

Summary
Studies in three mouse models of breast cancer identified profound discrepancies between cell autonomous and systemic Akt1 or Akt2 inducible deletion on breast cancer tumorigenesis and metastasis. While systemic Akt1 deletion, inhibits metastasis, cell autonomous Akt1 deletion does not. Single cell mRNA sequencing revealed that systemic Akt1 deletion maintains the pro-metastatic cluster within primary tumors but ablates pro-metastatic neutrophils. Systemic Akt1 deletion inhibits metastasis by impairing survival and mobilization of tumor-associated neutrophils. Importantly, either systemic or neutrophil-specific Akt1 deletion is sufficient to inhibit metastasis of Akt-proficient tumors. Thus, Akt1 specific inhibition could be therapeutic for breast cancer metastasis regardless of primary tumor origin. Systemic Akt2 deletion does not inhibit and exacerbates mammary tumorigenesis and metastasis, but cell autonomous Akt2 deletion prevents breast cancer tumorigenesis by ErbB2. Elevated circulating insulin level induced by Akt2 systemic deletion hyperactivates tumor Akt, exacerbating ErbB2 mediated tumorigenesis, curbed by pharmacological reduction of the elevated insulin.
Blurb
Chen et al. find that inducible systemic Akt1 deletion after tumor detection inhibits breast cancer metastasis of Akt-proficient tumor by inhibiting tumor-associated neutrophils. Inducible systemic Akt2 deletion does not inhibit breast cancer tumorigenesis and metastasis. Thus, specific Akt1 inhibition could be therapeutic for breast cancer metastasis regardless of tumor origin.
Graphical Abstract
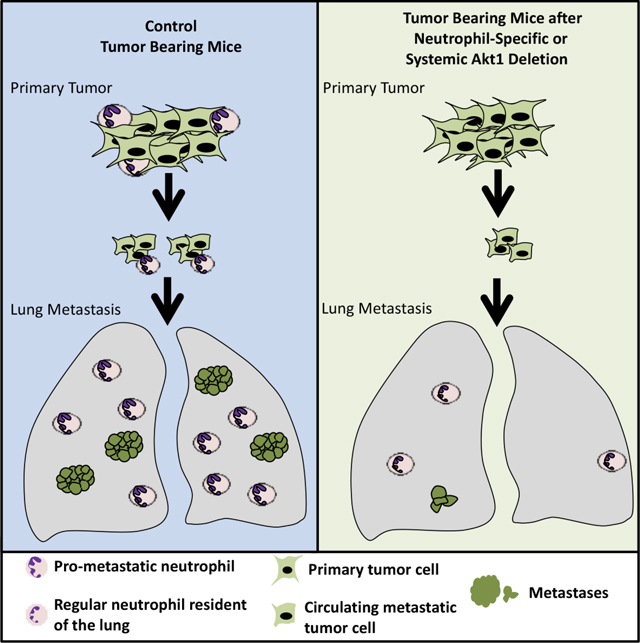
Introduction
The serine/threonine kinase Akt is frequently hyperactivated in breast cancer through multiple mechanisms, including PI3K activation, PTEN loss, and ErbB2/Her2/neu activation/amplification (Ciriello et al., 2015). However, previous studies regarding the roles of Akt isoforms in breast cancer did not provide a coherent understanding, and the results are controversial. The ablation of Akt1 in cell culture increased the migration and epithelial mesenchymal transition (EMT), whereas ablation of Akt2 decreased EMT (Irie et al., 2005). In a mouse model of breast cancer, the activation of Akt1 increased tumor development but decreased metastasis, whereas the activation of Akt2 did not increase tumor development but increased metastasis (Dillon et al., 2009; Hutchinson et al., 2004). Finally, in mouse models of breast cancer, the germ line deletion of Akt1 inhibits both tumor development (Ju et al., 2007; Maroulakou et al., 2007) and metastasis (Ju et al., 2007), whereas the germ line deletion of Akt2 enhances tumor development but appears to inhibit metastasis. (Maroulakou et al., 2007). However, these studies do not distinguish between the cell autonomous and non-cell autonomous effects of Akt isoforms on breast cancer. Furthermore, previous studies, which employed germ line deletions of Akt isoforms, do not emulate drug therapy because the mice are born with the deletions and thus could have potential developmental impacts on tumorigenesis. Therefore, we launched a comprehensive approach to examine the roles of Akt1 and Akt2 in mammary gland tumor initiation, progression and metastasis, and to understand their therapeutic implications. We employed three mouse models of breast cancer; two Her2 enriched models and one luminal B model. These studies uncovered several surprising and unexpected findings. First, unlike the germ line or systemic deletion of Akt2, after tumor onset, that does not inhibit and even exacerbates breast cancer tumorigenesis, cell autonomous Akt2 deletion completely inhibits breast cancer tumorigenesis at early stage. Second, unlike the cell autonomous deletion of Akt1 that does not inhibit metastasis, the systemic deletion of Akt1 after tumor onset inhibits metastasis. Thus, there are discrepancies between cell autonomous and systemic deletion of Akt isoforms after tumor onset with respect to breast cancer tumorigenesis and metastasis. We delineated the reasons for these discrepancies. We showed that the high circulating insulin levels as consequence of systemic Akt2 deletion hyperactivates the other Akt isoforms and overcomes the inhibitory effect of cell autonomous Akt2 deletion at early stages of breast cancer tumorigenesis. By delineating the reasons for the discrepancies between the effect of cell autonomous and systemic Akt1 deletion on metastasis, we uncovered a new mechanism by which systemic Akt1 deletion inhibits metastasis. Using single cell RNA sequencing (scRNAseq), we identified the metastatic cluster in MMTV- PyMT tumors, which remains intact after systemic Akt1 deletion. These results together with the finding that systemic Akt1 deletion inhibits metastasis of WT tumors, indicated that the effect of systemic Akt1 deletion on metastasis is not tumor intrinsic. The scRNAseq results showed that the tumor associated neutrophils are missing after systemic deletion of Akt1. Since tumor associated neutrophils play a pro-metastatic role in breast cancer (Mouchemore et al., 2018; Wculek and Malanchi, 2015), the results raised the possibility that the systemic effect of Akt1 deletion on metastasis is mediated by the deficiency of pro-metastatic neutrophils. Indeed, we showed that specific deletion of Akt1 in neutrophils is sufficient to inhibit metastasis of tumors in which Akt1 was not intrinsically deleted. These results showed that Akt1 specific inhibition can inhibit breast cancer metastasis regardless of the primary tumor type.
Results
We employed three mouse models of breast cancer; two Her2 enriched models and one luminal B model to distinguish between cell autonomous and systemic effects after tumor onset (Fig. S1). In each mouse model mice were euthanized at endpoint when tumors reached 2 cm in diameter to analyze metastasis. In this strategy quantification of metastasis is not a consequence of tumor growth.
The effects of Akt1 versus Akt2 cell autonomous deletion on primary tumors and metastasis driven by ErbB2.
In the cell autonomous mouse model (MMTV-Neu-IRES-Cre (NIC)), where ErbB2 and Cre recombinase are concomitantly expressed in the mammary cells (Ursini-Siegel et al., 2008) (Fig. 1A), we found that the cell autonomous deletion of Akt1 impaired tumor development and increased tumor-free survival (Fig. 1B), which is consistent with the effects of the germline deletion of Akt1 (Ju et al., 2007). Surprisingly, the cell autonomous deletion of Akt2 completely inhibited tumor development (Fig. 1B). The heterozygous deletion of Akt2 did not inhibit tumor development (Fig. 1B), indicating that the complete inhibition of tumor development by Akt2 deletion in the mammary gland is not due to transgene (ErbB2) silencing. This finding is in stark contrast to effects of the germ line or systemic deletion of Akt2, which did not inhibit tumor development, but rather exacerbated tumor development (Maroulakou et al., 2007) (Fig. 2). Immunoblot analysis showed that Akt1 was deleted in ErbB2-expressing mammary glands of MMTV-NIC;Akt1f/f mice, but no expression of ErbB2 or deletion of Akt2 was found in the mammary glands of MMTV-NIC;Akt2f/f mice (Fig. 1C). Thus, the expression of Akt2 seen in the mammary glands of these mice is not from cells that express the transgene but from normal mammary gland cells. Taken together, these results suggest that ErbB2 expression cannot be tolerated in the absence of Akt2 and that cells expressing ErbB2 in the absence of Akt2 are eliminated. The results also suggest that the germ line (Maroulakou et al., 2007) or systemic deletion of Akt2 (Fig. 2) enables ErbB2/Akt2−/− mammary gland tumor cells to proliferate and overcome the intolerance to ErbB2 expression in the absence of Akt2. To further verify these possibilities, we generated MMTV-NIC;R26LucLSL and MMTV-NIC;Akt2f/f;R26LucLSL mice, in which the luciferase (Luc) gene was inserted in the ubiquitously expressed Rosa26 (R26) locus and is expressed only if Cre recombinase is also expressed (Ventura et al., 2007) (Fig. 1D). The Cre recombinase is co-expressed with ErbB2 in MMTV-NIC mice as they are both driven by the same MMTV promoter. If the cells expressing MMTV-NIC survive, it is expected that luciferase will be expressed and detected in the mice by luminescence imaging. Our results showed luciferase expression only in the MMTV-NIC;R26LucLSL, MMTV-NIC;Akt1f/f;R26LucLSL, and MMTV-NIC;Akt2+/f;R26LucLSL mice but not in the MMTV-NIC;Akt2f/f;R26LucLSL mice (Fig. 1D). These results further established the notion that the expression of ErbB2 in the absence of Akt2 is not tolerated in mammary gland cells; thus, these cells are eliminated. It remains to be explained, however, why ErbB2 expression in the absence of Akt1 can be tolerated. One potential explanation is that ErbB2 expression cannot be tolerated when total Akt activity is reduced below a certain threshold level. Akt2 is expressed at the highest level and Akt1 is expressed at the lowest level at early stages of tumor development (Fig. 1E). Therefore, it is possible that the deletion of Akt2 reduces total Akt activity more than the deletion of Akt1 at early stages of tumor development. Further support for this assertion is shown and discussed below (Fig. 2H, 2I, and Fig. S3C).
Figure 1: The effect of the Akt1 or Akt2 cell autonomous deletion on ErbB2-mediated mammary primary tumors and metastasis.

A. The genotypes of mice and experimental strategy. Mice were euthanized at 5 weeks after tumor palpation, for immunoblotting, and at endpoint when tumors reached 2cm in diameter. B. Kaplan-Meier plots showing percent of tumor free mice as determined by the time tumor is palpated, of MMTV-NIC, MMTV-NIC-Akt1f/f, MMTV-NIC-Akt2f/f and MMTV-NIC-Akt2+/f mice. The number of mice is indicated. p<0.0001 MMTV-NIC versus MMTV-NIC-Akt1f/f and MMTV-NIC-Akt2f/f, p>0.05, MMTV-NIC versus MMTV-NIC-Akt1+/f using the log-rank test. C. Immunoblot showing ErbB2, ErbB3, Akt1 and Akt2 protein expression in the mammary glands of MMTV-NIC, MMTV-NIC;Akt1f/f and MMTV-NIC;Akt2f/f mice. Akt2 is not co-expressed with ErbB2 (see text). D. Luminescent imaging showing luciferase expression in MMTV-NIC;LSL-Luc, MMTV-NIC;Akt1f/f;LSL-Luc, MMTV-NIC;Akt2+/f;LSL-Luc, and MMTV-NIC;Akt2f/f;LSL-Luc mice. E. Representative immunoblot showing the expression of Akt1 and Akt2 during primary tumor development in MMTV-NIC mice. F. Table summarizing the incidence of lung metastasis in MMTV-NIC and MMTV-NIC;Akt1f/f mice. The mice were sacrificed at the primary tumor endpoint, and the lungs were scored for metastasis.
Figure 2: Consequences of the systemic deletion of Akt1 or Akt2 after tumor onset in MMTV-ErbB2 mice.

A. The genotypes of mice and experimental strategy. Mice were euthanized at two different time points. At 5 weeks after tumor palpation, to determine relative tumor growth and for immunoblotting, and at endpoint when tumors reached 2cm in diameter. B. Tumor volume at 5 weeks after tamoxifen injection. Data are presented as the means ±SEM. p<0.001 MMTV-ErbB2;Akt1f/f;R26CreERT2 vs. MMTVErbB2;R26CreERT2, p<0.025 MMTV-ErbB2;Akt2f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreERT2, using an unpaired t test. C. Percentage of Ki67-positive cells in tumor sections. Data are presented as the means ± SEM. P=0.017, MMTV-ErbB2;Akt1f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreER. P=0.006, MMTV-ErbB2;Akt2f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreERT2 using an unpaired t test. D. Kaplan-Meier tumor free survival curves as determined by tumor endpoint. P=0.0007, MMTV-ErbB2;Akt1f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreER. P=0.0018, MMTV-ErbB2;Akt2f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreERT2. E. Table summarizing the incidence of lung metastasis. F. Circulating levels of insulin in the absence of or after tamoxifen injection. Data are presented as the means ± SEM. P=0.0005, MMTV-ErbB2;Akt2f/f;R26CreERT2 vs. MMTV-ErbB2;R26CreERT2. G. Quantification of phosphorylated PRAS40, GSK3b, and Akt1(pSer473) relative to total PRAS40, GSK3b, and Akt1 in tumor extracts after tamoxifen injection into MMTV-ErbB2;R26CreERT2 or MMTV-ErbB2;Akt2f/f;R26CreERT2 mice. Data are presented as the means ± SEM. * P<0.03 using an unpaired t test. H. Kaplan-Meier plot showing percentage of tumor-free mice after tamoxifen injection into one-month-old MMTV-NIC;Akt2f/f;R26CreERT2 mice. I. Quantification of phosphorylated Akt1 (ser473), phosphorylated pan-Akt (ser473), phosphorylated PRAS40, and phosphorylated GSK3β, relative to total Akt1, total pan-Akt, total PRAS40, and total GSK3 in tumor extracts. Data are presented as the means ±SEM. * P<0.05 using an unpaired t test.
To analyze metastasis, the primary tumors were allowed to grow to endpoint (2cm in diameter), and subsequently the incidence of metastasis was determined. Thus, the incidence of metastasis is not a consequence of primary tumor growth. Interestingly, despite attenuating tumor development, the cell autonomous deletion of Akt1, unlike the germ line deletion of Akt1(Ju et al., 2007), did not inhibit the incidence of metastasis to the lung of tumor bearing mice (Fig. 1F).
Consequences of the systemic deletion of Akt1 or Akt2 after tumor onset in MMTV-ErbB2 mice.
The cell autonomous deletion of Akt1, unlike the Akt1 germ line deletion (Ju et al., 2007), did not affect tumor metastasis, raising the possibility that the effect of Akt1 on metastasis is systemic. To assess the effect of the systemic deletion of different Akt isoforms after tumor formation and to emulate single isoform inhibitor drug therapy, we used MMTV-ErbB2 mice (Muller et al., 1988), in which ErbB2 is overexpressed in the epithelial cells of the mammary gland. We bred MMTV-ErbB2 mice with either Akt1f/f;Rosa26(R26)CreERT2 mice or Akt2f/f;R26CreERT2 mice to generate MMTV-ErbB2;Akt1f/f;R26CreERT2 and MMTV-ErbB2;Akt2f/f;R26CreERT2 mice (Fig. 2A). As we have shown previously, the use of R26CreERT2 mice, in which CreERT2 was inserted in the ubiquitously expressed ROSA26 locus, enables the systemic deletion of Akt isoforms in adult mice after the injection of tamoxifen (Wang et al., 2016). After a latency period of 40–50 weeks, the mice developed palpable mammary tumors (approx. 0.1 cm3). We then performed the intraperitoneal (IP) injection of tamoxifen for only 5 consecutive days to induce Akt1 or Akt2 deletion (Fig. 2A). It should be emphasized that the deletion occurred after the injection of tamoxifen, and in order to eliminate any possible additional effect of tamoxifen, control MMTV-ErbB2;R26CreERT2 mice were also injected with tamoxifen for 5 consecutive days. One cohort of mice was euthanized after 5 weeks to determine primary tumor growth and the rest of the mice were euthanized when humane endpoint criteria were reached (when primary tumors reached 2cm in diameter) (Fig. 2A). At this time point the mice were analyzed for metastasis to the lung. The systemic Akt1 deletion markedly attenuated primary tumor growth (Fig. 2B) and proliferation as measured by Ki67 (Fig. 2C), indicating that the effect of Akt1 deletion on tumor growth is cytostatic, and thus extended the time to reach endpoint (tumor free survival) (Fig. 2D). Importantly, unlike the cell autonomous Akt1 deletion in mammary gland epithelial cells (Fig. 1F), which had no effect on tumor metastasis incidence, the systemic Akt1 deletion completely inhibited metastasis (Fig. 2E). It is important to note that metastasis was analyzed only when tumors reached end point in either control or experimental mice, and therefore the effect on metastasis is not a consequence of tumor growth. Taken together, the results strongly suggest that the inhibition of metastasis by the systemic deletion of Akt1 is not cell autonomous.
In contrast to the cell autonomous deletion of Akt2 and the systemic deletion of Akt1, the systemic deletion of Akt2 did not inhibit tumor growth (Fig. 2B, C), but rather increased tumor growth (Fig. 2B, C), decreased tumor free survival (Fig. 2D), and markedly increased the incidence of metastasis induced by ErbB2 (Fig. 2E). We speculated that the high level of circulating insulin induced by the systemic deletion of Akt2 (Fig. 2F) hyperactivates the other Akt isoforms and thus curbs the ability of the Akt2 deletion to inhibit metastasis. Consistently, we found that the systemic deletion of Akt2 markedly elevated Akt1 phosphorylation and total Akt activity, as measured by the phosphorylation of its substrates GSK3b and PRAS40. (Fig. 2G and Fig. S2A). Thus, the systemic deletion of Akt2 blunts the cell autonomous anti-tumorigenic activity of Akt2 (Fig. 1A) and is even pro-tumorigenic. To confirm this assertion, we crossed MMTV-NIC;Akt2f/f mice, which are resistant to tumorigenesis, with Akt2f/f;R26CreERT2 mice to generate MMTV-NIC;Akt2f/f;R26CreERT2 mice. Akt2 was systemically deleted in these animals at one month of age, which is 11 to 36 weeks before tumors were detected in MMTV-NIC mice (Fig. 1A). The systemic deletion of Akt2 abrogated the resistance to tumorigenesis exerted by the cell autonomous deletion of Akt2 (Fig. 2h). Importantly, the systemic deletion of Akt2 elevated Akt1 and possibly Akt3 activities and enabled the expression of ErbB2 in the absence of Akt2 in the mammary glands of MMTV-NIC;Akt2f/f;R26CreERT2 mice (Fig. 2I, and Fig. S2B), which is not expressed after cell autonomous deletion of Akt2 (Fig. 1C). Thus, although Akt2 is expressed at the highest level at early stages of mammary tumorigenesis (Fig. 1E) and its cell autonomous deletion could reduce substantially total Akt activity that disables ErbB2 expression, the systemic deletion of Akt2 enables ErbB2 expression because it hyperactivates the other Akt isoforms. These results provide strong additional support for our assertion that ErbB2 expression in the mammary gland is not tolerated when Akt activity is below a certain threshold level.
To further substantiate our assertion, we deleted Akt1 or Akt2 at late stages of tumor development when Akt1 expression is induced and Akt2 expression declines (Fig. 1E). If our assertion is correct, we should expect that cell autonomous deletion of Akt1 in late stage tumor cells would inhibit tumor growth better than Akt2 deletion and in contrast to the deletion early in tumor development. For this purpose, we isolated tumor cells derived from late stage tumors in MMTV-ErbB2;Akt1f/f;CreERT2 or MMTV-ErbB2;Akt2f/f;R26CreERT2 mice. The cells were orthotopically transplanted into nonobese diabetic (NOD)/Shi-scidIL-2Rγnull (NOG) mice. When the tumors were palpable, the mice were exposed to tamoxifen for 5 consecutive days to delete Akt1 or Akt2. The deletion of Akt1 markedly attenuated tumor growth (Fig. S3A), whereas the deletion of Akt2 attenuated tumor growth to a much lesser extent (Fig. S3B). The relative effect of Akt1 versus Akt2 on tumor growth is directly correlated with their relative individual expression in late stage tumors in which Akt1 expression is induced and Akt2 expression declines (Fig. 1E). Indeed, when compared to the Akt2 deletion, the deletion of Akt1 in MMTV-ErbB2 orthotopic tumors markedly decreased the total level of Akt, indicating that Akt1 is the predominant isoform at late stages of tumor development (Fig. S3C).
Consequences of the systemic deletion of Akt1 or Akt2 after tumor onset in MMTV-PyMT mice.
Polyoma virus middle T-antigen expression in the mammary gland of mouse mammary tumor virus-polyoma middle tumor antigen (MMTV-PyMT) mice induces several signaling pathways that are altered in human breast cancer, including the SRC and PI3K pathways. Specifically, the MMTV-PyMT mouse model will result in the development of multifocal mammary adenocarcinomas with a high incidence of metastatic lesions to the lymph nodes and lungs (Lin et al., 2003). Therefore, we employed this mouse model to study the effect of systemic Akt1 or Akt2 deletion on the incidence of metastasis. We generated MMTV-PyMT;R26CreERT2, MMTV-PyMT; Akt1f/f;R26CreERT2 and MMTV-PyMT;Akt2f/f;R26CreERT2 mice. After tumor onset, the systemic deletion of Akt1 or Akt2 was induced by tamoxifen injection. These mice were followed and subsequently sacrificed at the endpoint to assess metastasis (Fig. 3A). The Akt1 systemic deletion significantly increased tumor free survival, whereas the Akt2 systemic deletion did not (Fig. 3B). The systemic Akt1 deletion markedly reduced the number of metastatic nodules in the lungs but the systemic Akt2 deletion did not (Fig. 3C). To further establish that the effect of Akt1 on metastasis is not cell autonomous, we orthotopically implanted cells derived from tumors in MMTV-PyMT;Akt1f/f;R26CreERT2 and MMTV-PyMT;Akt2f/f;R26CreERT2 mice into NOG mice. After the tumors became palpable, the mice were treated with tamoxifen for 5 consecutive days. When tumors reached the endpoint, the mice were analyzed for metastases in the lungs. As shown in Fig. 3D and 3E, the cell autonomous deletion of neither Akt1 nor Akt2 changed the number of metastatic nodules, further supporting the systemic effect of Akt1 on metastasis. These results are consistent with the results obtained in MMTV-ErbB2 mice and further confirmed the discrepancy between the cell autonomous and systemic effects of Akt isoforms on mammary gland tumorigenesis. Indeed, similar to the results found in MMTV-ErbB2;Akt2f/f;R26CreERT2 mice, the systemic deletion of Akt2 in MMTV-PyMT;Akt2f/f;R26CreERT2 mice hyperactivated Akt1 and total Akt activity in the tumors (Fig. 3F and Fig. S4). The discrepancy between the inducible cell autonomous and non-cell autonomous effects of Akt1 and Akt2 deletions on tumor growth and metastasis again suggested that the inhibition of Akt1 might be superior to the inhibition of Pan-PI3K or Pan-Akt as a tumor therapy.
Figure 3: Consequences of systemic deletion of Akt1 or Akt2 after tumor onset in MMTV-PyMT mice.

A. The genotypes of mice and experimental strategy. B. Kaplan-Meier tumor free survival curves as determined by the tumor endpoint. p=0.0092, MMTV-PyMT;Akt1f/f;R26CreERT2 vs. MMTV-PyMT;R26CreERT2. C. Quantification of lung metastatic nodules. Data are presented as the means ± SEM. P=0.0066, MMTV-PyMT;Akt1f/f;R26CreERT2 vs. MMTV-PyMT;R26CreERT2 D. Quantification of lung metastatic nodules after orthotopic transplantation of MMTV-PyMT;Akt1f/f;R26CreERT2 cells into NOG mice in the presence or absence of Akt1. Data are presented as the means ± SEM. P=0.919, using an unpaired t test. E. Quantification of lung metastasis nodules after the orthotopic transplantation of MMTV-PyMT;Akt2f/f;R26CreERT2 cells into NOG mice in the presence or absence of Akt2. Data are presented as the means ± SEM. P=0.789, using an unpaired t test. F. Quantification of phosphorylated PRAS40, GSK3b, and Akt1 relative to total PRAS40, GSK3b, and Akt1 in tumor extracts after tamoxifen injection into MMTV-PyMT;R26CreERT2, MMTV-PyMT;Akt1f/f;R26CreERT2, and MMTV-PyMT;Akt2f/f;R26CreERT2 mice. Data are presented as the means ± SEM. * P<0.05, using an unpaired t test.
Systemic Akt1 deletion inhibits metastasis by impairing neutrophil mobilization.
To further delineate the systemic effects of Akt1 and Akt2 on mammary gland tumorigenesis and metastasis, we adopted Drop-seq technology for single cell RNA sequencing (scRNA-seq) as previously described (Macosko et al., 2015). Using this approach, we sequenced 7,791 cells over 5 biological replicates from the primary tumors of MMTV-PyMT mice and identified 17 clusters (Fig. 4A, Fig. S5A, and Table S1). Surprisingly, we identified nine distinct clusters of cells within the primary tumors based on the expression of PyMT (Fig. 4 and Fig. S5B), indicating that the tumors were very heterogeneous. Previous studies showed that keratin 14 (Krt14) is a marker for disseminating early metastatic cells, and its deletion suppresses metastasis (Cheung et al., 2016). Interestingly, within the nine distinct clusters of cells, we found one cluster (cluster 13) that expressed high levels of Krt14 (Fig. 4B). Cluster 13 also expressed other epithelial markers, such as Krt5, Krt7, Krt8, Krt17, and Krt18. However, cluster 13 also expressed a relatively high level of Vimentin (Vim), an EMT marker. Previous studies have shown that the high expression of Krt14 is also associated with the high expression of genes that regulate metastasis, such as Tenascin C (Tnc), Adam metallopeptidase (Adamts1), Caveolin 1 (Cav1), Jagged1 (Jag1), and Proepiregulin (Ereg) (Cheung et al., 2016). Indeed, cluster 13 expressed high levels of Tnc and Adamts1, as well as Cav1, Jag1, and Ereg (Fig. 4B and Table S1). Notably, cluster 11, which expressed high levels of vimentin, also expressed high levels of Ereg and Adamts1 and relatively high levels of Cav1 and Jag1 (Fig. 4B). However, high Krt14 expression, unlike in cluster 13, was not found in cluster 11. Paradoxically, cluster 13 expresses the highest level of E-cadherin (cdh1), whereas cluster 11 expresses the lowest level of cdh1 (Table S1). However, this is consistent with a recent report showing that E-cadherin is required for the survival of disseminating metastatic cells in this mouse model (Padmanaban et al., 2019).
Figure 4. Analysis of primary and metastatic tumors by scRNA-seq.

A. t-Distributed Stochastic Neighbor Embedding (t-SNE) plot of primary mammary gland tumors in MMTV-PyMT mice. Each cluster is characterized by a unique gene expression signature. A total of 7,791 primary breast tumor cells (N = 5) were used. Clusters 0, 1, 2, 3, 4, 5, 6, 11, and 13 are tumor cells expressing PyMT, whereas the remaining clusters are non-tumor cells. The clusters are color-coded. B. Dot plot showing expression of metastatic markers Ereg, Jag1, Cav1, Adamts1, Tnc, Vim, as well as Krt18, Krt17, Krt8, Krt7, Krt5, and Krt14 across the PyMT primary tumor clusters. C. tSNE plot of 3,979 metastatic tumor cells in the lung (N = 3). Clusters 0, 1, 4, 5 and 11 are PyMT-positive. D. Dot plot depicting expression of Prok2, Vegfa, Mmp9, Mmp8, S100a9, S100a8, and the prometastatic marker Krt14 across the clusters in the lung metastatic scRNA-seq analysis. E. Dot plot in the primary non-tumor clusters indicate that cluster 10 has a predominant gene expression profile of neutrophil markers Prok2, Vegfa, Mmp9, Mmp8, S100a9, S100a8. F. Graph showing the average number of pro-metastatic Krt14-positive cells for every 5,000 PyMT-positive tumor cells based on the scRNA-seq results. N(MMTV-PyMT) = 5, N(MMTV-PyMT; Akt1f/f) = 3, N(MMTV-PyMT; Akt2f/f) = 3. N. S = not significant (p>0.05). One-way analysis of variance (ANOVA) was used to calculate significance. G. Graph showing the average number of infiltrating neutrophils in each genotype. scRNA-seq reveals the absence of neutrophil cells in primary systemic Akt1 knockout tumors across all 3 biological replicates. N(MMTV-PyMT) = 5, N(MMTV-PyMT; Akt1f/f) = 3, N(MMTV-PyMT; Akt2f/f) = 3. N.S = not significant (p>0.05). One-way ANOVA was used to calculate significance. Error bars represent standard error.
Thereafter, we sequenced 3,979 single cell transcriptomes over 3 biological replicates from the macroscopic metastatic lesions in the lungs of MMTV-PyMT mice (Fig. 4C). The scRNA-seq results revealed five clusters (0, 1, 4, 5, and 11) that were derived from the primary tumors as determined by the high level of PyMT expression (Fig. S5C, and Table S2). Among the five clusters, only one, cluster 11, expressed high levels of Krt14 (Fig. 4D). After combining and re-clustering, the primary and metastatic scRNA-seq results, we found that cluster 19 (Fig. S6 A- C, and Table S4) consisted of cells from both the metastatic lung cluster 11 (Fig. 4C) and primary mammary tumor cluster 13 (Fig. 4A). This finding indicates that the last two clusters are similar and share a gene expression profile and traces a metastatic cluster found in the lung to a cluster in the primary tumors.
Micrometastases express high levels of Krt14, which are diminished in macrometastases (Cheung et al., 2016). Therefore, we analyzed the lungs of tumor-bearing mice that do not display macroscopic metastases. Consistently, we found only one cluster derived from the primary tumors, and this cluster expressed high levels of Krt14 and PyMT (Fig. S5D, E, and Table S3). Thus, among the distinct clusters in the primary tumors, we classified the high Krt14-expressing cluster as a pro-metastatic cluster.
Within the primary tumor, we also found nontumor cells that did not express PyMT, which include stromal cells, T cells, macrophages, and neutrophils (Fig. 4A). Importantly, a population of neutrophils was also found within the metastatic tumors in the lungs (Fig. 4C). Neutrophils play a pro-metastatic role in breast cancer (Mouchemore et al., 2018; Wculek and Malanchi, 2015), and a high neutrophil to lymphocyte ratio (NLR) is associated with worse overall survival and disease-free survival (Ethier et al., 2017). Consistent with the reported pro-metastatic role of neutrophils that provide a niche for the metastatic cells in the lungs, we found that neutrophils within lung metastases and primary tumors express, in addition to high Ly6g and Cxrc2, which are known neutrophils markers, relatively high levels of S100a8, S100a9, MMP8, MMP9, Bv8/Prok2 and vascular endothelial growth factor (Vegfa), which promote invasion and migration (Fig. 4D, E, and Fig. S5E).
After the systemic deletion of Akt1 or Akt2, analysis of the tumors using Drop-seq (3,194 cells over 3 replicates and 4,647 cells over 3 replicates, respectively) revealed that primary tumors had similar cell clusters as those in control mice, including a pro-metastatic cluster expressing high Krt14, which is co-segregated in control wild type (WT), Akt1-/-, and Akt2-/- primary tumors (Fig. S6A–C, and Table S5). Furthermore, the percentage of high Krt14-expressing cells in primary tumors after the systemic deletion of Akt1 or Akt2 was not significantly different from that in control primary tumors (Fig. 4C). These results suggest that the systemic Akt1 deletion did not change the relative presentation of the pro-metastatic cluster within the primary tumors.
While the presentation of a high Krt14-expressing cell cluster was similar in WT control mice and in mice after the systemic deletion of Akt1 or Akt2 (Fig. 4F, and Table S5), the presentation of neutrophils was completely diminished after Akt1 systemic deletion (Fig. 4G, and Fig. S6 D–E). These results raised the possibility that systemic Akt1 deletion inhibits metastasis by impairing neutrophil mobilization to the lung. To further assess this possibility, we examined whether systemic Akt1 deletion could affect pro-metastatic neutrophils in the lungs. The percentage of neutrophils in the lungs of non-tumor-bearing mice, as measured by anti-Ly6G staining, was not significantly different in control mice and in mice after either Akt1 or Akt2 systemic deletion (Fig. 5A). However, in tumor-bearing mice, the percentage of neutrophils in the lungs of control mice or in mice after Akt2 systemic deletion was markedly increased, whereas Akt1 systemic deletion did not increase the percentage of neutrophils in the lungs (Fig. 5B).
Figure 5. The effect of the systemic Akt1 or Akt2 deletion on neutrophil accumulation in the lungs of tumor-bearing mice.

A. Percentage of neutrophils in the lungs of nontumor-bearing mice after tamoxifen injection to systemically delete Akt1 or Akt2. The percentage of neutrophils was calculated by counting the number of Ly6G-positive cells relative to total hematoxylin and eosin-stained cells in lung tissue at endpoint sections as described in the Methods section. Data are presented as the means +/− SEM. P= 0.3, using unpaired t test. B. Quantification of lung neutrophils in control MMTV-PyMT mice and after systemic deletion of Akt1 or Akt2 at endpoint. Left: Representative lung section images stained with anti-Ly6G. Right: Quantification was performed after the primary tumors reached the endpoint. Data are presented as the means ± SEM. P=0.0066, MMTV-PyMT;Akt1f/f;R26CreERT2 vs. MMTV-PyMT;R26CreERT2 and p=0.518, MMTV-PyMT;Akt2f/f;R26CreERT2 vs. MMTV-PyMT;R26CreERT2 using an unpaired t test. Quantification of metastatic nodules (C) and neutrophils (D) in the lungs of R26CreERT2 and Akt1f/f;R26CreERT2 mice, which were orthotopically transplanted with MMTV-PyMT (WT) tumor cells and injected with tamoxifen at palpation. Upper panel shows schematic of experimental design. Quantification was performed when the primary tumors reached the endpoint. Data are presented as the means ±SEM. P=0.019 for metastatic nodules, and P=0.0034 for neutrophils using an unpaired t test. Quantification of metastatic nodules (E) and neutrophils (F) in the lungs of R26CreERT2 and Akt2f/f;R26CreERT2 mice, which were orthotopically transplanted with MMTV-PyMT (WT) tumor cells and injected with tamoxifen at palpation. Upper panel shows schematic of experimental design. Quantification was performed when the primary tumors reached the endpoint. Data are presented as the means ±SEM. P>0.05 for metastatic nodules and neutrophils using an unpaired t test.
If systemic Akt1 deletion inhibits metastasis by a systemic effect that impairs neutrophil mobilization to the lungs, then this deletion would also inhibit the metastasis of WT tumors. We therefore orthotopically implanted tumor cells derived from MMTV-PyMT mice into either R26CreERT2, Akt1f/f;R26CreERT2, or Akt2f/f;R26CreERT2 mice. When the tumors became palpable, the mice were injected with tamoxifen to systemically delete Akt1 or Akt2. When the tumors reached the endpoint, the mice were analyzed for metastasis. The systemic deletion of Akt1 markedly decreased the metastasis of WT tumors (Fig. 5C), which is directly correlated with the decrease in tumor-associated neutrophils in the lungs (Fig. 5D). However, when compared to WT control mice, the systemic deletion of Akt2 did not decrease the metastasis of WT tumors (Fig. 5E) and did not significantly affect the percentage of neutrophils in the lungs (Fig. 5F).
If the Akt1 deficiency in neutrophils determines decreased metastatic potential, then it is expected that the specific deletion of Akt1 in neutrophils could decrease metastasis. Therefore, we used MRP8-Cre mice, where specific deletion in neutrophils was documented (Abram et al., 2014; Passegue et al., 2004), to delete Akt1 specifically in neutrophils. We re-established the specific deletion in neutrophils and showed that Akt1 is deleted exclusively in neutrophils (Fig. S7 A, B). We orthotopically implanted E0771 mouse breast cancer cells into control MRP8-Cre, and MRP8-Cre;Akt1f/f mice. As shown in Fig. 6A, lung metastasis was diminished in MRP8-Cre;Akt1f/f mice compared to that of MRP8-Cre mice. Consistently, neutrophils were accumulated in the lungs of MRP8-Cre mice but markedly reduced in MRP8-Cre;Akt1f/f mice (Fig. 6B). Notably the percentage of neutrophils in the lungs after the systemic (Fig. 5D) or neutrophil specific deletion of Akt1 (Fig. 6B) is similar to the percentage of neutrophils in the non-tumor bearing mice (Fig. 5A). Thus, these results provide direct evidence that the systemic effect of the Akt1 deletion on metastasis is due to the effect on pro-metastatic or tumor-associated neutrophils. By contrast, the deletion of Akt2 specifically in neutrophils did not affect metastasis (Fig. S7C).
Figure 6. Consequences of Akt isoform deletion in the neutrophils of tumor-bearing mice.

A. The effect of Akt1 deletion on metastasis in MRP8-Cre mice after orthotopic transplantation of E0771 cells. Upper panel: Schematic of experimental design. Bottom panel: Quantification of metastasis. B. The effect of Akt1 deletion on neutrophils in the lungs of MRP8-Cre tumor-bearing mice (n=8). Upper panels show representative lung section images stained with anti-Ly6G. Data are presented as the means ±SEM. P<0.0001, for metastatic nodules and neutrophils using an unpaired t test. C. The effect of G-CSF on the survival of neutrophils isolated from control, and systemically deleted Akt1 and Akt2 tumor-bearing mice. Data are presented as the mean ± SEM. p=0.0055 for Akt1f/f;R26CreERT2 versus R26CreERT2 and p=0.099 for Akt2f/f;R26CreERT2 versus R26CreERT2. D. The effect of G-CSF on the level of MCL1 in neutrophils isolated from control and systemically deleted Akt1 and Akt2 tumor-bearing mice.
To understand the mechanism by which Akt1 affects tumor-associated neutrophils, we isolated neutrophils from tumor-bearing mice and exposed these cells to granulocyte colony stimulating factor (G-CSF) in vitro. G-CSF was shown to promote the survival of neutrophils and is required for the mobilization of tumor-associated neutrophils (Mouchemore et al., 2018). As shown in Fig. 6C, G-CSF increased the survival of neutrophils isolated from either control or Akt2-deficient tumor-bearing mice but not the survival of neutrophils isolated from Akt1-deficient tumor-bearing mice. These results suggest that, at least in part, Akt1 deficiency decreases the survival of tumor-associated neutrophils. In mouse models of mammary gland tumors, the inhibition of translation initiation factor eIF4E decreases metastasis to the lungs by decreasing the mobilization of neutrophils to these organs (Robichaud et al., 2018). This finding was attributed to the increase in neutrophil cell survival by eIF4E after exposure to G-CSF through the elevation of BCL2 and MCL1 (Robichaud et al., 2018). Since Akt, which is upstream of mTORC1 and eIF4E (Gingras et al., 1998), could also affect the level of MCL1 through the inhibition of GSK3 and the increase in its protein stability (Maurer et al., 2006), we examined the level of MCL1 after exposure to G-CSF. We found that MCL1 protein levels are induced by G-CSF in neutrophils derived from either control or Akt2-deficient tumor-bearing mice but not in neutrophils derived from Akt1-deficient tumor-bearing mice (Fig. 6D). Thus, Akt1 is required downstream of G-CSF to promote elevated MCL1 protein. Consistent with the posttranscriptional effect of Akt on MCL1, the mRNA levels of MCL1 did not change after Akt1 deletion (Fig. S7D). These results are consistent with other studies showing that MCL1 is particularly important for the survival of neutrophils and is induced by G-CSF (Dzhagalov et al., 2007). One possible reason why Akt1 and not Akt2 affects neutrophils in tumor-bearing mice is that Akt1 is the major expressed isoform in neutrophils of tumor bearing mice (Fig. S7E). Taken together, these results showed that the systemic effect of Akt1 deficiency on the inhibition of metastasis is due to the effect of Akt1 on the survival and mobilization of the neutrophils that promote metastasis.
Reducing insulin level after systemic Akt2 deletion inhibits mammary tumor development.
Our results suggested that the systemic deletion of Akt2 curbs the inhibition of mammary gland tumorigenesis because of the high circulating levels of insulin that hyperactivates the other Akt isoforms and other pro-oncogenic signaling pathways (Fig. 2 and Fig. 3). To further assess this possibility, we treated mice with the diabetic drug, metformin, to decrease insulin levels after systemic Akt2 deletion. However, metformin was not able to reduce insulin levels in these mice. We therefore fed the mice with a diet that includes the sodium-glucose co-transporter (SGLT2), inhibitor, the anti-diabetic drug, canagliflozin. SGLT2 is responsible for reabsorption of glucose in the kidney, and therefore limits the excretion of glucose through the urine (Tahara et al., 2013). Inhibition of SGLT2 by its selective inhibitor, canagliflozin, was shown to inhibit hyperglycemia and hyperinsulinemia without adverse consequences (Tahara et al., 2013). As shown in Fig. 2D and 2H, the systemic deletion of Akt2 in MMTV-ErbB2 and MMTV;NIC mice accelerated and exacerbated tumorigenesis, which was attributed to the high level of insulin. To affirm this possibility MMTV-ErbB2;Akt2f/f;R26CreERT2 or MMTV-NIC;Akt2f/f;R26CreERT2 mice were fed, after systemic Akt2 deletion, with a diet containing canagliflozin, with an average approximate dose of 40mg/kg BW/day. As shown in Fig. 7A, C canagliflozin substantially reduced the elevated insulin level observed after Akt2 systemic deletion. Consequently, the development of mammary gland tumorigenesis was markedly reduced when mice were treated with canagliflozin after systemic Akt2 deletion (Fig. 7B, D). Thus, systemic deletion of Akt2 does not inhibit and may even accelerate mammary gland tumorigenesis by elevating blood insulin levels. Reducing insulin levels after systemic Akt2 deletion inhibits the acceleration of mammary gland tumorigenesis by systemic Akt2 deletion.
Figure 7. Reducing circulating levels of insulin by SGLT2 inhibitor attenuates the mammary gland tumorigenesis after systemic Akt2 deletion.

MMTV-ErbB2;Akt2f/f;R26CreERT2 or MMTV-ErbB2;Akt2f/f;R26CreERT2 mice were injected with tamoxifen at 6 weeks of age and were subjected to either control chow diet or diet that includes canagliflozin (CANA). A,C. Insulin level at the indicated time points after chow diet or CANA diet. B,D. Kaplan-Meier curves showing percentage of tumor free mice after chow diet or CANA diet.
Discussion
The results described here underscore the importance of employing systemic deletion as a genetic proof of concept for cancer therapy, as cell autonomous deletion could otherwise be misleading. This notion was exemplified by the effect of the cell autonomous versus systemic deletion of Akt1 or Akt2 on breast cancer development and metastasis. Surprisingly, the cell autonomous deletion of Akt1 at tumor onset and after tumor formation did not inhibit metastasis, whereas systemic deletion of Akt1 markedly inhibited metastasis. We showed that the predominant mechanism by which systemic Akt1 deletion inhibits metastasis is by inhibiting the survival and mobilization of pro-metastatic neutrophils. These neutrophils express relatively high levels of prokineticin 2 (PROK2) and VEGFa, which promote angiogenesis, as well as the metalloproteases MMP8 and MMP9, which promote invasion. These effects would allow the extravasation and mobilization of neutrophils but could also promote cancer cell angiogenesis and invasion. Neutrophils also physically interact with cancer cells (Huh et al., 2010), and thus can increase the extravasation of disseminating cancer cells by expressing and secreting MMP8 and MMP9. More recently, it was shown that neutrophils actually escort disseminating cancer cells to the metastatic site (Szczerba et al., 2019); thus, it is possible that these cells also promote cancer cell extravasation. In addition, neutrophils form neutrophil extracellular traps (NETs) that stimulate migration and invasion and trap natural killer cells (Park et al., 2016). The pro-metastatic role of neutrophils was also recognized in human cancer patients, and a high NLR is associated with a poor prognosis (Mouchemore et al., 2018). The neutrophils derived from tumor-bearing mice or from cancer patients are distinct from normal neutrophils, as tumor-associated neutrophils lack immunosuppressive activity and have a higher migration capacity (Patel et al., 2018). Cancer cells promote the survival and mobilization of neutrophils by secreting G-CSF (Mellouli et al., 2010; Mouchemore et al., 2018). We showed that at least in vitro Akt1, and not Akt2, is required for the G-CSF-induced survival of neutrophils derived from tumor-bearing mice. The major mechanism by which G-CSF promotes the survival of neutrophils is by inducing the expression of the anti-apoptotic protein MCL1 (Dzhagalov et al., 2007). We showed that Akt1 deficiency prohibits the induction of MCL1 expression by G-CSF. We cannot completely exclude, however, other potential mechanisms by which the Akt1 deficiency impairs the survival and mobilization of tumor-associated neutrophils. Notably, we found that Akt1 is the major expressed Akt isoform in neutrophils of tumor bearing mice and this could explain why systemic deletion of Akt1and not Akt2 markedly affect the pro-metastatic neutrophils. Previous studies have shown the differential roles of Akt isoforms in neutrophil function. In contrast to our findings with respect to neutrophil function in tumor-bearing mice, the neutrophils derived from nontumor-bearing mice are more dependent on Akt2 than on Akt1 in response to stimulation by N-Formylmethionyl-leucyl-phenylalanine (fMLP) or phorbol myristate acetate (PMA) (Chen et al., 2010). This finding could be explained by the change in the phenotype of these neutrophils in response to tumor formation and by the different stimuli used. Consistently, we did not observe any change in neutrophils infiltration to the lung after systemic deletion of Akt1 in non-tumor bearing mice (Fig. 5A). Finally, It was speculated that in tumor bearing hosts there is a pressure to release neutrophils from the bone marrow prematurely and that these immature neutrophils can be converted to pro-tumorigenic pro-metastatic neutrophils (Coffelt et al., 2016). We identified cluster 18 in Fig. S6A and S6B, or cluster 21 in Fig. S6 D,F as neutrophils that are also present in the lung of tumor bearing mice but are missing after systemic deletion of Akt1 (Fig. 4G, and Fig. S6F). These neutrophils were identified as the pro-metastatic neutrophils and express the highest RNA level of Ly6g and Cxcr2, which are known neutrophil markers. However, we also identified cluster 14 in Fig. S6 D,F, as a potential neutrophil progenitor population. This cluster had the second highest RNA expression of Cxcr2 with no expression of Ly6g. Nevertheless, there is no difference in the percentage of cells in cluster 14 between control mice and Akt1 or Akt2 deleted mice (Fig. S6E). Although we cannot completely exclude the possibility that cells other than neutrophils in the tumor microenvironment could have an effect of metastasis after the deletion of Akt1, neutrophils are the only cell type that was eliminated in the tumor microenvironment. The recapitulation of Akt1 systemic deletion on metastasis by specific Akt1 deletion in neutrophils further support that this is the pre-dominant if not the exclusive mechanism by which systemic Akt1 deletion inhibits metastasis.
Our studies also show that the expression of ErbB2 in the absence of Akt2 only in mammary gland cells cannot be tolerated and these cells are likely eliminated. We hypothesized that ErbB2 expression cannot be tolerated in mammary gland cells in which Akt activity is below a certain threshold level. Akt2 is expressed at the highest level whereas Akt1 is expressed at the lowest level in early stages of mammary gland tumorigenesis. Thus, it is possible that total Akt activity is more reduced at this stage in the absence of Akt2 than in the absence of Akt1. However, systemic Akt2 deletion, which increases insulin levels and hyperactivates Akt1 and possibly Akt3, overcomes the intolerance of ErbB2 expression in the absence of Akt2 in the mammary gland. In late stage tumor growth, Akt1 expression is elevated, and Akt2 expression declines; thus, the growth of tumor cells derived from MMTV–ErbB2 mice is impaired to a much higher extent by Akt1 deletion than by Akt2 deletion. However, it remains to be determined how the cell autonomous deletion of Akt1, which is expressed at the highest level in late stages of tumor development, is tolerated in the presence of high ErbB2 expression. One possibility is that at late stages, the cells have already acquired additional lesions that enable the expression of ErbB2, despite low Akt activity.
The systemic deletion of Akt2 markedly increased tumor growth and metastasis in MMTV-ErbB2 mice and did not inhibit tumor growth and metastasis in MMTV-PyMT mice. This exacerbated effect on tumor growth was also observed with the germ line deletion of Akt2 (Maroulakou et al., 2007). Mechanistically, we showed that this is attributed to the high circulating levels of insulin as a consequence of systemic Akt2 deletion since reducing insulin levels after the systemic deletion of Akt2 inhibits tumorigenesis. Recently it was shown that reducing insulin level after treatment with pan-PI3K inhibitors, which elevate insulin level, increased their efficacy (Hopkins et al., 2018). Our results showed that downstream of PI3K inhibition, Akt2 inhibition is responsible for the elevated insulin.
As indicated in the introduction, the differential roles of Akt isoforms in breast cancer tumorigenesis and metastasis are complex and controversial. Previous studies with germ line deletion of Akt1 are consistent with our systemic deletion of Akt1 with respect to tumor development (Ju et al., 2007; Maroulakou et al., 2007) and metastasis (Ju et al., 2007). The germ line deletion of Akt2 in MMTV-ErbB2 mice, which increases tumor development and appears to inhibit metastasis (Maroulakou et al., 2007). Our results showed that systemic Akt2 deletion consistently accelerates tumor development in MMTV-ErbB2 mice, but in contrast to the germ line deletion the systemic Akt2 deletion also increased metastasis regardless of tumor growth (Fig. 2). It is not clear why unlike Akt2 systemic deletion after tumor onset, the germ line Akt2 deletion inhibits metastasis. However, this discrepancy could be, at least in part, because in the germ line deletion tumor onset occurred after the deletion, whereas the systemic deletion occurred after tumor onset. In addition, as indicated in the introduction, it cannot be completely ruled out that the germ line deletion might have potential developmental consequences that affect the outcomes with respect to metastasis. Interestingly, cell culture studies with human mammary epithelial cells showed that silencing Akt1 but not Akt2 increased cell migration and EMT (Irie et al., 2005). The in vitro studies with human mammary epithelial cells also suggest that while Akt1 is anti-migratory and inhibits EMT, Akt2 might be promoting EMT and pro-migratory (Irie et al., 2005). However, metastasis in vivo is not always related to EMT and migration in vitro. For instance, in MMTV-PyMT mouse model the identified metastatic cluster in the primary tumors (cluster 13, Fig.4A and Table S1) remains intact after Akt1 deletion. This cluster although expresses relatively high level of EMT genes also expresses relatively high level of E-cadherin (Table S1), suggesting that in vitro EMT analysis may not always reflect metastasis in vivo. Consistently, although the loss of E-cadherin increased invasion, E-cadherin is actually required for breast cancer metastasis largely to promote the survival and the seeding of the disseminating cells (Padmanaban et al., 2019). Notably Akt activity was reported to promote metastasis by phosphorylating and activating transcriptional regulator of EMT, Twist1 (Xue et al., 2012). A different report, however, showed that Akt1 specifically phosphorylates and degrades Twist1 (Li et al., 2016), and proposed that very high Akt1 activity might be pro-tumorigenic but negates metastasis, whereas lower Akt1 activity promotes metastasis. This is consistent with a gain of function studies where hyperactivating of Akt1 in the mammary gland of MMTV-ErbB2 mice, increased tumor growth but decreased metastasis (Dillon et al., 2009; Hutchinson et al., 2004). By contrast hyperactivation of Akt2 in the mammary gland of MMTV-ErbB2 mice increased metastasis (Dillon et al., 2009; Hutchinson et al., 2004). These results raised again the possibility that Akt1 and Akt2 have opposing effect on metastasis at the cell autonomous level. However, these studies do not take into consideration the systemic effect of Akt1 or Akt2 on metastasis. Our results clearly showed that the pre-dominant effect of systemic inducible Akt1 deletion, which emulates drug therapy, on breast cancer metastasis is largely not cell autonomous and is dependent on the tumor associated neutrophils.
Taken together, our results provide strong support for the use of systemic deletion after tumor onset as a proof of concept for cancer therapy. These results together with our previous results (Wang et al., 2016; Yu et al., 2015) provide support for using specific Akt1 inhibitors and avoiding Akt2 or pan-Akt inhibitors for cancer therapy. Furthermore, the effect of Akt1 systemic deletion on pro-metastatic neutrophils, but not on other functions of neutrophils, indicate that Akt1 specific inhibitors would be sufficient to selectively inhibit the pro-metastatic effect of neutrophils. Thus, specific Akt1 inhibition would inhibit breast cancer metastasis regardless of the origin of the primary tumor.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nissim Hay (nhay@uic.edu).
Materials Availability
The mice generated in this study are available upon request.
Data and Code Availability
The datasets generated in this study are available at GEO: GSE135096 Original data for figures in this study are accessible upon request and deposited to Mendeley Data: http://dx.doi.org/10.17632/77t55d5zjf.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains and mouse work
The MMTV-NIC mice and MMTV-Erbb2 mice were gifts from W.J. Muller (McGill University). The MMTV-PyMT and LSL-Luc mice were purchased from the Jackson Laboratory. The FVB/N WT mice were purchased from Charles River Laboratories. The R26CreERT2 knock-in mice (strain 01XAB) Akt1f/f, and Akt2f/f, Akt1f/f;R26RCreERT2 and Akt2f/f;R26RCreERT2 mice have been previously described (Wang et al., 2016). MMTV-PyMT;Akt1f/f;R26RCreERT2 and MMTV- PyMT;Akt2f/f;R26RCreERT2 mice were generated by crossing Akt1f/f;R26RCreERT2 or Akt2f/f;R26RCreERT2 mice with MMTV- PyMT mice. MMTV-Erbb2;Akt1f/f;R26RCreERT2 and MMTV-Erbb2;Akt2f/f;R26RCreERT2 were generated by crossing Akt1f/f;R26RCreERT2 or Akt2f/f;R26RCreERT2 mice with MMTV-Erbb2 mice. MMTV-NIC;Akt1f/f and MMTV-NIC;Akt2f/f mice were generated by crossing Akt1f/f or Akt2f/f with MMTV-NIC mice. MMTV-NIC;Akt2f/f;R26RCreERT2 mice were generated by crossing MMTV-NIC;Akt2f/f mice with Akt2f/f;R26RCreERT2 mice. MMTV-NIC;LSL-Luc mice were generated by crossing MMTV-NIC mice with LSL-Luc mice. MMTV-NIC;Akt1f/f;LSL-Luc mice and MMTV-NIC;Akt2f/f;LSL-Luc were generated by crossing of MMTV-NIC;LSL-Luc mice with Akt1f/f and Akt2f/f mice respectively. All mouse models were produced in an FVB/N background. Mice from other backgrounds were backcrossed to FVB/N mice for at least 10 generations. Cre recombinase activation was performed by the IP injection of 0.1 ml of 20 mg/ml of tamoxifen for 5 consecutive days. For IP injection, tamoxifen was dissolved in corn oil at a final concentration of 20 mg/ml via shaking at 37°C for 30 minutes as previously described (Wang et al., 2016). PCR, and western blot analysis of multiple tissues, including tumor tissues, were used to confirm the deletion of Akt isoforms. C57Bl6 MRP8-Cre-ires-GFP mice were purchased from the Jackson Laboratory and were crossed with Akt1f/f mice or Akt2f/f in C57Bl6 background to generate MRP8-Cre;Akt1f/f or MRP8-Cre;Akt1f/f mice. Both male and female mice were used for breeding. All experimental mice were females. NOG female mice were purchased from Jackson Laboratory. For the Canagliflozin diet experiments, the mice were injected with tamoxifen at 6 weeks of age and maintained on chow diet (Teklad #7012). On day 15, blood samples were collected from the tail vein using a heparinized micro-capillary tube to measure basal fed plasma glucose and insulin. Mice were then randomly divided into two groups. Canagliflozin (CANA) (MedChem Express, Monmouth Junction, NJ, USA) was administrated as a food additive at a concentration of 0.03% (w/w) into Teklad #7012 chow diet (Research Diets, Inc., New Brunswick, NJ, USA). Each group received control or CANA diet with ad libitum access to food until the day they are sacrificed. The average daily dose of canagliflozin (calculated from average daily food intake for mice and actual body weight) was approximately 40mg/kg BW/day, a dose effective in many studies (Naznin et al., 2017; Thrailkill et al., 2016; Watanabe et al., 2015). The body weight of each mouse was measured every week. Fed plasma glucose and insulin levels were measured every 4 weeks. Starting at 6-month of age, mice were palpated each week for tumor appearance and age of appearance was recorded. mice All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago (UIC), as required by the United States Animal Welfare Act and the policy of NIH.
Kaplan-Meier Plots
Kaplan-Meier plots were calculated for non-inducible deletion of Akt isoforms (Fig. 1B) as the time required for tumor onset and was shown as percent of tumor free mice. Percent of tumor free mice is also shown in Fig. 2H, Fig. 7B and Fig.7D. For the inducible deletion after tumor palpation, Kaplan -Meier plots were used to calculate the time that is required from palpation and tamoxifen administration to the time the tumor to reached endpoint (2cm diameter) is shown as tumor free survival (Fig. 2D, and Fig. 3B).
Primary tumor cell isolation
Mouse tumor tissues were dissected, washed in PBS supplemented with 5% P/S (penicillin/streptomycin), cut into small pieces (diameter~3 mm), and then digested in 1% collagenase IV in DMEM at 37°C for 30 minutes with shaking. The supernatant was discarded after centrifugation at 300 g for 5 minutes. The pellet was washed several times with PBS and passed through a 75 μm cell strainer to collect small cell clumps. The clumps were then transferred to plates containing DMEM supplemented with 10% FBS and 1% P/S in an incubator at 37°C with 5% CO2.
Tumor cell transplantation
Freshly isolated tumor cells (less than 3 days) were harvested and resuspended in PBS and Matrigel (Corning) in a 1:1 ratio with a final concentration of 0.1~1 × 106/100 μl. One hundred microliters of cells were transplanted to the fourth mammary gland fat pad of each side to recipient mice. Mice were monitored/palpated daily for 1 week to confirm successful tumor transplantation. After the tumor diameter reached 4 mM, the tumor size was measured every week until a certain time point or a humane endpoint. Tumor sizes were measured with a caliper and calculated by length*height*width*0.5. For transplantation 1×105 E0771 mouse breast cancer cells (obtained for ATCC, ATCC® CRL-3461) were orthotopically transplanted into the mammary glands of MRP8-Cre and MRP8-Cre;Akt1f/f mice as described above. When primary tumors reached 0.5cm3 the mice were analyzed for lung metastasis and neutrophils infiltration.
Bone marrow isolation
After muscle removal, the femurs and tibias were collected and rinsed with 70% ethanol followed by ice-cold PBS wash. The epiphyses were cut off. Then, a 10-cc syringe filled with RPMI medium supplemented with 10% FBS and 2 mM EDTA was used to flush the marrow cells from both ends with a 25-gauge needle into a 50 ml Falcon tube with a 40 μm cell strainer. After centrifugation at 1000 rpm for 5 min, the pellet was resuspended in 3 mL cold ammonium chloride potassium (ACK) lysis buffer for 1 min, centrifuged and washed with PBS and resuspended in the desired volume.
Neutrophil isolation
Bone marrow-derived neutrophils were isolated from mice using the following protocol adapted from Swamydas et al. 6. Bone marrow cells were obtained from mice by flushing the contents of the tibia and femur with complete RPMI medium supplemented with 2 mM EDTA using a 25-gauge needle. The cell suspension was run through a 100 μM cell strainer. The cells were pelleted at 430xg for 7 minutes at 4°C. Red blood cells were lysed by washing the cells with 20 mL of 0.2% NaCl for 20 seconds followed by the addition of 20 mL of 1.6% NaCl. After washing with PBS, the bone marrow cells were resuspended in ice-cold PBS and layered on the top of a Histopaque 1119/1077 (Sigma) gradient in a 15 mL Falcon tube. After centrifugation at 2000 rpm at room temperature for 30 minutes (without the break), the neutrophils were isolated by collecting the cells at the interface of the two Histopaque layers. The neutrophils were then washed twice with complete RPMI medium. Neutrophil viability and purity were assessed via trypan blue staining and flow cytometric analysis of Ly6G surface expression by using a PE-Ly6G antibody (Biolegend).
Cell culture
Freshly isolated mouse breast tumor cells were cultured in high glucose DMEM supplemented with 10% FBS and 1% P/S. Freshly isolated neutrophils were cultured in RPMI 1640 supplemented with 10% FBS and 1% P/S.
Neutrophil cell death assay
Freshly isolated neutrophils were cultured with or without G-CSF at a final concentration of 100 ng/ml for 24 hours. The neutrophils were then stained with Hoechst stain and propidium iodide (PI) for 30 minutes in an incubator followed by plate scanning with a Celigo Imaging Cytometer. The dead cell/live cell ratio was determined by the red cell number (PI stained)/blue cell number (Hoechst stained) ratio.
Measurement of glucose, insulin, and IL-6 levels
Glucose levels were measured with a glucometer test strip (Precision Xtra; Abbott Laboratories). Insulin levels were measured by Milliplex immunoassay (Millipore) according to the manufacturer’s instructions.
METHOD DETAILS
Tissue staining and immunohistochemistry
Breast tumor tissues were freshly collected and directly fixed in 10% formalin (Fisher Chemical). The lungs were inflated with PBS via the trachea and then removed from the ribcage, washed in PBS and dissected to lobules, before fixing in 10% formalin. After fixation for 24–48 hours (depending on tissue size) in formalin, the tissues were processed and embedded in paraffin blocks. The sections (5 μm) were stained with hematoxylin and eosin (H&E). For immunohistochemistry, antigen retrieval was performed by incubating the sections in 0.01 M sodium citrate (pH 6.0) at 95°C for 20 minutes, followed by cooling down to room temperature. The sections were treated with 0.3% H2O2 for 5 minutes to quench endogenous hydrogen peroxide. After blocking with normal serum, the sections were incubated with primary antibody at 4°C overnight. After incubation with the appropriate secondary antibodies from Vectastain ImmPress Kits (Vector Labs), a 3, 3 -diaminobenzidine (DAB) kit (Vector Labs) was applied to visualize the signal. The sections were then lightly counterstained with hematoxylin. To determine the lung metastatic incidence metastatic nodules were counted under the microscope after H&E staining. Infiltration of neutrophils to the lungs was quantified histology staining for Ly6G antibody. Pictures of 5 random fields were taken from each lung section. Quantification was performed via ImageJ. Lung neutrophil percentage = Ly6G positive cell number / total cell number in the lung.
Protein analysis by immunoblotting
The cells were harvested and washed with cold PBS and lysed in lysis buffer [20 mM HEPES, 1% Triton X-100 (TX-100), 150 mM NaCl, 1 mM EGTA, 1 mM EDTA] containing phosphatase inhibitors (10 mM sodium pyrophosphate, 20 mM β-glycerol-glycerophosphate, 100 mM sodium fluoride, 5 mM indoleacetic acid, 20 nM oleic acid) and a Pierce™ protease inhibitor mini tablet inhibitor and a Pierce™ phosphatase inhibitor mini tablet inhibitor (1 tablet per 10 mL lysis buffer). After sonication on ice, the solubilized proteins were collected by centrifugation, and the protein concentration was measured via a Bio-Rad protein assay. An equal amount of protein was aliquoted into Laemmli buffer, boiled for 5 minutes and processed by standard western blot procedures. The membranes were blocked with 5% skim milk in TBS with 0.1% Tween-20 for 1 hour at room temperature and incubated with specific primary antibodies at 4°C overnight. Enhanced chemiluminescence (ECL) western blot substrate was used for film development, and ImageJ was used for data quantification. The tissue samples collected from the animal were snap frozen in a dry ice-ethanol mixture and preserved at −80°C until needed for the experiment. The tissue samples for western blotting were homogenized in ice-cold lysis buffer and processed as stated above.
Single cell RNA-seq
Mouse tumor tissues were dissected, washed in PBS, cut into small pieces and then digested in 1% collagenase IV in DMEM at 37 for 45 minutes with shaking. Cell debris and red blood cells were removed with centrifuging and ACK lysis buffer. The cell pellets were washed with PBS and passed through a 40 μm cell strainer to form a single cell suspension and counted with trypan blue to determine cell number and viability. A sequencing library was constructed according to the Drop-Seq protocol(Butler et al., 2018; Macosko et al., 2015). Briefly, approximately 1.5–2.0×105 cells were resuspended in 1 mL 0.1% BSA PBS and loaded onto a microfluidic chip to isolate single cell droplets. The collected droplets were broken to release barcoded beads and resuspended in reverse transcriptase mix. After reverse transcription, the beads were treated with exonuclease I to remove unhybridized DNA, followed by PCR amplification. The amplified products were sent to the UIC Research Resources Center (RRC) to check the cDNA quality and quantity with TapeStation and Qubit. The libraries were constructed using the Nextera XT kit and then sequenced with the Illumina Nextseq 500 at RRC. The reading depth was approximately 40,000 per cell. Read 1 was 20 base pairs (bp) to determine the cell barcode and unique molecular identifier (UMI); read 2 was 63 bp to determine the cDNA region.
Bioinformatics data processing and analysis
Raw sequence data were filtered, trimmed and then aligned to the mouse genome (mm10). Uniquely mapped reads were grouped and counted to generate digital expression matrices and subjected to Seurat package (version v2.2.1) using R (version 3.3.2) to perform a single cell analysis(Butler et al., 2018; Macosko et al., 2015).
Quantification and Statistical Analysis
GraphPad Prism 6 was used for statistical analysis. Results are presented as mean ± SEM and the unpaired Student’s two-tailed t test or one-way ANOVA were used to calculate significance was used to determine the statistical significance (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Supplementary Material
Average expression of genes in each cluster in MMTV-PyMT primary breast tumor mice (related to Figure 4 A, B and Figure S5A).
Average expression of genes in each cluster in MMTV-PyMT lung metastasis tumor mice (related to Figure 4 and Figure S5C).
Average expression of genes in each cluster in MMTV-PyMT micro-metastatic lung tumor mice (related to Figure 4 and Figure S5 D,E).
Average expression of genes in each cluster in MMTV-PyMT primary and metastasis combined (related to Figure 4 and Figure S6 A–C).
Average expression of genes in the combined primary MMTV-PyMT, WT, Akt1 KO and Akt2 KO tumors (related to Figure 4F and Figure S6 A–C).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal rabbit anti-pan Akt (clone C67E7) | Cell Signaling Technology | Cat. # 4691 |
| Monoclonal rabbit anti-Akt1 (clone C73H10) | Cell Signaling Technology | Cat. # 2938 |
| Monoclonal rabbit anti-Akt2 (clone D6G47) | Cell Signaling Technology | Cat. # 3063 |
| Polyclonal rabbit anti-pSer473 pan Akt | Cell Signaling Technology | Cat. # 9271 |
| Polyclonal rabbit anti-pSer308 pan Akt | Cell Signaling Technology | Cat. # 9275 |
| Monoclonal rabbit anti-pSer473 Akt1 (clone D7F10) | Cell Signaling Technology | Cat. # 9018 |
| Monoclonal rabbit anti-pSer474 Akt2 (clone D3H2) | Cell Signaling Technology | Cat. # 8599 |
| Monoclonal rabbit anti-PRAS40 (clone D23C7) | Cell Signaling Technology | Cat. # 2691 |
| Monoclonal rabbit anti-phospho PRAS40 (clone C77D7) | Cell Signaling Technology | Cat. # 2997 |
| Monoclonal rabbit anti-GSK3-beta (clone D5C5Z) | Cell Signaling Technology | Cat. # 12456 |
| Monoclonal rabbit anti-p-GSK3-beta (clone D85E12) | Cell Signaling Technology | Cat. # 5558 |
| Monoclonal rabbit anti-GAPDH (clone 14C10) | Cell Signaling Technology | Cat. # 2118 |
| Monoclonal rabbit anti-b-Actin (clone 13E5) | Cell Signaling Technology | Cat. # 4970 |
| Monoclonal rat anti-Ly6G (clone 1A8) | Biolegend | Cat. # 127601 |
| Monoclonal rabbit anti-Ki67 (clone SP6) | Abcam | Cat. # ab16667 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Xylenes (Histological), Fisher Chemical | Fisher Scientific | Cat. # X3S-4 |
| Hematoxylin Solution, Harris Modified | Sigma-Aldrich | Cat. # HHS32–1L |
| Eosin Y solution, alcoholic | Sigma-Aldrich | Cat. # HT110116 |
| Recombinant Mouse G-CSF Protein | R&D Systems | Cat. # 414-CS-025 |
| Pierce™ ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat. # 32106 |
| Pierce™ Protease Inhibitor Mini Tablets | Thermo Fisher Scientific | Cat. # A32953 |
| Pierce™ Phosphtase Inhibitor Mini Tablets | Thermo Fisher Scientific | Cat. # A32957 |
| HyClone™ Dulbecco’s High Glucose Modified Eagles Medium | Fisher Scientific | Cat. #SH30022.01 |
| HyClone™ RPMI 1640 Media | Fisher Scientific | Cat. #SH30027.01 |
| HyClone™ Phosphate Buffered Saline (PBS) | Fisher Scientific | Cat. #SH30256.01 |
| Propidium iodide | Sigma-Aldrich | Cat. # P4170–25MG |
| Hoechst 33342, trihydrochloride, trihydrate | Thermo Fisher Scientific | Cat. # H3569 |
| Histopaque®−1077 | Sigma-Aldrich | Cat. # 10771–100ML |
| Histopaque®−1119 | Sigma-Aldrich | Cat. # 11191–100ML |
| Gibco™ ACK Lysing Buffer | Fisher Scientific | Cat. # A1049201 |
| Corning Matrigel Matrix | Corning Life Sciences | Cat. # 354230 |
| Collagenase, Type 4 | Worthington | Cat. #LS004188 |
| Tamoxifen | Sigma-Aldrich | Cat. #T5648–5G |
| Corn Oil | Sigma-Aldrich | Cat. #C8267–500ML |
| MK-2206 | Shelleck Chem | Cat. #S1078 |
| Canagliflozin (CANA) | MedChem Express, Monmouth Junction, NJ, USA | |
| MILLIPLEX MAP Mouse Metabolic Hormone Magnetic Bead Panel | Millipore Sigma | Cat. # MMHMAG44K |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE135096 |
| Original data for figures in this study | This paper | http://dx.doi.org/10.17632/77t55d5zjf.1 |
| Experimental Models: Cell Lines | ||
| E0771 mouse breast cancer cells | ATCC | ATCC® CRL-3461 |
| Experimental Models: Organisms/Strains | ||
| Mouse FVB/N: MMTV-NIC | W.J. Muller (McGill University) | N/A |
| Mouse FVB/N: MMTV-Erbb2 | W.J. Muller (McGill University) | N/A |
| Mouse FVB/N: Tg(MMTV-PyVT)634Mul/J (MMTV-PyMT) | The Jackson laboratory | JAX: 002374 |
| Mouse: FVB.129S6(B6)-Gt(ROSA)26Sortm1(Luc)Kael/J (LSL-Luc) | The Jackson laboratory | JAX:005125 |
| Mouse FVB/N: Rosa26(R26)CreERT2 knock-in mice (strain 01XAB) | Wang et al., 2016 | N/A |
| Mouse FVB/N: Akt1f/f | Wang et al., 2016 | N/A |
| Mouse FVB/N: Akt2f/f | Wang et al., 2016 | N/A |
| Mouse FVB/N:Akt1f/f;R26RCreERT2 | Wang et al., 2016 | N/A |
| Mouse FVB/N:Akt2f/f;R26RCreERT2 | Wang et al., 2016 | N/A |
| Mouse FVB/N: MMTV-NIC;Akt1f/f | This paper | N/A |
| Mouse FVB/N: MMTV-NIC;Akt2f/f | This paper | N/A |
| Mouse FVB/N: MMTV-Erbb2;Akt1f/f;R26RCreERT2 | This paper | N/A |
| Mouse FVB/N: MMTV-Erbb2;Akt2f/f;R26RCreERT2 | This paper | N/A |
| Mouse FVB/N: MMTV-NIC;Akt1f/f;LSL-Luc | This paper | N/A |
| Mouse FVB/N: MMTV-NIC;Akt2f/f;LSL-Luc | This paper | N/A |
| Mouse FVB/N: MMTV-PyMT;Akt1f/f;R26RCreERT2 | This paper | N/A |
| Mouse FVB/N: MMTV-PyMT;Akt2f/f;R26RCreERT2 | This paper | N/A |
| Mouse: MRP8-Cre-ires/GFP, Mrp8creTg | The Jackson laboratory | JAX: 021614 |
| Mouse: MRP8-Cre;Akt1f/f | This paper | N/A |
| Mouse: MRP8-Cre;Akt2f/f | This paper | N/A |
| Mouse: (NOD)/Shi-scidIL-2Rγnull (NOG) | The Jackson laboratory | JAX:016117 |
| Software and Algorithms | ||
| GraphPad Prism 6.0 | GraphPad Inc. | https://www.graphpad.com/scientificsoftware/prism/ |
| Image J | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Seurat | https://satijalab.org/seurat/ | |
| R-3.3.2 | R Core Team (2013). | https://cran.rproject.org/ |
Highlights.
Akt1 neutrophil specific deletion is sufficient to inhibit breast cancer metastasis.
Akt1 mammary gland specific deletion inhibits tumor growth but not metastasis.
Akt2 mammary gland specific deletion disables ErbB2 expression in the mammary gland.
High insulin after systemic Akt2 deletion prevents mammary tumorigenesis inhibition.
Acknowledgments
N.H. acknowledges the support from NIH grants R01AG016927, R01CA090764, and R01 CA206167, the VA merit award BX000733, and the VA research career scientist award IK6BX004602. X.C. acknowledges the support from the center for clinical and translational science. C.B acknowledges the support from F30CA228191. W.P. acknowledges the support from T32HL007829. M.M.A acknowledges the support from the center for clinical and translational science. M.V.F. acknowledges the support from NIH grants R01GM093827 and R35GM131707. We would like to thank Ling Jin for maintaining and genotyping the mice. N.H. would like to thank Louis Philipson (University of Chicago) for the suggestion to use SGLT2 inhibitors.
Footnotes
Declaration of Interests: The authors have no financial interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Tang H, Hay N, Xu J, and Ye RD (2010). Akt isoforms differentially regulate neutrophil functions. Blood 115, 4237–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KJ, Padmanaban V, Silvestri V, Schipper K, Cohen JD, Fairchild AN, Gorin MA, Verdone JE, Pienta KJ, Bader JS, and Ewald AJ (2016). Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc Natl Acad Sci U S A 113, E854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. (2015). Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffelt SB, Wellenstein MD, and de Visser KE (2016). Neutrophils in cancer: neutral no more. Nat Rev Cancer 16, 431–446. [DOI] [PubMed] [Google Scholar]
- Dillon RL, Marcotte R, Hennessy BT, Woodgett JR, Mills GB, and Muller WJ (2009). Akt1 and akt2 play distinct roles in the initiation and metastatic phases of mammary tumor progression. Cancer Res 69, 5057–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhagalov I, St John A, and He YW (2007). The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 109, 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethier JL, Desautels D, Templeton A, Shah PS, and Amir E. (2017). Prognostic role of neutrophil-to-lymphocyte ratio in breast cancer: a systematic review and meta-analysis. Breast Cancer Res 19, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, and Hay N. (1998). 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, Amadiume SC, Goncalves MD, Hodakoski C, Lundquist MR, et al. (2018). Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 560, 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SJ, Liang S, Sharma A, Dong C, and Robertson GP (2010). Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res 70, 6071–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Jin J, Cardiff RD, Woodgett JR, and Muller WJ (2004). Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res 64, 3171–3178. [DOI] [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, and Brugge JS (2005). Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol 171, 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju X, Katiyar S, Wang C, Liu M, Jiao X, Li S, Zhou J, Turner J, Lisanti MP, Russell RG, et al. (2007). Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci U S A 104, 7438–7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CW, Xia W, Lim SO, Hsu JL, Huo L, Wu Y, Li LY, Lai CC, Chang SS, Hsu YH, et al. (2016). AKT1 Inhibits Epithelial-to-Mesenchymal Transition in Breast Cancer through Phosphorylation-Dependent Twist1 Degradation. Cancer Res 76, 1451–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, and Pollard JW (2003). Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol 163, 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroulakou IG, Oemler W, Naber SP, and Tsichlis PN (2007). Akt1 ablation inhibits, whereas Akt2 ablation accelerates, the development of mammary adenocarcinomas in mouse mammary tumor virus (MMTV)-ErbB2/neu and MMTV-polyoma middle T transgenic mice. Cancer Res 67, 167–177. [DOI] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, and Green DR (2006). Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 21, 749–760. [DOI] [PubMed] [Google Scholar]
- Mellouli F, Ksouri H, Barbouche R, Maamer M, Hamed LB, Hmida S, Hassen AB, and Bejaoui M. (2010). Successful treatment of Fusarium solani ecthyma gangrenosum in a patient affected by leukocyte adhesion deficiency type 1 with granulocytes transfusions. BMC Dermatol 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchemore KA, Anderson RL, and Hamilton JA (2018). Neutrophils, G-CSF and their contribution to breast cancer metastasis. FEBS J 285, 665–679. [DOI] [PubMed] [Google Scholar]
- Muller WJ, Sinn E, Pattengale PK, Wallace R, and Leder P. (1988). Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 54, 105–115. [DOI] [PubMed] [Google Scholar]
- Naznin F, Sakoda H, Okada T, Tsubouchi H, Waise TM, Arakawa K, and Nakazato M. (2017). Canagliflozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced inflammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur J Pharmacol 794, 37–44. [DOI] [PubMed] [Google Scholar]
- Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, and Ewald AJ (2019). E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, et al. (2016). Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med 8, 361ra138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E, Wagner EF, and Weissman IL (2004). JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 119, 431–443. [DOI] [PubMed] [Google Scholar]
- Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, Lin C, Alicea-Torres K, Sehgal M, Nefedova Y, et al. (2018). Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol 19, 1236–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud N, Hsu BE, Istomine R, Alvarez F, Blagih J, Ma EH, Morales SV, Dai DL, Li G, Souleimanova M, et al. (2018). Translational control in the tumor microenvironment promotes lung metastasis: Phosphorylation of eIF4E in neutrophils. Proc Natl Acad Sci U S A 115, E2202–E2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R, Singer J, et al. (2019). Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557. [DOI] [PubMed] [Google Scholar]
- Tahara A, Kurosaki E, Yokono M, Yamajuku D, Kihara R, Hayashizaki Y, Takasu T, Imamura M, Li Q, Tomiyama H, et al. (2013). Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur J Pharmacol 715, 246–255. [DOI] [PubMed] [Google Scholar]
- Thrailkill KM, Clay Bunn R, Nyman JS, Rettiganti MR, Cockrell GE, Wahl EC, Uppuganti S, Lumpkin CK Jr., and Fowlkes JL (2016). SGLT2 inhibitor therapy improves blood glucose but does not prevent diabetic bone disease in diabetic DBA/2J male mice. Bone 82, 101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursini-Siegel J, Hardy WR, Zuo D, Lam SH, Sanguin-Gendreau V, Cardiff RD, Pawson T, and Muller WJ (2008). ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J 27, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, and Jacks T. (2007). Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu WN, Chen X, Peng XD, Jeon SM, Birnbaum MJ, Guzman G, and Hay N. (2016). Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 29, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Nakayama K, Taniuchi N, Horai Y, Kuriyama C, Ueta K, Arakawa K, Senbonmatsu T, and Shiotani M. (2015). Beneficial effects of canagliflozin in combination with pioglitazone on insulin sensitivity in rodent models of obese type 2 diabetes. PLoS One 10, e0116851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wculek SK, and Malanchi I. (2015). Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue G, Restuccia DF, Lan Q, Hynx D, Dirnhofer S, Hess D, Ruegg C, and Hemmings BA (2012). Akt/PKB-mediated phosphorylation of Twist1 promotes tumor metastasis via mediating cross-talk between PI3K/Akt and TGF-beta signaling axes. Cancer Discov 2, 248–259. [DOI] [PubMed] [Google Scholar]
- Yu WN, Nogueira V, Sobhakumari A, Patra KC, Bhaskar PT, and Hay N. (2015). Systemic Akt1 Deletion after Tumor Onset in p53(-/-) Mice Increases Lifespan and Regresses Thymic Lymphoma Emulating p53 Restoration. Cell Rep 12, 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Average expression of genes in each cluster in MMTV-PyMT primary breast tumor mice (related to Figure 4 A, B and Figure S5A).
Average expression of genes in each cluster in MMTV-PyMT lung metastasis tumor mice (related to Figure 4 and Figure S5C).
Average expression of genes in each cluster in MMTV-PyMT micro-metastatic lung tumor mice (related to Figure 4 and Figure S5 D,E).
Average expression of genes in each cluster in MMTV-PyMT primary and metastasis combined (related to Figure 4 and Figure S6 A–C).
Average expression of genes in the combined primary MMTV-PyMT, WT, Akt1 KO and Akt2 KO tumors (related to Figure 4F and Figure S6 A–C).
Data Availability Statement
The datasets generated in this study are available at GEO: GSE135096 Original data for figures in this study are accessible upon request and deposited to Mendeley Data: http://dx.doi.org/10.17632/77t55d5zjf.1