

Abstract
Bioorthogonal chemistries enable researchers to interrogate biomolecules in living systems. These reactions are highly selective and biocompatible and can be performed in many complex environments. However, like any organic transformation, there is no perfect bioorthogonal reaction. Choosing the “best fit” for a desired application is critical. Correspondingly, there must be a variety of chemistries—spanning a spectrum of rates and other features—to choose from. Over the past few years, significant strides have been made towards not only expanding the number of bioorthogonal chemistries, but also fine-tuning existing reactions for particular applications. In this Review, we highlight recent advances in bioorthogonal reaction development, focusing on how physical organic chemistry principles have guided probe design. The continued expansion of this toolset will provide more precisely tuned reagents for manipulating bonds in distinct environments.
I. Introduction
A comprehensive view of living systems requires tools and methods to probe biomolecules in their native habitats. Fluorescent proteins and other genetic tags have long been used in this capacity1. While powerful, such tools are not amenable to direct monitoring of non-proteinaceous targets, including small molecule metabolites. The need for more generalizable platforms spurred the development of bioorthogonal chemistries—reactions that are so selective that they can be used to covalently tag targets in live cells and, in some cases, living organisms. For decades, bioorthogonal reactions have been used to visualize and profile a broad spectrum of biomolecules. These studies have revealed fundamental new insights into various aspects of cell and organismal biology. Such studies also revealed limitations in the bioorthogonal toolkit that inspired the development of even more selective and finely tuned probes.
Central to all applications of bioorthogonal chemistry are reactions that are robust and compatible with living systems. The development of such transformations can be quite difficult. The solvent and temperature are fixed, and the reactions must proceed in the midst of a multitude of interfering functionality. The reactions often cannot be accelerated simply by “heating up” the subject or adding more reagent. Thus, the canonical rules for fine-tuning chemistries in round-bottom flasks often fail to translate to physiological environments, where few parameters can be varied2. Nonetheless, generations of chemists have been inspired to control bond formation in live cells and organisms. Their efforts have provided transformations that can be executed without detriment in living systems.
This article reviews the development of bioorthogonal reactions, with an emphasis on how mechanistic insights have driven the field. Like other areas of chemistry, no “one-size-fits- all” transformation exists. Rather, each reaction has its pros and cons, with limitations continuing to propel new advances. In the first section, we provide a brief history on bioorthogonal chemistry and introduce common tactics that facilitated early probe development. The bulk of the review then showcases recent achievements in bioorthogonal reaction design. We also highlight efforts to engineer bioorthogonal chemistries to be used concurrently for multi-component labeling. These and other innovations will continue to expand the collection of biocompatible and mutually orthogonal reagents.
II. Setting the stage: Classic bioorthogonal transformations
Chemists rely on robust and versatile reactions to craft complex molecules. Synthetic routes are drafted with reagent accessibilities, yields, and selectivities in mind. Ideally, the transformations are fast, selective, and applicable to a broad range of substrates. In reality, most reactions are not universally efficient and require tweaks to temperature, pH, or stoichiometry in different contexts. Catalysts and solvents are also varied to achieve optimal yields. Limitations in reaction scope often become the inspiration for new transformations. This iterative cycle of reaction discovery and refinement has provided a compendium of methods for controlling bond formation in various contexts. In some cases, hundreds, if not thousands, of specialized reagents have been developed to address shortcomings in reaction scope.
Iterative refinement has also been used to tune reagents for use in living systems, with certain considerations in mind (Figure 1)2,3. Bioorthogonal functional groups must toe the line of being kinetically and metabolically stable, yet prone to rapid reaction with complementary probes under physiological conditions. Such reactions must also be tolerant of water and other biological functional groups4–6. The constraints imposed by cells and tissues exclude many organic transformations. Several biological applications also demand reagents that impart a minimal steric “footprint”7,8. Thus, developing chemistries that feature small reagents is another important goal.
Figure 1.
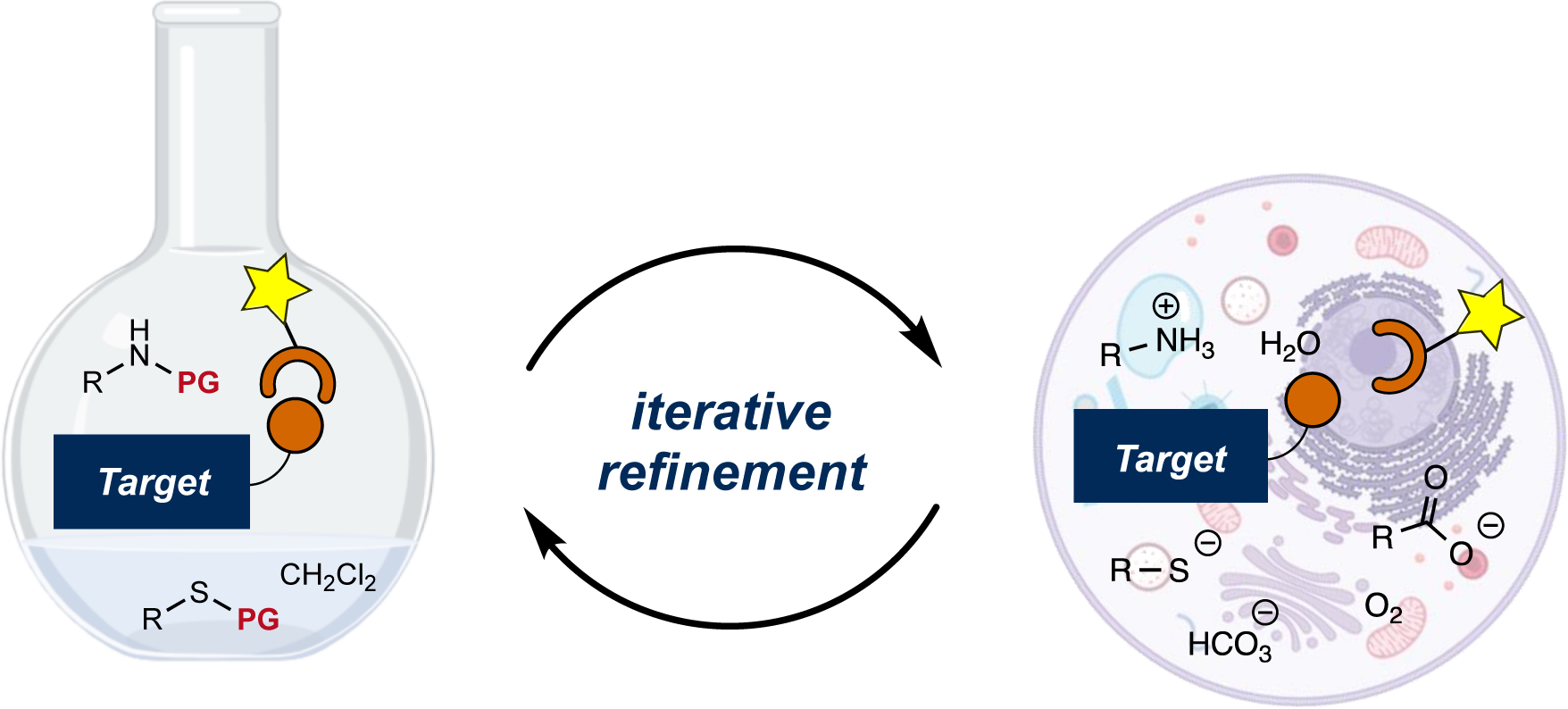
Translating reactions from round-bottom flasks to living systems. Reactants (orange circle and arc) used in cells and tissues must be compatible with cellular functionality. Challenging applications continue to drive the refinement of bioorthogonal reactions. Optimization in either flasks or cells invokes similar principles, although variables that are easily controlled in flasks are often invariant in living systems.
So, where does one begin? Hunting for unusual functional groups in heterologous organisms is a good starting point. Microbes and other species often have access to chemistries and functionality not present in mammalian cells. Thus, these motifs and chemistries can survive in living systems and are immediately “orthogonal”. A classic example is the alkyne, a motif that is present in numerous microbial metabolites9, but absent in higher eukaryotes. The alkyne has been applied as a bioorthogonal motif in numerous settings2. Popular bioorthogonal functional groups also comprise some unlikely candidates from synthetic chemistry10,11. Organic azides and strained alkynes were often viewed as too unstable for use in living systems. However, careful tuning provided reagents now recognized among the gold standards in the field. These examples provided important lessons for subsequent reagent development12. In this section, we provide a brief perspective on how some seemingly “misfit” functional groups became stalwarts of the two most common classes of bioorthogonal chemistry: polar reactions and cycloadditions. We highlight early obstacles and key advances in the development of these probes. Initial successes provided a roadmap for continued reagent refinement.
Polar reactions
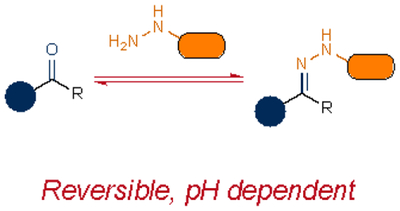
Aldehydes and ketones were among the first reagents employed as bioorthogonal labels. Such carbonyl groups were attractive for biomolecule tagging, given their small size and compatibility in living systems13. The electrophiles were also readily condensed with α-nucleophiles. Aldehyde and ketone condensations have been used to label a wide variety of biomolecules14–17. However, the reactions are pH sensitive and quite slow in physiological settings. Aniline catalysts18–21 can boost the rates, although the transformations remain difficult to execute in cellular environments.
While aldehydes and ketones have been less employed in cells, only a few other bioorthogonal functional groups rival their minimal size. Among the most influential has been the organic azide. This motif is abiotic and comprises just three atoms. Azides are remarkably inert in biological settings, but exhibit unique manifolds of reactivity. In one case, they can react with soft nucleophiles, including triarylphosphines (via Staudinger reduction). This reaction proceeds via an aza-ylide intermediate, which can be intercepted with neighboring electrophiles. Bertozzi and coworkers capitalized on this feature, installing an ester on the phosphine to trap the aza-ylide. The transformation ultimately linked the two reactants via an amide bond. This variant – termed the Staudinger ligation – was amenable to tagging azides in a variety of complex environments, including live cells22. Early applications featured glycans and post-translational modifications, although many other biomolecules have since been targeted23–27. The Staudinger ligation was also the first of its kind to be used in live rodents, a particularly demanding environment28.
The versatility of the Staudinger ligation propelled an entire field of reaction development. Many early studies were directed at improving ligation speed4,5. While the reaction was robust enough for certain in vivo applications, the slow rate proved challenging for imaging studies in rodents and higher organisms. Large boluses of reagent were required to drive covalent bond formation on reasonable time scales. Such quantities were not easily achieved, due to the limited water solubility of many phosphine probes. These shortcomings generally sidelined the Staudinger ligation in vivo, but proved a fruitful ground for inspiring other types of transformations with organic azides.
Cycloadditions
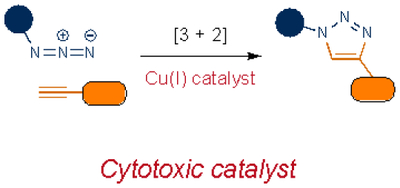
Cycloadditions are popular bioorthogonal transformations owing to their exquisite chemoselectivity. One of the earliest exploited was [3+2] cycloaddition with azides. In addition to being mild electrophiles, organic azides are 1,3-dipoles subject to react with alkynes. As noted earlier, alkynes are rare in higher eukaryotes, rendering them suitably orthogonal. However, azide-alkyne cycloadditions typically require high temperatures or pressures to proceed, conditions that are not biocompatible. A key breakthrough was the recognition that the cycloaddition could be accelerated via Cu(I). The copper-catalyzed azide-alkyne cycloaddition (CuAAC) is ubiquitous in chemical biology and other disciplines29,30, and is still inspiring new transformations31. Cu(I) cytotoxicity32–34 has precluded CuAAC application in vivo in some cases, although more biofriendly catalysts have been developed. Many feature water-soluble ligands that stabilize Cu(I) and prevent the formation of reactive oxygen species35–38. Polytriazole ligands, in particular, enabled CuAAC reactions to be conducted in live cells and zebrafish36.
The constraints posed by copper catalysts also inspired chemists to devise new strategies to ligate 1,3-dipoles. One of the most fruitful approaches relied on strain energy – bending the alkyne from its normal linear geometry39. The smallest of the stable cycloalkynes, cyclooctyne, was found to react with azides in the absence of a copper catalyst40. This work gave rise to an entire family of strain-promoted azide-alkyne cycloadditions (SPAACs). Such reactions have been widely used in complex biological settings, including plants41, C. elegans42, and mice43–46. While SPAAC reactions minimize toxicity in living systems, some of the alkynes react with biological thiols47. Moreover, the fastest rate constants with the most reactive strained alkynes plateau at rates of ~1 M−1 s−1, which are not suitable for some in vivo applications.
Other cycloadditions have been developed to address the need for fast-acting, biocompatible reagents. One notable class comprises inverse electron-demand Diels–Alder (IEDDA) cycloadditions. The IEDDA reaction between tetrazine and trans-cyclooctene (TCO), in particular, has gained prominence11,48–50. This reaction pair boasts rates up to 106 times faster than the most reactive SPAAC pair. Such rapid reactions have widespread use in cells and living organisms, as only small amounts of material are required to drive covalent bond formation. The kinetic profile of the tetrazine-TCO cycloaddition has enabled the exploration of new imaging platforms in vivo, including PET imaging51,52 and other radiolabels53–55. Tetrazine-TCO reactions have also been widely employed to tag biomolecules, release prodrugs56–59, and activate enzymes60–62.
III. Fine-tuning bioorthogonal reagents and reactions
Despite remarkable achievements in bioorthogonal reaction design over the past two decades, limitations remain. Many reagents are insufficiently stable for use in the most demanding biological settings. Others remain too slow to transition to in vivo applications. And while many of the reactions work well on single targets, most cannot be used in combination due to cross-reactivity concerns. Simultaneous tracking of multiple biomolecules remains a longstanding challenge. Collectively, these limitations underscore the need to not only develop new reactions, but also fine-tune existing chemistries. The next sections highlight recent approaches to address voids in the bioorthogonal toolbox and broaden the spectrum of reactivities (Table 1).
Table 1.
Tuning transformations for biological application. Bioorthogonal chemistries largely fall into two categories: polar reactions and cycloadditions. Examples of how limitations spurred the development of new reactions are shown.
| Reaction type | Overall transformation | Early example | Tuned variant |
|---|---|---|---|
| Polar | 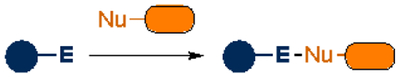 |
 |
 |
 |
 |
||
| Cycloaddition | 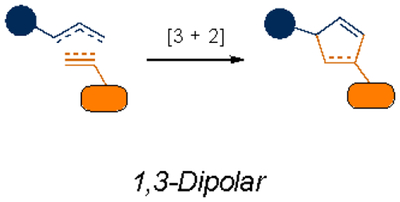 |
 |
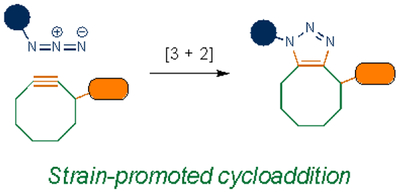 |
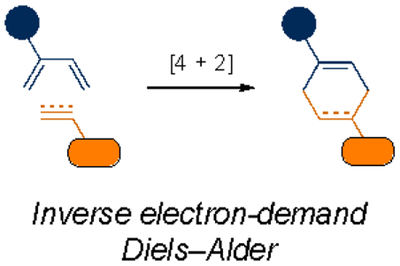 |
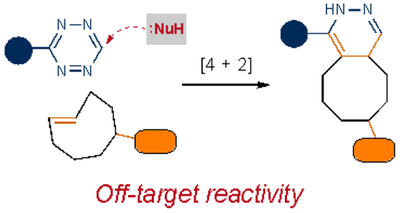 |
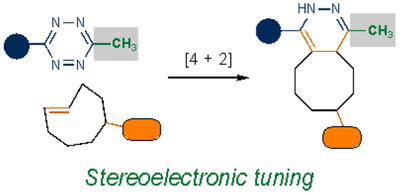 |
Tuning polar reagents and reactions
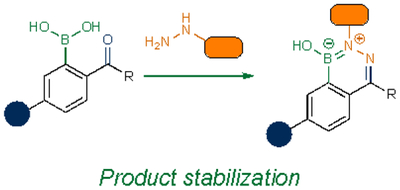
As noted previously, some of the earliest bioorthogonal transformations involved condensation reactions with carbonyls and α-nucleophiles. The reversible nature of these reactions presented challenges in biomolecule labeling. Efforts to forge more stable adducts have been undertaken, including the use of proximal stabilizing groups63–69. One example showcases boronic acids. These functional groups can coordinate ligation adducts, including hydrazones64 and oximes65,66, preventing hydrolysis. Boronic acids have also been used to stabilize ligation adducts from other α-nucleophiles67–69, and have recently been applied in tagging N-terminal cysteine residues on peptides and small proteins70,71.
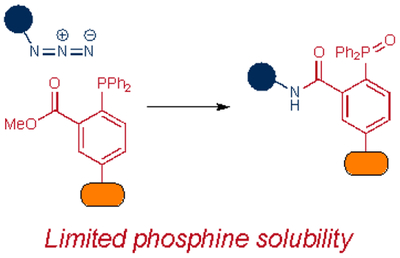
Polar reactions with azides have also been tuned for specific applications. The Staudinger ligation was an early target for optimization, with efforts focused on improving rates and reagent solubility. Introducing electron-withdrawing groups on the azide or electron-donating groups on the phosphine boosted the ligation speed. For example, electron-deficient aryl azides with fluorine72 and chlorine73 substituents exhibited faster rates compared to their unsubstituted counterparts. Interestingly, the key aza-ylide formed in these cases was a stable adduct, obviating the need for an electrophilic trap on the phosphine. The halogenated aryl azides have since been used to label glycans and proteins in live cells72,73. Generating suitable electron-rich phosphines for Staudinger ligation has been less straightforward. More nucleophilic scaffolds can reduce disulfide bonds in proteins and are prone to rapid oxidation. Despite this liability, numerous phosphine probes have been tuned for cellular and other applications74–76.
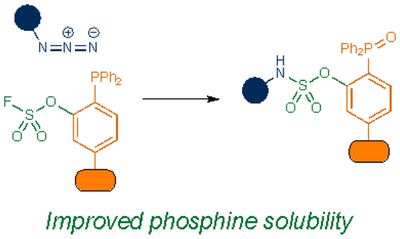
Phosphine reagents have also been tuned for improved biocompatibility. For example, Ren and co-workers synthesized phosphines comprising a fluorosulfate group77. This handle functions similarly to the ester trap in the original Staudinger ligation, by intercepting the aza-ylide. After ejection of fluoride and hydrolysis, the ligation provides an aryl sulfamate ester. The modified phosphine displayed markedly improved water solubility compared to initial Staudinger ligation probes. An additional benefit was that the sulfamate ester products mimic phosphate backbones, present in many biomolecules and metabolites.
Entirely different phosphorus nucleophiles have also been examined for azide ligation, with an eye towards forming mimics of biological functional groups. For example, phosphites78–80 and phosphonites81–83 react similarly with aryl azides to provide phosphoramidate and phosphonamidite adducts, respectively. These reactions can be used to selectively modify proteins and other biomolecules with a variety of probes.
Tuned phosphines have been exploited in other chemical contexts. Phosphines can react with Michael acceptors, forming stable phosphonium adducts. This transformation has been employed to label biomolecules. In one study, tris(2-carboxyethyl)phosphine (TCEP), a water-soluble reductant widely used in biology, was reacted with electron-deficient alkenes. This reaction was used to study protein glycosylation in live cells84 and crotonylation patterns on histone proteins85.
A more recent addition to the phosphine ligation kit comprises cyclopropenone derivatives (CpXs)86–89. In the case of cyclopropenones (X = O), the ligation proceeds via an initial Michael-type addition, followed by ring-opening to form a reactive ketene-ylide. The ketene can be trapped by pendant nucleophiles on the phosphine to furnish α,β-unsaturated carbonyls. Mono-substituted CpOs were subject to react with biologically relevant thiols86, but further tuning provided more bioinert scaffolds comprising dialkyl motifs87. These latter reagents can be used in live cells.
The reactivity of CpOs can be further modulated via heteroatom replacement, a common strategy for probe tuning in bioorthogonal chemistry. The sulfur variant of CpO – the cyclopropenethione (CpS) – also reacts rapidly with phosphines via thioketene-ylide intermediates88. Thiocarbonyl products are formed, which can be useful biophysical probes to study protein stability and function90,91. Nitrogen CpO heteroanalogs, cyclopropeniminium (CpN+) ions, were also explored as bioorthogonal reagents. Interestingly, CpN+ ions react with phosphines via a different mechanism than CpO or CpS derivatives. The initial Michael addition provides an enamine, which undergoes proton transfer instead of ring opening to give phosphonium bicycles89 (Figure 2). Thus, small changes to a core scaffold can have profound effects on reactivity.
Figure 2.
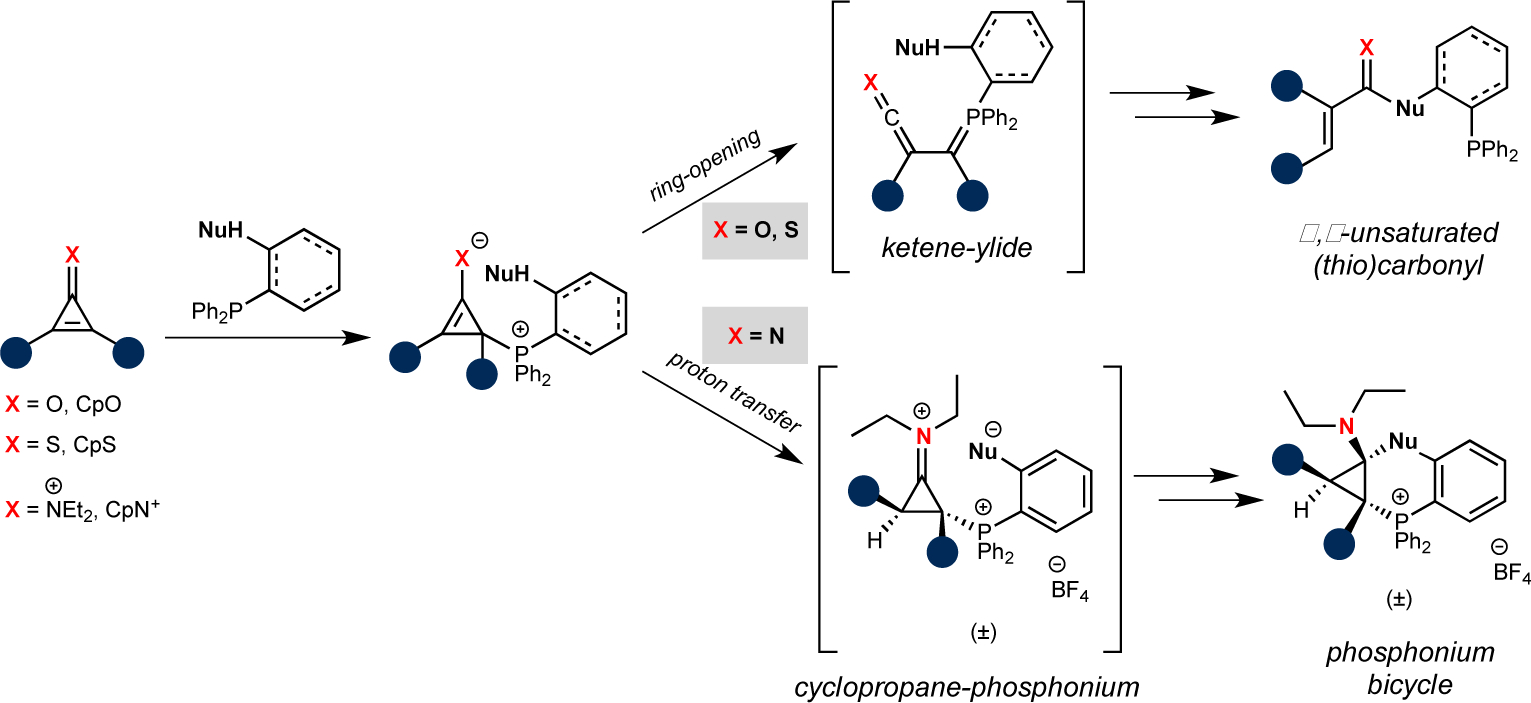
Cyclopropenone (CpX) derivatives exhibit unique reactivities. Analogs comprising oxygen or sulfur atoms react robustly with phosphines via ketene-ylide intermediates. Nitrogenous scaffolds react to form phosphonium bicycles.
Tuning dipoles for bioorthogonal cycloaddition
The azide-alkyne cycloaddition remains one of the most popular 1,3-dipolar cycloadditions for examining biological systems. The success of this ligation has served as inspiration for exploring new 1,3-dipoles (Figure 3A). One example includes nitrones92, reagents that can react more rapidly than azides with certain alkynes93–95. Nitrone-alkyne cycloadditions, similar to their azide-alkyne counterparts, can be accelerated by copper catalysis. Such reactions have been used to image sugar metabolism and receptor-ligand interactions in mammalian cells, as well as peptidoglycan structures in bacteria96–98.
Figure 3.
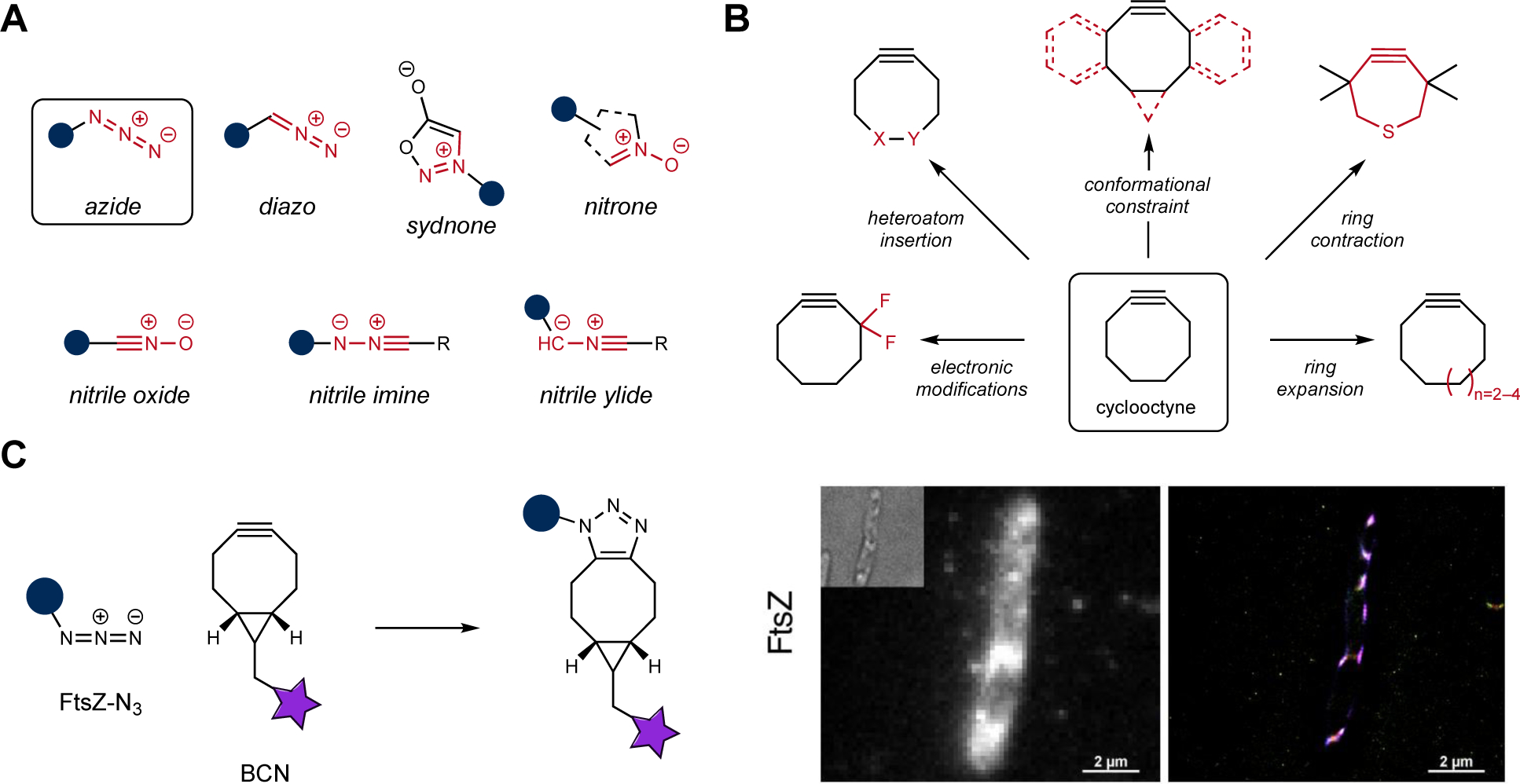
An expansive set of reagents for dipolar cycloaddition. (A) Several 1,3 dipoles are available for bioorthogonal labeling. Organic azides were among the first to be exploited for cycloadditions in cells. More recently, diazo, sydnone, and nitrone motifs have been developed for biomolecule labeling. Highly reactive dipoles (including nitrile oxides, imines, and ylides) are also viable ligation partners. They are typically produced “on demand” via photolysis or chemical uncaging. (B) Several classes of strained alkynes are available for bioorthogonal cycloaddition. These reagents derive from cyclooctyne, the first-reported variant. Extensive modifications to cyclooctyne provided a suite of reagents with altered reactivities. (C) Sample application of bioorthogonal cycloadditions. FtsZ, a bacterial protein involved in cellular division, was outfitted with an azide handle (left). Subsequent ligation with BCN-photoswitchable fluorophore conjugates enabled super-resolution microscopy in live E. coli (right). Images were adapted with permission from ref.148 The American Chemical Society
Another newcomer to the bioorthogonal dipole set is sydnone99. This 1,3-dipole reacts with alkynes to afford pyrazole adducts. Although initial applications required copper catalysis100, subsequent work identified scaffolds that were also capable of copper-free reactions with strained alkynes101. Sydnones have undergone further modification to modulate their reactivity for biological application. Chlorine substituents were found to increase reaction rates (30-fold) with a popular strained alkyne, bicyclo[6.1.0]nonyne (BCN)102. Fluorine substitution further boosted reaction rates of sydnones with a variety of strained alkynes103.
Close relatives of organic azides, diazo compounds have been similarly tuned as dipoles for bioorthogonal application104. The small size of the diazo group makes it attractive for biomolecule labeling, but such motifs were long thought to be too reactive for cellular use. However, diazo motifs are stable when in conjugation with aryl systems or electron-withdrawing groups (e.g., esters, amides)105,106. They ligate strained alkynes with second-order rate constants similar to azides. Diazo-cyclooctyne reactions have been used to tag cellular glycans among other targets107,108. Importantly, the resulting pyrazole adducts are stable to a number of biological nucleophiles109. Further diazo tuning has provided scaffolds that react with acyclic electron-deficient alkenes, a transformation that can be performed in the presence of azides110.
More reactive 1,3-dipoles have also been harnessed for bioorthogonal application. Most are masked until an external trigger (often light) is applied, enabling the reactant to be released “on demand”106,111–113. One such class of dipoles comprises nitrile imines. These motifs react robustly with strained alkenes such as norbornene114 and cyclopropene115,116, but they are prone to rapid hydrolysis in the absence of a ligation partner. Nitrile imines can be caged as tetrazoles or sydnones, functional groups that are more stable in biological environs. UV irradiation can liberate the reactive species. The starting tetrazole chromophores can be tuned for photolysis – and thus nitrile imine release – using different wavelengths of light117,118. This added layer of control has inspired the exploration of other “photo-click” reactions to expand the compendium of bioorthogonal chemistries119–121. Nitrile imines have also been extensively tuned via electronic122 and steric modification123.
Tuning dipolarophiles for bioorthogonal cycloaddition
Tuning bioorthogonal cycloadditions is perhaps best exemplified in the context of the strained cycloalkynes as dipolarophiles10,12. Many efforts have been directed at modifying ring strain, with an eye towards increasing reaction rates or in cellulo stability. Examples include modulation of ring size or conformation124–131, electronic perturbation132–135, installation of endocyclic heteroatoms136–138, and combinations thereof (Figure 3B)139,140. There have also been significant efforts to improve the water solubility of these relatively greasy probes. Towards this end, cycloalkynes featuring sulfamate backbones, as well as larger heterocyclic derivatives (up to 12-membered rings) have also been explored141–146. The heteroatom variants were generally more water soluble and stable. However, the benefits of ring expansion came at the cost of rate, a trade-off that is prevalent in bioorthogonal reagent tuning2. With the reactivity-stability axis in mind, one of the more impactful cycloalkynes has been BCN131. This scaffold has increased strain energy compared to the original cyclooctyne (due to the fused cyclopropane ring)147, but is remarkably stable in cellular environments. Coupled with its synthetic accessibility, BCN has become a staple strained alkyne for cycloaddition chemistries in a number of fields. In a recent example, BCN-fluorophore conjugates were used for super-resolution imaging in live cells (Figure 3C)148. FtsZ, a protein involved in bacterial cell division, was enzymatically outfitted with an azide. Subsequent treatment with cell-permeable BCN derivatives (comprising photoswitchable rhodamines) enabled protein localization patterns to be observed.
Efforts to modulate cycloalkyne reactivity have been bolstered by computation. Calculations can readily predict combinations of steric and electronic features to tune scaffolds for desired outcomes. One well-established approach for modulating cycloaddition partners relies on the Distortion/Interaction model149,150. This analysis computes the activation barrier for a given reaction by calculating the difference between a distortion energy (i.e., the energy required for reactants to adopt their ideal transition state geometries) and an interaction energy (i.e., favorable orbital overlap between the two reaction partners). The calculated activation energy is then correlated to a predicted rate constant, which can ultimately guide reagent tuning. One of the earliest demonstrations of the Distortion/Interaction model in bioorthogonal reagent design involved a series of biarylazacyclooctynone (BARAC) analogues. This study revealed structural features that impeded BARAC reactivity with azides and set the stage for the development of improved cyclooctynes151. Similar computational studies have been used to fine-tune sydnone-cycloalkyne reactions152 and other bioorthogonal cycloaddtions149,153,154.
Tuning dienophiles for bioorthogonal cycloaddition
Parallel developments in the realm of IEDDA have expanded the number of bioorthogonal chemistries in recent years. Reactions featuring TCO, in particular, have found widespread use in cells and in vivo11. Such transformations have also been the targets of extensive reagent tuning155–159. Many efforts have focused on pushing the kinetics of the tetrazine-TCO ligation (Figure 4). Increasing TCO strain was hypothesized to boost reaction rate, similar to the strained alkynes. Toward this end, Fox and coworkers designed scaffolds wherein TCO was forced to adopt a half-chair conformation (e.g., d-TCO155 and s-TCO155). Such motifs were predicted to be 5.6–5.9 kcal mol−1 higher in energy relative to the more stable crown conformation of TCO160. The more reactive d-TCO and s-TCO variants display bimolecular rate constants as high as 3 × 106 M−1 s−1.
Figure 4.
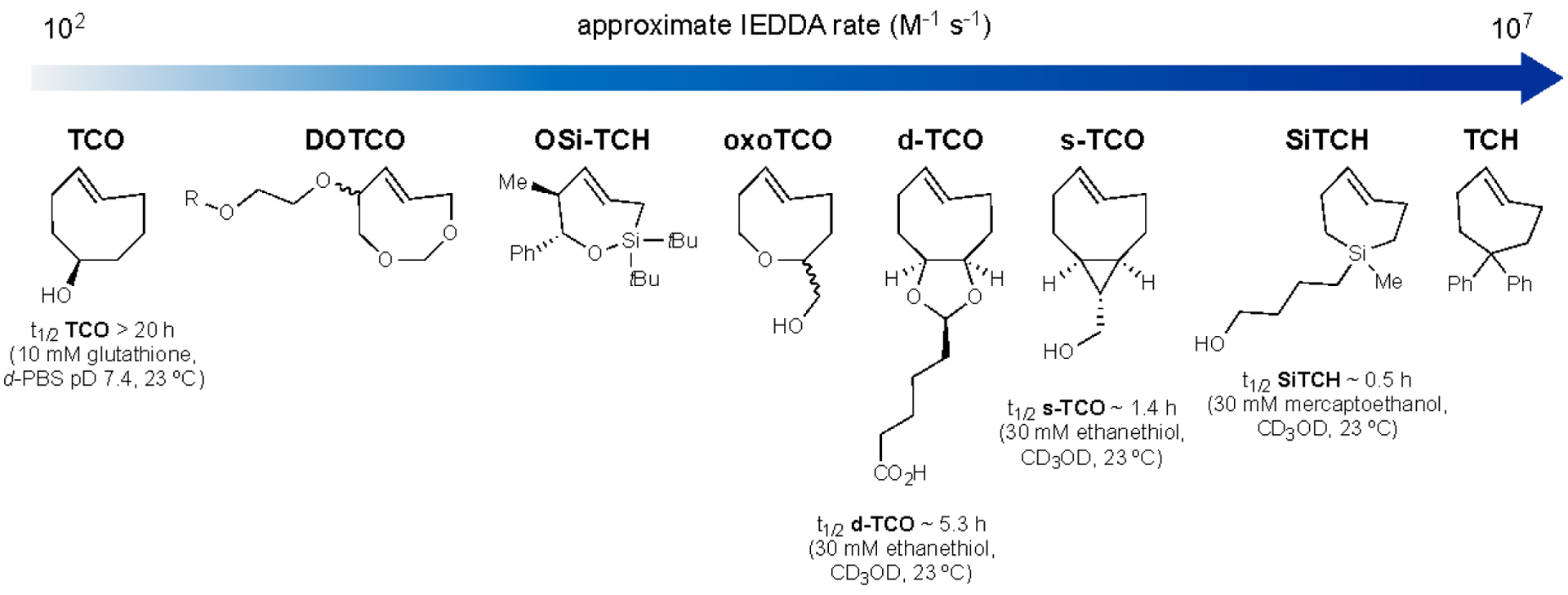
A collection of strained alkenes for IEDDA reactions. These reactions exhibit a range of reaction rates with tetrazines. trans-Cyclooctene (TCO) was the original member of this group. TCO has since been modified to access faster and more biocompatible reagents. Some of the scaffolds feature endocyclic heteroatoms (DOTCO156, OSi-TCH161, oxoTCO158), conformational constraints (d-TCO155, s-TCO155), and contracted rings (SiTCH154, TCH154). In general, the fastest reacting scaffolds exhibit the shortest half-lives in the presence of biological thiols.
Similar to the strained alkynes, ring contraction strategies have been employed in the context of strained alkenes. Recently, trans-cycloheptene (TCH) analogs have been reported as voracious dienophiles in IEDDA cycloadditions with tetrazines161,162. TCH readily isomerizes under ambient conditions but can be isolated as a stable complex with Ag(I). In addition, incorporation of an endocyclic silicon atom provided a more stable seven-membered cycloalkene, sila-trans-cycloheptene (SiTCH). The longer Si-C bond lengths in SiTCH relieved some ring strain, enabling facile isolation. Computational studies further revealed that the activation barrier for SiTCH reactivity with diphenyltetrazine was significantly lower compared to s-TCO, enabling rapid ligation. The second-order rate constant for SiTCH and a model tetrazine was 1.14 × 107 M−1 s−1, the fastest bioorthogonal ligation on record. Despite their impressive rates, though, TCH and SiTCH degrade rapidly in the presence of cellular thiols162.
1,3-Disubstituted cyclopropenes, alternative dienophiles for IEDDA reactions, have also been tuned for a variety of applications. We and others reported that these scaffolds react with tetrazines in biological environments163,164. Compared to TCO, the reaction between tetrazines and cyclopropenes is much slower. However, the small size and cellular stability of cyclopropenes have rendered them attractive for interrogating biomolecules in complex environments165. One notable example from the Chin lab showcased a cyclopropene-lysine analog to monitor nascent protein biosynthesis in Drosophila166.
The reactivity profile of the cyclopropene can be dramatically influenced by steric tuning. The Distortion/Interaction model predicted that increasing steric bulk at C3 on cyclopropene would drastically reduce its reactivity with tetrazines. Diminished reactivity was attributed to geometric constraints in the transition state. The poor orbital overlap between the cycloaddition partners was predicted to slow the ligation. Indeed, no reaction was observed even when 3,3-disbustituted cyclopropenes were subjected to a variety of functionalized tetrazines. Such cyclopropenes still reacted with nitrile imines, though, and this differential reactivity was exploited to label two proteins in tandem116.
Further cyclopropene tuning was achieved upon introduction of spirocycles at C3. Based on crystallographic data, cyclopropene 2 exhibited a reduced bond angle between the two C3-groups compared to parent cyclopropene 1 (Figure 5). The decreased bond angle drove the substituents away from the incoming nitrile imine, increasing ligation rates by 15-fold167. Interestingly, the spirocyclic cyclopropenes also exhibited reactivity with some tetrazines, and have been used to label cell-surface receptors in live cells168. Heteroatoms within the spirocycle further boosted cyclopropene reactivity by lowering LUMO energies169. Spirocyclic cyclopropenes have recently been outfitted with light-sensitive cages. Upon uncaging, the scaffolds become available for IEDDA ligation. These masked reagents can be used for biomolecule labeling with spatiotemporal control, but their syntheses remain challenging170,171.
Figure 5.
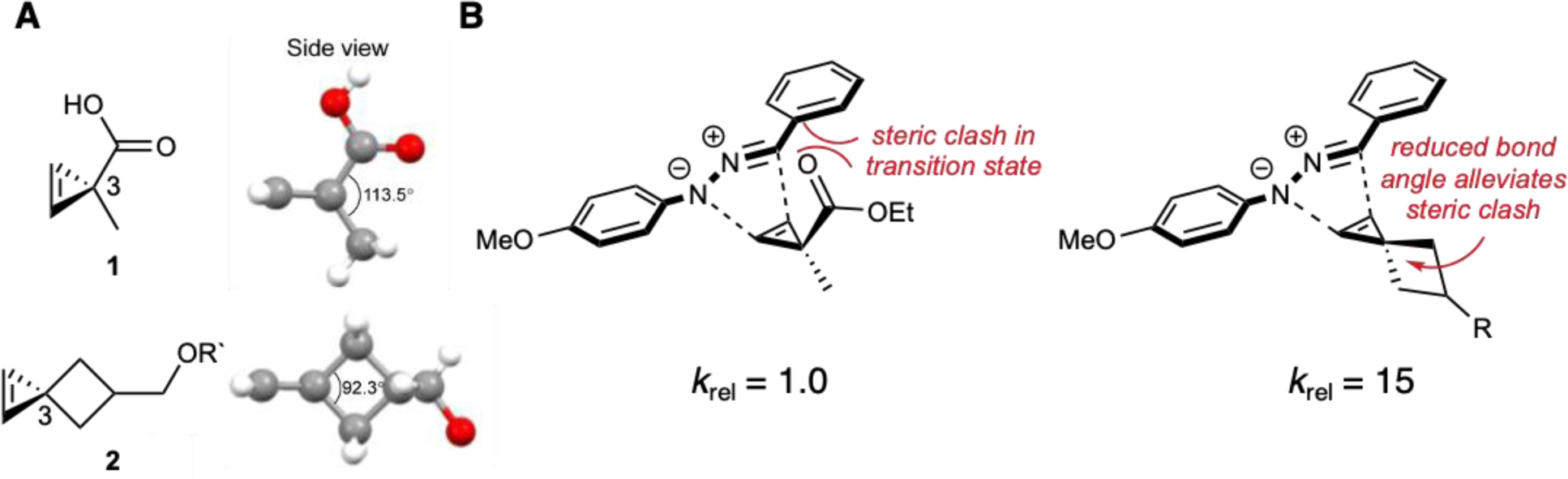
Steric modifications tune cyclopropene reactivity. (A) The angle between C3-substituents is reduced in spirocyclic cyclopropene 2 compared to parent compound 1. (B) The decreased bond angle minimizes steric clashes in the transition state, resulting in faster reactions with nitrile imines. Part A was adapted with permission from ref.167, The American Chemical Society.
Tetrazines have also been explored as reactants for other biocompatible cycloadditions. Recent work has featured transformations with isonitriles. These dienophiles react with tetrazines via [4+1] cycloaddition to provide imine products. With primary and secondary isonitriles, the imines undergo facile tautomerization and hydrolysis to liberate amines. Such isonitriles have proven useful as cages for amino fluorophores and small-molecule drugs172,173. Hydrolysis can be mitigated with appropriately tuned isonitriles174,175. For example, tertiary scaffolds react with tetrazines to form stable ligation adducts, as they cannot react further175. Because of their small size and versatility, isonitriles have been used to label biomolecules, including proteins176 and glycans177.
Simple vinyl groups have been shown to undergo IEDDA reactions with tetrazine probes. The small size of the vinyl motif is attractive for biomolecule tagging strategies. However, even with electron-rich vinyl reagents, most of the alkenes examined to date suffer from low aqueous solubility and slow rates178,179. Vinylboronic acids (VBAs) react significantly faster with tetrazines. The ligation liberates boronic acid, a major driving force for the reaction180. Even more rapid cycloadditions can be achieved with electron-rich VBAs and tetrazines outfitted with hydroxy groups. These latter groups coordinate the boronic acid motifs181,182. The VBA-tetrazine ligation has been used to profile the efficacy of proteasome inhibitors in live cells183. Many other cyclic184,185 and acyclic186 alkenes have been similarly explored as tetrazine ligation partners187.
Tuning dienes for bioorthogonal cycloaddition
The second half of the IEDDA reaction, the diene (most often, tetrazine), has also been thoroughly modified to achieve altered stability and reactivity profiles62,182,188–191. In one example, the stability of the tetrazine probe was modulated using electronic perturbations. An amino acid comprising a tetrazine motif was initially found to hydrolyze in the cellular milieu. The hydrolysis was facilitated by a labile secondary amine linkage. The electron-donating amine group also slowed IEDDA rates. Simple removal of the amine handle addressed both of these limitations.190 In another example, researchers synthesized a panel of tetrazines for amine uncaging with TCO. The tetrazines were screened with model enzymes bearing TCO-caged amino acids. The best tetrazines facilitated near quantitative uncaging in under 4 minutes with as little as 50 μM reagent62.
The rates that some tetrazine ligations achieve are unparalleled. Such rates, though, often come at the cost of probe stability. Many of the most reactive tetrazines are known to react with thiols, which can lead to off-target effects and high background labeling in cells192. To address this liability, some groups have drawn inspiration from caged bioorthogonal reagents. These efforts are focused on liberating the reactive tetrazine and take advantage of the redox properties of the motif. The reduced form, dihydrotetrazine, is unreactive towards dienophiles and stable in biological contexts. Dihydrotetrazines can then be oxidized to tetrazines in situ via enzymatic193, or photocatalytic193 approaches. Such strategies provide an avenue to employ even the most reactive tetrazines in biological environments. Caged tetrazines have also been employed to decorate electrode surfaces with biomolecules194.
The instability of some tetrazine motifs inspired pursuits of less electron-deficient dienes, including 1,2,4-triazines. Like its tetrazine counterpart, the triazine reacts via IEDDA with strained π-systems such as TCO and BCN195,196. While the rates of these reactions are markedly slower, the triazine is stable in the presence of biological thiols for over 24 hours at elevated pH. These scaffolds have since been installed in proteins195,197, as well as enzymatically appended onto DNA in vitro198,199. 1,2,4-Triazines have also been electronically tuned to achieve faster kinetic profiles200,201. For example, electron-withdrawing pyridinium groups have been used to improve cycloaddition rates. These scaffolds have been recently used to label mitochondria in live cells200. Subtle steric modifications to the triazine core also provided altered modes of reactivity with strained alkynes. In a recent study, the Distortion/Interaction model predicted that triazine substitutions at C3 and C6 would diminish reactivity with sterically encumbered strained alkynes202. Such reactions were predicted to remain facile with C5-substituted isomers, as steric clashes were minimized at the bond-forming centers (i.e., C3 and C6). The predictions were confirmed experimentally via simultaneous, dual labeling of two protein targets.
The number of dienes that can participate in IEDDA cycloadditions is also growing. A nitrogenous heterocycle, 4H-pyrazole, was recently shown to ligate the strained alkyne BCN203. At the outset, the pyrazole required further tuning to elicit robust reactivity. Addition of fluorine substituents was hypothesized to impart negative hyperconjugative effects on the pyrazole ring. This tuning would increase the antiaromatic character of the scaffold, resulting in destabilization of the pyrazole ring and more rapid ligation. Computational analyses verified that a gem-difluoro group decreased the LUMO values of the 4H-pyrazole, resulting in fast reactivity with BCN. Such antiaromaticity considerations along a reaction coordinate could be more generally exploited in bioorthogonal reaction design.
Another class of dienes comprises ortho-quinones. These motifs react with dienophiles via strain-promoted oxidation-controlled cyclooctyne–1,2-quinone (SPOCQ) cycloadditions204. Early iterations involved converting 1,2-catechols to the corresponding quinones using an exogenous oxidant205. SPOCQ reactions were orders of magnitude faster than azide-alkyne cycloadditions (second-order rate constants of ~500 M−1 s−1), on par with some tetrazine-TCO ligations. Later studies demonstrated that genetically encodable tyrosine tags could be selectively oxidized to quinones in situ using tyrosinase. These motifs could then be used to append small molecules for antibody-drug conjugate formation. The strategy enabled control over the location and number of warheads that were appended to the antibody206. ortho-Quinones were also found to react efficiently with strained alkenes, such as cyclopropenes207. However, the need to generate ortho-quinones in situ can impose constraints. ortho-Quinoline quinone methides were explored as more versatile alternatives, as these scaffolds can be generated in situ without any external triggers in biological environments. The motifs were found to react robustly with vinyl thioethers via hetero Diels–Alder cycloaddition. The unusual thioacetal adduct formed was found to be stable in aqueous solution at various pH values208,209.
IV. Combining mutually orthogonal reactions
As evident from above, the past decade has seen a surge in the number of transformations available for biological application. Despite the expanded toolkit, it remains challenging to apply more than one reaction at a time210. New reaction development has largely focused on labeling single targets. Identifying collections of compatible bioorthogonal chemistries would enable multicomponent labeling studies, and allow a broader set of biological processes to be examined. Finding such combinations of reactions has historically been challenging, as many popular reagents cross-react with one another. The search for orthogonal reactions has accelerated in recent years, aided by computational tools and reaction tuning.
Bioorthogonal reactions that feature unique mechanisms are well suited for multicomponent labeling studies. Reagents with distinct modes of reactivity can often mitigate cross-reactivity issues. For example, azide-alkyne cycloadditions can be used in tandem with hydrazine/ketone condensations211, various IEDDA reagents163,178,212–214, some 1,3-dipoles98,110, and other motifs180,215–217. Efforts to employ three mutually compatible bioorthogonal groups have also been pursued202,213,218–220. One recent example featured azide-, cyclopropene-, and alkyne-containing sugars to study the heterogeneity of glycan metabolism in plant cells219. The ligations could not be performed simultaneously, though, owing to cross-reactivities among the reaction partners. Cumbersome washes were also required between ligations, eroding temporal resolution. Only three studies to date have been able to achieve simultaneous triple labeling221–223. One notable example features two tetrazines, one that is sterically encumbered and reacts selectively with a small isonitrile. The second tetrazine ligates TCO in a typical IEDDA cycloaddition. The tetrazine reactions were combined with an azide/strained alkyne pair in a triple labeling experiment. Three model proteins, labeled with either a bulky tetrazine, a less encumbered tetrazine, or an azide were mixed and reacted with isonitrile-, TCO-, and strained-alkyne fluorophores. The matched adducts were detected by in-gel fluorescence, with no evidence of cross-reactivity (Figure 6)223. While this study showcased triple component labeling in a model context, the reagents should be applicable in other biological settings.
Figure 6.
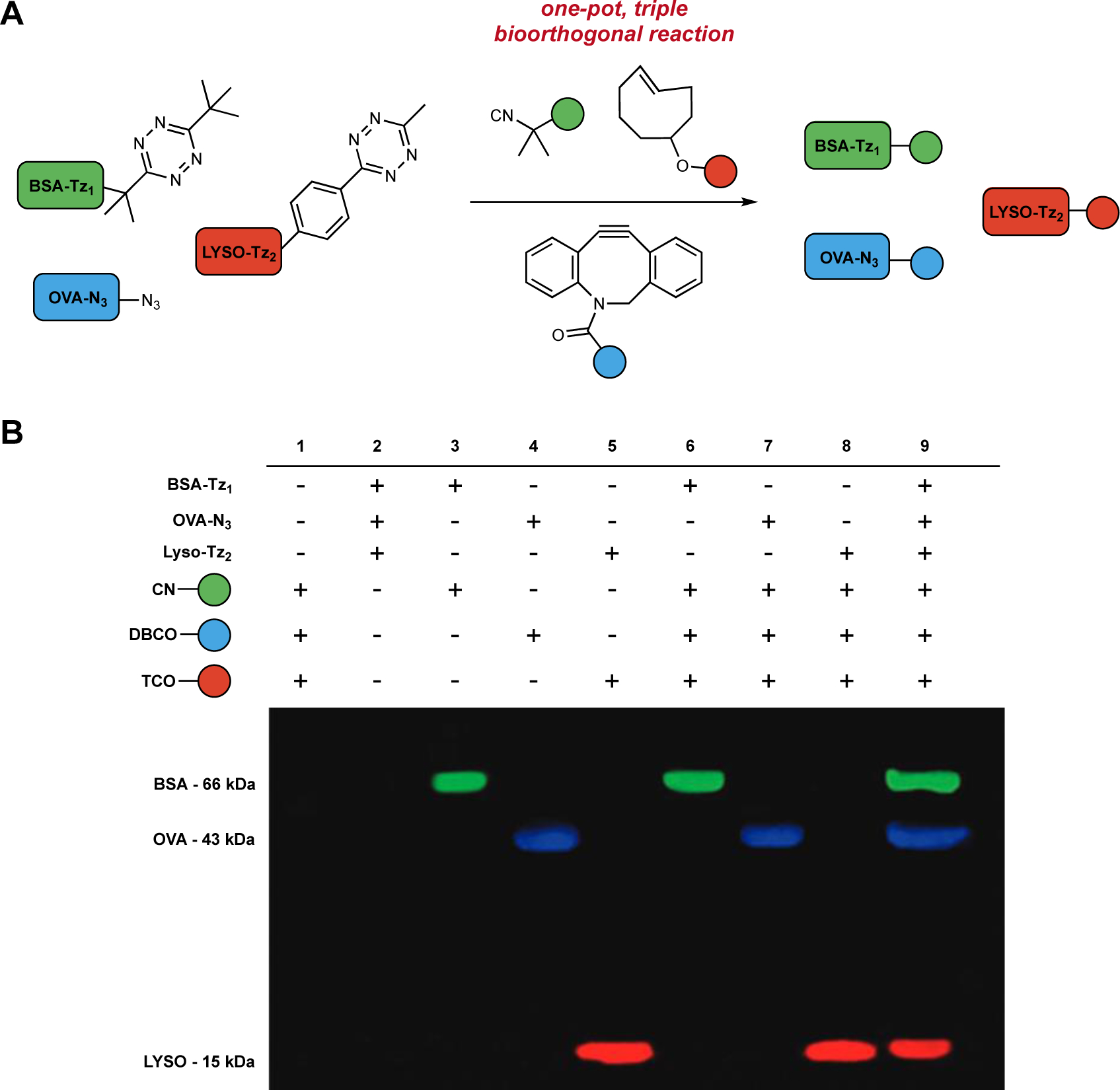
Collections of bioorthogonal chemistries for multicomponent labeling. (A) Three mechanistically distinct reactions – [4+1] tetrazine (Tz1)-isonitrile cycloaddition, [4+2] tetrazine (Tz2)-TCO cycloaddition, and [3+2] azide-alkyne cycloaddition – were used to simultaneously label three model proteins (BSA, OVA, and lysozyme). (B) Covalent protein adducts were visualized via gel electrophoresis followed by fluorescence scanning. No cross-reactivities were detected. Part B was adapted with permission from ref.223, Wiley-VCH.
V. Exploring new genres of reactivity
Identifying additional genres of bioorthogonal chemistry will continue to bolster multicomponent labeling studies. Recent efforts to explore new areas of chemical space – coupled with additional tuning of existing ligations – are proving fruitful. Boron reagents are gaining traction. In recent work, a diboron probe was found to react selectively with N-oxides. Importantly, the ligation proceeded efficiently inside cells, one of the harshest biological environments224. The unique mechanism of this reaction is further compatible with several existing bioorthogonal chemistries.
Perhaps the most noteworthy developments in polar reagent design involve sulfur(VI) fluorides. These motifs are remarkably stable in cellular environments, and react robustly with oxygen and nitrogen nucleophiles through sulfur(VI) fluoride exchange (SuFEx) chemistry225,226. SuFEx reagents have been used for drug design227,228, activity-based profiling229 and other protein targeting studies230. They have also been employed for examining protein-protein interactions via proximity-driven crosslinking231,232. SuFEx electrophiles also provide a mild and convenient method to introduce bioorthogonal azides onto a variety of amine targets, further exemplifying the utility of these motifs233.
Explorations into new cycloaddition platforms are also expanding the bioorthogonal toolkit. One example involves the quadricyclane ligation221. This reaction is a formal [2σ+2σ+2π] cycloaddition with Ni-bis(dithiolene) complexes, a rather unique mode of reactivity. Quadricyclanes are highly strained molecules (~80 kcal mol−1 of strain energy234), but can survive in aqueous conditions and in the presence of thiols for extended periods. Quadricyclanes have even been used for intracellular labeling applications via genetic code expansion235. The novelty of the quadricyclane reaction is likely to inspire continued exploration of other cycloaddition manifolds for bioorthogonal application.
Bioorthogonal platforms of reactivity have also expanded to include transition metals. These metals are virtually absent in the cellular milieu and can forge stable C-C bonds, making them attractive for selective labeling applications. Focused reviews on this topic have been covered recently, but a few examples are described below236,237. Water soluble Au(I) complexes have been developed to drive covalent bond formation in aqueous media238. Gold nanoparticles have also been employed for imaging in zebrafish239. More reactive Au(III) species can be harnessed for selective amidation reactions, although the metal complexes must be associated with scaffolding proteins240. Inspired by copper catalysis, several groups have optimized ligands to form water soluble, minimally toxic palladium complexes. Some have been used for Suzuki-Miyaura241,242 couplings in cells, along with Sonogashira reactions243,244, and alkynyl-carbamate deprotections245,246. Palladium nanoparticles247 and assemblies248 have also been developed for conducting various transformations in cellulo. Similar advances in ruthenium chemistry249–251 and iridium photocatalysis252 are enabling biomolecule targeting.
New developments in protein bioconjugation are also being leveraged for more general bioorthogonal application. One example features aryl diazonium ions253,254. These electrophiles react robustly with electron-rich aromatic side chains such as tyrosine. Site-specific modification with aryl diazoniums was historically challenging owing to the abundance of tyrosine residues in proteins. Further exploration revealed a selective reaction between 5-hydroxytryptophan (5HTP) and aryl diazonium reagents. The additional electron density in 5HTP enabled the use of less reactive diazonium reagents, preventing background labeling of other aromatic side chains. This reaction has since been used to label recombinant proteins and antibody derivatives255, and additional targets are anticipated. Further tuning of other protein-labeling strategies is likely to provide more bioorthogonal reagents256,257.
Conclusions
At its core, bioorthogonal chemistry is focused on controlling reactive functional groups in harsh environments. How does one design a pair of reagents to form a single adduct, in the confines of a cell or whole organism? The strict requirements imposed on these reactions have often forced researchers to explore unconventional handles. Historic work provided an initial set of reagents, many of which are still employed for examining biomolecules in vivo.
The toolbox has greatly expanded in recent years via iterative tuning of established probes. Some common themes from these studies have emerged. For example, the rates and stabilities of first-generation tools can be modulated for particular applications, drawing on common physical organic chemistry principles. The impacts of certain modifications can often be predicted computationally. Computational analyses can also be invaluable to the hunt for collections of mutually compatible transformations.
After decades of work on bioorthogonal chemistries, there is still no “one-size-fits-all” reaction. Rather, a spectrum of reactivities exists and the challenge lies in knowing how best to apply the probes. Recent successes in tuning highly reactive chemical handles suggest that other “fringe” functional groups can be harnessed for understanding biology. There is a further need for not just single-component reactions, but also collections of bioorthogonal chemistries that can be used in tandem210. The continued exploration of unique modes of reactivity256–260 will be useful in this regard. The strategies and examples highlighted in this Review provide a roadmap for continued expansion of the bioorthogonal toolkit, taking chemistries beyond flasks and into living systems.
Acknowledgements
S.S.N. is an Allergan Graduate Research Fellow. J.A.P. is a Cottrell Scholar, Alfred P. Sloan Fellow, and Dreyfus Scholar. This work was funded by the National Institutes of Health (R01 GM126226). We thank members of the Prescher laboratory for helpful discussions during the manuscript preparation.
References
- 1.Rodriguez EA et al. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci 42, 111–129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Row RD & Prescher JA Constructing New Bioorthogonal Reagents and Reactions. Acc. Chem. Res 51, 1073–1081 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian Y & Lin Q Fitness Factors for Bioorthogonal Chemical Probes. ACS Chem. Biol 14, 2489–2496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sletten EM & Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed 48, 6974–6998 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lang K & Chin JW Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev 114, 4764–4806 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Carrell T et al. in Cycloadditions in Bioorthogonal Chemistry. (eds. Vrabel M Carell T), 1–157 (Springer International Publishing, Switzerland, 2016). [Google Scholar]
- 7.Andresen M, Schmitz-Salue R & Jakobs S Short Tetracysteine Tags to β-Tubulin Demonstrate the Significance of Small Labels for Live Cell Imaging. Mol. Biol. Cell 15, 5616–5622 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grammel M & Hang HC Chemical reporters for biological discovery. Nat. Chem. Biol 9, 475–484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chai Q-Y, Yang Z, Lin H-W & Han B-N Alkynyl-Containing Peptides of Marine Origin. Mar. Drugs 14, 216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jewett JC & Bertozzi CR Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev 39, 1272–1279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Selvaraj R & Fox JM trans-Cyclooctene – a stable, voracious dienophile for bioorthogonal labeling. Curr. Opin. Chem. Biol 17, 753–760 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dommerholt J, Rutjes FPJT & van Delft FL Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem 374, 16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kölmel DK & Kool ET Oximes and Hydrazones in Bioconjugation: Mechanism and Catalysis. Chem. Rev 117, 10358–10376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahal LK, Yarema KJ & Bertozzi CR Engineering Chemical Reactivity on Cell Surfaces Through Oligosaccharide Biosynthesis. Science 276, 1125–1128 (1997). [DOI] [PubMed] [Google Scholar]
- 15.Datta D, Wang P, Carrico IS, Mayo SL & Tirrell DA A Designed Phenylalanyl-tRNA Synthetase Variant Allows Efficient in Vivo Incorporation of Aryl Ketone Functionality into Proteins. J. Am. Chem. Soc 124, 5652–5653 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Rabuka D, Rush JS, deHart GW, Wu P & Bertozzi CR Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc 7, 1052–1067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardisty RE, Kawasaki F, Sahakyan AB & Balasubramanian S Selective Chemical Labeling of Natural T Modifications in DNA. J. Am. Chem. Soc 137, 9270–9272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dirksen A, Dirksen S, Hackeng TM & Dawson PE Nucleophilic Catalysis of Hydrazone Formation and Transimination: Implications for Dynamic Covalent Chemistry. J. Am. Chem. Soc 128, 15602–15603 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Dirksen A, Hackeng TM & Dawson PE Nucleophilic Catalysis of Oxime Ligation. Angew. Chem. Int. Ed 45, 7581–7584 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Dirksen A & Dawson PE Rapid Oxime and Hydrazone Ligations with Aromatic Aldehydes for Biomolecular Labeling. Bioconjugate Chem. 19, 2543–2548 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsen D et al. Exceptionally rapid oxime and hydrazone formation promoted by catalytic amine buffers with low toxicity. Chem. Sci 9, 5252–5259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxon E & Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 287, 2007–2010 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Kiick KL, Saxon E, Tirrell DA & Bertozzi CR Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. U.S.A 99, 19–24 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saxon E et al. Investigating Cellular Metabolism of Synthetic Azidosugars with the Staudinger Ligation. J. Am. Chem. Soc 124, 14893–14902 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Cai J, Li X, Yue X & Taylor JS Nucleic Acid-Triggered Fluorescent Probe Activation by the Staudinger Reaction. J. Am. Chem. Soc 126, 16324–16325 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Hannoush RN & Sun J The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat. Chem. Biol 6, 498–506 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Chuh KN & Pratt MR Chemical methods for the proteome-wide identification of posttranslationally modified proteins. Curr. Opin. Chem. Biol 24, 27–37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prescher JA, Dube DH & Bertozzi CR Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Meldal M & Tornøe CW Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev 108, 2952–3015 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Thirumurugan P, Matosiuk D & Jozwiak K Click Chemistry for Drug Development and Diverse Chemical−Biology Applications. Chem. Rev 113, 4905–4979 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Lampkowski JS, Villa JK, Young TS & Young DD Development and Optimization of Glaser–Hay Bioconjugations. Angew. Chem. Int. Ed 54, 9343–9346 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Gaetke LM & Chow CK Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 189, 147–163 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Soares EV, Hebbelinck K & Soares HMVM Toxic effects caused by heavy metals in the yeast Saccharomyces cerevisiae: a comparative study. Can. J. Microbiol 49, 336–343 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Kennedy DC et al. Cellular Consequences of Copper Complexes Used To Catalyze Bioorthogonal Click Reactions. J. Am. Chem. Soc 133, 17993–18001 (2011). [DOI] [PubMed] [Google Scholar]
- 35.del Amo DS et al. Biocompatible Copper(I) Catalysts for in Vivo Imaging of Glycans. J. Am. Chem. Soc 132, 16893–16899 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Besanceney-Webler C et al. Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: A Comparative Study. Angew. Chem. Int. Ed 50, 8051–8056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uttamapinant C et al. Fast, Cell-Compatible Click Chemistry with Copper-Chelating Azides for Biomolecular Labeling. Angew. Chem. Int. Ed 51, 5852–5856 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang M et al. Biocompatible click chemistry enabled compartment-specific pH measurement inside E. coli. Nat. Commun 5, 4981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wittig G & Krebs A Zur Existenz niedergliedriger Cycloalkine, I. Chem. Ber 94, 3260–3275 (1961). [Google Scholar]
- 40.Agard NJ, Prescher JA & Bertozzi CR A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc 126, 15046–15047 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Rydahl MG, Hansen AR, Kračun SK & Mravec J Report on the Current Inventory of the Toolbox for Plant Cell Wall Analysis: Proteinaceous and Small Molecular Probes. Front. Plant Sci 9, 581 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuet KP et al. Cell-specific proteomic analysis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A 112, 2705–2710 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.tom Dieck S et al. Direct visualization of newly synthesized target proteins in situ. Nat. Methods 12, 411–414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ernst RJ et al. Genetic code expansion in the mouse brain. Nat. Chem. Biol 12, 776–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez-Castelao B et al. Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat. Biotechnol 35, 1196–1201 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Krogager TP et al. Labeling and identifying cell-type-specific proteomes in the mouse brain. Nat. Biotechnol 36, 156–159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Geel R, Prujin GJM, van Delft FL & Boelens WC Preventing Thiol-Yne Addition Improves the Specificity of Strain-Promoted Azide−Alkyne Cycloaddition. Bioconjugate Chem. 23, 392–398 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Blackman ML, Royzen M & Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc 130, 13518–13519 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor MT, Blackman ML, Dmitrenko O & Fox JM Design and Synthesis of Highly Reactive Dienophiles for the Tetrazine–trans-Cyclooctene Ligation. J. Am. Chem. Soc 133, 9646–9649 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Devaraj NK, Upadhyay R, Haun JB, Hilderbrand SA & Weissleder R Fast and Sensitive Pretargeted Labeling of Cancer Cells through a Tetrazine/trans‐Cyclooctene Cycloaddition. Angew. Chem. Int. Ed 48, 7013–7016 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang M et al. Conformationally Strained trans-Cyclooctene (sTCO) Enables the Rapid Construction of 18F-PET Probes via Tetrazine Ligation. Theranostics 6, 887–895 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Billaud EMF et al. Pretargeted PET Imaging Using a Bioorthogonal 18F‑Labeled trans-Cyclooctene in an Ovarian Carcinoma Model. Bioconjugate Chem. 28, 2915–2920 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Rossin R et al. Highly Reactive trans-Cyclooctene Tags with Improved Stability for Diels–Alder Chemistry in Living Systems. Bioconjugate Chem. 24, 1210–1217 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Läppchen T et al. DOTA-tetrazine probes with modified linkers for tumor pretargeting. Nucl. Med. Biol 55, 19–26 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Bilton HA, Ahmad Z, Janzen N, Czorny S & Valliant JF Preparation and Evaluation of 99mTc-labeled Tridentate Chelates for Pre-targeting Using Bioorthogonal Chemistry. J. Vis. Exp 120, e55188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Versteegen RM, Rossin R, ten Hoeve W, Janssen HM & Robillard MS Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem. Int. Ed 52, 14112–14116 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Rossin R et al. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun 9, 1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Versteegen RM et al. Click-to-Release from trans-Cyclooctenes: Mechanistic Insights and Expansion of Scope from Established Carbamate to Remarkable Ether Cleavage. Angew. Chem. Int. Ed 57, 10494–10499 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Mejia Oneto JM, Khan I, Seebald L & Royzen M In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug To Treat Soft Tissue Sarcoma. ACS Cent. Sci 2, 476–482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang G et al. Bioorthogonal Chemical Activation of Kinases in Living Systems. ACS Cent. Sci 2, 325–331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van der Gracht AMF et al. Chemical Control over T-Cell Activation in Vivo Using Deprotection of trans-Cyclooctene-Modified Epitopes. ACS Chem. Biol 13, 1569–1576 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fan X et al. Optimized Tetrazine Derivatives for Rapid Bioorthogonal Decaging in Living Cells. Angew. Chem. Int. Ed 55, 14046–14050 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Schmidt P, Zhou L, Tishinov K, Zimmerman K & Gillingham D Dialdehydes Lead to Exceptionally Fast Bioconjugations at Neutral pH by Virtue of a Cyclic Intermediate. Angew. Chem. Int. Ed 53, 10928–10931 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Dilek O, Lei Z, Mukherjee K & Bane S Rapid formation of a stable boron–nitrogen heterocycle in dilute, neutral aqueous solution for bioorthogonal coupling reactions. Chem. Commun 51, 16992–16995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmidt P, Stress C & Gillingham D Boronic acids facilitate rapid oxime condensations at neutral pH. Chem. Sci 6, 3329–3333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meadows MK, Roesner EK, Lynch VM, James TD & Anslyn EV Boronic Acid Mediated Coupling of Catechols and N-Hydroxylamines: A Bioorthogonal Reaction to Label Peptides. Org. Lett 19, 3179–3182 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Bandyopadhyay A, Cambray S & Gao J Fast Diazaborine Formation of Semicarbazide Enables Facile Labeling of Bacterial Pathogens. J. Am. Chem. Soc 139, 871–878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Akgun B et al. Synergic “Click” Boronate/Thiosemicarbazone System for Fast and Irreversible Bioorthogonal Conjugation in Live Cells. J. Am. Chem. Soc 139, 14285–14291 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Gu H et al. Formation of hydrazones and stabilized boron–nitrogen heterocycles in aqueous solution from carbohydrazides and ortho-formylphenylboronic acids. Org. Biomol. Chem 15, 7543–7548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bandyopadhyay A, Cambray S & Gao J Fast and selective labeling of N-terminal cysteines at neutral pH via thiazolidino boronate formation. Chem. Sci 7, 4589–4593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Faustino H, Silva MJSA, Veiros LF, Bernardes GJL & Gois PMP Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalisation. Chem. Sci 7, 5052–5058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sundhoro M, Jeon S, Park J, Ramström O & Yan M Perfluoroaryl Azide Staudinger Reaction: A Fast and Bioorthogonal Reaction. Angew. Chem. Int. Ed 56, 12117–12121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meguro T et al. Staudinger reaction using 2,6-dichlorophenyl azide derivatives for robust aza-ylide formation applicable to bioconjugation in living cells. Chem. Commun 54, 7904–7907 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Saneyoshi H et al. Triphenylphosphinecarboxamide: An Effective Reagent for the Reduction of Azides and Its Application to Nucleic Acid Detection. Org. Lett 16, 30–33 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Luo J, Liu Q, Morihiro K & Deiters A Small-molecule control of protein function through Staudinger reduction. Nat. Chem 8, 1027–1034 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lukasak B, Morihiro K & Deiters A Aryl Azides as Phosphine-Activated Switches for Small Molecule Function. Sci. Rep 9, 1470 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ren G, Zheng Q & Wang H Aryl Fluorosulfate Trapped Staudinger Reduction. Org. Lett 19, 1582–1585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Böhrsch V, Serwa R, Majkut P, Krause E & Hackenberger CPR Site-specific functionalisation of proteins by a Staudinger-type reaction using unsymmetrical phosphites. Chem. Commun 46, 3176–3178 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Serwa R et al. Site-specific PEGylation of proteins by a Staudinger-phosphite reaction. Chem. Sci 1, 596–602 (2010). [Google Scholar]
- 80.Nischan N, Kasper M-A, Mathew T & Hackenberger CPR Bis(arylmethyl)-substituted unsymmetrical phosphites for the synthesis of lipidated peptides via Staudinger-phosphite reactions. Org. Biomol. Chem 14, 7500–7508 (2016). [DOI] [PubMed] [Google Scholar]
- 81.Vallée MRJ et al. Staudinger-Phosphonite Reactions for the Chemoselective Transformation of Azido-Containing Peptides and Proteins. Org. Lett 13, 5440–5443 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Kasper M-A et al. Vinylphosphonites for Staudinger-induced chemoselective peptide cyclization and functionalization. Chem. Sci 10, 6322–6329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kasper M-A et al. Cysteine-Selective Phosphonamidate Electrophiles for Modular Protein Bioconjugations. Angew. Chem. Int. Ed 58, 11625–11630 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Lee Y-J, Kurra Y & Liu WR Phospha-Michael Addition as a New Click Reaction for Protein Functionalization. ChemBioChem 17, 456–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bos J & Muir TW A Chemical Probe for Protein Crotonylation. J. Am. Chem. Soc 140, 4757–4760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shih H-W & Prescher JA A Bioorthogonal Ligation of Cyclopropenones Mediated by Triarylphosphines. J. Am. Chem. Soc 137, 10036–10039 (2015). [DOI] [PubMed] [Google Scholar]
- 87.Row RD, Shih H-W, Alexander AT, Mehl RA & Prescher JA Cyclopropenones for Metabolic Targeting and Sequential Bioorthogonal Labeling. J. Am. Chem. Soc 139, 7370–7375 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Row RD & Prescher JA A Cyclopropenethione-Phosphine Ligation for Biomolecule Labeling. Org. Lett 20, 5614–5617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heiss TK & Prescher JA Cyclopropeniminium Ions Exhibit Unique Reactivity Profiles with Bioorthogonal Phosphines. J. Org. Chem 84, 7443–7448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Newberry RW, VanVeller B, Guzei IA & Raines RT n→π* Interactions of Amides and Thioamides: Implications for Protein Stability. J. Am. Chem. Soc 135, 7843–7846 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walters CR et al. The effects of thioamide backbone substitution on protein stability: a study in α-helical, β-sheet, and polyproline II helical contexts. Chem. Sci 8, 2868–2877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McKay CS, Moran J & Pezacki JP Nitrones as dipoles for rapid strain-promoted 1,3-dipolar cycloadditions with cyclooctynes. Chem. Commun 46, 931–933 (2010). [DOI] [PubMed] [Google Scholar]
- 93.McKay CS, Chigrinova M, Blake JA & Pezacki JP Kinetics studies of rapid strain-promoted [3 + 2]-cycloadditions of nitrones with biaryl-aza-cyclooctynone. Org. Biomol. Chem 10, 3066–3070 (2012). [DOI] [PubMed] [Google Scholar]
- 94.MacKenzie DA & Pezacki JP Kinetics studies of rapid strain-promoted [3+2] cycloadditions of nitrones with bicyclo[6.1.0]nonyne. Can. J. Chem 92, 337–340 (2014). [Google Scholar]
- 95.Gunawardene PN, Luo W, Polgar AM, Corrigan JF & Workentin MS Highly Electron-Deficient Pyridinium-Nitrones for Rapid and Tunable Inverse-Electron-Demand Strain-Promoted Alkyne-Nitrone Cycloaddition. Org. Lett 21, 5547–5551 (2019). [DOI] [PubMed] [Google Scholar]
- 96.McKay CS, Blake JA, Cheng J, Danielson DC & Pezacki JP Strain-promoted cycloadditions of cyclic nitrones with cyclooctynes for labeling human cancer cells. Chem. Commun 47, 10040–10042 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Sherratt AR et al. Copper-catalysed cycloaddition reactions of nitrones and alkynes for bioorthogonal labelling of living cells. RSC Adv. 4, 46966–46969 (2014). [Google Scholar]
- 98.Sherratt AR et al. Dual Strain-Promoted Alkyne–Nitrone Cycloadditions for Simultaneous Labeling of Bacterial Peptidoglycans. Bioconjugate Chem. 27, 1222–1226 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Decuypère E, Plougastel L, Audisio D & Taran F Sydnone–alkyne cycloaddition: applications in synthesis and bioconjugation. Chem. Commun 53, 11515–11527 (2017). [DOI] [PubMed] [Google Scholar]
- 100.Kolodych S et al. Discovery of Chemoselective and Biocompatible Reactions Using a High-Throughput Immunoassay Screening. Angew. Chem. Int. Ed 52, 12056–12060 (2013). [DOI] [PubMed] [Google Scholar]
- 101.Wallace S & Chin JW Strain-promoted sydnone bicyclo-[6.1.0]-nonyne cycloaddition. Chem. Sci 5, 1742–1744 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Plougastel L et al. 4-Halogeno-sydnones for fast strain promoted cycloaddition with bicyclo-[6.1.0]-nonyne. Chem. Commun 50, 9376–9378 (2014). [DOI] [PubMed] [Google Scholar]
- 103.Liu H et al. Ultrafast Click Chemistry with Fluorosydnones. Angew. Chem. Int. Ed 55, 12073–12077 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Mix KA, Aronoff MR & Raines RT Diazo Compounds: Versatile Tools for Chemical Biology. ACS Chem. Biol 11, 3233–3244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moran J, McKay CS & Pezacki JP Strain-promoted 1,3-dipolar cycloadditions of diazo compounds with cyclooctynes. Can. J. Chem 89, 148–151 (2011). [DOI] [PubMed] [Google Scholar]
- 106.Sanders BC et al. Metal-Free Sequential [3 + 2]-Dipolar Cycloadditions using Cyclooctynes and 1,3-Dipoles of Different Reactivity. J. Am. Chem. Soc 133, 949–957 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Josa-Culleré L, Wainman YA, Brindle KM & Leeper FJ Diazo group as a new chemical reporter for bioorthogonal labelling of biomolecules. RSC Adv. 4, 52241–52244 (2014). [Google Scholar]
- 108.Anderson KA, Aronoff MR, McGrath NA & Raines RT Diazo Groups Endure Metabolism and Enable Chemoselectivity in Cellulo. J. Am. Chem. Soc 137, 2412–2415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McGrath NA & Raines RT Diazo compounds as highly tunable reactants in 1,3-dipolar cycloaddition reactions with cycloalkynes. Chem. Sci 3, 3237–3240 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aronoff MR, Gold B & Raines RT 1,3-Dipolar Cycloadditions of Diazo Compounds in the Presence of Azides. Org. Lett 18, 1538–1541 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lim RKV & Lin Q Azirine ligation: fast and selective protein conjugation via photoinduced azirine–alkene cycloaddition. Chem. Commun 46, 7993–7995 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.King M & Wagner A Developments in the Field of Bioorthogonal Bond Forming Reactions—Past and Present Trends. Bioconjugate Chem. 25, 825–839 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Schäfer RJB et al. The Bioorthogonal Isonitrile–Chlorooxime Ligation. J. Am. Chem. Soc 141, 18644–18648 (2019). [DOI] [PubMed] [Google Scholar]
- 114.Yu Z, Lim RKV & Lin Q Synthesis of Macrocyclic Tetrazoles for Rapid Photoinduced Bioorthogonal 1,3‐Dipolar Cycloaddition Reactions. Chem. Eur. J 16, 13325–13329 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu Z, Pan Y, Wang Z, Wang J & Lin Q Genetically Encoded Cyclopropene Directs Rapid, Photoclick‐Chemistry‐Mediated Protein Labeling in Mammalian Cells. Angew. Chem. Int. Ed 51, 10600–10604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kamber DN et al. Isomeric Cyclopropenes Exhibit Unique Bioorthogonal Reactivities. J. Am. Chem. Soc 135, 13680–13683 (2013). [DOI] [PubMed] [Google Scholar]
- 117.An P, Yu Z & Lin Q Design of oligothiophene-based tetrazoles for laser-triggered photoclick chemistry in living cells. Chem. Commun 49, 9920–9922 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu Z, Ohulchanskyy TY, An P, Prasad PN & Lin Q Fluorogenic, Two-Photon-Triggered Photoclick Chemistry in Live Mammalian Cells. J. Am. Chem. Soc 135, 16766–16769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tasdelen MA & Yagci Y Light‐Induced Click Reactions. Angew. Chem. Int. Ed 52, 5930–5938 (2013). [DOI] [PubMed] [Google Scholar]
- 120.Li J et al. Visible Light-Initiated Bioorthogonal Photoclick Cycloaddition. J. Am. Chem. Soc 140, 14542–14546 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Zhang X et al. Photo-accelerated “click” reaction between diarylsydnones and ring-strained alkynes for bioorthogonal ligation. Chem. Commun 55, 7187–7190 (2019). [DOI] [PubMed] [Google Scholar]
- 122.Lim RKV & Lin Q Photoinducible Bioorthogonal Chemistry: A Spatiotemporally Controllable Tool to Visualize and Perturb Proteins in Live Cells. Acc. Chem. Res 44, 828–839 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.An P, Lewandowski TM, Erbay TG, Liu P & Lin Q Sterically Shielded, Stabilized Nitrile Imine for Rapid Bioorthogonal Protein Labeling in Live Cells. J. Am. Chem. Soc 140, 4860–4868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.de Almeida G, Sletten EM, Nakamura H, Palaniappan KK & Bertozzi CR Thiacycloalkynes for Copper-Free Click Chemistry. Angew. Chem. Int. Ed 51, 2443–2447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.King M, Baati R & Wagner A New tetramethylthiepinium (TMTI) for copper-free click chemistry. Chem. Commun 48, 9308–9309 (2012). [DOI] [PubMed] [Google Scholar]
- 126.de Almeida G, Townsend LC & Bertozzi CR Synthesis and Reactivity of Dibenzoselenacycloheptynes. Org. Lett 15, 3038–3041 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ning X, Guo J, Wolfert MA & Boons G-J Visualizing Metabolically Labeled Glycoconjugates of Living Cells by Copper‐Free and Fast Huisgen Cycloadditions. Angew. Chem. Int. Ed 47, 2253–2255 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stöckmann H et al. Development and evaluation of new cyclooctynes for cell surface glycan imaging in cancer cells. Chem. Sci 2, 932–936 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Friscourt F et al. Polar Dibenzocyclooctynes for Selective Labeling of Extracellular Glycoconjugates of Living Cells. J. Am. Chem. Soc 134, 5381–5389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Varga BR, Kállay M, Hegyi K, Béni S & Kele P A Non‐Fluorinated Monobenzocyclooctyne for Rapid Copper‐Free Click Reactions. Chem. Eur. J 18, 822–828 (2012). [DOI] [PubMed] [Google Scholar]
- 131.Dommerholt J et al. Readily Accessible Bicyclononynes for Bioorthogonal Labeling and Three‐Dimensional Imaging of Living Cells. Angew. Chem. Int. Ed 49, 9422–9425 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Agard NJ, Baskin JM, Prescher JA, Lo A & Bertozzi CR A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol 1, 644–648 (2006). [DOI] [PubMed] [Google Scholar]
- 133.Baskin JM et al. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A 104, 16793–16797 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sletten EM, Nakamura H, Jewett JC & Bertozzi CR Difluorobenzocyclooctyne: Synthesis, Reactivity, and Stabilization by β-Cyclodextrin. J. Am. Chem. Soc 132, 11799–11805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li W et al. Fluorodibenzocyclooctynes: A Trackable Click Reagent with Enhanced Reactivity. Chem. Eur. J 25, 10328–10332 (2019). [DOI] [PubMed] [Google Scholar]
- 136.Sletten EM & Bertozzi CR A Hydrophilic Azacyclooctyne for Cu-Free Click Chemistry. Org. Lett 10, 3097–3099 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jewett JC, Sletten EM & Bertozzi CR Rapid Cu-Free Click Chemistry with Readily Synthesized Biarylazacyclooctynones. J. Am. Chem. Soc 132, 3688–3690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Debets MF et al. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem. Commun 46, 97–99 (2010). [DOI] [PubMed] [Google Scholar]
- 139.Gold B, Shevchenko NE, Bonus N, Dudley GB & Alabugin IV Selective Transition State Stabilization via Hyperconjugative and Conjugative Assistance: Stereoelectronic Concept for Copper-Free Click Chemistry. J. Org. Chem 77, 75–89 (2012). [DOI] [PubMed] [Google Scholar]
- 140.Gold B, Batsomboon P, Dudley GB & Alabugin IV Alkynyl Crown Ethers as a Scaffold for Hyperconjugative Assistance in Noncatalyzed Azide–Alkyne Click Reactions: Ion Sensing through Enhanced Transition-State Stabilization. J. Org. Chem 79, 6221–6232 (2014). [DOI] [PubMed] [Google Scholar]
- 141.Kaneda K, Naruse R & Yamamoto S 2‑Aminobenzenesulfonamide-Containing Cyclononyne as Adjustable Click Reagent for Strain-Promoted Azide−Alkyne Cycloaddition. Org. Lett 19, 1096–1099 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Burke EG, Gold B, Hoang TT, Raines RT & Schomaker JM Fine-Tuning Strain and Electronic Activation of Strain-Promoted 1,3-Dipolar Cycloadditions with Endocyclic Sulfamates in SNO-OCTs. J. Am. Chem. Soc 139, 8029–8037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ni R, Mitsuda N, Kashiwagi T, Igawa K & Tomooka K Heteroatom-embedded Medium-Sized Cycloalkynes: Concise Synthesis, Structural Analysis, and Reactions. Angew. Chem. Int. Ed 54, 1190–1194 (2015). [DOI] [PubMed] [Google Scholar]
- 144.Del Grosso A et al. Strained alkynes derived from 2,2’-dihydroxy-1,1’-biaryls; synthesis and copper-free cycloaddition with azides. Org. Biomol. Chem 15, 4517–4521 (2017). [DOI] [PubMed] [Google Scholar]
- 145.Mistry A et al. Synthesis and cycloaddition reactions of strained alkynes derived from 2,2′-dihydroxy-1,1′-biaryls. Org. Biomol. Chem 16, 8965–8975 (2018). [DOI] [PubMed] [Google Scholar]
- 146.Harris T et al. Twisted Cycloalkynes and Remote Activation of “Click” Reactivity. Chem 3, 629–640 (2017). [Google Scholar]
- 147.Meier H, Schuh-Popitz C & Peiersen H Isolation of a Highly Strained Bicyclic Alkyne. Angew. Chem. Int. Ed 20, 270–271 (1981). [Google Scholar]
- 148.Ho SH & Tirrell DA Enzymatic Labeling of Bacterial Proteins for Super-resolution Imaging in Live Cells. ACS Cent. Sci 5, 1911–1919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu F, Liang Y & Houk KN Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities. Acc. Chem. Res 50, 2297–2308 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bickelhaupt FM & Houk KN Analyzing Reaction Rates with the Distortion/Interaction‐Activation Strain Model. Angew. Chem. Int. Ed 56, 10070–10086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gordon CG et al. Reactivity of Biarylazacyclooctynones in Copper-Free Click Chemistry. J. Am. Chem. Soc 134, 9199–9208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tao H et al. Origins of halogen effects in bioorthogonal sydnone cycloadditions. Chem. Commun 54, 5082–5085 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liu F, Paton RS, Kim S, Liang Y & Houk KN Diels–Alder reactivities of strained and unstrained cycloalkenes with normal and inverse-electron-demand dienes: Activation barriers and distortion/interaction analysis. J. Am. Chem. Soc 135, 15642–15649 (2013). [DOI] [PubMed] [Google Scholar]
- 154.Liu F, Liang Y & Houk KN Theoretical elucidation of the origins of substituent and strain effects on the rates of Diels–Alder reactions of 1,2,4,5-tetrazines. J. Am. Chem. Soc 136, 11483–11493 (2014). [DOI] [PubMed] [Google Scholar]
- 155.Darko A et al. Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation. Chem. Sci 5, 3770–3776 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kozma E et al. Hydrophilic trans-Cyclooctenylated Noncanonical Amino Acids for Fast Intracellular Protein Labeling. ChemBioChem 17, 1518–1524 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Knorr G, Kozma E, Herner A, Lemke EA & Kele P New Red-Emitting Tetrazine-Phenoxazine Fluorogenic Labels for Live-Cell Intracellular Bioorthogonal Labeling Schemes. Chem. Eur. J 22, 8972–8979 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Lambert WD et al. Computationally guided discovery of a reactive, hydrophilic trans-5-oxocene dienophile for bioorthogonal labeling. Org. Biomol. Chem 15, 6640–6644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Siegl SJ et al. Design and Synthesis of Aza-Bicyclononene Dienophiles for Rapid Fluorogenic Ligations. Chem. Eur. J 24, 2426–2432 (2018). [DOI] [PubMed] [Google Scholar]
- 160.Bach RD Ring Strain Energy in the Cyclooctyl System. The Effect of Strain Energy on [3 + 2] Cycloaddition Reactions with Azides. J. Am. Chem. Soc 131, 5233–5243 (2009). [DOI] [PubMed] [Google Scholar]
- 161.Santucci J III, Sanzone JR & Woerpel KA [4+2] Cycloadditions of Seven‐Membered‐Ring trans‐Alkenes: Decreasing Reactivity with Increasing Substitution of the Seven‐Membered Ring. Eur. J. Org. Chem 2016, 2933–2943 (2016). [Google Scholar]
- 162.Fang Y et al. Photochemical syntheses, transformations, and bioorthogonal chemistry of trans-cycloheptene and sila trans-cycloheptene Ag(I) complexes. Chem. Sci 9, 1953–1963 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Patterson DM, Nazarova LA, Xie B, Kamber DN & Prescher JA Functionalized Cyclopropenes as Bioorthogonal Chemical Reporters. J. Am. Chem. Soc 134, 18638–18643 (2012). [DOI] [PubMed] [Google Scholar]
- 164.Yang J, Šečkutė J, Cole CM & Devaraj NK Live-Cell Imaging of Cyclopropene Tags with Fluorogenic Tetrazine Cycloadditions. Angew. Chem. Int. Ed 51, 7476–7479 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ravasco JMJM, Monteiro CM & Trindade AF Cyclopropenes: a new tool for the study of biological systems. Org. Chem. Front 4, 1167–1198 (2017). [Google Scholar]
- 166.Elliot TS et al. Proteome labeling and protein identification in specific tissues and at specific developmental stages in an animal. Nat. Biotechnol 32, 465–472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yu Z & Lin Q Design of Spiro[2.3]hex-1-ene, a Genetically Encodable Double-Strained Alkene for Superfast Photoclick Chemistry. J. Am. Chem. Soc 136, 4153–4156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ramil CP et al. Spirohexene-Tetrazine Ligation Enables Bioorthogonal Labeling of Class B G Protein-Coupled Receptors in Live Cells. J. Am. Chem. Soc 139, 13376–13386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.An P, Wu H-Y, Lewandowski TM & Lin Q Hydrophilic azaspiroalkenes as robust bioorthogonal reporters. Chem. Commun 54, 14005–14008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kumar P, Jiang T, Li S, Zainul O & Laughlin ST Caged cyclopropenes for controlling bioorthogonal reactivity. Org. Biomol. Chem 16, 4081–4085 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Kumar P et al. Caged Cyclopropenes with Improved Tetrazine Ligation Kinetics. Org. Lett 21, 3721–3725 (2019). [DOI] [PubMed] [Google Scholar]
- 172.Tu J, Xu M, Parvez S, Peterson RT & Franzini RM Bioorthogonal Removal of 3‑Isocyanopropyl Groups Enables the Controlled Release of Fluorophores and Drugs in Vivo. J. Am. Chem. Soc 140, 8410–8414 (2018). [DOI] [PubMed] [Google Scholar]
- 173.Tu J et al. Isonitrile-responsive and bioorthogonally removable tetrazine protecting groups. Chem. Sci 11, 169–179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xu M, Deb T, Tu J & Franzini RM Tuning Isonitrile/Tetrazine Chemistry for Accelerated Deprotection and Formation of Stable Conjugates. J. Org. Chem 84, 15520–15529 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Stöckmann H, Neves AA, Stairs S, Brindle KM & Leeper FJ Exploring isonitrile-based click chemistry for ligation with biomolecules. Org. Biomol. Chem 9, 7303–7305 (2011). [DOI] [PubMed] [Google Scholar]
- 176.Chen Y et al. Addition of Isocyanide-containing Amino Acids to the Genetic Code for Protein Labeling and Activation. ACS Chem. Biol 14, 2793–2799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stairs S et al. Metabolic Glycan Imaging by Isonitrile–Tetrazine Click Chemistry. ChemBioChem 14, 1063–1067 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Niederwieser A et al. Two-Color Glycan Labeling of Live Cells by a Combination of Diels–Alder and Click Chemistry. Angew. Chem. Int. Ed 52, 4265–4268 (2013). [DOI] [PubMed] [Google Scholar]
- 179.Lee Y-J et al. Genetically encoded unstrained olefins for live cell labeling with tetrazine dyes. Chem. Commun 50, 13085–13088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Eising S, Lelivelt F & Bonger KM Vinylboronic Acids as Fast Reacting, Synthetically Accessible, and Stable Bioorthogonal Reactants in the Carboni–Lindsey Reaction. Angew. Chem. Int. Ed 55, 12243–12247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Eising S et al. Coordination-Assisted Bioorthogonal Chemistry: Orthogonal Tetrazine Ligation with Vinylboronic Acid and a Strained Alkene. ChemBioChem 19, 1648–1652 (2018). [DOI] [PubMed] [Google Scholar]
- 182.Eising S, Engwerda AHJ, Riedijk X, Bickelhaupt FM & Bonger KM Highly Stable and Selective Tetrazines for the Coordination-Assisted Bioorthogonal Ligation with Vinylboronic Acids. Bioconjugate Chem. 29, 3054–3059 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Eising S, van der Linden NGA, Kleinpenning F & Bonger KM Vinylboronic Acids as Efficient Bioorthogonal Reactants for Tetrazine Labeling in Living Cells. Bioconjugate Chem. 29, 982–986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Engelsma SB et al. Acylazetine as a Dienophile in Bioorthogonal Inverse Electron-Demand Diels−Alder Ligation. Org. Lett 16, 2744–2747 (2014). [DOI] [PubMed] [Google Scholar]
- 185.de Montes EG et al. Azabicyclic vinyl sulfones for residue-specific dual protein labelling. Chem. Sci 10, 4515–4522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Oliveira BL et al. A Minimal, Unstrained S-Allyl Handle for Pre-Targeting Diels–Alder Bioorthogonal Labeling in Live Cells. Angew. Chem. Int. Ed 55, 14683–14687 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Oliveira BL, Guo Z & Bernardes GJL Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev 46, 4811–5174 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Chen W, Wang D, Dai C, Hamelberg D & Wang B Clicking 1,2,4,5-tetrazine and cyclooctynes with tunable reaction rates. Chem. Commun 48, 1736–1738 (2012). [DOI] [PubMed] [Google Scholar]
- 189.Wang D et al. 3,6-Substituted-1,2,4,5-tetrazines: tuning reaction rates for staged labeling applications. Org. Biomol. Chem 12, 3950–3955 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Blizzard RJ et al. Ideal Bioorthogonal Reactions Using A Site-Specifically Encoded Tetrazine Amino Acid. J. Am. Chem. Soc 137, 10044–10047 (2015). [DOI] [PubMed] [Google Scholar]
- 191.Lambert WD et al. Installation of Minimal Tetrazines through Silver-Mediated Liebeskind–Srogl Coupling with Arylboronic Acids. J. Am. Chem. Soc 141, 17068–17074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Šečkutė J & Devaraj NK Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol 17, 761–767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhang H et al. Rapid Bioorthogonal Chemistry Turn-on through Enzymatic or Long Wavelength Photocatalytic Activation of Tetrazine Ligation. J. Am. Chem. Soc 138, 5978–5983 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ehret F, Wu H, Alexander SC & Devaraj NK Electrochemical Control of Rapid Bioorthogonal Tetrazine Ligations for Selective Functionalization of Microelectrodes. J. Am. Chem. Soc 137, 8876–8879 (2015). [DOI] [PubMed] [Google Scholar]
- 195.Kamber DN et al. 1,2,4-Triazines Are Versatile Bioorthogonal Reagents. J. Am. Chem. Soc 137, 8388–8391 (2015). [DOI] [PubMed] [Google Scholar]
- 196.Horner KA, Valette NM & Webb ME Strain-Promoted Reaction of 1,2,4-Triazines with Bicyclononynes. Chem. Eur. J 21, 14376–14381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Baalmann M, Ziegler MJ, Werther P, Wilhelm J & Wombacher R Enzymatic and Site-Specific Ligation of Minimal Size Tetrazines and Triazines to Proteins for Bioconjugation and Live-Cell Imaging. Bioconjugate Chem. 30, 1405–1414 (2019). [DOI] [PubMed] [Google Scholar]
- 198.Peewasan K & Wagenknecht H-A 1,2,4-Triazine-Modified 2′-Deoxyuridine Triphosphate for Efficient Bioorthogonal Fluorescent Labeling of DNA. ChemBioChem 18, 1473–1476 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Reisacher U et al. Triazine-Modified 7-Deaza-2’-Deoxyadenosines: Better Suited for Bioorthogonal Labeling of DNA by PCR than 2’-Deoxyuridines. Bioconjugate Chem. 30, 1773–1780 (2019). [DOI] [PubMed] [Google Scholar]
- 200.Siegl SJ, Dzijak R, Vázquez A, Pohl R & Vrabel M The discovery of pyridinium 1,2,4-triazines with enhanced performance in bioconjugation reactions. Chem. Sci 8, 3593–3598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kozhevnikov VN, Deary ME, Mantso T, Panayiotidis MI & Sims MT Iridium(III) complexes of 1,2,4-triazines as potential bioorthogonal reagents: metal coordination facilitates luminogenic reaction with strained cyclooctynes. Chem. Commun 55, 14283–14286 (2019). [DOI] [PubMed] [Google Scholar]
- 202.Kamber DN et al. Isomeric triazines exhibit unique profiles of bioorthogonal reactivity. Chem. Sci 10, 9109–9114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Levandowski BJ, Abularrage NS, Houk KN & Raines RT Hyperconjugative Antiaromaticity Activates 4H‑Pyrazoles as Inverse-Electron-Demand Diels−Alder Dienes. Org. Lett 21, 8492–8495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Bruins JJ, Albada B & van Delft F ortho‐Quinones and Analogues Thereof: Highly Reactive Intermediates for Fast and Selective Biofunctionalization. Chem. Eur. J 24, 4749–4756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Borrmann A et al. Strain-Promoted Oxidation-Controlled Cyclooctyne−1,2-Quinone Cycloaddition (SPOCQ) for Fast and Activatable Protein Conjugation. Bioconjugate Chem. 26, 257–261 (2015). [DOI] [PubMed] [Google Scholar]
- 206.Bruins JJ et al. Inducible, Site-Specific Protein Labeling by Tyrosine Oxidation−Strain-Promoted (4 + 2) Cycloaddition. Bioconjugate Chem. 28, 1189–1193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gahtory D, Sen R, Kuzmyn AR, Escorihuela J & Zuilhof H Strain-Promoted Cycloaddition of Cyclopropenes with o-Quinones: A Rapid Click Reaction. Angew. Chem. Int. Ed 57, 10118–10122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Li Q, Dong T, Liu X & Lei X A Bioorthogonal Ligation Enabled by Click Cycloaddition of o-Quinolinone Quinone Methide and Vinyl Thioether. J. Am. Chem. Soc 135, 4996–4999 (2013). [DOI] [PubMed] [Google Scholar]
- 209.Minard A, Liano D, Wang X & Antonio MD The unexplored potential of quinone methides in chemical biology. Bioorg. Med. Chem 27, 2298–2305 (2019). [DOI] [PubMed] [Google Scholar]
- 210.Shih H-W, Kamber DN & Prescher JA Building better bioorthogonal reactions. Curr. Opin. Chem. Biol 21, 103–111 (2014). [DOI] [PubMed] [Google Scholar]
- 211.Chang PV, Prescher JA, Hangauer MJ & Bertozzi CR Imaging Cell Surface Glycans with Bioorthogonal Chemical Reporters. J. Am. Chem. Soc 129, 8400–8401 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Karver MR, Weissleder R & Hilderbrand SA Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi‐Target Imaging. Angew. Chem. Int. Ed 51, 920–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Schoch J, Staudt M, Samanta A, Wiessler M & Jäschke A Site-Specific One-Pot Dual Labeling of DNA by Orthogonal Cycloaddition Chemistry. Bioconjugate Chem. 23, 1382–1386 (2012). [DOI] [PubMed] [Google Scholar]
- 214.Hudak JE, Alvarez D, Skelly A, von Adrian UH & Kasper DL Illuminating vital surface molecules of symbionts in health and disease. Nat. Microbiol 2, 17099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zhang X et al. Second Generation TQ-Ligation for Cell Organelle Imaging. ACS Chem. Biol 10, 1676–1683 (2015). [DOI] [PubMed] [Google Scholar]
- 216.Narayanam MK, Liang Y, Houk KN & Murphy JM Discovery of new mutually orthogonal bioorthogonal cycloaddition pairs through computational screening. Chem. Sci 7, 1257–1261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Shao Z et al. Bioorthogonal release of sulfonamides and mutually orthogonal liberation of two drugs. Chem. Commun 54, 14089–14092 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Winz M-L, Linder EC, Becker J & Jäschke A Site-specific one-pot triple click labeling for DNA and RNA. Chem. Commun 54, 11781–11784 (2018). [DOI] [PubMed] [Google Scholar]
- 219.Simon C et al. One, Two, Three: A Bioorthogonal Triple Labelling Strategy for Studying the Dynamics of Plant Cell Wall Formation In Vivo. Angew. Chem. Int. Ed 57, 16665–16671 (2018). [DOI] [PubMed] [Google Scholar]
- 220.Schart VF et al. Triple Orthogonal Labeling of Glycans by Applying Photoclick Chemistry. ChemBioChem 20, 166–171 (2019). [DOI] [PubMed] [Google Scholar]
- 221.Sletten EM & Bertozzi CR A Bioorthogonal Quadricyclane Ligation. J. Am. Chem. Soc 133, 17570–17573 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Italia JS et al. Mutually Orthogonal Nonsense-Suppression Systems and Conjugation Chemistries for Precise Protein Labeling at up to Three Distinct Sites. J. Am. Chem. Soc 141, 6204–6212 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Tu J et al. Stable, Reactive and Orthogonal Tetrazines: Dispersion Forces Promote the Cycloaddition with Isonitriles. Angew. Chem. Int. Ed 58, 9043–9048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim J & Bertozzi CR A Bioorthogonal Reaction of N-Oxide and Boron Reagents. Angew. Chem. Int. Ed 54, 15777–15781 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Dong J, Krasnova L, Finn MG & Sharpless KB Sulfur(VI) Fluoride Exchange (SuFEx): Another Good Reaction for Click Chemistry. Angew. Chem. Int. Ed 53, 9430–9448 (2014). [DOI] [PubMed] [Google Scholar]
- 226.Barrow AS et al. The growing applications of SuFEx click chemistry. Chem. Soc. Rev 48, 4731–4758 (2019). [DOI] [PubMed] [Google Scholar]
- 227.Zheng Q et al. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. U.S.A 116, 18808–18814 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mortenson DE et al. “Inverse Drug Discovery” Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J. Am. Chem. Soc 140, 200–210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Shannon A et al. Sulfonyl Fluoride Analogues as Activity‐Based Probes for Serine Proteases. ChemBioChem 13, 2327–2330 (2012). [DOI] [PubMed] [Google Scholar]
- 230.Hett EC et al. Rational Targeting of Active-Site Tyrosine Residues Using Sulfonyl Fluoride Probes. ACS Chem. Biol 10, 1094–1098 (2015). [DOI] [PubMed] [Google Scholar]
- 231.Wang N et al. Genetically Encoding Fluorosulfate-L-tyrosine To React with Lysine, Histidine, and Tyrosine via SuFEx in Proteins in Vivo. J. Am. Chem. Soc 140, 4995–4999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yang B et al. Proximity-enhanced SuFEx chemical cross-linker for specific and multitargeting cross-linking mass spectrometry. Proc. Natl. Acad. Sci. U.S.A 115, 11162–11167 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Meng G et al. Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89 (2019). [DOI] [PubMed] [Google Scholar]
- 234.Kabakoff DS, Bünzil J-CG, Oth JFM, Hammond WB & Berson JA Enthalpy and Kinetics of Isomerization of Quadricyclane to Norbornadiene. Strain Energy of Quadricyclane. J. Am. Chem. Soc 97, 1510–1512 (1975). [Google Scholar]
- 235.Tomlin FM et al. Site-specific incorporation of quadricyclane into a protein and photocleavage of the quadricyclane ligation adduct. Bioorg. Med. Chem 26, 5280–5290 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Martínez-Calvo M & Mascareñas JL Organometallic catalysis in biological media and living settings. Coord. Chem. Rev 359, 57–79 (2018). [Google Scholar]
- 237.Jang S-Y, Murale DP, Kim AD & Lee J-S Recent Developments in Metal‐Catalyzed Bio‐orthogonal Reactions for Biomolecule Tagging. ChemBioChem 20, 1498–1507 (2019). [DOI] [PubMed] [Google Scholar]
- 238.Vidal C, Tomás-Gamasa M, Destito P, López F & Mascareñas JL Concurrent and orthogonal gold(I) and ruthenium(II) catalysis inside living cells. Nat. Commun 9, 1913 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Pérez-López AM et al. Gold‐Triggered Uncaging Chemistry in Living Systems. Angew. Chem. Int. Ed 56, 12548–12552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Tsubokura K et al. In Vivo Gold Complex Catalysis within Live Mice. Angew. Chem. Int. Ed 56, 3579–3584 (2017). [DOI] [PubMed] [Google Scholar]
- 241.Chalker JM, Wood CSC & Davis BG A Convenient Catalyst for Aqueous and Protein Suzuki−Miyaura Cross-Coupling. J. Am. Chem. Soc 131, 16346–16347 (2009). [DOI] [PubMed] [Google Scholar]
- 242.Clavadetscher J, Indrigo E, Chankeshwara SV, Lilienkampf A & Bradley M In‐Cell Dual Drug Synthesis by Cancer‐Targeting Palladium Catalysts. Angew. Chem. Int. Ed 56, 6864–6868 (2017). [DOI] [PubMed] [Google Scholar]
- 243.Li N, Lim RKV, Edwardraja S & Lin Q Copper-Free Sonogashira Cross-Coupling for Functionalization of Alkyne-Encoded Proteins in Aqueous Medium and in Bacterial Cells. J. Am. Chem. Soc 133, 15316–15319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Li N, Ramil CP, Lim RKV & Lin Q A Genetically Encoded Alkyne Directs Palladium-Mediated Protein Labeling on Live Mammalian Cell Surface. ACS Chem. Biol 10, 379–384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Li J et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem 6, 352–361 (2014). [DOI] [PubMed] [Google Scholar]
- 246.Wang J et al. Chemical Remodeling of Cell-Surface Sialic Acids through a Palladium-Triggered Bioorthogonal Elimination Reaction. Angew. Chem. Int. Ed 54, 5364–5368 (2015). [DOI] [PubMed] [Google Scholar]
- 247.Yusop RM, Unciti-Broceta A, Johansson EMV, Sánchez-Martín RM & Bradley M Palladium-mediated intracellular chemistry. Nat. Chem 3, 239–243 (2011). [DOI] [PubMed] [Google Scholar]
- 248.Wang F, Zhang Y, Du Z, Ren J & Qu X Designed heterogeneous palladium catalysts for reversible light-controlled bioorthogonal catalysis in living cells. Nat. Commun 9, 1209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tomás-Gamasa M, Martínez-Calvo M, Couceiro JR & Mascareñas JL Transition metal catalysis in the mitochondria of living cells. Nat. Commun 7, 12538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lin YA, Chalker JM, Floyd N, Bernardes GJL & Davis BG Allyl Sulfides Are Privileged Substrates in Aqueous Cross-Metathesis: Application to Site-Selective Protein Modification. J. Am. Chem. Soc 130, 9642–9643 (2008). [DOI] [PubMed] [Google Scholar]
- 251.Lin YA et al. Rapid Cross-Metathesis for Reversible Protein Modifications via Chemical Access to Se-Allyl-selenocysteine in Proteins. J. Am. Chem. Soc 135, 12156–12159 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Bloom S et al. Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem 10, 205–211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hooker JM, Kovacs EW & Francis MB Interior Surface Modification of Bacteriophage MS2. J. Am. Chem. Soc 126, 3718–3719 (2004). [DOI] [PubMed] [Google Scholar]
- 254.Sengupta S & Chandrasekaran S Modifications of amino acids using arenediazonium salts. Org. Biomol. Chem 17, 8308–8329 (2019). [DOI] [PubMed] [Google Scholar]
- 255.Addy PS, Erickson SB, Italia JS & Chatterjee A A Chemoselective Rapid Azo-Coupling Reaction (CRACR) for Unclickable Bioconjugation. J. Am. Chem. Soc 139, 11670–11673 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lin S et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Taylor MT, Nelson JE, Suero M & Gaunt MJ A protein functionalization platform based on selective reactions at methionine residues. Nature 562, 563–568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Alvarez-Dorta D et al. Electrochemically Promoted Tyrosine-Click-Chemistry for Protein Labeling. J. Am. Chem. Soc 140, 17120–17126 (2018). [DOI] [PubMed] [Google Scholar]
- 259.Geng J et al. Radical polymerization inside living cells. Nat. Chem 11, 578–586 (2019). [DOI] [PubMed] [Google Scholar]
- 260.Vidal C, Tomás-Gamasa M, Gutiérrez-González A & Mascareñas JL Ruthenium-Catalyzed Redox Isomerizations inside Living Cells. J. Am. Chem. Soc 141, 5125–5129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]