

Abstract

σ-1 receptors (σ1R) modulate nociceptive signaling, driving the search for selective antagonists to take advantage of this promising target to treat pain. In this study, a new series of benzylpiperazinyl derivatives has been designed, synthesized, and characterized for their affinities toward σ1R and selectivity over the σ-2 receptor (σ2R). Notably, 3-cyclohexyl-1-{4-[(4-methoxyphenyl)methyl]piperazin-1-yl}propan-1-one (15) showed the highest σ1R receptor affinity (Ki σ1 = 1.6 nM) among the series with a significant improvement of the σ1R selectivity (Ki σ2/Ki σ1= 886) compared to the lead compound 8 (Ki σ2/Ki σ1= 432). Compound 15 was further tested in a mouse formalin assay of inflammatory pain and chronic nerve constriction injury (CCI) of neuropathic pain, where it produced dose-dependent (3–60 mg/kg, i.p.) antinociception and anti-allodynic effects. Moreover, compound 15 demonstrated no significant effects in a rotarod assay, suggesting that this σ1R antagonist did not produce sedation or impair locomotor responses. Overall, these results encourage the further development of our benzylpiperazine-based σ1R antagonists as potential therapeutics for chronic pain.
Keywords: Benzylpiperazines, σ receptors, σ-1 antagonist, analgesia, neuropathic pain
Introduction
The σ-1 receptor (σ1R) was initially categorized as an opioid receptor subtype because of the binding with the nonselective benzomorphan alazocine (SKF10,047).1 Subsequent studies have proven that naloxone did not possess antagonism at this receptor,2 and later molecular cloning and the X-ray crystallographic structure of the human σ1R revealed no homology with opioid receptors.3,4 Indeed, unlike opioid receptors, which possess the seven-transmembrane domain structure characteristics of G protein-coupled receptors, σ1R is a transmembrane protein that is present in numerous oligomeric states.4 The protomer contains one transmembrane domain, five α helices, and 10 β strands forming the ligand-binding pocket.4,5 Therefore, σ1R is now recognized as a unique chaperone protein mostly expressed at the endoplasmatic reticulum and is a highly conserved protein among different species with over 90% identical amino acid sequences.6
A second σ receptor subtype (named σ2R) was discovered and differentiated from the first subtype on the basis of size, tissue distribution, and ligand affinity.7,8 σ2R has been even more challenging to define than σ1R, and its crystal structure has not yet been reported. Indeed, the protein has just recently been cloned, with a sequence identical to the transmembrane protein 97 (TMEM97), a protein involved in cholesterol homeostasis.9 Since then, it is general practice to refer to this protein as σ2R/TMEM97. Like σ1R, σ2R/TMEM97 was initially miscategorized and correlated to the progesterone membrane component 1 (PGRMC1), which was thought to be the σ2R binding site.10 Finally, further studies clarified that σ2R/TMEM97 might still interact with PGRMC1 and LDLR (low-density lipoprotein receptor), forming a ternary complex that mediates LDL internalization and trafficking.11,12
Both σ receptor (σR) subtypes are present in several CNS areas and peripheral tissues such as the spleen and liver, as well as overexpressed on different human tumors.6,13 σ receptors are known to be expressed in key areas of pain control in the CNS such as the locus coeruleus, periaqueductal gray, and rostroventral medulla.14,15 In the CNS, these receptors are also very highly expressed in the dorsal root ganglia of the spinal cord, indicating a key role in the function of peripheral pain pathways.16,17 In agreement with their anatomical distribution, σRs modulate a broad range of body functions.18 Conversely, dysregulation of the physiological activities of σRs has been observed in several medical conditions, including drug addiction, neuropsychiatric disorders, cancer, and chronic pain.18−22 With an improving understanding of σRs, σR ligands have recently attracted increased attention from the scientific community for their potential as new medications to treat unmet medical needs,23−25 including the novel coronavirus disease 2019 (COVID-19) as recently reported.26,27 Notably, novel σR selective ligands are currently under evaluation in clinical trials as diagnostic agents (i.e., PET radiotracers) and therapeutic efficacy for some of the diseases mentioned above.28−30
Consistent with their molecular role as chaperones, the σRs interact with various proteins, modifying their function.31,32 Concerning their active modulatory activity in pain signaling, these protein targets include the μ-opioid receptor, ion channels, and the NMDA receptors.14 Significantly, σ1R antagonists block the activity of the σ1Rs, producing increased opioid and decreased NMDA receptor signaling, thereby enhancing antinociception by opioids and decreasing the hypersensitivity commonly associated with pathological pain.33 Interestingly, an increased number of ligands possessing different chemotypes and heterogeneous σRs binding profiles showed a significant antinociception effect in different preclinical in vivo pain models (Figure 1).34−41
Figure 1.
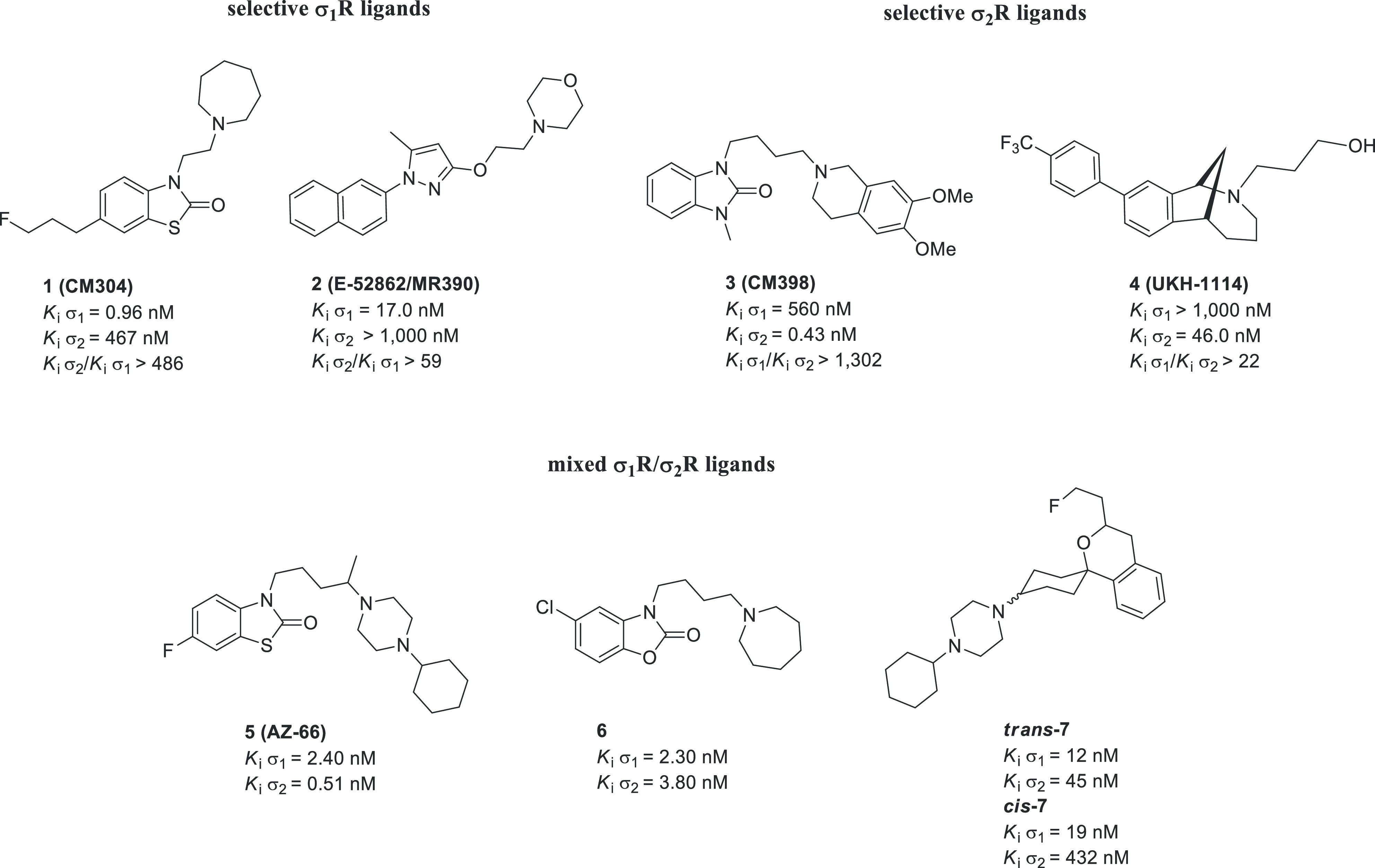
Structures of σRs ligands with antinociceptive activities: selective σ1R antagonists 1 and 2; selective σ2R ligands 3 and 4; mixed σ1R/σ2R ligands 5, 6, trans-7, and cis-7.
Continuing our efforts to discover selective σ1R ligands,42,43 in this paper, a set of new benzylpiperazines was synthesized and pharmacologically characterized for their analgesic effects in mice models of pain. Previously, we developed a series of bifunctional σRs ligands with in vitro antioxidant properties.44 These ligands were obtained by combining a preferred σR cyclic amino moiety, such as benzylpiperazine, with the 1,2-dithiolan-3-yl moiety belonging to the natural antioxidant compound α-lipoic acid (Figure 2A). The 4-methoxybenzylpiperazinyl derivative 8 was previously identified as a potent and selective ligand for the σ1R over σ2R/TMEM97 (Figure 2A). Moreover, previous structure–affinity relationships (SAfiRs) suggested that the introduction of a para-substituent at the secondary hydrophobic domain (HYD2) improved the affinity and selectivity at σ1R.44 On the contrary, little exploration of both the chain linker and the primary hydrophobic domain (HYD1) was performed. With this in mind, we used compound 8 as our lead molecule to develop new benzylpiperazine derivatives as potentially more potent and selective σ1R ligands over σ2R (Figure 2B).
Figure 2.

(A) 2D and 3D structures of lead compound 8. (B) Glennon’s σ1R pharmacophoric features (i.e., primary hydrophobic, blue; basic nitrogen, green; secondary hydrophonic, red) and the general structure of newly benzylpiperazine derivatives.
Similar to 8, the newly synthesized compounds fulfilled Glennon’s pharmacophore model in which two distal hydrophobic regions and a central positive ionizable nitrogen give the essential features for the binding at σ1R (Figure 2A,B).45 Due to the promising outcomes obtained with previous benzylpiperazines, in this new series, we maintained the 4-methoxybenzylpiperazinyl moiety as the HYD2 and we modified the other distal hydrophobic region and the linker portion (i.e., 13–16 and 20–22). Specifically, the substitution of a phenyl, phenoxy, or cyclohexyl group in place of a lipoyl one as the HYD1 was carried out. Additionally, to explore the importance of an additional H-bond donor group for the target binding, the 4-methoxy substituent was replaced with a hydroxymethyl one (24).
Results and Discussion
Chemistry
Reagents and conditions for preparing the final compounds 13–16, 20–22, and 24 are summarized in Schemes 1 and 2. Precisely, 4-methoxybenzylpiperazinyl analogues 13–16 and 20–22 were synthesized starting from the suitable activated acids 9–12 or acyl chlorides 17–19, according to the two pathways reported in Scheme 1. In the first case, acids 9–12 were activated by a reaction with 1,1′-carbonyldiimidazole (CDI) in dry dichloromethane (DCM) to give acyl imidazole intermediates (not isolated), which were then reacted with 1-(4-methoxybenzyl)piperazine in a protective nitrogen atmosphere to afford final amides 13–16 in good yields (36–68%). Amides 20–22 were obtained directly by the coupling of 1-(4-methoxybenzyl)piperazine with the corresponding organic halides (17–19) in dry tetrahydrofuran (THF) and using triethylamine as a base catalyst.
Scheme 1. Reaction Pathways for Compounds 13–16 and 20–22.

Reagents and conditions: (a) CDI, dry DCM, RT, then 1-(4-methoxybenzyl)piperazine, 0 °C for 30 min, then RT, 1–2 h, 33–68%; (b) TEA, dry THF, 0 °C for 30 min, then RT, 1–3 days, 61–67%.
Scheme 2. Reaction Pathways for Compounds 23 and 24.

Reagents and conditions: (a) CDI, dry DCM, room temperature, then piperazine, 0 °C for 30 min, then RT, 1 h, 75%; (b) 4-(chloromethyl)benzyl alcohol, K2CO3, KI, DCM, MW (150 W), 120 °C, 2 h, 90%.
The final compound 24 was prepared following the two-step reaction depicted in Scheme 2. The acid derivative 9 was activated by CDI and converted into amide intermediate 23 by reaction with piperazine. Subsequently, treatment with the commercially available 4-(chloromethyl)benzyl alcohol, with K2CO3 and KI, and using microwaves (MW) irradiation gave the final benzylpiperazine derivative 24 in excellent yield (90%).
Final compounds were characterized as a free base and submitted as such for the in vitro binding assay. Compound 15 has been converted into oxalate salt for in vivo behavioral studies.
σR Binding Properties and SAfiRs
The affinities of the newly synthesized benzylpiperazine derivatives (13–16, 20–22, and 24) for the σ1 and σ2 receptors were evaluated in radioligand binding assay using [3H]-pentazocine and [3H]-DTG, respectively, as radioligands in the presence of haloperidol to determine the nonspecific binding. All tested compounds displayed higher selectivity ratio values (Ki σ2/Ki σ1) than the reference ligand, haloperidol (Table 1). Moreover, compounds 15 and 24 showed improved or similar σ1R selectivity (Ki σ2/Ki σ1= 886 and 423, respectively) compared to the lead compound 8 (Ki σ2/Ki σ1= 432).
Table 1. σRs Binding Affinities for 13–16, 20–22, and 24.
|
Ki (nM) ± SDa |
|||
|---|---|---|---|
| compound | σ1R | σ2R | Ki σ2/Ki σ1 |
| 8b | 5.7 ± 0.1 | 2,460 ± 85 | 432 |
| 13 | 18.1 ± 0.44 | 1,162 ± 40 | 64 |
| 14 | 13.3 ± 0.22 | 1,644 ± 37 | 124 |
| 15 | 1.6 ± 0.05 | 1,418 ± 18 | 886 |
| 16 | 11.3 ± 0.26 | 3,968 ± 130 | 351 |
| 20 | 102 ± 0.6 | 4,367 ± 33 | 43 |
| 21 | 8.8 ± 0.22 | 3,253 ± 40 | 370 |
| 22 | 145 ± 0.5 | 23,190 ± 146 | 160 |
| 24 | 6.1 ± 0.1 | 2,583 ± 54 | 423 |
| haloperidol | 1.6 ± 0.1 | 17.6 ± 0.1 | 11 |
Ki values are expressed as mean ± SD of three independent experiments.
Data from ref (44).
Regarding the σ1R affinity, a clear trend based on the length of the linker chain between the distal phenyl ring and the central amide group can be observed in analogues 13–14, 16, 21, and 22 (i.e., ethylene > vinylene ≃ propylene > butylene ≫ methylene). Indeed, chain shortening from four (13) to two methylene units (21) led to an increase in σ1R affinity (Ki σ1 = 18.1 and 8.8 nM, respectively); however, a further shortening to only one methylene unit (22) was detrimental (Ki σ1 = 145 nM). Thus, the length of the ethylene chain in compound 21 produced optimal σ1R affinity (Ki = 8.8 nM) and selectivity (370-fold) among this set. A phenyl ring instead of a lipoyl one was tolerated concerning the HYD1 domain, although a loss of selectivity was observed (8 vs 13), whereas the introduction of a phenoxy group (20) gave the worst result. Further replacement with a cyclohexyl ring resulted in a remarkable improvement of both the affinity and selectivity for σ1R (15 vs 21).
Finally, an additional H-bond donor group (24) in the secondary hydrophobic domain improved neither the σ1R affinity nor the selectivity significantly (24 vs 8). Compound 15, which showed the best binding profile among the series (Ki σ1 = 1.6 nM; Ki σ2 = 1,418 nM; Ki σ2/Ki σ1 = 886), was selected for a more in-depth in vivo pharmacological evaluation.
Focally Induced Inflammatory Nociception
Due to the lack of reliable in vitro assays to establish the agonist/antagonist properties of σRs ligands, the intrinsic functional activity of 15 was assessed by using a behavioral model of nociception. Consistent with the evidence of σ1R modulation of nociceptive signaling,46 σ1R antagonists ameliorate pain responses in a focally induced inflammatory nociception model such as the formalin assay.15,47,48 Compound 15 showed significant dose-dependent antinociception in this assay (Figure 3A), consistent with action as a putative σ1R antagonist.
Figure 3.

Formalin-induced inflammation assay: (A) compound 15 demonstrates a dose-dependent increase in % antinociception and (B) decreases summed time spent licking of formalin-treated paw in a dose-dependent manner compared to the vehicle control and the selective σ1R antagonist (CM304). n = 10 for all points.
Compound 15 showed a similar efficacy at the highest dose in reducing time spent licking the injected paw compared to the positive control CM304 (1), a well-characterized selective σ1R antagonist (Figure 3B),35 with an ED50 (and 95% C.I.) value of 12.7 (9.9–16.6) mg/kg, i.p. These results confirm previous reports from Romero et al. in 2012, Gris et al. in 2014, and Cirino et al. in 2019,35,49,50 where σ1R antagonists blocked peripheral nociception associated with inflammatory pain responses.
Focally Induced Neuropathy
On the basis of the observed antinociceptive effect exerted by 15 in the formalin assay, we further characterized 15 in a representative model of neuropathic pain. We selected the chronic constriction injury (CCI) model as a widely used and validated assay to produce allodynia.51−54 Compound 15 demonstrated significant dose-dependent anti-allodynic effects (F(5, 184) = 21.17; p < 0.0001; two-way ANOVA with Tukey’s post hoc test; Figure 4), with significant increases in withdrawal thresholds at 20, 60, and 80 min post-injection of a 60 mg/kg, i.p. dose (p < 0.05; Tukey’s post hoc test). These effects were comparable to the results of the positive control gabapentin (Figure 4). CM304, the reference selective σ1R antagonist, demonstrated anti-allodynic effects that are comparable to those of gabapentin in a time-dependent manner. The anti-allodynic effects of CM304 peaked at 40 min but began to diminish at 60 min. The current results are consistent with a mechanistic interpretation of anti-allodynia through the σ1R antagonism. Notably, CCI produces a focal injury of the sciatic nerve that has been demonstrated to enhance the labeling of spinal σ1Rs in a manner enhancing nociceptive signaling.55 Currently, these studies with compound 15 verify previous findings stating that noxious stimuli are attenuated by σ1R antagonists.35,48,56
Figure 4.
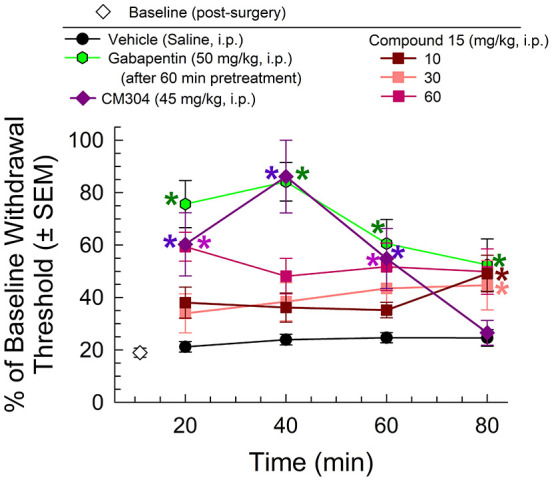
Chronic constriction injury model testing: mechanical allodynia produced from sciatic nerve constriction were reduced after compound 15 treatment, similar to the positive control (gabapentin) with a longer duration of action than the reference compound CM304. n = 8–13 for all groups. * = significantly different from vehicle controls; p < 0.05.
Induced Locomotor Activity
Treatments for neuropathic pain may be complicated by concordant sedation and impairment of motor function, as demonstrated by gabapentin.57 To eliminate the potential complication of impaired locomotion or sedation, the effect of compound 15 on elicited locomotor activity was assessed using the rotarod assay.35 The positive control and κ-opioid receptor agonist trans-(±)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl]benzeneacetamide hydrochloride (U50,488, 10 mg/kg, i.p.) significantly impaired evoked locomotor activity compared to the vehicle control (F(4, 301) = 26.02; p < 0.0001; two-way ANOVA with Tukey’s post hoc test; Figure 5) up to 60 min after administration. In contrast, at doses proving effective in the pain assays, compound 15 did not significantly impair evoked locomotor activity, although the 30 mg/kg, i.p. dose produced a singular increase in locomotor performance 60 min post-administration (p = 0.003). Although the mechanism of σRs involvement in motor coordination and sedation has not yet been fully defined, the current results confirm the recent finding by Cirino et al.,35 showing that selective σ1R antagonists fail to produce sedative effects or impair evoked locomotor activity in rodents, confirming their analgesic properties.
Figure 5.
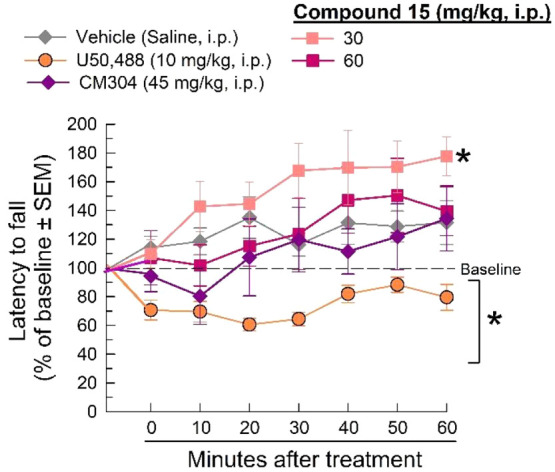
Sedation and evoked, coordinated locomotor function were assessed using the rotarod apparatus following the administration of either saline (i.p.), U50,488 (10 mg/kg, ip), CM304 (45 mg/kg, i.p.), or compound 15 (30 and 60 mg/kg, i.p.). * = significantly different from baseline response (p < 0.05). n = 8–12.
Conclusions
This work has described the design and synthesis of new benzylpiperazinyl derivatives possessing high affinities for σ1R (Ki σ1 = 1.6–145 nM) and selectivity over σ2R (Ki σ2/Ki σ1 = 43–886). Following Glennon’s structural features criteria necessary for σ1R binding, we discovered compound 15 as a potent and selective σ1R ligand. Especially, the use of hydrophobic cyclohexyl or phenyl groups and the 4-methoxybenzylpiperazinyl moiety (HYD1 and HYD2, respectively) linked by three-carbon units linker (i.e., 15, 16, and 21) was an excellent combination to obtain optimal σRs binding profiles. Importantly, behavioral pharmacology studies showed that 15 produced significant antinociceptive and anti-allodynic effects in preclinical mouse models of pain without impaired locomotor activity, supporting the development of benzylpiperazine-based σ1R antagonists as potential therapeutics for chronic pain.
Methods
Chemistry
Melting points were performed in an IA9200 electrothermal apparatus equipped with a digital thermometer in glass capillary tubes and are uncorrected. The elemental analyses for C, H, and N were within ±0.4% of the theoretical values and were recorded on a Carlo Erba elemental analyzer Mod 1108 apparatus. Infrared spectra were determined in KBr disks (solid samples) or NaCl plates (oil samples) on a PerkinElmer 1600 Series FT-IR spectrometer. The 1H NMR and 13C NMR spectra of intermediate and final compounds were recorded with a Varian Inova Unity (200 MHz) spectrometer and a Varian Inova Unity (500 MHz) spectrometer using a DMSO-d6 solution. The chemical shifts are reported in δ values (ppm), using tetramethylsilane (TMS) as the internal standard; the coupling constants (J) are given in hertz (Hz). The signal multiplicities are characterized as s (singlet), d (doublet), t (triplet), or m (multiplet). Microwave irradiation experiments were carried out with a CEM Discovery instrument using closed Pyrex glass tubes with Teflon-coated septa. Thin-layer chromatography (TLC) on Merck plates (aluminum sheet coated with silica gel 60 F254) was used to monitor the progress of reactions and to test the purity (≥95%) of all the synthesized compounds, and spots were visualized under UV (λ = 254 and 366 nm) or in an iodine chamber. The purification of synthesized compounds by column chromatography was performed using Merck silica-gel 60 (230–400 mesh). All chemicals and solvents were purchased from commercial vendors and were of reagent grade.
General Procedure for the Synthesis of 4-(Methoxyphenyl)methylpiperazine Derivatives 13–16
1,1′-Carbonyldiimidazole (1.0 equiv) was mixed to a stirred solution of suitable acid 9–12 (1.0 equiv) in dry DCM (6 mL) at room temperature. Then, after no gas evolution was observed, the mixture thus obtained was added dropwise to a stirred solution of 1-(4-methoxybenzyl)piperazine (1.1 equiv) in dry DCM (6 mL) at 0 °C under a nitrogen atmosphere. The reaction was carried out for 30 min at 0 °C and then for 1–2 h at room temperature. The mixture was washed with 10% aqueous NaCl solution (4 × 10 mL) and H2O (2 × 10 mL). The organic layer was dried over anhydrous sodium sulfate and then evaporated under reduced pressure to obtain a crude, which was purified as specified for each final product.
1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-5-phenylpentan-1-one (13)
The yellow crude oil was purified by flash column chromatography using ethyl acetate/methanol (9.5:0.5, v/v) as an eluent to afford 13 (0.547 g, 60.4%) as a colorless oil. IR (neat, selected lines): cm–1 3447, 2945, 1646, 1508, 1458, 1242, 748. 1H NMR (200 MHz, DMSO-d6): δ 7.00–7.35 (m, 5H + 2H, aromatic), 6.80–6.95 (m, 2H, aromatic), 3.73 (s, 3H, OCH3), 3.35–3.50 (m, 4H + 2H, piperazine + ArCH2N), 2.57 (t, J = 7.0 Hz, 2H, NCOCH2CH2), 2.10–2.40 (m, 4H + 2H, piperazine + CH2CH2C6H5), 1.35–1.65 (m, 4H, CH2CH2CH2CH2). 13C NMR (126 MHz, DMSO-d6): δ 170.4, 158.3, 142.1, 130.1, 129.6, 128.3, 128.2, 125.6, 113.6, 61.3, 55.0, 52.7, 52.2, 44.9, 41.0, 34.9, 32.1, 30.6, 24.4. Anal. calcd for C23H30N2O2: C, 75.37; H, 8.25; N, 7.64. Found: C, 75.15; H, 8.32; N, 7.56.
1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-4-phenylbutan-1-one (14)
The crude was purified by recrystallization from ethanol/water (1:2, v/v) to afford 14 (0.431 g, 54.5%) as white crystals. Mp: 91.0–93.9 °C. IR (KBr, selected lines): cm–1 3028, 2952, 1640, 1509, 1240. 1H NMR (200 MHz, DMSO-d6): δ 7.37–7.10 (m, 5H + 2H, aromatic), 6.95–6.80 (m, 2H, aromatic), 3.72 (s, 3H, OCH3), 2.57 (s, 2H, ArCH2N), 3.50–3.20 (m, 4H, piperazine), 3.40 (t, J = 7.2 Hz, 2H, NCOCH2CH2), 2.40–2.20 (m, 4H + 2H, piperazine + CH2CH2C6H5), 1.85–1.60 (m, 2H, CH2CH2CH2). 13C NMR (50 MHz, DMSO-d6): δ 170.2, 158.4, 141.9, 130.2, 129.6, 128.3, 125.8, 113.6, 61.3, 55.0, 52.8, 52.3, 44.9, 41.0, 34.7, 31.7, 26.8. Anal. calcd for C22H28N2O2: C, 74.97; H, 8.01; N, 7.95. Found: C, 75.11; H, 8.22; N, 7.83.
3-Cyclohexyl-1-{4-[(4-methoxyphenyl)methyl]piperazin-1-yl}propan-1-one (15)
Compound 15 was prepared by the general procedure described for the synthesis of derivatives 13–16 using dry THF instead of dry DCM as a solvent. The yellow crude oil was purified by flash column chromatography using ethyl acetate/methanol (9.7:0.3, v/v) as an eluent to afford 15 (0.360 g, 54.4%) as a white solid. Mp: 69.6–72.5 °C. IR (KBr, selected lines): cm–1 3064, 3029, 2828, 1643, 1277, 1037, 737. 1H NMR (free base, 200 MHz, DMSO-d6): δ 7.25–7.15 (m, 2H, aromatic), 6.95–6.85 (m, 2H, aromatic), 3.73 (s, 3H, OCH3), 3.50–3.20 (m, 4H, piperazine), 3.40 (s, 2H, ArCH2N), 2.40–2.10 (m, 4H + 2H, piperazine + NCOCH2CH2), 1.75–1.45 (m, 5H, cyclohexane), 1.40–1.00 (m, 2H + 4H, CH2CH2C6H11 + cyclohexane), 1.00–0.70 (m, 2H, cyclohexane). 13C NMR (oxalate salt, 126 MHz, DMSO-d6): δ 171.1, 163.2, 159.4, 131.7, 125.0, 114.0, 59.7, 55.2, 51.5, 51.1, 43.2, 36.8, 32.7, 32.2, 29.7, 26.2, 25.8. Anal. calcd for C21H32N2O2: C, 73.22; H, 9.36; N, 8.13. Found: C, 73.02; H, 9.13; N, 8.32.
(2E)-1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-3-phenylprop-2-en-1-one (16)
The yellow crude oil was purified by flash column chromatography using ethyl acetate/methanol (9.7:0.3, v/v) as an eluent to afford 16 (0.139 g, 32.8%) as a light yellow solid. Mp: 120.8–122.6 °C. IR (KBr, selected lines): cm–1 2986, 1651, 1605, 1455, 1236. 1H NMR (200 MHz, DMSO-d6): δ 7.80–7.60 (m, 2H, aromatic), 7.55–7.31 (m, 3H + 1H, aromatic + COCH=CH), 7.30–7.12 (m, 2H + 1H, aromatic + COCH=CH), 6.95–6.80 (m, 2H, aromatic), 3.74 (s, 3H, OCH3), 3.80–3.25 (m, 4H, piperazine), 3.43 (s, 2H, ArCH2N), 2.55–2.20 (m, 4H, piperazine). 13C NMR (126 MHz, DMSO-d6): δ 164.4, 158.3, 141.5, 135.1, 130.2, 129.6, 129.5, 128.7, 128.0, 118.2, 113.6, 61.2, 55.0, 53.1, 52.2, 45.1, 41.7. Anal. calcd for C21H24N2O2: C, 74.97; H, 7.19; N, 8.33. Found: C, 74.78; H, 7.32; N, 8.11.
General Procedure for the Synthesis of 4-(Methoxyphenyl)methylpiperazine Derivatives 20–22
A mixture of 1-(4-methoxybenzyl)piperazine (1.0 equiv) and triethylamine (1.0 equiv) in dry THF (5 mL) was prepared and left under stirring for 10 min at 0 °C. Subsequently, the appropriate acyl chloride (17–19, 1.0 equiv) was added to the obtained solution, and the reaction was carried out at 0 °C for 30 min and then at room temperature for 1–3 days. At the end of the reaction time, the solvent was evaporated to dryness under a vacuum. The crude product thus obtained was solubilized in DCM and then washed with a water solution of Na2CO3 0.1 M (2 × 20 mL) and NaCl 10% (20 mL). The organic layer was dried over anhydrous sodium sulfate and evaporated under reduced pressure to obtain a residue, which was purified as specified for each final product.
1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-4-phenoxybutan-1-one (20)
The yellow crude oil was triturated with light petroleum ether at 40–60 °C to give a white solid, which was collected, washed with petroleum ether, and dried. The crude thus obtained was purified by recrystallization from ethanol/water (1:2, v/v) to afford 20 (0.277 g, 59.8%) as white crystals. Mp: 88.3–91.3 °C. IR (KBr, selected lines): cm–1 3058, 2945, 1647, 1252, 757. 1H NMR (200 MHz, DMSO-d6): δ 7.35–7.15 (m, 2H + 2H, aromatic), 7.00–6.80 (m, 2H + 3H, aromatic), 3.96 (t, J = 6.4 Hz, 2H, CH2CH2OPh), 3.73 (s, 3H, OCH3), 3.50–3.20 (m, 4H, piperazine), 3.39 (s, 2H, ArCH2N), 2.45 (t, J = 7.2 Hz, 2H, NCOCH2CH2), 2.35–2.20 (m, 4H, piperazine), 2.00–1.80 (m, 2H, CH2CH2CH2). 13C NMR (50 MHz, DMSO-d6): δ 170.0, 158.5, 158.4, 130.2, 129.6, 129.5, 120.5, 114.4, 113.6, 66.7, 61.3, 55.0, 52.7, 52.3, 44.9, 41.1, 28.6, 24.5. Anal. calcd for C22H28N2O3: C, 71.71; H, 7.66; N, 7.60. Found: C, 71.94; H, 7.75; N, 7.73.
1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-3-phenylpropan-1-one (21)
The yellow crude oil was purified by flash column chromatography using ethyl acetate/methanol (9.5:0.5, v/v) as an eluent to afford 21 (0.289 g, 67.9%) as a colorless oil. IR (neat, selected lines): cm–1 3482, 2934, 2806, 1637, 1512, 1245, 1032, 999, 701. 1H NMR (200 MHz, DMSO-d6): δ 7.35–7.10 (m, 5H + 2H, aromatic), 6.95–6.80 (m, 2H, aromatic), 3.73 (s, 3H, OCH3), 3.55–3.20 (m, 4H, piperazine), 3.38 (s, 2H, ArCH2N), 2.79 (t, J = 7.8 Hz, 2H, NCOCH2CH2), 2.58 (t, J = 7.8 Hz, 2H, CH2CH2C6H5), 2.40–2.15 (m, 4H, piperazine). 13C NMR (126 MHz, DMSO-d6): δ 169.8, 158.3, 141.4, 130.1, 129.6, 128.4, 128.2, 125.9, 113.6, 61.3, 55.0, 52.6, 52.2, 44.9, 41.1, 33.9, 30.8. Anal. calcd for C21H26N2O2: C, 74.52; H, 7.74; N, 8.28. Found: C, 74.41; H, 7.60; N, 8.10.
1-{4-[(4-Methoxyphenyl)methyl]piperazin-1-yl}-2-phenylethan-1-one (22)
The yellow crude oil was purified by flash column chromatography using ethyl acetate/methanol (9.5:0.5, v/v) as an eluent to afford 22 (0.252 g, 61.7%) as a white solid. Mp: 97.6–99.8 °C. IR (KBr, selected lines): cm–1 3032, 3018, 2924, 2802, 1647, 1438, 1235, 1036, 794. 1H NMR (200 MHz, DMSO-d6): δ 7.35–7.10 (m, 5H + 2H, aromatic), 6.93–6.80 (m, 2H, aromatic), 3.73 (s, 3H, OCH3), 3.69 (s, 2H, NCOCH2C6H5), 3.50–3.30 (m, 4H, piperazine), 3.38 (s, 2H, ArCH2N), 2.30–2.15 (m, 4H, piperazine). 13C NMR (50 MHz, DMSO-d6): δ 168.8, 158.4, 135.9, 130.2, 129.6, 129.0, 128.4, 126.4, 113.6, 61.3, 55.0, 52.7, 52.1, 45.5, 41.3. Anal. calcd for C20H24N2O2: C, 74.04; H, 7.46; N, 8.64. Found: C, 73.87; H, 7.27; N, 8.55.
5-Phenyl-1-(piperazin-1-yl)pentan-1-one (23)
1,1′-Carbonyldiimidazole (0.910 g, 5.61 mmol) was added to a solution of 5-phenyl-valeric acid (9) (0.80 g, 4.49 mmol) in dry DCM (8 mL) at room temperature. Then, after no gas evolution was observed, the mixture thus obtained was added dropwise to a stirred solution of piperazine (1.93 g, 22.44 mmol) in dry DCM (10 mL) at 0 °C, under a nitrogen atmosphere. The reaction was carried out for 30 min at 0 °C and for 1 h at room temperature. The mixture was washed with 10% aqueous NaCl solution (4 × 10 mL) and H2O (2 × 10 mL). The organic layer was dried over anhydrous sodium sulfate and evaporated to dryness under reduced pressure to obtain 23 (0.82 g, 73.8%) as a pure yellow oil and used for the next step without further purification. IR (KBr, selected lines): cm–1 3460, 2936, 1652, 1455, 701. 1H NMR (200 MHz, DMSO-d6): δ 7.34–7.09 (m, 5H, aromatic), 3.60–2.80 (m, 4H, piperazine), 2.68–2.46 (m, 4H + 2H, piperazine + NCOCH2CH2), 2.28 (t, J = 7.2 Hz, 2H, CH2CH2C6H5), 1.68–1.48 (m, 4H, CH2CH2CH2CH2). Anal. calcd for C15H22N2O: C, 73.13; H, 9.00; N, 11.37. Found: C, 73.00; H, 9.15; N, 11.17.
1-(4-{[4-(Hydroxymethyl)phenyl]methyl}piperazin-1-yl)-5-phenylpentan-1-one (24)
A mixture of compound 16 (0.765 g, 2.98 mmol), K2CO3 (0.619 g, 4.48 mmol), a catalytic amount of KI, and [4-(chloromethyl)phenyl]methanol (0.146 g, 3.58 mmol) in DCM (2 mL) was placed in a 10 mL Pyrex glass tube, sealed with a Teflon-coated septum. The mixture was heated and stirred at 120 °C under microwave irradiations for 2 h (run time 2 min, microwave max power 150W, max pressure 150 Psi). Subsequently, the reaction mixture was washed with 10% aqueous NaCl solution (4 × 10 mL) and H2O (2 × 10 mL). The organic layer was dried over anhydrous sodium sulfate and evaporated to dryness under reduced pressure to obtain a yellow oil. The purification of the crude product was performed by flash column chromatography using ethyl acetate/methanol (9:1, v/v) as an eluent to afford 24 (0.54 g, 91.7%) as a light yellow oil. IR (KBr, selected lines): cm–1 3420, 3024, 2933, 1636, 1458, 1346, 1231, 1000, 701. 1H NMR (200 MHz, DMSO-d6): δ 7.35–7.10 (m, 5H + 4H, aromatic), 5.16 (t, J = 5.7 Hz, 1H, CH2OH), 4.47 (d, J = 5.7 Hz, 2H, CH2OH), 3.50–3.15 (m, 4H, piperazine), 3.44 (s, 2H, ArCH2N), 2.57 (t, J = 7.2 Hz, 2H, NCOCH2CH2), 2.40–2.10 (m, 2H + 4H, CH2CH2C6H5 + piperazine), 1.70–1.35 (m, 4H, CH2CH2CH2CH2). 13C NMR (126 MHz, DMSO-d6): δ 170.4, 142.1, 141.3, 136.1, 128.7, 128.3, 128.2, 126.4, 125.6, 62.7, 61.7, 52.9, 52.3, 44.9, 41.0, 34.9, 32.1, 30.5, 24.4. Anal. calcd for C23H30N2O2: C, 75.37; H, 8.25; N, 7.64. Found: C, 75.20; H, 8.09; N, 7.50.
σRs Binding Assays
Binding tests were performed following known reported protocols.39,44 The binding assay for σ1R was carried out using guinea pig brain membrane homogenates according to DeHaven-Hudkins et al.,58 while the binding assay for σ2R was performed following experimental procedures described by Mach et al.59 Inhibitory constants (Ki) were calculated using the radioligand binding analysis software EBDA/Ligand (Elsevier/Biosoft).
Behavioral Pharmacology
Animals
Adult male C57BL/6J and CD-1 mice housed five to a cage (8–12 weeks of age) were used. C57BL/6J mice were used for evoked locomotor rotarod and formalin assays.35,60,61 Antinociception was confirmed with the use of CD-1 mice in the CCI nerve assay. The CD-1 strain has been well-validated for antinociceptive62 and mechanical anti-allodynic testing.63,64 All test compounds were administered using the intraperitoneal (i.p.) route. All animal studies reported herein adhere to ARRIVE guidelines.65 Animals were randomly assigned, and researchers were blinded to group treatments. Animals were housed on a 12:12 h light/dark cycle (lights off at 7:00 pm) with ad libitum access to food and water except during experimental sessions. All procedures were preapproved by the Institutional Animal Care and Use Committee (University of Florida) and conducted in accordance with the 2011 NIH Guide for the Care and Use of Laboratory Animals.
Formalin Test
The efficacy of compound 15 to ameliorate inflammatory nociception was achieved with the use of C57BL/6J mice in the formalin assay as previously described.53 After a 10 min pretreatment (i.p.) of vehicle control (saline), CM304 (3–30 mg/kg, i.p.), or compound 15 (3–30 mg/kg, i.p.), an intraplantar (i.pl.) injection of 5% formalin (2.5 μg in 15 μL) was administered into the right hind paw. Time spent licking the right hind paw was recorded in 5 min intervals for 60 min following injection. The last 55 min of assessment was used to determine the inflammatory response stimulus. Data were analyzed as the summed duration of licking hind paw.
CCI Assay
CCI was introduced in CD-1 mice that were first anesthetized with isoflurane as described by Hoot et al.66 and Cirino et al.35 to induce mechanical allodynia.51−54 After anesthetization, mice were subjected to surgery where an incision was made along the surface of the biceps femoris of the right hind paw.66 Blunt forceps were used to split the muscle and expose the right sciatic nerve. The tips of two 0.1–10 μL pipet tips facing opposite directions were passed under the sciatic nerve to allow for easy passing of two sutures under the nerve, 1 mm apart. The sutures were tied loosely around the nerve and knotted twice, and the skin was closed with 29 mm skin staples. Mice were given a 7 day recovery period prior to the baseline von Frey testing as described below to confirm the induction of hyperalgesia in each mouse. Animals demonstrating allodynia or a response to lower pressure were deemed to have neuropathic pain. Allodynic mice were then administered (i.p.) either vehicle (saline), morphine (10 mg/kg, i.p.), gabapentin (50 mg/kg, i.p.), CM304 (45 mg/kg, i.p.), or compound 15 (10–60 mg/kg, i.p.). Note that gabapentin was tested 1 h postinjection to circumvent known sedative effects that may confound the assay.67 Each mouse was then tested for the modulation of tactile allodynia every 20 min up until 80 min post-injection with the use of von Frey testing. The assessment of mechanical allodynia was performed to measure compound 15’s efficacy against CCI-induced allodynia as described.51−54 Mice were habituated on a mesh platform for 1 h prior to testing. Filaments of increasing pressure (0.4–6 g) were applied and then held to the plantar surface of both the injured and uninjured hind paws of mice for approximately 1–2 s prior to drug administration to record baseline responses to a peripheral stimulus. The filaments were applied with increasing strengths, and threshold responses were defined as two hind paw responses per trial of the same filament strength.
Control or test compounds were administered (i.p.), and paw-withdrawal thresholds were again recorded from 20 to 80 min postinjection. Each hind paw was tested in a counterbalanced manner. Each time was measured in triplicate and then averaged. Responsiveness was a clear withdrawal, shaking, or licking of the paw. To account for variability between mice, data are presented as the percent of baseline paw withdrawal thresholds following filament stimulation of the ipsilateral hind paw. The following equation was used: % anti-allodynia = 100 × ([mean paw withdrawal force {g} in control group – paw withdrawal force {g} of each mouse]/mean paw withdrawal force [g] in control group).
Rotarod Assay
The rotarod coordination assay was used to assess effects on evoked locomotor activity in C57BL/6J mice administered vehicle (saline, i.p.), morphine (10 mg/kg, i.p.), U50,488 (10 mg/kg, i.p.), CM304 (45 mg/kg, i.p.), or compound 15 (30–60 mg/kg, i.p.) using methods described previously.68,69 Seven habituation trials were performed where the last habituation trial was used as an initial baseline of performance. The mice were administered (i.p.) test agents and then evaluated every 10 min in accelerated speed trials (180 s max latency at 0–20 rpm) over a 60 min period. The latency to fall was measured in seconds. Data are reported as the mean percent change from each mouse’s initial baseline latency to fall. Decreased latencies to fall in the rotarod test indicate impaired motor coordination or sedation
Statistical Analysis
All data are presented as mean ± SEM. Significance is indicated as *p < 0.05 and was analyzed using two-way ANOVA with Tukey’s post hoc analysis. Statistical analysis was performed with the use of GraphPad Prism 9.0 software. Dose response lines were analyzed by linear or nonlinear regression modeling and ED50 values (dose yielding 50% effect) along with 95% confidence limits using each individual data points. The rotarod data are expressed as the % change from baseline performance for each animal’s baseline response.
Acknowledgments
The authors would like to thank Dr. Christopher R. McCurdy for providing the selective σ1R antagonist CM304.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.1c00106.
Figures of 1H and 13C NMR spectra of compounds 13–16, 20–22, 23, and 24 (PDF)
Author Contributions
G.R., J.P.M., and S.I. participated in research design. G.R., F.B., and V.P. synthesized, purified, and characterized all compounds. E.A. conducted in vitro binding experiments. L.L.W. conducted in vivo pharmacology experiments. M.N.M. and L.S. contributed reagents, materials, and analysis tools. M.N.M., O.P., L.W., and J.P.M. performed data analysis. S.I. wrote the original draft. G.R., L.L.W., J.P.M., and S.I. wrote and contributed to the writing of the manuscript. All authors reviewed and approved the final version of the manuscript.
This work was supported, in part, by grants from PON R&I funds 2014–2020, CUP: E66C18001320007, AIM1872330, activity 1 (S.I.) and from the Department of Defense CMRMP PR161310/P1 (J.P.M.). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
The authors declare no competing financial interest.
Supplementary Material
References
- Gilbert P. E.; Martin W. R. (1976) The effects of morphine and nalorphine-like drugs in the nondependent, morphine-dependent and cyclazocine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 198, 66–82. [PubMed] [Google Scholar]
- Vaupel D. B. (1983) Naltrexone fails to antagonize the sigma effects of PCP and SKF 10,047 in the dog. Eur. J. Pharmacol. 92, 269–274. 10.1016/0014-2999(83)90297-2. [DOI] [PubMed] [Google Scholar]
- Hanner M.; Moebius F. F.; Flandorfer A.; Knaus H. G.; Striessnig J.; Kempner E.; Glossmann H. (1996) Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. U. S. A. 93, 8072–8077. 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Zheng S.; Gurpinar E.; Koehl A.; Manglik A.; Kruse A. C. (2016) Crystal structure of the human σ1 receptor. Nature 532, 527–530. 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H. R.; Kruse A. C. (2019) The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 40, 636–654. 10.1016/j.tips.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekuda R.; Prasad P. D.; Fei Y. J.; Leibach F. H.; Ganapathy V. (1996) Cloning and functional expression of the human type 1 sigma receptor (hSigmaR1). Biochem. Biophys. Res. Commun. 229, 553–5588. 10.1006/bbrc.1996.1842. [DOI] [PubMed] [Google Scholar]
- Bowen W. D.; Hellewell S. B.; McGarry K. A. (1989) Evidence for a multi-site model of the rat brain sigma receptor. Eur. J. Pharmacol. 163, 309–318. 10.1016/0014-2999(89)90200-8. [DOI] [PubMed] [Google Scholar]
- Hellewell S. B.; Bowen W. D. (1990) A sigma-like binding site in rat pheochromocytoma (PC12) cells: decreased affinity for (+)-benzomorphans and lower molecular weight suggest a different sigma receptor form from that of guinea pig brain. Brain Res. 527, 244–253. 10.1016/0006-8993(90)91143-5. [DOI] [PubMed] [Google Scholar]
- Alon A.; Schmidt H. R.; Wood M. D.; Sahn J. J.; Martin S. F.; Kruse A. C. (2017) Identification of the gene that codes for the σ(2) receptor. Proc. Natl. Acad. Sci. U. S. A. 114, 7160–7165. 10.1073/pnas.1705154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zeng C.; Chu W.; Pan F.; Rothfuss J. M.; Zhang F.; Tu Z.; Zhou D.; Zeng D.; Vangveravong S. H.; et al. (2011) Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat. Commun. 2, 380. 10.1038/ncomms1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riad A.; Zeng C.; Weng C. C.; Winters H.; Xu K.; Makvandi M.; Metz T.; Carlin S.; Mach R. H. (2018) Sigma-2 Receptor/TMEM97 and PGRMC-1 Increase the rate of internalization of LDL by LDL receptor through the formation of a ternary complex. Sci. Rep. 8, 16845. 10.1038/s41598-018-35430-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu U. B.; Mavlyutov T. A.; Chu M. L.; Yang H.; Schulman A.; Mesangeau C.; McCurdy C. R.; Guo L. W.; Ruoho A. E. (2015) The sigma-2 receptor and progesterone receptor membrane component 1 are different binding sites derived from independent genes. EBioMedicine 2, 1806–1813. 10.1016/j.ebiom.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilner B. J.; John C. S.; Bowen W. D. (1995) Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 55, 408–413. [PubMed] [Google Scholar]
- Cobos E. J.; Entrena J. M.; Nieto F. R.; Cendán C. M.; Pozo E. (2008) Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr. Neuropharmacol. 6, 344–366. 10.2174/157015908787386113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Fernández C.; Entrena J. M.; Baeyens J. M.; Cobos E. J. (2017) Sigma-1 Receptor antagonists: a new class of neuromodulatory analgesics. Adv. Exp. Med. Biol. 964, 109–132. 10.1007/978-3-319-50174-1_9. [DOI] [PubMed] [Google Scholar]
- Sánchez-Fernández C.; Montilla-García Á.; González-Cano R.; Nieto F. R.; Romero L.; Artacho-Cordón A.; Montes R.; Fernández-Pastor B.; Merlos M.; Baeyens J. M.; Entrena J. M.; Cobos E. J. (2014) Modulation of peripheral μ-opioid analgesia by σ1 receptors. J. Pharmacol. Exp. Ther. 348, 32–45. 10.1124/jpet.113.208272. [DOI] [PubMed] [Google Scholar]
- Bravo-Caparrós I.; Ruiz-Cantero M. C.; Perazzoli G.; Cronin S. J. F.; Vela J. M.; Hamed M. F.; Penninger J. M.; Baeyens J. M.; Cobos E. J.; Nieto F. R. (2020) Sigma-1 receptors control neuropathic pain and macrophage infiltration into the dorsal root ganglion after peripheral nerve injury. FASEB J. 34, 5951–5966. 10.1096/fj.201901921R. [DOI] [PubMed] [Google Scholar]
- Rousseaux C. G.; Greene S. F. (2016) Sigma receptors [σRs]: biology in normal and diseased states. J. Recept. Signal Transduction Res. 36, 327–388. 10.3109/10799893.2015.1015737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambo D. O.; Lebowitz J. J.; Khoshbouei H. (2018) The sigma-1 receptor as a regulator of dopamine neurotransmission: a potential therapeutic target for methamphetamine addiction. Pharmacol. Ther. 186, 152–167. 10.1016/j.pharmthera.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. S.; Lu H. L.; Zhang L. J.; Wu Z. (2014) Sigma-2 receptor ligands and their perspectives in cancer diagnosis and therapy. Med. Res. Rev. 34, 532–566. 10.1002/med.21297. [DOI] [PubMed] [Google Scholar]
- Jia J.; Cheng J.; Wang C.; Zhen X. (2018) Sigma-1 receptor-modulated neuroinflammation in neurological diseases. Front. Cell. Neurosci. 12, 314. 10.3389/fncel.2018.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos M.; Romero L.; Zamanillo D.; Plata-Salamán C.; Vela J. M. (2017) Sigma-1 receptor and pain. Handb. Exp. Pharmacol. 244, 131–161. 10.1007/164_2017_9. [DOI] [PubMed] [Google Scholar]
- Intagliata S.; Alsharif W. F.; Mesangeau C.; Fazio N.; Seminerio M.; Xu Y.-T.; Matsumoto R. R.; McCurdy C. R. (2019) Benzimidazolone-based selective σ2 receptor ligands: Synthesis and pharmacological evaluation. Eur. J. Med. Chem. 165, 250–257. 10.1016/j.ejmech.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Waarde A.; Rybczynska A. A.; Ramakrishnan N. K.; Ishiwata K.; Elsinga P. H.; Dierckx R. A. (2015) Potential applications for sigma receptor ligands in cancer diagnosis and therapy. Biochim. Biophys. Acta, Biomembr. 1848, 2703–2714. 10.1016/j.bbamem.2014.08.022. [DOI] [PubMed] [Google Scholar]
- Nicholson H. E.; Alsharif W. F.; Comeau A. B.; Mesangeau C.; Intagliata S.; Mottinelli M.; McCurdy C. R.; Bowen W. D. (2019) Divergent cytotoxic and metabolically stimulative functions of sigma-2 receptors: structure-activity relationships of 6-acetyl-3-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)benzo[d]oxazol-2(3H)-one (SN79) derivatives. J. Pharmacol. Exp. Ther. 368, 272–281. 10.1124/jpet.118.253484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O’Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; et al. (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Hiatt J.; Bouhaddou M.; Rezelj V. V.; Ulferts S.; Braberg H.; Jureka A. S.; Obernier K.; Guo J. Z.; Batra J.; et al. (2020) Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 370, eabe9403 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agha H.; McCurdy C. R. (2021) In vitro and in vivo sigma 1 receptor imaging studies in different disease states. RSC Med. Chem. 12, 154–177. 10.1039/D0MD00186D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C.; Riad A.; Mach R. H. (2020) The biological function of sigma-2 receptor/TMEM97 and its utility in PET imaging studies in cancer. Cancers 12, 1877. 10.3390/cancers12071877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz H. P.; Stoll K. D. (2004) Clinical trials with sigma ligands. Pharmacopsychiatry 37 (Suppl 3), 214–220. 10.1055/s-2004-832680. [DOI] [PubMed] [Google Scholar]
- Penke B.; Fulop L.; Szucs M.; Frecska E. (2017) The role of sigma-1 receptor, an intracellular chaperone in neurodegenerative diseases. Curr. Neuropharmacol. 16 (1), 97–116. 10.2174/1570159X15666170529104323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Su T. P. (2007) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M.; Sánchez-Blázquez P.; Herrero-Labrador R.; Martínez-Murillo R.; Merlos M.; Vela J. M.; Garzón J. (2015) The σ1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxid. Redox Signaling 22, 799–818. 10.1089/ars.2014.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz J. L.; Cuberes R.; Berrocal J.; Contijoch M.; Christmann U.; Fernández A.; Port A.; Holenz J.; Buschmann H.; Laggner C.; et al. (2012) Synthesis and biological evaluation of the 1-arylpyrazole class of σ(1) receptor antagonists: identification of 4-{2-[5-methyl-1-(naphthalen-2-yl)-1H-pyrazol-3-yloxy]ethyl}morpholine (S1RA, E-52862). J. Med. Chem. 55, 8211–8224. 10.1021/jm3007323. [DOI] [PubMed] [Google Scholar]
- Cirino T. J.; Eans S. O.; Medina J. M.; Wilson L. L.; Mottinelli M.; Intagliata S.; McCurdy C. R.; McLaughlin J. P. (2019) Characterization of sigma 1 receptor antagonist CM-304 and its analog, AZ-66: novel therapeutics against allodynia and induced pain. Front. Pharmacol. 10, 678. 10.3389/fphar.2019.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déciga-Campos M.; Melo-Hernández L. A.; Torres-Gómez H.; Wünsch B.; Schepmann D.; González-Trujano M. E.; Espinosa-Juárez J.; López-Muñoz F. J.; Navarrete-Vázquez G. (2020) Design and synthesis of N-(benzylpiperidinyl)-4-fluorobenzamide: a haloperidol analog that reduces neuropathic nociception via σ(1) receptor antagonism. Life Sci. 245, 117348. 10.1016/j.lfs.2020.117348. [DOI] [PubMed] [Google Scholar]
- Intagliata S.; Sharma A.; King T. I.; Mesangeau C.; Seminerio M.; Chin F. T.; Wilson L. L.; Matsumoto R. R.; McLaughlin J. P.; Avery B. A.; McCurdy C. R. (2020) Discovery of a highly selective sigma-2 receptor ligand, 1-(4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-3-methyl-1H-benzo[d]imidazol-2(3H)-one (CM398), with drug-like properties and antinociceptive effects in vivo. AAPS J. 22, 94. 10.1208/s12248-020-00472-x. [DOI] [PubMed] [Google Scholar]
- Sahn J. J.; Mejia G. L.; Ray P. R.; Martin S. F.; Price T. J. (2017) Sigma 2 receptor/Tmem97 agonists produce long lasting antineuropathic pain effects in mice. ACS Chem. Neurosci. 8, 1801–1811. 10.1021/acschemneuro.7b00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo G.; Prezzavento O.; Intagliata S.; Pittalà V.; Modica M. N.; Marrazzo A.; Turnaturi R.; Parenti C.; Chiechio S.; Arena E.; et al. (2019) Synthesis, in vitro and in vivo characterization of new benzoxazole and benzothiazole-based sigma receptor ligands. Eur. J. Med. Chem. 174, 226–235. 10.1016/j.ejmech.2019.04.056. [DOI] [PubMed] [Google Scholar]
- Bergkemper M.; Kronenberg E.; Thum S.; Börgel F.; Daniliuc C.; Schepmann D.; Nieto F. R.; Brust P.; Reinoso R. F.; Alvarez I.; Wünsch B. (2018) Synthesis, receptor affinity, and antiallodynic activity of spirocyclic σ receptor ligands with exocyclic amino moiety. J. Med. Chem. 61, 9666–9690. 10.1021/acs.jmedchem.8b01183. [DOI] [PubMed] [Google Scholar]
- Prezzavento O.; Arena E.; Sánchez-Fernández C.; Turnaturi R.; Parenti C.; Marrazzo A.; Catalano R.; Amata E.; Pasquinucci L.; Cobos E. J. (2017) (+)-and (−)-Phenazocine enantiomers: Evaluation of their dual opioid agonist/σ(1) antagonist properties and antinociceptive effects. Eur. J. Med. Chem. 125, 603–610. 10.1016/j.ejmech.2016.09.077. [DOI] [PubMed] [Google Scholar]
- Intagliata S.; Agha H.; Kopajtic T. A.; Katz J. L.; Kamble S. H.; Sharma A.; Avery B. A.; McCurdy C. R. (2020) Exploring 1-adamantanamine as an alternative amine moiety for metabolically labile azepane ring in newly synthesized benzo[d]thiazol-2(3H)one σ receptor ligands. Med. Chem. Res. 29, 1697–1706. 10.1007/s00044-020-02597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa R.; Kamble S. H.; Kanumuri R. S.; King T. I.; Berthold E. C.; Intagliata S.; Sharma A.; McCurdy C. R. (2020) Bioanalytical method development and pharmacokinetics of MCI-92, a sigma-1 receptor ligand. J. Pharm. Biomed. Anal. 191, 113610. 10.1016/j.jpba.2020.113610. [DOI] [PubMed] [Google Scholar]
- Arena E.; Cacciatore I.; Cerasa L. S.; Turkez H.; Pittalà V.; Pasquinucci L.; Marrazzo A.; Parenti C.; Di Stefano A.; Prezzavento O. (2016) New bifunctional antioxidant/σ1 agonist ligands: Preliminary chemico-physical and biological evaluation. Bioorg. Med. Chem. 24, 3149–56. 10.1016/j.bmc.2016.05.045. [DOI] [PubMed] [Google Scholar]
- Glennon R. A.; Ablordeppey S. Y.; Ismaiel A. M.; el-Ashmawy M. B.; Fischer J. B.; Howie K. B. (1994) Structural features important for sigma 1 receptor binding. J. Med. Chem. 37, 1214–1219. 10.1021/jm00034a020. [DOI] [PubMed] [Google Scholar]
- Cendán C. M.; Pujalte J. M.; Portillo-Salido E.; Montoliu L.; Baeyens J. M. (2005) Formalin-induced pain is reduced in sigma(1) receptor knockout mice. Eur. J. Pharmacol. 511, 73–74. 10.1016/j.ejphar.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Cendán C. M.; Pujalte J. M.; Portillo-Salido E.; Baeyens J. M. (2005) Antinociceptive effects of haloperidol and its metabolites in the formalin test in mice. Psychopharmacology 182, 485–93. 10.1007/s00213-005-0127-z. [DOI] [PubMed] [Google Scholar]
- Vidal-Torres A.; Fernández-Pastor B.; Carceller A.; Vela J. M.; Merlos M.; Zamanillo D. (2014) Effects of the selective sigma-1 receptor antagonist S1RA on formalin-induced pain behavior and neurotransmitter release in the spinal cord in rats. J. Neurochem. 129, 484–94. 10.1111/jnc.12648. [DOI] [PubMed] [Google Scholar]
- Romero L.; Zamanillo D.; Nadal X.; Sánchez-Arroyos R.; Rivera-Arconada I.; Dordal A.; Montero A.; Muro A.; Bura A.; Segalés C.; et al. (2012) Pharmacological properties of S1RA, a new sigma-1 receptor antagonist that inhibits neuropathic pain and activity-induced spinal sensitization. Br. J. Pharmacol. 166, 2289–306. 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris G.; Merlos M.; Vela J. M.; Zamanillo D.; Portillo-Salido E. (2014) S1RA, a selective sigma-1 receptor antagonist, inhibits inflammatory pain in the carrageenan and complete Freund’s adjuvant models in mice. Behav. Pharmacol. 25, 226–235. 10.1097/FBP.0000000000000038. [DOI] [PubMed] [Google Scholar]
- Bennett G. J.; Xie Y. K. (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107. 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Pitcher G. M.; Ritchie J.; Henry J. L. (1999) Nerve constriction in the rat: model of neuropathic, surgical and central pain. Pain 83, 37–46. 10.1016/S0304-3959(99)00085-8. [DOI] [PubMed] [Google Scholar]
- Cheng H. Y.; Pitcher G. M.; Laviolette S. R.; Whishaw I. Q.; Tong K. I.; Kockeritz L. K.; Wada T.; Joza N. A.; Crackower M.; Goncalves J.; et al. (2002) DREAM is a critical transcriptional repressor for pain modulation. Cell 108, 31–43. 10.1016/S0092-8674(01)00629-8. [DOI] [PubMed] [Google Scholar]
- Xu M.; Petraschka M.; McLaughlin J. P.; Westenbroek R. E.; Caron M. G.; Lefkowitz R. J.; Czyzyk T. A.; Pintar J. E.; Terman G. W.; Chavkin C. (2004) Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J. Neurosci. 24, 4576–4584. 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh D. H.; Kim H. W.; Yoon S. Y.; Seo H. S.; Kwon Y. B.; Kim K. W.; Han H. J.; Beitz A. J.; Na H. S.; Lee J. H. (2008) Intrathecal injection of the sigma(1) receptor antagonist BD1047 blocks both mechanical allodynia and increases in spinal NR1 expression during the induction phase of rodent neuropathic pain. Anesthesiology 109, 879–89. 10.1097/ALN.0b013e3181895a83. [DOI] [PubMed] [Google Scholar]
- Bravo-Caparrós I.; Perazzoli G.; Yeste S.; Cikes D.; Baeyens J. M.; Cobos E. J.; Nieto F. R. (2019) Sigma-1 receptor inhibition reduces neuropathic pain induced by partial sciatic nerve transection in mice by opioid-dependent and -independent mechanisms. Front. Pharmacol. 10, 613. 10.3389/fphar.2019.00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evoy K. E.; Morrison M. D.; Saklad S. R. (2017) Abuse and misuse of pregabalin and gabapentin. Drugs 77, 403–426. 10.1007/s40265-017-0700-x. [DOI] [PubMed] [Google Scholar]
- DeHaven-Hudkins D. L.; Fleissner L. C.; Ford-Rice F. Y. (1992) Characterization of the binding of [3H](+)-pentazocine to σ recognition sites in guinea pig brain. Eur. J. Pharmacol., Mol. Pharmacol. Sect. 227, 371–378. 10.1016/0922-4106(92)90153-M. [DOI] [PubMed] [Google Scholar]
- Mach R. H.; Smith C. R.; Childers S. R. (1995) Ibogaine possesses a selective affinity for σ2 receptors. Life Sci. 57, PL57–PL62. 10.1016/0024-3205(95)00301-L. [DOI] [PubMed] [Google Scholar]
- Mogil J. S.; Kest B.; Sadowski B.; Belknap J. K. (1996) Differential genetic mediation of sensitivity to morphine in genetic models of opiate antinociception: influence of nociceptive assay. J. Pharmacol. Exp. Ther. 276 (2), 532–544. [PubMed] [Google Scholar]
- Wilson S. G.; Smith S. B.; Chesler E. J.; Melton K. A.; Haas J. J.; Mitton B.; Strasburg K.; Hubert L.; Rodriguez-Zas S. L.; Mogil J. S. (2003) The heritability of antinociception: common pharmacogenetic mediation of five neurochemically distinct analgesics. J. Pharmacol. Exp. Ther. 304, 547–559. 10.1124/jpet.102.041889. [DOI] [PubMed] [Google Scholar]
- Mogil J. S.; Smith S. B.; O’Reilly M. K.; Plourde G. (2005) Influence of nociception and stress-induced antinociception on genetic variation in isoflurane anesthetic potency among mouse strains. Anesthesiology 103, 751–8. 10.1097/00000542-200510000-00013. [DOI] [PubMed] [Google Scholar]
- LaCroix-Fralish M. L.; Rutkowski M. D.; Weinstein J. N.; Mogil J. S.; Deleo J. A. (2005) The magnitude of mechanical allodynia in a rodent model of lumbar radiculopathy is dependent on strain and sex. Spine 30, 1821–1827. 10.1097/01.brs.0000174122.63291.38. [DOI] [PubMed] [Google Scholar]
- Feehan A. K.; Morgenweck J.; Zhang X.; Amgott-Kwan A. T.; Zadina J. E. (2017) Novel endomorphin analogs are more potent and longer-lasting analgesics in neuropathic, inflammatory, postoperative, and visceral pain relative to morphine. J. Pain 18, 1526–1541. 10.1016/j.jpain.2017.08.007. [DOI] [PubMed] [Google Scholar]
- Kilkenny C.; Browne W. J.; Cuthill I.; Emerson M.; Altman D. G. (2010) Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. J. Pharmacol. Pharmacother. 1 (2), 94–99. 10.4103/0976-500X.72351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoot M. R.; Sim-Selley L. J.; Poklis J. L.; Abdullah R. A.; Scoggins K. L.; Selley D. E.; Dewey W. L. (2010) Chronic constriction injury reduces cannabinoid receptor 1 activity in the rostral anterior cingulate cortex of mice. Brain Res. 1339, 18–25. 10.1016/j.brainres.2010.03.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad N.; Subhan F.; Islam N. U.; Shahid M.; Rahman F. U.; Sewell R. D. E. (2017) Gabapentin and its salicylaldehyde derivative alleviate allodynia and hypoalgesia in a cisplatin-induced neuropathic pain model. Eur. J. Pharmacol. 814, 302–312. 10.1016/j.ejphar.2017.08.040. [DOI] [PubMed] [Google Scholar]
- Eans S. O.; Ganno M. L.; Mizrachi E.; Houghten R. A.; Dooley C. T.; McLaughlin J. P.; Nefzi A. (2015) Parallel synthesis of hexahydrodiimidazodiazepines heterocyclic peptidomimetics and their in vitro and in vivo activities at μ (MOR), δ (DOR), and κ (KOR) opioid receptors. J. Med. Chem. 58, 4905–4917. 10.1021/jm501637c. [DOI] [PubMed] [Google Scholar]
- Reilley K. J.; Giulianotti M.; Dooley C. T.; Nefzi A.; McLaughlin J. P.; Houghten R. A. (2010) Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 12, 318–329. 10.1208/s12248-010-9191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.