

Abstract
An overview of the oxidative deamination of N-acetylneuraminic acid derivatives (Neu5Ac) leading to the formation of ketodeoxynonulosonic acid (KDN), its stereoisomers and glycosides is presented. A brief historical introduction to the deamination is given, followed by a description of recent advances in reaction conditions, which have allowed application of the process to Neu5Ac thioglycosides, and that have enabled the range of nucleophiles incorporated in the course of the reaction to be extended beyond the original acetate and azide. Recent advances resulting in derivatization of the Neu55Ac 4-position concomitant with replacement of the acetamido group, via the presumed intermediacy of a vinyl diazonium ion, are then described. The literature on the mechanism of the deamination reaction is next considered leading to the presentation of an overall mechanistic framework that accounts for all observations to date. Finally, the application of the deamination reaction to complex Neu5Ac-based oligosaccharides and other aminosugars is presented.
1. Introduction
Deamination of aliphatic amines via nitrosylation of their amide derivatives followed by thermal extrusion of nitrogen with formation of the corresponding esters was described by White in the 1950’s.1-4 Thus, in a series of papers White first established methods for the synthesis of N-alkyl-N-nitrosoamides using multiple reagents such as acidified aqueous sodium nitrite, sodium nitrite-acetic anhydride, dinitrogen trioxide, nitrosyl chloride, nitrosyl bromide, and dinitrogen tetroxide, ultimately settling on the latter as the optimal reagent for further studies. Subsequent reports focused on the N→O migration of the acyl group to form a diazo intermediate, which eliminated nitrogen under thermal conditions giving rise to the corresponding esters. The deamination reaction was studied from multiple angles to ascertain the influence of the alkyl and acyl groups, solvent, and temperature, eventually leading White to propose a mechanism accounting for the formation of the various products.3 As determined with amides derived from chiral amines, the reaction proceeds primarily with retention of configuration suggesting the possible involvement of contact ion pairs following the initial N→O acyl migration and expulsion of molecular nitrogen (Scheme 1). The method and related deamination reactions of aminosugars were systematically reviewed by Williams in 1975.5
Scheme 1.
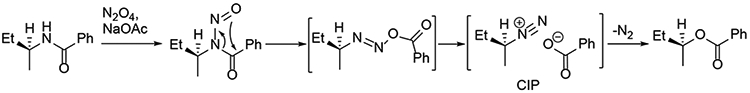
White’s Deamination Reaction.
2. Oxidative Deamination of N-Acetylneuraminic Acid
2.1. Ogura’s Deamination
White’s deamination conditions were employed by Ogura and coworkers for the synthesis of 3-deoxy-D-glycero-D-galacto-2-nonulosonic acid (KDN) starting from the methyl N-acetyl-4,7,8,9-tetra-O-acetyl-β-neuraminosidate (1), a protected form of N-acetylneuraminic acid (Neu5Ac).6 Thus, nitrosylation of 1 with dinitrogen tetroxide generated the intermediate nitrosoamide 2, which then underwent the thermal decomposition to give compound 4, albeit in a low yield of 17%, along with a rearranged product 5 (20%) and an inseparable mixture of 6 (27%) and 7 (27%) resulting from elimination (Scheme 2). It was proposed that the acetoxy group was transferred in an intramolecular fashion from the acetoxydiazene 3 to C5 producing 4 with retention of configuration. The deamination product 4 was then converted to KDN 8 in 23% yield over seven steps. The rearranged product 5 was considered to arise from transfer of the acetoxy group in 3 to the 6-position with concomitant migration of the ring oxygen to C5. Albeit low yielding, these findings unlocked the possibility of deamination of neuraminic acid derivatives, paving way for syntheses of KDN and other sialic acid derivatives, which were otherwise available only from the biological resources or by enzymatic methods.7
Scheme 2.
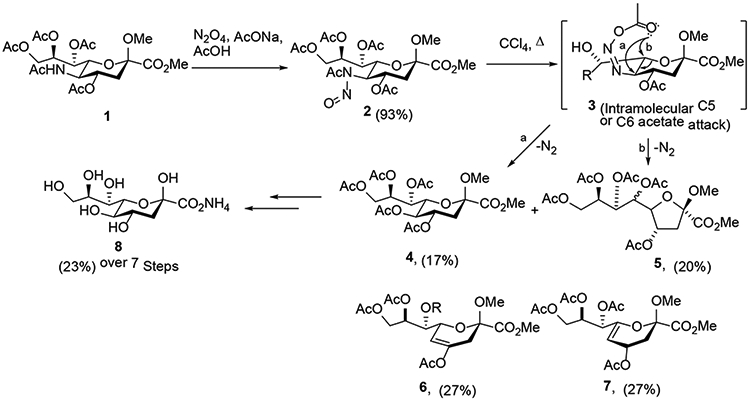
Preparation and Thermal Rearrangement of a N-Acetyl-N-nitrosoneuraminic Acid (2) Derivative by Ogura and coworkers.
2.2. Zbiral’s Deamination
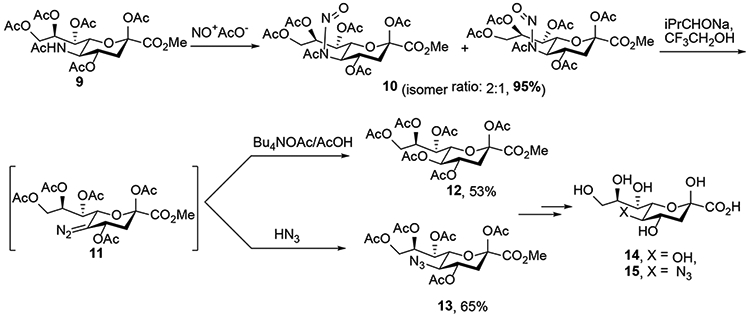
In 1990, Schreiner and Zbiral disclosed improved conditions for the deamination reaction replacing Ogura’s low-yielding thermal rearrangement by treatment of the nitrosoamide with the mildly basic sodium trifluoroethoxide (CF3CH2ONa) to generate the diazo intermediate, all while avoiding Zemplén deacetylation of the multiple ester groups.8 Working with the pyranosyl ester 9, Zbiral and Schreiner used nitrosyl acetate, generated in situ from sodium acetate and dinitrogen tetroxide, to prepare the nitrosoacetamido 10 in 95% yield, which, was determined to be a 2:1 E:Z mixture by NMR spectroscopy. Migratory deacylation and formation of the unstable diazo intermediate 11 was followed by addition of tetrabutylammonium acetate and acetic acid or of hydrogen azide led to displacement of the diazo group from 11 and formation of acetate 12 (53%) or azide 13 (65%), respectively. Intermediate 10 was also subjected to thermal decomposition in toluene to prepare the ring-contracted furanosyl derivative, and the elimination products analogous to those reported by Ogura.6 Removal of the protecting groups completed the synthesis of KDN 14 and 5-azido-5-deacetamido-neuraminic acid (15). Zbiral’s deamination conditions, with the higher yields, were a considerable improvement on those used by Ogura and coworkers, and consequently were adopted by other groups for replacement of the acetamido group in Neu5Ac derivatives and still remain a method of choice.
Nonetheless yet further improved conditions were required to extend the scope of the reaction beyond the simple glycosides employed in the Ogura and Zbiral laboratories. In particular, the conditions were not compatible with thioglycosides, since the nitrosylium ion NO+ is known to activate thioglycosides.9 Further, the initial range of nucleophiles described was limited to acetic acid and hydrazoic acid.
2.3. Navuluri and Crich’s Deamination
With this in mind Navuluri and Crich studied the use of commercially available nitrosyl tetrafluoroborate for nitrosylation of 9 to give 10.10 Further, after the addition of acetic acid to afford the KDN derivative, they employed an ozonolytic work-up step to destroy the standard elimination by-products and thereby simplify the purification of 12, which was subsequently employed to good effect in the formation of equatorially selective KDN donors 18 (β) and 19 (α), and then of various KDN glycosides (Scheme 4).
Scheme 4.
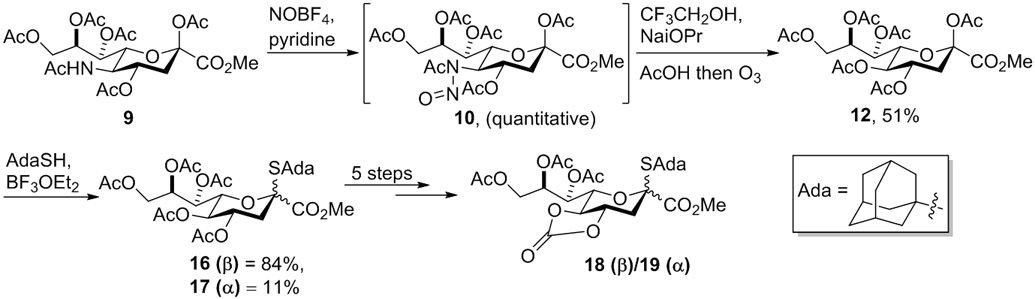
Oxidative Deamination by Navuluri and Crich.
In a further improvement of the method Navuluri and Crich reported that the NOBF4/pyridine conditions for nitrosylation were compatible with the adamantanyl thioglycoside, enabling its installation prior to the deamination step, in contrast to the previous reports.11,9 The key to compatibility with the thioglycoside moiety was the use of pyridine, which complexed with the extremely reactive nitrosonium ion forming the N-nitrosopyridinium complex and so moderating it’s reactivity. In this manner oxidative deamination thioglycoside of 20 afforded the KDN adamantanyl thioglycoside 16 in 58% yield, suitable for direct use in glycosylation reactions (Scheme 5). Navuluri and Crich also used a variety of nucleophiles in the displacement step and so prepared a number of novel C5 modified sialyl derivatives (Scheme 5). Thus, the use of thioacetic acid as a nucleophile produced the 5-thioacetyl derivative 22 in 63% yield, whereas that of the hydrofluoric acid-pyridine complex availed the 5-deoxy-5-fluoro-3-deoxy-D-nonulosonic acid derivative 23 in 53% yield. Reaction with the HF•pyridine complex also generated 11% of the 5-isopropoxy KDN derivative 24 due to attack by the isopropanol present in the reaction mixture in large excess. The yield of the fluoride derivative 23 was further increased to 63% by suppressing the formation of the isopropyl ether byproduct 24 by employing a pre-formed CF3CH2ONa salt in combination with 18-crown-6, thereby completely excluding isopropanol from the reaction mixture. Nevertheless, the observation of the isopropoxy displacement product 24 hinted that even alcohols might serve as nucleophile. Indeed, the reaction conditions could be tweaked by introduction of propargyl alcohol as a nucleophile in combination with non-nucleophilic HBF4 when the 5-propargyl ether derivative 25 was isolated in 66% yield.
Scheme 5.
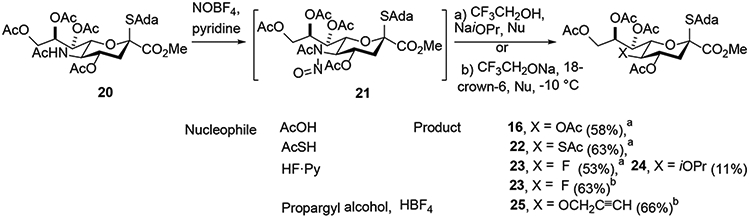
Oxidative Deamination of Adamantanyl Thioglycoside 20 and Use of Assorted Nucleophiles.
2.4. Application to the Synthesis of Pseudaminic Acid and Legionaminic Acid and Their Analogs
Aside from Neu5Ac and KDN, numerous other nonulosonic acid derivatives have been isolated mainly from pathogenic bacteria.12,13 These include multiple derivatives of pseudaminic acid and legionaminic acid differing in glycosylation and amide substitution patterns that are represented by 5,7-diacetamido-3,5,7,9-tetradeoxy-β-L-glycero-L-manno-2-non-ulosonic acid and 5,7-diacetamido-3,5,7,9-tetradeoxy-β-D-glycero-D-galacto-non-2-ulosonic acid, respectively. The presence of such sialyl derivatives on the surface of many pathogenic Gram-negative bacteria, such as Campylobacter jejuni, Helicobacter pylori, Pseudomonas aeruginosa, Shigella bodyii, and Acinetobacter baumanii, suggests an important role of these bacteria in pathogenesis.14-18 However, the exact role played by these sugars is still mostly unclear due to the difficulties in accessing these compounds and their glycosides. Therefore, synthesis of these sialyl derivatives has attracted considerable attention from synthetic chemists.
2.4.1. Synthesis of Pseudaminic Acid by Keifel, Payne and Coworkers.
A key step in the synthesis of pseudaminic acid and its derivatives by Kiefel and coworkers was the conversion of 1 to the KDN derivative 4 in 57% yield (Scheme 6).18 This conversion was achieved by nitrosylation with sodium nitrite in a mixture of acetic anhydride and acetic acid followed by heating to 50 °C, and was conducted on a scale of 2.3 g. Subsequent elaboration of 4 toward pseudaminic acid required differentiation of the five acetylated hydroxyl groups.19 This was achieved in three steps by Zemplén deacetylation, installation of an acetonide on the 8,9- diol, and regioselective monosilylation on the most reactive hydroxyl group of the so-formed triol to attain to compound 26. Triflation of the remaining free hydroxyl groups in compound 26 was followed by displacement with azide to obtain the key 5,7-diazido-intermediate 27, from which the synthesis of 28 was successfully completed.20
Scheme 6.
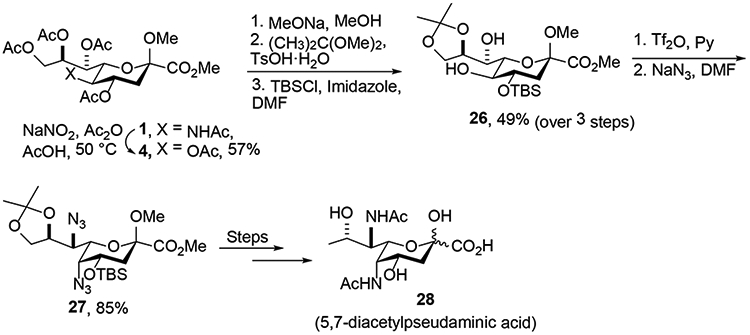
Synthesis of 5,7-Diacetylpseudaminic Acid (28) by the Keifel and Payne Groups.
2.4.2. Synthesis of Pseudaminic Acid and its Glycosides by the Crich Group
The Crich laboratory began their studies toward thesynthesis of pseudaminic acid with the preparation of a model pseudaminic acid donor that featured the Zbiral type deamination of 21 employing nitrosyl tetrafluorborate in pyridine for the critical nitrosylation step, and the use of levulinic acid as proton source and nucleophile (Scheme 7).21 This reaction, which afforded the 5-O-levulinoyl KDN derivative 29 in 49% yield and with retention of configuration, was conducted on a scale of 1 g. Selective cleavage of the levulinate in the presence of multiple acetate esters was then achieved by the action of hydrazine hydrate and acetic acid. Triflation, followed by azide displacement subsequently afforded the model compound 31 with which to study the influence of the axial azide group on glycosylation. The use of levulinic acid as nucleophile, while predictable on the basis of the widespread application of acetic acid in this context, generated an unusual byproduct 30 in addition to the simple substitution product 29, which is further commented below. It was also demonstrated in the course of this study that triflic acid could be used as proton source and nucleophile in the deamination reaction, enabling direct formation of the KDN 5-O-triflate 32 in 34% yield as a single isomer. Finally, and following the precedent from the Zbiral laboratory, hydrazoic acid was employed as nucleophile leading to the equatorial 5-azido derivative 33 in 51% yield (Scheme 7). Lessons learnt from this model study were then applied to the synthesis of an actual pseudaminic acid donor.22 Thus, Neu5Ac was converted through a number of steps to the acetamide 34, which was nitrosylated, subjected to sodium trifluoroethoxide, and finally exposed to levulinic acid at −10 °C. On a scale of 5 g, this reaction afforded the equatorial levulinate 36 in 55% yield (Scheme 8). This reaction is noteworthy for the scale on which it was conducted and for the use of the naphthylmethyl (Nap) ether as protecting group at the 7-position. It is of interest that when the deamination was conducted on the 9-deoxy derivative 39 under conditions mimicking those employed by Kiefel and coworkers, the product 41 was obtained in 38% yield following removal of the levulinoyl group, but as a 1:1 mixture of diastereoisomers. The poor stereoselectivity of this reaction, when contrasted with the conversion of 1 to 4 in the Kiefel synthesis,19 conducted under similar conditions, emphasizes the sensitivity of the deamination process to the protecting groups and deoxygenation of the side chain, which is discussed in greater detail below. With a satisfactory synthesis of 36 in hand Dhakal and Crich were able to complete their synthesis of the pseudaminic acid donor 37 and demonstrate its use for the first time in the highly stereoselective synthesis of equatorial pseudaminic acid glycosides.
Scheme 7.
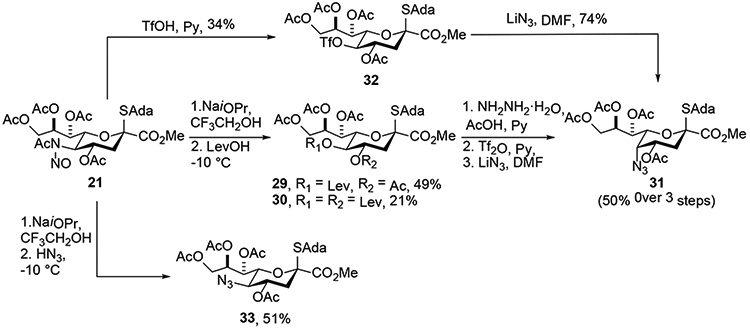
Synthesis of 5-Azido-5-deacetamidosialyl- and 5-epi-Azido-5-deacetamidosialyl Thioglycoside Donors 31 and 33.
Scheme 8.
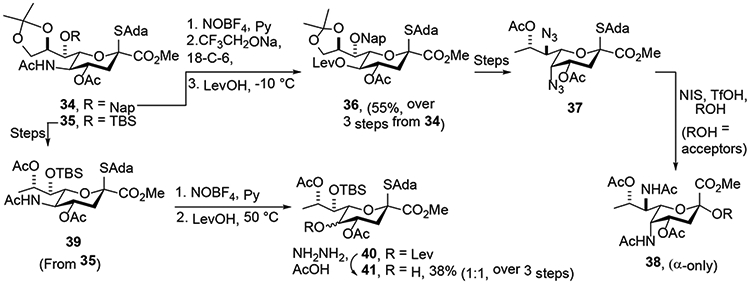
Synthesis of Pseudaminic acid donor (37): Influence of Deoxygenation on Stereoselectivity
2.5. Use of Thiophenols and Phenols as Nucleophile
Seeking to further extend the range of compatible nucleophiles for the substitution reaction, Crich and coworkers examined the use of phenols and thiophenols.23 Albeit only a limited number were examined, thiophenols followed the standard pattern giving the products of substitution with retention of configuration (42, 44 and 45), and lesser amounts of a 4-deoxy product 43 (Scheme 9). With phenols on the other hand a more complex and nucleophile specific spectrum of products was observed. Thus, phenol itself afforded the enol ether 46 and the disubstitution product 47 in 31 and 33% yield, respectively (Scheme 10). β-Naphthol on the other hand afforded a new skeletal type demonstrated by X-ray crystallography to be the cis-fused tricyclic product 48, which was isolated in 57% yield (Scheme 10). Analogous products 50-52 were obtained in comparable yields when 6-hydroxyquinoline, 5-hydroxyindole, and 3,5-dimethoxyphenol were applied as nucleophile.
Scheme 9.
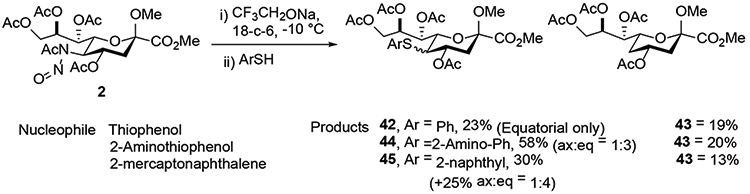
Thiophenols as Nucleophiles in the Zbiral Reaction.
Scheme 10.
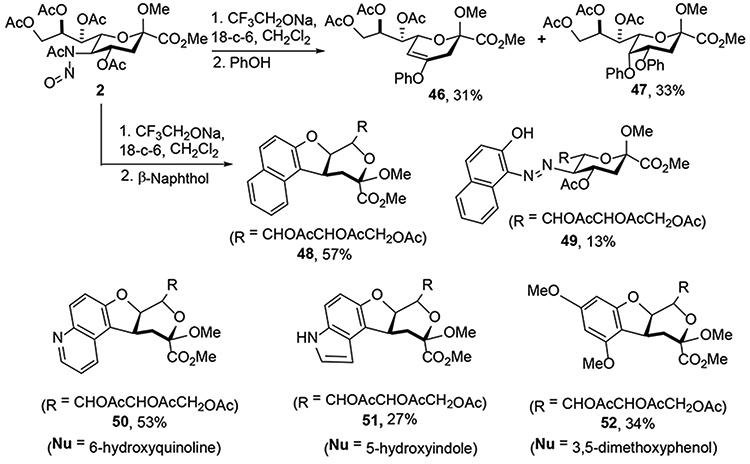
Phenols as Nucleophiles in the Zbiral Reaction.
2.6. Use of Hydroxylamine Derivatives and Aniline as Nucleophile
Looking to extend the range of nucleophiles even further Crich and coworkers examined the use of N-hydroxybenzotriazole, N-hydroxyphthalimide, acetohydroxamic, and finally aniline.24 N-Hydroxybenzotriazole performed similarly to acetic acid and gave the simple substitution product 53 (Scheme 11). With N-hydroxyphthalimide the situation was more complex and the disubstitution product 55 was obtained in 32% yield, alongside the simple substitution product 54, which was isolated in 36% yield. It will be noted that this disubstitution product is analogous to that (30) seen with levulinic acid (Scheme 7), but has a different configuration to that seen with phenol (47) (Scheme 10) as discussed in more detail below. With acetohydroxamic acid a cyclic product (56) was obtained in 21% yield that recalls the adduct 48 isolated from the reaction with β-naphthol (Scheme 10). A similar cyclic product on the β-face of the pyranoside ring, the aziridine 57, was obtained in 49% yield when aniline was employed as nucleophile.24
Scheme 11.
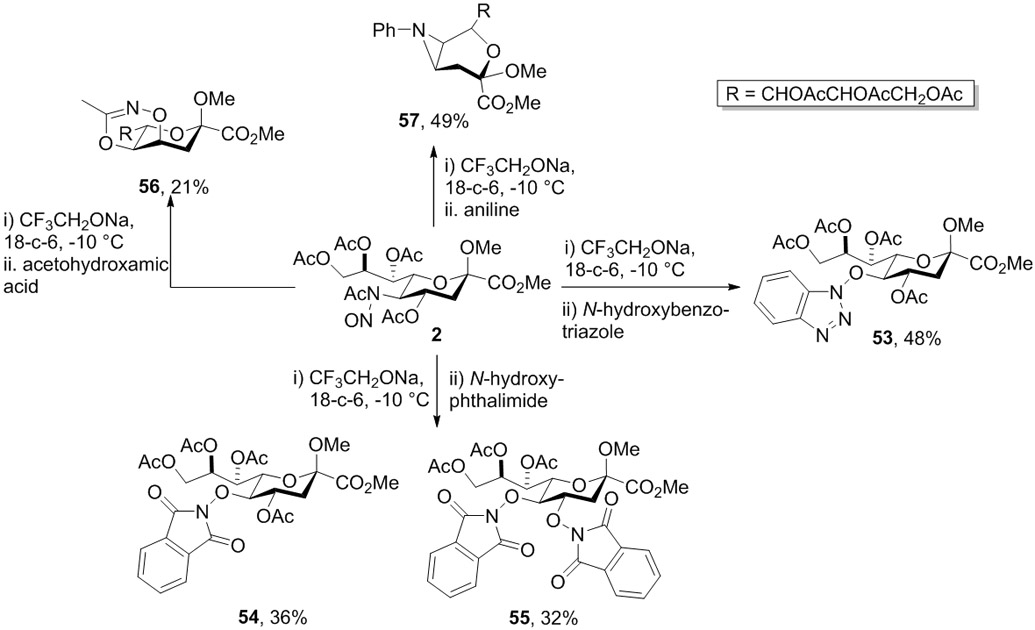
Exploration of Further Nucleophiles by the Crich Group
2.7. Mechanism of the Reaction
With regard to the mechanism of the deamination reaction, the key issue is the origin of the retention of configuration achieved in most reported examples. Ogura and coworkers6 (Scheme 2) proposed that intramolecular acetoxy transfer to the C-5 or C-6 position on thermal decomposition of the acetoxydiazene 3, leads to the formation of KDN or the ring contraction product, respectively, whereas a carbenium ion was presumed as intermediate en route to the alkenes 6 and 7. Schreiner and Zbiral,8 on the other hand invoked formation of a diazoalkane 11 (Scheme 3) followed by formation of an acetoxonium ion resulting from participation of an acetate group, with subsequent ring opening by the nucleophile to form the equatorially substituted derivatives. Schreiner and Zbiral further suggested that the anti-configured nitrosoacetamide decomposes into the two alkene byproducts, whereas the syn-isomer produces the furanoside ring. Neither group, however, conducted experiments to test their hypotheses. In an attempt to shed light on these issues Buda and Crich conducted an extensive series of experiments focusing on the influence of protecting groups.25
Scheme 3.
Schreiner and Zbiral’s Deamination Protocol.
2.7.1. Effect of Protecting Groups
To assess the effects exerted by the protecting groups from different positions and to evaluate the possibility of stereodirecting participation by esters at the 4- or 7-positions through dioxalenium or dioxenium ions, several N-acetylneuraminic acid derivatives bearing different protecting groups were prepared and subjected to the Zbiral-type reaction conditions.25 The products obtained, yields, and equatorial versus axial selectivity for each substrate are summarized in Figure 1. The retention of configuration observed in compounds 58, 59, and 60 carrying either a benzyl or a silyl ether at O4 or O7 in place of the typical acetyl groups eliminates stereodirecting ester participation through five or six-membered intermediates from consideration. This observation was further supported by the formation of the 4-O and 7-O-tert-butoxycarbonyl protected systems 61 and 62, which were not accompanied by any of the cyclic carbonates typically observed from participation by a Boc group. In a similar vein, no lactone formation accompanied the tert-butyl ester 63 under the standard deamination conditions. In contrast, when both the O4 and O7 acetyl groups were replaced with benzyl ethers, partial erosion of selectivity was observed with the product 64 isolated as 4.5:1 equatorial:axial mixture. Finally, with the 4,7-di-O-benzyl-8,9-O-isopropylidene system, lacking ester groups altogether, all selectivity was lost and the product 65 isolated as a 1:1 mixture of diastereoisomers. To this list should be added the 9-deoxy derivative 40 (Scheme 8), which also did not exhibit the usual preference for substitution with retention of configuration, albeit at a higher temperature. It is also noteworthy that the crystalline N-nitrosoacetamide 66 when stored at 4 °C for several weeks underwent decomposition to the product 67, isolated in 38% yield, with complete inversion of configuration. This observation contrasts to the 40% yield of the more typical KDN product 65 obtained as a mixture of anomers from the same donor under the standard conditions in solution.
Figure 1.
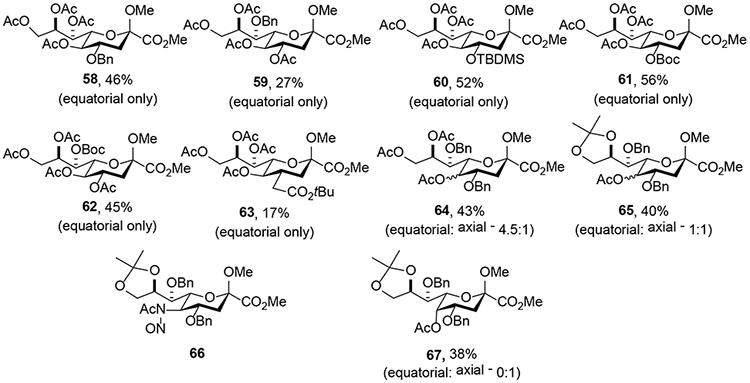
Effect of Protecting Groups on the Stereoselectivity of the Deamination Reaction with Acetic Acid as Nucleophile.
2.7.2. Isotopic labeling studies
Isotopic labeling was used to further probe the reaction mechanism.23,25 Oxidative deamination of 2 using deuterated reagents, CF3CH2OD, iPrOD and CH3CO2D generated a single product 4-d with retention of configuration and complete incorporation of deuterium at the H5-position (Scheme 12). The use of acetic acid 99% enriched in 13C at the carbonyl C as a nucleophile with the methyl glycoside 2 as substrate afforded compound 13C1-4 in 53% yield, clearly demonstrating the substitution to be an intermolecular process at least under the Zbiral-type conditions (Scheme 12). In contrast, with the thioglycoside 21 as substrate the product 13C1-16 was found by mass spectrometry be a 2.5:1 mixture of mono- and di-isotopomers. Extensive NMR experimentation revealed the second 13C-labelled acetate group in the di-isotopomer to have been incorporated at the 4-position. This conclusion was confirmed when levulinic acid was employed as nucleophile with 21 as substrate leading to the isolation of the minor disubstitution product 30 (Scheme 7). The anomer 68 of 21 afforded only the product 13C1-17 of substitution at the 5-position.
Scheme 12.
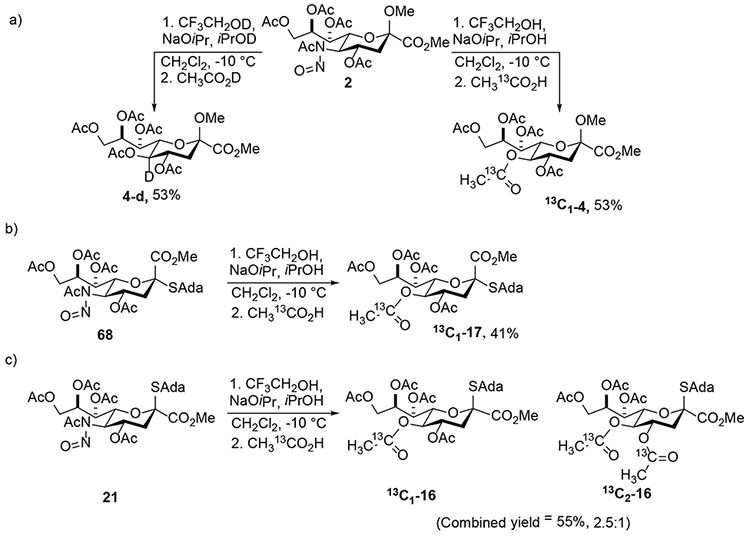
Isotopic Labeling Studies.
2.7.3. Overall Mechanism
This ensemble of results led to the proposal of an overall mechanism as detailed in Scheme 13.25 The mechanism begins with selective deacetylation of 69, by the weak nucleophile CF3CH2ONa generating the hydroxydiazine 70, which eliminates a water molecule to give the diazonium ion 71. This intermediate 71 is in equilibrium with its conjugate diazoalkane 72, as confirmed by the deuterium-labelling study. Notably, the incorporation of deuterium at the 5-position with retention of configuration suggests that protonation of the diazoalkane 72 takes place preferentially from the axial direction to give diazonium ion 71 directly in the chair conformation consistent with the principles of stereoelectronic control. In the next step of the reaction carbenium ion 75 and/or oxonium ion 76 are formed from the diazonium ion 71 on extrusion of nitrogen. Alternatively, expulsion of N2 either from 71 or 72 potentially yields an intermediate carbene 73, which undergoes stabilization by the ring oxygen in the form of the oxabicyclo[3.1.0]hexyl ylide 74. Protonation of carbene 73 or of the ylide 74 would then result in either the carbenium ion 75 or the oxabicyclo[3.1.0]hexyl oxonium ion 76. Nucleophilic attack at C5- of the oxabicyclo[3.1.0]hexyl oxonium ion 76 then generates substitution product 77 with inversion of configuration, leading overall to substitution with retention of configuration. Competing nucleophilic attack on C6 of the bicyclic oxonium ion 76 would lead to ring contraction and formation of the furanoside 78 observed by the early workers in the field. In contrast to stereoselective attack on the oxonium ion 76, nucleophilic attack on the carbenium ion 75 would be expected to be less selective and afford mixtures of stereoisomers. It was suggested that the relative contributions of 75 and 76 to what maybe delocalized non-classical structures are influenced by the protecting group array (Scheme 13). Thus, the presence of electron withdrawing groups such as acetates would significantly destabilize the carbenium ion 75 and so favor the bicyclic oxabicyclo[3.1.0]hexyl oxonium ion 76 resulting overall in equatorially selective attackby what amounts to a double inversion mechanism. On the other hand, with less electron withdrawing benzyl ethers or silyl ethers the carbenium ion 75 plays a greater role and selectivity is correspondingly reduced. The alkenes 80 and 81 are formed from the diazonium ion (71) stage with elimination of N2 or from the elimination at the bicyclic carbenium ion- 75 oxonium ion 76 stage. The formation of the cyclic oxonium ion 76 from the diazonium ion 71, considered to be an important element in the usual overall retention of configuration, is best viewed as an endothermic reaction step proceeding with a late (cation 75-like) transition state. As such the stereoelectronic considerations which, at first sight, argue against such a mechanism, because of the poor overlap of the ring oxygen lone pairs with the C5-N2 σ* orbital, do not come into play.
Scheme 13.
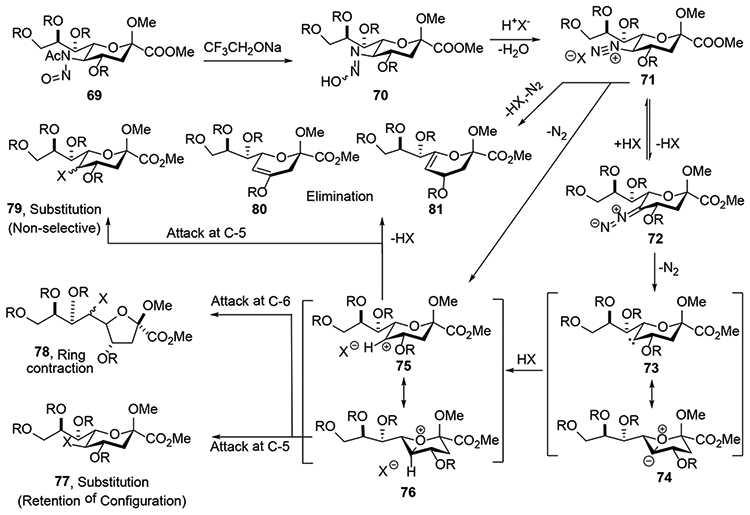
Buda and Crich’s mechanism.
Slow decomposition of crystalline 66 (Figure 1) at 4 °C in the crystal and without the addition of external reagents is considered to take place by an intramolecular pathway and a contact ion pair by an Ogura-like mechanism. Presumably, the confines of the crystal lattice are such the CIP is formed on the α-face resulting in the overall decomposition with inversion of configuration.
2.7.4. Mechanism of Substitution at the 4-Position
The products arising from the use of phenols as nucleophile are considered to arise from the elimination of the 4-acetoxy group at the level of the intermediate diazoalkene 72 giving rise to a vinyl diazonium ion 82.23 This potent electrophile then undergoes Michael addition at the 4-position leading to a new diazoalkane. After protonation of this diazoalkane nitrogen is displaced from the diazonium ion to yield the disubstituted products. With β-naphthol, the Michael addition takes place on the top face of the vinyl diazonium ion, presumably on the conformer 82-2HO, with C-C bond formation and is irreversible thereby defining the kinetic mode of attack. This Michael addition is followed by intramolecular protonation of the resultant diazoalkane and substitution by the phenolic hydroxyl group to give the tricyclic product 48 (Scheme 14). With phenol on the other hand initial Michael attack on the β-face of the 82-OH2, half-chair is considered to be reversible, resulting in eventual thermodynamic attack on the α-face resulting in diazoalkane 85 (Scheme 15). This diazoalkane is protonated on the α-face, consistent with the isotopic labelling studies conducted with deuterioacetic acid (Scheme 12) giving a contact ion pair 86, primed to undergo collapse with inversion of configuration at the 5- position leading to the observed product 47 (Scheme 10).23
Scheme 14.
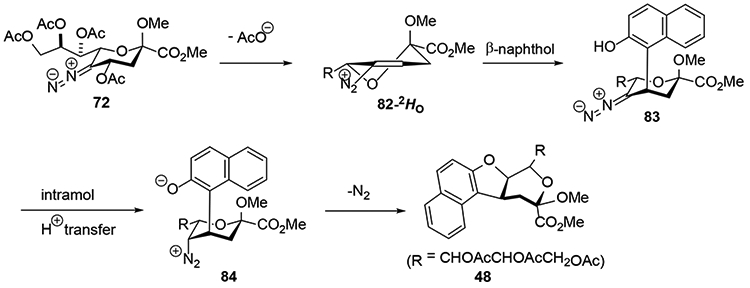
Mechanism of Substitution with β-naphthol.
Scheme 15.
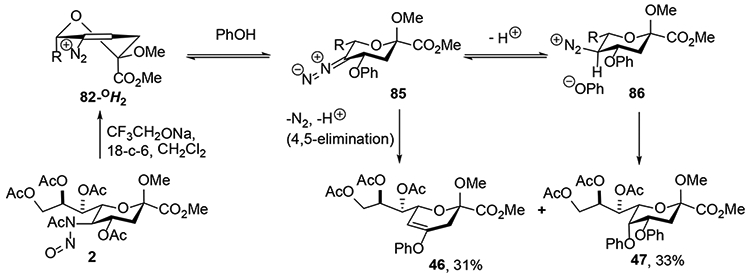
Mechanism of Substitution with Phenol
The essential difference between the classical reaction manifold observed with acetic acid, thioacetic acid, HF-pyridine, alcohols in the presence of fluoroboric acid,11 thiophenols, N-hydroxybenzotriazole, and N-hydroxyphthalimide, and the new reaction manifold leading to substitution of the acetamido group and the acetoxy group at the 4-position is considered to be the pKa of the nucleophile. Thus, with nucleophiles of pKa <~8 the classical reaction manifold is followed, whereas with nucleophiles of pKa >~8 the diazonium ion - diazoalkane equilibrium is considered to favor the latter resulting in the elimination of the 4-acetoxy group and formation of the vinyl diazonium ion 88 (Scheme 16).23, 24 This hypothesis is consistent with the literature pKa of the methyldiazonium ion (~10) and the acidification caused by the adjacent electron-withdrawing C-O bonds.23
Scheme 16.
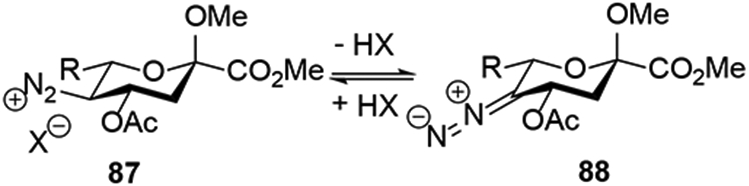
Key equilibrium.
It will be noted that following protonation of the diazoalkane 89 and formation of a contact ion pair encompassing the diazonium ion 90 and the conjugate base of the nucleophile, the stereochemical outcome of the reaction is again nucleophile dependent. With carboxylic acids, HF, thiophenol, N-hydroxybenzotriazole, and N-hydroxyphthalimide, the stability of the counter- ion in the CIP is such that immediate collapse with loss of nitrogen and formation of an axial C- Nu bond is not typically observed, rather intervention by the ring oxygen and the oxonium 91 takes place ultimately giving the equatorial product (Scheme 17). The exception to this rule is the formation of the axial acetate 67 on decomposition of nitrosoamide 66 in the crystal, when diffusional escape of the counterion from the CIP is prevented by the crystal lattice. On the other hand, with less stabilized counterions such as phenate, diffusional equilibration of the CIP is retarded to the extent that recombination within the CIP is the favored pathway (Scheme 17).
Scheme 17.
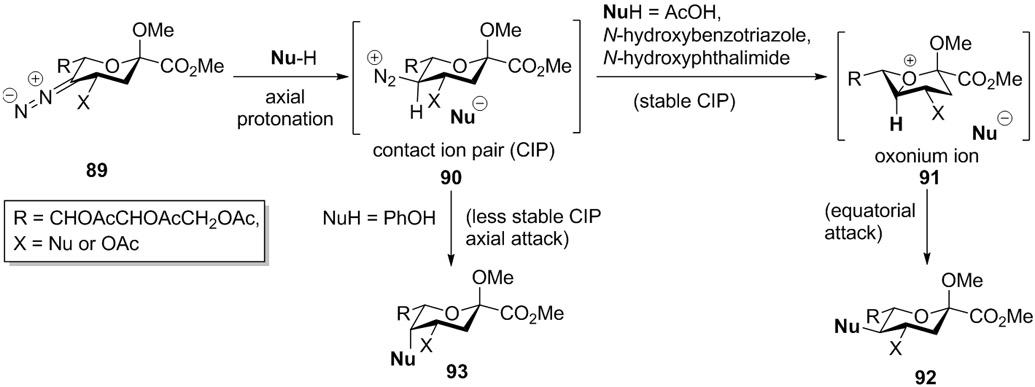
Stability of contact ion pair (CIP) and preferential attack of the nucleophile.
2.7.5. Participation from an Anomeric Thioether Group
The curious case of C4-acetoxy displacement alongside substitution at the C5-position, observed in the course of the labeling experiment with 13C acetic acid as nucleophile on β-thioadamantyl neuraminic acid derivative 21 (Scheme 12), and confirmed by the use of levulinic acid as nucleophile, is proposed to proceed via participation of anomeric thiol group (Scheme 18).25 Formation of the diazo intermediate 94 is followed with an SN2 displacement of the C4- acetoxy group making a bicyclic positively charged species 95, which is then attacked by the labeled acetic acid in SN2 fashion to generate intermediate 96, with overall retention of configuration. The intermediate 96 then follows usual C5-elimination-substitution mechanism pathway to afford the di-substituted derivative 13C2-16.
Scheme 18.
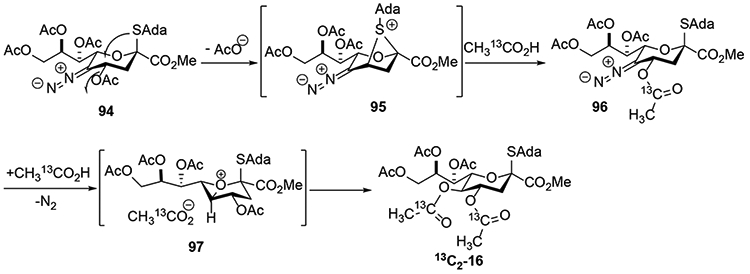
Acetoxy Substitution at C4-Position via Participation of the Anomeric Thioglycoside.
3. Related Deamination Reactions of Other Aminosugars
The oxidative deamination reaction is not limited to Neu5Ac and its variants. Indeed, even before the application of the process to Neu5Ac by the Ogura and Zbiral and groups, deamination of 2-amino-2-deoxy and 4-amino-4-deoxy sugars had been studied by various laboratories, as covered in Williams’ review.5 This early work is nevertheless commented here because it provides additional support for the mechanism proposed in Scheme 13 and especially for the proposed oxonium ion intermediate. Thus, the earlier workers proposed related bicyclic oxonium ions in substitution reactions at the 4-position of pyranoside rings and in the deamination of 2-aminoglucopyranose derivatives (Scheme 19).26-28 For example, the deamination of methyl 4-glucosamine 98 with nitrous acid gave methyl α-glucopyranoside as the major product with retention of configuration, along with β-L-altrofuranoside 101, 4,5-anhydro-D-galactose 102, and D-glucofuranose 104 as minor products,26 leading to the postulation of oxabicyclo[3.1.0]hexyl oxonium ion 99 as intermediate. According to this reasonable hypothesis equatorial attack of water on oxonium ion 99 affords the major compound 100, whereas the furanoside 101 arises from attack of water at the 5-position. To explain the formation of D-glucofuranose 104 it was suggested that the 99 undergoes opening of the furanose ring leading to the of 4,5-anhydrogalactose 102. Attack of aldehyde 102 or its hydrate 103 then provides a mechanism for the formation of 104. In another example, displacement of the 4-O-sulfonyl group from the methyl rhamnoside 105 by azide ion at elevated temperature led to the formation of a ring-contracted 5-azido-5,6-dideoxy- -talofuranose derivative 107 supposedly through formation of oxabicyclo[3.1.0]hexyl oxonium ion 106 as intermediate.27 In yet another example, treatment of N-acetyl-N-nitroso-α/β-D-glucosamine tetraacetate 108 with 50% aqueous acetone, resulted in contraction of the pyranosyl ring and formation of triacetyl-2,5-anhydro-D-manno derivative 110, which was isolated in the form of the various acetals 111.28 Attack from the ring oxygen on the incipient carbenium at C2 was invoked with formation of a bicyclic oxonium ion 109. Opening of this bicyclic ion by a lone pair of electrons on the glycosidic oxygen completes the ring contraction and leads to the isolation of 111. Interestingly, the ring contraction reaction is condition dependent as when the α-anomer of N-acetyl-N-nitroso-D-glucosamine tetraacetate 108 was allowed to stand in chloroform β-D-glucosyl pentaacetate 113 was formed.28 As the analogous process did not occur with the β-anomer it was suggested that the oxabicyclo[3.1.0]hexyl oxonium ion 109 was opened by participation from the anomeric acetate generating 112, which is then attacked by the acetate at C-1 position from the β-face resulting in 113 with migration of the anomeric acetate. When the deamination reaction is applied to a 1,5-anhydroglucitol 114, lacking the glycosidic bond, no ring contraction is observed and the major product arises from substitution ; 115 forms with retention of configuration.
Scheme 19.
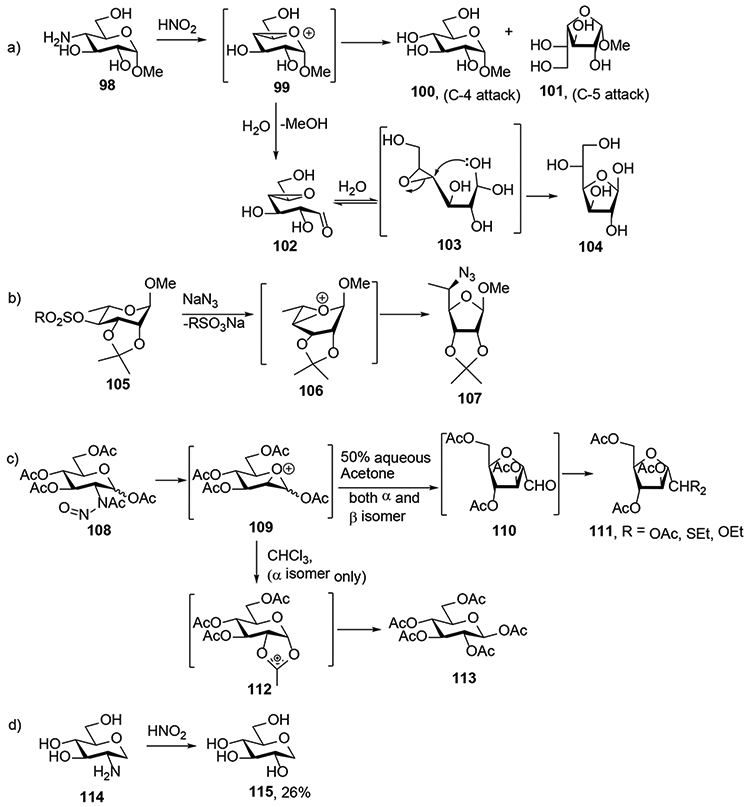
Literature Reports Invoking Bicyclic Oxonium Ion Intermediate.
4. Application to Complex Substrates
Finally, we highlight the fact that, while the oxidative deamination reaction has mostly been applied to simple alkyl and acetoxy glycosides, it is also compatible with far more complex substrates as long as the reaction medium is adequately buffered.11 Thus, it was demonstrated by Navuluri and Crich that the deamination could be applied to complex di-, tri-, and tetrasaccharides derived from Neu5Ac, provided care was taken to use the nitronium tetrafluoroborate salt as nitrosylating agent in the presence of pyridine for the first step of the protocol. Multiple different nucleophiles were applied in what is an effective method for the conversion of Neu5Ac-based oligosaccharides to the corresponding KDN-based oligosaccahrides (Scheme 20).11 Of particular note was the application of the process to the double deamination of a 1,9-linked disialoside 126, which afforded a moderate 42% yield of 127, provided that a mixture of acetic acid and tetrabutylammonium acetate was used in the final reaction step (Scheme 21).11
Scheme 20.
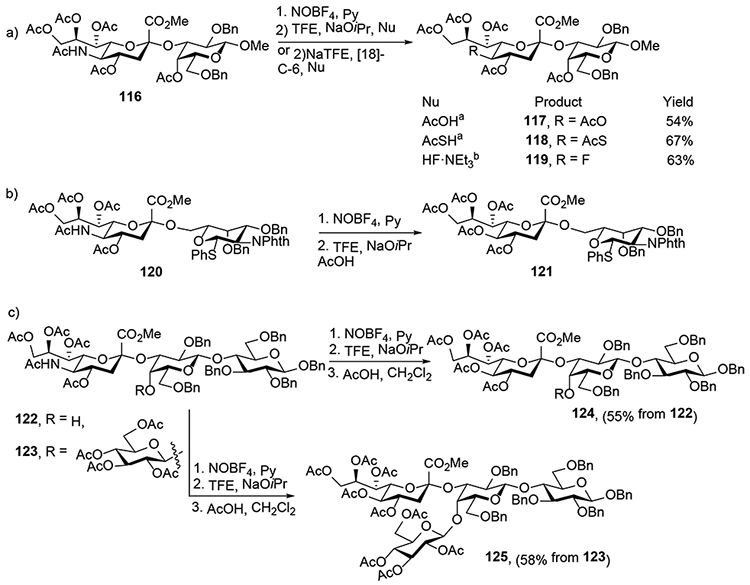
Late Stage Oxidative Deamination on Di-, Tri-, and Tetrasaccharides.
Scheme 21.
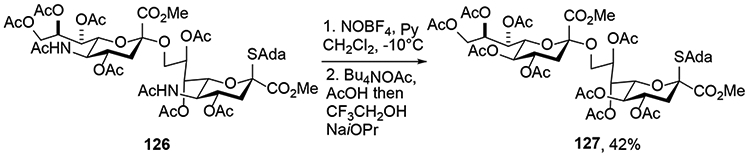
Double Deamination of a 1,9-linked disialoside.
Although, the deamination reaction has been popular in the synthesis of sialic acid derivatives, it has been seldom used to carry out the oxidative deamination of other molecules beyond the simple 2- and 4-glucosamine derivatives discussed above. Thus, the recent application of the reaction to the 4’’-amine of the structurally unusual aminoglycoside antibiotic apramycin 128 (Scheme 22) is noteworthy.29 This reaction, which was conducted under the Zbiral conditions and which took place with retention of configuration consistent with the example of 4-aminoglucose (Scheme 19), afforded saccharocin 132, a biosynthetic precursor of apramycin itself.
Scheme 22.
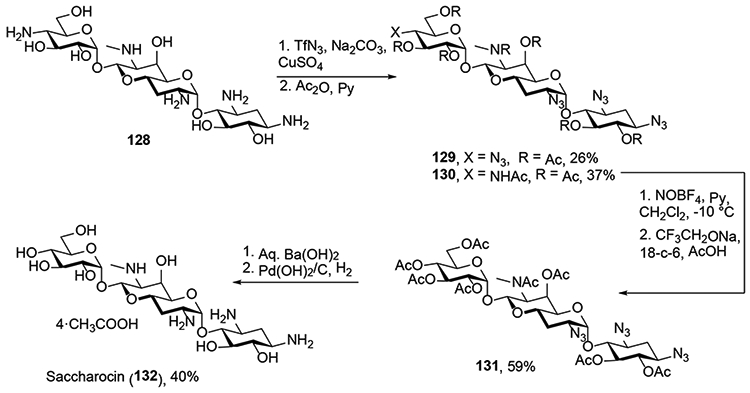
Oxidative Deamination of an Apramycin Derivative and Synthesis of Saccharocin.
5. Conclusion
The deamination reaction has come a long way since its inception. It has played a crucial role in the synthesis of neuraminic acid variants, particularly KDN and pseudaminic acid. The range of compatible nucleophiles has been considerably extended beyond the initial acetic acid and hydrazoic acid to enable the replacement of the C5-NHAc by a range of C5-X bonds, all with retention of configuration. Furthermore recent work has uncovered a new reaction manifold that permits concomitant replacement of the C4-O bond by a range of C-X bonds, including C-C bonds in some instances, thereby opening up new vistas for the production of unusual sialic acid derivatives. The development of modified conditions for the generation of the necessary intermediate N-nitrosoacetamide, involving simple buffering of the nitrosylation step with pyridine, have opened the possibility of conducting the process in the presence of thioglycosides and on complex, sensitive oligosaccharides. These methodological advances have been paralleled by advances in the understanding of the reaction mechanism including the manner in which protecting groups influence the stereochemical outcome of the reaction, and that in which the pKa of the added nucleophile affects the overall reaction outcome, including the involvement of the 4-position and the stereochemical outcome at the 5-position. These multiple advances portend a bright future for the oxidative deamination of Neu5Ac and its relatives, limited only by the imagination of organic and carbohydrate chemists.
Acknowledgement.
We thank the NIH (GM62160) for support of our work in this area.
References
- 1.White EH, J. Am. Chem. Soc, 1955, 77, 6008. [Google Scholar]
- 2.White EH, J. Am. Chem. Soc, 1955, 77, 6011. [Google Scholar]
- 3.White EH, J. Am. Chem. Soc, 1955, 77, 6014. [Google Scholar]
- 4.White EH, J. Am. Chem. Soc, 1961, 83, 1179. [Google Scholar]
- 5.Williams JM, Adv. Carbohydr. Chem. Biochem 1975, 31, 9. [Google Scholar]
- 6.Shirai R, Nakamura M, Hara S, Takayanagi H and Ogura H, Tetrahedron Lett. 1988, 29, 4449. [Google Scholar]
- 7.Nadano D, Iwasaki M, Endo S, Kitajima K, Inoue S, and Inoue Y, J. Biol. Chem, 1986, 261, 11550–11557. [PubMed] [Google Scholar]
- 8.Schreiner E and Zbiral EA, Liebigs Ann. Chem, 1990, 581. [Google Scholar]
- 9.Pozsgay V and Jennings HJ, J. Org. Chem 1987, 52, 4635. [Google Scholar]
- 10.Crich D and Navuluri C, Angew. Chem. Int. Ed, 2010, 49, 3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navuluri C and Crich D, Angew. Chem. Int. Ed, 2013, 52, 11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Varki A, Glycobiology, 1992, 2, 25; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Angata T and Varki A, Chem. Rev, 2002, 102, 439; [DOI] [PubMed] [Google Scholar]; c) Chen X and Varki A, ACS Chem. Biol, 2010, 5, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Knirel YA, Shashkov AS, Tsvetkov YE, Jansson P-E and Zaehringer U, Adv. Carbohydr. Chem. Biochem, 2003, 58, 371; [DOI] [PubMed] [Google Scholar]; b) Knirel YA, Shevelev SD and Perepelov AV, Mendeleev Commun., 2011, 21, 173; [Google Scholar]; c) Zunk M and Kiefel MJ, RSC Adv.2014, 4, 3413. [Google Scholar]
- 14.a) Thibault P, Logan SM, Kelly JF, Brisson J-R, Ewing CP, Trust TJ and Guerry P, J. Biol. Chem, 2001, 276, 34862; [DOI] [PubMed] [Google Scholar]; b) Logan SM, Hui JPM, Vinogradov E, Aubry AJ, Melanson JE, Kelly JF, Notha H and Soo EC, FEBS J., 2009, 276, 1014; [DOI] [PubMed] [Google Scholar]
- 15.c) Schirm M, Schoenhofen IC, Logan SM, Waldron KC and Thibault P, Anal. Chem, 2005, 77, 7774; [DOI] [PubMed] [Google Scholar]; b) Schirm M, Soo EC, Aubry AJ, Austin J, Thibault P and Logan SM, Mol. Microbiol, 2003, 48, 1579. [DOI] [PubMed] [Google Scholar]
- 16.a) Knirel YA, Vinogradov EV, L’vov VL, Kocharova NA, Shashkov AS, Dmitriev BA and Kochetkov NK, Carbohydr. Res, 1984, 133, C5; [DOI] [PubMed] [Google Scholar]; b) Knirel YA, Kocharova NA, Shashkov AS, Dmitriev BA, Kochetkov NK, Stanislavsky ES and Mashilova GM, Eur. J. Biochem, 1987, 163, 639; [DOI] [PubMed] [Google Scholar]
- 17.a) Knirel YA, Vinogradov EV, Shashkov AS, Kochetkov NK, L’vov VL and Dmitriev BA, Carbohydr. Res, 1985, 141, C1; [DOI] [PubMed] [Google Scholar]; b) L’vov VL, Shashkov AS and Dmitriev BA Bioorg. Khim,1987, 13, 223; [PubMed] [Google Scholar]
- 18.a) Kenyon JJ, Notaro A, Hsu LY, De Castro C and Hall RM, Sci Rep., 2017, 7, 11357; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kenyon JJ, Marzaioli AM, De Castro C and Hall RM, Glycobiology, 2015, 25, 644; [DOI] [PubMed] [Google Scholar]; c) Tsvetkov YE, Shashkov AS, Knirel YA and Zähringer U, Carbohydr. Res, 2001, 331, 233. [DOI] [PubMed] [Google Scholar]
- 19.Zunk M, Williams JT, Carter J and Kiefel MJ, Org. Biomol. Chem, 2014, 12, 2918. [DOI] [PubMed] [Google Scholar]
- 20.Williams JT, Corcilius L, Kiefel MJ and Payne RJ, J. Org. Chem, 2016, 81, 2607. [DOI] [PubMed] [Google Scholar]
- 21.a) Dhakal B, Buda S, and Crich D, J. Org. Chem 2016, 81, 10617; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Popik O, Dhakal B and Crich D, J. Org. Chem, 2017, 82, 6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhakal B and Crich D, J. Am. Chem. Soc, 2018, 140, 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawsawi M, Wickramasinghe A and Crich D, J. Org. Chem 2019, 84, 14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hawsawi M, Pirrone MG, Wickramasinghe A and Crich D, Carbohydr. Res 2020, 490, 107921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buda S and Crich D, J. Am. Chem. Soc 2016, 138, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng Ying Kin NMK; Williams JM; Horsington AJ Chem. Soc. C, 1971, 1578. [Google Scholar]
- 27.a) Stevens CL, Glinski RP, Taylor KG, Blumbergs P and Sirokman F, J. Am. Chem. Soc 1966, 88, 2073; [Google Scholar]; b) Hanessian, S. Chem. Commun, 1966, 796; [Google Scholar]; c) Stevens CL, Glinski RP, Taylor KG, and Sirokman F J. Org. Chem, 1970, 35, 592. [Google Scholar]
- 28.Llewellyn JW, and Williams JM, Carbohydr. Res, 1973, 28, 339. [Google Scholar]
- 29.Sarpe VA, Pirrone MG, Haldimann K, Hobbie SN, Vasella A and Crich D, Med. Chem. Commun 2019, 10, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]