

Abstract
Objective:
Thalamic atrophy is among the earliest brain changes detected in patients with multiple sclerosis (MS) and the degree of thalamic atrophy is a strong predictor of disability progression. The causes of thalamic atrophy are not fully understood. Here, we investigate the contributions of thalamic demyelinated lesions, thalamic neuronal loss, and cerebral white matter (WM) lesions to thalamic volume.
Methods:
We used postmortem in situ magnetic resonance imaging (MRI) scans of 95 subjects with MS to correlate thalamic lesion volumes with global MRI metrics. We histologically characterized thalamic demyelination patterns and compared neuronal loss and neuritic pathology in the thalami with the extremes of volume.
Results:
Grossly apparent thalamic discolorations in cm-thick brain slices were T2/fluid-attenuated inversion recovery (FLAIR) hyperintense, T1-hypointense, and appeared as perivascular demyelinated lesions with dystrophic neurons/axons. Subependymal demyelinated lesions with axonal loss and microglial/macrophage activation were also observed. The 12 subjects with the least thalamic volume had a 17.6% reduction of median neuronal density in the dorsomedial/ventrolateral and pulvinar nuclei compared with the 14 subjects with the greatest thalamic volume (p = 0.03). After correcting for age, disease duration, sex, and T2 lesion volume, the total (p = 0.20), ovoid (p = 0.31), or subependymal (p = 0.44) MRI thalamic lesion volumes correlated with thalamic volume. Thalamic volume correlated with cerebral T2 lesion volume (Spearman’s rho = −0.65, p < 0.001; p < 0.0001 after correcting for age, disease duration, and sex).
Interpretation:
Our findings suggest the degeneration of efferent/afferent thalamic projections and/or a neurodegenerative process as greater contributors to thalamic atrophy than thalamic demyelinating lesions.
Multiple sclerosis (MS) is an important cause of non-traumatic disability in young adults and affects 1 million individuals in the United States.1 MS is characterized by focal demyelinating lesions, axonal pathology, and brain atrophy representing features of inflammation and neurodegeneration. Grey matter (GM) atrophy, correlates better with physical disability, cognitive impairment, and fatigue than white matter (WM) lesion burden.2,3
Preferential thalamic volume loss relative to cortical and other deep GM structures is often a feature of individuals with clinically isolated syndromes and radiologically isolated syndromes and is associated with increased risk for developing MS.4–9 Thalamic volume is a candidate marker for neurodegeneration as lower values confer a greater risk of physical disability worsening independent of the disease course (clinically isolated syndrome; relapsing–remitting [RR], secondary progressive [SP], or primary progressive [PP] MS).10 Isolated thalamic atrophy also predicts the likelihood of achieving “no evidence of disease activity” at 2 years.11
Thalamic atrophy has received increasing attention as an outcome measure in clinical trials given its association with cognitive impairment, physical disability, and fatigue.12–14 Thalamic volume loss has been examined in post hoc analyses of disease modifying therapies (DMTs; fingolimod and natalizumab) and was incorporated as a secondary outcome prospectively (eg, phase III trials of ozanimod).15–17 Patients on highly efficacious DMTs (eg, monoclonal antibodies) also have lower rates of thalamic atrophy compared to patients on lower efficacy DMTs (eg, interferon-beta and glatiramer acetate).15
The cause of thalamic volume loss is not completely understood. Potential mechanisms of thalamic volume loss include intrinsic injury via (1) inflammatory-mediated demyelination in the thalamus, (2) primary neurodegeneration of axons and neurons, or (3) antero−/retrograde axonal degeneration caused by demyelination or transection of thalamic projection fibers. Using magnetic resonance imaging (MRI), thalamic volume loss has been speculated to be due to either focal thalamic demyelinating lesions or global WM lesion burden.18,19
Prior pathological studies have described thalamic lesions adjacent to the ependymal surface/periventricular surface in the medial and anterior thalamic nuclei, with decreased neuronal density, reduced neuronal size, and abnormal neuronal shape, without apoptosis of neurons.20–22 Several studies using ultra-high field MRI find that a majority of patients with MS have thalamic lesions.18,23
Using a cohort of 95 subjects with MS with postmortem in situ MRIs, we sought to investigate the relationship among thalamic lesion volume, global cerebral WM lesion volume, and thalamic volume. We hypothesized that thalamic volume could correlate with thalamic lesions, loss of neurons independent of demyelination, or injury to afferent/efferent tracts (eg, cerebral WM T2 lesion burden). We characterized the extent of thalamic demyelination and microglial/macrophage activation using immunocytochemistry using fixed tissue from the cohort. We also determined the extent of neuronal injury and loss in thalamic lesions as well as in subjects with the extremes of thalamic volume.
Methods
Postmortem Program
Samples for the study were obtained from the Cleveland Clinic MS Brain Donation Program, as previously described.24 The program entails a postmortem in situ MRI within 6 hours of death followed by brain and spinal cord removal and fixation. Inclusion criteria include: diagnosis of MS, death within 75 miles of the center, and feasibility to complete MRI within 6 hours of death. Cases with prolonged hypoxia or >72 hours of mechanical ventilation, recent central nervous system (CNS) infection, human immunodeficiency virus, or hepatitis B/C were excluded. The brain donation program is approved by the Cleveland Clinic Institutional Review Board and samples were obtained either after obtaining informed consent from patients before their death or by their next of kin following death (explained in detail in prior publications).2,24–26 Time from death to start of postmortem in situ MRI was available for 70 of 95 cases and to the start of brain fixation for 95 of 95 cases (Table 1).
TABLE 1.
Postmortem Demographics (n = 95)
| Female | 61 (64%) | |
| Race | White | 83 (87%) |
| African American | 11 (12%) | |
| Hispanic | 1 (1%) | |
| Course | SP | 74 (72%) |
| PP | 14 (15%) | |
| RR | 7 (7%) | |
| Age at death, mean yr (SD) | 58.5 (±12.1) | |
| Disease duration, median yr (IQR) | 25.0 (15.0–34.0) | |
| Time from death to MRI, median h (IQR) | 4.4 (3.1–6.0) | |
| Time from death to brain fixation, median h (IQR) | 7.0 (5.7–8.9) | |
IQR = interquartile range; MRI = magnetic resonance imaging; PP = primary progressive; RR = relapsing remitting; SP = secondary progressive.
Tissue Processing and Immunohistochemistry
Brain and spinal cord were removed as previously described.24 One cerebral hemisphere was fixed in 4% paraformaldehyde (PFA). Alternate slices of the opposite hemisphere were fixed in 4% PFA or flash frozen. Gross examination of all brain slices was conducted looking for thalamic lesions. The thalamic portion of thalamocapsular lesions were also counted as thalamic lesions. Slices containing grossly apparent discolorations were identified at the level of the dorsomedial/ventrolateral or pulvinar nuclei given their relatively large size of the nuclei and defined borders to ensure location within the thalamus. The tissue blocks were removed and sectioned at a thickness of 30 μm using a sliding microtome.
Methods for staining these free floating sections were followed as previously described.27 Sections were incubated with primary antibodies for 2 to 3 days at 4°C. Primary antibodies included: proteolipid protein (PLP; AA3 rat clone, a gift from Wendy Macklin, University of Denver, CO, 1:250 dilution) for myelin, HuR (Santa Cruz Biotechnology, Dallas, TX, mouse IgG, SC-5261, 3A2 clone, 1:500 dilution) for neurons, major histocompatibility complex class II (MHCII, HLA-DR CR3/43, Dako, Santa Clara, CA, mouse monoclonal, M0775, 1:500 dilution) for activated microglia, and SMI31 (mouse IgG1k, Biolegend, San Diego, CA, 801601, 1:2500 dilution) and SMI32 (mouse monoclonal, Biolegend, 801701, 1:2500 dilution) for axons and dendrites. Sections were incubated in secondary antibodies (1:1000 biotinylated goat anti-rat and mouse IgG heavy/light chain Vector BA-9401 and BA-9200 [Burlingame, CA], respectively) for 1 hour at room temperature using a horseradish peroxidase – 3,3′-diaminobenzidine (DAB) immunohistochemistry protocol. Confocal microscopy was performed on a Zeiss LSM 800 with a 40× objective and numerical aperture of 1.4 and secondary antibodies for immunofluorescence included Alexa-Fluor goat anti-mouse 488 A11029 and goat anti-rat 594 A11007 (Life Technologies Corporation, Eugene, OR).
To characterize neuronal density (HuR) in thalamic demyelinated lesions and surrounding nondemyelinated normal appearing grey matter (NAGM), fixed blocks were selected by identifying grossly apparent thalamic lesions. Subjects with available and intact thalamic tissue at the level of the dorsomedial/ventrolateral or pulvinar nuclear groups with sizeable lesions (to permit 5 regions of interest [ROIs] at 20× magnification per section) were selected. Sixteen tissue blocks from 15 subjects were used to quantify neuronal density in demyelinated perivascular regions and adjacent NAGM. Fourteen blocks from 13 subjects in subependymal regions and adjacent NAGM were stained to quantify differences in neuronal populations but not included in the analysis as neuronal populations were not consistently found in subependymal regions. Five ROIs in demyelinated or NAGM regions were imaged at 20× magnification per 30 μm free floating section obtained from tissue blocks. Neurons were automatically thresholded (size >200 μm2) and counted using an in-house script using National Institutes of Health (NIH) Fiji (https://fiji.sc)28; neuronal density was reported as the number of neurons counted per area measured (mm2).
Axonal density (SMI31 + SMI32) was quantified at 40× magnification with 5 ROIs using 30 μm free floating sections along 23 subependymal demyelinated lesions and adjacent normal appearing subependymal GM. Images were processed to remove the layer of subependymal cells and automatically thresholded using an in-house Fiji script to report percent area occupied by the DAB stain.
Neuronal density and percent area occupied by activated microglia/macrophages (major histocompatibility complex [MHC] class II) were performed in thalamic NAGM regions (dorsomedial/ventrolateral and pulvinar nuclei) in 5 ROIs in cases with the least and most thalamic volume.
MRI Acquisition
Postmortem in situ imaging was obtained on 1.5 (n = 31) and 3 Tesla (n = 65) MRI with the following sequences: T1-weighted 3D Magnetization Prepared Rapid Acquisition of Gradient Echo (MPRAGE), T2-weighted image, fluid-attenuated inversion recovery (FLAIR), and paired proton density-weighted images with and without magnetization transfer pulse.24 After MRI, subjects were transported to the autopsy suite for brain and spinal cord removal and processing.
MRI Analysis
T1 and T2-lesion volumes were calculated using a semiautomated technique, as previously described.29 Volumetric measures were done after lesion filling. Thalamic volume was calculated using Advanced Normalization Tools (ANTs; www.picsl.upenn.edu/software/ants/) with postprocessing on T1 MPRAGE and normalized to intracranial volume. Thalamic lesion volumes were manually segmented in 93 of the 95 cases (2 cases omitted due to artifact and poor resolution of the thalamus) using a multimodal approach: hyperintense on T2/FLAIR or hypointense on T1 MPRAGE with the later having greater resolution. We classified thalamic lesions as subependymal or ovoid. “Periventricular” lesions, which are perivascular by histology or described as having a central vein sign radiographically, were classified as ovoid in this study, distinct from subependymal lesions. The thalamic portion of thalamocapsular lesions were also classified as “ovoid.” Brain parenchymal (75/95 subjects), GM, and WM fractions (72/95 subjects) were obtained using Statistical Parametric Mapping (SPM; www.fil.ion.ucl.ac.uk/spm). Cortical thickness (temporal, frontal, global, parietal, and occipital) was obtained using cortical longitudinal atrophy detection algorithm (CLADA30) in 44 of 95 subjects and a custom in-house method using ANTs was used for upper cervical cord cross-sectional area (27/95 subjects); not all subjects were analyzed due to image quality (eg, temperature effects) or coverage (upper cervical cord).
Statistical Methods
Statistical analysis was performed using R Statistical Software (RStudio version 1.2.5003/R version 3.6.3).31 Histopathologic comparisons between thalamic demyelinated (perivascular and subependymal) and NAGM (adjacent to perivascular and subependymal areas) ROIs for neuronal density were performed using analysis of variance (ANOVA) with Tukey’s honestly significant difference for multiple comparisons of means (95% family-wise confidence interval [CI]). Axonal density was compared between demyelinated and normal appearing subependymal regions using a paired t-test at a 95% CI. Linear models were designed to test the contribution of different MRI measures to thalamic volume while correcting for sex and age with statistical significance set at p = 0.05. Subjects with the least (15th percentile) and most (85th percentile) thalamic volume were identified to compare differences in the number of grossly apparent lesions, and differences in neuronal density (size >200 μm2; Wilcoxon rank sum test with continuity correction) and microglial/macrophage activation (Student’s t-test) in the NAGM.
Results
Postmortem Cohort
Characteristics of the postmortem cohort (n = 95) are provided in Table 1. Subjects were predominantly white (n = 83; 87%), women (n = 61; 64%) with mean age of 58.5 years (SD 12.1; range = 27–97 years). Most subjects were diagnosed with secondary progressive MS (n = 74; 72%) with a long disease duration (median = 25.0 years; interquartile range [IQR] = 15.0–34.0 years). Subjects received their postmortem in situ MRI at median of 4.4 hours after death and brain fixation was started at a median of 7.0 hours after death. Figure 1 outlines a flowchart of the number of subjects used for the studies below.
FIGURE 1:

Flowchart of the number of subjects and tissue specimens used for the study. Subsets of tissue sections were used to stain for demyelinating lesions and characterize lesions based on microglial/macrophage activity and neuronal/axonal density. Subjects were also selected for extremes of thalamic volume identified on magnetic resonance imaging (MRI; 15th and 85th percentiles).
Gross Features of Thalamic Lesions
We first examined cm-thick brain slices for thalamic lesions. Sixty-five of 95 postmortem brains (68%) had discoloration of the thalamus (Fig 2A). Of 107 grossly apparent lesions >0.5 cm (maximum diameter), 99 (93%) were confirmed on MRI (hyperintense on T2/FLAIR or hypointense on T1 MPRAGE). Lesions <0.5 cm were less frequently identified by MRI (17/46; 37%). Forty-nine grossly apparent lesions from 43 subjects were removed and sectioned. All 49 macroscopically apparent thalamic discolorations in cm-thick brain slices were perivascular demyelinated lesions (Figs 2B and 3). We separated the cohort into subjects with the most (85th percentile, n = 14) and least MRI thalamic volume (15th percentile, n = 12), which represented a 1.7-fold difference in volume. There was no difference between the thalami with the most and least volume in the number of grossly apparent lesions that were 0.5 cm or larger (20 vs 15 lesions, respectively; p = 0.49) or smaller than 0.5 cm (7 vs 3 lesions, respectively; n = 0.47).
FIGURE 2:

Representative case of a subject with grossly apparent hyperpigmented thalamic lesions on a 1 cm fixed coronal slice (A); enlarged lesion shown below. Both ovoid (B) and subependymal (C) lesions are noted using PLP immunohistochemistry for myelin; asterisk (*) denotes location of vessel. Magnetic resonance imaging (MRI) coronal images, with thalamus depicted with a white outline, (D) depict fluid-attenuated inversion recovery (FLAIR) and T1-Magnetization Prepared Rapid Acquisition of Gradient Echo (MPRAGE) sequences as well as ovoid (blue) and subependymal (Subep.; green) manually segmented lesions. The heart-shaped hyperpigmented lesion straddling the dorsomedial and ventrolateral nuclei is indicated with an arrow and an enlarged inset of the lesion is below (A). The lesion is demyelinated (B) and labeled as ovoid/perivascular (“Ovoid”) on MRI T1 MPRAGE (D). Subependymal lesions (C) were not identified on gross examination (A) and subtle on MRI (D; labeled “Subep.”). PV, perivascular; PLP, proteolipid protein.
FIGURE 3:
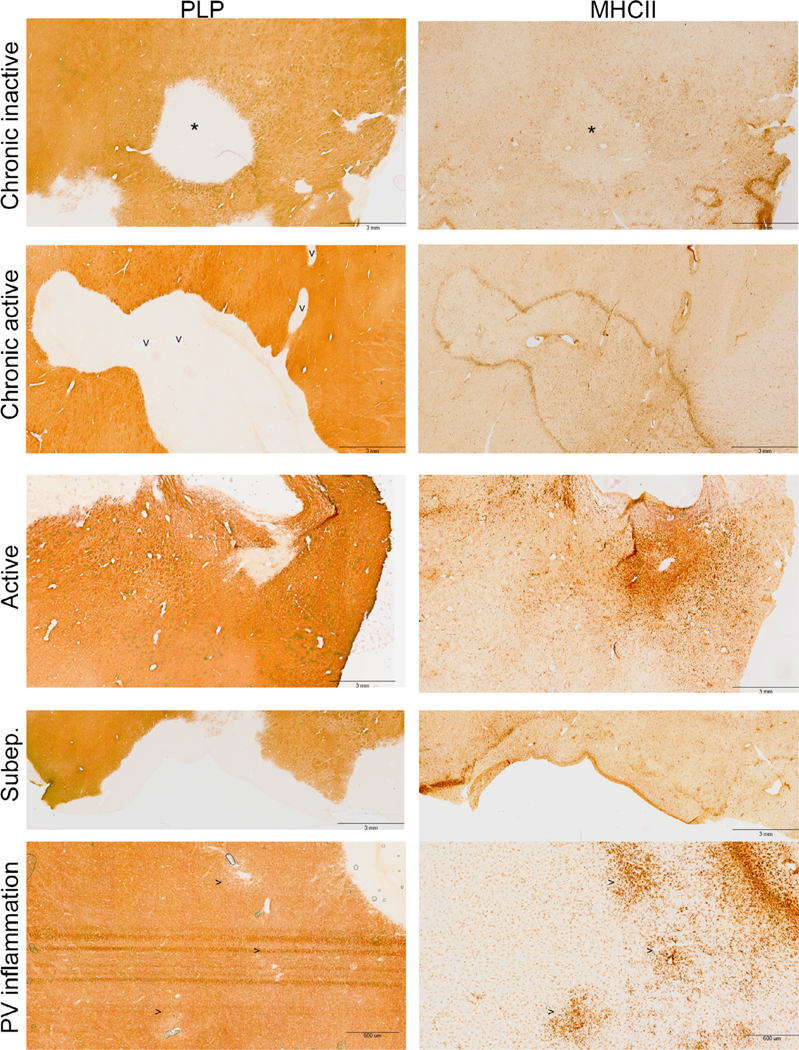
Patterns of thalamic lesions characterized by immunohistochemistochemical staining for myelin (PLP) and activated microglia/macrophages (MHCII). Lesions top to bottom: chronic inactive (hypocellular center with asterisk*), chronic active (rim of hypercellularity; vessels labeled with “V”), active (hypercellular), and subependymal (Subep.) demyelination, areas of perivascular inflammation without demyelination (vessels labeled “>”). Scale bars are 3 mm for all images except for “PV inflammation” (600 μm). MHC, major histocompatibility complex; NAGM, normal appearing grey matter; PLP, proteolipid protein; PV, perivascular.
Histological Features of Thalamic Lesions
Two patterns of demyelination were identified in immunostained sections: perivascular areas of demyelination (ovoid/perivascular) and thin band of myelin loss lining the ventricular surface (subependymal; see Figs 2 and 3). All of the grossly apparent hyperpigmented lesions in cm-thick slices were ovoid-shaped; subependymal lesions were not grossly visible but were observed in sections stained for myelin proteins. Subependymal lesions only spanned a few millimeters from the ventricular surface, whereas all ovoid lesions were larger and had a central vessel.
Sixty-five demyelinated perivascular lesions from 14 subjects were further characterized by microglial/macrophage activation patterns by MHC class II (MHCII; representative images in Fig 3). Five (7.7%) were active lesions with abundant MHCII-positive cells throughout the demyelinated area, 18 (27.7%) were chronic active lesions with a rim of MHCII-positive cells at the lesion border, 24 (36.9%) were chronic inactive lesions with little MHCII accumulation within or at the border of the demyelinated area, and 18 (27.7%) were small foci of perivascular microglial activation without demyelination. Subependymal regions with demyelination were identified in 23 subjects to quantify axonal changes (below). All of the 23 subependymal lesions were observed to have increased MHCII-positive cells along the ventricular surface; 4 of 23 had a clearly demarcated rim of MHCII-positive cells at the NAGM border of the lesion.
Morphologically, neurons seemed dystrophic in lesions compared to a more typical polygonal appearance in normal appearing tissue. This finding was more marked in subependymal regions compared to perivascular areas (Fig 4). Neuronal density was similar between demyelinated perivascular lesions compared to adjacent NAGM (46.2 vs 46.0 neurons/mm2; p = 0.95). Neuronal density in perivascular demyelinated lesions and adjacent NAGM were not different either at the level of the dorsomedial/ventrolateral nuclei (95% CI = −17.2 to 2.0) or the pulvinar nucleus (95% CI = −18.0 to 23.0). Comparison of neuronal density along subependymal demyelinated versus myelinated areas was unreliable as thalamic neurons did not consistently border the ependymal surface.
FIGURE 4:

Representative images of neuronal morphology (HuR) in perivascular (top left) and subependymal (bottom left) areas of demyelination. Neurons appeared more dystrophic in areas of subependymal demyelination (labeled “^”) compared to perivascular. Adjacent areas of NAGM to perivascular and subependymal regions are shown on panels to the right. Scale bar represents 100 μm. Inset in top right of images show magnified representative neuronal cell bodies. NAGM, normal appearing grey matter.
Decreased axonal/dendritic density (SMI31/32) and dystrophy was more noticeable along subependymal lesions (Fig 5 above). The longitudinal orientation of axons in subependymal regions permitted quantification of axonal density in all 23 lesions; axonal density was 49.2% lower (95% CI = 45.3–53.0; p < 0.0001) in demyelinated versus normal appearing subependymal GM. Axons were relatively preserved in perivascular lesions and the multiple orientations of WM tracts prevented reliable quantification; however, some axons and dendrites appeared swollen (Fig 5 below).
FIGURE 5:

Representative images of axonal/dendritic morphology (SMI31 and SMI32) in thalamic perivascular and subependymal areas of demyelination and normal appearing grey matter (NAGM). Above panels represent 3,3 ′-diaminobenzidine (DAB) immunohistochemistry of perivascular (top left) and subependymal (bottom left) areas of demyelination and adjacent NAGM. A blood vessel (V) and the third ventricle (asterisk “*”) are labeled. Demyelinated areas have reduced axonal/dendritic density, particularly in subependymal areas compared to perivascular. Areas of axonal swelling/blebs are apparent. Below panel are confocal micrographs of demyelinated (left) and normal appearing (right) thalamus. Some axons appear swollen and irregular (white arrow) in demyelinated regions.
MRI Features of Thalamic Lesions
Thalamic lesions were identified as areas of T2/FLAIR hyperintensity with associated T1 hypointensity in 93 of 95 subjects (2 subjects were not analyzed for thalamic lesions due to poor resolution/artifact). Thalamic lesions were manually segmented and the 2 lesion subtypes identified on histopathology were also evident on MRI. Similarly lesions were classified as: (1) ovoid lesions within thalamic GM (eg, perivascular and periventricular), and (2) subependymal lesions (usually a thin rim) lining the lateral or third ventricles (see Fig 2). Ovoid lesions were found in the majority of subjects (n = 81; 86%), whereas subependymal lesions were only present in 28 (29%) with significantly smaller lesion volumes. Median (IQR) volumes (cm3) of lesions were as follows: ovoid 0.17 ± 0.32, subependymal (0.00 ± 0.06), and total 0.24 ± 0.40 (range = 0–3.1 cm3).
After adjusting for age, disease duration, sex, and T2LV, neither total (standardized coefficient [β] = 0.11, p = 0.20), ovoid (β = 0.09, p = 0.31), nor subependymal (β = 0.07, p = 0.44), thalamic lesion volumes correlated with thalamic volume (range = 7.7–21.9 cm3; median = 13.4 cm3).
Neuronal and Microglial Differences in Extremes of Thalamic Volume
As thalamic lesion burden did not correlate with thalamic volume, we speculated cerebral WM lesions may cause neuronal loss in NAGM as a consequence of secondary neurodegeneration. We evaluated neuronal density in thalamic NAGM in subjects with ≤15th percentile thalamic volume to subjects with ≥85th percentile to determine whether lower thalamic volume correlated with decreased neuronal density. Table 2 summarizes differences between MRI features for subjects in these 2 extremes; there was a 1.7-fold lower median thalamic volume in subjects with the least volume.
TABLE 2.
Extremes of Thalamic Volume in Subjects Used for Analysis of Neuronal Densities
| Least thalamic volume (n = 12) | Greatest thalamic volume (n = 14) | P | |
|---|---|---|---|
| Age | 54.0 [39.8–60.3] | 56.0 [52.0–64.0] | 0.35 |
| Female sex | 7 | 12 | |
| Race | |||
| White | 7 | 13 | |
| African American | 4 | 1 | |
| Hispanic | 1 | ||
| Disease duration | 19.5 [12.8–25.5] | 16.0 [13.5–28.3] | 0.70 |
| Disease course | RR0 | RR2 | |
| SP 11 | SP 9 | ||
| PP 1 | PP 3 | ||
| Thalamic volume, cm3 | 9.9 [9.2–10.2] | 17.1 [16.6–18.1] | <0.0001 |
| Thalamic lesion volume, cm3 | 0.85 [0.38–1.27] | 0.51 [0.14–0.66] | 0.35 |
| BPF | 0.64 [0.53–0.68] | 0.81 [0.77–0.84] | 0.0001 |
| T2LV, cm3 | 98.1 [74.1–115.7] | 11.0 [3.0–22.0] | <0.0001 |
| T1LV, cm3 | 20.5 [14.4–35.5] | 5.2 [1.6–8.8] | 0.002 |
| WMF | 0.29 [0.26–0.30] | 0.32 [0.31–0.33] | 0.005 |
| GMF | 0.41 [0.35.42] | 0.48 [0.45–0.49] | 0.003 |
BPF = brain parenchymal fraction; GMF = grey matter fraction; T1LV = T1 lesion volume; T2LV = T2 lesion volume; PP = primary progressive; RR = relapsing remitting; SP = secondary progressive; WMF = white matter fraction.
Neuronal densities (count per mm2 of tissue; 5 ROIs in NAGM per 30 μm section; HuR antibody) were compared between subjects with the least (n = 12) or greatest (n = 14) thalamic volume (see Table 2). Median (IQR) neuronal density was 17.6% lower in subjects with the least volume (37.9 [29.8–46.1]) compared to the greatest volume (45.2 [33.6–56.8]) in all measured neurons >200 μm2 (Wilcoxon rank sum test with continuity correction p = 0.03). Similarly, the median density of smaller neurons (200–400 μm2) was 26.1% reduced in subjects with the least (13.3 [7.7–18.9]) compared to the most volume (17.3 [9.6–25.1]; p = 0.0004). Median density of larger neurons (>400 μm2) were not significantly different between subjects with the least volume compared to the most (p = 0.66).
For the area occupied by activated microglia/macrophages (MHC class II), 11 subjects with the most thalamic volume and 12 subjects with the least volume were examined. Five ROIs in NAGM were thresholded for determining the percent area occupied by MHCII staining. The areas occupied by activated microglia/macrophages were similar in thalami with the most and least thalamic volume (9.2% vs 10.1%; p = 0.19).
Extra-Thalamic MRI Correlates of Thalamic Volume
In the cohort of subjects with extremes of thalamic volume used for comparison of neuronal and activated microglial/macrophage density in NAGM, we also compared MRI metrics (see Table 2). We found brain parenchymal fraction (BPF), and WM/GM fractions were significantly lower in subjects with the least thalamic volume and T1 and T2 lesion volume (T1LV and T2LV) were significantly higher in subjects with the least thalamic volume.
Thalamic volume correlated with T2LV (Fig 6; adjusted r2 = 0.23 [p < 0.0001], Spearman’s rho = −0.65 [p < 0.0001]). Extra-thalamic measures were significantly associated with thalamic volume: BPF (rho = 0.60; p < 0.0001), temporal cortical thickness (CT; rho = 0.64; p < 0.0001), occipital CT (rho = 0.44; p = 0.003), T1LV (rho = −0.49; p < 0.0001), global CT (rho = 0.61; p < 0.0001), frontal CT (rho = 0.63; p = 0.0001), and parietal CT (rho = 0.58; p < 0.0001); upper cervical cord cross sectional area approached significance (rho = 0.38; p = 0.05).
FIGURE 6:

Thalamic volume plotted along with white matter T2 lesion volume. Linear model shown with a best-fit line (blue) and a 95% confidence interval (shaded in in grey). A significant correlation was found between with T2 lesion volume and thalamic volume, adjusted r2 = 0.23 (p < 0.0001), Spearman’s rho = − 0.65 (p < 0.0001). [Color figure can be viewed at www.annalsofneurology.org]
Thalamic volume was modeled with either T2LV or thalamic lesion volume after adjusting for covariates: thalamic volume ~12.40–0.04*T2LV + 0.09 * age – 0.08*disease duration +0.55*thalamic lesion volume – 1.41*male sex; adjusted r2 = 0.41. T2LV was a significant predictor (standardized β = −0.55; p < 0.0001), whereas thalamic lesion volume was not (p = 0.20). Covariates age (β = 0.39; p = 0.0001), male sex (β = −0.26; p = 0.003), and disease duration (β = −0.34; p = 0.0007) were also significant.
Discussion
Thalamic volume loss correlates with physical disability and cognitive impairment in MS and is becoming increasingly incorporated as an outcome measure in clinical trials. The underlying pathology inherent to the thalamus in MS and the mechanisms that lead to thalamic volume loss are poorly understood. In our study of MRI and pathological data on thalami from a postmortem cohort of subjects with advanced MS, we evaluate the contribution of thalamic lesions and extra-thalamic injury to thalamic volume and further characterized thalamic lesions and neuronal changes in subjects with extremes of volume. We detected several types of thalamic demyelination, which was associated with neuronal and axonal dystrophic changes in perivascular lesions and axonal loss in subependymal lesions. However, measures of global white and grey matter injury (particularly T2LV) correlated with thalamic volume better than with thalamic lesions. Additionally, neuronal density in NAGM was reduced at least in proportion, and perhaps in excess to thalamic volume loss.
Thalamic lesions showed several interesting features. Similar to prior pathological studies, we found areas of ovoid (perivascular) and subependymal demyelination. We also found all thalamic discolorations in cm-thick slices were ovoid perivascular demyelinated lesions upon pathological examination of tissue sections. Dilated superficial thalamic veins were observed on some postmortem MRIs and perivascular lesions in the anterior and dorsomedial nuclei were likely branches of the thalamostriate, anterior thalamic, and superficial thalamic veins.32,33 In contrast to others, we did not find differences in neuronal density in demyelinated perivascular lesions and adjacent NAGM in dorsomedial, ventrolateral, and pulvinar nuclei.20,21 Qualitatively, some neurons appeared to be dystrophic (less polygonal cell bodies), particularly in subependymal lesions relative to perivascular lesions, which could be a harbinger for neuronal loss. This finding could be explained by differences in vulnerability of neuronal populations among regions, neuronal subtypes, or the mechanism of demyelination.34 There is significant axonal/dendritic loss and axonal dystrophy in subependymal areas raising the potential for excitotoxic elements in the cerebrospinal fluid (CSF) mediating injury. Axons in perivascular lesions appeared relatively preserved but occasional dystrophic axons were found.
Most perivascular lesions identified with MHCII for activated microglia/macrophages were either chronic inactive or chronic active, but some areas of active and perivascular inflammation without demyelination were also observed. Subependymal lesions invariably had increased MHCII along the ventricular surface further supporting a link to the CSF; and some rims were detected along the lesion edge bordering NAGM. Although “active” lesions could presumably be a confounder in thalamic volume if associated with edema and cellular infiltration, the proportion of active lesions was 7.7% and the lesions were small. In contrast, MRI thalamic lesion volume ranged from 0 to 3.1 cm3 with thalamic volume ranging from 7.7 to 21.9 cm3; a subject with the greatest thalamic volume (21.9 cm3) also had one of the largest thalamic lesion volumes (2.7 cm3).
Although cortical lesions were not evaluated in this study, potential mechanisms for deep GM demyelination, may be similar to cortical demyelination with thalamic subependymal lesions paralleling cortical subpial lesions and perivascular lesions mimicking WM lesions. The mechanism for subependymal/subpial demyelination may involve toxin/chemokine infiltration from the CSF, whereas perivascular lesions are mediated by recruitment of peripheral immune cells. Common mediators that lead to cortical and deep GM lesions is supported by the correlation between deep GM and cortical demyelination lesion burden.9,18 Patterns of perivascular demyelinating lesions in the cortex have been linked to the location of vascular territories that lie at the leukocortical junction, draining superficial layers, or extending through all 6 layers.35 Our observation of prominent thalamic veins on MRI and the distribution of perivascular lesions suggests a similar role of vasculature in mediating deep GM demyelination.
Histologically, thalamic neuronal density in MS compared to controls has been estimated to be decreased by 22%.20 We expected no statistical difference in neuronal density in subjects with the greatest and least volume if the loss of neurons occurred in proportion to loss of thalamic volume. Conversely, if neurons were lost in excess or were relatively preserved in relation to loss of thalamic volume, we would observe statistical differences. We compared neuronal densities in thalami with the least and most volume and found an overall 17.6% reduction in neuronal densities in those thalami with the least volume, suggesting neurons were lost in excess to volume loss. Loss of larger neurons (>400 μm2) seemed to be proportional to thalamic volume loss, whereas loss of smaller thalamic neurons (200–400 μm2) was greater. These findings may indicate smaller interneurons could be preferentially lost relative to the larger relay neurons. Although activated microglia/macrophages in NAGM were abundant, there were no differences between subjects with the greatest or least thalamic volume.
In a linear regression, significant correlates of thalamic volume included age, male sex, and T2 lesion volume, and not thalamic lesion volume (total, ovoid, and subependymal). As the thalamus has extensive afferent and efferent connections, volume loss likely occurs due to secondary neurodegeneration associated with WM lesions along these tracts causing loss of neurons and axons in the thalamus. The concept of thalamic nuclear volume correlating with projecting tracts has been noted recently with lateral geniculate volume correlating with ganglion cell inner plexiform layer using optical coherence tomography.36 As the uniform parallel axonal orientation in the corpus callosum facilitates quantification, axonal loss in the corpus callosum has been demonstrated to correlate with cerebral WM lesions.37 There is also a potential for lesions not routinely imaged (eg, cortical or spinal cord) or primary neurodegeneration as contributors to thalamic volume as its atrophy can be an early feature of MS.
Our study has several limitations. The cohort of patients with advanced MS may not capture mechanisms present with early thalamic volume loss. The contribution of pathologic processes in addition to MS is difficult to estimate, both related to aging as well as hypoxia at the time of death. We adjusted analysis for age and attempted to minimize hypoxic effects by excluding those with prolonged hypoxia. We also attempted to limit death-related MRI changes by imaging patients as quickly as possible within 6 hours of death. We have acquired tissue over nearly 3 decades and processed tissue for each subject with short and long fixation and freezing to allow for different immunohistological and genomic studies; this limits analysis of the entirety of a structure, such as the thalamus for each case to perform stereological pathologic studies. Thalamic demyelinating lesions could have deleterious effects to synapses and dendrites that were not measured in our study. The high density of axons with multiple orientations within the thalamus and around perivascular lesions made their quantification unreliable; we quantified axons in subependymal regions with a lower density and longitudinal orientation. We are unable to identify “active” MS lesions on MRI as the use of gadolinium for breakdown of the blood brain barrier is not possible in a postmortem cohort. The small size of active lesions identified on histology make co-registering with MRI challenging. Advanced imaging methods, including greater field strength, that improve detection of thalamic lesions and the study of individual thalamic nuclear groups may further delineate the role of deep GM pathology and its correlation with clinical disability.
Our results suggest thalamic volume, due to its extensive connectivity via afferent/efferent tracts, is better explained by WM lesion burden than by thalamic demyelination. Ultimately, identifying mechanisms that contribute to primary or secondary neurodegeneration will direct attention to therapeutics that may slow or reverse these processes or to more sensitive imaging biomarkers for determination of therapeutic effectiveness. As it is difficult to measure what is “lost” due to thalamic atrophy by immunohistochemistry, future directions include elaborating cell-specific genomic changes (eg, neuronal subtypes) in subjects with thalamic volume extremes and identifying the relationship of thalamic nuclear-specific changes with clinical disability.
Acknowledgments
K.R.M. is funded by National Multiple Sclerosis Society Clinician Scientist Development Award FAN-1507-05606 and the NIH NINDS K23 Career Development Award 1K23NS109328.
Postmortem program funded by NIH/NINDS R35 to B.D.T. and Sanofi Genzyme.
We thank Dr Xiaofeng Wang in the Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic Foundation, for reviewing our biostatistical analyses.
Footnotes
Potential Conflicts of Interest
The authors declared no conflict of interest.
References
- 1.Wallin MT. The prevalence of MS in the US: A population-based healthcare database approach: US MS prevalence workgroup. European Committee for Treatment and Research in Multiple Sclerosis; October 26, 2017 2017; Paris, France. [Google Scholar]
- 2.Trapp BD, Peterson J, Ransohoff RM, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med 1998;338:278–285. [DOI] [PubMed] [Google Scholar]
- 3.Nourbakhsh B, Azevedo C, Maghzi AH, et al. Subcortical grey matter volumes predict subsequent walking function in early multiple sclerosis. J Neurol Sci 2016;366:229–233. [DOI] [PubMed] [Google Scholar]
- 4.Azevedo CJ, Overton E, Khadka S, et al. Early CNS neurodegeneration in radiologically isolated syndrome. Neurol Neuroimmunol Neuroinflamm 2015;2:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nourbakhsh B, Azevedo C, Nunan-Saah J, et al. Longitudinal associations between brain structural changes and fatigue in early MS. Mult Scler Relat Disord 2016;5:29–33. [DOI] [PubMed] [Google Scholar]
- 6.Hyncicova E, Vyhnalek M, Kalina A, et al. Cognitive impairment and structural brain changes in patients with clinically isolated syndrome at high risk for multiple sclerosis. J Neurol 2017;264:482–493. [DOI] [PubMed] [Google Scholar]
- 7.Steckova T, Hlustik P, Sladkova V, et al. Thalamic atrophy and cognitive impairment in clinically isolated syndrome and multiple sclerosis. J Neurol Sci 2014;342:62–68. [DOI] [PubMed] [Google Scholar]
- 8.Zivadinov R, Havrdova E, Bergsland N, et al. Thalamic atrophy is associated with development of clinically definite multiple sclerosis. Radiology 2013;268:831–841. [DOI] [PubMed] [Google Scholar]
- 9.Mesaros S, Rocca MA, Absinta M, et al. Evidence of thalamic gray matter loss in pediatric multiple sclerosis. Neurology 2008;70: 1107–1112. [DOI] [PubMed] [Google Scholar]
- 10.Eshaghi A, Prados F, Brownlee WJ, et al. Deep gray matter volume loss drives disability worsening in multiple sclerosis. Ann Neurol 2018;83:210–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanninen K, Viitala M, Paavilainen T, et al. Thalamic atrophy without whole brain atrophy is associated with absence of 2-year NEDA in multiple sclerosis. Front Neurol 2019;10:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calabrese M, Rinaldi F, Grossi P, et al. Basal ganglia and frontal/parietal cortical atrophy is associated with fatigue in relapsing-remitting multiple sclerosis. Mult Scler 2010;16:1220–1228. [DOI] [PubMed] [Google Scholar]
- 13.Kletenik I, Alvarez E, Honce JM, et al. Subjective cognitive concern in multiple sclerosis is associated with reduced thalamic and cortical gray matter volumes. Mult Scler J Exp Transl Clin 2019;5: 2055217319827618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klaren RE, Hubbard EA, Motl RW, et al. Objectively measured physical activity is associated with brain volumetric measurements in multiple sclerosis. Behav Neurol 2015;2015:482536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sotirchos ES, Gonzalez-Caldito N, Dewey BE, et al. Effect of disease-modifying therapies on subcortical gray matter atrophy in multiple sclerosis. Mult Scler 2019;11:1352458519826364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15:373–381. [DOI] [PubMed] [Google Scholar]
- 17.Gaetano L, Haring DA, Radue EW, et al. Fingolimod effect on gray matter, thalamus, and white matter in patients with multiple sclerosis. Neurology 2018;90:e1324–e1332. [DOI] [PubMed] [Google Scholar]
- 18.Harrison DM, Oh J, Roy S, et al. Thalamic lesions in multiple sclerosis by 7T MRI: clinical implications and relationship to cortical pathology. Mult Scler 2015;21:1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louapre C, Govindarajan ST, Gianni C, et al. Heterogeneous pathological processes account for thalamic degeneration in multiple sclerosis: insights from 7 T imaging. Mult Scler 2017;1:1352458517726382. [DOI] [PubMed] [Google Scholar]
- 20.Cifelli A, Arridge M, Jezzard P, et al. Thalamic neurodegeneration in multiple sclerosis. Ann Neurol 2002;52:650–653. [DOI] [PubMed] [Google Scholar]
- 21.Vercellino M, Masera S, Lorenzatti M, et al. Demyelination, inflammation, and neurodegeneration in multiple sclerosis deep gray matter. J Neuropathol Exp Neurol 2009;68:489–502. [DOI] [PubMed] [Google Scholar]
- 22.Vercellino M, Plano F, Votta B, et al. Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol 2005;64:1101–1107. [DOI] [PubMed] [Google Scholar]
- 23.van de Pavert SH, Muhlert N, Sethi V, et al. DIR-visible grey matter lesions and atrophy in multiple sclerosis: partners in crime? J Neurol Neurosurg Psychiatry 2016;87:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dutta R, Mahajan KR, Nakamura K, et al. Comprehensive autopsy program for individuals with multiple sclerosis. JoVE 2019;149: e59511. [DOI] [PubMed] [Google Scholar]
- 25.Trapp BD, Vignos M, Dudman J, et al. Cortical neuronal densities and cerebral white matter demyelination in multiple sclerosis: a retrospective study. Lancet Neurol 2018;17:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakamura K, Chen JT, Ontaneda D, et al. T1−/T2-weighted ratio differs in demyelinated cortex in multiple sclerosis. Ann Neurol 2017; 82:635–639. [DOI] [PubMed] [Google Scholar]
- 27.Bo L, Mork S, Kong PA, et al. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J Neuroimmunol 1994;51:135–146. [DOI] [PubMed] [Google Scholar]
- 28.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fisher E, Chang A, Fox RJ, et al. Imaging correlates of axonal swelling in chronic multiple sclerosis brains. Ann Neurol 2007;62: 219–228. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura K, Fox R, Fisher E. CLADA: cortical longitudinal atrophy detection algorithm. Neuroimage 2011;54:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2020. [Google Scholar]
- 32.Gogia B, Kumar VA, Chavali LS, et al. MRI venous architecture of the thalamus. J Neurol Sci 2016;370:88–93. [DOI] [PubMed] [Google Scholar]
- 33.Giudicelli G, Salamon G. The veins of the thalamus. Neuroradiology 1970;1:92–98. [Google Scholar]
- 34.Schirmer L, Velmeshev D, Holmqvist S, et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019;573: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kidd D, Barkhof F, McConnell R, et al. Cortical lesions in multiple sclerosis. Brain 1999;122:17–26. [DOI] [PubMed] [Google Scholar]
- 36.Papadopoulou A, Gaetano L, Pfister A, et al. Damage of the lateral geniculate nucleus in MS: assessing the missing node of the visual pathway. Neurology 2019;92:e2240–e2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evangelou N, Konz D, Esiri MM, et al. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain 2000;123:1845–1849. [DOI] [PubMed] [Google Scholar]