

Abstract
The cytosolic DNA-sensing cGAS–STING pathway was originally characterized as a key innate immune mediator responsible for the induction of antiviral genes in response to foreign DNA species in the cytosol. Mounting evidence, however, points to a complex role for cGAS and STING in cancer. Two recent reports, by Ranoa et al. (Cancer Research, 2018; https://doi.org/10.1158/0008-5472.CAN-18-1972) and Nassour et al. (Nature 2019;565:659–663), dissect the function of this pathway during the early steps of cellular transformation and shed light on the complexity and context-dependence of cGAS–STING signaling in cancer.
As the first line of host defense, the innate immune system detects invading pathogens and initiates downstream immune responses. Viral DNA is first sensed by cyclic GMP-AMP (cGAMP) synthase (cGAS), which in turn activates the endoplasmic reticulum (ER)-membrane adaptor stimulator of interferon genes (STING) through the production of the second messenger, cGAMP [1,2]. Activated STING subsequently translocates from the ER to ER–Golgi intermediate compartments, leading to induction of multiple downstream transcriptional signaling cascades, including the interferon regulatory factor 3 (IRF3) and nuclear factor-κB (NF-κB) pathways [3]. However, cGAS can also sense genomic self-DNA when it is aberrantly located in the cytosol. This occurs as a result of nuclear DNA damage or rupture of chromosome-containing micronuclei that are commonly found in genomically unstable cancer cells [4-7]. As a result, cytosolic DNA sensing was found in some contexts to inhibit [8-11] or promote [7,11-14] tumor progression.
In line with its role in proinflammatory signaling, STING activation has been shown to exert an inhibitory effect on primary tumor growth and on the proliferation of normal cells that acquire abnormal karyotypes. This effect is in part tumor cell-extrinsic, through activation of cell-mediated immunity in response to type I interferon signaling, or cell-intrinsic, through the induction of cellular senescence [2,3,10]. Whereas the former has received considerable attention, epithelial cell-intrinsic consequences of STING activation have remained poorly understood.
Two recent papers have attempted to further dissect the cellular pathways downstream of STING and their role in homeostasis and in maintaining genome stability [8,9]. In the first, Ranoa et al. studied the cell-cycle phenotype of STING depletion in primarily p53-proficient tumor cells and fibroblasts derived from humans and mouse embryos [8]. In line with in vivo transplantation results, they found that cells exhibit increased proliferation in the absence of STING. This was accompanied by a reduction in the G2/M cell fraction and increased aneuploidy, tetraploidization, and chromosome damage. Because STING ultimately functions through transcriptional activation, and to gain an insight into the mechanism by which STING affects cell-cycle progression, the researchers focused on genes whose upregulation after irradiation is STING-dependent. In addition to genes related to inflammation, genes associated with the cell cycle were enriched in STING-depleted cells.
The investigators subsequently focused on CDKN1A (p21), a cyclin-dependent kinase inhibitor that signals downstream of NF-κB and p53 to promote cell-cycle arrest in response to DNA damage. Inhibition of NF-κB was found to attenuate p21 upregulation in response to DNA damage, whereas p53 deficiency fully abrogated p21 signaling. Based on these data, the researchers proposed a model whereby STING promotes chromosome stability through the activation of p21 in a p53- and NF-κB-dependent manner (Figure 1A).
Figure 1. The Consequences of cGAS–STING Activation Are Highly Dependent on the Status of Downstream Pathways.
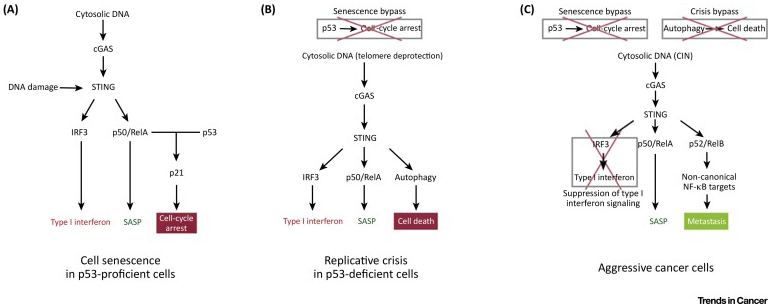
(A) In normal proliferating cells, DNA damage and cytosolic DNA arising from chromosome segregation defects activate the cGAS–STING pathway, which in conjunction with p53 signaling promotes p21-dependent cell-cycle arrest. In addition, cytosolic DNA sensing promotes robust type I interferon signaling, leading to activation of cell-mediated immunity. (B) In cells that manage to bypass senescence, STING activation can still promote cell death in response to telomeric damage through the activation of macroautophagy, a process referred to as replicative crisis. (C) In chromosomally unstable cancer cells, however, STING signaling has been shown to promote aggressive tumor cell behavior and metastasis without inducing type I interferon signaling, suggesting that the consequences of cytosolic DNA signaling in cancer are highly dependent on the status of downstream signaling pathways. Abbreviations: cGAS, cyclic GMP-AMP (cGAMP) synthase; CIN, chromosomal instability; IRF3, interferon regulatory factor 3; SASP, senescence-associated secretory phenotype; STING, stimulator of interferon genes.
In the second report, Nassour et al. identified another means by which STING protects against the proliferation of cells with chromosome damage [9]. By following human lung fibroblasts (cell lines IMR90 and WI38) in which the p53 and Rb pathways have been inactivated, cells underwent replicative crisis after ~85–105 rounds of population doublings. It is noteworthy that this senescence-independent process represents a second layer of defense against malignant transformation by killing cells with widespread chromosome damage. The authors found that genomic damage resulting from telomere shortening led to the formation of cytosolic DNA, which in turn engages cGAS and STING signaling. Unlike intrachromosomal DNA damage, that directly promoted apoptosis, telomeric crisis activated macroautophagy and cell death in a cytosolic DNA- and cGAS–STING-dependent manner. Indeed, depletion of either cGAS or STING enabled cells with complex karyotypes to propagate undeterred (Figure 1B).
The results described in these two reports reveal the essentiality of intact pathways downstream of cytosolic DNA signaling in safeguarding against cellular transformation. Equally, they highlight the importance of the cellular context in determining the consequences of cGAS–STING pathway activation. In the presence of functional p53, cells are likely to undergo senescence before reaching replicative crisis, explaining why cGAS and STING depletion promotes fibroblast proliferation after as few as five passages [10]. However, in the event of senescence bypass, transformation of cells with abnormal chromosomes is prevented through cell death mediated by macroautophagy in response to STING activation. In these contexts the effects of cGAS or STING depletion are not observed until at least 40–50 passages.
These results shed light on the apparently paradoxical roles of the cGAS–STING pathway in cancer [8,9,4,7,11-14]. Although loss of cGAS or STING might serve as a mechanism to abrogate key safeguards against transformation and immune recognition in a minority of tumors, a significant proportion of human cancers and cancer-derived cell lines have intact cGAS and STING proteins. Their function has been linked to poor prognosis and metastatic progression in many cancer types [7,11,12,15] (Figure 1C). Perhaps not surprisingly, the major cellular pathways associated with cytosolic DNA signaling, such as chromosomal instability (CIN), autophagy, and the senescence-associated secretory phenotype (SASP), have all been shown to have opposing roles in cancer – they are tumor-suppressive during the early stages of tumorigenesis and tumor-promoting in the later stages of disease progression. Therefore, the net effect of cytosolic DNA signaling in cancer is crucially dependent on the integrity of its downstream pathways. Genetic events that enable senescence bypass or abrogation of autophagy would alter the consequences of cGAS–STING activation on cellular fitness and can lead to diametrically opposite effects. For instance, abrogation of this pathway had no effect on in vitro proliferation of p53-defective MDA-MB-231 breast cancer cells, which are characterized by high levels of CIN [7]. It is also important to note that these results do not directly implicate STING in the process of chromosome segregation during mitosis, but only in preventing downstream tolerance for genomic instability arising in normal cells and assuming properly functioning checkpoints that enable senescence or cell death during replicative crisis.
This body of work represents a step forward in our understanding of the various pathways activated in response to cytosolic DNA. Historically, cGAS and STING have been primarily examined through the lens of type I interferon induction; from this perspective it was widely assumed that this pathway was completely lost in cancer cells. Invoking a cell-intrinsic role in cell-cycle regulation and autophagy adds a layer of complexity and expands the scope as well as the biological consequences of cytosolic DNA sensing. Key questions remain to be answered. Among them are a better insight into the mechanisms by which STING activation promotes autophagy, how tumor cells suppress type I interferon signaling while preserving other STING-dependent pathways, and a more comprehensive delineation of the contexts in which cGAS and STING act synonymously versus independently. For instance, in the case of cell-cycle regulation, depletion of STING – but not of cGAS – impacted on the G1/S transition in response to chromosome damage [8]. This supports recent evidence for cGAS-independent STING signaling in response to nuclear DNA damage [16]. On the other hand, autophagy was equally sensitive to disruption of cGAS or STING [9]. A better understanding of cytosolic DNA signaling pathways in their proper contexts will be crucial for our ability to harness this biology toward a therapeutic benefit.
Footnotes
Disclaimer Statement
S.F.B. has consulted for Sanofi and also holds a patent that that includes targeting CIN and the cGAS–STING pathway in advanced cancers.
References
- 1.Sun L et al. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa H and Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cai X et al. (2014) The cGAS–cGAMP–STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296 [DOI] [PubMed] [Google Scholar]
- 4.Ahn J et al. (2014) Inflammation-driven carcinogenesis is mediated through STING. Nat. Commun 5, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harding SM et al. (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackenzie KJ et al. (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bakhoum SF et al. (2018) Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranoa DRE et al. (2018) STING promotes homeostasis via regulation of cell proliferation and chromosomal stability. Cancer Res. Published online November 27, 2018. 10.1158/0008-5472.CAN-18-1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nassour J et al. (2019) Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H et al. (2017) cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U. S. A 114, E4612–E4620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dou Z et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q et al. (2016) Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu H et al. (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 [DOI] [PubMed] [Google Scholar]
- 14.Liang H et al. (2017) Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun 8, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An X et al. (2018) An analysis of the expression and association with immune cell infiltration of the cGAS/STING pathway in pan-cancer. Mol. Ther. Nucleic Acids 14, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunphy G et al. (2018) Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol. Cell 71, 745–760 [DOI] [PMC free article] [PubMed] [Google Scholar]