretion of MCP-1 from omental adipocytes.
Figure 6. Metformin prevent omentum metastasis by decreasing MCP-1 secretion from adipocytes.
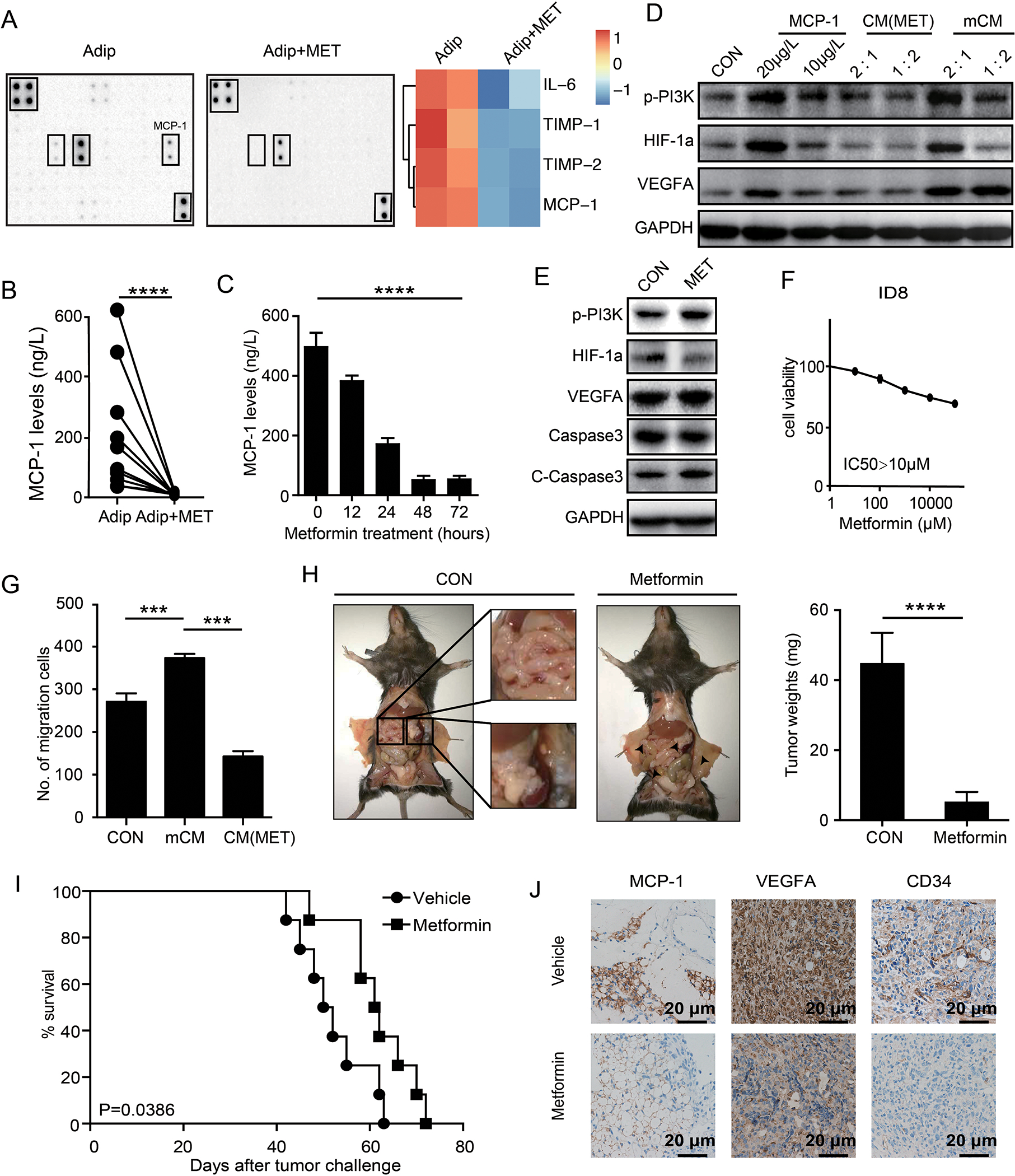
(A) The cytokine protein array analysis of the mCM from adipocytes and adipocytes pre-treated with metformin for 12 hours. Heat map of the expression levels of adipocytes is displayed on the right panel. (B) Quantification of MCP-1 levels in primary omental adipocytes from ovarian cancer patients with or without metformin treatments. (n=3 technical replicates for three independent experiments) (C) Quantification of MCP-1 levels in primary omental adipocytes treated with various time-points as indicated. (n=3 technical replicates for three independent experiments) (D) Western blot of p-PI3K, HIF-1α and VEGF-A treated as indicated in ID8 cells. 1:2 and 2:1 refer to the ratios the conditional medium mixed with complete medium. (n=3 independent experiments) (E) Western blot of indicated proteins after treatments with or without metformin in ID8 cells. (n=3 independent experiments) (F) Dose response curves of metformin in ID8 cell lines treated with varying concentrations of metformin for 72 hr. (n=3 technical replicates for three independent experiments) (G) Bar graph of the migrations of ID8 cells towards complete medium, conditional medium from the adipocytes and conditional medium (mCM) from the adipocytes pre-treated with metformin (CM (MET)). (n=3 technical replicates for three independent experiments) (H) Gross anatomy of tumor burdens in WT mice treated with or without metformin for 4 weeks. The white squares show mesentery and omentum macrometastases in vehicle treated mice, and the white arrows indicated micrometastases in metformin treated mice. The quantification bar graph of tumor weights is displayed on the right panel. (I) The Kaplan-Meier (K-M) survival curves of the overall survival of the mice in (G). (J) Representative immunohistochemical straining of MCP-1, VEGF-A and CD34 in tumors from mice in (G). Scale bar corresponds to 50 μm.
Discussion
The interaction of the tumor and microenvironment niche is increasingly drawing attention. Tumor cells dynamically interact with multiple normal cells such as fibroblasts, immune cells and adipocytes, etc. [10, 26]. Of all cells present in the microenvironment, adipocytes are the least well studied. In our study, we confirmed that resection of the omentum dramatically impaired the peritoneal metastasis of ovarian cancer cells in mice models, highlighting the role of adipocytes in fostering ovarian tumor cell metastasis.
Increasing evidence shows that dysfunctional adipose releasing IL-6, IL-8, TNF-α, MCP-1 and several other cytokines is closely related to the progression of various cancers. These cytokines take part in epithelial mesenchymal transition, angiogenesis, apoptosis resistance and metastasis in cancer progression [27, 28]. Since there is a complex regulatory network and overlap of cytokine between adipocytes and cancer cells, by subtracting the cytokines secreted by SKOV3 cells, we identified that MCP-1 might well account for the observed tumor-promoting phenotype of adipocytes. Indeed, recombinant MCP-1 recapitulates the promotion effects observed in the presence of adipocytes. Further, neutralization of MCP-1 in CM abrogated ovarian cancer migration, invasion, and partially inhibited chemo-resistance. Moreover, blockade of the MCP-1/CCR-2 axis with a CCR2 antagonist or by knockdown of CCR2 inhibits the proinvasive effect of adipocytes. Although MCP-1 could be secreted by cancer cells [29], we found MCP-lwas exclusively secreted from omentum adipocytes but not ovarian cancer cells, which is consistent with the previous report that MCP-1 is significantly under-expressed or absent in HGOSC [30]. Therefore, omentum adipocytes are the main source of CCL2 in the abdomen, which also facilitated ovarian cancer omentum specific metastasis.
MCP-1 stimulates various types of cellular signaling for cancer progression [31]. Mechanistically, through microarray, high-throughput functional protein assay (RPPA), and data mining with multiple datasets, we demonstrated the associations between MCP-1/CCR-2 and activation of mTOR pathway, along with its downstream effectors HIF-1α and VEGF-A. This hypothesis is further supported by the fact MCP-1 antibody (Figure 5A) and CCR2 antagonist (Supplementary figure 7) prevented the activation of the mTOR and HIF-1α/VEGF-A pathway by CM and MCP-1, supporting the MCP-1/CCR-2 mediated activation of the mTOR pathway and pro-invasion ability. Consistent with our findings, MIZUTANI et al. also reported that MCP-1 increases AKT/ mTOR phosphorylation in prostate cancer cells [32]. VEGF-A has been implicated in angiogenesis, tumor invasion and metastasis and is associated with poor prognosis. Thus, it is likely that PI3K/AKT/mTOR and its downstream target HIF-1α through upregulating VEGF-A contribute to the effects of MCP-1 on metastasis to the omentum.
Theoretically, MCP-1 contributes to the progression of ovarian cancer via two major mechanisms: MCP-1 as a direct promoting factor for CCR-2 cancer cells and the attraction of tumor associated-macrophage (TAM) along with consequent regulation of tumor immune microenvironment. In our system, ID8 omental metastases were diminished and overall survivals were prolonged in MCP-1−/− mice, but not in CCR-2−/− mice, supporting the direct effect of MCP-1 on CCR-2 expressed cancer cells, rather than an indirect effect through the tumor microenvironment, playing a crucial role in facilitating omentus metastasis. This finding is constant with the report that MCP-1 promotes invasion and adhesion of SKOV-3 ovarian cancer cells in a manner unrelated to TAMs [33]. Actually, the reported roles of MCP-1 as a factor conferring immunity against the tumors are still controversial. First, it is still debatable whether MCP-1 directly affects macrophage polarization [34]. Moreover, except macrophage, CCR-2 is broadly expressed in a variety of tissue types, such as T cells, B cells, natural killer cells, which contribute to the paradoxical effects of MCP-1/CCR-2 activation in cancer development [35]. Third, Sica, etc reported that human ovarian carcinoma cells do not express CCR-2 in macrophages [14]. Although the overall survival was not prolonged, a mild tumor suppression was observed in CCR-2−/− mice (Figure 3B), indicating the indirect role of MCP-1 through the TAMs on omentum metastasis is not fully excluded. So, further investigation, like in macrophages specific CCR-2 KO mice, are warranted to identify if CCR-2 in macrophages is the potential key target of MCP-1 modulating ovarian cancer metastasis.
CCR-2 is reported expressed in certain types of tumor cells, such as prostate cancer [36] and ovarian cancers [37]. Binding MCP-1 to CCR-2 directly stimulates their proliferation and migration, greatly contributing to tumorigenesis [33]. Further, high expression of CCR-2 associates with aggressive phenotype and poor overall survival in gastric cancer or prostate cells [36, 37]. Consistent with these studies, we found that approximately 50% of cases showed moderate to intense CCR-2 staining. More importantly, CCR-2 expression is highly heterogeneous in ovarian cancer. Tumor cells located at the invasive front of ovarian cancer displayed higher CCR-2 expression. Moreover, compared with primary tumors, CCR-2 is much higher in omentum metastasis. All of these support the important role of CCR-2 in ovarian cancer metastasis.
Metformin use has been associated with decreased cancer risks and improved prognosis of different cancer types recently [38–41]. We confirmed that metformin may decrease ovarian cancer omental metastasis through or partially through its inhibitory effects on MCP-1 secretion from adipocytes. Even after omentectomy, adipocytes remain in the abdomen, such as in the mesentery, which may contribute to the high risk (58%) of abdomen relapse in ovarian cancer [42]. Thus metformin could potentially benefit ovarian cancer patients even after omentectomy.
We have demonstrated that the interaction between MCP-1 secreted by adipocytes and CCR-2 expressed in ovarian cancer cells facilitates migration and omentum metastasis in ovarian cancer cell line models, xenografts, and transgenic mouse models. Importantly, the therapeutic effects of the blockade of the MCP-1/CCR-2 axis using anti-MCP-1 antibodies or CCR-2 antagonists alone or in combination with other agents have been tested in breast cancer or prostate cancer in clinical trials, and positive results have been reported [43, 44]. These studies provide additional impetus for studies of therapeutic strategy targeting at MCP-1/CCR-2 pathway, and we anticipate that CCL2/CCR-2 axis may represent a promising therapeutic target to prevent ovarian cancer progression.
Materials and methods
Cell culture
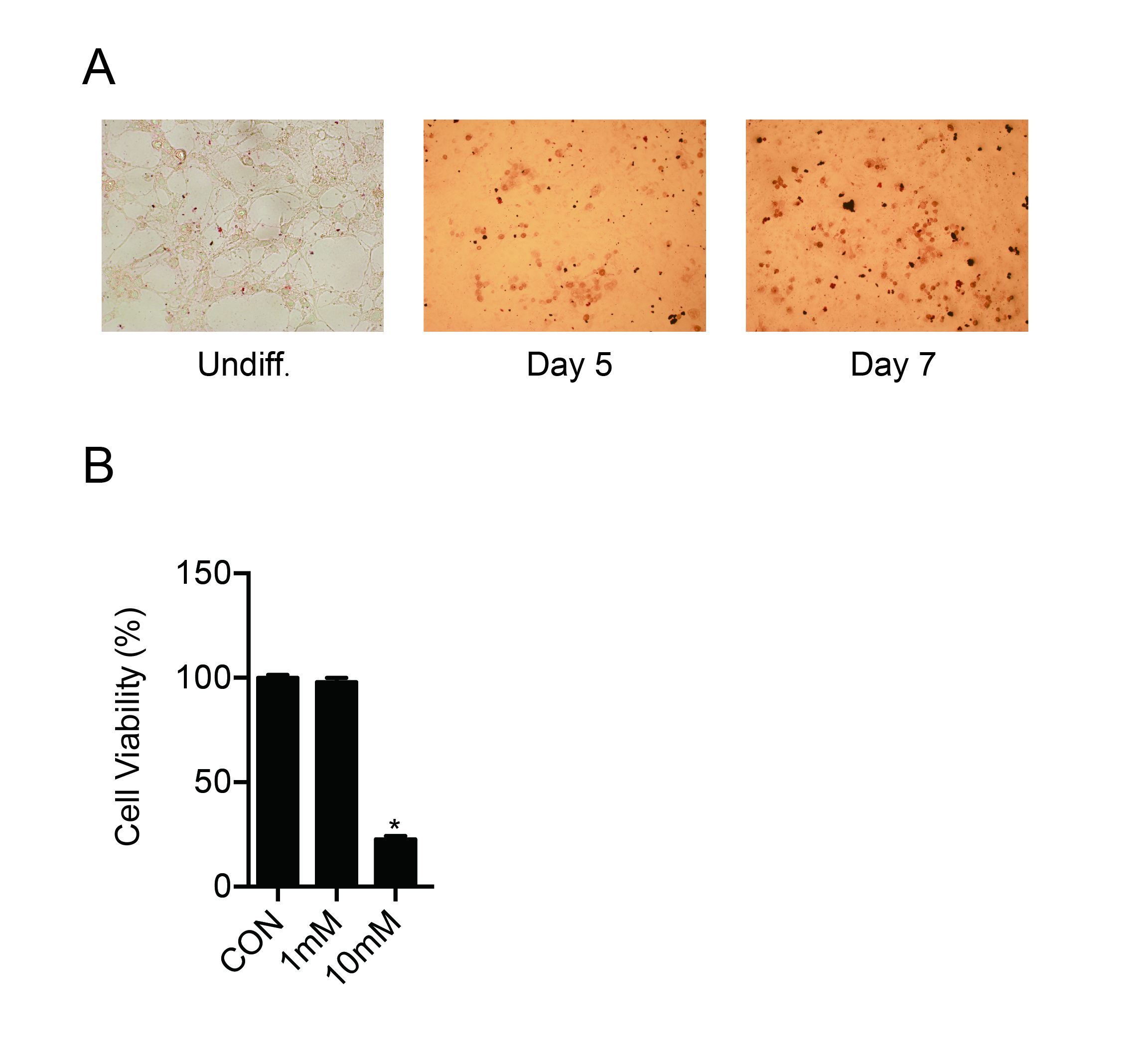
SKOV3, A2780 ovarian cancer cell lines and mouse 3T3-L1 preadipocyte cell line were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). The SKOV3-ERFP cells ectopically expressing ERFP, were derived from SKOV3. The SKOV3-M1 and SKOV3-M2 cells is derived from SKOV3 omentum metastasis in vivo. C13 and OV2008 cell lines were gifts from Prof. Benjamin K. Tsang at the Ottawa Health Research Institute, Ottawa, Canada. ID8 cell line was provided by K. Roby (Department of Anatomy and Cell Biology, University of Kansas). The SKOV3, SKOV3-ERFP, and SKOV3-M1/2 cells were cultured in McCoy’s 5A (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco). C13, OV2008, A2780 cells were cultured in RPMI-1640 (Gibco) supplemented with 10% FBS. ID8 was cultured in DMEM (Gibco) supplemented with 2% FBS and Insulin-Transferrin-Selenium (ITS, Gibco). Mouse 3T3-L1 cell line was cultured in DMEM (Gibco) supplemented with 2% FBS and followed with inducing medium I (DMEM, 2% FBS, 0.5 μM IBMX, 1 μM Dexamethasone, and 10 mg/L insulin) and inducing medium II (DMEM, 2% FBS, 10 mg/L insulin). Then, the induced adipocyte-like 3T3-L1 cells were cultured in DMEM (Gibco) supplemented with 2% FBS.
Animal models
All manipulations were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Tongji Hospital. Female, 8 weeks old NOD/SCID mice, Balb/c-null, and C57BL/6 mice were purchased from Beijing HFK Bioscience and housed in laminar flow cabinets under specific pathogen-free conditions. MCP1−/− (004434-B6.129S4-Ccl2tm1Rol/J) and CCR-2−/− (004999 - B6.129S4-Ccr2tm1Ifc/J) mice were purchase from Jackson Labs.
Ovarian cancer peritoneal metastasis mouse model
SKOV3-ERFP (3×106) cells were injected intraperitoneally into NOD/SCID mice. 35 days later, mice were sacrificed for fluorescence stereozoom microscope examination.
Ovarian cancer peritoneal metastasis model after omentectomy
Omentectomies and sham-operations were performed in NOD/SCID mice. One week later, SKOV3 cells (3×106) were injected intraperitoneally. 35 days later, mice were sacrificed for evaluation.
Ovarian cancer peritoneal metastasis model after MCP-1 neutralization antibody treatment
MCP-1 neutralization antibodies (5mg/kg body weight) were injected intraperitoneally (twice a week) after SKOV3 tumor-bearing model was established as mentioned above. 35 days later, mice were sacrificed for evaluation.
C13 xenograft model with or without adipocytes
C13 cells (3×106) alone or in combination with adipocytes (1×104) were subcutaneously injected into the right and left flank of Balb/c-null mice respectively. Tumor sizes were determined every 2 days and calculated as length × (square of width)/2.
Ovarian cancer allograft models in WT, MCP1−/− mice and CCR-2−/− mice
The ID8 cells (5×106) were injected intraperitoneally to C57 WT, MCP1−/− and CCR-2−/− female mice (n=8). WT mice and all target gene knockout mice were C57BL/6 background. For metformin efficacy experiments, ID8 cells (5×106) were injected intraperitoneally to C57 WT mice. Weight and abdominal girth of mice were monitored every two days. After sacrifice, tumor size was measure by a caliper as W2*L/2. Tumor from WT and MCP1−/− was collected and infiltrated in RNA Later (Thermo) for RNA microarray analysis, and paraformaldehyde for further hematoxylin-eosin (H&E) staining and immunohistochemical studies. In order to adequately power biological validation experiments, five to eight knockout and WT mice were used throughout.
Ovarian cancer peritoneal metastasis model after metformin treatment
Metformin (100 mg/kg/day, Beyotime, s1741–1g) was given by oral gavage for 4 weeks after ID8 tumor-bearing model was established as mentioned above. Tumors were collected and infiltrated in paraformaldehyde for further hematoxylin-eosin (H&E) staining and immunohistochemical studies. In order to adequately power biological validation experiments, five to eight mice for each group were used throughout.
Adipocytes derived conditional medium
Human adipocytes were acquired from fresh omentum of ovarian cancer patients undergoing surgery. Fresh omentum was minced with scissors, digested by gentleMACS (Miltenyi) [45] to obtain single cells. After centrifuge with condition of 100rpm, the suspension separated into 4 layers, which are cell pallets, conditional medium, adipocytes and free fat oil. And then we carefully isolated adipocytes, counted and cultured in six-well plates with DF12 (Gibco) supplemented with 2% FBS (Gibco) for 1×106 adipocytes. 48 hours later, the supernatant was harvested as conditioned medium of human primary cultured adipocytes (hCM). hCM were filtered through 200-mesh sieves to remove cell debris before use.
3T3-L1 cells were chemically induced by DMEM with 10% FBS (Gibco), 0.5mM IBMX (TOPSCIENCE, T1713), 1μM Dexamethasone (TOPSCIENCE, T1076), 5μg/ml insulin (Bovine, Sigma I-5500) for 2 days and DMEM with 10% FBS (Gibco), 10μg/ml insulin (Bovine, Sigma I-5500) for 2 days to differentiate into adipocyte-like cells. Then the supernatant of induced adipocyte-like 3T3-L1 cells was collected at 24 hours as mouse conditioned medium (mCM). After pretreatment with metformin (1 mM, Beyotime, catalog number s1741–1g) for 12 hours, the induced adipocyte-like 3T3-L1 cells were cultured with complete medium for 24 hours, and the supernatant collected as CM (MET).
In vitro migration and invasion assays
Transwell inserts (6.5 mm diameter, 8.0 μm pore size) were placed in each of 24-well plates with or without matirgel (dilution with McCoy’s 5A 1:5, BD, #354230) for migration and invasion assays respectively according to previously reported procedures [46]. 1×104 SKOV3 or 4×104 ID8 cells, respectively, were seeded on the upper chamber, while complete medium with or without adipocytes (1×104), human/murine MCP-1 recombinant protein (50ng/L, 500ng/L, 5μg/L or 20 μg/L, Peprotech, #300–04 or 250–10), mCM or hCM, CCR-2 antagonist (RS 102895 hydrochloride, 2 μg/L, Tocris, #2089) or human/murine MCP-1 neutralization antibody (1 mg/L, R&D, #MAB679 and AF-479-NA) as indicated were placed in the lower chamber. 12 hours later, the inserts were removed and migrated cells were stained with 0.05% crystal violet.
Cytokine protein arrays
1×106 primary adipocytes were cultured by complete medium with or without 1 mM metformin for 12 hours. SKOV3 cells were cultured by complete medium for 48 hours.
Cytokine protein analysis of the supernatant mentioned above was performed according the protocol provided in the Raybio C-Series Human Obesity Antibody Array C1 (Raybio, catalog number AAH-ADI-1–4). The estimated protein concentrations were normalized by a median polish method and corrected for protein loading using the average expression levels. The label of all cytokines were displayed in supplementary table.
Tissue Samples
47 paired primary ovarian cancer tissue and omentum metastases, 2 normal ovary tissue and 3 borderline tumor samples were acquired in Tongji Hospital of Huazhong University of Science and Technology (Wuhan, China) prior to preoperative radiotherapy or chemotherapy. The study was approved by ethical committee of Tongji Hospital and signed informed consent were obtained from all of patients.
Enzyme-linked immunosorbent assay (ELISA)
MCP-1 levels were quantified by Human CCL-2/MCP-1 Quantikine ELISA Kit (R&D, DCP00) in the supernatant of 1×106 primary adipocytes, SKOV3 G3, C13, SKOV3, A2780, OV2008 cell lines and primary ovarian cancer cells. MCP levels in the supernatant of ID8 cells treated by 1mM metformin for 12 hours, 24 hours, 48 hours or 72 hours.
Immunohistochemical analysis
Immunohistochemical staining was performed according to previously reported procedures [47]. Sectioned ovarian cancer slices were heated, deparaffinization, rehydrated and antigen retrieved. After blocking the slices with BSA (5%, Solarbio), we applied primary antibodies against CCR-2 (dilution 1:50, catalog number 176390, Abcam), phosphorylated AKT (dilution 1:100, catalog number 4060, Cell Signaling Technology), phosphorylated mTOR (dilution 1:50, catalog number 109268, Abcam), HIF-1α (dilution 1:50, catalog number 20960–1-AP, Proteintech), VEGF-A (dilution 1:50, catalog number 19003–1-AP, Proteintech) and CD34 (dilution 1:50, catalog number 8536, Abcam). Then, the slices were exposed to the conjugating secondary antibody at the recommended concentrations followed with DAB+ chromogen. Subsequently following a final dehydration, images were acquired.
GTEx and National Cell Institute 60 (NCI60) analysis
Gene expression data of different tissue in abnominal cavity was downloaded form GTExPortal (https://www.gtexportal.org/home/gene/CCL2). mRNA expression data of NCI60 was downloaded from Cellminer (http://discover.nci.nih.gov/cenminer/). Cell lines were assigned into high or low CCR-2 groups using median expression as the cutoff value. Gene ontology (GO) analysis of whole transcriptional expression was performed between the two groups.
Western blot
Western blot assays were performed as previously reported[48]. After blocking with 5% BSA for 1 hour, the membranes were incubated with primary antibodies against phosphorylated phosphatidylinositol 3-kinase PI3Kp85 (dilution 1:1000, catalog number 4228, Cell Signaling Technology), PI3Kp85 (dilution 1:1000, catalog number 4257s, Cell Signaling Technology), phosphorylated AKT (dilution 1:1000, catalog number 4060, Cell Signaling Technology), AKT (dilution 1:1000, catalog number 4691, Cell Signaling Technology), phosphorylated mechanistic target of rapamycin (mTOR, dilution 1:1000, catalog number 109268, Abcam), mTOR (dilution 1:500, catalog number A2445, ABclonal), HIF-1α (dilution 1:500, catalog number 20960–1-AP, Proteintech), VEGF-A (dilution 1:500, catalog number 19003–1-AP, Proteintech), CCR-2 (dilution 1:500, catalog number A2855, ABclonal), Cleaved-caspase 3 and caspase 3 (dilution 1:500, catalog number A11319) overnight at 4°C. Then, the membranes were incubated with the corresponding 1:2000 horseradish peroxidase (HRP)-linked secondary antibodies (Antigen), and the signals were detected by the enhanced ECL system (Pierce).
Whole transcript expression array and GEO dataset analysis
Total RNAs of metastases from WT mice or MCP-1−/− mice (3:3) were submitted for analysis with Affymetrix MTA2.0 chips, original graphics were scanned with Affymetrix Scanner 3000. For GEO datasets of TCGA ([49], n=489), PMID17290060 ([50], n=114), GSE17260, GSE26193, GSE9891, GSE32062, data were downloaded from Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo). Raw data were subjected to intensity normalization using affy package in R (Bioconductor), followed by log transformation and quantile normalization. Normalized data were checked for quality and determined to be free of outliers by analysis using box plots, density plots and MA plots. Differential expression genes between high or low CCR-2 expression tumors (medium expression as the cutoff value) were calculated using a linear model provided by the limma package, then a preranked gene list of all expressed genes ordered by log2 fold change were constructed. To identify the potentially altered pathways in the high CCR2 patients, we performed gene set enrichment analysis (GSEA) using the package “clusterProfiler” in R. P-values<0.05 were used as significant.
Human phospho-RTK array kit
Phospho-kinase Assay Kit (ARY003B, R&D System) was used to detect levels of protein phosphorylation after culture with MCP-1 for 24 hours in C13 and SKOV3 cells according to the manufacturer’s instructions.
RPPA
SKOV3 cells were culture with or without MCP-1 (10 μg/L or 20μg/L) for 24. Protein lysates were analyzed by RPPA in MDACC CCSG (The Cancer Center Support Grant) supported RPPA Core. Antibodies and approaches are described at the RPPA website (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html).
Statistical analysis
All data are presented as the mean ± SEM. Prism software (GraphPad, La Jolla, CA, USA) was used to display all graphics. SPSS (version 17.0) was used for all statistical analysis. Two-sided Student’s t test was used to compare differences between two groups of cells in vitro. P<0.05 is considered significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank our patients for donating biospecimens and clinical data. We thank Dr. X.Jin and Dr. Z. Yang for donating the SKOV3-ERFP and SKOV3 G3 cell lines. We thank Prof. Benjamin K. Tsang for donating the C13 and OV2008 cell lines.
Funding
This study was supported by the National Key R&D Program of China (2016YFC1303100), the National Basic Research Program of China (973 Program, 2015CB553903), Nature and Science Foundation of China (81272859, 81572569, 81402163, 81402164, 81501530, 81671394, 81370469), the International S&T Cooperation Program of China (No. 2013DFA31400). GBM has support from NIH P50CA217685, Ovarian Cancer Research Foundation, and a kind gift from the Adelson Medical Research Fund.
Footnotes
Competing interests
The authors declare that they have no competing interests directly with this project. GBM is a SAB member or Consultant with AstraZeneca, Catena Pharmaceuticals, Critical Outcome Technologies, ImmunoMET, Ionis, Signalchem Lifesciences, Symphogen, Takeda/Millennium Pharmaceuticals, and Tarveda, has Stock Options or Financial arrangements with Catena Pharmaceuticals, ImmunoMet, SignalChem, Spindle Top Ventures, Tarveda, has Licensed Technology with a HRD assay to Myriad Genetics and a DSP patent with Nanostring and has sponsored Research with Adelson Medical Research Foundation, AstraZeneca, Breast Cancer Research Foundation, Immunomet, Ionis, Komen Research Foundation, Nanostring, Ovarian Cancer Research Foundation, Pfizer, Prospect Creek Foundation, and Takeda/Millennium Pharmaceuticals.
Reference
- 1.Torre LA, Trabert B, DeSantis CE, Miller KD, G Samimi CD Runowicz, et al. , Ovarian cancer statistics, 2018. CA Cancer J Clin, 2018. 68(4): p. 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castadot P, Magne N, Berghmans T, Drowart A, Baeyens L, Smets D, et al. , [Ovarian metastasis and lung adenocarcinoma: a case report]. Cancer Radiother, 2005. 9(3): p. 183–6. [DOI] [PubMed] [Google Scholar]
- 3.Lim MC, Kang S, Lee KS, Han SS, Park SJ, Seo SS, et al. , The clinical significance of hepatic parenchymal metastasis in patients with primary epithelial ovarian cancer. Gynecol Oncol, 2009. 112(1): p. 28–34. [DOI] [PubMed] [Google Scholar]
- 4.Naora H and Montell DJ, Ovarian cancer metastasis: integrating insights from disparate model organisms. Nat Rev Cancer, 2005. 5(5): p. 355–66. [DOI] [PubMed] [Google Scholar]
- 5.Tarin D, Price JE, G Kettlewell M, G Souter R, Vass AC, and Crossley B, Mechanisms of human tumor metastasis studied in patients with peritoneovenous shunts. Cancer Res, 1984. 44(8): p. 3584–92. [PubMed] [Google Scholar]
- 6.Banerjee S and Kaye SB, New strategies in the treatment of ovarian cancer: current clinical perspectives and future potential. Clin Cancer Res, 2013. 19(5): p. 961–8. [DOI] [PubMed] [Google Scholar]
- 7.Robinson E, Fisher N, Stamelos V, Redman C, and Richardson A, New strategies for the treatment of ovarian cancer. Biochem Soc Trans, 2014. 42(1): p. 125–9. [DOI] [PubMed] [Google Scholar]
- 8.Bae HS, Kim HJ, Hong JH, Lee JK, Lee NW, and Y Song J, Obesity and epithelial ovarian cancer survival: a systematic review and meta-analysis. J Ovarian Res, 2014. 7: p. 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang HS, Yoon C, Myung SK, and Park SM, Effect of obesity on survival of women with epithelial ovarian cancer: a systematic review and meta-analysis of observational studies. Int J Gynecol Cancer, 2011. 21(9): p. 1525–32. [DOI] [PubMed] [Google Scholar]
- 10.Hoy AJ, Balaban S, and Saunders DN, Adipocyte-Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol Med, 2017. 23(5): p. 381–392. [DOI] [PubMed] [Google Scholar]
- 11.Xu XJ, Gauthier MS, Hess DT, Apovian CM, Cacicedo JM, Gokce N, et al. , Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res, 2012. 53(4): p. 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Visser M, Bouter LM, McQuillan GM, Wener MH, and Harris TB, Elevated C-reactive protein levels in overweight and obese adults. JAMA,1999. 282(22): p. 2131–5. [DOI] [PubMed] [Google Scholar]
- 13.Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, et al. , Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab, 2014. 19(1): p. 162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sica A, Saccani A, Bottazzi B, Bemasconi S, Allavena P, Gaetano B, et al. , Defective expression of the monocyte chemotactic protein-1 receptor CCR2 in macrophages associated with human ovarian carcinoma. J Immunol, 2000. 164(2): p. 733–8. [DOI] [PubMed] [Google Scholar]
- 15.Milliken D, Scotton C, Raju S, F Balkwill, and J. Wilson, Analysis of chemokines and chemokine receptor expression in ovarian cancer ascites. Clin Cancer Res, 2002. 8(4): p. 1108–14. [PubMed] [Google Scholar]
- 16.Kojima R, Taniguchi H, Tsuzuki A, Nakamura K, Sakakura Y, and Ito M, Hypertonicity-induced expression of monocyte chemoattractant protein-1 through a novel cis-acting element and MAPK signaling pathways. J Immunol, 2010. 184(9): p. 5253–62. [DOI] [PubMed] [Google Scholar]
- 17.Pequeux C, Raymond-Letron I, Blacher S, Boudou F, Adlanmerini M, Fouque MJ, et al. , Stromal estrogen receptor-alpha promotes tumor growth by normalizing an increased angiogenesis. Cancer Res, 2012. 72(12): p. 3010–9. [DOI] [PubMed] [Google Scholar]
- 18.Tabuso M, Homer-Vanniasinkam S, Adya R, and Arasaradnam RP, Role of tissue microenvironment resident adipocytes in colon cancer. World J Gastroenterol, 2017. 23(32): p. 5829–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vande Broek I, Asosingh K, Vanderkerken K, Straetmans N, Van Camp B, and Van Riet I, Chemokine receptor CCR2 is expressed by human multiple myeloma cells and mediates migration to bone marrow stromal cell-produced monocyte chemotactic proteins MCP-1, −2 and −3. Br J Cancer, 2003. 88(6): p. 855–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsuyada A, Chow A, Wu J, G Somlo P Chu, S. Loera, et al. , CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res, 2012. 72(11): p. 2768–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang WB, Jokar I, Zou A, Lambert D, Dendukuri P, and Cheng N, CCL2/CCR2 chemokine signaling coordinates survival and motility of breast cancer cells through Smad3 protein- and p42/44 mitogen-activated protein kinase (MAPK)-dependent mechanisms. J Biol Chem, 2012. 287(43): p. 36593–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He YY, Du MR, Guo PF, He XJ, Zhou WH, Zhu XY, et al. , Regulation of C-C motif chemokine ligand 2 and its receptor in human decidual stromal cells by pregnancy-associated hormones in early gestation. Hum Reprod, 2007. 22(10): p. 2733–42. [DOI] [PubMed] [Google Scholar]
- 23.Guru SK, Pathania AS, Kumar S, Ramesh D, Kumar M, Rana S, et al. , Secalonic Acid-D Represses HIF1alpha/VEGF-Mediated Angiogenesis by Regulating the Akt/mTOR/p70S6K Signaling Cascade. Cancer Res, 2015. 75(14): p. 2886–96. [DOI] [PubMed] [Google Scholar]
- 24.Lu CH, Hung YJ, and Hsieh PS, Additional effect of metformin and celecoxib against lipid dysregulation and adipose tissue inflammation in high-fat fed rats with insulin resistance and fatty liver. Eur J Pharmacol, 2016. 789: p. 60–67. [DOI] [PubMed] [Google Scholar]
- 25.Al-Wahab Z, Mert I, Tebbe C, Chhina J, Hijaz M, Morris RT, et al. , Metformin prevents aggressive ovarian cancer growth driven by high-energy diet: similarity with calorie restriction. Oncotarget, 2015. 6(13): p. 10908–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez GM, Galpin KJC, McCloskey CW, and Vanderhyden BC, The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers (Basel), 2018. 10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YX, Zhu N, Zhang CJ, Wang YK, T Wu H, Li Q, et al. , Friend or foe: Multiple roles of adipose tissue in cancer formation and progression. J Cell Physiol, 2019. [DOI] [PubMed] [Google Scholar]
- 28.Yu H, Dilbaz S, Cossmann J, Hoang AC, Diedrich V, Herwig A, et al. , Breast milk alkylglycerols sustain beige adipocytes through adipose tissue macrophages. J Clin Invest, 2019. 130: p. 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen H, He M, Lin R, Zhan M, Xu S, Huang X, et al. , PLEK2 promotes gallbladder cancer invasion and metastasis through EGFR/CCL2 pathway. J Exp Clin Cancer Res, 2019. 38(1): p. 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojnarowicz P, Gambaro K, de Ladurantaye M, Quinn MC, Provencher D, Mes-Masson AM, et al. , Overexpressing the CCL2 chemokine in an epithelial ovarian cancer cell line results in latency of in vivo tumourigenicity. Oncogenesis, 2012. 1: p. e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yumimoto K, Sugiyama S, Mimori K, and Nakayama KI, Potentials of C-C motif chemokine 2-C-C chemokine receptor type 2 blockers including propagermanium as anticancer agents. Cancer Sci, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizutani K, Roca H, Varsos Z, and Pienta KJ, Possible mechanism of CCL2-induced Akt activation in prostate cancer cells. Anticancer Res, 2009. 29(8): p. 3109–13. [PubMed] [Google Scholar]
- 33.Furukawa S, Soeda S, Kiko Y, Suzuki O, Hashimoto Y, Watanabe T, et al. , MCP-1 promotes invasion and adhesion of human ovarian cancer cells. Anticancer Res, 2013. 33(11): p. 4785–90. [PubMed] [Google Scholar]
- 34.Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. , CCL2 shapes macrophage polarization by GM-CSF andM-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol, 2014. 192(8): p. 3858–67. [DOI] [PubMed] [Google Scholar]
- 35.Manome Y, Wen PY, Hershowitz A, Tanaka T, Rollins BJ, Kufe DW, et al. , Monocyte chemoattractant protein-1 (MCP-1) gene transduction: an effective tumor vaccine strategy for non-intracranial tumors. Cancer Immunol Immunother, 1995. 41(4): p. 227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu GX, Zhang X, Li S, Koiiche RD, Sindsceii JH, and Song H, Monocyte chemotactic protein-1 and CC chemokine receptor 2 polymorphisms and prognosis of renal cell carcinoma. Tumour Biol, 2013. 34(5): p. 2741–6. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Lu Y, and Pienta KJ, Multiple roles of chemokine (C-C motif) ligand 2 in promoting prostate cancer growth. J Natl Cancer Inst, 2010. 102(8): p. 522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Algire C, Moiseeva O, Deschenes-Simard X, Amrein L, Petruccelli L, Birman E, et al. , Metformin reduces endogenous reactive oxygen species and associatedDNA damage. Cancer Prev Res (Phila), 2012. 5(4): p. 536–43. [DOI] [PubMed] [Google Scholar]
- 39.Sikka A, Kaur M, Agarwal C, G Deep, and R. Agarwal, Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle, 2012. 11 (7): p. 1374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rattan R, Graham RP, Maguire JL, Giri S, and Shridhar V, Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia, 2011. 13(5): p. 483–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero IL, Mukherjee A, Kenny HA, Litchfield LM, and Lengyel E, Molecular pathways: trafficking of metabolic resources in the tumor microenvironment. Clin Cancer Res, 2015. 21(4): p. 680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giuliani M, Gui B, Valentini AL, G SE DI, Micco M, Rodolfino E, et al. , Early detection of recurrence or progression disease in patients with ovarian cancer after primary debulking surgery: a diagnostic challenge still unresolved. Correlation between CTfindings and CA 125 levels. Minerva Ginecol, 2017. [DOI] [PubMed] [Google Scholar]
- 43.Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, et al. , Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs, 2013. 31(3): p. 760–8. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. , Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut, 2017. 66(1): p. 157–167. [DOI] [PubMed] [Google Scholar]
- 45.Baldan V, Griffiths R, Hawkins RE, and Gilham DE, Efficient and reproducible generation of tumour-infiltrating lymphocytes for renal cell carcinoma. Br J Cancer, 2015. 112(9): p. 1510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Chen JN, Yang L, Li J, Wu W, Huang M, et al. , Tumor-contacted Neutrophils Promote Metastasis by a CD90-TIMP-1 Juxtacrine-Paracrine Loop. Clin Cancer Res, 2018. [DOI] [PubMed] [Google Scholar]
- 47.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, et al. , Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med, 2011. 17(11): p. 1498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu TP, Huang MD, Xia R, Liu XX, Sun M, Yin L, et al. , Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fbronectin1 expression. J Hematol Oncol, 2014. 7: p. 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Research N, Integrated genomic analyses of ovarian carcinoma. Nature, 2011. 474(7353): p. 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teng PN, Wang G, Hood BL, Conrads KA, Hamilton CA, Maxwell GL, et al. , Identification of candidate circulating cisplatin-resistant biomarkers from epithelial ovarian carcinoma cell secretomes. Br J Cancer, 2014. 110(1): p. 123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.