

ABSTRACT
The current study aims to investigate the significance of N6-methyladenosine (m6A) methylation-related genes in the clinical prognosis of hepatocellular carcinoma (HCC) using bioinformatics analyses based on The Cancer Genome Atlas (TCGA) database. Transcriptome data and corresponding clinical data on m6A methylation-related genes (including 15 genes) were obtained from TCGA database. Differential expression of 15 genes was identified. Survival curves of subgroups based on m6A methylation-related gene expression levels were plotted. We selected potential predictive genes and analyzed their prognostic values using bioinformatics methods. Eleven genes (METTL3, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, FTO, KIAA1429, HNRNPC, HNRNPA2B1, and RBM15) were found to be overexpressed in HCC. Of these, five genes had worse survival (P < 0.05). There was a significant difference in the survival rate between subgroups with different expression levels of m6A. We selected five potential predictors (METTL3, KIAA1429, ZC3H13, YTHDF1, and YTHDF2) that met the independent predictive value. ZC3H13 was upregulated in patients with high cancer risk, whereas METTL3, KIAA1429, YTHDF1, and YTHDF2 were downregulated. In summary, we found that the expression levels of m6A methylation-related genes were different in patients with HCC and correlated with survival and prognosis. This implies that m6A methylation-related genes may be promising prognostic indicators or therapeutic targets for HCC.
KEYWORDS: Hepatocellular Carcinoma, N6-methyladenosine methylation, TCGA
Graphical abstract
Introduction
Hepatocellular carcinoma (HCC) is the most common primary form of liver cancer. In the past few decades, HCC has become the fifth most common cancer with a second highest mortality rate and a poor survival outcome worldwide [1]. Several risk factors have been linked to HCC [2]; however, its prognostic predictions are yet to be fully elucidated.
RNA modification is a post-transcriptional regulation that influences RNA stability and degradation. More than 150 RNA modifications have been identified, which are widely distributed in various types of RNA, such as mRNA, tRNA, rRNA, sncRNA, and lncRNA. However, the biological value of most RNA modifications remain unexplored due to technical limitations. N6-methyladenosine (m6A) which refers to the methylation modification of the sixth nitrogen (N) atom of adenine (A) accounts for more than 60% of RNA modifications and affects almost all RNA metabolic activities, such as splicing, transport, translation, and degradation [3].
The modification level of transcript m6A is dynamically regulated by methyltransferase (writer), binding protein (reader), and demethylase (erasers). METTL3, METTL14, KIAA1429, RBM15, WTAP, and ZC3H13 have been shown to act as m6A methyltransferases (‘writers’) [4,5]. HNRNPC, HNRNPA2B1, and YT521-B homology (YTH) domain family members, including YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3 have been identified as the ‘readers’ of m6A and modulate mRNA metabolic activities [6–8].ALKBH5 and FTO, key demethylases specifically removing m6A from target mRNAs, have been identified as the ‘erasers’ of m6A [9,10].
m6A affects multiple aspects of mRNA metabolism and regulates gene translation. Dysregulation of m6A is thus assumed to be relate to various biological processes, including cancer progression. Moreover, these genes do not function alone, but can interact with each other. However, whether m6A and its key modulators play a specific role in inhibiting or promoting cancers remain inconclusive to date. Although great progress has been made in finding biomarkers for tumor prognosis, less than 1% of biomarkers are used in clinical practice [11]. Furthermore, most investigations on m6A methylation-related genes are single or small combination studies. At present, there is no comprehensive study on the role of m6A methylation-related genes in HCC. Therefore, it is important to assess the relationship between m6A methylation-related genes and HCC at the genetic level. The present study analyzed associations between m6A methylation-related gene expression and clinical prognosis of patients, in an attempt to find novel prognostic biomarkers and therapeutic targets for HCC.
Methods and materials
Data collection
Transcriptome data and corresponding clinical data of HCC were obtained from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/).mRNA expression data of 374 tumors and 50 normal tissue, as well asclinical information including age, gender, grade, clinical stage, and TNM stage of 348 HCC patients were collected.
Bioinformatic analysis/statistics
The differential expression of 15 m6A methylation-related genes (METTL3, METTL14, WTAP, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, FTO, KIAA1429, ALKBH5, FTO, HNRNPC, HNRNPA2B1, and RBM15) in HCC and normal control samples were evaluated using R software (Version 3.8; http://www.bioconductor.org/packages/release/bioc/html). The heatmap of these genes were plotted using R software. Pearson correlation analyses were performed to identify gene-to-gene correlation. Kaplan–Meier (KM) survival analysis was performed to assess the effect of each gene on survivalbased on UALCAN website (http://ualcan.path.uab.edu/index.html). The P-value of 0.05 was considered the significant threshold in all tests.
PCA and survival analyses of subgroups
Consensus clustering is a class discovery technique for the detection of unknown possible clusters consisting of items with similar intrinsic features [12]. Based on comprehensive expression of the 15 genes, we identified distinct subgroups of 374 tumor samples with R’s ConsensusClusterPlus package, using principal component analysis (PCA) to verify the results of the grouping. Survival curves between two subgroups were plotted using the KM method.
Prognostic value of m6A methylation-related genes
The 15 genes in question were analyzed by univariate Cox regression, in which candidate genes were selected if they satisfied the screening condition of P < 0.05. Thereafter, we utilized LASSO regression for high-dimensional data to select the most useful prognostic factors using the ‘glment’ package in R software [13]. Five genes were selected and their related risk score were also calculated. Patients were divided into high-risk and low-risk groups based on the median expression of m6A methylation-related genes. The relationship between m6A related genes and survival rates was analyzed by the KM survival approach. Log-rank tests were employed to calculate the P-value of KM survival curves. The receiver operating characteristic (ROC) curve was drawn to test the accuracy of the model. Univariate and multivariate Cox regression analyses were performed to identify prognostic factors for HCC. The heatmap of m6A methylation-related genes and clinical risk factors were plotted.
Results
TCGA dataset and patients’ characteristics
HCC tissue (374) and 50 adjacent normal tissue from TCGA were enrolled in the current study. A total of 348 patients (238 males and 110 females) were included following exclusion of samples having incomplete clinical data. The average age of patients was 58.80 years. Clinical data included age, gender, grade, clinical stage, and TNM stage.
Expression of m6A methylation-related genes in HCC
We constructed a gene expression heatmap with 15 m6Amethylation-related genes to get an overview for the expression in HCC, according to TCGA, 11 genes (METTL3, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, FTO, KIAA1429, HNRNPC, HNRNPA2B1, and RBM15) showed significant upregulation in tumors compared with that in adjacent normal tissue in HCC (Figure 1(a)).
Figure 1.
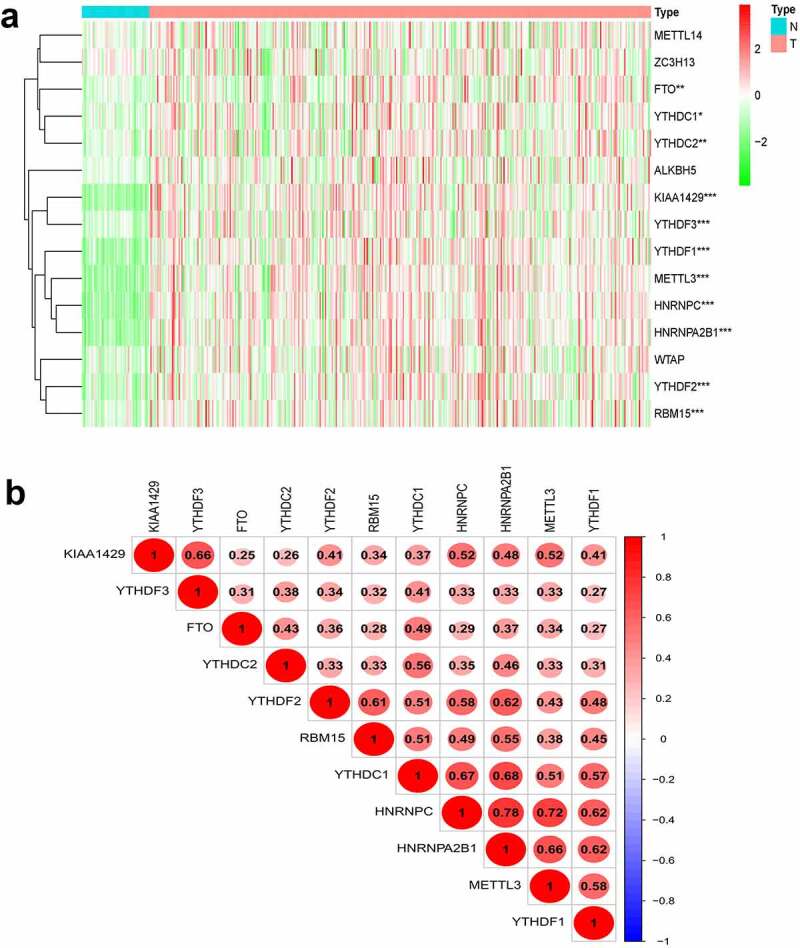
Expression, correlation, and prognostic information of m6A methylation-related genes. (a) Heatmaps of m6A methylation-related genes expressed in tumors and adjacent normal tissue. (***P < 0.001, ** P < 0.01, *P < 0.05) (b) Correlation matrix of interaction in m6A methylation-related genes. Correlation coefficients are plotted with negative correlation (blue) and positive correlation (red).
As shown in Figure 1(b), Pearson correlation analysis demonstrated that all 11 genes showed a positive correlation with each other. The highest correlation was observed for HNRNPC and HNRNPA2B1 with a correlation coefficient of 0.78. HNRNPC and METTL3 as well as YTHDC1 and HNRNPA2B1 were also strongly correlated, and their correlation coefficients were 0.72 and 0.68, respectively.
Survival analysis of m6A methylation-related genes
UALCAN (http://ualcan.path.uab.edu/index.html) online tool was used to identify survival data of 11 genes. It was found that patients with high expression of five genes (HNRNPA2B1, HNRNPC, METTL3, YTHDF1, and YTHDF2) had a significantly worse survival (P < 0.05) (Figure 2(a–e)) while the remaining six genes showed no significant difference (P > 0.05).
Figure 2.
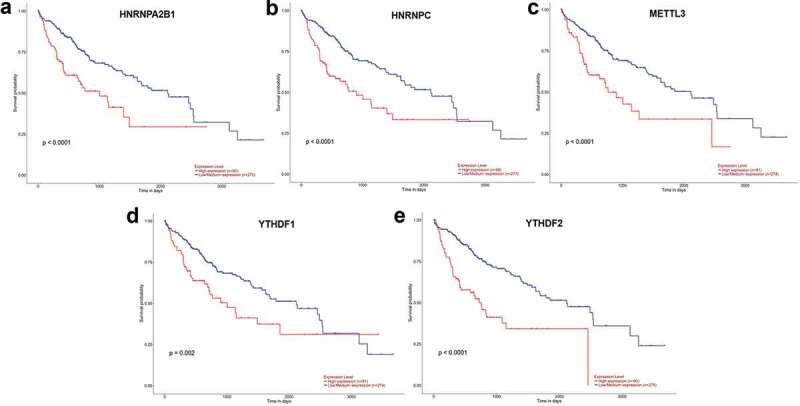
(a–e) Prognostic information for five of 11 genes, which had a significantly worse survival rate (P < 0.05).
Based on m6A methylation-related gene expression levels, we identified distinct subgroups of 374 tumor samples using R’s ConsensusClusterPlus package. And we calculated cluster-consensus and item-consensus results. The output displayed k (2 to 4) subgroups, shown in Figure 3(a–c). We found that k = 2 achieved adequate selection. All patients were successfully categorized into two subgroups in terms of the most stable k value (Figure 3(a–d)).
Figure 3.
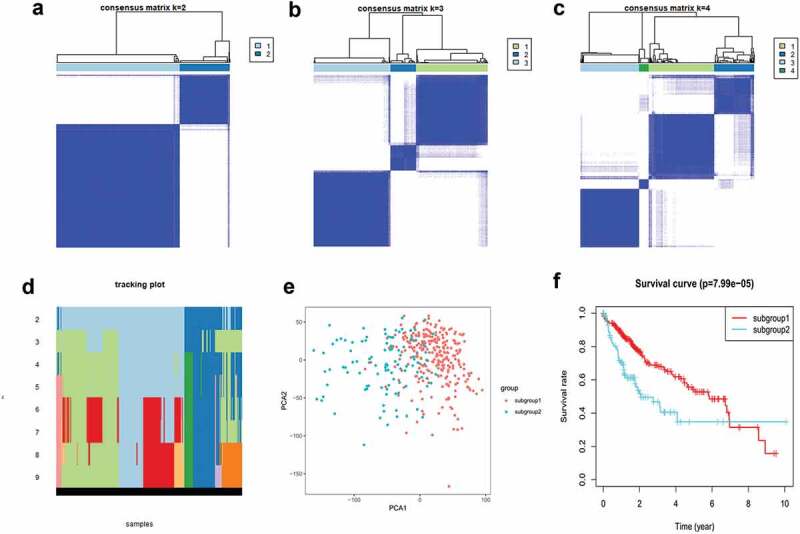
Identification and analysis of two subgroups of 374 tumor samples that exhibited distinct m6A expression. (a–c) Consensus clustering matrix for k = 2, 3, and 4. (d) Tracking plot for k = 2 to 9. (e) Principal component analysis (PCA) of the two subgroups. (f) Kaplan–Meier survival plots of the two subgroups.
As shown in Figure 3(e), subgroup1 represented a high level of gene expression, while subgroup2 did not. The horizontal axis represents the first principal component, while the vertical represents the second. Principal component analysis (PCA) showed that subgroup1 can assemble together and so can subgroup2. These results indicated that our grouping was accurate. Overall survival analysis of differentially expressed genes indicated that survival duration of subgroup1 significantly improved (P < 0.05) (Figure 3(f)).
Construction of LASSO model
We used univariate Cox regression to analyze 15 genes, and 10 candidate genes were selected with P < 0.05 as a screening condition (Figure 4(a)). The LASSO Cox regression model was used to select the most predictive genes as prognostic indicators. λ was selected when the median of the sum of squared residuals was the smallest. Five potential predictors (Figure 4(b–c)). METTL3, KIAA1429, ZC3H13, YTHDF1, and YTHDF2 were identified as prognostic factors for HCC. The risk score of five genes was also calculated for further univariate and multivariate Cox regression analyses.
Figure 4.
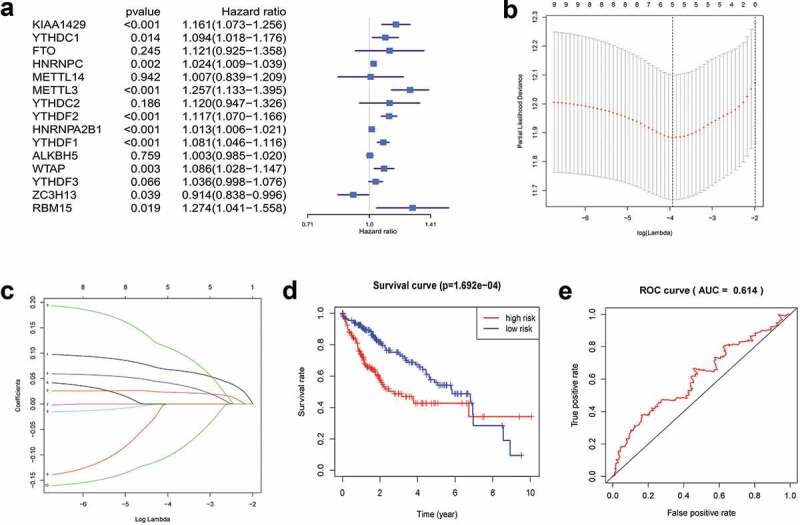
Gene selection and survival analysis in HCC prognosis prediction. (a) Forest plots for hazard ratios (HRs) of survival‑associated m6A methylation-related genes in HCC. (b) Partial likelihood deviance versus log (λ)was drawn using LASSO Cox regression model. (c) Coefficients of selected features are shown by lambda parameter. (d) Kaplan–Meier survival plots of the two groups. (e) ROC curves of the survival model in HCC (AUC = 61.4%).
Patients were divided into high-risk and low-risk groups based on the combined model with cutoff values at the median expression of the five candidate genes. The low-risk group showed consistent better prognosis than high-risk groups. The survival curve was plotted by the KM method (Figure 4(d)). We also compared the prognostic efficiency of risk factors through ROC curves. The results showed that areas under the curve (AUC) were 61.4% (Figure 4(e)), indicating that m6A methylation-related genes could serve as biomarkers in prognosis of HCC.
Prognostic value of the 5 m6A methylation-related genes
Univariate analysis showed that T stage, clinical stage, grade, and risk score of m6A methylation-related genes affected the prognosis of patients (P < 0.05). Age, gender, grade, M stage, and N stage did not correlate with the prognosis of HCC (P > 0.05) (Figure 5(a)). The results of multivariate regression analysis showed that the risk score of m6A methylation-related genes was an independent prognostic factor in HCC (P < 0.05) (Figure 5(b)).
Figure 5.
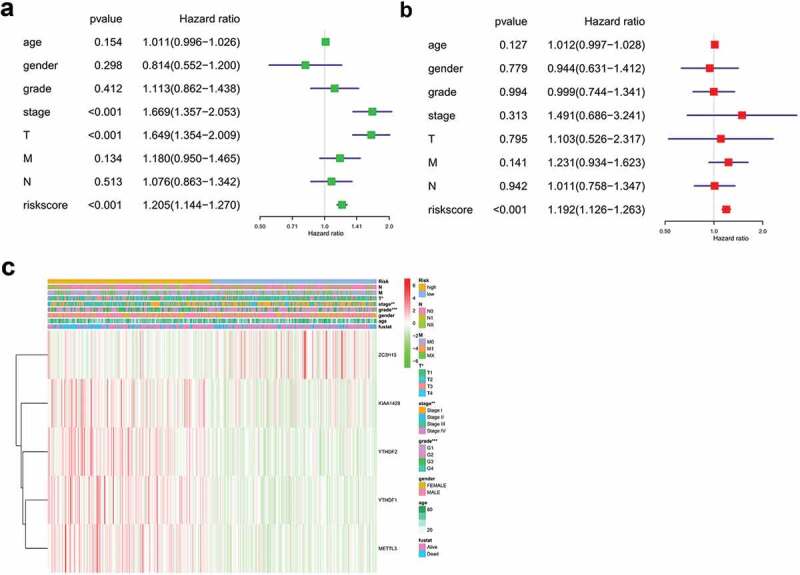
Forest plot and heatmap of m6A methylation-related genes and clinical risk factors. (a) Forest plot of univariate Cox regression analysis in HCC. (b) Forest plot of multivariate Cox regression analysis in HCC. (c) Heatmap of m6A methylation-related genes and clinical risk factors.(***P < 0.001, ** P < 0.01, *P < 0.05).
As shown in Figure 5(c), protective genesZC3H13 had a tendency to be upregulated in low-risk patients, whereas METTL3, YTHDF1, YTHDF2, and KIAA1429 had a tendency to be highly expressed in high-risk patients. T stage, clinical stage, and grade were linked with the degree of risk, while M stage, N stage, gender, and age had no significance (P > 0.05).
Discussion
Hepatocellular carcinoma has become a fundamental public health concern worldwide. To identify more useful prognostic biomarkers for HCC, using bioinformatics based on TCGA, we established 15 gene signatures (including METTL3, METTL14, KIAA1429, RBM15, ZC3H13, WTAP, YTHDF1, YTHDF2, YTHDF3, YTHDC1, YTHDC2, HNRNPC, HNRNPA2B1, ALKBH5, and FTO) for HCC prognosis prediction. Eleven m6A methylation-related genes were up-regulated in HCC and all showed a positive correlation with each other. Five genes (HNRNPA2B1, HNRNPC, METTL3, YTHDF1, and YTHDF2) were linked to significantly worse survival. Our study demonstrated that m6A methylation-related genes were widely distributed in tumor tissue, indicating their important roles in HCC prognosis prediction. In addition, m6A methylation-related genes were strongly associated with each other in regulatory networks, suggesting their cooperation in cancer development. Furthermore, HNRNPA2B1, HNRNPC, METTL3, YTHDF1, and YTHDF2 may have deleterious effects on patients with HCC due to their association with worse survival. These findings imply that m6A modulators are potential targets for HCC treatment. In the following paragraphs, we will briefly discuss the relationship between these genes and HCC one by one.
Consensus cluster uses a variety of different clustering methods as inputs, so as to find a more suitable clustering method than each individual method. Subgrouping tumors helps to develop personalized treatments for individual patients. Based on gene expression levels, data were divided into two subgroups using R’s ConsensusClusterPlus package. Principal component analysis showed a separation between subgroup1 and subgroup2. Overall survival analysis indicated that survival duration of subgroup1 significantly improved, suggesting that survival time correlated with comprehensive expression level of m6A methylation-related genes.
The LASSO algorithm analyzes all independent variables simultaneously and selects the most influential variables [14]. Thus, far more accurate than the traditional regression methods. According to LASSO Cox analysis, five of 15 genes (METTL3, YTHDF1, YTHDF2, KIAA1429, and ZC3H13) were identified as prognostic factors for HCC. The predictive power of m6A methylation-related genes on HCC prognosis was evaluated by the ROC curve. The results demonstrated that m6A methylation-related genes were involved in the survival of HCC. The risk score of m6A methylation-related genes (METTL3, YTHDF1, YTHDF2, KIAA1429, and ZC3H13) might be a powerful biomarker for HCC survival. High expression levels of METTL3, YTHDF1, YTHDF2, and KIAA1429 predicted a poor prognosis, whereas ZC3H13 can be regarded as protective genes. T stage, clinical stage, and grade were linked with the degree of risk. However, precise biological behaviors of these five genes in HCC remain to be interpreted. We conducted a comprehensive biological analysis on the 15 most important m6A methylation-related genes, which was more comprehensive than previous studies on the influence of a single gene on disease. As interactions between m6A methylation-related genes exist, our study more accurately reflected their influence on HCC.
Heterogeneous nuclear ribonucleoprotein A2B1 (HNRNPA2B1) is an m6A reader which promotes miRNA biogenesis. Previous studies showed that HNRNPA2B1 was highly expressed in a variety of human cancers, such as prostate cancer [15], pancreatic cancer [16], and hepatocellular carcinoma [17]. Higher expression levels of HNRNPA2B1 have been reported in HCC [18]. Zhou et al. [19] reported that HNRNPAB induces epithelial–mesenchymal transition (EMT) and promotes metastasis of hepatocellular carcinoma, which was consistent with our findings.
Heterogeneous nuclear ribonucleoprotein C (HNRNPC) is an RNA-binding protein and well known for its regulatory roles in RNA splicing, 3′ end processing [20], and translation [21]. Overexpression of HNRNPC has been observed in multiple tumors, including glioblastomas [22], melanomas [23], and hepatocellular carcinomas [24]. However, the role of HNRNPC in HCC is still poorly documented. Our study provided a reference for further research.
Methyltransferase-like 3 (METTL3) determines the levels and distribution of target-specific m6A modifications [25]. Knockdown of METTL3 remarkably reduced the level of m6A in mRNAs3. METTL3 has been demonstrated to participate in tumorigenesis and the progression of several cancers [26]. For example, METTL3 promotes the progression of breast cancer by inhibiting tumor suppressor let-7 g [27]. Visvanathan et al. reported that upregulation of METTL3 was associated with worse survival in glioblastoma cells [28]. Chen et al. reported that METTL3 served as an oncogene and contributed to the progression of HCC and lung metastasis [29]. Upregulation of METTL3 contributed to cancer metastasis and predicted poor prognosis in patients with HCC [30]. However, Aravalli et al. observed that knockout of METTL3 remarkably suppressed HCC tumorigenesis and development [31]. The main reason for the dual role of METTL3 in cancer regulation may account for different targeted pathways and cancer heterogeneity.
YTH domain family 1 and 2 (YTHDF1and YTHDF2) are located in the cytoplasmic compartment [32,33]. YTHDF1 interacts with translation initiation factors to promote translation.YTHDF2 regulates the stability of target mRNAs [34]. The binding of ribosome to m6A-modified RNA and the translation of RNA can be reduced by knocking down YTHDF1 [35]. Previous studies have demonstrated that YTHDF1 and YTHDF2 are highly expressed in hepatocellular carcinoma, affecting cell cycle and metabolism of tumor cells, and the prognosis of high YTHDF1 expression in patients was poor [36]. Our survival analysis results were in agreement with those of previous studies.
KIAA1429 is a component of m6A ‘writers.’ Knocking down of KIAA1429 led to a considerable reduction of m6A in mRNA, suggesting that KIAA1429 was essential for the methyltransferase complex. Qian et al.found that KIAA1429 was highly expressed in breast cancer tissue, but frequently down-regulated in non-cancerous breast tissue [37]. A previous study demonstrated that KIAA1429 facilitated migration and invasion of HCC by inhibiting ID2 via upregulation of m6A modification [38]. These studies were consistent in that KIAA1429 played an important role in cancer progression and might potentially prevent or treat cancers.
Zinc finger CCCH domain-containing protein 13 (ZC3H13) acts as m6A methyltransferases (‘writers’). It usually functions by interacting with other m6A writer complex subunits [39]. Knockdown of ZC3H13 in mouse embryonic stem cells significantly decreases global m6A level on mRNA [40]. However, a paucity of evidence for liver malignancy research exists. Our study suggests that ZC3H13 has a tendency to be upregulated in low-risk patients, indicating that the prognosis of HCC may be improved by regulating ZC3H13 expression.
Overall, combination of m6A methylation-related genes with clinical parameters may have better predictive efficacy than a single biomarker. In recent years, m6A methylation-related genes have shown great potential in prognosis prediction of cancer. Our study preliminarily demonstrated that expression levels of m6A methylation-related genes play important roles in progression of HCC and may act as a prognostic predictor for this disease. However, there was a limitation to the present study. Our study was based on an individual source from TCGA, without validation from independent cohorts. More studies are needed for further clarification of these findings.
Conclusion
The expression of m6A methylation-related genes highly correlates with clinical features of HCC and may predict its prognosis as well as guide individualized therapy in clinical practice. Our study provides important evidence for future detection of the role of m6A methylation in HCC.
Acknowledgements
The authors would like to express their gratitude to Dr. Hua-Yu Wu for his expert advice and kind help in some biochemical analyses. The authors would like to thank the staff members of Department of Gastroenterology, the Second Affiliated Hospital of Guangxi Medical University for their technical assistance and kind encouragement.
Funding Statement
No funding was received.
Highlights
The expression levels of m6A methylation-related genes were correlated with prognosis of HCC.
Five m6A methylation-related genes were upheld the independent predictive value of HCC.
m6A methylation-related genes can be regarded as prognostic indicators.
Authors’ contributions
JL and GLS designed the study and wrote the original manuscript. MBQ and YRO downloaded and analyzed the data. SLP and JAH supervised the study and edited the manuscript.
Availability of data and materials
The datasets analyzed during the present study are available from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/).
Disclosure statement
The authors declare that they have no competing interests.
References
- [1].EASL Clinical Practice . Guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1): 182–236. [DOI] [PubMed] [Google Scholar]
- [2].Pascual S, Miralles C, Bernabe JM, et al. Surveillance and diagnosis of hepatocellular carcinoma: A systematic review. World J Clin Cases. 2019;7(16):2269–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Batista PJ, Molinie B, Wang J, et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhou J, Wan J, Gao X, et al. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kern SE.Why your new cancer biomarker may never work: recurrent patterns and remarkable diversity in biomarker failures. Cancer Res. 2012;72(23):6097–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sauerbrei W, Royston P, Binder H. Selection of important variables and determination of functional form for continuous predictors in multivariable model building. Stat Med. 2007;26(30):5512–5528. [DOI] [PubMed] [Google Scholar]
- [14].Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
- [15].Stockley J, Villasevil ME, Nixon C, et al. The RNA-binding protein hnRNPA2 regulates beta-catenin protein expression and is overexpressed in prostate cancer. RNA Biol. 2014;11(6):755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Siveke JT. The increasing diversity of KRAS signaling in pancreatic cancer. Gastroenterology. 2014;147(4):736–739. [DOI] [PubMed] [Google Scholar]
- [17].Shilo A, Ben Hur V, Denichenko P, et al. Splicing factor hnRNP A2 activates the Ras-MAPK-ERK pathway by controlling A-Raf splicing in hepatocellular carcinoma development. RNA. 2014;20(4):505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cui H, Wu F, Sun Y, et al. Up-regulation and subcellular localization of hnRNP A2/B1 in the development of hepatocellular carcinoma. BMC Cancer. 2010;10:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou ZJ, Dai Z, Zhou SL, et al. HNRNPAB induces epithelial-mesenchymal transition and promotes metastasis of hepatocellular carcinoma by transcriptionally activating SNAIL. Cancer Res. 2014;74(10):2750–2762. [DOI] [PubMed] [Google Scholar]
- [20].Gruber AJ, Schmidt R, Gruber AR, et al. A comprehensive analysis of 3ʹ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016;26(8):1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee EK, Kim HH, Kuwano Y, et al. hnRNP C promotes APP translation by competing with FMRP for APP mRNA recruitment to P bodies. Nat Struct Mol Biol. 2010;17(6):732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Park YM, Hwang SJ, Masuda K, et al. Heterogeneous nuclear ribonucleoprotein C1/C2 controls the metastatic potential of glioblastoma by regulating PDCD4. Mol Cell Biol. 2012;32(20):4237–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mulnix RE, Pitman RT, Retzer A, et al. hnRNP C1/C2 and Pur-beta proteins mediate induction of senescence by oligonucleotides homologous to the telomere overhang. Onco Targets Ther. 2013;7:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sun W, Xing B, Sun Y, et al. Proteome analysis of hepatocellular carcinoma by two-dimensional difference gel electrophoresis: novel protein markers in hepatocellular carcinoma tissues. Mol Cell Proteomics. 2007;6(10):1798–1808. [DOI] [PubMed] [Google Scholar]
- [25].Sorci M, Ianniello Z, Cruciani S, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018;9(8):796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lin S, Choe J, Du P, et al. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cai X, Wang X, Cao C, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19. [DOI] [PubMed] [Google Scholar]
- [28].Visvanathan A, Patil V, Arora A, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–533. [DOI] [PubMed] [Google Scholar]
- [29].Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology (Baltimore, Md). 2018;67(6):2254–2270. [DOI] [PubMed] [Google Scholar]
- [30].Ma JZ, Yang F, Zhou CC, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology (Baltimore, Md). 2017;65(2):529–543. [DOI] [PubMed] [Google Scholar]
- [31].Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch Toxicol. 2013;87(2):227–247. [DOI] [PubMed] [Google Scholar]
- [32].Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. [DOI] [PubMed] [Google Scholar]
- [33].Li A, Chen YS, Ping XL, et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xu C, Wang X, Liu K, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–929. [DOI] [PubMed] [Google Scholar]
- [35].Wang X, Zhao BS, Roundtree IA, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhao X, Chen Y, Mao Q, et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomarkers. 2018;21(4):859–868. [DOI] [PubMed] [Google Scholar]
- [37].Qian JY, Gao J, Sun X, et al. KIAA1429 acts as an oncogenic factor in breast cancer by regulating CDK1 in an N6-methyladenosine-independent manner. Oncogene. 2019;38(33):6123–6141. [DOI] [PubMed] [Google Scholar]
- [38].Cheng X, Li M, Rao X, et al. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 2019;12:3421–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guo J, Tang HW, Li J, et al. Xio is a component of the Drosophila sex determination pathway and RNA N(6)-methyladenosine methyltransferase complex. Proc Natl Acad Sci USA. 2018;115(14):3674–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wen J, Lv R, Ma H, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028–38.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed during the present study are available from The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/).