

Abstract

We describe a general catalytic methodology for the enantioselective dearomative alkylation of pyridine derivatives with Grignard reagents, allowing direct access to nearly enantiopure chiral dihydro-4-pyridones with yields up to 98%. The methodology involves dearomatization of in situ-formed N-acylpyridinium salts, employing alkyl organomagnesium reagents as nucleophiles and a chiral copper (I) complex as the catalyst. Computational and mechanistic studies provide insights into the origin of the reactivity and enantioselectivity of the catalytic process.
Keywords: dihydro-4-pyridones, dearomatization, organomagnesium reagents, acylpyridiniums, copper catalysis
Pyridine and its derivatives are among the most significant heterocyclic units. Not only are they found in the structures of many natural products and pharmaceuticals but they also serve as building blocks for constructing other common scaffolds such as six-membered aza-heterocycles.1 Prime examples are dihydropyridine and piperidine motives, which are ubiquitous in numerous alkaloids and bioactive molecules. As a result, extensive work has been directed toward the synthesis of chiral dihydropyridine and piperidine derivatives.2,3
Several methodologies relying on the conjugate addition of organometallics to pyridine and dihydropyridones (commonly synthesized from the corresponding pyridine-based precursors) have been reported to date.4 At the same time, elegant strategies that depend on the direct use of pyridinium salts as substrates in addition reactions have been reported as well.5,6 Among literature precedence, the strategy that makes use of Grignard reagents and relies on chiral pyridinium salts to control the stereochemistry of the addition step has found the broadest application and still remains the preferred option (Scheme 1A).5 On the other hand, catalytic examples that make use of prochiral acylpyridinium ions are especially attractive in terms of atom economy and cost-efficiency. As a result, several catalytic methods that utilize prochiral acylpyridinium ions have also been reported,6 including nickel-catalyzed arylations6h,6i and copper-catalyzed alkylations,6j both using organozinc reagents (Scheme 1B). While highly enantiopure products can be obtained using these strategies, both reports are restricted to unsubstituted pyridine or 4-methoxypyridine and make use of expensive zinc reagents. Furthermore, the alkylation protocol suffers from a limited scope of nucleophiles and low yields.6j
Scheme 1. State of the Art in Asymmetric Dearomatization of Acylpyridinium Ions with Organometallics.

Grignard reagents are one of the most attractive organometallics in terms of their availability, cost-efficiency, and reactivity. However, the high reactivity of both acylpyridinium ions and Grignard reagents leads to fast non-catalyzed background reactions, which results in racemic products and is difficult to outcompete for a chiral catalyst. As a result, the only known example that makes use of Grignard reagents relies on stereoselective synthesis using acylpyridinium ions and a chiral auxiliary. Recently, we reported the enantioselective catalytic synthesis of chiral dihydro-4-pyridones via asymmetric addition of Grignards to pyridones, the low reactivity of which is beneficial to achieve asymmetric induction in catalytic conditions (Scheme 1C).4i Despite good yields and excellent enantioselectivities, this procedure has its own drawbacks: the substrate must be prepared from 4-MeO-pyridines in two low yielding additional steps and, as in the case of the abovementioned catalytic additions of organozinc reagents, this methodology is limited to a single pyridone substrate.
Herein, we disclose a highly enantioselective catalytic dearomatization of in situ-generated N-acylpyridinium salts using highly reactive Grignard reagents and a chiral copper catalyst. What sets this method apart is the use of readily available Grignard reagents, a broader scope that includes substituted pyridines, and operational simplicity. This allows straightforward access to enantioenriched derivatives of dihydropyridone, enabling the further generation of multiple stereocenters.
We started off our investigations using 4-methoxypyridine 1a as the starting material, phenyl chloroformate as the acylating agent, and EtMgBr as the Grignard reagent (Table 1). Anticipating a background reaction between the highly reactive acylpyridinium ion and the Grignard reagent, we chose −78 °C as the reaction temperature for the optimization studies. First, we investigated the rate of the uncatalyzed reaction in different solvents, namely, THF, Et2O, and toluene. In all cases, full conversion of 1a to the addition product 2a was observed (entry 1), indicating that a catalyst with very high turnover frequency is needed to outcompete the uncatalyzed addition reaction. Based on our recent experience in combining copper catalysis with Grignard reagents,4i,7 we employed a Cu (I) salt as a potential catalyst for this reaction. In the presence of CuBr·SMe2 (5 mol %), full conversion to the product was obtained in THF, but to our surprise, no product was formed when performing the reaction in toluene or Et2O (entries 3–4). A subsequent ligand screening in combination with CuBr·SMe2 involved a variety of commercially available chiral ligands L1–L7, including bidentate ferrocenyl- and biaryl-based diphosphines and monodentate phosphoramidite. The only chiral ligand that showed promising results in combination with the Cu (I) salt was diphosphine ligand L1. While using L1 in combination with a copper salt in THF still results in racemic 2a, some enantioenrichment (37% ee) was observed when the reaction was carried out in 2-Me-THF (entries 5 and 6). Moreover, when the reaction was performed in other solvents (Et2O, CH2Cl2, and toluene, entries 7–9), the L1-Cu (I) catalyst system provided good results with full conversion to product 2a and enantiomeric excess (ee) values between 88 and 94%. In contrast, the catalytic system with bidentate ligands L2–L6 provided racemic products, whereas monodentate L7 gave no conversion. Consequently, we adopted the following reaction conditions as the optimal ones for further studies: L1 (6 mol %), CuBr·SMe2 (5 mol %), and Grignard reagent (2.0 equiv) in toluene at −78 °C for 12 h (entry 8).
Table 1. Optimization of Reaction Conditions for the Addition of EtMgBr to 4-Methoxypyridine 1aa.

| entry | L-Cu (I)b | solvent | Conv. (%)c | ee (%)d |
|---|---|---|---|---|
| 1 | THF, Et2O, toluene | full | ||
| 2 | Cu (I) | THF | full | 0 |
| 3 | Cu (I) | toluene | 0 | |
| 4 | Cu (I) | Et2O | 0 | |
| 5 | L1-Cu (I) | THF | full | 0 |
| 6 | L1-Cu (I) | 2-Me-THF | full | 37 |
| 7 | L1-Cu (I) | Et2O | full | 88 |
| 8 | L1-Cu (I) | toluene | full | 94 |
| 9 | L1-Cu (I) | CH2Cl2 | full | 93 |
Reaction conditions: 4-methoxypyridine 1a (0.2 mmol), phenylchloroformate (2.0 equiv), and EtMgBr (2.0 equiv), in solvent (2 mL) at −78 °C, for 12 h.
Ligand L (6 mol %), CuBr·SMe2 (5 mol %).
Conversions were determined by 1H NMR.
The ee was determined by HPLC on a chiral stationary phase.
With the optimized conditions in hand, we investigated the scope of the reaction with respect to the acylating reagents (Scheme 2). With the exception of isopropyl chloroformate, for which the corresponding product 2f was not formed, all tested acylating reagents, including aliphatic and aromatic chloroformates, provided high isolated yields (82 to 98%) and enantioselectivities (81 to >99% ee).
Scheme 2. Scope of Acylating Reagents.

Reaction conditions are the same as in Table 1 using different acylating agents. The reported yields correspond to isolated yields.
The reaction using benzyl chloroformate, for which product (2d) was obtained with the highest ee at −78 °C (>99%), was also attempted at room temperature. The increase in the reaction temperature was detrimental for the stereoselectivity, with the enantiomeric purity decreasing from >99% to 52%.
Based on the described results, benzyl chloroformate was selected as the acylating reagent for the investigation of the scope of Grignard reagents. We were pleased to find that a wide variety of alkyl Grignards, including β- and γ-branched reagents, were tolerated and gave the corresponding products (2d, 2j–m) with good yields and high ees (Scheme 3). The addition of secondary Grignard reagents (cyclopentyl and isopropyl magnesium bromide), however, led to the racemic products. Various functionalized Grignard reagents were also evaluated which delivered their corresponding products (2n–2q) with moderate to high yields (58–94%) and ees exceeding 98%.
Scheme 3. Scope of Grignard Reagents.

Reaction conditions are the same as in Table 1 using different Grignard reagents. The reported yields correspond to isolated yields.
The next important question to assess was whether derivatives of 4-methoxypyridine are amenable to our catalytic system because if successful, this would represent a straightforward route to chiral precursors that are highly valuable for generating molecules with multiple stereocenters. Literature precedence reveals that the substituted derivatives of 4-methoxypyridine are often not successful candidates for nucleophilic additions due to a diminished tendency to form pyridinium ions, which is crucial for the reactivity toward nucleophiles. We opted for the addition of EtMgBr to 4-methoxy-2-methylpyridine 3a as a model reaction under the optimal reaction conditions found for substrate 1a (Table 1, entry 8). However, under these conditions, no conversion was observed. Suspecting that the steric hindrance introduced by the additional substituent might impede the pyridinium salt formation, we decided to exchange benzyl chloroformate for methyl chloroformate to stimulate the formation of pyridinium ions.
Optimization of the reaction conditions with this substrate revealed that a synthetically useful yield (66%) of the desired addition product with 97% ee can be obtained with CH2Cl2 as the solvent (Table 2, compare entry 3 with entries 2 and 4–7).
Table 2. Optimization of Reaction Conditions for the Addition of EtMgBr to 3aa.

| entry | R | solvent | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | Bn | toluene | 0 | Nd |
| 2 | Me | THF | <10 | 13 |
| 3 | Me | CH2Cl2 | 66 | 97 |
| 4 | Me | Et2O | 15 | 82 |
| 5 | Me | 2-Me-THF | 15 | 31 |
| 6 | Me | toluene | <5 | Nd |
| 7 | Me | MTBE | 30 | 90 |
| 8 | Ph | CH2Cl2 | 0 | Nd |
| 9 | Bn | CH2Cl2 | 0 | Nd |
Reaction conditions: 3a (0.2 mmol), chloroformate reagent (2.0 equiv), EtMgBr (2.0 equiv), ligand L1 (6 mol %), and CuBr·SMe2 (5 mol %) in solvent (2 mL).
The reported yields correspond to isolated yields.
The ee was determined by HPLC on a chiral stationary phase.
On the other hand, in the same solvent, no substrate conversion was observed for benzyl and phenyl acylpyridinium salts (entries 8–9). Encouraged by these results, we synthesized several 2-substituted 4-methoxypyridine substrates and evaluated them in the reaction with EtMgBr (Scheme 4), obtaining addition products with various alkyl substituents (4a–d) with decent yields (51–66%) and good to high enantioselectivities (80–97%).
Scheme 4. Scope of 4-Methoxypyridine Derivatives as Substrates.

Reaction conditions are the same as in Table 2, using different substituted 4-methoxypyridines. The reported yields correspond to isolated yields. bL1 (12 mol %) and CuBr·SMe2 (10 mol %) were used.
Also, 4-methoxyquinoline 3e shows potential for this catalytic system, providing product 4e with 75% yield and 97% ee, whereas 2-phenyl- and 2-bromo-4-methoxypyridines 3f and 3g did not result in any conversion. Finally, a substituent at the 3-position of the methoxypyridine was also tolerated, providing the corresponding product 4h with 62% yield and 82% ee.
To demonstrate the utility of our catalytic system, a gram-scale reaction was performed, using only 1 mol % of the catalyst, maintaining 68% yield and 97% ee in the formation of product 4a (Scheme 5a). Furthermore, the products accessed in this work are important building blocks for the synthesis of alkaloids. For example, our method allows access to compound 2q, which upon deprotection gives product 5, a key compound in the synthesis of Barrenazine A and B (Scheme 5b).8 Other examples are the synthesis of alkaloid precursor 6 with 92% yield as single trans diastereoisomer after hydrogenation9 and the synthesis of product 7, a crucial precursor to access the natural product (−)-epimyrtine (Scheme 5c).10
Scheme 5. Synthetic Applications of the Methodology.

To gain more insights into the origin of the stereoselectivity, we have initially conducted density functional theory calculations.11 In line with the experimental data, we posit that the overall transformation consists of two stages: (i) formation of a pyridinium ion upon acylation and (ii) interaction of the resulting salt with the chiral copper complex, leading to the final chiral product.
For the computational studies, we selected CuBr in combination with L1 as the chiral copper complex, 4-methoxy-2-methylpyridine 3a activated with methyl chloroformate as the substrate, and EtMgBr·2Et2O as the Grignard reagent. Prior to the exploration of the reaction of the acylpyridinium salt with the Grignard reagent in the presence of the copper catalyst, we explored the direct addition of the Grignard reagent to II. We found that the activation energy for the direct addition of the Grignard reagent to II is 14.70 kcal/mol (Figure S3). Then, we explored the copper salt speciation in the presence of ligand L1 and the Grignard reagent.
We found that both the formation of the L1–CuBr complex from [Cu2Br2(SMe2)2] and the subsequent transmetallation upon addition of EtMgBr leading to organocopper species L1–CuEt are highly exergonic processes (ΔG = −37.90 and −39.21 kcal/mol, respectively, Figure S4). With this in mind, we envisioned that the resulting organocopper species L1–CuEt can coordinate to II either via the carbonyl moiety, yielding III, or via the pyridine ring, specifically at position C-5-C-6, rendering IV. These two species are very close in energy and therefore likely in equilibrium. For the formation of IV, the coordination of the metallic center to the substrate is accompanied by the release of one of the phosphine groups at the ligand from the copper center in order to allow copper to accommodate the pyridinium substrate in its first coordination sphere (Scheme 6a). Although decoordination of the phosphine moiety is enthalpically disfavored, this is partially balanced by the coordination of the substrate to the copper center. At this point, the delivery of the Et group from the copper complex to the substrate can take place from complexes III or IV. However, the path from III toward the product is energetically more demanding than that from IV (Scheme S1). Therefore, continuing with complex IV, the approach of the copper can take place on either side of the two faces of the pyridine ring. Our calculations show that the proS diastereomeric complex is 2.04 kcal/mol more stable than the proR counterpart. Once species IV-proS is formed, it can further progress via nucleophilic addition of the ethyl moiety to form species V. The release of Et is facilitated by the approach of the dangling phosphorous to the metallic center, which ensures that the copper center remains tetracoordinated in this step. The evolution of IV-proS toward V-S involves a transition state with an activation energy of 7.75 kcal/mol, whereas the analogous transition state in the case of IV-proR involves an activation energy of 22.05 kcal/mol, rendering it non-competitive under the working conditions. The striking energy difference between these two transition states can be rationalized from the analysis of their structures (Scheme 6c). In TS-(IV-V)-R, one of the P–Cu bonds is significantly elongated (dCu–P-5 = 3.38 Å) in order to accommodate the substrate. The reason behind the need of dissociation of the phosphorous atom resides in the clash between one of the phenyl rings of the ligand and the substrate. On the contrary, in the TS-(IV-V)-S, the phenyl group is now replaced by a hydrogen atom, thus significantly reducing the steric interaction between the substrate and the substituents in the ligand. Furthermore, the weakening of one of the phosphorous–copper bonds at TS-(IV-V)-R results in slightly shortened Cu–C bond distances (dCu–C-5 = 2.13 Å and dCu–C-6 = 2.14 Å), creating an earlier and also energetically more demanding transition state. Once Et is transferred from the organocopper to the 4-methoxy-2-methyl-pyridine, the counterion can behave as a new ligand, yielding V (R: −78.21 and S: −71.21 kcal/mol). In V, the interaction between the metal and the substrate at position 5 (Scheme 6a) is very weak, particularly in V-S (dCu–C-5(V-R) = 2.16 Å and dCu–C-5(V-S) = 2.40 Å), and the breaking of this complex is very favorable (ΔGL1–CuCl = −82.46 kcal/mol). The release of the L1–CuCl complex ensures a new catalytic cycle.
Scheme 6. Proposed Mechanism for the Cu-Catalyzed Enantioselective Addition of Grignard Reagents to Substrate 1a.
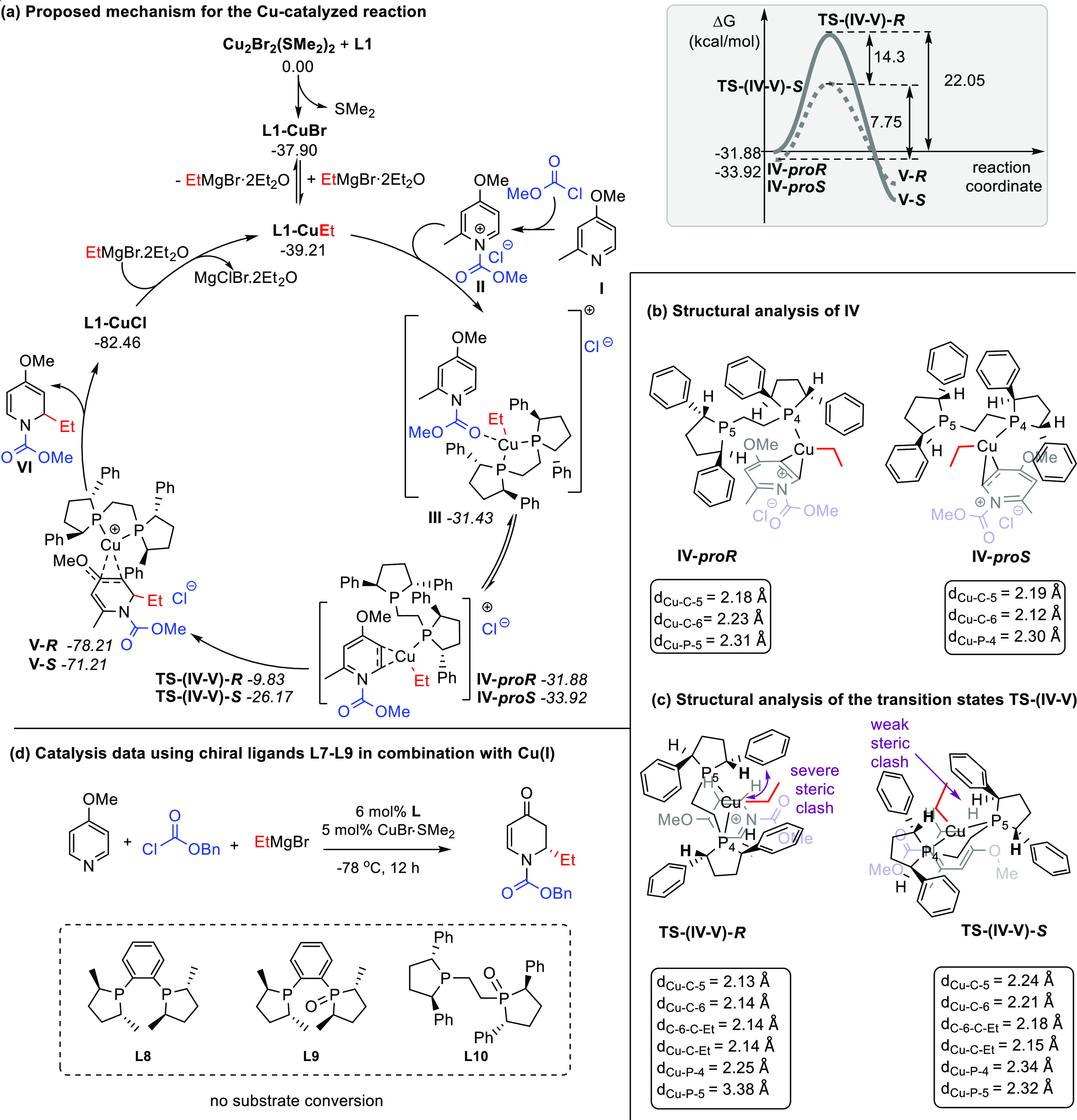
Calculations were performed at the PCM(CH2Cl2)12/B3LYP-D3/def2tzvpp//B3LYP-D3/def2svpp13 computational level using the Gaussian 09 program.14 The thermochemistry was obtained at 1 atm and 298.15 K. The pyridinium species and the L1 ligand are depicted in black, the protecting group in blue, and the ethyl group in red. (a) Left: proposed reaction mechanism. Right: reaction profile of the diastereoselective-determining step. (b) Structural analysis of the minima IV (proR and proS). (c) Structural analysis of the transition states TS-(IV-V). For visualization purposes, additional color coding is used in sections (b) and (c), showing the pyridinium species in light gray and blue, the L1 ligand in black, and the ethyl group in red. (d) Results of the catalytic addition of EtMgBr to 1a using Cu(I) salt in combination with chiral ligands L8–L10 under optimized reaction conditions.
The computational data highlight the importance of the bidentate character of ligand L1, as well as the presence of a flexible linkage between the two phosphine moieties in its structure for the reactivity of the catalytic system and its enantiodifferentiating ability. To corroborate these results experimentally, we synthesized three chiral ligands L8–L10 (Scheme 6d) that lack either the flexible linkage (L8 and L9) or have a less pronounced bidentate character (L9 and L10). These ligands were tested in combination with Cu(I) salt in the reaction of the synthesis of 2d under optimized reaction conditions. In stark contrast to the results obtained with L1 (Scheme 3), no substrate conversion was observed when ligands L8–L10 were used. Next, we carried out NMR spectroscopic studies of the copper complexes formed upon mixing of the copper salt with chiral ligands L9 and L10, aiming to see the binding mode of these mono-oxidized ligands to copper (Figures S6 and S7 in Supporting Information). The results obtained confirmed the bidentate complexation of the rigid mono-oxidized ligand L9 to the copper, while mono-oxidized ligand L10 with the flexible link behaved as a monodentate ligand. In an attempt to further understand the influence of the binding angle and the ligand rigidity, we recomputed the stereo-determining step using L8 (Scheme S2) and found that enantiodiscrimination is in principle possible, but that the difference in energy between the paths leading to the R and S enantiomers of the product is smaller than that in the case when using our optimal catalyst (with ligand L1). The less pronounced enantiodiscrimination is caused by the rigidity of the ligand that impedes the release of one of the phosphine arms. Importantly, the energy of the complexes IV with this ligand, as well as the subsequent transition states, is quite high, explaining the fruitless attempts to make this reaction work experimentally and emphasizing the importance of flexible bidentate ligands for the current transformation.
In conclusion, we have developed highly efficient catalytic asymmetric addition of Grignard reagents to in situ-formed N-acylpyridinium salts, with high yields and ees. The reaction is operationally simple and tolerates a wide range of pyridine derivatives and Grignard reagents and thus has a potential for applications in the synthesis of alkaloids and other complex building blocks. Finally, mechanistic studies revealed that certain structural motives, namely, the bidentate character and the flexible linkage between two binding arms, are responsible for the reactivity of the catalyst and for the transfer of chiral information from the catalyst to the final product.
Acknowledgments
Financial support from the European Research Council (S.R.H. grant no. 773264, LACOPAROM), The Netherlands Organization for Scientific Research (NWO-VICI to S.R.H.), and the China Scholarship Council (CSC, to Y.G.) are acknowledged. M.C.R. thanks the Centro de Supercomputación de Galicia (CESGA) for the free allocation of computational resources and the Xunta de Galicia (Galicia, Spain) for financial support through the ED481B-Axudas de apoio á etapa de formación posdoutoral (modalidade A) fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c01544.
General experimental protocols, mechanistic studies, and full characterization of all the species reported; 1H NMR and 13C NMR spectra of all the reported compounds; HPLC traces and HRMS data of new compounds; computational details; electronic energies; stability and imaginary frequencies of each stationary point; and Cartesian coordinates (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; b Kiuru P.; Yli-Kauhaluoma J.. Pyridine and its Derivatives in Heterocycles in Natural Product Synthesis; Majumdar K. C., Chattopadhyay S. K., Eds.; Wiley-VCH: Weinheim, 2011. Vol. 8, Chapter 8, pp. 267–297; [Google Scholar]; c Escolano C.; Amat M.; Bosch J. Chiral Oxazolopiperidone Lactams: Versatile Intermediates for the Enantioselective Synthesis of Piperidine-Containing Natural Products. Chem.—Eur. J. 2006, 12, 8198–8207. 10.1002/chem.200600813. [DOI] [PubMed] [Google Scholar]; d Ma X.; Gang D. R. The Lycopodium Alkaloids. Nat. Prod. Rep. 2004, 21, 752–772. 10.1039/b409720n. [DOI] [PubMed] [Google Scholar]; e Stout D. M.; Meyers A. I. Recent Advances in the Chemistry of Dihydropyridines. Chem. Rev. 1982, 82, 223–243. 10.1021/cr00048a004. [DOI] [Google Scholar]
- a Sánchez-Roselló M.; Aceña J. L.; Simón-Fuentes A.; del Pozo C. A General Overview of the Organocatalytic Intramolecular aza-Michael reaction. Chem. Soc. Rev. 2014, 43, 7430–7453. 10.1039/c4cs00156g. [DOI] [PubMed] [Google Scholar]; b Kobayashi S.; Komiyama S.; Ishitani H. The First Enantioselective Aza-Diels-Alder Reactions of Imino Dienophiles on Use of a Chiral Zirconium Catalyst. Angew. Chem., Int. Ed. 1998, 37, 979–981. . [DOI] [PubMed] [Google Scholar]; c Yu J.; Shi F.; Gong L.-Z. Brønsted-Acid-Catalyzed Asymmetric Multicomponent Reactions for the Facile Synthesis of Highly Enantioenriched Structurally Diverse Nitrogenous Heterocycles. Acc. Chem. Res. 2011, 44, 1156–1171. 10.1021/ar2000343. [DOI] [PubMed] [Google Scholar]; d Hussain M.; Banchelin T. S.-L.; Andersson H.; Olsson R.; Almqvist F. Enantioselective Synthesis of Substituted Piperidines by Addition of Aryl Grignard Reagents to Pyridine N-Oxides. Org. Lett. 2013, 15, 54–57. 10.1021/ol303085q. [DOI] [PubMed] [Google Scholar]; e Andersson H.; Gustafsson M.; Boström D.; Olsson R.; Almqvist F. The Regio- and Stereoselective Synthesis of trans-2,3-Dihydropyridine-N-oxides and Piperidines. Angew. Chem., Int. Ed. 2009, 48, 3288–3291. 10.1002/anie.200900189. [DOI] [PubMed] [Google Scholar]; f Gribble M. W. Jr; Liu R. Y.; Buchwald S. L. Evidence for Simultaneous Dearomatization of Two Aromatic Rings under Mild Conditions in Cu(I)-Catalyzed Direct Asymmetric Dearomatization of Pyridine. J. Am. Chem. Soc. 2020, 142, 11252–11269. 10.1021/jacs.0c04486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zheng C.; You S.-L. Transfer Hydrogenation with Hantzsch Esters and Related Organic Hydride Donors. Chem. Soc. Rev. 2012, 41, 2498–2518. 10.1039/c1cs15268h. [DOI] [PubMed] [Google Scholar]; b Yang Z.-P.; Wu Q.-F.; Shao W.; You S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. 10.1021/jacs.5b10440. [DOI] [PubMed] [Google Scholar]; c Ding Q.; Zhou X.; Fan R. Recent Advances in Dearomatization of Heteroaromatic Compounds. Org. Biomol. Chem. 2014, 12, 4807–4815. 10.1039/c4ob00371c. [DOI] [PubMed] [Google Scholar]; d Gribble M. W. Jr; Guo S.; Buchwald S. L. Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc. 2018, 140, 5057–5060. 10.1021/jacs.8b02568. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bertuzzi G.; Bernardi L.; Fochi M. Nucleophilic Dearomatization of Activated Pyridines. Catalysts 2018, 8, 632–665. 10.3390/catal8120632. [DOI] [Google Scholar]
- a Shintani R.; Tokunaga N.; Doi H.; Hayashi T. A New Entry of Nucleophiles in Rhodium-Catalyzed Asymmetric 1,4-Addition Reactions: Addition of Organozinc Reagents for the Synthesis of 2-Aryl-4-Piperidones. J. Am. Chem. Soc. 2004, 126, 6240–6241. 10.1021/ja048825m. [DOI] [PubMed] [Google Scholar]; b Müller D.; Alexakis A. Copper-Catalyzed Asymmetric 1,4-Addition of Alkenyl Alanes to N-Substituted-2-3-Dehydro-4-Piperidones. Org. Lett. 2012, 14, 1842–1845. 10.1021/ol3004436. [DOI] [PubMed] [Google Scholar]; c Jagt R. B. C.; de Vries J. G.; Feringa B. L.; Minnaard A. J. Enantioselective Synthesis of 2-Aryl-4-Piperidones via Rhodium/Phosphoramidite-Catalyzed Conjugate Addition of Arylboroxines. Org. Lett. 2005, 7, 2433–2435. 10.1021/ol050734m. [DOI] [PubMed] [Google Scholar]; d Xu Q.; Zhang R.; Zhang T.; Shi M. Asymmetric 1,4-Addition of Arylboronic Acids to 2,3-Dihydro-4-pyridones Catalyzed by Axially Chiral NHC–Pd(II) Complexes. J. Org. Chem. 2010, 75, 3935–3937. 10.1021/jo1006224. [DOI] [PubMed] [Google Scholar]; e Sebesta R.; Pizzuti M. G.; Boersma A. J.; Minnaard A. J.; Feringa B. L. Catalytic Enantioselective Conjugate Addition of Dialkylzinc Reagents to N-Substituted-2,3-Dehydro-4-Piperidones. Chem. Commun. 2005, 1711–3. 10.1039/b417727d. [DOI] [PubMed] [Google Scholar]; f Pizzuti M. G.; Minnaard A. J.; Feringa B. L. Catalytic Asymmetric Synthesis of the Alkaloid (+)-Myrtine. Org. Biomol. Chem. 2008, 6, 3464–3466. 10.1039/b807575a. [DOI] [PubMed] [Google Scholar]; g Guo F.; Dhakal R. C.; Dieter R. K. Conjugate Addition Reactions of N-Carbamoyl-4-Pyridones and 2,3-Dihydropyridones with Grignard Reagents in the Absence of Cu(I) salts. J. Org. Chem. 2013, 78, 8451–8464. 10.1021/jo400936z. [DOI] [PubMed] [Google Scholar]; h Dieter R. K.; Guo F. Conjugate Addition Reactions of N-Carbamoyl-4-Pyridones with Organometallic Reagents. J. Org. Chem. 2009, 74, 3843–3848. 10.1021/jo900327q. [DOI] [PubMed] [Google Scholar]; i Guo Y.; Harutyunyan S. R. Highly Enantioselective Catalytic Addition of Grignard Reagents to N-Heterocyclic Acceptors. Angew. Chem., Int. Ed. 2019, 58, 12950–12954. 10.1002/anie.201906237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tsukanov S. V.; Marks L. R.; Comins D. L. Studies Toward the Synthesis of Lepadiformine A. J. Org. Chem. 2016, 81, 10433–10443. 10.1021/acs.joc.6b01514. [DOI] [PubMed] [Google Scholar]; b Tsukanov S. V.; Comins D. L. Concise Total Synthesis of the Frog Alkaloid (−)-205 B. Angew. Chem., Int. Ed. 2011, 50, 8626–8628. 10.1002/anie.201103596. [DOI] [PubMed] [Google Scholar]; c Comins D. L.; Kuethe J. T.; Hong H.; Lakner F. J.; Concolino T. E.; Rheingold A. L. Diastereoselective Addition of Prochiral Metallo Enolates to Chiral 1-Acylpyridinium Salts. J. Am. Chem. Soc. 1999, 121, 2651–2652. 10.1021/ja990024+. [DOI] [Google Scholar]; d Comins D. L.; Joseph S. P.; Goehring R. R. Asymmetric Synthesis of 2-Alkyl(Aryl)-2,3-dihydro-4-pyridones by Addition of Grignard Reagents to Chiral 1-Acyl-4-methoxypyridinium Salts. J. Am. Chem. Soc. 1994, 116, 4719–4728. 10.1021/ja00090a019. [DOI] [Google Scholar]; e Charette A. B.; Grenon M.; Lemire A.; Pourashraf M.; Martel J. Practical and Highly Regio- and Stereoselective Synthesis of 2-Substituted Dihydropyridines and Piperidines: Application to the Synthesis of (−)-Coniine. J. Am. Chem. Soc. 2001, 123, 11829–11830. 10.1021/ja017136x. [DOI] [PubMed] [Google Scholar]; f Legault C.; Charette A. B. Complexation Promoted Additions to N-Benzoyliminopyridinium Ylides. A Novel and Highly Regioselective Approach to Polysubstituted Piperidines. J. Am. Chem. Soc. 2003, 125, 6360–6361. 10.1021/ja0348647. [DOI] [PubMed] [Google Scholar]; g Hoesl C. E.; Maurus M.; Pabel J.; Polborn K.; Wanner K. T. Generation of Chiral N-Acylpyridinium Ions by Means of Silyl Triflates and their Diastereoselective Trapping Reactions: Formation of N-Acyldihydropyridines and N-Acyldihydropyridones. Tetrahedron 2002, 58, 6757–6770. 10.1016/s0040-4020(02)00675-0. [DOI] [Google Scholar]
- a García Mancheño O.; Asmus S.; Zurro M.; Fischer T. Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis. Angew. Chem., Int. Ed. 2015, 54, 8823–8827. 10.1002/anie.201502708. [DOI] [PubMed] [Google Scholar]; b Sun Z.; Yu S.; Ding Z.; Ma D. Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu–Bis(oxazoline) Complexes. J. Am. Chem. Soc. 2007, 129, 9300–9301. 10.1021/ja0734849. [DOI] [PubMed] [Google Scholar]; c Ichikawa E.; Suzuki M.; Yabu K.; Albert M.; Kanai M.; Shibasaki M. New Entries in Lewis Acid–Lewis Base Bifunctional Asymmetric Catalyst: Catalytic Enantioselective Reissert Reaction of Pyridine Derivatives. J. Am. Chem. Soc. 2004, 126, 11808–11809. 10.1021/ja045966f. [DOI] [PubMed] [Google Scholar]; d Bertuzzi G.; Sinisi A.; Caruana L.; Mazzanti A.; Fochi M.; Bernardi L. Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity. ACS Catal. 2016, 6, 6473–6477. 10.1021/acscatal.6b01962. [DOI] [Google Scholar]; e Nadeau C.; Aly S.; Belyk K. Rhodium-Catalyzed Enantioselective Addition of Boronic Acids to N-Benzylnicotinate Salts. J. Am. Chem. Soc. 2011, 133, 2878–2880. 10.1021/ja111540g. [DOI] [PubMed] [Google Scholar]; f Robinson D. J.; Spurlin S. P.; Gorden J. D.; Karimov R. R. Enantioselective Synthesis of Dihydropyridines Containing Quaternary Stereocenters through Dearomatization of Pyridinium Salts. ACS Catal. 2020, 10, 51–55. 10.1021/acscatal.9b03874. [DOI] [Google Scholar]; h Chau S. T.; Lutz J. P.; Wu K.; Doyle A. G. Nickel-Catalyzed Enantioselective Arylation of Pyridinium Ions: Harnessing an Iminium Ion Activation Mode. Angew. Chem., Int. Ed. 2013, 52, 9153–9156. 10.1002/anie.201303994. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Lutz J. P.; Chau S. T.; Doyle A. G. Nickel-Catalyzed Enantioselective Arylation of Pyridine. Chem. Sci. 2016, 7, 4105–4109. 10.1039/c6sc00702c. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Ángeles Fernández-Ibáñez M.; Maciá B.; Pizzuti M. G.; Minnaard A. J.; Feringa B. L. Catalytic Enantioselective Addition of Dialkylzinc Reagents to N-Acylpyridinium Salts. Angew. Chem., Int. Ed. 2009, 48, 9339–9341. 10.1002/anie.200904981. [DOI] [PubMed] [Google Scholar]
- a Rodríguez-Fernández M.; Yan X.; Collados J. F.; White P. B.; Harutyunyan S. R. Lewis Acid Enabled Copper-Catalyzed Asymmetric Synthesis of Chiral β-Substituted Amides. J. Am. Chem. Soc. 2017, 139, 14224–14231. 10.1021/jacs.7b07344. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yan X.; Harutyunyan S. R. Catalytic Enantioselective Addition of Organometallics to Unprotected Carboxylic Acids. Nat. Commun. 2019, 10, 3402. 10.1038/s41467-019-11345-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yan X.; Ge L.; Castiñeira Reis M.; Harutyunyan S. R. Nucleophilic Dearomatization of N-Heteroaromatics Enabled by Lewis Acids and Copper Catalysis. J. Am. Chem. Soc. 2020, 142, 20247–20256. 10.1021/jacs.0c09974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez M. M.; Sarandeses L. A.; Sestelo J. P. Enantioselective synthesis of (−)-barrenazines A and B. Tetrahedron Lett. 2007, 48, 8536–8539. 10.1016/j.tetlet.2007.09.145. [DOI] [Google Scholar]
- Gouault N.; Le Roch M.; de Campos Pinto G.; David M. Total Synthesis of Dendrobate Alkaloid (+)-241D, Isosolenopsin and Isosolenopsin A: Application of a Gold-Catalyzed Cyclization. Org. Biomol. Chem. 2012, 10, 5541–5546. 10.1039/c2ob25685a. [DOI] [PubMed] [Google Scholar]
- a Suárez-Castillo O. R.; Montiel-Ortega L. A.; Meléndez-Rodríguez M.; Sánchez-Zavala M. Cleavage of Alkoxycarbonyl Protecting Groups from Carbamates by t-BuNH2. Tetrahedron Lett. 2007, 48, 17–20. 10.1016/j.tetlet.2006.11.024. [DOI] [Google Scholar]; b Yang Y. Building Polyfunctional Piperidines: a Stereoselective Strategy of a Three-component Mannich reaction Inspired by Biosynthesis and Applications in the Synthesis of Natural Alkaloids (+)-241D; (−)-241D; Isosolenopsin A and (−)-Epimyrtine. RSC Adv. 2015, 5, 18894–18908. 10.1039/c4ra14418j. [DOI] [Google Scholar]
- Kohn W.; Sham L. J. Self-Consistent Equations Including Exchange and Correlation Effects. J. Phys. Rev. 1965, 140, A1133. 10.1103/physrev.140.a1133. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. Density-functional Thermochemistry. III. The role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Becke A. D. A New Mixing of Hartree-Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]; c Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; d Weigend F.; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 09; Gaussian, Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.