

Abstract
We analyzed gene expression in peripheral blood mononuclear cells (PBMCs) from patients with systemic lupus erythematosus (SLE) using public databases. The goal was to identify lupus biomarkers by determining whether differentially expressed genes are mediated by methylation, miRNA, or SNP. Two cDNA microarrays were subjected to integration analysis, and we calculated the mutually differentially expressed genes (|log2fold change (FC)| > 1, P < 0.05). These genes were analyzed using gene otology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and protein-protein interaction (PPI) networks. The differences in methylation sites for two methylation chips were calculated and the differentially methylated sites were annotated. These genes were compared to the differentially expressed genes. We obtained 135 differentially expressed microRNAs from the microRNA-chip results using PBMCs from SLE and healthy individuals. Predictive microRNA target genes were identified using GO, KEGG pathways, and PPI networks. The target genes identified were compared to the differentially expressed genes. We downloaded Chinese SLE genome-wide association study data from SLE-related literature, analyzed the loci with a P value < 0.05, and used annotated SLE-associated SNPs. We selected the genes corresponding to an SNP located on an exon and determined the intersection with the differentially expressed genes. We found 18 differentially expressed genes in both cDNA microarrays. The methylation chips had 50 corresponding methylation sites. On the basis of these results, we identified two genes, IFI44 and IFI44L. We further identified 135 differentially expressed microRNAs predicted to affect 5766 target genes. Two identified genes were in common with the differentially expressed genes. Finally, SNP annotated genes and cDNA chip genes overlap with identified MX1. Therefore, we used existing data to analyze the causes of differential gene expression in SLE, introducing new methods for determining biomarkers and therapeutic targets.
Keywords: SLE, bioinformatics, methylation, miRNA, SNP
Introduction
Systemic lupus erythematosus (SLE) is a multi-organ autoimmune chronic disease [1] occurring primarily in reproductive-aged women. The clinical manifestations are diverse and systemic organs can be affected. Severe lupus therefore often has a poor prognosis. The SLE prevalence in the Caucasian population is lower than that in other ethnicities. A higher prevalence rate is observed among Asians and African Americans, whereas the highest prevalence is found in the Caribbean [2]. The pathogenesis of SLE is complex and likely influenced by genetics, environment, and hormone levels. Sm and dsDNA antibodies are SLE-specific antibodies, with a positive expression rate of only 30%. A large percentage of connective tissue disease patients will evolve into SLE. Thus, early diagnosis of SLE can benefit patients, but this is often challenging. To provide screening markers and new therapeutic targets for SLE prevention and diagnosis, it is essential to understand the pathogenesis of the disease at the molecular level.
Gene expression regulation is a complex event, and transcription initiation is the primary control point for gene expression [3]. Gene transcription regulation elements involve specific sequences of DNA. Regulation of these factors affects RNA polymerase activity. In addition to the regulation of transcription initiation levels, other processes such as gene activation, post-transcriptional and post-translational processing, as well as translation can modulate gene expression.
Methylation is catalyzed by methyltransferase, which selectively adds methyl groups to cytosine of DNA to form 5-methylcytosine, and it is commonly found in the 5’-CG-3’ sequence. Sequence-specific methylation-binding protein (MBD/MeCP) binds to the methylated CpG island in the promoter region, preventing transcriptional factors from acting on the promoter, thereby inhibiting gene transcription processes [4]. Decreased DNA methylation in lymphocytes is associated with disease activity in SLE patients [5].
MicroRNAs (miRNAs) are endogenous, small RNAs approximately 20 to 24 nucleotides in length that are associated with disease activity [6]. Single stranded RNA precursors are processed by the Dicer enzyme, which has a variety of important regulatory effects in the cell. The 5’ mature phosphate and the 3’ hydroxyl group are the distinguishing marks of functional mRNA. It can be targeted for degradation by a miRNA fragment of the same length. miRNAs can be combined with sequence complementary mRNAs, and sometimes even with specific DNA fragments, thus silencing the gene.
Single nucleotide polymorphisms (SNPs) refer to DNA sequence polymorphisms at the genomic level due to individual nucleotide changes [7]. It is the most common variant of human genetic variation, accounting for more than 90% of all known genetic polymorphisms [8]. Multiple human leukocyte antigen (HLA) genes located at 6p21.1-6p21.3 were shown to be the most potent genetic risk factors for SLE in European and Chinese populations [9].
With the rapid development of bioinformatics, the use of gene expression profiling to explore the relationship between differential gene expression and disease phenotypes has grown [10,11]. A number of sequencing microarray studies have been applied to SLE, but the data have not been utilized or explored fully [12,13]. This study analyzed the possible causes of differential gene expression in SLE from the three aspects of DNA methylation, miRNA modification, and SNPs utilizing microarray data.
Materials and methods
Data collection
1. cDNA array: two data sets were used in this study, one derived from NCBI GEO and one from EMBL-EBI (accession numbers GSE81622 and MTAB145, respectively). The differentially expressed genes were from two independent SLE PBMCs of 93 specimens (55 SLE and 38 healthy individuals). These data sets were created from the same microarray platforms generated by GPL10558 Illumina HumanHT-12 V4.0 expression BeadChip. GSE81622 was uploaded to NCBI GEO by UT Southwestern Medical Center, United States. The subjects were ethnically Chinese and the degree of disease activity is expressed as the SLE Disease Activity Index (SLEDAI). The Cambridge Institute of Medical Research in the UK uploaded MTAB145 to EMBL-EBI. The subjects included 12 Caucasians and 1 Asian. The experimental platforms are GPL10558, Illumina, HumanHT-12, V4.0 expression BeadChip. Total RNA was extracted from PBMCs isolated from SLE patients and healthy controls, and it was assessed through microarrays. 2. Methylation data set: The methylated data set was downloaded from the GEO of NCBI (GSE82221 and GSE76056) with 77 specimens (42 SLE and 35 NC). The test platform is GPL13534 Illumina HumanMethylation450 BeadChip (HumanMethylation450_15017482). Two GEO data sets were used in this study. GSE82221, uploaded to NCBI GEO by the UT Southwestern Medical Center, had ethnic Chinese subjects. GSE76056 was uploaded by the University of Hong Kong. The study subjects were Chinese individuals living in Hong Kong. Their experimental platforms were GPL13534 Illumina HumanMethylation450 BeadChip (HumanMethylation450_15017482), and the peripheral blood was extracted for methylation sequencing. 3. miRNA data: Since we did not find relevant miRNA chip in the GEO database, we analyzed the chip data in the literature [14]. According to the literature, eight cases of SLE samples and seven NC samples were detected by array analysis. 4. SNP immunochip data: In the literature [15], 2645 cases and 4058 controls from Korean (KR), Han Chinese (HC), and Malaysian Chinese (MC) populations were genotyped using the Illumina ImmunoChip array at the Oklahoma Medical Research Foundation (OMRF). Quality controlled ImmunoChip genotype data from Chinese SLE specimens were downloaded from the SLE literature, which included 480 SLE and 486 normal samples, after 10 SLE and 7 normal samples were removed. These samples were removed because they were obtained from individuals with a genetic relationship matrix of more than 25%.
Differential expression gene analysis
The original probe level data (CEL file) in the R package [16] was passed through the powerful multi-array averaging algorithm RMA. Through background correction, a quantile normalization procedure and the set of probe values was summed to achieve the expression value. The methylated data set was downloaded from NCBI’s GEO. When multiple probe sets were mapped to the same gene, the mean of the probe set value was used as the expression value. The Limma package determines differentially expressed genes [17]. We used |log2fold change (FC)| > 1, P < 0.05, and FDR < 0.05 as the criteria for the differential expression of genes in SLE and NC PBMCs. We used FDR to correct the P-value.
Functional enrichment analysis
The online software database [18] for annotation, visualization, and integrated discovery (DAVID, https://david.ncifcrf.gov/) was used to perform Gene Ontology (GO) annotation analysis. Different gene expression levels were identified in the two-chip overlap recognition input from the DAVID co-expression analysis, with P < 0.05 considered to be significantly enriched. DAVID uses a number of publicly available bio-databases to identify any interactions for the input gene list such as molecular functions, biological processes, and cellular components. KOBAS 3.0 Online Software (http://kobas.cbi.pku.edu.cn/Home.do) was used for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis [19], with a cutoff of P < 0.05 for significant enrichment. The differentially expressed genes were uploaded to the search tool for the Gene/Protein Interactions Search (STRING), version 9.1 (http://string-db.org/), to construct the interaction network. The STRING software operator developed and operated by EMBL, SIB, and UZH, is a database containing all known and predicted protein interactions. Interactions with direct (physical) and indirect (functional) associations were derived from studies reporting protein interactions, genome analysis and prediction, high-throughput experiments, and co-expression studies.
Methylation analysis
Quality control of methylation levels in normal and SLE samples was based on two peaks near 0 and 1. Methylation levels were low around 0 and high near 1. We performed normalization of the beta values (i.e., M/(M + UM), where M is methylated signal intensity and UM is unmethylated signal intensity. After quantile normalization, the median value is in a straight line to eliminate differences between chips. The methylation levels of normal and SLE samples were assessed. The differentially methylated sites for the corresponding gene from normal and SLE tissues were calculated through the minfi R packet (qval < 0.05). The differential methylation sites were annotated through the GPL13534 platform file. The genes corresponding to the different methylation sites were obtained. Cluster analysis of differentially methylated loci was assessed through the pheatmap R package. Fifty corresponding genes with consistent site methylation were analyzed through GO, KEGG pathways, and protein-protein interaction (PPI) networks. Furthermore, differential analysis of the cDNA microarray was performed.
miRNA analysis
We utilized bioinformatics for 135 miRNAs concluded in the literature [14] using four different algorithms: miRanda-mirSVR (http://www.microrna.org/), miRDB (http://mirdb.org/miRDB/), miRTarBase (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index), and TargetScan 6.2 (http://www.targetscan.org/). Pathway analysis and gene function analysis were assessed through GO (P < 0.05 for the filter) and KEGG pathway analysis (P < 0.05 for the filter). The interaction between proteins was constructed through the STRING tool for the network of key genes and screening the protein interaction network. Next, we predicted the cross analysis for the miRNA target genes and evaluated cDNA gene chip differences.
SNP analysis
In the Illumina ImmunoChip array of Chinese individuals [15], subjects that had more than 10% missing genotypes were excluded from the analysis. In addition, SNPs that had more than 5% missing genotypes, were out of Hardy-Weinberg equilibrium (P < 0.0001 in controls), or had less than 0.5% minor allele frequency (MAF) were also excluded from the analysis. In the quality control step, SNPs with overlapping clusters were filtered out. We analyzed the quality-controlled SNPs in the Illumina ImmunoChip array data, with P < 0.05 loci. We used ANNOVAR software to annotate SLE-associated SNPs selected from the annotation of gene loci exons. We found 199 genes that may be significantly associated with SLE. Finally, gene analysis was performed based on differences from the cDNA chip.
Results
Differential gene screening
GSE81622 had 90 genes with statistically significant differences, including 56 up-regulated and 34 down-regulated genes (|logFC| > 1 and P < 0.05). MTAB145 demonstrated statistically significant differences in 70 genes, including 48 up-regulated and 22 down-regulated genes (|logFC| > 1 and P < 0.05). The 90 differentially expressed genes from GSE81622 were compared to the 70 differentially expressed genes from MTAB145, yielding 18 genes in common (see Figure 1; IFI27, USP18, IFI44L, IFI44, EPSTI1, MX1, HP, OAS3, RNASE3, ORM1, IFIT1, DEFA4, LCN2, CAMP, BPI, DEFA1, CA1 and ALAS2, logFC and P values are shown in Table 1).
Figure 1.
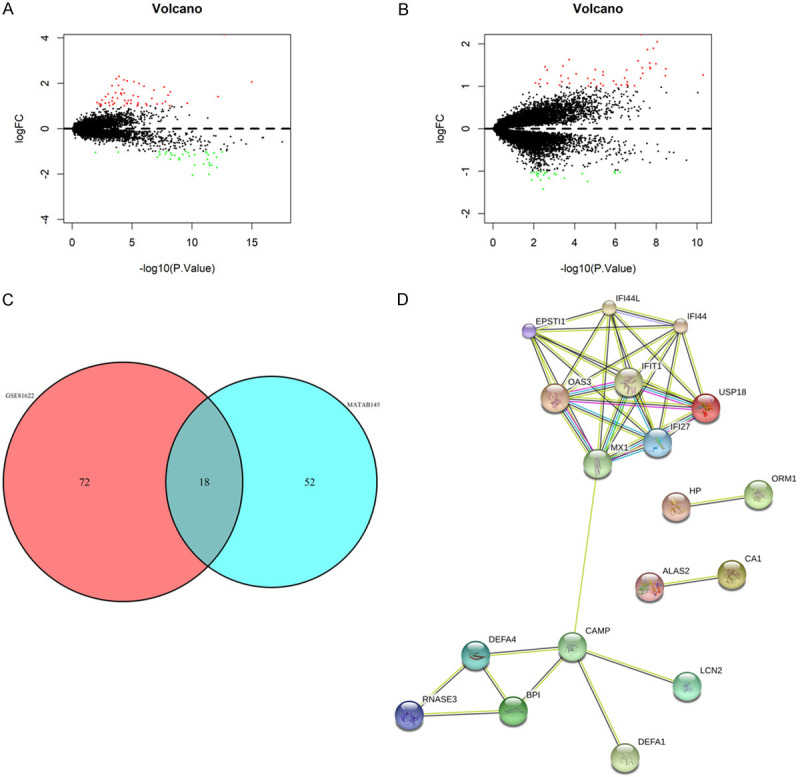
A. The volcano figure of GSE81622. The horizontal axis is -log10 (P value), and the vertical axis is log2 (FC). Each point represents a gene. Ninety genes with significantly differentially expressed genes were screened out, and 56 up-regulated genes and 34 down-regulated genes were identified (|logFC| > 1, P < 0.05). B. The volcano figure of MTAB145. The horizontal axis is -log10 (P value), and the vertical axis is log2 (FC). Each point represents a gene. Screening 70 statistically significant differentially expressed genes yielded 48 up-regulated and 22 down-regulated genes (|logFC| > 1 and P < 0.05). C. Venn diagram of GSE81622 and MTAB145. It shows that the two chips share 18 genes. D. Gene protein-protein interaction networks for 18 differentially expressed genes. MX1 connected eight nodes.
Table 1.
Expression of differentially expressed genes in cDNA microarray
| Gene | GSE81622 | MTAB145 | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| logFC | P-value | FDR | logFC | P-value | FDR | |
| IFI27 | 4.146808761 | 2.00E-13 | 3.38E-10 | 1.449551354 | 2.02E-08 | 2.02E-08 |
| USP18 | 1.012344931 | 7.18E-09 | 1.26E-06 | 1.530429631 | 1.04E-08 | 1.04E-08 |
| IFI44 | 1.476222698 | 9.94E-09 | 1.64E-06 | 1.898491118 | 1.36E-08 | 1.36E-08 |
| IFI44L | 1.834553348 | 2.09E-08 | 3.10E-06 | 2.051325192 | 9.02E-09 | 9.02E-09 |
| EPSTI1 | 1.103100053 | 2.98E-08 | 4.12E-06 | 1.413536751 | 3.36E-09 | 3.36E-09 |
| MX1 | 1.02422664 | 5.89E-07 | 5.05E-05 | 1.865771862 | 2.69E-08 | 2.69E-08 |
| HP | 1.41004245 | 3.02E-06 | 0.0001926 | 1.389449066 | 9.59E-06 | 9.59E-06 |
| OAS3 | 1.054949925 | 3.13E-06 | 0.0001986 | 1.301830305 | 4.62E-08 | 4.62E-08 |
| RNASE3 | 1.462443976 | 7.83E-06 | 0.0004219 | 1.107798678 | 0.0004248 | 0.0004248 |
| ORM1 | 1.095193294 | 2.59E-05 | 0.00105 | 1.290680308 | 8.38E-05 | 8.38E-05 |
| IFIT1 | 1.248862368 | 4.07E-05 | 0.0014683 | 1.43575638 | 4.85E-08 | 4.85E-08 |
| DEFA4 | 2.144391991 | 4.81E-05 | 0.00166 | 1.068182139 | 0.0021415 | 0.0021415 |
| LCN2 | 1.506994716 | 7.59E-05 | 0.002417 | 1.381762964 | 0.0021815 | 0.0021815 |
| CAMP | 1.90506454 | 0.0001377 | 0.003722 | 1.630292052 | 0.0001853 | 0.0001853 |
| BPI | 1.368480036 | 0.0004171 | 0.0085143 | 1.189265692 | 2.29E-05 | 2.29E-05 |
| DEFA1 | 1.887848952 | 0.000466 | 0.0092576 | 1.080519806 | 0.0081768 | 0.0081768 |
| CA1 | 1.414096877 | 0.0016177 | 0.0228452 | 1.057748117 | 0.0056329 | 0.0056329 |
| ALAS2 | 1.156245494 | 0.0081077 | 0.0713169 | 1.460402929 | 0.0027871 | 0.0027871 |
Note: logFc: the log of fold change; FDR: false discovery rate.
GO, KEGG, and PPI analysis
We used DAVID to assess the 18 differentially expressed genes through GO enrichment analysis (P < 0.05 as a filter). We found 62 related GOs, as shown in Table 2. The common differentially expressed genes from the two microarrays were not sufficient for KEGG pathway analysis, and KEGG analysis for common pathways did not find any associated pathways. Using the PPI STRING software analysis, we found 18 stress proteins, including MX1, and eight connected nodes (Figure 1). The circle represents the gene, the line represents the protein interactions between genes, and the inner circle represents the protein structure (small nodes: proteins of unknown three-dimensional structure; large nodes: three-dimensional structures known or predicted). The color of the thread represents different evidence types for protein interactions (the red line represents the existence of mixed evidence; the green line represents neighborhood evidence; the blue line represents concurrent evidence; the purple line represents experimental evidence; the yellow line represents evidence from text mining; the light blue line represents evidence from public databases; the black line represents evidence from co-expression data).
Table 2.
Top 10 of GO analysis of differentially expressed genes
| Term | Count | P-value | Genes in test set |
|---|---|---|---|
| GO: 6952~defense response | 9 | 1.36E-09 | ORM1, DEFA4, MX1, HP, LCN2, BPI, RNASE3, DEFA1, CAMP |
| GO: 51707~response to other organism | 7 | 2.02E-08 | DEFA4, MX1, IFI44, BPI, RNASE3, DEFA1, CAMP |
| GO: 42742~defense response to bacterium | 5 | 7.14E-08 | DEFA4, BPI, RNASE3, DEFA1, CAMP |
| GO: 9607~response to biotic stimulus | 7 | 9.44E-08 | DEFA4, MX1, IFI44, BPI, RNASE3, DEFA1, CAMP |
| GO: 31640~killing of cells of another organism | 3 | 6.96E-07 | DEFA4, DEFA1, CAMP |
| GO: 6950~response to stress | 10 | 9.51E-07 | ORM1, ALAS2, DEFA4, MX1, HP, LCN2, BPI, RNASE3, DEFA1, CAMP |
| GO: 50896~response to stimulus | 13 | 1.09E-06 | ORM1, ALAS2, DEFA4, MX1, HP, IFI44, RNASE3, DEFA1, IFI44L, OAS3, LCN2, BPI, CAMP |
| GO: 9617~response to bacterium | 5 | 1.93E-06 | DEFA4, BPI, RNASE3, DEFA1, CAMP |
| GO: 6879~cellular iron ion homeostasis | 3 | 2.90E-06 | ALAS2, HP, LCN2 |
| GO: 1906~cell killing | 3 | 3.53E-06 | DEFA4, DEFA1, CAMP |
Note: Term: enrichment GO; Count: the number of genes that fall on term; P-Value: statistical P value of enrichment.
Differential methylation sites and corresponding genes
Quality control and normalization for normal and SLE sample methylation levels were done through the minfi R package [20]. Differences in methylation sites were calculated between normal and SLE tissue (qval < 0.05) to obtain different methylation sites for the two chips (Figures 2, 3). The different methylation sites were annotated. Next, through the GPL13534 platform file, genes corresponding to the different methylation sites were obtained. When comparing the genes with different methylation sites from GSE76056 and GSE82221, 84 genes were common between the two datasets. However, the methylation levels were not consistent for all 84 genes. For some genes, the GSE76056 methylation levels were high whereas the GSE82221 methylation levels were low. After eliminating genes with differing methylation levels, 50 genes remained with a consistent degree of methylation. The number of differentially methylated regions in the TSS1500, TSS200, 5’UTR, first exon, gene body, and 3’ UTR of these 50 genes were 11, 7, 12, 7, 29, and 2, respectively, while the number of methylated regions in the CpG island, N shelf, N shore, S shore, and S Shelf were 15, 1, 9, 8, and 2, respectively. GO, KEGG, and PPI analysis were performed for these genes. DAVID was used to perform GO enrichment analysis for the 50 differentially expressed genes with P < 0.05 used as a filter condition. We found four GO enrichment related functions (Table 3). We used KOBAS 3.0 for KEGG pathway enrichment analysis for the 50 genes with P < 0.05 as a filter condition. We found three related KEGG pathways (Table 4). Using STRING software analysis of PPI (Figure 4), we found 23 stress proteins, including ISG15 and RSAD2, connected by 12 nodes. The genes identified in both the methylation chip and cDNA microarray include IFI44 (methylation site: cg07107453) and IFI44L (methylation sites: cg00458211, cg13304609, cg06872964, cg03607951, and cg05696877).
Figure 2.

GSE76056 cluster analysis chart. Vertical coordinates represent differentially methylated sites, and the abscissa represents the sample. There were 315 differentially methylated loci corresponding to 88 genes.
Figure 3.

GSE82221 cluster analysis chart. There were 95,910 different loci corresponding to 15,895 genes.
Table 3.
The differentially methylated sites corresponding to 50 genes for GO analysis
| Term | P-value | Corr P-value | Count | Genes in test set |
|---|---|---|---|---|
| GO: 9615~response to virus | 7.73E-14 | 8.31E-11 | 11 | HERC5, PLSCR1, RSAD2, DDX58, PRKRA, AHRR, EIF2AK2, IFI44, ISG15, LAP3, TRIM11 |
| GO: 9607~response to biotic stimulus | 1.29E-09 | 5.97E-07 | 12 | HERC5, PLSCR1, RSAD2, HSPA1L, DDX58, PRKRA, AHRR, EIF2AK2, IFI44, ISG15, LAP3, TRIM11 |
| GO: 51707~response to other organism | 1.67E-09 | 5.97E-07 | 11 | HERC5, PLSCR1, RSAD2, DDX58, PRKRA, AHRR, EIF2AK2, IFI44, ISG15, LAP3, TRIM11 |
| GO: 51704~multi-organism processc | 6.31E-07 | 1.70E-04 | 12 | HERC5, PLSCR1, RSAD2, DDX58, PRKRA, TAP2, AHRR, EIF2AK2, IFI44, ISG15, LAP3, TRIM11 |
Note: Term: Enriched GO; P-Value: statistical P value of enrichment; Count: The number of genes that fall on Term; Corr P-value: correct P-value.
Table 4.
The differentially methylated sites corresponded to 50 genes for KEGG analysis
| Term | Count | P-value | Genes |
|---|---|---|---|
| hsa04974: Protein digestion and absorption | 3 | 0.021912576 | COL22A1, ATP1A1, COL11A2 |
| hsa05164: Influenza A | 3 | 0.081691414 | DDX58, RSAD2, EIF2AK2 |
| hsa05168: Herpes simplex infection | 3 | 0.097346053 | DDX58, TAP2, EIF2AK2 |
Note: Term: Enriched KEGG; Count: The number of genes that fall on Term; P-Value: statistical P value of Enrichment.
Figure 4.

A. Analysis of 50 differentially methylated loci corresponding to GO. Four related GO enrichment functions were found. B. Protein-protein interaction map for 50 differentially methylated loci. A total of 23 prominent proteins were found, of which ISG15 and RSAD2 were the most important because of the highest number of interactions (12 nodes joined).
MiRNA corresponding genes, GO analysis, and KEGG analysis
Through miRanda, miRTarBase, and TargetScan network tools, we obtained 5766 target gene projections for the specified microRNAs from the literature (14). Through DAVID for GO function enrichment analysis of target genes (P < 0.05 used as a filter), we found 847 related terms (top 10 shown in Table 5; Figure 5). We used KOBAS 3.0 to perform KEGG pathway enrichment analysis of the miRNA target genes (P < 0.05). We found 177 related KEGG pathways (top 10 shown in Table 6). The String tool was used to obtain the target genes and protein-protein interactions. Key genes were identified through screening the network and generating a protein interaction network diagram (Figure 5). Genetic relationships were generated by assessing the network node number, obtaining the core genes, and drawing the core gene histogram (Figure 5).
Table 5.
Top 10 target gene GO enrichment results
| Term | Count | P-value |
|---|---|---|
| GO: 0005515~protein binding | 1449 | 3.78E-65 |
| GO: 0005654~nucleoplasm | 543 | 6.09E-37 |
| GO: 0005737~cytoplasm | 875 | 6.09E-36 |
| GO: 0005829~cytosol | 612 | 8.80E-35 |
| GO: 0045944~positive regulation of transcription from RNA polymerase II promoter | 259 | 6.19E-34 |
| GO: 0005634~nucleus | 892 | 7.30E-34 |
| GO: 0000122~negative regulation of transcription from RNA polymerase II promoter | 198 | 2.77E-28 |
| GO: 0045893~positive regulation of transcription, DNA-template | 153 | 1.23E-25 |
| GO: 0016020~membrane | 391 | 3.31E-18 |
| GO: 0003682~chromatin binding | 112 | 5.71E-18 |
Note: Enriched GO Count: the number of genes that fall on term; P-Value: statistical P-value of enrichment.
Figure 5.

A. miRNA target gene GO enrichment results. We found 847 GO enrichment functions. The abscissa is the enriched GO and the ordinate is the number and ratio of the target genes. Different colors represent different GO categories: molecular function (green), biological process (red), and cellular component (blue). B. Histogram of core genes in target genes. The number of connections between a coordinate gene and other genes, the coordinate is the gene name, and height represents the number of gene connections.
Table 6.
The top 10 results of MiRNA predicts target gene KEGG enrichment
| Pathway ID | Description | P-value | Number of DERNAs |
|---|---|---|---|
| hsa05200 | Pathways in cancer | 7.57E-44 | 121 |
| hsa05205 | Proteoglycans in cancer | 5.78E-34 | 78 |
| hsa04151 | PI3K-Akt signaling pathway | 1.29E-32 | 96 |
| hsa04144 | Endocytosis | 2.85E-32 | 84 |
| hsa04010 | MAPK signaling pathway | 4.32E-32 | 83 |
| hsa05206 | MicroRNAs in cancer | 1.03E-28 | 84 |
| hsa04014 | Ras signaling pathway | 1.09E-26 | 71 |
| hsa04722 | Neurotrophin signaling pathway | 2.01E-24 | 51 |
| hsa04068 | FoxO signaling pathway | 3.73E-24 | 53 |
| hsa05161 | Hepatitis B | 1.79E-23 | 54 |
Note: P-value: statistical p value of enrichment; Number of DERNAS: differential gene number.
SNP analysis
We downloaded the data based on Chinese SLE patients from the SLE related literature (15), including 490 SLE samples and 493 normal samples. The data were processed, and P < 0.05 was selected in the GWAS analysis. Through ANNOVAR software to annotate the SNPs associated with SLE, we selected the UTR5, exonic, and UTR3 sites from the annotation results (Top 10 exonic sites as shown in Table 7). We obtained 31, 199, and 128 results, respectively. These were associated with SLE and located on the gene UTR5, exons, and UTR3, which may be significantly associated with SLE. The 199 genes were located on the exons of 169 genes, the 31 genes were located in the UTR5 of 30 genes. The 94 genes were located in the UTR3 of 128 genes. Finally, the intersection between SNPs and cDNA chip genes yielded the MX1 gene.
Table 7.
Characteristics of SNP loci located in the top 10 exon regions of genes
| Chr | Start | End | RNP ID | Allele1 | Allele2 | P-value | Odds ratio | 95% CI | Func.refgene | Gene.refgene |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 138196065 | 138196066 | rs2230926 | G | T | 7.82E-08 | 2.67 | 1.84-3.86 | exonic | TNFAIP3 |
| 6 | 32609103 | 32609104 | rs9272689 | G | A | 2.51E-05 | 1.47 | 1.23-1.76 | exonic | HLA-DQA1 |
| 6 | 35260529 | 35260530 | rs1557568 | C | T | 6.45E-05 | 1.46 | 1.21-1.75 | exonic | ZNF76 |
| 10 | 50025395 | 50025396 | rs7097397 | G | A | 1.37E-04 | 1.43 | 1.19-1.72 | exonic | WDFY4 |
| 11 | 10715123 | 10715124 | rs10770136 | T | C | 6.35E-04 | 0.61 | 0.46-0.81 | exonic | MRVI1 |
| 2 | 103011083 | 103011084 | rs12619169 | A | G | 7.18E-04 | 3.6 | 1.63-7.93 | exonic | IL18R1 |
| 8 | 57078932 | 57078933 | rs35883156 | T | G | 7.82E-04 | 0.53 | 0.36-0.77 | exonic | PLAG1 |
| 1 | 114515716 | 114515717 | rs2358996 | A | G | 9.70E-04 | 0.74 | 0.62-0.89 | exonic | HIPK1 |
| 6 | 32609285 | 32609286 | rs1048087 | T | C | 1.16E-03 | 0.75 | 0.62-0.89 | exonic | HLA-DQA1 |
| 19 | 47258841 | 47258842 | rs2287717 | T | C | 1.91E-03 | 0.62 | 0.45-0.84 | exonic | FKRP |
Note: Chr: chromosome; Rs: P-value: GWAS statistical significance; Func.refgene: Relative gene location; Gene. refgene: gene.
Discussion
SLE is an inflammatory disease involving multiple organs that has a genetic component. The male to female ratio is approximately 1:9 [21]. Differentially expressed genes have been characterized for SLE [22], and while they play an important role, the cause of the differential expression is unclear. Bioinformatics analysis can provide the basis for genetic variations in the expression levels in order to understand these causes. A total of 18 differentially expressed genes, corresponding to 50 methylation sites, and 199 disease-related SNP loci located on exons, were identified. From these results, two genes may be associated with methylation, two genes may be regulated by miRNA, and one differentially expressed gene may be related to SNPs. This study is the first systematic analysis based on bioinformatics for SLE morbidity key gene research. It provides evidence and information for clinical research and subsequent experiments. It also makes greater use of existing data, which will permit the identification of more reliable peripheral blood biomarkers in SLE.
Bioinformatics is a combination of biological and computer science [23]. We utilized a public database of gene chip data to explore genetic variations. In this study, we strictly adhered to the rules and standards in the selection of chip data, through multiple samples, genes, and microarray data to make the results more credible and to decrease the error rate. In a GSE81622 data analysis study, we previously assessed 90 genes to determine key genes or gene groups. We also utilized data from the public database hosted by the European Bioinformatics Institute (EMBL-EBI) to select subjects, experimental platforms, and the method of microarray data analysis. This search yielded one dataset (MTAB145) that we used to identify differentially expressed genes common to both datasets. GSE81622 was uploaded in 2016, and the subjects included SLE, LN, and normal controls. MTAB145 is the result of experimental data obtained in 2010, which involves microarray experiments in patients with simultaneous vasculitis. The gender matching of normal subjects (13 M/12 F) and SLE (13 F) in MTAB145 was different, but the gender matching between SLE and healthy individuals in GSE81622 was more balanced. The average age of SLE patients in GSE81622 was 29.2, while the average age in MTAB145 was 47 years, in spite of NI matched. The disease activity scores of SLE patients selected from the two datasets were SLEDAI and BILAG, respectively. GSE81622 was obtained through the Illumina HumanHT-12 V4.0 expression Bead Chip, and MTAB145 was conducted through Affymetrix Human Genome U133 Plus 2 arrays. All of these factors may have led to the differences between the two datasets. Moreover, the experimental error of each experiment may have also played a role. This further supports the reliability of the 18 genes identified in this study. Because two centers used subjects with racial differences in our cDNA chip selection, we showed that the 18 genes in SLE morbidity played an important role depending on the patient’s ethnicity. Bing et al. [24] integrated four GEO datasets from chip results of monocytes, PBMCs, and whole blood cells. They determined that IFI6, IFI27, IFI44L, OAS1, OAS2, EIF2AK2, PLSCR1, STAT1, RNASE2, and GSTO1 are trademark SLE genes. Our study also identified a subset of these results (IFI27, IFI44L, and RNASE2), although we used a slightly different platform. By using SLE PBMCs, our research is more targeted. Studies have reported that epithelial stromal interaction (EPSTI1) [25], 1, 2’-5’ oligoadenylate synthetase 3 (OSA3) [26], tetratricopeptide repeats 1 (IFIT1) [27], LCN2 [28], and lipocalin are highly expressed in SLE. BPI is the target antigen of ANCA in SLE [29], and it is used to identify propylthiouracil (PTU)-induced lupus-like syndrome (LLS) and SLE [30]. Differences in the expression of USP18 and DEFA4 in SLE patients [26] could provide a better explanation for the difference in the SLE ratio between men and women. This study also identified genes that have not been reported in SLE such as HP, ORM1, CAMP, DEFA1, CA1, and ALAS2. The majority of studies on these genes were conducted in tumors or inflammatory diseases such as type 1 diabetes. However, whether they are also involved in the pathogenesis of SLE requires further study.
DNA methylation changes gene expression through promoter hypo-methylation, which can lead to transcriptional activation causing overexpression. SLE lymphocytes have low methylation levels overall [31], and long-term exposure to methylation drugs (such as hydralazine and procainamide) can induce lupus changes [32]. In the two methylation datasets, the disease activity of patients with SLE varied. The SLE SLEDAI score was an average of 12 points in GSE82221, while the SLEDAI average score was only 3.667 in GSE76056. There were also differences in the disease duration between the two datasets, GSE82221 (4 months) and 2.5 in GSE76056 (2.5 years). Both datasets (GSE82221 and GSE76056) were analyzed through the Illumina Human Methylation 450 K Bead Chip (Human Methylation 450_15017482). The difference in the results between the two methylated microarrays may be due to different experimental design. Taken together, there are still 50 genes that correspond to the extent of methylation, and we believe that the results (GSE82221 and GSE76056) are more reliable. To ascertain if the methylation regulation of the 50 genes can lead to changes in the phenotype of genetic conditions, we identified differentially expressed genes through two methods and found two genes: IFL44 and IFL44L. Mok et al. [33] found 19 methylation loci associated with lupus nephritis and verified the low methylation status for IFI44 (cg01079652) in CD4+ T cells. By integrating the results from two methylation chips, we found low methylation of IFI44L, which is consistent with previous studies. cDNA chip results indicate a high expression of IFI44L, allowing the speculation that IFI44L has low methylation during gene expression. One gene may have multiple methylation sites, and different methylation sites may have different methylation levels. Coit et al. [34] separated neutrophils from 15 women diagnosed with SLE and 15 NC PBMCs and detected abnormally low methylation for cg05696877, cg06872964, cg13304609, and cg03607951, which corresponds to the gene for IFI44L as well as cg01079652, corresponding to the gene for IFI44. A multicenter study found that two sites in the IFI44L promoter region, site1 (Chr1: 79 085 222) and site2 (Chr1: 79 085 250; cg06872964) had significant methylation differences between SLE and NC. Therefore, IFI44L can be a highly sensitive and specific diagnostic indicator for SLE [35]. After integrating the chip data, we found altered methylation sites in IFI44L, in addition to cg06872964 (cg00458211, cg13304609, cg03607951, and cg05696877). The methylation site in IFI44 has not been reported.
miRNA are non-coding small RNA of messenger RNA molecules that function as important transcription factors [36]. We analyzed the microarray data from Chen et al. [14], which included eight SLE female patients from the Division of Clinical Immunology, University of Debrecen. All SLE patients were treated with methyl prednisolone therapy at an average dose of 4 mg per day. The highest treatment dose was not more than 8 mg. SLE patients did not show clinical activity, and the SLEDAI score was below 4 points. Our analysis found 135 differentially expressed miRNA corresponding to 5766 target genes. The GO function enrichment analysis revealed 847 related GOs, and the first three were protein binding, nucleoplasm, and cytoplasm. KEGG analysis revealed 177 pathways, including ‘pathways in cancer’ as the most significant. A meta-analysis [37] showed a connection between SLE and malignant tumors. SLE can increase the risk of non-Hodgkin’s lymphoma, leukemia, and Hodgkin’s lymphoma (HL), as well as cancer of the larynx, lung, liver, vulva and vagina, thyroid, and some non-hematologic malignancies. However, it also reduces the risk of skin melanoma. Moreover, hormone and immunosuppressive agents used for treating lupus have antitumor effects, further confirming the importance of similar or identical pathways in cancer and SLE. miRNA changes the phenotype of the disease by regulating gene expression, and further study is necessary to determine whether these pathways are regulated.
The genes obtained by cDNA chip included IFI44L and OAS3 repeated in two parts, but miRNA generally negatively regulates gene expression. Previous studies have shown that IFI44L and OAS3 expression is higher in SLE patients than in healthy individuals. Yet, in this study, the miRNA is also highly expressed, suggesting some contradictions. In this study, IFI44L was predicted by hsa-miR-15b-3p. Previous results (14) suggest that miR-15b-3p is expressed at a level 16 times higher in SLE patients (the miR-15b-3p FC is greater than 4) than in NC patients. OAS3 is predicted by the hsa-miR-143-3p gene, and the same 8-fold increase in expression in SLE (FC greater than 4) compared to NC was also reported by Chen et al. However, no studies have been conducted to verify the expression of these two miRNAs in SLE. The miRNA chip is designed such that individual and race differences might have a significant influence on the results. Therefore, further experimental verification is essential.
SNP is the most common factor in human genetic variation, accounting for more than 90% of all known genetic polymorphisms. Morris et al. [38] suggest that the genetic basis of SLE in Asians has increased in prevalence. Genome-wide association studies (39) have found that over 60 genetic loci confer a risk for SLE, but most of the genetic contributions to SLE remain unknown [39]. Han et al. [40] presented the first GWAS study in Han Chinese by genotyping 1047 cases and 1205 controls through Illumina Human 610-Quad Bead Chips, which identified nine new susceptibility loci. This study only identified differences in SNPs and corresponding genes between SLE and NC. However, whether these SNPs can lead to gene transcription or protein translation change is unknown and requires further investigation. SNPs in the coding region (coding SNP, cSNP) are relatively small, because the mutation rate in the exon is only one-fifth that of the surrounding sequence. However, it is important in the study of hereditary diseases. Yang et al. [41] genotyped 320 (27 males, 293 females) SLE patients through the Illumina 610-Quad Human Bead Chip with SNPs reaching 620, 901. After the GWAS analysis, they chose two SNPs for further validation. They determined that allelic expression analysis from PBMCs showed significantly lower expression levels from the risk allele in SNP rs1128334, which is located in the UTR3 of ETS. They also demonstrated that rs7097397 in WDFY4 changes an arginine residue to glutamine (R1816Q) in this protein. SLE patients with NCF1 gene polymorphisms have decreased oxidative stress [42], suggesting that NCF1 gene exon polymorphism leads to changes in protein coding and expression. We found SNPs located in exons by analyzing the SNPs in the literature [15]. It is presumed that the SNPs located in the exon region may lead to changes in phenotype and match with the cDNA gene. We identified MX1 as an altered gene through the corresponding SNP for rs469390 (P = 4.44 e-02, OR = 4.44, 95% CI = 0.66-0.99). The MX1 promoter rs2071430 G/T polymorphism is associated with SLE susceptibility [43]. However, there are no studies to date on the presence of SNP abnormalities in exons.
Here, we identified 18 differentially expressed genes by integrating the cDNA chip data. These genes can be further studied to ascertain their role in the pathogenesis of SLE, provide guidance on early diagnosis and disease prediction, and suggest therapeutic targets for SLE. Since we only integrated the two central cDNA chips, both of which exhibit heterogeneity for the Asian and European population, we cannot exclude possible racial differences in gene expression. This important factor still requires further experiments to clarify its impact. We identified genes that overlap in expression from the two cDNA microarrays. The results generated a small number of genes obtained by the intersection of methylation chips, miRNAs, and SNPs. However, the genes are highly reliable because they were validated by two chips. We searched for “SLE and miRNA” in the GEO database and found no relevant miRNA microarray. Liu et al. [44] obtained miRNA microarray data from three SLE and NC patients, resulting in 29 differentially expressed miRNAs (P < 0.05, logFC > 1). Although the results were different from those of previous studies, they suggested that race, individual differences, and test methods led to the different results. This study was too small to be included in our analysis. In this study, we analyzed the literature from the past five years, which could lead to a biased outcome. For example, Dai et al. [45] was excluded from our analysis due to our criteria in publication year and experimental methods. Therefore, there is still a need for in-depth analysis of the sample, and for experimental verification. The differentially expressed genes were analyzed through DNA methylation, miRNA, and SNPs. All the data were objective and accurate, which provides a basis for the mechanism of differential gene expression. The next step will be to test the identified genes.
In conclusion, we mined existing publicly available data to analyze the reasons for differentially expressed genes in SLE. The combination of these datasets provides the basis for experimental verification. The results from this analysis might provide biomarkers and new therapeutic targets for SLE.
Acknowledgements
This work was supported by grants from Natural Science Foundation of Hunan Province (806268104034).
Disclosure of conflict of interest
None.
References
- 1.Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–2121. doi: 10.1056/NEJMra1100359. [DOI] [PubMed] [Google Scholar]
- 2.Yap DY, Chan TM. Lupus nephritis in Asia: clinical features and management. Kidney Dis (Basel) 2015;1:100–109. doi: 10.1159/000430458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouda E, Stapon A, Garcia-Diaz M. Mechanisms of mammalian mitochondrial transcription. Protein Sci. 2019;28:1594–1605. doi: 10.1002/pro.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu X, Li F, Yang B, Liang J, Qin H, Xu J. Effects of ultraviolet B exposure on DNA methylation in patients with systemic lupus erythematosus. Exp Ther Med. 2013;5:1219–1225. doi: 10.3892/etm.2013.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deng C, Kaplan MJ, Yang J, Ray D, Zhang Z, McCune WJ, Hanash SM, Richardson BC. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 6.Rissland OS, Subtelny AO, Wang M, Lugowski A, Nicholson B, Laver JD, Sidhu SS, Smibert CA, Lipshitz HD, Bartel DP. The influence of microRNAs and poly(A) tail length on endogenous mRNA-protein complexes. Genome Biol. 2017;18:211. doi: 10.1186/s13059-017-1330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mooney MA, Nigg JT, McWeeney SK, Wilmot B. Functional and genomic context in pathway analysis of GWAS data. Trends Genet. 2014;30:390–400. doi: 10.1016/j.tig.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theodoratou E, Timofeeva M, Li X, Meng X, Ioannidis JPA. Nature, nurture, and cancer risks: genetic and nutritional contributions to cancer. Annu Rev Nutr. 2017;37:293–320. doi: 10.1146/annurev-nutr-071715-051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan M, Yu B, Wan J, Zhang X, Wu Z, Zhong Q, Zhang W, Zou H. Identification of BANK1 polymorphisms by unlabelled probe high resolution melting: association with systemic lupus erythematosus susceptibility and autoantibody production in Han Chinese. Rheumatology (Oxford) 2011;50:473–480. doi: 10.1093/rheumatology/keq353. [DOI] [PubMed] [Google Scholar]
- 10.van Dam S, Vosa U, van der Graaf A, Franke L, de Magalhaes JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018;19:575–592. doi: 10.1093/bib/bbw139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linz U. Commentary on effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466) Cancer. 2010;116:1844–1846. doi: 10.1002/cncr.24950. [DOI] [PubMed] [Google Scholar]
- 12.Omidi F, Hosseini SA, Ahmadi A, Hassanzadeh K, Rajaei S, Cesaire HM, Hosseini V. Discovering the signature of a lupus-related microRNA profile in the Gene Expression Omnibus repository. Lupus. 2020;29:1321–1335. doi: 10.1177/0961203320944473. [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Chen W, Zheng F, Tang D, Liu D, Wang G, Xu Y, Yin L, Zhang X, Dai Y. Reconstruction and analysis of the aberrant lncRNA-miRNA-mRNA network in systemic lupus erythematosus. Lupus. 2020;29:398–406. doi: 10.1177/0961203320908927. [DOI] [PubMed] [Google Scholar]
- 14.Chen JQ, Papp G, Poliska S, Szabo K, Tarr T, Balint BL, Szodoray P, Zeher M. MicroRNA expression profiles identify disease-specific alterations in systemic lupus erythematosus and primary Sjogren’s syndrome. PLoS One. 2017;12:e0174585. doi: 10.1371/journal.pone.0174585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, Ma J, Qi YY, Kim-Howard X, Motghare P, Bhattarai K, Adler A, Bang SY, Lee HS, Kim TH, Kang YM, Suh CH, Chung WT, Park YB, Choe JY, Shim SC, Kochi Y, Suzuki A, Kubo M, Sumida T, Yamamoto K, Lee SS, Kim YJ, Han BG, Dozmorov M, Kaufman KM, Wren JD, Harley JB, Shen N, Chua KH, Zhang H, Bae SC, Nath SK. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48:323–330. doi: 10.1038/ng.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Diboun I, Wernisch L, Orengo CA, Koltzenburg M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics. 2006;7:252. doi: 10.1186/1471-2164-7-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan TC, Fang H, Magder LS, Petri MA. Differences between male and female systemic lupus erythematosus in a multiethnic population. J Rheumatol. 2012;39:759–769. doi: 10.3899/jrheum.111061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghodke-Puranik Y, Niewold TB. Immunogenetics of systemic lupus erythematosus: a comprehensive review. J Autoimmun. 2015;64:125–136. doi: 10.1016/j.jaut.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogeweg P. The roots of bioinformatics in theoretical biology. PLoS Comput Biol. 2011;7:e1002021. doi: 10.1371/journal.pcbi.1002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bing PF, Xia W, Wang L, Zhang YH, Lei SF, Deng FY. Common marker genes identified from various sample types for systemic lupus erythematosus. PLoS One. 2016;11:e0156234. doi: 10.1371/journal.pone.0156234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii T, Onda H, Tanigawa A, Ohshima S, Fujiwara H, Mima T, Katada Y, Deguchi H, Suemura M, Miyake T, Miyatake K, Kawase I, Zhao H, Tomiyama Y, Saeki Y, Nojima H. Isolation and expression profiling of genes upregulated in the peripheral blood cells of systemic lupus erythematosus patients. DNA Res. 2005;12:429–439. doi: 10.1093/dnares/dsi020. [DOI] [PubMed] [Google Scholar]
- 26.Fan H, Zhao G, Ren D, Liu F, Dong G, Hou Y. Gender differences of B cell signature related to estrogen-induced IFI44L/BAFF in systemic lupus erythematosus. Immunol Lett. 2017;181:71–78. doi: 10.1016/j.imlet.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Ye S, Pang H, Gu YY, Hua J, Chen XG, Bao CD, Wang Y, Zhang W, Qian J, Tsao BP, Hahn BH, Chen SL, Rao ZH, Shen N. Protein interaction for an interferon-inducible systemic lupus associated gene, IFIT1. Rheumatology (Oxford) 2003;42:1155–1163. doi: 10.1093/rheumatology/keg315. [DOI] [PubMed] [Google Scholar]
- 28.Torres-Salido MT, Cortes-Hernandez J, Vidal X, Pedrosa A, Vilardell-Tarres M, Ordi-Ros J. Neutrophil gelatinase-associated lipocalin as a biomarker for lupus nephritis. Nephrol Dial Transplant. 2014;29:1740–1749. doi: 10.1093/ndt/gfu062. [DOI] [PubMed] [Google Scholar]
- 29.Cooper T, Savige J, Nassis L, Paspaliaris B, Neeson P, Neil J, Knight KR, Daskalakis M, Doery JC. Clinical associations and characterisation of antineutrophil cytoplasmic antibodies directed against bactericidal/permeability-increasing protein and azurocidin. Rheumatol Int. 2000;19:129–136. doi: 10.1007/s002960050116. [DOI] [PubMed] [Google Scholar]
- 30.Gajic-Veljic M, Bonaci-Nikolic B, Lekic B, Skiljevic D, Ciric J, Zoric S, Stojimirovic B, Nikolic M. Importance of low serum DNase I activity and polyspecific anti-neutrophil cytoplasmic antibodies in propylthiouracil-induced lupus-like syndrome. Rheumatology (Oxford) 2015;54:2061–2070. doi: 10.1093/rheumatology/kev243. [DOI] [PubMed] [Google Scholar]
- 31.Hedrich CM, Mabert K, Rauen T, Tsokos GC. DNA methylation in systemic lupus erythematosus. Epigenomics. 2017;9:505–525. doi: 10.2217/epi-2016-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balada E, Castro-Marrero J, Felip L, Ordi-Ros J, Vilardell-Tarres M. Associations between the expression of epigenetically regulated genes and the expression of DNMTs and MBDs in systemic lupus erythematosus. PLoS One. 2012;7:e45897. doi: 10.1371/journal.pone.0045897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mok A, Solomon O, Nayak RR, Coit P, Quach HL, Nititham J, Sawalha AH, Barcellos LF, Criswell LA, Chung SA. Genome-wide profiling identifies associations between lupus nephritis and differential methylation of genes regulating tissue hypoxia and type 1 interferon responses. Lupus Sci Med. 2016;3:e000183. doi: 10.1136/lupus-2016-000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, Kaplan MJ, Sawalha AH. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun. 2015;58:59–66. doi: 10.1016/j.jaut.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, Liu Y, Jiang J, Luo S, Tan Y, Wu H, Renauer P, Del Mar Ayala Gutierrez M, Castillo Palma MJ, Ortega Castro R, Fernandez-Roldan C, Raya E, Faria R, Carvalho C, Alarcon-Riquelme ME, Xiang Z, Chen J, Li F, Ling G, Zhao H, Liao X, Lin Y, Sawalha AH, Lu Q. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. 2016;75:1998–2006. doi: 10.1136/annrheumdis-2015-208410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehta A, Baltimore D. MicroRNAs as regulatory elements in immune system logic. Nat Rev Immunol. 2016;16:279–294. doi: 10.1038/nri.2016.40. [DOI] [PubMed] [Google Scholar]
- 37.Cao L, Tong H, Xu G, Liu P, Meng H, Wang J, Zhao X, Tang Y, Jin J. Systemic lupus erythematous and malignancy risk: a meta-analysis. PLoS One. 2015;10:e0122964. doi: 10.1371/journal.pone.0122964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris DL, Sheng Y, Zhang Y, Wang YF, Zhu Z, Tombleson P, Chen L, Cunninghame Graham DS, Bentham J, Roberts AL, Chen R, Zuo X, Wang T, Wen L, Yang C, Liu L, Yang L, Li F, Huang Y, Yin X, Yang S, Ronnblom L, Furnrohr BG, Voll RE, Schett G, Costedoat-Chalumeau N, Gaffney PM, Lau YL, Zhang X, Yang W, Cui Y, Vyse TJ. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat Genet. 2016;48:940–946. doi: 10.1038/ng.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almlof JC, Alexsson A, Imgenberg-Kreuz J, Sylwan L, Backlin C, Leonard D, Nordmark G, Tandre K, Eloranta ML, Padyukov L, Bengtsson C, Jonsen A, Dahlqvist SR, Sjowall C, Bengtsson AA, Gunnarsson I, Svenungsson E, Ronnblom L, Sandling JK, Syvanen AC. Novel risk genes for systemic lupus erythematosus predicted by random forest classification. Sci Rep. 2017;7:6236. doi: 10.1038/s41598-017-06516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, Xu JH, Cai ZM, Huang W, Zhao GP, Xie HF, Fang H, Lu QJ, Xu JH, Li XP, Pan YF, Deng DQ, Zeng FQ, Ye ZZ, Zhang XY, Wang QW, Hao F, Ma L, Zuo XB, Zhou FS, Du WH, Cheng YL, Yang JQ, Shen SK, Li J, Sheng YJ, Zuo XX, Zhu WF, Gao F, Zhang PL, Guo Q, Li B, Gao M, Xiao FL, Quan C, Zhang C, Zhang Z, Zhu KJ, Li Y, Hu DY, Lu WS, Huang JL, Liu SX, Li H, Ren YQ, Wang ZX, Yang CJ, Wang PG, Zhou WM, Lv YM, Zhang AP, Zhang SQ, Lin D, Li Y, Low HQ, Shen M, Zhai ZF, Wang Y, Zhang FY, Yang S, Liu JJ, Zhang XJ. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 41.Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC, Chan TM, Wong RW, Lee KW, Mok MY, Wong SN, Leung AM, Li XP, Avihingsanon Y, Wong CM, Lee TL, Ho MH, Lee PP, Chang YK, Li PH, Li RJ, Zhang L, Wong WH, Ng IO, Lau CS, Sham PC, Lau YL Asian Lupus Genetics Consortium. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsson LM, Johansson AC, Gullstrand B, Jonsen A, Saevarsdottir S, Ronnblom L, Leonard D, Wettero J, Sjowall C, Svenungsson E, Gunnarsson I, Bengtsson AA, Holmdahl R. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. 2017;76:1607–1613. doi: 10.1136/annrheumdis-2017-211287. [DOI] [PubMed] [Google Scholar]
- 43.AlFadhli S, Al-Mutairi M, Al Tameemi B, Nizam R. Influence of MX1 promoter rs2071430 G/T polymorphism on susceptibility to systemic lupus erythematosus. Clin Rheumatol. 2016;35:623–629. doi: 10.1007/s10067-016-3179-z. [DOI] [PubMed] [Google Scholar]
- 44.Liu D, Zhao H, Zhao S, Wang X. MicroRNA expression profiles of peripheral blood mononuclear cells in patients with systemic lupus erythematosus. Acta Histochem. 2014;116:891–897. doi: 10.1016/j.acthis.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007;16:939–946. doi: 10.1177/0961203307084158. [DOI] [PubMed] [Google Scholar]