

Abstract
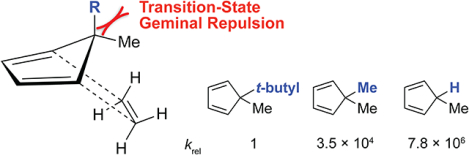
We have experimentally and computationally explored the sluggish Diels–Alder reactivities of the geminally substituted 5,5-dimethylcyclopentadiene and 5,5-dimethyl-2,3-diazacyclopentadiene (4,4-dimethyl-4H-pyrazole) scaffolds. We found that geminal dimethylation of 1,2,3,4-tetramethylcyclopentadiene to 1,2,3,4,5,5-hexamethylcyclopentadiene decreases the Diels–Alder reactivity towards maleimide by 954-fold. Quantum mechanical calculations revealed that the decreased Diels–Alder reactivities of gem-dimethyl substituted cyclopentadienes and 2,3-diazacyclopentadienes are not a consequence of unfavorable steric interactions between the diene and dienophile as reported previously, but a consequence of the increased repulsion within the gem-dimethyl group in the transition state. The findings have implications for the use of cyclopentadienes in “click” chemistry.
Keywords: Diels–Alder, Click Chemistry, Kinetics, Cycloaddition, Density Functional Theory
Graphical Abstract
1. Introduction
The rapid reactivity and excellent yield of cyclopentadienes in normal-electron demand Diels–Alder reactions with electron-deficient dienophiles are ideal for “click” chemistry applications.1 The Diels–Alder reactivities of cyclopentadienes and 2,3-diazacyclopentadienes (4H-pyrazoles) are tunable through hyperconjugative interactions of the diene π-system with the substituents at the saturated center.2–5 Hyperconjugative σ-donors stabilize the diene by inducing aromatic 6π-electron delocalization, whereas hyperconjugative σ-acceptors destabilize the diene by inducing antiaromatic 4π-electron delocalization.6,7 The computationally generated magnetic, energetic, and geometric measures of aromaticity for cyclopentadiene and 5,5-dimethylcyclopentadiene listed in Table 1 imply that the cyclic hyperconjugative electron delocalization is less effective in 5,5-dimethylcyclopentadiene than in cyclopentadiene.6,8 This decreased electron delocalization should promote Diels–Alder reactivity, but the literature indicates otherwise.9
Table 1.
| NICS(0) | Λ | ASE | BI | |
|---|---|---|---|---|
 |
−3.1 | −3.5 | 2.60 | 29 |
 |
−2.2 | −2.4 | 0.85 | 22 |
NICS(0), nuclear independent chemical shift; Λ, diamagnetic susceptibility exaltation; ASE, aromatic stabilization energy (kcal/mol); BI, Bird Index.
Diels–Alder reactions of 5,5-dimethylcyclopentadiene have been reported with highly reactive electron-deficient or strained dienophiles such as benzyne,10 2-chloroacrylonitrile,11 maleic anhydride,9,12 and maleimides.12–14 5,5-Dimethylcyclopentadiene does not dimerize, even upon heating to 200 °C.9,15 This stability is in contrast to cyclopentadiene, which is highly reactive as a Diels–Alder diene towards a range of dienophiles and dimerizes readily at room temperature.16,17
Rouse and Tyler proposed that the geminal dimethyl group of 5,5-dimethylcyclopentadiene “exerts a marked steric hindrance to the approach of dienophiles.”9 Yet, studies on the π-facial selectivity of 5-methylcyclopentadiene suggest that the methyl substituent does not significantly hinder the approach of the dienophile.3,18 Scheme 1 shows that the Diels–Alder reaction between 5-methylcyclopentadiene and 4-phenyl-1,2,4-triazole-3,5-dione forms a mixture of syn and anti cycloadducts with a modest preference for the syn cycloadduct.18 This observation is consistent with a computational study from the Houk group on the π-facial selectivity of 5-substituted cyclopentadienes.3 They reported that the syn Diels–Alder reaction of 5-methylcyclopentadiene is kinetically favored by ~1 kcal/mol over the anti cycloaddition when ethylene is the dienophile.
Scheme 1.
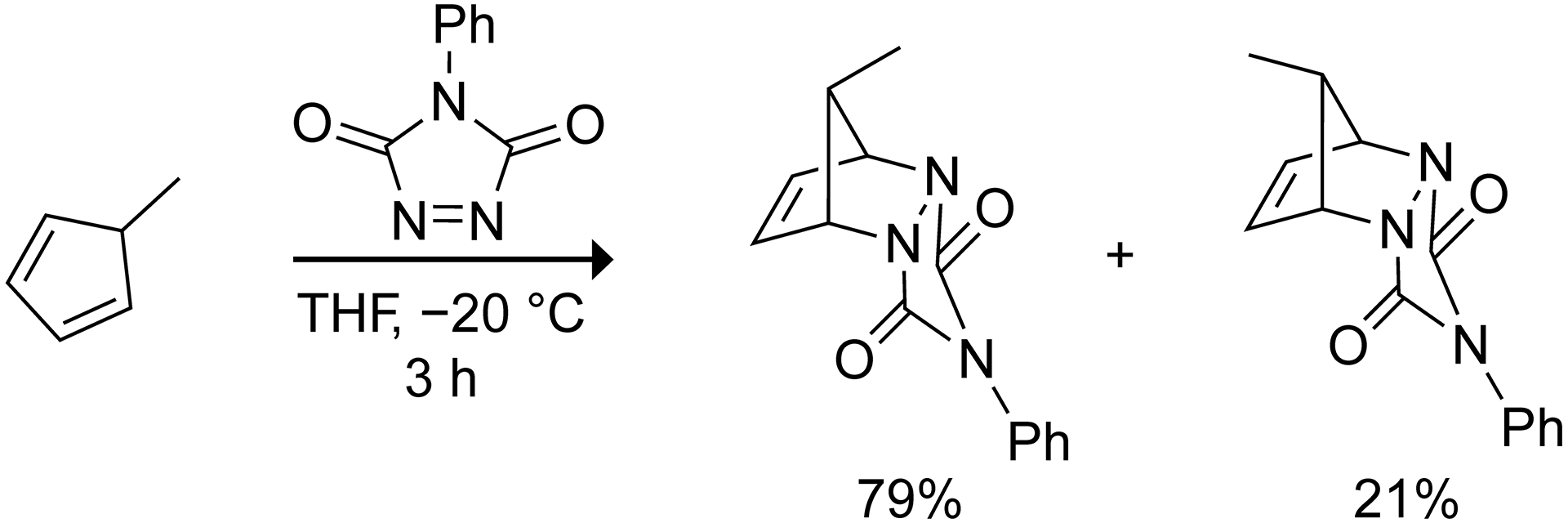
π-Facial selectivity in the Diels–Alder reaction of 5-methylcyclopentadiene with 4-phenyl-1,2,4-triazole-3,5-dione.18
Geminally substituted 5,5-dimethyl-2,3-diazacyclopentadienes (4,4-dimethyl-4H-pyrrazoles) react poorly as Diels–Alder dienes. Even with highly reactive strained dienophiles, Diels–Alder reactions of 5,5-dimethyl-2,3-diazacyclopentadienes do not readily proceed unless promoted with an acid catalyst.4,19–22 To understand the poor Diels–Alder reactivities of geminally substituted 5,5-dimethylcyclopentadienes and 5,5-dimethyl-2,3- diazacyclopentadienes, we have computationally and experimentally studied the effect of the gem-dimethyl substitution on reactivity. Our results provide new insight on the basis for the sluggish reactivity of these dienes.
2. Results and discussion
To quantify the effect of geminal dimethylation on Diels–Alder reactivity, we experimentally determined the second-order rate constants for the Diels–Alder reactions of 1,2,3,4-tetramethylcyclopentadiene (Me4Cp), 1,2,3,4,5-pentamethylcyclopentadiene (Me5Cp), and 1,2,3,4,5,5-hexamethylcyclopentadiene (Me6Cp) with maleimide. Second-order reaction kinetics were assessed with UV–vis spectroscopy for Me4Cp and Me5Cp. NMR spectroscopy was used for Me6Cp because the reaction was too slow to observe the reaction rates otherwise. We note that an experimental comparison between the Diels–Alder reactivity of a 5,5-dimethyl-2,3-diazacyclopentadiene and a 2,3-diazacyclopentadiene without substituents at the saturated center is not possible because unsubstituted 2,3-diazacyclopentadienes rapidly isomerize into 1H-pyrazoles.23 The experimental second-order rate constants for the Diels–Alder reactions of Me4Cp, Me5Cp, and Me6Cp with maleimide as the dienophile are 124, 85, and 0.13 M−1s−1, respectively (Scheme 2). Thus, the presence of a gem-dimethyl group on the saturated center reduces the Diels–Alder reactivity of the diene by approximately 3 orders of magnitude.
Scheme 2.
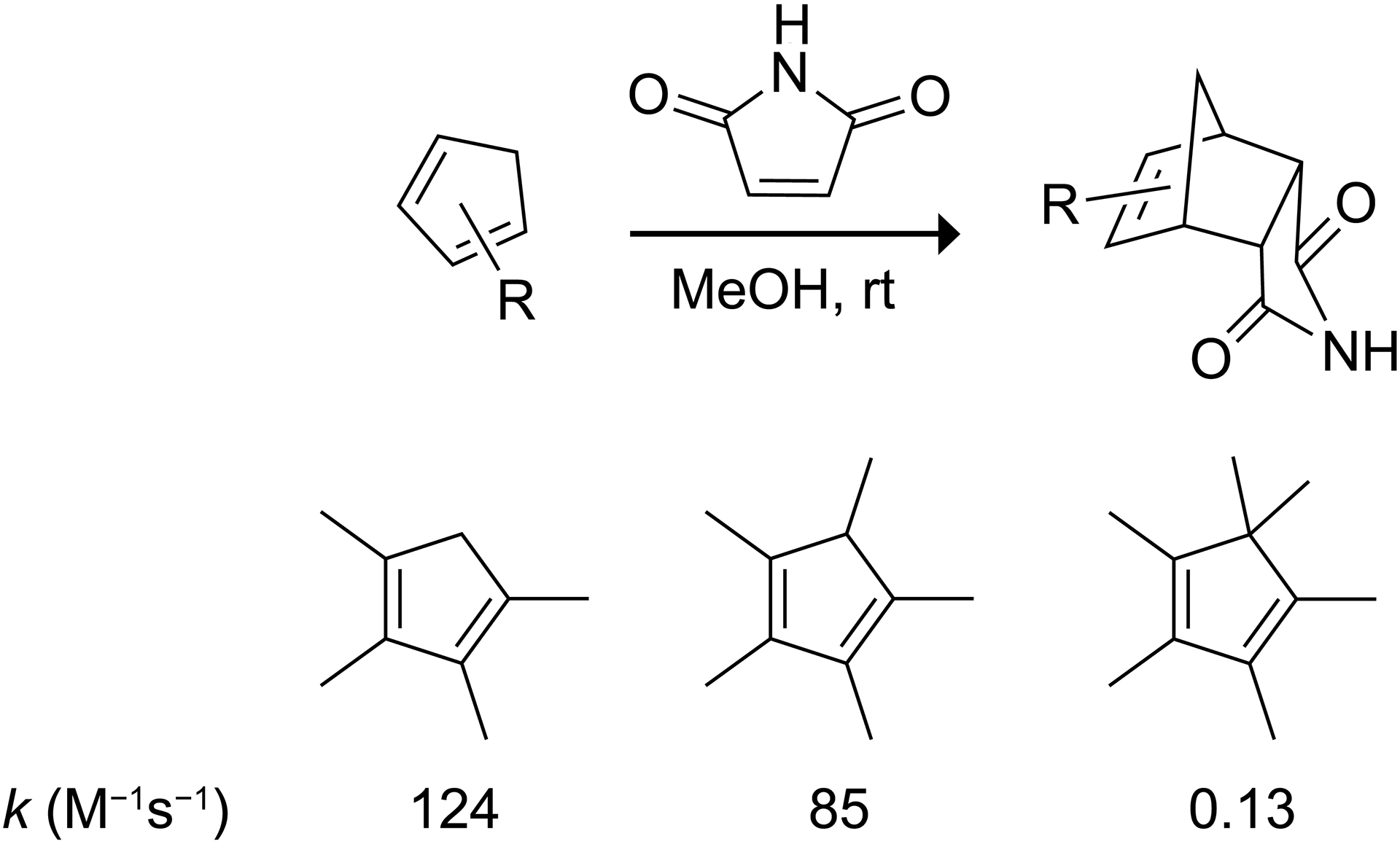
Second-order rate constants for the Diels–Alder reactions of Me4Cp, Me5Cp, and Me6Cp with maleimide.
To understand the origin of the decreased Diels–Alder reactivity in 5,5-dimethylcyclopentadienes and 5,5-dimethyl-2,3-diazacyclopentadienes, we studied computationally the reactions of these dienes and their unsubstituted counterparts with ethylene. Calculations were performed at the M06-2X/6-311++G(d,p)//M06-2X/6-31+G(d) level of theory in Gaussian16 Rev. C.01.24,25 The ground state structures of cyclopentadiene, 5,5-dimethylcyclopentadiene, 2,3-diazacyclopentadiene, and 5,5-dimethyl-2,3-diazacyclopentadiene are shown in Fig. 1. The angle between the geminal substituents is represented by θ2. Geminal dimethylation increases θ2 from 106.4° in cyclopentadiene to110.1° in 5,5-dimethylcyclopentadiene and from 107.6° in 2,3-diazacyclopentadiene to 111.2° in 5,5-dimethyl-2,3-diazacyclopentadiene. The increase in θ2 results in a decrease in the angle between the ring substituents of C5, which is represented by θ1. Specifically, θ1 decreases from 103.2° in cyclopentadiene to 101.8° in 5,5-dimethylcyclopentadiene, and from 97.5° in 2,3-diazacyclopentadiene to 96.3° in 5,5-dimethyl- 2,3-diazacyclopentadiene. The decrease in θ1 decreases the distance between the diene termini of cyclopentadiene and 2,3-diazacyclopentadiene, which is represented by r, by 0.02 and 0.01 Å, respectively. These structural changes upon geminal dimethylation are consistent with the Thorpe–Ingold effect.26–28
Fig. 1.
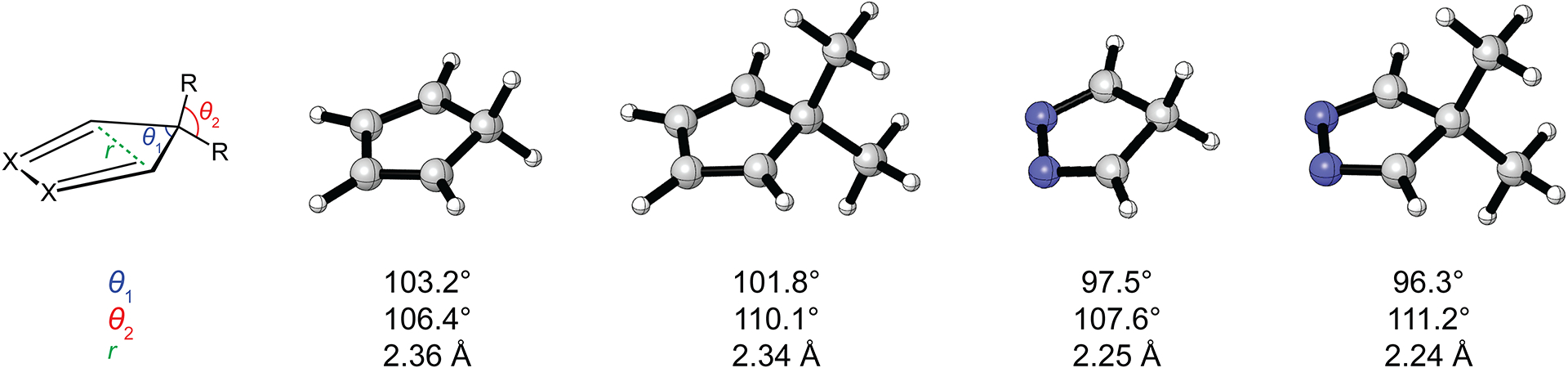
M06-2X/6-31+G(d)-optimized ground state structures of (left to right) cyclopentadiene, 5,5-dimethylcyclopentadiene, 2,3-diazacyclopentadiene, and 5,5-dimethyl-2,3-diazacyclopentadiene.
The transition state structures and Gibbs free energies of activation (ΔG‡) for the Diels–Alder reactions of cyclopentadiene (TS-1), 5,5-dimethylcyclopentadiene (TS-2), 2,3-diazacyclopentadiene (TS-3), and 5,5-dimethyl-2,3-diazacyclopentadiene (TS-4) with ethylene are shown in Fig. 2. Geminal dimethylation decreases the Diels–Alder reactivity of cyclopentadiene and 2,3-diazacyclopentadiene by 310- and 35-fold, respectively.29 Geminal dimethylation has little influence on the reaction energies.
Fig. 2.
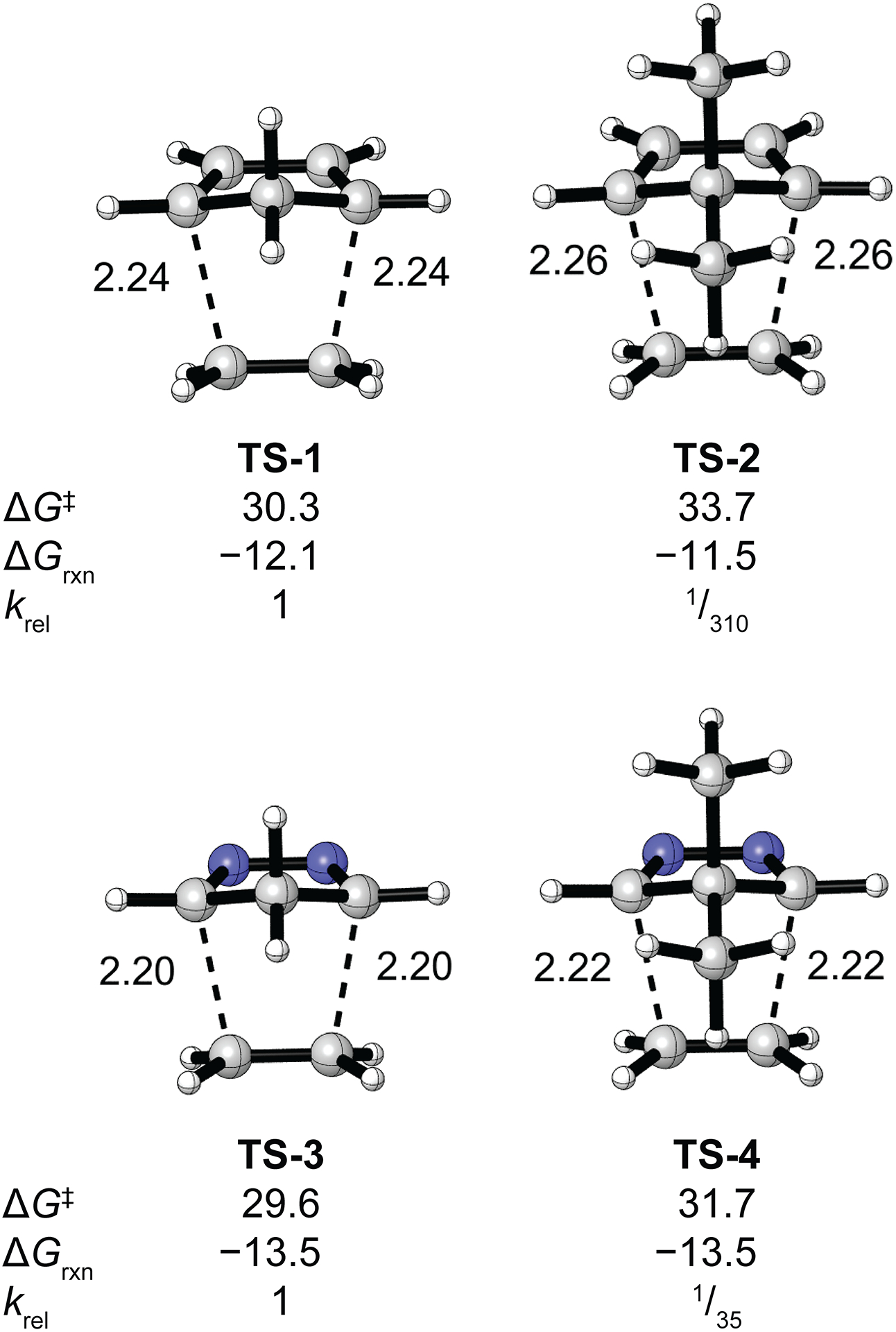
Transition state structures and Gibbs free activation (ΔG‡) and reaction (ΔGrxn) energies in kcal/mol for the Diels–Alder reactions of ethylene with cyclopentadiene (TS-1), 5,5-dimethylcyclopentadiene (TS-2), 2,3-diazacyclopentadiene (TS-3), and 5,5-dimethyl-2,3-diazacyclopentadiene (TS-4).
To understand the origin of the decreased reactivity upon the geminal dimethylation of the cyclopentadiene and 2,3-diazacyclopentadiene scaffolds, we analyzed the Diels–Alder reactions of cyclopentadiene, 5,5-dimethylcyclopentadiene, 2,3-diazacyclopentadiene, and 5,5-dimethyl-2,3-diazacyclopentadiene with the distortion/interaction–activation strain model.30 This model dissects the electronic energies into the distortion energies and interaction energies. The distortion energies represent the energy required to deform the ground state structures of the reactants into the geometries of the transition state. The interaction energies are the energies of interactions that occur between diene and dienophile during the course of bond formation. These interactions include steric (i.e., Pauli-repulsion), orbital, electrostatic, and dispersive interactions. The transition states of the gem-dimethyl substituted dienes occur later than those of the unsubstituted dienes. To account for the different timing of the transition state structures, we performed the distortion/interaction–activation strain analysis along the intrinsic reaction coordinate (IRC) defined by the average length of the forming bonds.31 The results from the analysis are shown in Fig. 3. The distortion/interaction–activation strain analysis indicates that the decrease in reactivity upon geminal dimethylation of the dienes is related to an increase in the distortion energies.
Fig. 3.
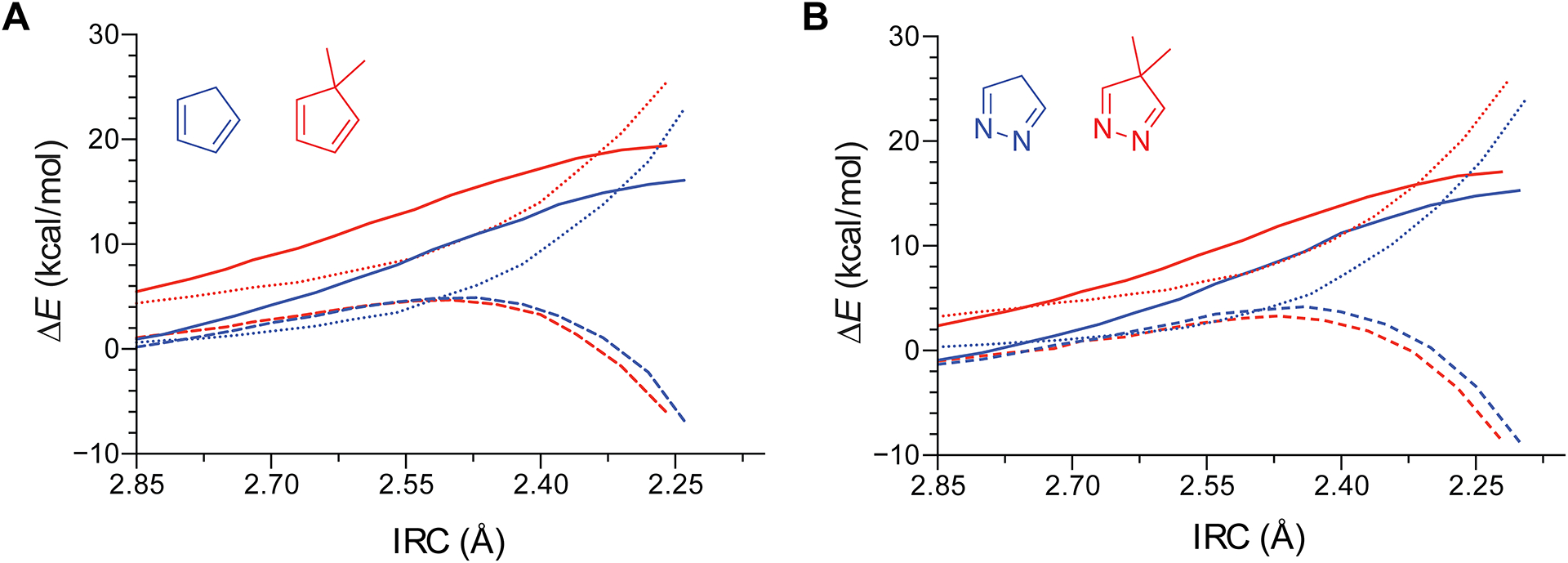
Distortion/interaction–activation strain analysis for the Diels–Alder reactions of ethylene with cyclopentadiene and 5,5-dimethylcyclopentadiene (A), and 2,3-diazacyclopentadiene and 5,5-dimethyl-2,3-diazacyclopentadiene (B). The analysis was carried out along the intrinsic reaction coordinate (IRC) defined by the average distance of the forming bonds in Ångstroms. (Solid lines: Electronic energies; Short dashed lines: Distortion energies; Long dashed lines: Interaction energies).
The distortion/interaction–activation strain analysis revealed that the gem-dimethyl–substituted dienes require more energy to deform into their transition state geometries than do the unsubstituted dienes. The values of θ2 from the ground state and transition state structures of the geminally substituted dienes are listed in Fig. 4. The values of θ2 decrease as ethylene approaches the geminally substituted dienes. For 5,5-dimethylcyclopentadiene and 5,5-dimethyl-2,3-diazacyclopentadiene, θ2 decreases by −4.3° and −3.7°, respectively. The compression of θ2 during the reaction amplifies the geminal repulsion within the gem-dimethyl group. This increase in geminal repulsion destabilizes the geminally substituted dienes and provides an explanation for their higher distortion energies and decreased Diels–Alder reactivities relative to the unsubstituted dienes.
Fig. 4.
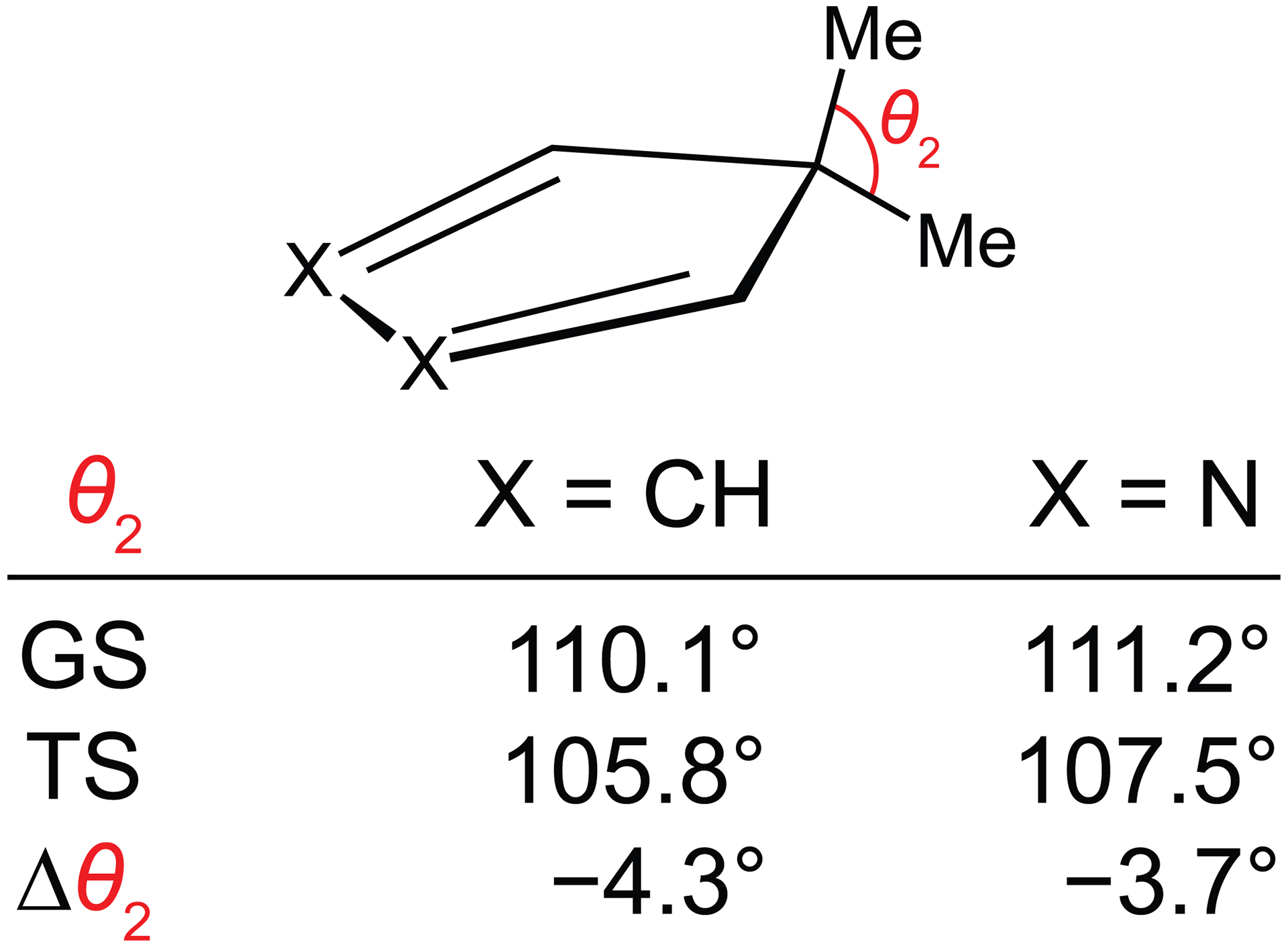
Calculated values of θ2 for the ground state (GS) and transition state (TS) during the Diels–Alder reactions with ethylene of 5,5-dimethylcyclopentadiene and 5,5-dimethyl-2,3-diazacyclopentadiene.
To study the effects of geminal repulsion on the Diels–Alder reactivity further, we computationally investigated the Diels–Alder reactions of 5-methylcyclopentadiene, 5,5-dimethylcyclopentadiene and 5-methyl-5-t-butyl-cyclopentadiene with ethylene. The transition state structures with the Gibbs free activation and reaction energies for these Diels–Alder reactions are shown in Fig. 5. Replacing the methyl group in 5,5-dimethylcyclopentadiene with a hydrogen atom decreases the activation energy by 3.2 kcal/mol, which corresponds to a rate increase of 220-fold. Notably, the computed Gibbs free energies of activation for the Diels–Alder reactions of 5-methylcyclopentadiene and cyclopentadiene with ethylene are nearly identical, differing by only 0.2 kcal/mol. In contrast to a hydrogen atom, replacing the methyl group in 5,5-dimethylcyclopentadiene with a t-butyl group increases the activation energy by 6.2 kcal/mol, which corresponds to a rate decrease of 35,000-fold. Between the ground state and transition state geometries, the geminal repulsion increases as θ2 in 5-methyl-5-t-butyl-cyclopentadiene compresses from 112.6° to 107.1°. Overall, the increased geminal repulsion from replacing the hydrogen atom in 5-methylcyclopentadiene with a t-butyl group results in a predicted 7.8 million-fold decrease in reactivity.
Fig. 5.
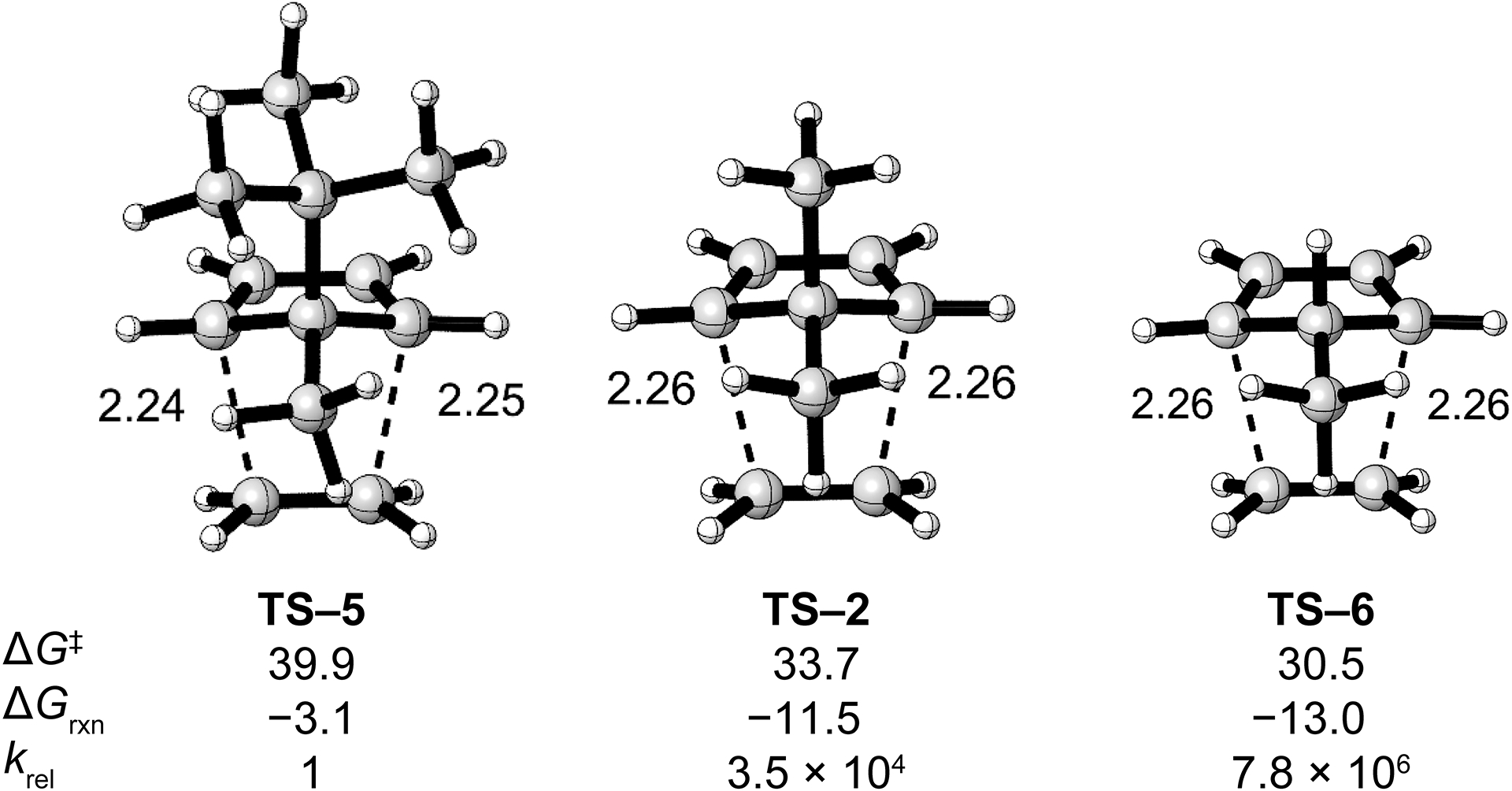
Transition state structures and Gibbs free activation (ΔG‡) and reaction (ΔGrxn) energies in kcal/mol for the Diels–Alder reactions of 5-methyl-5-t-butyl-cyclopentadiene (TS–5), 5,5-dimethylcyclopentadiene (TS–2), and 5-methylcyclopentadiene (TS–6) with ethylene.
3. Conclusions
The effect of gem-dimethyl substitution on the Diels–Alder reactivity of the cyclopentadiene scaffold was investigated by experiment and computation. Experimentally, we found that the Diels–Alder reactivity of 1,2,3,4-tetramethylcyclopentadiene towards maleimide decreased by 954-fold upon geminal dimethylation. A distortion/interaction–activation strain analysis revealed that the poor Diels–Alder reactivities of geminally substituted 5,5-dimethylcyclopentadienes and 5,5-dimethyl-2,3-diazacyclopentadienes result from the increased geminal repulsion between the gem-dimethyl group in the transition state.
Geminally substituted peralkylcyclopentadienes are often used as “click” reagents because they do not readily dimerize or isomerize.32 We conclude that switching from gem-dimethyl substitution to 1,2,3,4,5-pentaalkylcyclopentadienes will result in faster Diels–Alder reactions while discouraging dimerization or isomerization side reactions.
4. Experimental section
4.1. Computational methods
Calculations were performed in Gaussian 16 Rev. C.01 using the M06-2X functional.24,25 Geometries were obtained with the 6-31+G(d) basis set. Energetics were calculated using the 6-311++G(d,p) basis set. Normal mode analysis of each structure was used to verify that each stationary point is a first-order saddle point for transition state structures and a minimum for the ground state structures.
4.2. General experimental
All chemicals were from commercial sources and were used without further purification. NMR spectra were acquired with an Avance Neo 400 spectrometer from Bruker (Billerica, MA). UV–vis experiments were carried out with a Cary 60 UV–vis spectrometer from Agilent Technologies (Santa Clara, CA) with measurements every 0.1 s for kinetic experiments. The phrase “concentrated under reduced pressure” refers to the removal of solvents and other volatile materials using a rotary evaporator at water aspirator pressure (<20 Torr) while maintaining a water-bath temperature of 40 °C. Residual solvent was removed from samples by the vacuum (<0.1 Torr) achieved by a mechanical belt-drive oil pump. All procedures were performed in air at ambient temperature (~22 °C) and pressure (1.0 atm) unless indicated otherwise.
4.3. Reaction of maleimide with 1,2,3,4-tetramethylcyclopentadiene (Me4Cp)
Stock solutions of Me4Cp (200 μM) and maleimide (200 μM) were prepared in MeOH. Aliquots (0.5 mL) of each reactant were mixed, and the absorbance at 280 nm was monitored until no maleimide remained. The reaction was carried out in triplicate. A plot of [maleimide]−1 versus time was used to calculate the second-order rate constant.
4.4. Reaction of maleimide with 1,2,3,4,5-pentamethylcyclopentadiene (Me5Cp)
Stock solutions of Me5Cp (200 μM) and maleimide (200 μM) were prepared in MeOH. Aliquots (0.5 mL) of each reactant were mixed, and the absorbance at 280 nm was monitored until no maleimide remained. The reaction was carried out in triplicate. A plot of [maleimide]−1 versus time was used to calculate the second-order rate constant.
4.5. Reaction of maleimide with 1,2,3,4,5,5-hexamethylcyclopentadiene (Me6Cp)
Stock solutions of Me6Cp (20 mM) and maleimide (20 mM) were prepared in MeOD. Aliquots (0.3 mL) of each reactant were mixed, and 1H NMR spectra were obtained every 1 min for 30 min. The reaction was carried out in triplicate. A plot of [maleimide]−1 versus time was used to calculate the second-order rate constant.
4.6. Synthesis of 1,2,3,4,5,5-hexamethylcyclopentadiene (Me6Cp)
Lithium pentamethylcyclopentadienide (2.905 g, 20.5 mmol) was transferred to a flask that had been purged with vaccum and filled with Ar(g) three times. Under an Ar(g) atmosphere, diethyl ether (30 mL) was added with stirring. MeI (1.56 mL, 3.55 g, 25 mmol) was added to the stirring solution dropwise and stirring was continued for 18 h. The reaction mixture was filtered to remove the precipitated LiI, and the filtrate was concentrated under reduced pressure to yield a dark orange oil. Distillation of the oil gave 39.9 mg (0.266 mmol, 1.3%) of 1,2,3,4,5,5-hexamethylcyclopentadiene as a yellow oil. Note: The low yield was due largely to decomposition of the product during distillation. 1H NMR (400 MHz, CDCl3, δ): 1.79 (s, 6H), 1.75 (s, 6H), 0.91 (s, 6H). 13C NMR (101 MHz, CDCl3, δ): 142.51, 131.94, 52.15, 21.77, 11.01, 9.50.
Supplementary Material
Acknowledgement
B.J.L. was supported by postdoctoral fellowship F32 GM137543 (NIH). N.S.A was supported by a graduate research fellowship from the NSF (grant no. 1745302). This work was supported by Grant R01 GM044783 (NIH). Computational resources were provided by the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges at the Pittsburgh Supercomputing Center through allocation TG-CHE190066. XSEDE is supported by Grant ACI-1548562 (NSF).33
References
- [1].Levandowski BJ, Raines RT, Click chemistry with cyclopentadiene, Chem. Rev. (2021), doi: 10.1021/acs.chemrev.0c01055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Levandowski BJ, Zou L, Houk KN, Schleyer hyperconjugative aromaticity and Diels–Alder reactivity of 5-substituted cyclopentadienes, J. Comput. Chem. 37 (2016) 117–123. [DOI] [PubMed] [Google Scholar]
- [3].Levandowski BJ, Zou L, Houk KN, Hyperconjugative aromaticity and antiaromaticity control the reactivities and π-facial stereoselectivities of 5-substituted cyclopentadiene Diels–Alder cycloadditions, J. Org. Chem. 83 (2018) 14658–14666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levandowski BJ, Abularrage NS, Houk KN, Raines RT, Hyperconjugative antiaromaticity activates 4-pyrazoles as inverse-electron-demand Diels–Alder dienes, Org. Lett. 21 (2019) 8492–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abularrage NS, Levandowski BJ, Raines RT, Synthesis and Diels–Alder reactivity of 4-fluoro-4-methyl-4H-pyrazoles, IJMS 21 (2020) 3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nyulászi L, P.v.R. Schleyer, Hyperconjugative π-aromaticity: How to make cyclopentadiene aromatic, J. Am. Chem. Soc. 121 (1999) 6872–6875. [Google Scholar]
- [7].Fernández I, Wu JI, Schleyer P.v.R., Substituent effects on “hyperconjugative” aromaticity and antiaromaticity in planar cyclopolyenes, Org. Lett. 15 (2013) 2990–2993. [DOI] [PubMed] [Google Scholar]
- [8].Dauben HJ, Wilson JD, Laity JL, Diamagnetic susceptibility exaltation as a criterion of aromaticity, J. Am. Chem. Soc. 90 (1968) 811–813. [Google Scholar]
- [9].Rouse RS, Tyler WE III, 5,5-Dimethylcyclopentadiene, J. Org. Chem. 26 (1961) 3525–3527. [Google Scholar]
- [10].Warrener R, Harrison P, π-Bond screening in benzonorbornadienes: The role of 7-substituents in governing the facial selectivity for the Diels–Alder reaction of benzonorbornadienes with 3,6-di(2-pyridyl)-s-tetrazine, Molecules 6 (2001) 353–369. [Google Scholar]
- [11].Jefford CW, Wallace TW, Can NTH, Rimbault CG, Synthesis of 7,7-dimethylnorbornadiene, J. Org. Chem. 44 (1979) 689–691. [Google Scholar]
- [12].Wilcox CF, Mesirov M, The preparation of 5,5-dimethylcyclopentadiene, J. Org. Chem. 25 (1960) 1841–1844. [Google Scholar]
- [13].McLean S, Haynes P, Substitution in the cyclopentadienide anion series, Tetrahedron 21 (1965) 2313–2327. [Google Scholar]
- [14].Schultz KP, Spivey DW, Kirkbride Loya E, Kellon JE, Taylor LM, McConville MR, Photochemical locking and unlocking of an acyl nitroso dienophile in the Diels–Alder reaction, Tetrahedron Lett. 57 (2016) 1296–1299. [Google Scholar]
- [15].McLean S, Findlay DM, Thermal and photochemical rearrangements of methyl-substituted cyclopentadienes, Can. J. Chem. 48 (1970) 3107–3109. [Google Scholar]
- [16].Raistrick B, Sapiro RH, Newitt DM, 360. Liquid-phase reactions at high pressures. Part V. The polymerisation of cyclopentadiene and α-dicyclopentadiene, J. Chem. Soc. (1939) 1761–1769. [Google Scholar]
- [17].Levandowski BJ, Houk KN, Theoretical analysis of reactivity patterns in Diels–Alder reactions of cyclopentadiene, cyclohexadiene, and cycloheptadiene with symmetrical and unsymmetrical dienophiles, J. Org. Chem. 80 (2015) 3530–3537. [DOI] [PubMed] [Google Scholar]
- [18].Letourneau JE, Wellman MA, Burnell DJ, Diels–Alder reactions of 5-alkyl-1,3-cyclopentadienes, J. Org. Chem. 62 (1997) 7272–7277. [DOI] [PubMed] [Google Scholar]
- [19].Beck K, Höhn A, Hünig S, Prokschy F, Azobrücken aus Azinen I Isopyrazole als elektronenarme Diene zur Synthese von 2,3-Diazabicyclo[2.2.1]heptenen, Chem. Ber. 117 (1984) 517–533. [Google Scholar]
- [20].Beck K, Hünig S, Klärner F-G, Kraft P, Artschwager-Perl U, Azobrücken aus Azinen, VII1) Diels–Alder-Reaktionen mit inversem Elektronenbedarf zwischen Isopyrazolen und Cycloalkenen sowie Cycloalkadienen.—Ein Vergleich von Säure-Katalyse und Beschleunigung durch Druck, Chem. Ber. 120 (1987) 2041–2051. [Google Scholar]
- [21].Adam W, Harrer HM, Nau WM, Peters K, Electronic substituent effects on the acid-catalyzed [4+ + 2] cycloaddition of isopyrazoles with cyclopentadiene and the photochemical and thermal denitrogenation of the resulting 1,4-diaryl-7,7-dimethyl-2,3-diazabicyclo[2.2.1]hept-2-ene azoalkanes to bicyclo[2.1.0]pentanes, J. Org. Chem. 59 (1994) 3786–3797. [Google Scholar]
- [22].Adam W, Ammon H, Nau WM, Peters K, 4-Halo-4H-pyrazoles: Cycloaddition with cyclopentadiene to azoalkanes of the 2,3-diazabicyclo[2.2.1]hept-2-ene type versus electrophilic addition with cyclopentene, J. Org. Chem. 59 (1994) 7067–7071. [Google Scholar]
- [23].Sammes MP, Katritzky AR, The 4H-pyrazoles, Adv. Heterocycl. Chem. 34 (1983) 53–78. [Google Scholar]
- [24].Zhao Y, Truhlar DG, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc. 120 (2008) 215–241. [Google Scholar]
- [25].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams F Ding F Lipparini F Egidi J Goings B Peng A Petrone T Henderson D Ranasinghe VG Zakrzewski J Gao N Rega G Zheng W Liang M Hada M Ehara K Toyota R Fukuda J Hasegawa M Ishida T Nakajima Y Honda O Kitao H Nakai T Vreven K Throssell JA Montgomery JE Jr. Peralta F Ogliaro MJ Bearpark JJ Heyd EN Brothers KN Kudin VN Staroverov TA Keith R Kobayashi J Normand K Raghavachari AP Rendell JC Burant SS Iyengar J Tomasi M Cossi JM Millam M Klene C Adamo R Cammi JW Ochterski RL Martin K Morokuma O Farkas JB Foresman DJ Fox Gaussian 16 Rev. C.01, Wallingford, CT, 2016. [Google Scholar]
- [26].Beesley RM, Ingold CK, Thorpe JF, CXIX.—The formation and stability of spiro-compounds. Part I. spiro-Compounds from cyclohexane, J. Chem. Soc., Trans. 107 (1915) 1080–1106. [Google Scholar]
- [27].Jung ME, Piizzi G, gem-Disubstituent effect: Theoretical basis and synthetic applications, Chem. Rev. 105 (2005) 1735–1766. [DOI] [PubMed] [Google Scholar]
- [28].Levine MN, Raines RT, Trimethyl lock: A trigger for molecule release in chemistry, biology, and pharmacology, Chem. Sci. 3 (2012) 2412–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Relative rates were calculated with the Arrhenius equation.
- [30].Bickelhaupt FM, Houk KN, Analyzing reaction rates with the distortion/interaction–activation strain model, Angew. Chem. Int. Ed. 56 (2017) 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Levandowski BJ, Hamlin TA, Bickelhaupt FM, Houk KN, Role of orbital interactions and activation strain (distortion energies) on reactivities in the normal and inverse electron-demand cycloadditions of strained and unstrained cycloalkenes, J. Org. Chem. 82 (2017) 8668–8675. [DOI] [PubMed] [Google Scholar]
- [32].St Amant AH, Lemen D, Florinas S, Mao S, Fazenbaker C, Zhong H, Wu H, Gao C, Christie RJ, Read de Alaniz J, Tuning the Diels–Alder reaction for bioconjugation to maleimide drug-linkers, Bioconjugate Chem. 29 (2018) 2406–2414. [DOI] [PubMed] [Google Scholar]
- [33].Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, Roskies R, Scott JR, Wilkins-Diehr N, XSEDE: Accelerating scientific discovery, Comput. Sci. Eng. 16 (2014) 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.