

Abstract
Increasing studies have reported that long noncoding RNAs (lncRNAs) play critical roles in the initiation and progression of carcinogenesis. However, the underlying regulatory mechanisms of lncRNA-related competing endogenous RNA (ceRNA) network in colorectal cancer (CRC) are not fully understood. In the present study, we systematically analyzed the expression levels and prognostic values of dysregulated microRNAs (miRNAs) in human CRC to identify novel survival-related lncRNA-miRNA-mRNA ceRNA regulatory network. As a result, 28 dysregulated miRNAs were obtained, and hsa-miR-195-5p was identified as a key oncogene in human CRC based on analyses of expression levels and prognostic values. By means of stepwise prediction and validation, two upstream lncRNAs (NEAT1, XIST) and eight downstream mRNAs (ACOX1, CYP26B1, IRF4, ITPR1, LITAF, PHLPP2, RECK, and TPM2) were identified as key genes that interact with hsa-miR-195-5p. A ceRNA regulatory network consisted of these key genes was constructed, and Gene Set Enrichment Analysis (GSEA) indicated the possible association of key mRNAs with CRC onset and progression. Importantly, immune infiltration analysis revealed that the ceRNA network was remarkably associated with infiltration abundance of multiple immune cells and expression levels of immune checkpoints. These findings indicate that NEAT1 and XIST are potential prognostic factors that affect CRC onset and progression by targeting miR-195-5p.
Keywords: Colorectal cancer, competing endogenous RNA, long noncoding RNA, microRNA, bioinformatic analysis, immune infiltration
Introduction
Malignant tumor, with more than 18.0 million newly diagnosed cases and 9.0 million deaths in 2018, has become the main threat to public health around the world [1]. Among them, colorectal cancer (CRC) is one of the most frequently diagnosed digestive malignancies, which remains the third leading cause of cancer-related death worldwide [1,2]. Early diagnosis and timely radical resection are critical for CRC. The 5-year survival rate is approximately 90% when the localized disease was diagnosed at an early stage. However, the 5-year survival rate of patients with distant metastasis dropped sharply to only 12% [3]. Metastasis and recurrence are the major causes of death in CRC patients. It is estimated that more than 50% of CRC patients will die from metastasis-related complications, which might be due to the lack of specific and sensitive biomarkers [4]. To make matters worse, available epidemiological data indicate that the incidence rates of human CRC have increased faster than any other types of cancer around the world [5]. Despite advances in surgical technology and targeted therapy, the prognosis of patients with CRC remains dismal because of recurrence and distant metastasis. Therefore, exploring the molecular mechanisms underlying colorectal carcinogenesis is imperative and meaningful to provide novel prognostic and therapeutic biomarkers for clinical CRC treatment.
The occurrence of CRC is a complex multi-step process involving accumulation of various genetic and epigenetic alternations [6]. In recent years, increasing evidence has indicated that long noncoding RNA (lncRNA) plays a significant role in the development of multiple types of malignant tumor, including CRC [7-10]. LncRNA is a class of non-coding transcripts with more than 200 nucleotides in length [11]. Salmena et al [12] first proposed the competing endogenous RNA (ceRNA) hypothesis that genes could achieve crosstalk among each other by forming a regulatory network. The ceRNA hypothesis postulates that lncRNAs could act as endogenous miRNA sponges and inhibit miRNA function, thereby modulating miRNA-mediated target inhibition. In other words, the expression level of miRNA could be negatively correlated with its upstream lncRNA and downstream mRNA simultaneously [13]. Therefore, ceRNA crosstalk is a vital mechanism underlying the complex pathogenesis and multistage progression of human CRC, but our knowledge about the lncRNA related ceRNA network in human CRC remains limited, exploring the ceRNA regulatory network might be a potential pointcut for identifying novel early diagnostic and therapeutic targets of CRC and deserves further research.
In the present study, dysregulated miRNAs in human CRC were identified by mining the gene expression profiles obtained from the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) database. Subsequently, two miRNAs were identified as key miRNA for further study through comprehensive consideration of the expression levels and prognostic values of these miRNAs in human CRC. Then, the upstream lncRNAs and downstream mRNAs of the key miRNAs were predicted by using multiple bioinformatic tools, and a ceRNA regulatory network was constructed and visualized by using Cytoscape software. In addition, the Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of the ceRNA regulatory network were performed by using the DAVID database. Ultimately, the expression levels and prognostic values of the upstream lncRNAs and downstream mRNAs in the ceRNA regulatory network were assessed, and a novel ceRNA network in which each RNA was significantly correlated with the prognosis of patients with CRC was constructed successfully. Furthermore, we used the Gene Set Enrichment Analysis (GSEA) software package to explore the pathway enrichment of this survival-related ceRNA regulatory network. Finally, Tumor Immune Estimation Resource (TIMER) database was used to estimate the correlations between ceRNA network and immune infiltration level in CRC microenvironment (Figure 1). Through our efforts, it is hoped that the results of our research will contribute to enhancing our understanding of the regulatory mechanisms underlying the pathogenesis of human CRC and finding new therapeutics for clinical CRC treatment.
Figure 1.
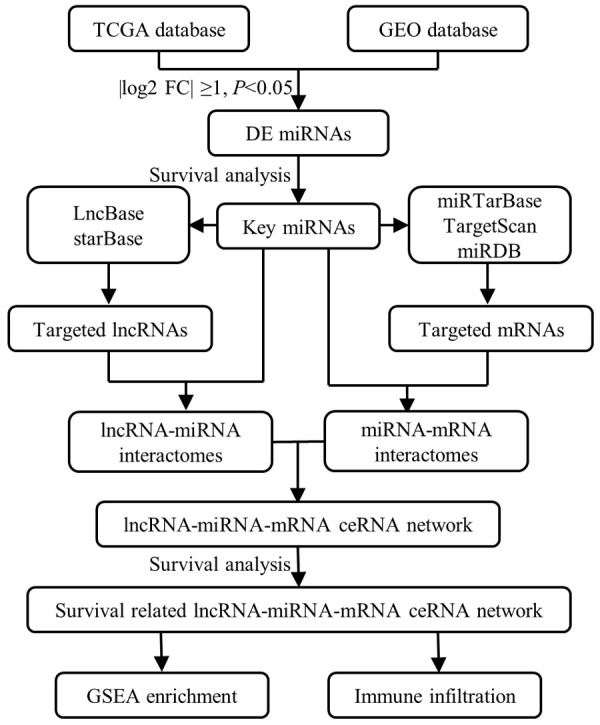
Flow chart of the construction of ceRNA network and regulatory axes in CRC.
Materials and methods
Data download and preprocessing
To screen out the dysregulated miRNAs in human CRC, human miRNA expression profile GSE108153 (including 21 cancer samples and 21 adjacent normal samples) [14] was downloaded from the Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo) [15], which was based on the GPL19730 platform (Agilent-046064 Unrestricted_Human_miRNA_V19.0_Microarray). Meanwhile, to enhance the reliability of our results, miRNA expression profiles (including 9 normal samples and 539 cancer samples) and clinical information (including 547 cases) of patients with CRC were obtained from The Cancer Genome Atlas (TCGA, https://gdc-portal.nci.nih.gov/) database.
Identification of dysregulated miRNAs
First, we converted the miRNA probe ID in the matrix files to mature miRNA name based on the annotation document of its platform files through the Perl language (https://www.perl.org/). Subsequently, the “Limma” package in R software (V 3.6.2; http://www.r-project.org/) was used to identify differentially expressed miRNAs between CRC samples and normal samples both in miRNA expression profiles obtained from GEO and TCGA databases. Adjusted P-value of < 0.05 and absolute value of log2 fold change (log2 FC) ≥1 were considered as the cut-off criteria [16]. Finally, the differentially expressed miRNAs dysregulated in both GEO and TCGA datasets were chosen for subsequent analysis by using the intersect function of the Venn Diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/).
Identification of key miRNAs
To identify the key miRNAs associated with the prognosis of CRC patients, the expression levels and prognostic values of the dysregulated miRNAs were assessed by using clinical information obtained from the TCGA database. The dysregulated miRNAs not only differentially expressed between the CRC cancer samples and normal samples, but also associated with the prognosis of CRC patients were identified as key miRNAs in human CRC and were chosen for subsequent research, according to the criteria of a P-value < 0.05. In addition, we further validated the expression pattern of key miRNAs in multiple types of cancer by using the Gene Expression Display Server (GEDS, http://bioinfo.life.hust.edu.cn/web/GEDS/) database, a web-based tool for quantification, comparison, and visualization of gene expression data based on The Cancer Genome Atlas, Genotype-Tissue Expression (GETx), Cancer Cell Line Encyclopedia (CCLE), and MD Anderson Cell Lines Project (MCLP) databases [17].
Construction of ceRNA regulatory network and functional enrichment analysis
The starBase database (http://starbase.sysu.edu.cn/) is an online tool that allows researchers to perform Pan-Cancer analysis on RNA-RNA and RBP-RNA interactions [18]. The LncBase V2 database (http://carolina.imis.athena-innovation.gr/) is a web-based tool which caters information regarding cell type specific miRNA-lncRNA interaction and enables users to easily identify interactions in 66 different cell types, spanning 36 tissues for human and mouse [19]. The upstream lncRNAs which interacted with the key miRNAs were predicted by using these two databases. Only those lncRNAs predicted by both two databases and interacted with all key miRNAs simultaneously were identified as upstream lncRNAs. Subsequently, the downstream mRNAs which interacted with the key miRNAs were predicted by using miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/) [20], targetScan (http://www.targetscan.org) [21], and miRDB (http://www.mirdb.org/) [22]. To enhance the reliability of the predicted interaction relationships, only the predicted mRNAs presented in all three databases were identified as the downstream mRNAs. Ultimately, the lncRNA-miRNA-mRNA regulatory network was constructed and visualized by Cytoscape software (v3.7.2, http://www.cytoscape.org/) [23]. Furthermore, the functional enrichment analysis of this ceRNA regulatory was performed by using the Database for Annotation, Visualization, and Integrated Discovery (DAVID v6.8 https://david.ncifcrf.gov/home.jsp) [24] bioinformatics database. Only the terms with a P-value < 0.05 were considered as statistically significant.
Gene expression validation and survival analysis
To further enhance the reliability of the predicted interaction relationships, the expression levels of upstream lncRNAs were assessed by using the Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/detail.php), a web-based tool that provides fast and customizable functionalities including differential expression analysis, profiling plotting, correlation analysis, and patient survival analysis based on TCGA and Genotype-Tissue Expression data [25]. The expression levels of downstream mRNAs were assessed by using the gene expression profiles in human CRC obtained from the TCGA database. Subsequently, the prognostic values of the upstream lncRNAs and downstream mRNAs were validated in human CRC by using the PrognoScan (http://dna00.bio.kyutech.ac.jp/PrognoScan/) database, a web-based tool for assessing the biological relationship between gene expression and prognosis of multiple types of cancer [26]. Only genes that complied with the ceRNA hypothesis and associated with the prognosis of patients with CRC were chosen for subsequent analysis.
Identification of survival related ceRNA regulatory network and functional annotation
Based on the prediction and evaluation above, a novel lncRNA-miRNA-mRNA regulatory network associated with the prognosis of CRC patients was constructed and visualized by Cytoscape software. In addition, to further explore the underlying role of this survival-related ceRNA regulatory network in the process of human CRC, functional annotation of genes in the ceRNA regulatory network was analyzed by the Gene Set Enrichment Analysis (GSEA, https://www.gsea-msigdb.org/) database, with an initial database of 1,325 biologically defined gene sets [27]. The enriched terms with P-value of < 0.05 were considered statistically significant.
Correlation of ceRNA network with immune infiltration level in human CRC
The Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/) is an online database established for systematical analysis of immune infiltrates across multiple cancer types [28]. We applied the TIMER database to assess the correlations of gene expression in the ceRNA regulatory network with the level of immune infiltrates in human CRC. In addition, the correlations between the ceRNA regulatory network and abundance of T-cell infiltration in human CRC were confirmed by using the GEPIA database.
Statistical analysis
Most of the statistical analyses were carried out using the bioinformatic tools mentioned above, while the rest were carried out using R software (v 3.6.2). Identification of differentially expressed genes was assessed by Student’s t-test, and Benjamini-Hochberg False Discovery Rate (FDR) method was applied to adjust the P-value. Kaplan-Meier curve analysis with log-rank test was used to evaluate the prognostic values of miRNAs in human CRC. Spearman correlation coefficients were used to evaluate the correlations. A P-value < 0.05 was considered statistically different.
Results
Identification of dysregulated miRNAs in human CRC
As shown in the volcano plot and heatmap, differentially expressed miRNAs in GEO and TCGA datasets were identified separately according to the predefined threshold (Figure 2A-D). In the GSE108153 dataset, a total of 49 dysregulated miRNAs were identified. Meanwhile, a total of 504 dysregulated miRNAs were also screened out in the TCGA dataset. A total of 28 dysregulated miRNAs were selected for subsequent analyses by using the intersect function of the Venn Diagram (Figure 2E).
Figure 2.
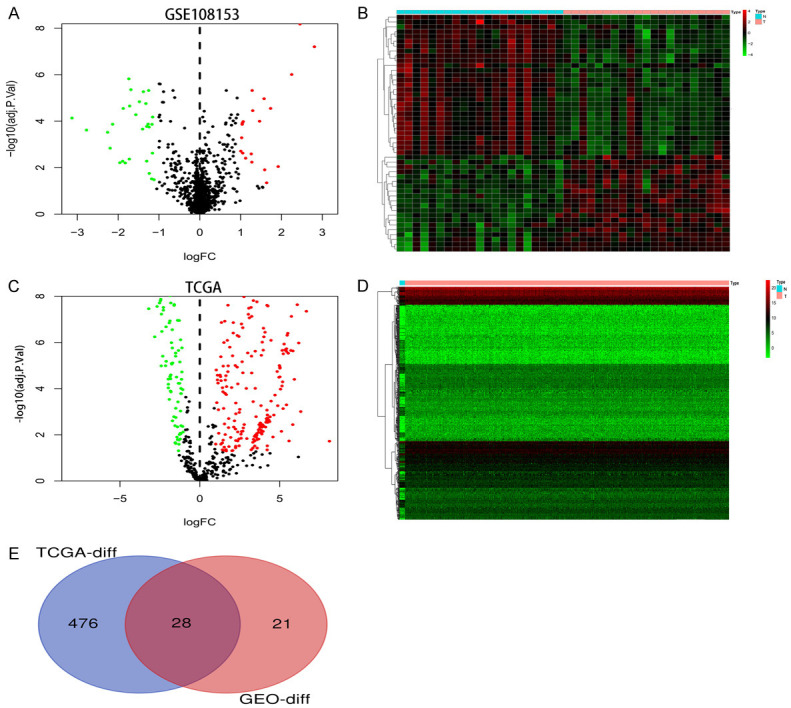
Identification of differentially expressed miRNA in human CRC. A, C. The volcano of differentially expressed miRNA in GSE108153 and TCGA datasets. The green dots and red dots represent the significantly downregulated and upregulated miRNAs, respectively. The black dots represent miRNAs with no significant difference. B, D. The heat map of differentially expressed miRNA in GSE108153 and TCGA datasets. E. The intersection of differentially expressed miRNAs in GSE108153 and TCGA datasets, respectively.
Identification of key miRNAs
To evaluate the prognostic values of dysregulated miRNAs for overall survival in human CRC, clinical information, including survival status and survival time of 547 CRC patients, were obtained from the TCGA database. The results showed that five of the dysregulated miRNAs (miR-195-5p, miR-10b-5p, miR-21-3p, miR-125b-5p, and miR-145-5p) were significantly associated with the prognosis of CRC patients (P < 0.05; Figure 3A-E). Meanwhile, we compared the expression levels of these survival-related miRNAs between cancer samples and normal samples in the TCGA dataset. After combining the prognostic value and expression level, we found that only two of them (miR-195-5p and miR-10b-5p) showed both high expression and poor prognosis in human CRC (P < 0.05; Figure 3F-J). To further enhance the reliability of our screening results, we verified the two dysregulated miRNAs in the GEDS database. As shown in Figure 3K and 3L, miR-195-5p and miR-10b-5p were dysregulated in multiple cancer types, including CRC. Thus, those two miRNAs were chosen for subsequent analyses.
Figure 3.

Identification of key miRNAs in CRC. A-E. The Kaplan-Meier curve of the key miRNAs. F-J. The expression box plot of the key miRNAs. K, L. The expression box plot of hsa-miR-9-5p and hsa-miR-10b-5p in multiple types of cancers.
Construction of ceRNA regulatory network
The upstream lncRNAs that could potentially bind to the key miRNAs were predicted through starBase and LncBase V2 databases, and a total of three upstream lncRNAs were predicted to regulate the two key miRNAs simultaneously (Figure 4A). Subsequently, to identify downstream mRNAs that could potentially be targeted by those two key miRNAs, we applied miRTarBase, targetScan, and miRDB to improve the credibility of the predicted results. As a result, a total of 287 downstream mRNAs were predicted to be targeted by miR-195-5p, while there were 22 downstream mRNAs could potentially be targeted by miR-10b-5p (Figure 4B, 4C). Based on the prediction above, a ceRNA regulatory network comprised of six lncRNA-miRNA pairs and 310 miRNA-mRNA pairs was established and visualized by Cytoscape software (Figure 4D).
Figure 4.

Identification of lncRNA-miRNA-mRNA regulatory network in CRC. A. The intersection of upstream lncRNAs targeted by hsa-miR-9-5p and hsa-miR-10b-5p in LncBase v2 and starBase databases. B, C. The intersection of downstream mRNAs targeted by hsa-miR-9-5p and hsa-miR-10b-5p in miRTarBase, Targetscan, and miRDB databases. D. The lncRNA-miRNA-mRNA regulatory network constructed by Cytoscape software. The green round rectangle in the network represents miRNA. The pink ellipse in the network represents mRNA. The red diamond in the network represents lncRNA.
Functional analysis for the ceRNA regulatory network
We used the DAVID database to further explore the biological functions of the ceRNA regulatory network. A total of 222 GO terms and 45 KEGG pathways were enriched, and the top 10 enriched GO terms of each group and the top 20 enriched KEGG pathways according to P-value were displayed (Tables 1, 2). In the biological processes (BP) group, the ceRNA regulatory network is mainly enriched in the process of regulation of transcription from RNA polymerase II promoter, regulation of cell migration, regulation of cell adhesion, and regulation of the apoptotic process. In the cellular component (CC) group, the ceRNA regulatory network is mainly enriched in nucleoplasm, cytoplasm, and nucleus. In the molecular function (MF) group, the ceRNA regulatory network is mainly enriched in protein binding, beta-catenin binding, and ubiquitin-protein ligase activity. These results indicated that the ceRNA network was significantly enriched in terms correlated with cell proliferation, cell migration, and cell apoptosis. The KEGG enrichment analysis revealed that the ceRNA regulatory network was significantly enriched in multiple cancer-related pathways such as PI3K-Akt signaling pathway, FoxO signaling pathway, signaling pathways regulating pluripotency of stem cells, and AMPK signaling pathway.
Table 1.
Gene Ontology analysis of the ceRNA regulatory network
| Category | Term | Count | P Value |
|---|---|---|---|
| GOTERM_BP_DIRECT | GO: 0045944~positive regulation of transcription from RNA polymerase II promoter | 40 | 1.07E-06 |
| GOTERM_BP_DIRECT | GO: 0006468~protein phosphorylation | 24 | 4.97E-06 |
| GOTERM_BP_DIRECT | GO: 0016055~Wnt signaling pathway | 14 | 2.35E-05 |
| GOTERM_BP_DIRECT | GO: 0000122~negative regulation of transcription from RNA polymerase II promoter | 28 | 1.43E-04 |
| GOTERM_BP_DIRECT | GO: 0030336~negative regulation of cell migration | 9 | 2.44E-04 |
| GOTERM_BP_DIRECT | GO: 0035264~multicellular organism growth | 8 | 4.71E-04 |
| GOTERM_BP_DIRECT | GO: 0001657~ureteric bud development | 6 | 4.74E-04 |
| GOTERM_BP_DIRECT | GO: 0030512~negative regulation of transforming growth factor beta receptor signaling pathway | 7 | 8.30E-04 |
| GOTERM_BP_DIRECT | GO: 0045785~positive regulation of cell adhesion | 6 | 8.48E-04 |
| GOTERM_BP_DIRECT | GO: 0043066~negative regulation of apoptotic process | 19 | 0.001023 |
| GOTERM_CC_DIRECT | GO: 0005654~nucleoplasm | 91 | 2.72E-11 |
| GOTERM_CC_DIRECT | GO: 0005737~cytoplasm | 134 | 1.46E-09 |
| GOTERM_CC_DIRECT | GO: 0005829~cytosol | 91 | 1.90E-07 |
| GOTERM_CC_DIRECT | GO: 0005634~nucleus | 128 | 8.32E-07 |
| GOTERM_CC_DIRECT | GO: 0016604~nuclear body | 5 | 0.001925 |
| GOTERM_CC_DIRECT | GO: 0035068~micro-ribonucleoprotein complex | 3 | 0.003777 |
| GOTERM_CC_DIRECT | GO: 0005770~late endosome | 8 | 0.0038 |
| GOTERM_CC_DIRECT | GO: 0070578~RISC-loading complex | 3 | 0.005232 |
| GOTERM_CC_DIRECT | GO: 0005913~cell-cell adherens junction | 13 | 0.006775 |
| GOTERM_CC_DIRECT | GO: 0000421~autophagosome membrane | 4 | 0.007488 |
| GOTERM_MF_DIRECT | GO: 0005515~protein binding | 215 | 2.47E-15 |
| GOTERM_MF_DIRECT | GO: 0008013~beta-catenin binding | 11 | 1.25E-06 |
| GOTERM_MF_DIRECT | GO: 0061630~ubiquitin protein ligase activity | 14 | 1.93E-05 |
| GOTERM_MF_DIRECT | GO: 0048185~activin binding | 5 | 3.64E-05 |
| GOTERM_MF_DIRECT | GO: 0003682~chromatin binding | 20 | 4.27E-05 |
| GOTERM_MF_DIRECT | GO: 0004672~protein kinase activity | 17 | 4.50E-04 |
| GOTERM_MF_DIRECT | GO: 0030165~PDZ domain binding | 8 | 6.52E-04 |
| GOTERM_MF_DIRECT | GO: 0004674~protein serine/threonine kinase activity | 17 | 7.43E-04 |
| GOTERM_MF_DIRECT | GO: 0019901~protein kinase binding | 17 | 7.43E-04 |
| GOTERM_MF_DIRECT | GO: 0043565~sequence-specific DNA binding | 20 | 0.001444 |
Table 2.
Kyoto Encyclopedia of Genes and Genomes pathway analysis of the ceRNA regulatory network
| Category | Term | Count | P Value |
|---|---|---|---|
| KEGG_PATHWAY | hsa05221: Acute myeloid leukemia | 8 | 9.08E-05 |
| KEGG_PATHWAY | hsa05212: Pancreatic cancer | 8 | 2.36E-04 |
| KEGG_PATHWAY | hsa05211: Renal cell carcinoma | 8 | 2.60E-04 |
| KEGG_PATHWAY | hsa05205: Proteoglycans in cancer | 13 | 4.60E-04 |
| KEGG_PATHWAY | hsa05200: Pathways in cancer | 19 | 5.18E-04 |
| KEGG_PATHWAY | hsa04151: PI3K-Akt signaling pathway | 17 | 9.45E-04 |
| KEGG_PATHWAY | hsa05222: Small cell lung cancer | 8 | 0.0012152 |
| KEGG_PATHWAY | hsa04114: Oocyte meiosis | 9 | 0.0013139 |
| KEGG_PATHWAY | hsa04910: Insulin signaling pathway | 10 | 0.001344 |
| KEGG_PATHWAY | hsa04914: Progesterone-mediated oocyte maturation | 8 | 0.0013934 |
| KEGG_PATHWAY | hsa04110: Cell cycle | 9 | 0.0026574 |
| KEGG_PATHWAY | hsa04068: FoxO signaling pathway | 9 | 0.0042832 |
| KEGG_PATHWAY | hsa04550: Signaling pathways regulating pluripotency of stem cells | 9 | 0.0055743 |
| KEGG_PATHWAY | hsa05210: Colorectal cancer | 6 | 0.0067139 |
| KEGG_PATHWAY | hsa04722: Neurotrophin signaling pathway | 8 | 0.0083948 |
| KEGG_PATHWAY | hsa04152: AMPK signaling pathway | 8 | 0.0095582 |
| KEGG_PATHWAY | hsa05220: Chronic myeloid leukemia | 6 | 0.012461 |
| KEGG_PATHWAY | hsa05203: Viral carcinogenesis | 10 | 0.0175239 |
| KEGG_PATHWAY | hsa05213: Endometrial cancer | 5 | 0.0176717 |
| KEGG_PATHWAY | hsa04931: Insulin resistance | 7 | 0.0179023 |
Identification and validation of key lncRNA and mRNA
Based on the ceRNA hypothesis, the miRNA could be negatively correlated with its upstream lncRNA and downstream mRNA simultaneously. Therefore, we assessed the expression levels and prognostic values of the predicted lncRNAs for overall survival in patients with CRC by using GEPIA and PrognoScan databases, respectively. Only two lncRNAs (NEAT1 and XIST) showed both low expression and improved prognosis in patients with CRC (Figure 5A, 5B). As for downstream mRNA, we collected gene expression profiles in human CRC from the TCGA database to assess the expression levels of the predicted mRNA. Meanwhile, the prognostic values of those mRNAs for overall survival in patients with CRC were also evaluated using the PrognoScan database. A total of 1964 downregulated genes were identified in human CRC from the TCGA database, and we then integrated the 1964 downregulated genes with the downstream mRNAs in the ceRNA regulatory network. As a result, no specific gene was identified to be targeted by miR-10b-5p, while there were 16 specific genes could potentially be targeted by miR-195-5p (Figure 5C, 5D).
Figure 5.

Identification of survival-related lncRNA-miRNA-mRNA regulatory network in CRC. A, B. The expression levels and prognostic values of upstream lncRNA NEAT1 and XIST. C, D. The intersection of downstream mRNAs and down-regulated mRNAs in TCGA datasets, respectively. E-L. The expression levels and prognostic values of downstream mRNAs.
Subsequently, survival analysis found that eight of the 16 specific genes (ACOX1, CYP26B1, IRF4, ITPR1, LITAF, PHLPP2, RECK, and TPM2) were significantly associated with the prognosis of patients with CRC. The expression boxplot and survival curve of those mRNAs are displayed in Figure 5E-L. Those two lncRNAs and eight mRNAs were chosen for further analysis.
Integration of survival-related ceRNA regulatory network and GSEA enrichment analysis
Based on the validation above, a survival-related ceRNA regulatory network including two lncRNA-miRNA pairs (NEATI-miR-195-5p and XIST-miR-195-5p) and eight miRNA-mRNA pairs (miR-195-5p-ACOX1/CYP26B1/IRF4/ITPR1/LITAF/PHLPP2/RECK/TPM2) was established (Figure 6A). Each component in the ceRNA regulatory network had significant prognostic values in human CRC, and fully complied with the rule of the ceRNA hypothesis. After that, we used the GSEA software package to further explore the biological function of the survival-related ceRNA regulatory network. As shown in Figure 6B, GSEA enrichment analysis indicated that the survival-related ceRNA regulatory network was significantly enriched in multiple cancer-related pathways, such as the JAK-STAT signaling pathway, mTOR signaling pathway, TGF-β signaling pathway, Wnt signaling pathway, ERBB signaling pathway, and VEGF signaling pathway.
Figure 6.

Construction of survival-related lncRNA-miRNA-mRNA regulatory network in human CRC. A. The ceRNA regulatory network associated with the prognosis of patients with CRC. The red diamond represents key lncRNA. The green round rectangle represents key miRNA. The pink ellipse represents key mRNA. B. GSEA enrichment analysis of mRNAs in the ceRNA regulatory network.
Correlation of ceRNA network with immune infiltration level in human CRC
The correlation between the survival-related ceRNA regulatory network and tumor-infiltrating immune cells in the CRC microenvironment was assessed by using TIMER. As shown in Figure 7A-H, we found that gene expression in the ceRNA regulatory network was significantly associated with infiltrating immune cells, including B cells, CD4+/CD8+ T cells, macrophages, meutrophils, and dendritic cells, in the human CRC microenvironment. Subsequently, the correlations between the ceRNA regulatory network and the abundance of T cells infiltration in human CRC were further evaluated by using the GEPIA database, and the results indicated that the survival-related ceRNA regulatory network was closely associated with the abundance of central memory T cells (r = 0.75, P = 0), effector T cells (r = 0.73, P = 0), effector Treg T-cells (r = 0.74, P = 0), exhausted T cells (r = 0.68, P = 0), naïve T cells (r = 0.72, P = 0), resident memory T cells (r = 0.69, P = 0), resting Treg T-cells (r = 0.76, P = 3.9 e-69), and Th1-like cells (r = 0.72, P = 0) (Figure 8A-H). We also found that the ceRNA regulatory network was closely associated with the expression of multiple well-known immune checkpoints, such as PDL1 (r = 0.48, P = 1.6 e-16), CTLA-4 (r = 0.60, P = 1.3 e-37), TIM-3 (r = 0.71, P = 1.1 e-56), and LAG-3 (r = 0.46, P = 2 e-20), suggesting that the ceRNA regulatory network participates in the process of immune checkpoint mediated T cell failure (Figure 8I-L). Therefore, these findings further support that this survival-related ceRNA regulatory network was closely correlated with immune infiltration level in human CRC, suggesting that the ceRNA regulatory network might be an important participant in immune escape in the CRC microenvironment.
Figure 7.

Association of ceRNA network expression with immune infiltration level in CRC. A-H. Association of mRNA expression in the survival-related ceRNA regulatory network with immune infiltration level in CRC.
Figure 8.

Associations of ceRNA network expression with T cell immune infiltration abundance and immune checkpoint expression level in CRC. A-H. Expression of mRNAs in the survival-related ceRNA network was significantly correlated with immune infiltration abundance of T cells. I-L. Expression of mRNAs in the survival-related ceRNA network was significantly correlated with the expression level of the immune checkpoint.
Discussion
Accumulating studies have reported that lncRNA acts as a post-transcriptional regulator that regulates downstream mRNA expression by sponging miRNA, and participates in multiple processes of tumorigenesis, including CRC. For instance, lncRNA CRNDE has been reported to function as a sponge for miR-181a-5p, thereby promoting the progression and chemoresistance of CRC cells through modulating the expression levels of TCF4 and the activity of Wnt/β-catenin signaling [29]. Previous research reported that lncRNA SNHG1 promotes CRC cell growth by sponging miR-154-5p to regulate the expression level of CCND2, which plays a critical role in cell cycle progression [30]. LncRNA MIR17HG was reported to increase the expression level of NF-κB/RELA through competitively sponging miR-375, thereby promoting tumorigenesis and metastasis in CRC cells. In addition, MIR17HG was reported to upregulate the expression of PD-L1, suggesting that it may participant in immune escape in the CRC microenvironment [31]. Nevertheless, the clinical utility of lncRNA-related ceRNA regulatory network is limited at present. Exploring the lncRNA related ceRNA regulatory network will help to provide a comprehensive perspective of the molecular mechanisms of gene interaction and regulation in human CRC.
In the present study, dysregulated miRNAs with prognostic significance in human CRC were identified in cancer samples compared with adjacent non-cancer samples from TCGA and GEO databases. Then, a novel lncRNA-miRNA-mRNA regulatory network involved in CRC was constructed through stepwise prediction and validation. Functional enrichment analysis revealed that this novel ceRNA regulatory network was significantly enriched in some cancer-related pathways, such as PI3K-Akt signaling pathway [32], FoxO signaling pathway [33], Wnt signaling pathway [34], and AMPK signaling pathway [35]. To enhance the reliability of our study, we further assessed the expression levels and prognostic values of genes in the ceRNA regulatory network and constructed a survival-related ceRNA regulatory network; each constituent in the network has prognostic significance in human CRC, which might provide novel diagnostic and therapeutic targets for clinical treatment of CRC.
LncRNAs are defined as transcripts that are > 200 nucleotides in length and have no ability to code protein [36]. Previous studies have reported that lncRNA participates in multiple biological processes and tumorigenesis by sponging miRNA [37]. In the present study, we found that lncRNA NEAT1 and XIST were not only dysregulated in CRC, but also significantly associated with poor prognosis, and these two lncRNAs were identified as key lncRNA in CRC. Studies have shown that expression of lncRNA XIST was dysregulated in CRC cell lines and tissues. XIST knockdown inhibited the cell migration and invasion of CRC cells in vitro and in vivo by regulating the processes of epithelial-mesenchymal transition (EMT), an important step in tumor progression and metastasis [38,39]. In addition, dysregulated XIST has been found to be involved in chemoresistance of CRC to Doxorubicin, with the silencing of XIST significantly enhancing the anti-tumor effect of Doxorubicin in CRC [40]. There is also increasing evidence that lncRNA NEAT1 was dysregulated in human CRC tissues and was significantly correlated with worse overall survival in CRC patients. NEAT1 regulates the Wnt/β-catenin signaling pathway by targeting DDX5, thereby facilitating CRC cell migration and invasion in vitro and in vivo [41]. Furthermore, NEAT1 also has been found to participate in chemoresistance of CRC, and NEAT1 knockdown increased the sensitivity of 5-fluorouracil and promoted apoptosis in CRC cells [42]. These studies above improved the reliability of our research finding that dysregulation of lncRNA XIST and NEAT1 was closely correlated with the pathogenesis of CRC, which is valuable for further study.
MiRNAs are defined as endogenous non-coding RNAs that are 21-25 nucleotides in length and suppress protein-coding gene expression by binding to the 3’-untranslated regions (UTRs) of mRNAs [43]. Dysregulation of miRNA has been reported to be associated with the onset and progression of human CRC. For instance, expression of miR-330 was downregulated in CRC samples compared with normal control, and CRC patients with low miR-330 expression showed poorer overall survival than those with high miR-330 expression. As of mechanism, miR-330 suppresses cell proliferation, migration, invasion, and angiogenesis by reducing the phosphorylation of AKT and STAT3 via targeting HMGA2 in vitro [44]. Moreover, expression of miR-128-3p in tumor samples was closely associated with oxaliplatin sensitivity in human CRC, and forced-expression of miR-128-3p significantly improved the sensitivity of CRC cells to oxaliplatin [45]. In the present study, we found that miR-195-5p was sponged by XIST and NEAT1, it was significantly associated with the prognosis of patients with CRC, and miR-195-5p was identified as a potential biomarker in CRC. Studies have shown that expression of miR-195-5p was dysregulated in CRC, and miR-195-5p acts as an independent risk factor for overall survival in CRC [46]. Importantly, miR-195-5p has been proved to regulate the stemness and chemoresistance of CRC cells through inhibiting Notch signaling pathway, and forced-expression of miR-195-5p significantly improved the sensitivity of CRC cells to 5-fluorouracil [47]. Intriguingly, we also found that the role of NEAT1/XIST-miR-195-5p regulatory axes in human cancer has been proved by previous studies, which partially enhanced the reliability of our results [48,49]. However, studies focused on the significance of NEAT1/XIST-miR-195-5p regulatory axes in CRC remains largely limited, and they are worthy of further exploration.
We further analyzed and proved the downstream mRNAs that were targeted by the key miRNA, and eight survival-related mRNAs were identified as key mRNAs in CRC that were regulated by miR-195-5p. In agreement with our results, previous studies have reported that these key mRNAs participate in the initiation and progression of multiple types of cancers, including CRC. IRF4, as a member of the interferon regulating factor family, is downregulated by PIP5K1A in human CRC [50]. PHLPP2 belongs to the phosphokinase family that has been found to be down-regulated in CRC, and CRC patients with high PHLPP2 expression showed a better prognosis than those with low PHLPP2 expression [51]. TPM2 has been found to be down-regulated in CRC cell lines and tissues, and reduction of TPM2 was associated with increased migration and proliferation of CRC cells in vitro [52]. RECK is a unique matrix metalloproteinase (MMP) regulator that has been found to be lowly expressed in CRC tissues and cell lines. RECK knockdown enhanced the migratory and invasive rates of CRC cells [53]. ITPR1 is an isoform of inositol 1,4,5-trisphosphate receptor (ITPR), and there is increasing evidence that ITPR expression was associated with aggressiveness of CRC cells [54]. CYP26B1 belongs to the CYP26 enzyme hydroxylate retinoic acid family that has been reported to be dysregulated in CRC, and CYP26B1 expression was significantly correlated with the clinical outcome of patients with CRC [55]. ACOX1, which catalyzes the initial step for peroxisomal β-oxidation, has been reported to associate with peroxisomal disorders and carcinogenesis in liver cancer [56]. LITAF has been reported to function as a tumor suppressor and is frequently down-regulated in multiple cancer types because its expression is regulated by the tumor suppressor protein, p53 [57]. However, there is currently no research that focused on the significance of ACOX1 and LITAF in human CRC, so they are worthy of further study. Furthermore, GSEA analysis revealed that all of these key mRNAs were significantly enriched in well-known tumorigenesis-related pathways, such as the JAK-STAT signaling pathway [58], TGF-β signaling pathway [59], ERBB signaling pathway [60], and VEGF signaling pathway [61]. These reports partially improved the credibility of our research, and further research on the significance of the novel ceRNA network in CRC is valuable.
Tumor microenvironment (TME), a complex system composed of tumor cells, stromal cells, immune cells, fibroblasts, blood vessels, and cytokines, has been reported to regulate some well-known oncogenic process including apoptosis, angiogenesis, hypoxia, and immune escape [62]. Immune cells are important components of TME, which induced the host immune response by secreting cytokines, thereby inhibiting or promoting the progression of tumor cells [63]. For example, the regulatory T cells (Tregs) secrete immunosuppressive cytokines TGF-β and interleukin-10 (IL-10) to protect cancer cells from cytotoxic T cell response and induce immune tolerance, while CD8+ T cells secrete interleukin-2 (IL-2), tumor necrosis factor α (TNF-α) and interferon-γ (IFN-γ) to kill cancer cells in the TME [64]. Immunotherapy has shown encouraging results as an emerging therapy for some cancers. However, existing evidence suggested that most CRC patients were insensitive to immunotherapy compared with esophageal cancer and lung cancer. Therefore, exploring the infiltration abundance and function of immune cells is important to enhance the efficacy of immunotherapy in CRC. In the present study, we found that the ceRNA regulatory network is significantly associated with the infiltration of immune cells, including CD4+/CD8+ T cells and macrophages. Importantly, we found that the ceRNA regulatory network is significantly correlated with the expression of some well-known immune checkpoints. Accumulating studies have reported that tumor-infiltrating lymphocytes, including CD4+ and CD8+ T cells, are important components of immune cells with anti-tumor functions. Furthermore, immune cells are highly heterogeneous in TME compared with noncancerous areas, and infiltration of tumor-infiltrating lymphocytes was associated with improved survival in some cases [63]. Studies also have indicated that ligands of immune checkpoints are highly expressed in cancer cells and TME, leading to T cells’ functional exhaustion and T cell failure, thereby inducing immune escape of CRC cells in the TME [65]. Based on previous studies and our results above, we speculate that the survival-related ceRNA regulatory network identified by our study may play an important role in immune escape of cancer cells in CRC TME. Further study on this survival-related ceRNA network will help to improve the efficacy of immunotherapy in CRC.
Some limitations inevitably existed in the present study. First and foremost, all conclusions of our study were based on online databases such as TCGA and GEO. Further studies will be needed to validate our findings, and to elucidate whether and how the survival-related ceRNA regulatory network regulates CRC. Second, we did not evaluate the subtype of CRC, such as mismatch repair (MMR) and microsatellite instability (MSI) status, which may affect the expression profiles and prognosis of CRC patients. Third, we cannot obtain all the clinicopathologic data of each patient, and the survival analysis was performed by online database automatically, which may lead to bias.
In conclusion, we successfully constructed a survival-related ceRNA regulatory network in CRC by means of stepwise prediction and validation, and each component of the ceRNA regulatory network was significantly correlated with the prognosis of patients with CRC. More importantly, immune infiltration analysis revealed that the survival-related ceRNA regulatory network was remarkably associated with infiltration abundance of multiple immune cells and expression levels of immune checkpoints. Our findings provide valuable clues into improving the efficacy of targeted therapy and immunotherapy for human CRC.
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 3.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL, Siegel RL. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363–385. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- 4.Van Cutsem E, Grothey A. Regorafenib for metastatic colorectal cancer - authors’ reply. Lancet. 2013;381:1538–1539. doi: 10.1016/S0140-6736(13)60978-2. [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y, Bian Y, Zhang Q, Hu J, Li L, Yang M, Qian H, Yu L, Liu B, Qian X. Construction and analysis of dysregulated lncRNA-associated ceRNA network in colorectal cancer. J Cell Biochem. 2019;120:9250–9263. doi: 10.1002/jcb.28201. [DOI] [PubMed] [Google Scholar]
- 6.Donadon M, Ribero D, Morris-Stiff G, Abdalla EK, Vauthey JN. New paradigm in the management of liver-only metastases from colorectal cancer. Gastrointest Cancer Res. 2007;1:20–27. [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, Liu J, Luo XJ, Meng Q, Pu HY, Wang YN, Hu PS, Liu ZX, Zeng ZL, Zhao Q, Deng R, Zhu XF, Ju HQ, Xu RH. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18:174. doi: 10.1186/s12943-019-1105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bian Z, Zhang J, Li M, Feng Y, Wang X, Zhang J, Yao S, Jin G, Du J, Han W, Yin Y, Huang S, Fei B, Zou J, Huang Z. LncRNA-FEZF1-AS1 promotes tumor proliferation and metastasis in colorectal cancer by regulating PKM2 signaling. Clin Cancer Res. 2018;24:4808–4819. doi: 10.1158/1078-0432.CCR-17-2967. [DOI] [PubMed] [Google Scholar]
- 9.Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:3850–3856. [PubMed] [Google Scholar]
- 10.Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, Li S, Zhao JC, Yu J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35:2746–2755. doi: 10.1038/onc.2015.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Wu Z, Fu X, Han W. Long noncoding RNAs: insights from biological features and functions to diseases. Med Res Rev. 2013;33:517–553. doi: 10.1002/med.21254. [DOI] [PubMed] [Google Scholar]
- 12.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang L, Du WW, Yang X, Chen K, Ghanekar A, Levy G, Yang W, Yee AJ, Lu WY, Xuan JW, Gao Z, Xie F, He C, Deng Z, Yang BB. Versican 3’-untranslated region (3’-UTR) functions as a ceRNA in inducing the development of hepatocellular carcinoma by regulating miRNA activity. FASEB J. 2013;27:907–919. doi: 10.1096/fj.12-220905. [DOI] [PubMed] [Google Scholar]
- 14.Lu JH, Zuo ZX, Wang W, Zhao Q, Qiu MZ, Luo HY, Chen ZH, Mo HY, Wang F, Yang DD, Wang Y, Wei XL, Wu QN, Ju HQ, Xu RH, Zeng ZL. A two-microRNA-based signature predicts first-line chemotherapy outcomes in advanced colorectal cancer patients. Cell Death Discov. 2018;4:116. doi: 10.1038/s41420-018-0133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia M, Liu CJ, Zhang Q, Guo AY. GEDS: a gene expression display server for mRNAs, miRNAs and proteins. Cells. 2019;8:675. doi: 10.3390/cells8070675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li JH, Liu S, Zhou H, Qu LH, Yang JH. StarBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paraskevopoulou MD, Vlachos IS, Karagkouni D, Georgakilas G, Kanellos I, Vergoulis T, Zagganas K, Tsanakas P, Floros E, Dalamagas T, Hatzigeorgiou AG. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44:D231–D238. doi: 10.1093/nar/gkv1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, Chiew MY, Tai CS, Wei TY, Tsai TR, Huang HT, Wang CY, Wu HY, Ho SY, Chen PR, Chuang CH, Hsieh PJ, Wu YS, Chen WL, Li MJ, Wu YC, Huang XY, Ng FL, Buddhakosai W, Huang PC, Lan KC, Huang CY, Weng SL, Cheng YN, Liang C, Hsu WL, Huang HD. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46:D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48:D127–D131. doi: 10.1093/nar/gkz757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, Li B, Liu XS. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–e110. doi: 10.1158/0008-5472.CAN-17-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han P, Li JW, Zhang BM, Lv JC, Li YM, Gu XY, Yu ZW, Jia YH, Bai XF, Li L, Liu YL, Cui BB. The lncRNA CRNDE promotes colorectal cancer cell proliferation and chemoresistance via miR-181a-5p-mediated regulation of Wnt/β-catenin signaling. Mol Cancer. 2017;16:9. doi: 10.1186/s12943-017-0583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M, Chen X, Lin K, Zeng K, Liu X, Pan B, Xu X, Xu T, Hu X, Sun L, He B, Pan Y, Sun H, Wang S. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol Cancer. 2018;17:141. doi: 10.1186/s12943-018-0894-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Meng Q, Li X, Yang H, Xu J, Gao N, Sun H, Wu S, Familiari G, Relucenti M, Zhu H, Wu J, Chen R. Long noncoding RNA MIR17HG promotes colorectal cancer progression via miR-17-5p. Cancer Res. 2019;79:4882–4895. doi: 10.1158/0008-5472.CAN-18-3880. [DOI] [PubMed] [Google Scholar]
- 32.Luo J, Yao JF, Deng XF, Zheng XD, Jia M, Wang YQ, Huang Y, Zhu JH. 14, 15-EET induces breast cancer cell EMT and cisplatin resistance by up-regulating integrin αvβ3 and activating FAK/PI3K/AKT signaling. J Exp Clin Cancer Res. 2018;37:23. doi: 10.1186/s13046-018-0694-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farhan M, Wang H, Gaur U, Little PJ, Xu J, Zheng W. FOXO signaling pathways as therapeutic targets in cancer. Int J Biol Sci. 2017;13:815–827. doi: 10.7150/ijbs.20052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–1473. doi: 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu F, Cui WQ, Wei Y, Cui J, Qiu J, Hu LL, Gong WY, Dong JC, Liu BJ. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J Exp Clin Cancer Res. 2018;37:207. doi: 10.1186/s13046-018-0878-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22:5–7. doi: 10.1038/nsmb.2942. [DOI] [PubMed] [Google Scholar]
- 37.Augoff K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol Cancer. 2012;11:5. doi: 10.1186/1476-4598-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen DL, Chen LZ, Lu YX, Zhang DS, Zeng ZL, Pan ZZ, Huang P, Wang FH, Li YH, Ju HQ, Xu RH. Long noncoding RNA XIST expedites metastasis and modulates epithelial-mesenchymal transition in colorectal cancer. Cell Death Dis. 2017;8:e3011. doi: 10.1038/cddis.2017.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Zhu J, Zhang R, Yang D, Li J, Yan X, Jin K, Li W, Liu X, Zhao J, Shang W, Yu T. Knockdown of long non-coding RNA XIST inhibited doxorubicin resistance in colorectal cancer by upregulation of miR-124 and downregulation of SGK1. Cell Physiol Biochem. 2018;51:113–128. doi: 10.1159/000495168. [DOI] [PubMed] [Google Scholar]
- 41.Zhang M, Weng W, Zhang Q, Wu Y, Ni S, Tan C, Xu M, Sun H, Liu C, Wei P, Du X. The lncRNA NEAT1 activates Wnt/β-catenin signaling and promotes colorectal cancer progression via interacting with DDX5. J Hematol Oncol. 2018;11:113. doi: 10.1186/s13045-018-0656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Jiang G, Ren W, Wang B, Yang C, Li M. LncRNA NEAT1 regulates 5-Fu sensitivity, apoptosis and invasion in colorectal cancer through the MiR-150-5p/CPSF4 axis. Onco Targets Ther. 2020;13:6373–6383. doi: 10.2147/OTT.S239432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol. 2010;11:252–263. doi: 10.1038/nrm2868. [DOI] [PubMed] [Google Scholar]
- 44.Mansoori B, Mohammadi A, Naghizadeh S, Gjerstorff M, Shanehbandi D, Shirjang S, Najafi S, Holmskov U, Khaze V, Duijf PHG, Baradaran B. miR-330 suppresses EMT and induces apoptosis by downregulating HMGA2 in human colorectal cancer. J Cell Physiol. 2020;235:920–931. doi: 10.1002/jcp.29007. [DOI] [PubMed] [Google Scholar]
- 45.Liu T, Zhang X, Du L, Wang Y, Liu X, Tian H, Wang L, Li P, Zhao Y, Duan W, Xie Y, Sun Z, Wang C. Exosome-transmitted miR-128-3p increase chemosensitivity of oxaliplatin-resistant colorectal cancer. Mol Cancer. 2019;18:43. doi: 10.1186/s12943-019-0981-7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Sun M, Song H, Wang S, Zhang C, Zheng L, Chen F, Shi D, Chen Y, Yang C, Xiang Z, Liu Q, Wei C, Xiong B. Integrated analysis identifies microRNA-195 as a suppressor of Hippo-YAP pathway in colorectal cancer. J Hematol Oncol. 2017;10:79. doi: 10.1186/s13045-017-0445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Y, Wang M, Hu H, Huang Q, Chen Y, Wang G. Overcoming stemness and chemoresistance in colorectal cancer through miR-195-5p-modulated inhibition of notch signaling. Int J Biol Macromol. 2018;117:445–453. doi: 10.1016/j.ijbiomac.2018.05.151. [DOI] [PubMed] [Google Scholar]
- 48.Cheng H, Malhotra A. Evaluation of potential of long noncoding RNA NEAT1 in colorectal cancer. J Environ Pathol Toxicol Oncol. 2020;39:101–111. doi: 10.1615/JEnvironPatholToxicolOncol.2020032508. [DOI] [PubMed] [Google Scholar]
- 49.Yang C, Wu K, Wang S, Wei G. Long non-coding RNA XIST promotes osteosarcoma progression by targeting YAP via miR-195-5p. J Cell Biochem. 2018;119:5646–5656. doi: 10.1002/jcb.26743. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Q, Zhang C, Ma JX, Ren H, Sun Y, Xu JZ. Circular RNA PIP5K1A promotes colon cancer development through inhibiting miR-1273a. World J Gastroenterol. 2019;25:5300–5309. doi: 10.3748/wjg.v25.i35.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu SH, Xu XY, Sun CB, Wen FF, He S, Gao XQ, Liu YH, Liu L. Expression of PHLPP2 correlates with clinicopathologic characteristics and prognosis in colorectal cancer. Int J Clin Exp Pathol. 2019;12:2909–2919. [PMC free article] [PubMed] [Google Scholar]
- 52.Cui J, Cai Y, Hu Y, Huang Z, Luo Y, Kaz AM, Yang Z, Chen D, Fan X, Grady WM, Wang J. Epigenetic silencing of TPM2 contributes to colorectal cancer progression upon RhoA activation. Tumour Biol. 2016;37:12477–12483. doi: 10.1007/s13277-016-5103-1. [DOI] [PubMed] [Google Scholar]
- 53.Wei LJ, Bai DM, Wang ZY, Liu BC. MicroRNA-375 accelerates the invasion and migration of colorectal cancer through targeting RECK. Eur Rev Med Pharmacol Sci. 2019;23:4738–4745. doi: 10.26355/eurrev_201906_18055. [DOI] [PubMed] [Google Scholar]
- 54.Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, Iwakiri Y, Nathanson MH, Yamaguchi K. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium. 2010;48:315–323. doi: 10.1016/j.ceca.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown GT, Cash BG, Blihoghe D, Johansson P, Alnabulsi A, Murray GI. The expression and prognostic significance of retinoic acid metabolising enzymes in colorectal cancer. PLoS One. 2014;9:e90776. doi: 10.1371/journal.pone.0090776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng FM, Chen WB, Qin T, Lv LN, Feng B, Lu YL, Li ZQ, Wang XC, Tao LJ, Li HW, Li SY. ACOX1 destabilizes p73 to suppress intrinsic apoptosis pathway and regulates sensitivity to doxorubicin in lymphoma cells. BMB Rep. 2019;52:566–571. doi: 10.5483/BMBRep.2019.52.9.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zou J, Guo P, Lv N, Huang D. Lipopolysaccharide-induced tumor necrosis factor-α factor enhances inflammation and is associated with cancer (Review) Mol Med Rep. 2015;12:6399–6404. doi: 10.3892/mmr.2015.4243. [DOI] [PubMed] [Google Scholar]
- 58.Jiang L, Zhao XH, Mao YL, Wang JF, Zheng HJ, You QS. Long non-coding RNA RP11-468E2.5 curtails colorectal cancer cell proliferation and stimulates apoptosis via the JAK/STAT signaling pathway by targeting STAT5 and STAT6. J Exp Clin Cancer Res. 2019;38:465. doi: 10.1186/s13046-019-1428-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y, Cao H, Jiao Z, Pakala SB, Sirigiri DN, Li W, Kumar R, Mishra L. Carcinoembryonic antigen interacts with TGF-{beta} receptor and inhibits TGF-{beta} signaling in colorectal cancers. Cancer Res. 2010;70:8159–8168. doi: 10.1158/0008-5472.CAN-10-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong W, Cao Z, Pang Y, Feng T, Tian H. CARF, as an oncogene, promotes colorectal cancer stemness by activating ERBB signaling pathway. Onco Targets Ther. 2019;12:9041–9051. doi: 10.2147/OTT.S225733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun W, Wang X, Li J, You C, Lu P, Feng H, Kong Y, Zhang H, Liu Y, Jiao R, Chen X, Ba Y. MicroRNA-181a promotes angiogenesis in colorectal cancer by targeting SRCIN1 to promote the SRC/VEGF signaling pathway. Cell Death Dis. 2018;9:438. doi: 10.1038/s41419-018-0490-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Casey SC, Amedei A, Aquilano K, Azmi AS, Benencia F, Bhakta D, Bilsland AE, Boosani CS, Chen S, Ciriolo MR, Crawford S, Fujii H, Georgakilas AG, Guha G, Halicka D, Helferich WG, Heneberg P, Honoki K, Keith WN, Kerkar SP, Mohammed SI, Niccolai E, Nowsheen S, Vasantha Rupasinghe HP, Samadi A, Singh N, Talib WH, Venkateswaran V, Whelan RL, Yang X, Felsher DW. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin Cancer Biol. 2015;35(Suppl):S199–S223. doi: 10.1016/j.semcancer.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ge P, Wang W, Li L, Zhang G, Gao Z, Tang Z, Dang X, Wu Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed Pharmacother. 2019;118:109228. doi: 10.1016/j.biopha.2019.109228. [DOI] [PubMed] [Google Scholar]
- 64.Xiong Y, Wang Y, Tiruthani K. Tumor immune microenvironment and nano-immunotherapeutics in colorectal cancer. Nanomedicine. 2019;21:102034. doi: 10.1016/j.nano.2019.102034. [DOI] [PubMed] [Google Scholar]
- 65.Emambux S, Tachon G, Junca A, Tougeron D. Results and challenges of immune checkpoint inhibitors in colorectal cancer. Expert Opin Biol Ther. 2018;18:561–573. doi: 10.1080/14712598.2018.1445222. [DOI] [PubMed] [Google Scholar]