

ABSTRACT
The multi-attribute method (MAM) has garnered attention as a new quality control method of therapeutic monoclonal antibodies (mAbs). MAM analysis allows multiple relative quantifications of several structural attributes of therapeutic mAbs; however, some issues remain to be addressed in its procedures especially for sample preparation. The goal of this study was to optimize the sample preparation method for MAM analysis of mAbs. Using a model mAb, we compared five sample preparation methods based on sequence coverage, peptide redundancy, missed cleavage and chemical deamidation. It was found that low pH buffer and short digestion time reduced artificial deamidation. The desalting process after carboxymethylation was essential to obtaining high sequence coverage by a short digestion time. The generation of missed cleavage peptides was also improved by using a trypsin/lysyl endopeptidase (Lys-C) mixture. Next, we evaluated the usefulness of our method as a part of MAM analysis. Finally, 17 glycopeptides, 2 deamidated peptides and N- and C-terminal peptides of the heavy chain were successfully monitored with acceptable mass accuracy and coefficient of variation (CV, %) of the relative peak area. On the other hand, 4 oxidated peptides indicated the unavoidable slightly higher inter-assay CV (%) of the peak area ratio due to the instability in the MS sample solution. Collectively, we demonstrated that our method was applicable as an easy and reliable sample preparation method for MAM analysis, and the variation in the relative peak area could be influenced by the modification type rather than by the amount of each peptide.
KEYWORDS: Multi-attribute method, monoclonal antibody, peptide mapping, quality attribute, relative quantification
Graphical Abstract
Introduction
Over the past decade, therapeutic monoclonal antibodies (mAbs) have become one of the most attractive biological therapeutics. As of 2019, more than 60 therapeutic mAbs have already been approved in the United States, the European Union, and Japan [1]. Novel modalities that include therapeutic mAbs related to SARS-CoV2 and biosimilars related to mAbs are being actively developed, and the development of mAbs is expected to continue in the future [2–5]. In the development of therapeutic mAbs, it is essential to characterize the structural aspects because certain structural characteristics are strongly associated with safety and efficacy. However, the rapid structural characterization of mAbs has been continuously challenging due to their excessive structural complexity. Currently, a panel of physicochemical and biological methods has been used to evaluate the quality attributes of mAbs [6]. The conventional methods include high-performance liquid chromatography (HPLC), sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), capillary gel electrophoresis and liquid chromatography/mass spectrometry (LC/MS). Most of these conventional methods usually only indirectly monitor one of the characteristics of mAbs, and therefore, it is costly and time consuming to perform a comprehensive characterization of mAbs.
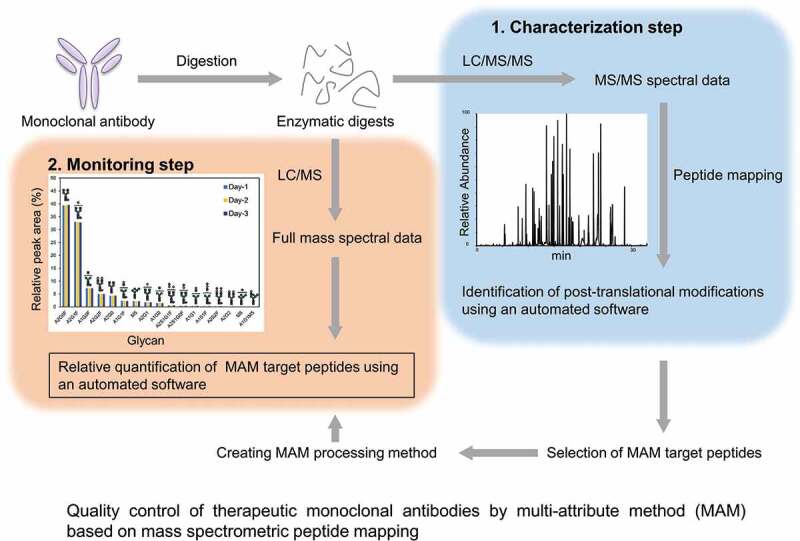
The multi-attribute method (MAM) approach is based on mass spectrometric peptide mapping and has garnered attention as a new and alternative method based on conventional methods. In MAM, the analyte, i.e., a therapeutic protein, is first digested into peptides, and the sample is prepared for peptide mapping. The enzymatic digests are analyzed by liquid chromatography/tandem mass spectrometry (LC/MS/MS) using ultra-high-performance liquid chromatography (UHPLC) and high-resolution and accurate mass spectrometry (HRMS). Then, the mass spectrometric data are applied to a database search analysis using peptide mapping software (Figure 1, characterization step), and the resulting data are used to create a processing method for monitoring the MAM target peptides. In this step, it is important that the target peptides are selected in accordance with the quality control strategy of each drug, and most of the actual target peptides would be mainly peptides with several post-translational modifications (PTMs), including the fragment crystallizable (Fc) region-glycosylation, deamidation, isomerization, oxidation, glycation, N-terminal pyroglutamination, and C-terminal Lys cleavage. Data for MAM monitoring are acquired by a full mass scan using only UHPLC/HRMS, and finally, the relative quantification of target peptides is performed by using software for MAM (Figure 1, monitoring step). If unknown peaks are detected, then the peaks can be further analyzed for identification by LC/MS/MS. The relative amount of each peptide is usually estimated based on the peak area intensity. Recently, several MAM workflows have been provided by MS and software vendors and have gradually been used by Chemistry, Manufacturing and Control (CMC) employees [7]. Previously, Rogers et al. demonstrated that an MAM approach using an Orbitrap-type mass spectrometer and software for identification and quantification has the potential to replace partial conventional chromatographic and electrophoretic methods currently used in the quality control of therapeutic mAbs [8]. Bomans et al. showed an MAM approach that combines high-throughput sample preparation and LC/MS using a Q-TOF-type mass spectrometer and applied this approach to monitor Fc-glycosylation, oxidation, deamidation, isomerization and glycation [9]. Furthermore, a regulatory agency has also a keen interest in the quality control of protein therapeutics by MAM [10]. MAM can provide detailed qualitative and quantitative information about the structural characteristics of mAbs and may enable the improved productivity and the reduced risk and cost of the development stage. Therefore, MAM might be useful as an alternative method to some conventional methods such as glycosylation profiling and evaluating charge variants.
Figure 1.
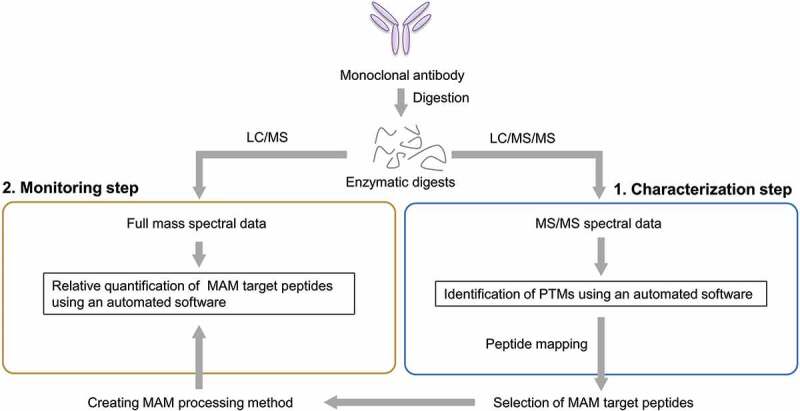
Typical MAM flowchart. MAM is usually carried out in two steps: a characterization step and a monitoring step. First, mAb is digested with a protease, such as trypsin, for peptide mapping. The enzymatic digests are analyzed by liquid chromatography/tandem mass spectrometry. Using the acquired mass spectrometric data, modified peptides are comprehensively identified. Target peptides are selected in accordance with the quality control strategy, and the characterization data are used to create a processing method for monitoring the MAM target peptides. Next, MAM monitoring data are acquired by a full mass scan analysis, and the relative quantification of the target peptides is performed based on the peak area intensity.
However, an issue that remains to be addressed in MAM analysis is the sample preparation process. The major problem is artificial modifications generated during sample preparation. This issue remains a hurdle for applying MAM to the quality control of therapeutic mAbs during the commercial production process. A previous report demonstrated that Asn in certain motifs, such as Asn-Gly, are susceptible to deamidation during enzymatic digestion [11]. Generally, in the case of peptide mapping in identification tests, insufficient attention could be directed toward artificial chemical modifications because the main purpose of peptide mapping is to confirm the amino acid sequence of the protein. However, MAM is categorized as a quantitative analysis method to evaluate the profile of molecular variants, and it is critical to reduce artificial modifications during sample preparation for the reliable quantification of peptides with PTMs.
In the current study, the sample preparation method for MAM analysis was proposed by referring to a previous report by Ren et al [12]. This proposed method was compared with previously reported methods and sample preparation kits using performance metrics, namely, sequence coverage, peptide redundancy, missed cleavage and chemical deamidations. Next, the usefulness of the proposed method was evaluated as a part of the MAM analysis. This study is the first report to compare several sample preparation methods for peptide mapping. We believe that our sample preparation method is useful for providing reliable data using MAM for the characterization and quality control of therapeutic mAbs.
Materials and methods
Materials
Commercially available trastuzumab was used as the model mAb. Formic acid (FA), acetonitrile, 0.1% FA and 0.1% FA/acetonitrile were obtained from Kanto Chemical (Tokyo, Japan). Tris-HCl and sodium monoiodoacetate (MIA), and guanidine-HCl (GuHCl) and dithiothreitol (DTT) were purchased from Sigma (MO, USA) and Thermo Fisher Scientific (CA, USA), respectively.
Sample preparation methods
Method 1
Trastuzumab (5 μg) was dissolved in 150 μL of 0.25 M Tris-HCl buffer (pH 7.5 or pH 8.5) containing 7.5 M GuHCl. After adding 3.0 μL of 0.5 M DTT, the solution was incubated at room temperature for 30 min. Then, 0.5 M MIA (7.0 μL) was added to the solution, and the mixture was incubated at room temperature for 15 min in the dark. The reaction was stopped by the addition of 0.5 M DTT (4.0 μL). The carboxymethylated proteins were buffer exchanged into a 0.1 M Tris-HCl buffer at pH 7.5 or pH 8.5 (digestion buffer) using an NAP-5 column (GE Healthcare, Buckinghamshire, UK) and incubated with trypsin (Trypsin Gold, MS Grade, Promega, WI, USA) and/or trypsin/Lys-C mixtures (Trypsin/Lys-C Mix, MS Grade, Promega) at 37°C. The total amount of enzyme was 2.0 μg, and the enzymes were prepared in three conditions: trypsin and Lys-C at a ratio of 1:1, 3:1 or trypsin only. The digestion time was performed under four conditions: 30 min, 1 h, 2 h and 16 h. The final digest was quenched with the addition of 5 μL of 20% FA.
The tryptic digests were desalted using an Oasis HLB μElution plate (Waters, MA, USA), as indicated in Table 3. The elution was dried by SpeedVac and dissolved in 50 μL 0.1% FA for LC/MS analysis.
Table 3.
The detail multi-attribute method procedure for Method 1.
| Denaturation | Reduction/Carboxymethylation | Tryptic digestion | Peptide purification |
|---|---|---|---|
| mAb sample (5 µg)a ↓←GuHClb (150 μL) Denatured sample (150 μL) |
Denatured sample solution ↓←Reducing agentc (3 µL) ↓←Incubation at room temperature for 30 min ↓←Alkylation agentd (7 μL) ↓←Incubation at room temperature for 15 min (in dark) ↓←Reducing agentc (4 µL) Reduced and carbamidomethylated protein (RCM) sample solution (164 μL) |
RCM sample solution ↓←Buffer exchanged with NAP5 ↓←Eluted with digestion buffer (600 μL) ↓←Digesting solutione (10 + 10 μL) ↓←Incubation at 37°C for 30 min Tryptic digestion (620 μL) |
Tryptic digest ↓←Washed with 90% ACN 0.1% FA (200 μL, 50% ACN 0.1% FA (200 μL) and 0.1% FA (200 μL)×3 ↓←Sample applied to Oasis HLB µElution plate ↓← Washed with 0.1% FA (200 µL) ↓←Eluted with 0.1% FA 50% ACN (100 µL) Dried using Speed Vac. Dissolved in 0.1% FA (50 μL) LC/MS sample (50 μL) |
a, mAb sample was diluted to 5 mg/mL using PBS and dried using Speed Vac.
b, 7.5 M guanidine/HCl, 0.25 M Tris-HCl (pH7.5).
c, 500 mM dithiothreitol.
d, 500 mM monoiodoacetate.
e, 0.1 mg/ml trypsin solution and 0.1 mg/ml trypsin and Lys-C mixture solution.
Method 2
Trastuzumab (5 μg) was dissolved in 50 μL of 0.5 M Tris-HCl at pH 8.6 containing 7 M GuHCl and 5 mM EDTA. After adding 2.0 μL of 1 M DTT, the solution was incubated at 65°C for 30 min. Then, 1 M MIA (4.8 μL) was added to the solution, and the mixture was incubated at room temperature for 40 min in the dark. After the reaction was stopped by the addition of 1 M DTT (1.2 μL), the mixture was desalted using a PD10 column and freeze-dried. The carboxymethylated proteins were dissolved in 100 μL of 50 mM Tris-HCl buffer (pH 8.5) and incubated with 50 ng of Trypsin Gold at 37°C for 16 h. The enzymatic digest was desalted using an Oasis HLB μElution plate. The elution was dried and dissolved in 50 μL of FA solution.
Method 3
MPEX PTS Reagents (GL Sciences, Tokyo, Japan) were used as Method 3. Trastuzumab (5 μg) was dissolved in 20 μL MPEX B. After adding 1.0 μL of 0.1 M DTT in MPEX Reagent A, the solution was incubated at room temperature for 30 min. MPEX Reagent A (1.0 μL) containing 0.55 M MIA was added to the solution, and the mixture was incubated at room temperature for 30 min in the dark. After incubation, MPEX Reagent A (77 μL) and 2.0 μL of (2 mg/mL) trypsin only, trypsin/Lys-C (1:1) or trypsin/Lys-C (3:1) were added, and the solution was incubated at 37°C. The digestion times were 30 min, 1 h, 2 h and 16 h. MPEX Reagent C (100 μL) were added to the sample solution after digestion. The mixture was acidified by 1.0 μL of MPEX Reagent D and vortexed vigorously. After centrifugation of the mixture, the aqueous phase containing peptides was collected. The resulting peptides were desalted using an Oasis HLB μElution plate. The elution was dried and dissolved in 50 μL of FA solution.
Methods 4 and 5
The AccuMAPTM Low pH Protein Digestion Kit (Promega, Madison, WI, USA) and SMART DigestTM Trypsin Kit (Thermo Fisher Scientific) were used in Methods 4 and 5. The digestion procedure was performed according to the corresponding kit protocol.
In Method 5, after digestion, reductive alkylation was performed as follows: DTT was added to a final concentration of 10 mM, and the solution was incubated at 57°C for 30 min. MIA was added to a final concentration of 20 mM, and the mixture was incubated at room temperature for 30 min in the dark. The reaction was stopped by adding 20% FA (1.0 μL) and 11 mM DTT. The enzymatic digest was desalted using an Oasis HLB μElution plate. The elution was dried and dissolved in 50 μL of FA solution.
LC/MS
LC/MS analysis was performed on UHPLC using a Vanquish UHPLC System coupled to an MS using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). The UHPLC was equipped with an ACQUITY UPLC CSH C18 column (1.7 µm particle size, 2.1 mm inner diameter (I.D.), 150 mm length; Waters, Manchester, UK) utilizing a column temperature of 45°C. Milli-Q® water containing 0.1% FA and acetonitrile containing 0.1% FA were used as mobile phases A and B, respectively. The flow rate was set to 300 μL/min, and the gradient programme was as follows: an isocratic flow at 2% B for 2 min and a linear gradient from 2% B to 40% B for 35 min, 40% B to 90% B for 1 min followed by an isocratic flow at 90% B for 5 min and 90% B to 2% B for 2 min, and finally re-equilibrated at 2% B for another 15 min. The total run time per sample was 65 min. The mass spectrometric conditions were as follows: electrospray voltage, 3.5 kV in positive ion mode; source temperature, 375°C; full mass scan range, m/z 350–2000; full mass scan Orbitrap resolution, 120,000; collision energy for data-dependent higher-energy collisional dissociation-MS/MS experiment using ion trap, 28% and MS/MS isolation window, 2 u. Internal mass calibration was performed using a lock mass of m/z 391.284 and m/z 445.120.
Data analysis
Peptide mapping conditions
The raw MS files were subjected to BioPharma Finder™ 3.1 (Thermo Fisher Scientific) for peptide mapping. The peptide identifications were performed by database searching against trastuzumab sequence-based accurate mass of a full mass scan and assignments of product ions in MS/MS spectra. The search parameters were as follows: a mass tolerance of ±5 ppm, confidence score of >95 and peak area of >1000. Carboxymethylation (+58.005 Da) was set as a static modification of Cys residues. Oxidation (+15.995 Da) of Met and Trp, deamidations (+0.984 Da) of Asn and Gln, pyroglutaminated Glu (−18.011 Da) of N-terminal Glu, residual C-terminal Lys (+128.095 Da) and several glycosylations. Targeted modified peptides were extracted as wbpf form files for preparing the processing file for MAM monitoring.
Conditions for MAM monitoring
The wbpf form file was subjected to MAM monitoring software, Chromeleon 7.2.10. For MAM monitoring, single mass scan data were acquired by LC/MS using full mass scan analysis. The parameters to sort target peptides from the single mass scan data were set as follows: mass tolerance: ±5 ppm, peak width: 1 spectrum, and extracted ions: 3 isotopic ions against a target m/z value. The integrated peak area of the extracted ions was calculated, and the relative peak area of the monitoring peptide was calculated using the integrated peak area of multiple target m/z values by the following formula:
For example, the relative peak area ratio of the pyroglutaminated N-terminal peptide was calculated as the percentage of the peak area of the doubly charged ion (m/z 932.50) from the pyroglutaminated ion against the total peak area of the doubly and triply charged ions (m/z 941.51 and m/z 628.01) from the unmodified peptide and the doubly charged ion (m/z 932.50) from the pyroglutaminated ion.
Calculations of performance metrics of peptide mapping
The performance metrics of peptide mapping were calculated by the following formulas:
| (i) |
| (ii) |
| (iii) |
| (iv) |
Results
Optimization of the sample preparation method
Using trastuzumab as a model mAb, we drafted the sample preparation method for MAM (Method 1) by referring to the previously reported method (reference method) of Ren et al [12]. Both method conditions are summarized in Table 1. In Method 1, trastuzumab was denatured, reduced and carboxymethylated, and the buffer was exchanged into the digestion buffer using an NAP™-5 column. These steps were the same as the reference method; however, the denaturation buffer volume in Method 1 was only 150 μl, whereas that of the reference method was 500 μl. The volume was changed due to a reduction in protein diffusion in the NAP™-5 column, which improved protein recovery during the buffer exchange process. Another distinction between the two methods was the enzyme composition. After the buffer exchange step, the sample was digested with a mixture of trypsin and lysyl endopeptidase (Lys-C) (trypsin/Lys-C = 3:1) in Method 1, although trypsin was only used in the reference method. In addition, the enzyme concentration during digestion of Method 1 was ten times that of the reference method. We thought that these changes contributed to reducing the redundancy of peptides (peptide redundancy) observed in the peptide mapping.
Table 1.
The operating condition of the sample preparation methods compared in this study.
| Parameter |
Method 1 (Proposed method) |
Method 2 |
Method 3 |
Method 4 |
Method 5 |
Reference method |
|---|---|---|---|---|---|---|
| Denaturing reagent |
GuHCl |
GuHCl |
SDC and SLS |
GuHCl |
Not applicable |
GuHCl |
| RCM (temp.) |
DTT/MIA (room temp.) |
DTT/MIA (65°C) |
DTT/MIA (room temp.) |
TCEP/IAA (37°C) |
(After digestion) DTT/MIA (57°C) |
DTT/MIA (room temp.) |
| Treatment after RCM | Desalting/buffer exchange by NAP5 column | Desalting by PD10 column and lyophilization | Dilution | Dilution | Not applicable | Desalting/buffer exchange by NAP5 column |
| Enzyme | Trypsin:Lys-C (3:1) | Trypsin | Trypsin:Lys-C (3:1) | Trypsin:Lys-Ca (1:1) | Immobilized trypsin | Trypsin |
| The amount of enzyme per unit weight of protein | 0.4 | 0.01 | 0.4 | 4 | unknown | 0.04 |
| Digestion buffer (pH) | Tris-HCl (7.5) | Tris-HCl (8.5) | ABC (8.5~) | Ammonium acetate (5.5–7.0) | Unknown (7.0) | Tris-HCl (7.5) |
| Digestion temp. | 37°C | 37°C | 37°C | 37°C | 70°C | 37°C |
| Digestion time | 30 min | 16 h | 16 h | 4 h | 45 min | 30 min |
| Required time (days) | 1 | 3 | 2 | 1 | 1 | 1 |
a, Low pH resistant recombinant Lys-C. RCM, reduction/carboxymethylation or carbamidomethylation; GuHCl, guanidine hydrochloride; SDC, sodium deoxycholate; SLS, sodium N-dodecanoylsarcosinate; DTT, dithiothreitol; MIA, sodium monoiodoacetate; temp., temperature; TCEP, tris(2-carboxyethyl)phosphine hydrochloride; IAA, iodoacetamide; Lys-C, lysyl endopeptidase; ABC, ammonium bicarbonate.
Next, we compared Method 1 with four different methods: a traditional method (Method 2), a commercial kit for proteomics (Method 3), and two commercial kits for peptide mapping (Methods 4 and 5). The method utilities were evaluated by the scores based on four performance metrics: (i) sequence coverage, (ii) peptide redundancy, (iii) zero-missed (undigested) cleavage ratio and (iv) deamidation level on Asn55 in the IYPTN55GYTR peptide from the heavy chain (H-chain). The calculation methods of each parameter are described in the Materials and Methods section. Higher sequence coverage is important to enable comprehensive monitoring. Lower peptide redundancy and a higher zero-missed cleavage ratio in the peptide map are important to simplify the selection of monitoring peptides because it is methodologically not easy to monitor multiple peptides for a certain modification. The deamidation level of Asn55 in the H-chain is also evaluated as a model of artificial modifications during sample preparation because it is well known that the Asn in the Asn-Gly sequence is prone to deamidation [13,14] (Figure 2). Because Asn55 is in the complementarity determining region (CDR) of trastuzumab, where the deamidation level needs to be controlled, this peptide can also be a model that is measured by MAM during the quality control of mAb products. The scores are summarized in Table 2. The weights of scores were set to 10:8:3:10 against (i):(ii):(iii):(iv) by considering the impact on the comprehensiveness, ease and reliability of the relative quantification. The remarkable points against the metrics of Methods 1–5 are described below.
Figure 2.

The amino acid sequences of the heavy and light chains from trastuzumab. Bolds indicate Lys (K) and Arg (R). The C-terminal side of Lys was cleaved by trypsin and Lys-C; the C-terminal side of Arg was cleaved by trypsin. The amino acid sequence was confirmed according to the WHO International Nonproprietary Names (INNs) list. Trastuzumab emtansine, WHO Drug Information, Vol. 25, No. 1, 2011, Recommended INN: List 65, P89.
Table 2.
Comparison of the proposed method (Method 1), traditional method (Method 2), commercial kit for proteomics (Method 3), and two commercial kits for peptide mapping (Methods 4 and 5).
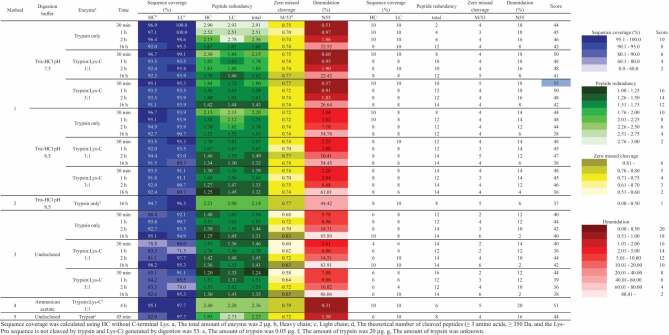 |
Sequence coverage was calculated using HC without C-terminal Lys. a, The total amount of enzyme was 2 µg. b, Heavy chain; c, Light chain; d, The theoretical number of cleaved peptides (≥ 3 amino acids, ≥ 350 Da, and the Lys-Pro sequence is not cleaved by trypsin and Lys-C) generated by digestion was 53. e, The amount of trypsin was 0.05 µg. f, The amount of trypsin was 20 µg. g, The amount of trypsin was unknown.
Sequence coverage (%)
The peptides were identified according to precursor ion mass error tolerance (within ±5 ppm) and confidence score (>0.99) in the BioPharma Finder™ 3.1 software. When the model trastuzumab was treated by Method 1 using digestion buffer at pH 7.5 or pH 8.5 for different digestion times (30 min, 1 h, 2 h and 16 h), the sequence coverage decreased over time (Table 2). The highest sequence coverage was observed in the digestion time of 30 min (H-chain, 96.9%; light chain, 100%), which was digested by trypsin only at pH 7.5. The reason why a shorter digestion time was better in this result may be that short peptides, which were generated by the complete digestion, were undetectable in peptide mapping. Interestingly, the treatment by Method 3 indicated that the sequence coverage increased over time, which contrasts with that of the treatment by Method 1, suggesting a slow digestion speed in Method 3. The peptide maps by Methods 2, 4 and 5 showed satisfactory coverages. No differences in the sequence coverage was observed between the enzymatic conditions of the trypsin only and trypsin/Lys-C mixture digestions.
Peptide redundancy
The trends of peptide redundancy of Methods 1 and 3 were similar. Both peptide redundancies decreased by increasing the digestion time. The redundancies also decreased in the order of the enzymatic conditions as follows: trypsin only>trypsin:Lys-C (1:1)>trypsin:Lys-C (3:1), as shown in Figure 3(a). In addition, using Method 1, the peptide redundancy was lower when the digestion buffer was at pH 8.5 than when the digestion buffer was at pH 7.5. Collectively, the conditions with the lowest peptide redundancy in Method 1 were as follows: pH of digestion buffer, 8.5; enzymatic conditions, trypsin:Lys-C (3:1); and digestion time, 16 h. Method 2 (traditional method) showed a high peptide redundancy (>2) even though there was enough digestion time. This result may be caused by a lower amount of enzyme. The peptide redundancy of Methods 4 and 5 were relatively higher than that of the other methods, suggesting insufficient digestion in Methods 4 and 5.
Figure 3.

Comparison of performance metrics between the five sample preparation methods.
(a) peptide redundancy, (b) deamidation level.
Zero-missed cleavage ratio
The theoretical number of tryptic peptides from trastuzumab is 53 (≥ 3 amino acids, ≥ 350 Da, and the Lys-Pro sequence is not cleaved by trypsin or Lys-C). We calculated the percentage of the detected zero-missed cleavage ratio against the 53 peptides. As a result, the zero-missed cleavage ratio values of Methods 1, 2, 4 and 5 were more than 70%. However, the zero-missed cleavage ratio value of Method 3 with a short digestion time (30 min – 1 h) were in the range of 50–70%, suggesting a slow digestion speed of Method 3.
Deamidation
These metrics showed remarkable differences among the 5 methods (Figure 3(b)). Deamidation occurs during the production process of mAbs as well as the sample preparation step for MAM. Therefore, to evaluate the true value of the deamidation level of the sample, it is important to avoid deamidation during sample preparation. In our study, the highest deamidation value of 86.06%, which may also include artificial deamidation, was observed by Method 3. In Method 1, using digestion buffer pH 7.5, the deamidation level increased from 0.5–0.6% to 22.4–26.6% with increasing digestion time. The same trend was observed using the digestion buffer at pH 8.5. By comparing the digestion buffer at pH 7.5 and pH 8.5, the deamidation levels when using the buffer at pH 8.5 were 5 times higher than those at pH 7.5. On the other hand, there were no differences among the three digestion conditions (trypsin only, trypsin:Lys-C (1:1), trypsin:Lys-C (3:1)). The values of Methods 2 and 3 were higher than those of Method 1 for the same digestion time. Method 4, which was characterized by a digestion at a low pH level, showed the lowest deamidation levels (0.3%) among the five methods. Method 5 also had a relatively low deamidation level. Considering these results, the most important factors for the suppression of deamidation were digestion time and pH of the digestion buffer.
Method evaluation based on scores
Total scores of all data were calculated according to the preset score for each metric (Table 2). The highest score was 53 for Method 1 using the digestion buffer at pH 7.5, enzymatic condition of trypsin:Lys-C (3:1), and digestion time of 30 min. Therefore, Method 1 was determined to be suitable as the sample preparation method (proposed method) for MAM analysis. The details of the proposed method are summarized in Table 3.
Evaluation of usefulness in MAM analysis
Extraction of monitoring peptides
To evaluate the usefulness of the proposed method, the tryptic digest from trastuzumab was analyzed by data-dependent LC/MS/MS for peptide characterization. Figure 4 shows the base peak chromatogram from the full mass scan in the data-dependent scan. The main peaks were detected in the range from 2 min to 30 min. The peptides were identified according to precursor ion mass error tolerance (within ±5 ppm), confidence score (>0.95) and peak area (>1000) of BioPharma Finder™ 3.1 software. We selected only the more intense unmodified peptides out of the redundant unmodified peptides to simplify the peptide map. As a result, 39 unmodified peptides were extracted. The sequence coverages of the H-chain and light chain (L-chain) were 92.7% and 93.0%, respectively (Figure 5 and Table S1). Here, some modified peptides that have the same amino acid sequence as unmodified peptides were also found in the characterization process. In the modified peptides, we finally selected the glycosylated peptides, deamidated or oxidated peptides, N-terminal pyroglutaminated peptide and C-terminal peptide with residual Lys as model MAM monitoring peptides (Table 4). The modification sites were located at 3 amino acids in the variable domain of the H-chain (Vh domain), 2 amino acids in one of the constant domains of the H-chain (Ch2 domain) and 2 amino acids in the Ch3 domain, and 2 amino acids in the variable domain of the L-chain (Vl domain) (Figure 6).
Figure 4.
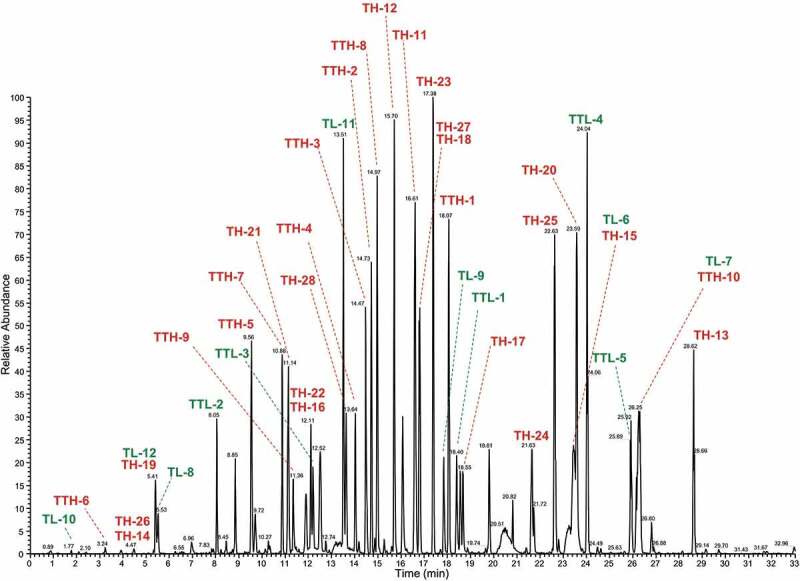
The base peak chromatogram of tryptic digests from trastuzumab prepared by our proposed sample preparation method. TH and TL were the peptides from the constant region of trastuzumab. TTH and TTL were the peptides containing the CDR region of trastuzumab.
Figure 5.

Coverage map of the peptide map obtained by the characterization stage of MAM analysis. Sequence coverage of the H-chain was calculated using the sequence without the C-terminal Lys. The colored gradation indicates the relative intensity against the intensity of the most intense peptide in the peptide map. TH and TL were peptides containing a constant region of trastuzumab. TTH and TTL are peptides containing the CDR region of trastuzumab.
Table 4.
The modified peptides available for MAM monitoring.
| Peptide name | Charge state | Observed m/z | Measured mass | Calculated exact mass | Mass error (ppm) |
Location | Peptide sequencea | Modification | Site |
|---|---|---|---|---|---|---|---|---|---|
| TTHb-1 | 2 | 941.505 | 1880.996 | 1880.996 | 0.250 | E1-R19 | EVQLVESGGGLVQPGGSLR | Unmodified | |
| 3 | 628.006 | 1880.995 | 1880.996 | −0.159 | |||||
| 2 | 932.501 | 1862.986 | 1862.987 | −0.075 | EVQLVESGGGLVQPGGSLR | Pyroglutamination | E1 | ||
| TTH-5 | 1 | 1084.542 | 1083.534 | 1083.535 | −0.434 | I51-R59 | IYPTNGYTR | Unmodified | |
| 2 | 542.775 | 1083.535 | 1083.535 | 0.148 | |||||
| 2 | 543.267 | 1084.519 | 1084.519 | 0.148 | IYPTNGYTR | Deamidation | N55 | ||
| TTH-8 | 1 | 1310.652 | 1309.645 | 1309.645 | −0.130 | N77-R87 | NTAYLQMNSLR | Unmodified | |
| 2 | 655.830 | 1309.645 | 1309.645 | 0.428 | |||||
| 2 | 663.828 | 1325.641 | 1325.640 | 1.252 | NTAYLQMNSLR | Oxidation | M83 | ||
| THc-16 | 1 | 835.435 | 834.428 | 834.427 | 0.755 | D252-R258 | DTLMISR | Unmodified | |
| 2 | 418.221 | 834.427 | 834.427 | 0.431 | |||||
| 1 | 851.430 | 850.423 | 850.422 | 0.976 | DTLMISR | Oxidation | M255 | ||
| 2 | 426.218 | 850.422 | 850.422 | 0.423 | Oxidation | ||||
| TH-19 | 2 | 836.4086 | 1670.803 | 1670.801 | 0.874 | T292-R304 | TKPREEQYNSTYR | Unmodified | |
| 3 | 557.941 | 1670.802 | 1670.801 | 0.353 | |||||
| 4 | 418.708 | 1670.801 | 1670.801 | −0.168 | |||||
| 2 | 1558.675 | 3115.335 | 3115.335 | 0.083 | TKPREEQYNSTYR | Glycosylationd (A2G0F) | N300 | ||
| 3 | 1039.453 | 3115.338 | 3115.335 | 0.896 | |||||
| 4 | 779.843 | 3115.342 | 3115.335 | 2.285 | |||||
| 2 | 1639.701 | 3277.387 | 3277.388 | −0.348 | Glycosylation (A2G1F) | ||||
| 3 | 1093.471 | 3277.390 | 3277.388 | 0.516 | |||||
| 4 | 820.356 | 3277.394 | 3277.388 | 1.745 | |||||
| 3 | 971.760 | 2912.258 | 2912.256 | 0.546 | Glycosylation (A1G0F) | ||||
| 4 | 729.072 | 2912.259 | 2912.256 | 1.003 | |||||
| 2 | 1720.730 | 3439.446 | 3439.441 | 1.471 | Glycosylation (A2G2F) | ||||
| 3 | 1147.488 | 3439.441 | 3439.441 | 0.084 | |||||
| 4 | 860.868 | 3439.445 | 3439.441 | 1.023 | |||||
| 3 | 990.767 | 2969.280 | 2969.277 | 0.940 | Glycosylation (A2G0) | ||||
| 4 | 743.328 | 2969.281 | 2969.277 | 1.320 | |||||
| 3 | 1025.776 | 3074.307 | 3074.309 | −0.475 | Glycosylation (A1G1F) | ||||
| 4 | 769.585 | 3074.309 | 3074.309 | 0.250 | |||||
| 3 | 963.416 | 2887.225 | 2887.224 | 0.447 | Glycosylation (M5) | ||||
| 4 | 722.814 | 2887.227 | 2887.224 | 1.011 | |||||
| 3 | 1044.785 | 3131.333 | 3131.330 | 0.923 | Glycosylation (A2G1) | ||||
| 4 | 783.841 | 3131.333 | 3131.330 | 1.060 | |||||
| 3 | 923.074 | 2766.201 | 2766.198 | 1.009 | Glycosylation (A1G0) | ||||
| 4 | 692.557 | 2766.200 | 2766.198 | 0.622 | |||||
| TH-19 | 3 | 1244.519 | 3730.535 | 3730.536 | −0.298 | T292-R304 | TKPREEQYNSTYR | Glycosylation (A2S1G1F) | N300 |
| 4 | 933.642 | 3730.539 | 3730.536 | 0.783 | |||||
| 3 | 1190.502 | 3568.485 | 3568.483 | 0.586 | Glycosylation (A2S1G0F) | ||||
| 4 | 893.129 | 3568.485 | 3568.483 | 0.538 | |||||
| 3 | 977.092 | 2928.254 | 2928.251 | 1.089 | Glycosylation (A1G1) | ||||
| 4 | 733.071 | 2928.253 | 2928.251 | 0.792 | |||||
| 3 | 1122.809 | 3365.405 | 3365.404 | 0.175 | Glycosylation (A1S1F) | ||||
| 4 | 842.360 | 3365.410 | 3365.404 | 1.819 | |||||
| 3 | 1341.549 | 4021.625 | 4021.632 | −1.619 | Glycosylation (A2S2F) | ||||
| 4 | 1006.415 | 4021.632 | 4021.632 | −0.070 | |||||
| 3 | 1098.802 | 3293.384 | 3293.383 | 0.452 | Glycosylation (A2G2) | ||||
| 4 | 824.354 | 3293.387 | 3293.383 | 1.312 | |||||
| 3 | 1017.433 | 3049.278 | 3049.277 | 0.259 | Glycosylation (M6) | ||||
| 4 | 763.327 | 3049.279 | 3049.277 | 0.498 | |||||
| 3 | 1085.126 | 3252.357 | 3252.356 | 0.335 | Glycosylation (A1G1M5) | ||||
| 4 | 814.097 | 3252.358 | 3252.356 | 0.529 | |||||
| TTH-22 | 3 | 781.731 | 2342.171 | 2342.169 | 0.722 | G344-K363 | GQPREPQVYTLPPSREEMTK | Unmodified | |
| 4 | 586.550 | 2342.171 | 2342.169 | 0.862 | |||||
| 3 | 787.062 | 2358.165 | 2358.164 | 0.547 | GQPREPQVYTLPPSREEMTK | Oxidation | M361 | ||
| 4 | 590.549 | 2358.165 | 2358.164 | 0.645 | |||||
| TH-28 | 1 | 788.452 | 787.444 | 787.444 | 0.292 | S443-K450 | SLSLSPGK | Unmodified | |
| 2 | 394.729 | 787.444 | 787.444 | −0.432 | |||||
| 1 | 660.357 | 659.350 | 659.349 | 1.259 | SLSLSPG | C-terminal Lys clipping | G449 | ||
| TTLb-1 | 2 | 1305.630 | 2609.245 | 2609.243 | 0.981 | D1-R24 | DIQMTQSPSSLSASVGDRVTIT(C)R | Unmodified | (C23) |
| 3 | 870.757 | 2609.248 | 2609.243 | 2.104 | |||||
| 2 | 1313.626 | 2625.237 | 2625.237 | −0.206 | DIQMTQSPSSLSASVGDRVTIT(C)R | Oxidation, (Carboxymethylation) | M4 (C23) | ||
| 3 | 876.087 | 2625.238 | 2625.237 | 0.110 | |||||
| TTL-3 | 2 | 1144.095 | 2286.175 | 2286.176 | −0.280 | A25-K45 | ASQDVNTAVAWYQQKPGKAPK | Unmodified | |
| 3 | 763.066 | 2286.177 | 2286.176 | 0.739 | |||||
| 4 | 572.552 | 2286.179 | 2286.176 | 1.234 | |||||
| 2 | 1144.587 | 2287.160 | 2287.160 | 0.245 | ASQDVNTAVAWYQQKPGKAPK | Deamidation | N30 | ||
| 3 | 763.395 | 2287.163 | 2287.160 | 1.264 | |||||
| 4 | 572.798 | 2287.164 | 2287.160 | 1.758 |
a, The underlined amino acid residue is the modification site. b, TTH and TTL are the peptides containing the CDR region of trastuzumab. c, TH is the peptide containing the constant region of trastuzumab. d, The deduced glycan structures are shown in Figure 5.
Figure 6.

The modification sites of the modified peptides used as model monitoring peptides.
Analytical performance evaluation
Considering sensitivity and repeatability, the analytical performance evaluation was designed as follows: mass accuracy (±5 ppm), intra-assay CV (%) (repeatability) of the peak area (<15%) and intra-assay CV (%) of the retention time (<5%) against the most intense ion for each monitoring modified peptide. In this study, MAM analyses of tryptic digests from a single trastuzumab lot were carried out in triplicate per day and repeated for 3 days (Figure S1). Therefore, the analytical performance evaluation was carried out using the mass spectrometric data obtained by these MAM analyses. As a result, the mass accuracy of the 9 peptides was in the range from −2.48 ppm to 2.11 ppm (Table S2). The worst CV (%) of the peak area ratio and retention time were 14.78% for TH-22 and 0.29% for TH-19 (Table S3). These results demonstrated the acceptable sensitivity and repeatability of our method.
Evaluation of MAM analysis using our sample preparation method
The average relative peak area percentages of 17 glycopeptides from all experiments are shown in Figure 7. The average relative peak area of glycopeptides having three major glycans, A2G0F, A2G1F and A2G2F, was 39.46% (CV, 0.26%), 32.87% (CV, 0.41%) and 5.01% (CV, 1.12%), respectively, which agrees with a previous report [15]. Minor components, such as M6 and A1G1M5 (hybrid-type glycan), were also detected successfully. Surprisingly, the inter-assay CV (%) of all monitored glycopeptides was <7%. This result implied that our method was applicable for monitoring even a small amount of modified peptides including glycopeptides. The relative peak area percentages of all modified peptides are summarized in Table 5. The average values from the inter-assays of N-terminal pyroglutamation and C-terminal truncation for the H-chain were 1.83% (CV, 2.20%) and 98.72% (CV, 0.03%), respectively. The degree of deamidation at Asn55 in the H-chain was only 0.94% (CV, 5.85%). On the other hand, the deamidation level at Asn30 in the L-chain was higher than the deamidation level at Asn55 in the H-chain (inter-assay average, 12.68%; CV, 0.93%), suggesting a difference in the initial deamidation level at each site. The inter-assay averages of oxidations at Met83, Met255 and Met361 in the H-chain and Met4 in the L-chain were 3.01% (CV, 25.33%), 7.13% (CV, 16.29%), 4.23% (CV, 20.88%) and 4.37% (CV, 33.36%), respectively. Importantly, the inter-assay CV (intermediate precision) (%) values of oxidated peptides were slightly higher than those of other peptides (Figure 8). The levels of oxidation in the results from the 5 methods in Table 1 were also evaluated; however, remarkable differences were not observed. Given that a single lot of trastuzumab was used in this study, artificial oxidation may have occurred during the desalting of tryptic digests or during sample vial storage. To address the cause, the long-term stability of the peptides in an acidic solution related to chemical modifications was evaluated by analyzing two degraded samples. The first sample was stored in 0.1% FA solution at 37°C for 10 days and then additionally stored at 8°C for 30 days in the dark. The second sample was stored in 0.1% FA solution at 8°C for 40 days in the dark. As shown in Table S4, the oxidated peptides were increased in both samples. It was indicated that the instability in an acidic solution such as the LC/MS sample solution containing FA may be related to the higher CV (%) values of oxidated peptides. Additionally, the N-terminal pyroglutamated peptide was increased in degraded sample-1, indicating that heat stress influenced the formation of pyroglutamine. These results implied the importance of possible rapid analysis and storage of digested peptides at a low temperature.
Figure 7.

The results of the relative quantification of glycopeptides. The relative peak area (%) was calculated as the ratio of each glycopeptide against the total peak area, including unglycosylated peptide and all glycosylated peptides. Green circle, mannose; blue square, N-acetylglucosamine; yellow circle, galactose; red triangle, fucose; purple diamond, N-acetylneuraminic acid.
Table 5.
Summary of the MAM monitoring of the modified peptides.
| H-chain/ L-chain |
Peptide name | Relative peak area percentage |
Total average | CVb | Modification (site) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day 1 |
Day 2 |
Day 3 |
|||||||||||||||||
| 1 | 2 | 3 | Average | CVa | 1 | 2 | 3 | Average | CVa | 1 | 2 | 3 | Average | CVa | |||||
| H-chain | TTHd-1 | 1.83 | 1.80 | 1.83 | 1.82 | 0.82 | 1.80 | 1.86 | 1.92 | 1.86 | 3.35 | 1.81 | 1.80 | 1.81 | 1.80 | 0.46 | 1.83 | 2.20 | Pyroglutamination (E1) |
| TTH-5 | 0.97 | 1.00 | 0.96 | 0.97 | 1.99 | 0.97 | 0.97 | 0.97 | 0.97 | 0.39 | 0.88 | 0.86 | 0.86 | 0.87 | 1.10 | 0.94 | 5.85 | Deamidation (N55) | |
| TTH-8 | 2.99 | 2.83 | 2.51 | 2.78 | 8.93 | 4.03 | 3.87 | 3.97 | 3.96 | 2.03 | 2.12 | 2.28 | 2.44 | 2.28 | 7.08 | 3.01 | 25.33 | Oxidation (M83) | |
| THe-16 | 7.33 | 6.93 | 6.34 | 6.87 | 7.23 | 8.63 | 8.50 | 8.50 | 8.55 | 0.87 | 5.67 | 6.04 | 6.26 | 5.99 | 4.93 | 7.13 | 16.29 | Oxidation (M255) | |
| TH-19 | 39.43 | 39.28 | 39.42 | 39.38 | 0.21 | 39.43 | 39.64 | 39.50 | 39.52 | 0.27 | 39.59 | 39.43 | 39.43 | 39.48 | 0.24 | 39.46 | 0.26 | A2G0Fc (N300) | |
| TH-19 | 33.00 | 32.76 | 32.70 | 32.82 | 0.49 | 32.99 | 32.85 | 33.10 | 32.98 | 0.39 | 32.76 | 32.82 | 32.88 | 32.82 | 0.17 | 32.87 | 0.41 | A2G1Fc (N300) | |
| TH-19 | 7.22 | 7.48 | 7.45 | 7.38 | 1.97 | 7.16 | 7.31 | 7.17 | 7.22 | 1.19 | 7.27 | 7.28 | 7.24 | 7.27 | 0.27 | 7.29 | 1.56 | A1G0Fc (N300) | |
| TH-19 | 5.02 | 4.94 | 4.97 | 4.98 | 0.82 | 4.98 | 5.10 | 4.97 | 5.02 | 1.48 | 5.01 | 5.07 | 5.07 | 5.05 | 0.72 | 5.01 | 1.12 | A2G2Fc (N300) | |
| TH-19 | 4.30 | 4.34 | 4.28 | 4.31 | 0.66 | 4.33 | 4.24 | 4.31 | 4.29 | 1.13 | 4.37 | 4.38 | 4.33 | 4.36 | 0.57 | 4.32 | 1.01 | A2G0c (N300) | |
| TH-19 | 2.35 | 2.42 | 2.41 | 2.40 | 1.73 | 2.35 | 2.37 | 2.27 | 2.33 | 2.29 | 2.34 | 2.33 | 2.36 | 2.34 | 0.65 | 2.36 | 1.95 | A1G1Fc (N300) | |
| TH-19 | 2.27 | 2.24 | 2.24 | 2.25 | 0.86 | 2.23 | 2.18 | 2.19 | 2.20 | 1.33 | 2.19 | 2.21 | 2.19 | 2.20 | 0.62 | 2.22 | 1.43 | M5c (N300) | |
| TH-19 | 1.80 | 1.78 | 1.75 | 1.78 | 1.21 | 1.74 | 1.75 | 1.78 | 1.76 | 1.27 | 1.81 | 1.79 | 1.78 | 1.79 | 0.81 | 1.78 | 1.34 | A2G1c (N300) | |
| TH-19 | 1.47 | 1.49 | 1.50 | 1.49 | 0.89 | 1.50 | 1.42 | 1.47 | 1.47 | 2.75 | 1.46 | 1.47 | 1.52 | 1.48 | 2.24 | 1.48 | 1.93 | A1G0c (N300) | |
| TH-19 | 0.58 | 0.59 | 0.60 | 0.59 | 2.22 | 0.62 | 0.57 | 0.59 | 0.60 | 3.67 | 0.58 | 0.59 | 0.58 | 0.58 | 0.88 | 0.59 | 2.38 | A2S1G1Fc (N300) | |
| TH-19 | 0.42 | 0.43 | 0.44 | 0.43 | 1.52 | 0.45 | 0.43 | 0.44 | 0.44 | 2.36 | 0.44 | 0.42 | 0.43 | 0.43 | 2.04 | 0.43 | 1.89 | A2S1G0Fc (N300) | |
| TH-19 | 0.34 | 0.35 | 0.34 | 0.35 | 1.55 | 0.33 | 0.32 | 0.34 | 0.33 | 3.80 | 0.34 | 0.34 | 0.34 | 0.34 | 0.19 | 0.34 | 2.92 | A1G1c (N300) | |
| TH-19 | 0.20 | 0.21 | 0.20 | 0.20 | 2.45 | 0.21 | 0.20 | 0.20 | 0.21 | 3.17 | 0.20 | 0.21 | 0.20 | 0.20 | 1.16 | 0.20 | 2.24 | A1S1Fc (N300) | |
| TH-19 | 0.16 | 0.19 | 0.20 | 0.18 | 10.54 | 0.20 | 0.20 | 0.20 | 0.20 | 0.83 | 0.19 | 0.20 | 0.19 | 0.19 | 2.50 | 0.19 | 6.21 | A2S2Fc (N300) | |
| TH-19 | 0.16 | 0.18 | 0.17 | 0.17 | 5.08 | 0.17 | 0.15 | 0.17 | 0.16 | 5.99 | 0.16 | 0.15 | 0.17 | 0.16 | 4.86 | 0.16 | 5.06 | A2G2c (N300) | |
| TH-19 | 0.13 | 0.12 | 0.12 | 0.12 | 3.64 | 0.13 | 0.13 | 0.13 | 0.13 | 1.68 | 0.12 | 0.13 | 0.13 | 0.13 | 2.08 | 0.13 | 3.53 | M6c (N300) | |
| TH-19 | 0.11 | 0.11 | 0.11 | 0.11 | 0.69 | 0.11 | 0.10 | 0.10 | 0.11 | 2.06 | 0.10 | 0.10 | 0.10 | 0.10 | 0.99 | 0.10 | 2.95 | A1G1M5c (N300) | |
| TH-22 | 4.23 | 4.12 | 3.82 | 4.06 | 5.27 | 5.11 | 5.44 | 5.39 | 5.31 | 3.36 | 3.18 | 3.31 | 3.50 | 3.33 | 4.73 | 4.23 | 20.88 | Oxidation (M361) | |
| TH-28 | 98.66 | 98.68 | 98.74 | 98.69 | 0.04 | 98.71 | 98.71 | 98.74 | 98.72 | 0.02 | 98.74 | 98.74 | 98.74 | 98.74 | <0.01 | 98.72 | 0.03 | C-terminal Lys clipping (G449) | |
| L-chain | TTLd-1 | 3.84 | 4.09 | 3.63 | 3.85 | 5.98 | 6.09 | 6.43 | 6.20 | 6.24 | 2.77 | 2.87 | 3.16 | 3.01 | 3.01 | 4.89 | 4.37 | 33.36 | Oxidation (M4) |
| TTLe-3 | 12.54 | 12.59 | 12.77 | 12.63 | 0.94 | 12.77 | 12.62 | 12.66 | 12.69 | 0.62 | 12.79 | 12.52 | 12.84 | 12.72 | 1.37 | 12.68 | 0.93 | Deamidation (N30) | |
Three individual preparations of digested peptides were prepared and analyzed each day. a, Intra-assay CV (%); b, Inter-assay CV (%); c, Glycosylation; d, TTH and TTL are the peptides containing the CDR region of trastuzumab; e, TH and TL are the peptides containing the constant region of trastuzumab
Figure 8.
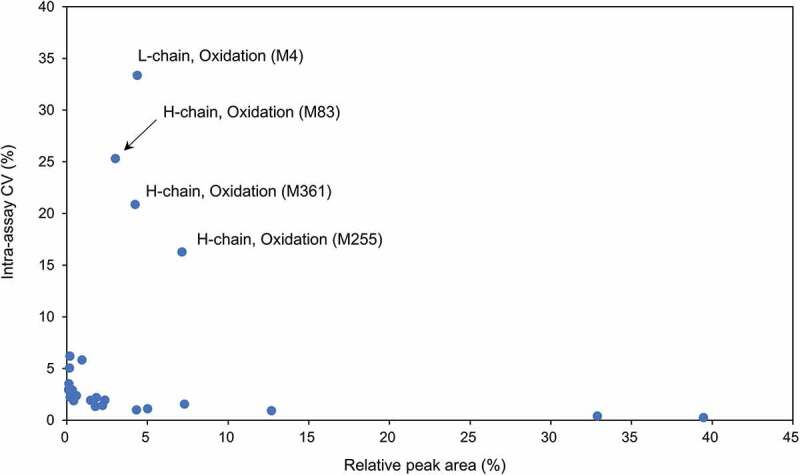
The relationship between the total relative peak area and inter-assay CV (%) of the modified peptides.
Discussion
In this study, sequence coverage, peptide redundancy, zero-missed cleavage ratio and deamidation level on Asn55 of the H-chain were used as indicators to evaluate the appropriateness of the sample preparation methods. When comparing the digestion buffers with pH 7.5 and pH 8.5, the use of the digestion buffer with pH 7.5 was effective in reducing the deamidation level, while there was a tendency toward a higher peptide redundancy. To reduce the redundancy, in other words, improving the digestion efficacy, digestion by a combination of trypsin and Lys-C was critical. More practically, the peptide redundancy when using trypsin/Lys-C (3:1) was half the peptide redundancy than that when using trypsin only. What is more notable was that high sequence coverage was successfully achieved by short digestion times of only 30 min. This result is consistent with the findings of a previous report [12]. In usual peptide mapping, the reaction mixture after reduction and carboxymethylation (RCM) is often diluted to maintain enzymatic activities in the digestion process; however, some RCM buffer reagents could delay the digestion by inhibiting enzyme activities. We thought that desalting after RCM may be an essential procedure for short-term digestion. It is known that one of the most susceptible artificial modifications during peptide mapping is deamidation [14]. In the peptide mapping of trastuzumab, the most susceptible deamidation site was at Asn55 in the H-chain. Surprisingly, the deamidation level was less than 1.0% by using Method 1 (proposed method) with the digestion buffer at pH 7.5. It was demonstrated that the proposed method enables MAM analysis without being affected by artificial deamidation. Interestingly, Method 4 showed the lowest deamidation level, only 0.3%. However, this method also showed the worst peptide redundancy score, suggesting there are many missed cleavage peptides. Therefore, it should be noted that the selection of monitoring peptides in the MAM analysis using Method 4 could become complicated. The completely cleaved glycopeptide (EEQYNSTYR) was detected easily in peptide mapping using Method 3.
In this study, we demonstrated that MAM analysis of the 25 modified peptides from trastuzumab was possible. Importantly, we noticed that the inter-assay CV (%) values of the peptides with an oxidated Met were clearly higher than those of other peptides, but the intra-assay CV (%) values were acceptable. A long-term stability test revealed that the higher CV (%) values of oxidated peptides might be caused by the instability in acidic solution. Considering that acidic solutions containing FA or trifluoroacetic acid (TFA) are used as general MS sample solutions, it might be difficult to improve the CV (%) value of oxidated peptides, while it could be important to evaluate the usefulness of antioxidants. Therefore, this result indicated that variations in the relative peak area could be influenced by the modification type, rather than by the amount of each peptide. Optionally, the assessment of stressed samples prepared under several conditions such as heat and pH variations may be useful for identifying susceptible chemical modification sites and selecting peptides to be considered as monitoring peptides.
As an analytical performance evaluation of the MAM analysis, we selected 3 performance metrics: mass accuracy, CV (%) of peak area and CV (%) of retention time. The performance metrics and their criteria for system check should be selected for the purpose of MAM monitoring each modified peptide. Our analytical performance evaluation method may also be able to be used for assessments of system suitability and sample suitability for the implementation of MAM analyses during quality control.
mAbs undergo several modifications during manufacturing or storage. Previous studies have reported that certain components of Fc-glycans are involved in structural fluctuations and immune effector functions, such as antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity [16–18]. Therefore, it could be important to monitor glycopeptides to ensure the safety and efficacy of mAb products. Other modifications are also related to mAb efficacy. For example, deamidation in CDR causes a significant reduction in the antigen binding affinity [19]. Oxidation of Met and Trp also result in reduced binding affinity to antigens and Fc receptors [20–22]. In addition, chemical modifications can potentially increase in immunogenicity [23]. These reports indicate the importance of site-specific modification monitoring. We demonstrated that the monitoring of multiple deamidated or oxidized peptides is easily, reliably and simultaneously achieved by using the proposed method. Isomerization and glycation also impact biological functions [24–26]; therefore, these modifications can also become monitoring candidates. However, it is difficult to identify them. For example, particular MS/MS measurements such as electron transfer dissociation (ETD)-MS/MS analysis are needed for the structural determination of isoaspartic acid (isoAsp) in peptides because ETD-MS/MS generates the diagnostic fragment ion of isoAsp [27,28]. The glycation site is mainly Lys, and glycation can inhibit digestion using trypsin [26]. We think that the reliable and robust monitoring of peptides with both modifications is a future task.
MAM can be used during multiple stages of developing mAb products, e.g., process characterization during the establishment of the manufacturing process, in-process control for the commercial production process or specifications. Recently, more extensive analyses of quality attributes are required because the development of mAbs using the Quality by Design approach and/or production using novel technology such as continuous manufacturing, which needs more precise control during the manufacturing process, are increasing. Thus, an MAM that enables the efficient simultaneous analysis of multiple quality attributes will be useful as a platform analytical technology for these products. However, rapid sample preparation of mAb during manufacturing process is difficult; therefore, establishing a process analytical technology (PAT) [29] by combining appropriate sampling method and MAM approach requires further investigation.
Conclusions
We established an optimized sample preparation method for MAM analysis. We found that digestion with low pH buffer and desalting processes after RCM were critical to reduce artificial deamidation and obtain a peptide map with high sequence coverage. In addition, the use of a trypsin/Lys-C mixture was effective in improving the generation of missed cleavage peptides. By our optimized sample preparation method, the simultaneous monitoring of several modified peptides was successfully achieved with acceptable mass accuracy and inter-assay CV (%) of relative peak area. In this study, we demonstrated that our method was applicable as an easy and reliable sample preparation method for MAM analysis, and variation in the relative peak area could be influenced by the modification type rather than by the amount of each peptide.
Funding Statement
This work was supported in part by the [Japan Agency for Medical Research and Development (AMED)] under Grants [numbers JP20ae0101059 and JP20mk0101152]; Japan Agency for Medical Research and Development [JP20ae0101059]; Japan Agency for Medical Research and Development [JP20mk0101152].
Highlights
An easy and reliable sample preparation method for MAM analysis was optimized.
A low pH buffer and short digestion time reduced artificial deamidation.
The desalting process was essential for short time digestion.
Variation in the relative peak area was influenced by the modification type.
Disclosure statement
The authors have no conflicts of interest to declare.
References
- [1].Kaplon H, Muralidharan M, Schneider Z, et al. Antibodies to watch in 2020. MAbs. 2020;12:1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sakanaka C.Antibody therapeutics: bench to bedside. Yakugaku Zasshi. 2017;137:817–822. [DOI] [PubMed] [Google Scholar]
- [3].Grilo AL, Mantalaris A.. The increasingly human and profitable monoclonal antibody market. Trends Biotechnol. 2019;37:9–16. [DOI] [PubMed] [Google Scholar]
- [4].Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu X, Han M, Li T, et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci U S A. 2020;117:10970–10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xu Y, Wang D, Mason B, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies. MAbs. 2019;11:239–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang Y, Guo J. Characterization and QC of biopharmaceuticals by MS-based ‘multi-attribute method’: advantages and challenges. Bioanalysis. 2017;9:499–502. [DOI] [PubMed] [Google Scholar]
- [8].Rogers RS, Nightlinger NS, Livingston B, et al. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. MAbs. 2015;7:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bomans K, Haberger M, Bonnington L, et al. Monitoring of antibody modifications by semi-automated liquid chromatography mass spectrometry peptide mapping. Am Pharm Rev. 2016;19:16–21. [Google Scholar]
- [10].Rogstad S, Yan H, Wang X, et al. Multi-attribute method for quality control of therapeutic proteins. Anal Chem. 2019;91:14170–14177. [DOI] [PubMed] [Google Scholar]
- [11].Chelius D, Rehder DS, Bondarenko PV. Identification and characterization of deamidation sites in the conserved regions of human immunoglobulin gamma antibodies. Anal Chem. 2005;77:6004–6011. [DOI] [PubMed] [Google Scholar]
- [12].Ren D, Pipes GD, Liu D, et al. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal Biochem. 2009;392:12–21. [DOI] [PubMed] [Google Scholar]
- [13].Bults P, Bischoff R, Bakker H, et al. MS/MS-based monitoring of in vivo protein biotransformation: quantitative determination of trastuzumab and its deamidation products in human plasma. Anal Chem. 2016;88:1871–1877. [DOI] [PubMed] [Google Scholar]
- [14].Krokhin OV, Antonovici M, Ens W, et al. Deamidation of -Asn-Gly- sequences during sample preparation for proteomics: consequences for MALDI and HPLC-MALDI analysis. Anal Chem. 2006;78:6645–6650. [DOI] [PubMed] [Google Scholar]
- [15].Segu Z, Stone T, Berdugo C, et al. A rapid method for relative quantification of N-glycans from a therapeutic monoclonal antibody during trastuzumab biosimilar development. MAbs. 2020;12:1750794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li W, Zhu Z, Chen W, et al. Crystallizable fragment glycoengineering for therapeutic antibodies development. Front Immunol. 2017;8:1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Aoyama M, Hashii N, Tsukimura W, et al. Effects of terminal galactose residues in mannose alpha1-6 arm of Fc-glycan on the effector functions of therapeutic monoclonal antibodies. MAbs. 2019;11:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vlasak J, Bussat MC, Wang S, et al. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem. 2009;392:145–154. [DOI] [PubMed] [Google Scholar]
- [20].Bertolotti-Ciarlet A, Wang W, Lownes R, et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol Immunol. 2009;46:1878–1882. [DOI] [PubMed] [Google Scholar]
- [21].Pan H, Chen K, Chu L, et al. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009;18:424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wei Z, Feng J, Lin HY, et al. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal Chem. 2007;79:2797–2805. [DOI] [PubMed] [Google Scholar]
- [23].Hermeling S, Crommelin DJ, Schellekens H, et al. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res. 2004;21:897–903. [DOI] [PubMed] [Google Scholar]
- [24].Huang L, Lu J, Wroblewski VJ, et al. In vivo deamidation characterization of monoclonal antibody by LC/MS/MS. Anal Chem. 2005;77:1432–1439. [DOI] [PubMed] [Google Scholar]
- [25].Cacia J, Keck R, Presta LG, et al. Isomerization of an aspartic acid residue in the complementarity-determining regions of a recombinant antibody to human IgE: identification and effect on binding affinity. Biochemistry. 1996;35:1897–1903. [DOI] [PubMed] [Google Scholar]
- [26].Wei B, Berning K, Quan C, et al. Glycation of antibodies: modification, methods and potential effects on biological functions. MAbs. 2017;9:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chan WY, Chan TW, O’Connor PB. Electron transfer dissociation with supplemental activation to differentiate aspartic and isoaspartic residues in doubly charged peptide cations. J Am Soc Mass Spectrom. 2010;21:1012–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Eakin CM, Miller A, Kerr J, et al. Assessing analytical methods to monitor isoAsp formation in monoclonal antibodies. Front Pharmacol. 2014;5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].International conference on harmonization of technical requirements for registration of pharmaceuticals for human use. ICH Harmonized Tripartite Guidline, Pharmaceutical Dvelopment Q8 R2, August2009.