

ABSTRACT
Commercial nitric acid (HNO3) and ammonia (NH3) are mostly produced through the Ostwald process and the Haber-Bosch process, respectively. However, high energy demand and enormous greenhouse gas accompy these processes. The development of economical and green ways to synthesize HNO3 and NH3 is highly desirable for solving the global energy and environmental crisis. Here, we present two energy-efficient and environmentally friendly strategies to synthesize HNO3 and NH3 at distributed sources, including the electrocatalytic oxidation of N2 in air to HNO3 and the electrocatalytic reduction of residual 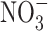 contamination in water to NH3. The isotope-labeling studies combined with theoretical calculation reveal the reaction path of the two proposed strategies, confirming the origin of the electrochemical products. Importantly, the electrooxidation-generated
contamination in water to NH3. The isotope-labeling studies combined with theoretical calculation reveal the reaction path of the two proposed strategies, confirming the origin of the electrochemical products. Importantly, the electrooxidation-generated  ions may also serve as reactants for the electroreduction synthesis of NH3 in the future. Our work may open avenues for energy-efficient and green production of HNO3 and NH3 at distributed sources.
ions may also serve as reactants for the electroreduction synthesis of NH3 in the future. Our work may open avenues for energy-efficient and green production of HNO3 and NH3 at distributed sources.
Keywords: ammonia, nitric acid, distributed sources, electrocatalysis, green synthesis
INTRODUCTION
Transformations of nitrogen into reactive forms, particularly nitric acid (HNO3) and ammonia (NH3), are vital to living organisms and industrial processes [1–4]. The worldwide production of HNO3 and NH3 was 50 and 150 million metric tons, respectively, in 2017 [5]. Commercial HNO3 is produced through the catalytic oxidation of NH3 (Ostwald process), but this approach is energy-intensive [5]. On the other hand, NH3 is mostly manufactured using the Haber-Bosch method [6–9], which consumes ∼2% of global power and discharges ∼1.5% of global greenhouse gas [10–16]. Moreover, the large-scale chemical plants, and hence centralized HNO3 and NH3 sources, induce a serious waste of fossil fuels during the transportation process [17]. Therefore, searching novel solutions that allow energy-efficient and environmentally friendly synthesis of HNO3 and NH3 at distributed sources are urgently needed [18,19]. Electrocatalysis represents a potentially alternative strategy [20]. However, present electrocatalysis is focused on preparing NH3 through the reduction of pure dinitrogen, which is produced by the energy-intensive air-separation process [21–24]. Thus, it is of great interest to develop novel electrochemical routes to achieving green synthesis of HNO3 and NH3 under benign conditions.
Herein, we present two electrochemical strategies. Strategy I is the electrocatalytic oxidation of N2 to HNO3 by using air as the nitrogen source. Strategy II is the electrochemical reduction preparation of NH3(aq) from residual nitrate ion ( ) contamination in water [25,26]:
) contamination in water [25,26]:
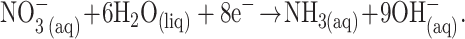 |
(1) |
We found that N2 is electro-oxidized into HNO3 over platinum foil with ∼1.23% Faradaic efficiency at +2.19 V vs. RHE (the reversible hydrogen electrode) and the waste of  is electro-reduced with approximately 33.6% NH3(aq) selectivity at −0.65 V vs. RHE over Co3O4 nanorod arrays. Our results demonstrate how the electrochemical methods of Strategy I and Strategy II produce HNO3 and NH3 at distributed sources. These findings provide a new avenue for producing the reactive nitrogen species in an ‘economic’ and ‘clean’ way, especially once the electrocatalysis reaction is driven by renewable energy [27].
is electro-reduced with approximately 33.6% NH3(aq) selectivity at −0.65 V vs. RHE over Co3O4 nanorod arrays. Our results demonstrate how the electrochemical methods of Strategy I and Strategy II produce HNO3 and NH3 at distributed sources. These findings provide a new avenue for producing the reactive nitrogen species in an ‘economic’ and ‘clean’ way, especially once the electrocatalysis reaction is driven by renewable energy [27].
RESULTS AND DISCUSSION
An H-type cell divided by a proton-exchange membrane was used for the electrocatalytic tests of Strategy I (Fig. 1a). To explore the catalytic behavior of N2 electrooxidation (Strategy I), air was bubbled onto the anode. H2O in electrolyte (0.3 M K2SO4) and N2 in air combine with platinum foil as an electrocatalyst to form HNO3. We tested the linear sweep voltammetry curves of platinum foil in Ar- and air-saturated electrolyte under ambient conditions (Fig. 1b). All potentials in this work were recorded and converted to the RHE scale. As the potential moves above +2.13 V, the current density is distinguishably enhanced under air-saturated electrolyte, revealing that N2 in air can be catalysed into oxidative products. The produced 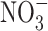 and
and 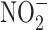 are quantified based on the standard method [28] by using ultraviolet-visible (UV-Vis) spectrophotometry (Supplementary Fig. S1, available as Supplementary Data at NSR online). Anion chromatography was also adopted to confirm the accuracy of UV-Vis spectrophotometry for detecting the yield of
are quantified based on the standard method [28] by using ultraviolet-visible (UV-Vis) spectrophotometry (Supplementary Fig. S1, available as Supplementary Data at NSR online). Anion chromatography was also adopted to confirm the accuracy of UV-Vis spectrophotometry for detecting the yield of 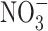 (Supplementary Table S1, available as Supplementary Data at NSR online). The effect of the anode potentials on the yields of oxidative products and the corresponding Faradaic efficiencies were investigated (Fig. 1c). Although the gap in the current density between Ar and air condition keeps enlarging with the increase in potential, the highest Faradaic efficiency of 1.23% for
(Supplementary Table S1, available as Supplementary Data at NSR online). The effect of the anode potentials on the yields of oxidative products and the corresponding Faradaic efficiencies were investigated (Fig. 1c). Although the gap in the current density between Ar and air condition keeps enlarging with the increase in potential, the highest Faradaic efficiency of 1.23% for  was obtained at +2.19 V. The yields of
was obtained at +2.19 V. The yields of 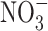 and
and  at +2.19 V achieve 0.06 and 0.0004 μmol h−1 cm−2, respectively, and remained almost unchanged with the increase in potential (Fig. 1c and Supplementary Fig. S2, available as Supplementary Data at NSR online). So, the optimal operating potential for N2 electrooxidation over platinum electrocatalyst in this work was +2.19 V. Furthermore, some control experiments, including Ar-saturated electrolyte with a potentiostatic test (+2.19 V) and air-saturated electrolyte without external potential, were performed. Undetected oxidative products in both cases further confirmed the electrocatalytic oxidation of N2 in air to
at +2.19 V achieve 0.06 and 0.0004 μmol h−1 cm−2, respectively, and remained almost unchanged with the increase in potential (Fig. 1c and Supplementary Fig. S2, available as Supplementary Data at NSR online). So, the optimal operating potential for N2 electrooxidation over platinum electrocatalyst in this work was +2.19 V. Furthermore, some control experiments, including Ar-saturated electrolyte with a potentiostatic test (+2.19 V) and air-saturated electrolyte without external potential, were performed. Undetected oxidative products in both cases further confirmed the electrocatalytic oxidation of N2 in air to 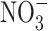 and
and 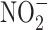 as designed in Strategy I (Supplementary Table S2, available as Supplementary Data at NSR online). For practical application, the durability of the catalyst is crucial. After consecutive recycling tests, the catalytic performance showed no obvious decline, demonstrating the high stability of platinum foil toward the electrooxidation of N2 (Fig. 1d). As shown in Fig. 1e, Strategy I goes through multiple processes. We calculated the free energy of N2 electrooxidation over the Pt (200) plane based on the X-ray diffraction (XRD) result (Supplementary Fig. S3, available as Supplementary Data at NSR online). First, the N2 molecule was chemically absorbed by the platinum to form N2* with a total energy change of −0.097 eV, indicating that this reaction can take place spontaneously. N2* reacted with OH− to produce N2OH* and then N2OH* was dehydrogenated to N2O* or continued to react with OH− to produce N2O2H2* with a larger reaction energy. Both N2O* and N2O2H2* will evolve into NO* with the intermediate of N2O2H* and NOH*. Note that NO* could be directly desorbed from the catalyst surface and then oxidized into HNO3 and HNO2 in solution (Equation (2)). Meanwhile, NO* can be oxidized to NO2* with the intermediate of NO2H*. With further increase in potential, NO2* desorbed from the catalyst surface. Finally, NO2 was transformed to HNO3 in solution through Equation (3). Based on these results, we can deduce the reaction path from N2 in air to HNO3 via the as-proposed Strategy I:
as designed in Strategy I (Supplementary Table S2, available as Supplementary Data at NSR online). For practical application, the durability of the catalyst is crucial. After consecutive recycling tests, the catalytic performance showed no obvious decline, demonstrating the high stability of platinum foil toward the electrooxidation of N2 (Fig. 1d). As shown in Fig. 1e, Strategy I goes through multiple processes. We calculated the free energy of N2 electrooxidation over the Pt (200) plane based on the X-ray diffraction (XRD) result (Supplementary Fig. S3, available as Supplementary Data at NSR online). First, the N2 molecule was chemically absorbed by the platinum to form N2* with a total energy change of −0.097 eV, indicating that this reaction can take place spontaneously. N2* reacted with OH− to produce N2OH* and then N2OH* was dehydrogenated to N2O* or continued to react with OH− to produce N2O2H2* with a larger reaction energy. Both N2O* and N2O2H2* will evolve into NO* with the intermediate of N2O2H* and NOH*. Note that NO* could be directly desorbed from the catalyst surface and then oxidized into HNO3 and HNO2 in solution (Equation (2)). Meanwhile, NO* can be oxidized to NO2* with the intermediate of NO2H*. With further increase in potential, NO2* desorbed from the catalyst surface. Finally, NO2 was transformed to HNO3 in solution through Equation (3). Based on these results, we can deduce the reaction path from N2 in air to HNO3 via the as-proposed Strategy I:
 |
(2) |
Figure 1.

(a) Schematic illustration for as-proposed Strategy I. (b) Linear sweep voltammetric curves of platinum foil in Ar-saturated (blue line) and air-saturated (red line) electrolyte. (c) Yields of  and Faradaic efficiencies after potentiostatic test at given potentials. (d) The cycling tests of N2 electrooxidation over the same piece of Pt electrode. (e) Calculated free-energy diagram for N2 electrooxidation over platinum foil. Ball-and-stick model in (b) and (e): blue ball (N atom), red ball (O atom), pink ball (H atom) and green ball (Pt atom).
and Faradaic efficiencies after potentiostatic test at given potentials. (d) The cycling tests of N2 electrooxidation over the same piece of Pt electrode. (e) Calculated free-energy diagram for N2 electrooxidation over platinum foil. Ball-and-stick model in (b) and (e): blue ball (N atom), red ball (O atom), pink ball (H atom) and green ball (Pt atom).
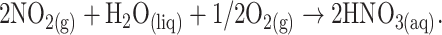 |
(3) |
To demonstrate Strategy II, involving electroreducing residual  contamination in water, KNO3 was added to a cathode cell and reduced into NH3(aq) (Fig. 2a). Presently, the main sources of nitrate for the as-proposed Strategy II are residual contamination in water, including industrial wastewater, domestic sewage, animal waste and nitrogen fertilizers. In the future, if the efficiency of Strategy I can be further improved, the electrooxidation generation of
contamination in water, KNO3 was added to a cathode cell and reduced into NH3(aq) (Fig. 2a). Presently, the main sources of nitrate for the as-proposed Strategy II are residual contamination in water, including industrial wastewater, domestic sewage, animal waste and nitrogen fertilizers. In the future, if the efficiency of Strategy I can be further improved, the electrooxidation generation of  may also serve as a reactant for Strategy II. We constructed Co3O4 nanorod arrays supported on Ti mesh as a model electrocatalyst (Supplementary Fig. S4, available as Supplementary Data at NSR online). The linear sweep voltammetry curves of the Co3O4 electrode in 0.1 M K2SO4 electrolyte with and without KNO3 were performed under ambient conditions (Fig. 2b). The current density was obviously enhanced in the presence of KNO3, indicating that
may also serve as a reactant for Strategy II. We constructed Co3O4 nanorod arrays supported on Ti mesh as a model electrocatalyst (Supplementary Fig. S4, available as Supplementary Data at NSR online). The linear sweep voltammetry curves of the Co3O4 electrode in 0.1 M K2SO4 electrolyte with and without KNO3 were performed under ambient conditions (Fig. 2b). The current density was obviously enhanced in the presence of KNO3, indicating that  in solution can be catalysed into reductive products. The yields of NH3(aq) [28] and
in solution can be catalysed into reductive products. The yields of NH3(aq) [28] and  were quantified based on UV-Vis spectrophotometers (Supplementary Fig. S5, available as Supplementary Data at NSR online). The cation chromatography method was also adopted to confirm the accuracy of UV-Vis spectrophotometry for detecting the yield of NH3(aq) (Supplementary Table S3, available as Supplementary Data at NSR online). Following the potentiostatic test conducted at −0.65 V vs. RHE, which corresponded to 100 mA cm−2 of current density (Fig. 2b), the selectivity of different reductive products is displayed in Fig. 2c. The selectivity of NH3(aq) was 33.6% over the Co3O4 electrode, and the other reductive products included
were quantified based on UV-Vis spectrophotometers (Supplementary Fig. S5, available as Supplementary Data at NSR online). The cation chromatography method was also adopted to confirm the accuracy of UV-Vis spectrophotometry for detecting the yield of NH3(aq) (Supplementary Table S3, available as Supplementary Data at NSR online). Following the potentiostatic test conducted at −0.65 V vs. RHE, which corresponded to 100 mA cm−2 of current density (Fig. 2b), the selectivity of different reductive products is displayed in Fig. 2c. The selectivity of NH3(aq) was 33.6% over the Co3O4 electrode, and the other reductive products included  , NOx, N2 and N2H4, etc. [29–31]. Furthermore, the Co3O4 electrode supported on the Ti substrate was conducted under a potentiostatic test (–0.65 V) in 0.1 M K2SO4 electrolyte without KNO3. The negligible NH3(aq) in the final solution further confirms the electrocatalytic reduction of
, NOx, N2 and N2H4, etc. [29–31]. Furthermore, the Co3O4 electrode supported on the Ti substrate was conducted under a potentiostatic test (–0.65 V) in 0.1 M K2SO4 electrolyte without KNO3. The negligible NH3(aq) in the final solution further confirms the electrocatalytic reduction of 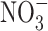 to NH3(aq) via Strategy II (Supplementary Fig. S6, available as Supplementary Data at NSR online). The bare Ti substrate showed a much smaller yield of NH3(aq) (0.029 mmol h−1 cm−2) compared to that of Co3O4 supported on the Ti substrate (0.854 mmol h−1 cm−2), indicating the high activity of Co3O4 for the electrocatalytic reduction of
to NH3(aq) via Strategy II (Supplementary Fig. S6, available as Supplementary Data at NSR online). The bare Ti substrate showed a much smaller yield of NH3(aq) (0.029 mmol h−1 cm−2) compared to that of Co3O4 supported on the Ti substrate (0.854 mmol h−1 cm−2), indicating the high activity of Co3O4 for the electrocatalytic reduction of  (Supplementary Fig. S6, available as Supplementary Data at NSR online). During the consecutive recycling test, the catalytic activities displayed almost no decline (Fig. 2d). And the Co3O4 electrode was maintained well after the electroreduction test (Supplementary Fig. S7, available as Supplementary Data at NSR online). These results demonstrate the high durability of the Co3O4 electrode for
(Supplementary Fig. S6, available as Supplementary Data at NSR online). During the consecutive recycling test, the catalytic activities displayed almost no decline (Fig. 2d). And the Co3O4 electrode was maintained well after the electroreduction test (Supplementary Fig. S7, available as Supplementary Data at NSR online). These results demonstrate the high durability of the Co3O4 electrode for  electroreduction. Note that the electroreduction of
electroreduction. Note that the electroreduction of  was reported in previous work, but they focused on the degradation of residual
was reported in previous work, but they focused on the degradation of residual  in water into environmentally friendly products [32–34]. We herein propose utilization of the waste of
in water into environmentally friendly products [32–34]. We herein propose utilization of the waste of  , provided by environmental contaminations, to produce the high value-added NH3(aq) via electroreduction.
, provided by environmental contaminations, to produce the high value-added NH3(aq) via electroreduction.
Figure 2.

(a) Schematic illustration for as-proposed Strategy II. (b) Linear sweep voltammetric curves of Co3O4 electrode in 0.1 M K2SO4 electrolyte with and without KNO3. (c) Selectivity of 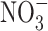 electroreduction products at −0.65 V vs. RHE. (d) The cycling tests of
electroreduction products at −0.65 V vs. RHE. (d) The cycling tests of 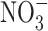 reduction over the same piece of Co3O4 electrode.
reduction over the same piece of Co3O4 electrode.
To confirm the origin of the 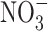 generated from N2 electrooxidation, we designed an isotopic-labeling study using 15N2 (>99 atom% 15N) and 14N2 (with the natural abundance of 0.36 atom% 15N) as the feeding gas for Strategy I [35]. As seen in Fig. 3a, the sample using 14N2 as the feeding gas produced 0.44% 15
generated from N2 electrooxidation, we designed an isotopic-labeling study using 15N2 (>99 atom% 15N) and 14N2 (with the natural abundance of 0.36 atom% 15N) as the feeding gas for Strategy I [35]. As seen in Fig. 3a, the sample using 14N2 as the feeding gas produced 0.44% 15 , while the isotopic-labeled sample showed 17.40% abundance of 15
, while the isotopic-labeled sample showed 17.40% abundance of 15 —much higher than the natural abundance of 15N. The concentration difference of 15N for the isotopic-labeled sample between the 15N2 reactant and the 15
—much higher than the natural abundance of 15N. The concentration difference of 15N for the isotopic-labeled sample between the 15N2 reactant and the 15 product may arise from the leaking and/or residue of air in the reactor. These results clearly confirm that the N element of the generated
product may arise from the leaking and/or residue of air in the reactor. These results clearly confirm that the N element of the generated  via Strategy I came from N2. We also performed an isotopic-labeling study using K15NO3 (20.3 atom% 15N) and K14NO3 (0.36 atom% 15N) as the reactants to explore the origin of the NH3(aq). It can be seen that the isotopic-labeled sample exhibited 18.98% 15NH3(aq) but the sample using K14NO3 without an isotopic label as a reference showed only 0.36% 15NH3(aq) (Fig. 3b). These results demonstrate that the N element of the formed NH3(aq) via Strategy II originated from the
via Strategy I came from N2. We also performed an isotopic-labeling study using K15NO3 (20.3 atom% 15N) and K14NO3 (0.36 atom% 15N) as the reactants to explore the origin of the NH3(aq). It can be seen that the isotopic-labeled sample exhibited 18.98% 15NH3(aq) but the sample using K14NO3 without an isotopic label as a reference showed only 0.36% 15NH3(aq) (Fig. 3b). These results demonstrate that the N element of the formed NH3(aq) via Strategy II originated from the  species.
species.
Figure 3.

(a) The isotopic mass spectrometry of 15 with 15N2 and 14N2 as the feeding gas for Strategy I. (b) The isotopic mass spectrometry of 15NH3(aq) with 15KNO3 and 14KNO3 as the reactants for Strategy II.
with 15N2 and 14N2 as the feeding gas for Strategy I. (b) The isotopic mass spectrometry of 15NH3(aq) with 15KNO3 and 14KNO3 as the reactants for Strategy II.
CONCLUSIONS
In summary, we present two energy-efficient and environmentally friendly strategies to prepare HNO3 and NH3 at distributed sources. Strategy I is the electrocatalytic oxidation of N2 in air to HNO3 and platinum foil is adopted as the model catalyst, showing a generation rate of 0.06 μmol h−1 cm−2 for HNO3 at +2.19 V. Strategy II is the electrocatalytic reduction of residual  contamination in water to NH3(aq) and Co3O4 nanorod arrays supported on a Ti mesh, exhibiting a selectivity of 33.6% for NH3(aq) at −0.65 V. Combined with the theoretical calculation, the reaction path from N2 in air to HNO3 via the as-proposed Strategy I was deduced. The isotope-labeling studies using 15N2 and K15NO3 confirmed the origin of the electrochemical products. Moreover, the platinum electrode and Co3O4 electrode showed high durability for the electrocatalytic oxidation of N2 and the electrocatalytic reduction of
contamination in water to NH3(aq) and Co3O4 nanorod arrays supported on a Ti mesh, exhibiting a selectivity of 33.6% for NH3(aq) at −0.65 V. Combined with the theoretical calculation, the reaction path from N2 in air to HNO3 via the as-proposed Strategy I was deduced. The isotope-labeling studies using 15N2 and K15NO3 confirmed the origin of the electrochemical products. Moreover, the platinum electrode and Co3O4 electrode showed high durability for the electrocatalytic oxidation of N2 and the electrocatalytic reduction of  , respectively. In the future,
, respectively. In the future, 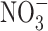 from Strategy I may also serve as a reactant for the electroreduction synthesis of NH3. Our results presented here provide new avenues for energy-efficient and green production of HNO3 and NH3 at distributed sources.
from Strategy I may also serve as a reactant for the electroreduction synthesis of NH3. Our results presented here provide new avenues for energy-efficient and green production of HNO3 and NH3 at distributed sources.
METHODS
Preparation of Co3O4 cathode for  reduction via Strategy II
reduction via Strategy II
In a typical process, 2 mmol Co(NO3)2·6H2O, 10 mmol urea and 8 mmol NH4F were dissolved in 36 mL distilled water under stirring for 5 min. The aqueous solution was moved to a 50-mL Teflon-lined autoclave and then a piece of Ti mesh (1 × 3 cm2) was immersed in the above solution. The autoclave was sealed and heated at 120°C for 9 h, followed by cooling down to ambient temperature. The sample was washed using distilled water and ethanol six times and then dried in a vacuum oven overnight. The dried sample was annealed at 300°C for 2 h in air to acquire the final product of Co3O4 nanorod arrays supported on a Ti mesh.
Characterization
The scanning electron microscopy images were acquired from a Hitachi S-4800 scanning electron microscope. Transmission electron microscopy and high-resolution transmission electron microscopy images were taken using a JEOL-2100F system. The XRD was measured using a Bruker D8 Focus Diffraction System with a Cu Kα source (λ = 0.154178 nm). X-ray photoelectron spectrum analysis was recorded via a PHI 5000 Versaprobe system using monochromatic
Al Kα radiation. All binding energies were revised according to the C 1-s peak at 284.8 eV. The ultraviolet-visible (UV-Vis) absorbance spectra were measured on a Beijing Purkinje General T6 new century spectrophotometer. Anion chromatography was performed on an ICS-1100, Thermo. Cation chromatography was conducted on an ICS-900, Thermo. The concentration of 15N isotope labeling was established by isotopic mass spectrometry (MAT-271). The pH values of the electrolytes were determined using a pH-meter (LE438 pH electrode, Mettler Toledo, USA).
Electrochemical measurements
Electrochemical measurements were conducted using an electrochemical workstation (CHI 660D, Chenhua, Shanghai). A typical H-type electrolytic cell divided by a proton-exchange membrane (Nafion 117) was used. Except for special instructions, all potentials were recorded against the RHE. The potentials against the saturated calomel electrode (SCE) were translated to those against the RHE using the following equation: E (vs. RHE) = E (vs. SCE) + 0.2415 + 0.059 × pH. All the polarization curves were the steady lines after many cycles and the current density was normalized to the geometric surface area.
For the electrooxidation of N2 to HNO3 via Strategy I, Pt plates (1 × 1 cm2) were used as both the working electrode and the counter electrode, the reference electrode was SCE and 0.3 M K2SO4 solution (70 mL) was adopted as the electrolyte. Air (99.99% purity) was bubbled into the anodic compartment with a flow rate of 10 mL min−1 in the whole electrochemical process. The linear sweep voltammetry was performed at a rate of 10 mV s−1 and the potentiostatic test was tested at a different anodic voltage for 20 h with the electrolyte agitated at a stirring rate of ∼350 rpm. An absorption flask containing 5 mL K2SO4 solution (0.3 M) was connected to the gas outlet of an anodic half cell to avoid the loss of products due to air bubbling. After the electrooxidation measurements, the components of the mixed solutions in the anodic compartment and absorption flask were both analysed. For comparison, the electrooxidation measurements were also measured with all the testing conditions consistent with aforementioned N2 oxidation except that air was replaced by Ar or the external potential was removed.
The electroreduction of  to NH3 via Strategy II was carried out in a three-electrode configuration with as-prepared Co3O4 (1 × 1 cm2) electrode, SCE and platinum foil as working electrode, reference electrode and counter electrode, respectively; 0.1 M K2SO4 solution (80 mL) was used as the electrolyte and evenly distributed to the cathode and anode compartment. KNO3 (100 g L−1) was added into the cathode compartment as a reactant. Prior to the
to NH3 via Strategy II was carried out in a three-electrode configuration with as-prepared Co3O4 (1 × 1 cm2) electrode, SCE and platinum foil as working electrode, reference electrode and counter electrode, respectively; 0.1 M K2SO4 solution (80 mL) was used as the electrolyte and evenly distributed to the cathode and anode compartment. KNO3 (100 g L−1) was added into the cathode compartment as a reactant. Prior to the  electroreduction test, the cathode electrolyte was purged with Ar (99.99% purity) for 30 min. The linear sweep voltammetry was performed at a rate of 20 mV s−1 and the potentiostatic test was conducted at −0.65 V for 3 h at a stirring rate of ∼350 rpm. For comparison, the electroreduction measurements were also conducted with all the testing conditions consistent with the aforementioned
electroreduction test, the cathode electrolyte was purged with Ar (99.99% purity) for 30 min. The linear sweep voltammetry was performed at a rate of 20 mV s−1 and the potentiostatic test was conducted at −0.65 V for 3 h at a stirring rate of ∼350 rpm. For comparison, the electroreduction measurements were also conducted with all the testing conditions consistent with the aforementioned  reduction except that the Co3O4 cathode was replaced by the Ti mesh or the Co3O4 cathode was immersed in electrolyte (0.1 M K2SO4) without the addition of KNO3.
reduction except that the Co3O4 cathode was replaced by the Ti mesh or the Co3O4 cathode was immersed in electrolyte (0.1 M K2SO4) without the addition of KNO3.
Ion-concentration detection methods
The electrolytes pre and post test were first diluted to appropriate concentration and then tested using a UV-Vis spectrophotometer to quantify the concentration. The concentrations of nitrate-N, nitrite-N and ammonia-N were estimated by UV-Vis spectrophotometry according to the standard method. The specific approaches are as follows.
Determination of nitrate-N
First, a certain amount of electrolyte was taken out of the electrolytic cell and diluted to 5 mL to the detection range. Then, 0.1 mL 1 M HCl and 0.01 mL 0.8 wt% sulfamic acid solution were added to the aforementioned solution. The absorption spectrum was tested using an ultraviolet-visible spectrophotometer and the absorption intensities at wavelengths of 220 and 275 nm were recorded. The final absorbance value was calculated using the equation: A = A220nm – 2A275nm. The concentration–absorbance curve was made using a series of standard potassium nitrate solutions and the potassium nitrate crystal was dried at 105–110°C for 2 h in advance.
Determination of nitrite-N
A mixture of p-aminobenzenesulfonamide (4 g), N-(1-Naphthyl)ethylenediamine dihydrochloride (0.2 g), ultrapure water (50 mL) and phosphoric acid (10 mL, ρ = 1.70 g/mL) was used as a color reagent. A certain amount of electrolyte was taken from the electrolytic cell and diluted to 5 mL to the detection range. Next, 0.1 mL color reagent was added into the aforementioned 5-mL solution and mixed to uniformity, and the absorption intensity at a wavelength of 540 nm was recorded after sitting for 20 min. The concentration–absorbance curve was calibrated using a series of standard sodium nitrite solutions.
Determination of ammonia-N
Ammonia-N was determined using Nessler's reagent as the color reagent. First, a certain amount of electrolyte was taken from the electrolytic cell and diluted to 5 mL to the detection range. Next, 0.1 mL potassium sodium tartrate solution (ρ = 500 g/L) was added and mixed thoroughly, then 0.1 mL Nessler's reagent was put into the solution. The absorption intensity at a wavelength of 420 nm was recorded after sitting for 20 min. The concentration–absorbance curve was made using a series of standard ammonium chloride solutions and the ammonium chloride crystal was dried at 105°C for 2 h in advance.
The calibration curves of nitrate-N and nitrite-N for the electrooxidation of N2 are shown in Supplementary Fig. S1, available as Supplementary Data at NSR online (0.3 M K2SO4 as the background solution). The calibration curves of nitrate-N, nitrite-N and ammonia-N for the electroreduction of 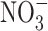 are shown in Supplementary Fig. S5, available as Supplementary Data at NSR online (ultrapure water as the background solution).
are shown in Supplementary Fig. S5, available as Supplementary Data at NSR online (ultrapure water as the background solution).
All the results obtained using UV-Vis spectrophotometry were compared with those of ion chromatography (Supplementary Tables S1 and S3, available as Supplementary Data at NSR online). The pre-treatment of the cation chromatography measurement of ammonia-N after the electroreduction test was as follows. First, the electrolyte was put into a round flask and then the pH was adjusted to ∼8 by the addition of NaOH. The solution was then heated to ∼100°C and went through condensation until the 90% electrolyte was distilled. The distilled solution was collected using 35 mL ultrapure water and used for the cation chromatography measurement.
Calculation of the yield, selectivity and Faradaic efficiency
For the N2 electrooxidation experiments via Strategy I, the yield of  and
and  was calculated using Equation (4) and Equation (5), respectively:
was calculated using Equation (4) and Equation (5), respectively:
 |
(4) |
 |
(5) |
The Faradaic efficiency was calculated according to the charge consumed for synthesizing the NO gas and the total charge passed through the electrode using Equation (6):
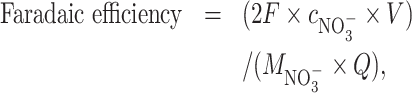 |
(6) |
where cNO3− is the concentration of NOx−, V is the volume of the electrolyte in the anode compartment, MNO3− is the molar mass of  , MNO2− is the molar mass of
, MNO2− is the molar mass of  , t is the electrolysis time, S is the geometric area of the Pt plate, F is the Faradaic constant (96 485 C mol−1) and Q is the total charge passing the electrode. (Note that NO and NO2 gas consumed two and four electrons, respectively. We prudently chose NO gas as the product to calculate the Faradaic efficiency.)
, t is the electrolysis time, S is the geometric area of the Pt plate, F is the Faradaic constant (96 485 C mol−1) and Q is the total charge passing the electrode. (Note that NO and NO2 gas consumed two and four electrons, respectively. We prudently chose NO gas as the product to calculate the Faradaic efficiency.)
For the  electroreduction experiments via Strategy II, the yield of NH3(aq) was calculated using Equation (7):
electroreduction experiments via Strategy II, the yield of NH3(aq) was calculated using Equation (7):
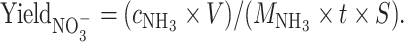 |
(7) |
The conversion of  was obtained from Equation (8):
was obtained from Equation (8):
 |
(8) |
The selectivity of product was acquired by Equation (9):
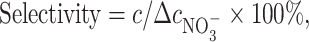 |
(9) |
where cNH3 is the concentration of NH3(aq), ΔcNO3− is the concentration difference of  before and after electrolysis, c0 is the initial concentration of
before and after electrolysis, c0 is the initial concentration of  , and c is the concentration of products, including NH3(aq),
, and c is the concentration of products, including NH3(aq), 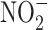 .
.
15N isotope-labeling experiment
The isotope-labeling reactants of 15N2 (>99 atom% 15N) and K15NO3 (20.3 atom% 15N) were purchased from the Shanghai Research Institute of Chemical Industry Co. Ltd. The isotope-labeling concentration of products via both Strategy I and Strategy II was established by isotopic mass spectrometry from the Shanghai Engineering Research Center of Stable Isotope.
For isotope labeling the N2 electrooxidation, we carried out the batch experiments using 15N2 as the feeding gas for five successive times and collected all the electrolytes together after electrolysis. In order to increase the concentration of 15 for isotopic mass spectrometry, the collected electrolytes were first alkalified to pH ∼7 by adding 1 M KOH solution and then concentrated by 10 times via distilling at 70°C. For comparison, 14N2 with natural-abundance 15N (0.36 atom%) was used as a feeding gas to replace 15N2 with other conditions being consistent.
for isotopic mass spectrometry, the collected electrolytes were first alkalified to pH ∼7 by adding 1 M KOH solution and then concentrated by 10 times via distilling at 70°C. For comparison, 14N2 with natural-abundance 15N (0.36 atom%) was used as a feeding gas to replace 15N2 with other conditions being consistent.
For isotope labeling the  electroreduction, the pH of final electrolyte was adjusted to ∼3 using 1 M HCl solution before isotopic mass spectrometry. For comparison, K14NO3 with natural-abundance 15N (0.36 atom%) was used as the reactant to replace K15NO3 with the other conditions being consistent.
electroreduction, the pH of final electrolyte was adjusted to ∼3 using 1 M HCl solution before isotopic mass spectrometry. For comparison, K14NO3 with natural-abundance 15N (0.36 atom%) was used as the reactant to replace K15NO3 with the other conditions being consistent.
Theoretical simulation
Although the XRD pattern of platinum after the electrooxidation process (not shown here) showed no obvious new species, the high oxidation state of PtOH or PtOx might exist in an amorphous state. Considering the complexity of the electrochemical oxidation process, herein, we only adopted Pt as a model for simulation. The theoretical calculations were conducted using density functional theory with the Perdew–Burke–Ernzerbof form of generalized gradient approximation functional [36]. The plane wave energy was cut off at 400 eV. The Vienna abinitio simulation package was used [37,38]. The Fermi scheme was used for electron occupancy with an energy smearing of 0.1 eV. The first Brillouin zone was adopted in the Monkhorst−Pack grid [39]. The 3 × 3 × 1 k-point mesh was taken for the surface calculation. The energy of 1.0 × 10−6 eV atom−1 and force of 0.01 eV Å−1 were set as the convergence criterion for geometry optimization. The spin polarization was considered in all calculations. To accurately describe the van der Waals (vdW) interaction, the non-local van der Waals density functional (vdW-DF) was employed in our work [40,41].
The Pt (200) surface was obtained by cutting the Pt bulk along the {200} direction based on the XRD result (Supplementary Fig. S3, available as Supplementary Data at NSR online). The thickness of the surface slab was chosen to be a three-layer slab. In all structural optimization calculations, the bottom atoms were frozen, while the other atoms were allowed to relax. A vacuum layer as large as 15 Å was used along the c direction normal to the surface to avoid periodic interactions. For the nitrogen oxidation reaction, the reaction energies of the elementary reactions were employed to estimate the activity of the catalyst.
The free-energy changes of the nitrogen oxidation were calculated to show the reaction trend. The change in free energy (ΔG) of the per reaction step from the initial state to the final state of the reaction was calculated as:
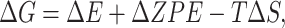 |
where ΔE is the change in the total reaction energy obtained from the density functional theory (DFT) calculations and ΔZPE is the change in the zero-point energy. T is the temperature (298.15 K) and ΔS is the change in entropy. The free energy of H+ is equal to that of half H2 according to the computational hydrogen electrode model proposed by Nørskov [42], while the OH− is estimated by H2O → OH− + H+. The zero-point energy and the entropies of the adsorbed nitrogen oxidation species were taken from the vibrational frequencies.
Supplementary Material
Acknowledgements
The characterizations were made at the Analysis & Testing Center in Tianjin University and the 15N isotope-labeling experiments were conducted at the Shanghai Engineering Research Center of Stable Isotope.
FUNDING
This work was supported by the National Natural Science Foundation of China (21871206, 21701122 and 21422104) and the Natural Science Foundation of Tianjin City (17JCJQJC44700).
Conflict of interest statement. None declared.
REFERENCES
- 1. Hoffman BM, Lukoyanov D, Yang ZYet al.. Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev 2014; 114: 4041–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive Earth's biogeochemical cycles. Science 2008; 320: 1034–9. [DOI] [PubMed] [Google Scholar]
- 3. Chirik PJ. Nitrogen fixation: one electron at a time. Nat Chem 2009; 1: 520–2. [DOI] [PubMed] [Google Scholar]
- 4. Li L, Wang Y, Vanka Set al.. Nitrogen photofixation over III-nitride nanowires assisted by ruthenium clusters of low atomicity. Angew Chem Int Ed 2017; 56: 8701–5. [DOI] [PubMed] [Google Scholar]
- 5. Chen JG, Crooks RM, Seefeldt LCet al.. Beyond fossil fuel-driven nitrogen transformations. Science 2018; 360: eaar6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Honkala K, Hellman A, Remediakis INet al.. Ammonia synthesis from first-principles calculations. Science 2005; 307: 555–8. [DOI] [PubMed] [Google Scholar]
- 7. Smil V. Detonator of the population explosion. Nature 1999; 400: 415. [Google Scholar]
- 8. Huang P, Liu W, He Zet al.. Single atom accelerates ammonia photosynthesis. Sci China Chem 2018; 61: 1187–96. [Google Scholar]
- 9. Wang X, Wang W, Qiao Met al.. Atomically dispersed Au1 catalyst towards efficient electrochemical synthesis of ammonia. Sci Bull 2018; 63: 1246–53. [DOI] [PubMed] [Google Scholar]
- 10. Kitano M, Inoue Y, Yamazaki Yet al.. Ammonia synthesis using a stable electride as an electron donor and reversible hydrogen store. Nat Chem 2012; 4: 934–40. [DOI] [PubMed] [Google Scholar]
- 11. Smith BE. Nitrogenase reveals its inner secrets. Science 2002; 297: 1654–5. [DOI] [PubMed] [Google Scholar]
- 12. van Kessel MA, Speth DR, Albertsen Met al.. Complete nitrification by a single microorganism. Nature 2015; 528: 555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang L, Xia M, Wang Het al.. Greening ammonia toward the solar ammonia refinery. Joule 2018; 2: 1055–74. [Google Scholar]
- 14. Smil V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. Cambridge, MA: MIT Press, 2004. [Google Scholar]
- 15. Li H, Shang J, Ai Zet al.. Efficient visible light nitrogen fixation with BiOBr nanosheets of oxygen vacancies on the exposed {001} facets. J Am Chem Soc 2015; 137: 6393–9. [DOI] [PubMed] [Google Scholar]
- 16. Qiu W, Xie XY, Qiu Jet al.. High-performance artificial nitrogen fixation at ambient conditions using a metal-free electrocatalyst. Nat Commun 2018; 9: 3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Medford AJ, Hatzell MC. Photon-driven nitrogen fixation: current progress, thermodynamic considerations, and future outlook. ACS Catal 2017; 7: 2624–43. [Google Scholar]
- 18. Chen S, Perathoner S, Ampelli Cet al.. Electrocatalytic synthesis of ammonia at room temperature and atmospheric pressure from water and nitrogen on a carbon-nanotube-based electrocatalyst. Angew Chem Int Ed 2017; 56: 2699–703. [DOI] [PubMed] [Google Scholar]
- 19. Zhang N, Jalil A, Wu Det al.. Refining defect states in W18O49 by Mo doping: a strategy for tuning N2 activation towards solar-driven nitrogen fixation. J Am Chem Soc 2018; 140: 9434–43. [DOI] [PubMed] [Google Scholar]
- 20. Rosca V, Duca M, de Groot MTet al.. Nitrogen cycle electrocatalysis. Chem Rev 2009; 109: 2209–44. [DOI] [PubMed] [Google Scholar]
- 21. Shi MM, Bao D, Wulan BRet al.. Au sub-nanoclusters on TiO2 toward highly efficient and selective electrocatalyst for N2 conversion to NH3 at ambient conditions. Adv Mater 2017; 29: 1606550. [DOI] [PubMed] [Google Scholar]
- 22. Pickett CJ, Talarmin J. Electrosynthesis of ammonia. Nature 1985; 317: 652–3. [Google Scholar]
- 23. Bao D, Zhang Q, Meng FLet al.. Electrochemical reduction of N2 under ambient conditions for artificial N2 fixation and renewable energy storage using N2/NH3 cycle. Adv Mater 2017; 29: 1604799. [DOI] [PubMed] [Google Scholar]
- 24. Zhou F, Azofra LM, Ali Met al.. Electro-synthesis of ammonia from nitrogen at ambient temperature and pressure in ionic liquids. Energy Environ Sci 2017; 10: 2516–20. [Google Scholar]
- 25. Showers WJ, Genna B, McDade Tet al.. Nitrate contamination in groundwater on an urbanized dairy farm. Environ Sci Technol 2008; 42: 4683–8. [DOI] [PubMed] [Google Scholar]
- 26. Castoldi L, Matarrese R, Morandi Set al.. New insights on the adsorption, thermal decomposition and reduction of NOx over Pt- and Ba-based catalysts. Appl Catal B 2018; 224: 249–63. [Google Scholar]
- 27. Service RF. Liquid sunshine. Science 2018; 361: 120–3. [DOI] [PubMed] [Google Scholar]
- 28. APHA, AWWA, WPCF . Standard Methods for the Examination of Water and Wastewater. Washington, DC: American Public Health Association, 1998. [Google Scholar]
- 29. Duca M, Koper MTM. Powering denitrification: the perspectives of electrocatalytic nitrate reduction. Energy Environ Sci 2012; 5: 9726–42. [Google Scholar]
- 30. Pérez-Gallent E, Figueiredo MC, Katsounaros Iet al.. Electrocatalytic reduction of nitrate on copper single crystals in acidic and alkaline solutions. Electrochim Acta 2017; 227: 77–84. [Google Scholar]
- 31. Reyter D, Belanger D, Roue L. Nitrate removal by a paired electrolysis on copper and Ti/IrO2 coupled electrodes-influence of the anode/cathode surface area ratio. Water Res 2010; 44: 1918–26. [DOI] [PubMed] [Google Scholar]
- 32. Dash BP, Chaudhari S. Electrochemical denitrificaton of simulated ground water. Water Res 2005; 39: 4065–72. [DOI] [PubMed] [Google Scholar]
- 33. Martinez-Huitle CA, Brillas E. Electrochemical alternatives for drinking water disinfection. Angew Chem Int Ed 2008; 47: 1998–2005. [DOI] [PubMed] [Google Scholar]
- 34. Martínez J, Ortiz A, Ortiz I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates. Appl Catal B 2017; 207: 42–59. [Google Scholar]
- 35. Ardenkjær-Larsen JH, Fridlund B, Gram Aet al.. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc Natl Acad Sci USA 2003; 100: 10158–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett 1996; 77: 3865–8. [DOI] [PubMed] [Google Scholar]
- 37. Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 1996; 6: 15–50. [DOI] [PubMed] [Google Scholar]
- 38. Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B 1993; 47: 558–61. [DOI] [PubMed] [Google Scholar]
- 39. Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B 1976; 13: 5188–92. [Google Scholar]
- 40. Dion M, Rydberg H, Schröder Eet al.. Van der Waals density functional for general geometries. Phys Rev Lett 2004; 92: 246401. [DOI] [PubMed] [Google Scholar]
- 41. Kliměs J, Bowler DR, Michaelides A. Chemical accuracy for the van der Waals density functional. J Phys Condens Matter 2010; 22: 022201. [DOI] [PubMed] [Google Scholar]
- 42. Nørskov JK, Rossmeisl J, Logadottir Aet al.. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B 2004; 108: 17886–92. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.