

Abstract
Effective transgene expression in mammalian cells relies on successful delivery, cytoplasmic trafficking, and nuclear translocation of the delivered vector, but delivery is impeded by several formidable physicochemical barriers on the surface of and within the target cell. Although methods to overcome cellular exclusion and endosomal entrapment have been studied extensively, strategies to overcome inefficient nuclear entry and subsequent intranuclear barriers to effective transient gene expression have only been sparsely explored. In particular, the role of nuclear packaging of DNA with histone proteins, which governs endogenous gene expression, has not been extensively elucidated in the case of exogenously delivered plasmids. In this work, a parallel screen of small molecule inhibitors of chromatin-modifying enzymes resulted in the identification of class I/II HDACs, sirtuins, LSD1, HATs, and the methyltransferases EZH2 and MLL as targets whose inhibition led to the enhancement of transgene expression following polymer-mediated. Delivery of plasmid DNA. Quantitative PCR studies revealed that HDAC inhibition enhances the amount of plasmid DNA delivered to the nucleus in UMUC3 human bladder cancer cells. Native chromatin immunoprecipitation (N-ChIP)-qPCR experiments in CHO-K1 cells indicated that plasmids indeed interact with intracellular core Histone H3, and inhibitors of HDAC and LSD1 proteins are able to modulate this interaction. Pair-wise treatments of effective inhibitors led to synergistic enhancement of transgene expression to varying extents in both cell types. Our results demonstrate that the ability to modulate enzymes that play a role in epigenetic processes can enhance the efficacy of non-viral gene delivery, resulting in significant implications for gene therapy and industrial biotechnology.
INTRODUCTION
Transgene delivery and subsequent protein expression is a promising therapeutic approach for several diseases and is also employed to facilitate high-throughput protein production in biotechnology[1]. Non-viral gene delivery using plasmid DNA (pDNA) is an attractive alternative to viral vectors, but often fails to yield high levels of transgene expression. Several barriers severely limit the efficacy of non-viral transgene expression both at the organism and cellular levels[2–5]. At the cellular level, transport of pDNA into the nucleus governs the efficacy of subsequent transgene expression, and the nucleus is widely considered as the “final frontier” for the effective delivery of plasmid DNA to cells[6, 7].
The immense genetic material in human cells (6 billion base pairs) is packaged within nuclei only ~6μm in diameter. This is accomplished through highly regulated condensation by histone proteins, scaffolding proteins, regulator proteins, and non-coding RNA into a structure known as chromatin. DNA-histone interactions play a pivotal role in the epigenetic regulation of gene expression in cells. In addition to DNA methyltransferases, which facilitate methylation of promoter DNA and generally have an inhibitory effect on gene expression [8], a number of enzymes can modify histones post-translationally, which, in turn, affects chromatin structure and, ultimately, gene expression. Activating and repressive chromatin states can switch from one to the other. Once switched, a state can persist over many cycles of DNA replication and mitosis. Chromatin-mediated memory is a mode of epigenetics, where daughter cells inherit changes in gene expression that are not driven by changes in the nucleic acid sequence. Epigenetic chromatin modifications are involved in a number of cellular processes, including development [9], metabolic regulation [10], and aging [11]. Improper epigenetic regulation can lead to diseases such as cancer [12] and neurodegenerative disorders [13].
The “histone code theory” classifies proteins as writers (enzymes that carry out histone modifications), erasers (those that remove functional modifications), and readers (those that recognize the “epigenetic mark” of these modifications and recruit other proteins for chromatin remodeling) [14, 15]. Enzymes that catalyze histone post-translational modifications (PTMs) such as lysine (de)acetylation, serine, threonine, and tyrosine (de)phosphorylation, and lysine and arginine (de)methylation mark histones for reader enzymes to modulate a change in chromatin structure and gene expression. Acetylation and phosphorylation on histones are known to activate gene expression [13]. Histone methylation, on the other hand, can have activating or repressing effects on local gene expression; for instance, di- or tri- methylation of lysine 4 on the core histone H3 (H3K4me2 or H3K4me3, respectively) activates transcription, while trimethylation of H3K9 (H3K9me3) and H3K27me3 promotes repression of transcription [13].
Although many epigenetic modifications are well understood in the context of genomic DNA, a thorough understanding of how native histones regulate the expression of exogenously delivered DNA (e.g. plasmid DNA or pDNA) is largely lacking. However, some evidence of the role of intranuclear proteins, including histones, can be obtained from previous studies which show that sodium butyrate, a general inhibitor of histone deacetylases [16], and our studies which show that class I HDAC inhibitors Entinostat [17] and trichostatin A [18] enhance transgene expression following pDNA delivery to mammalian cells. Additionally, DNA methyltransferase inhibition with 5-azacytidine has been shown to enhance transgene expression [19]. Further relevant to histone/plasmid interactions in the nucleus, Kay et al. demonstrated that exogenously delivered plasmids and their derivative minicircles can interact with histones in target cells, with histone modifications understood generally to increase or decrease gene expression in chromatin mostly correctly predicting plasmid expression in their mouse liver lysate system [20].
In this study, we screened a commercially available library of 89 small-molecule inhibitors of histone modifiers and PTM-binding proteins in order to identify key targets that influence non-viral transgene expression in two highly distinct mammalian cell lines: CHO-K1 and UMUC3 CHO-K1 is an adherent cell line derived from the rodent Cricetulus griseus (Chinese hamster) [https://www.atcc.org/products/all/CCL-61.aspx]. These cells are widely used in basic research and industrial production of pharmaceutical proteins [21]. UMUC3 is a human urinary bladder cancer-derived line that is widely used in basic research and drug development. CHO-K1 has a relatively low number of chromosomes (21 autosomes, 1 sex chromosome) compared to other mammalian lines [22, 23], a lower fraction (37.79%) of repetitious elements compared to human cells (46%)[24], and overrepresentation of translation, metabolism and protein modification-related gene functions [24]. UMUC3 and CHO-K1 have broad genomic distributions of epigenetic chromatin modifications including DNA methylation, expression-associated histone H3K4me3, K27ac, and K36me3, and silencing-associated H3K9me3, and K27me3 [23, 25]. However, global histone proteomics investigations suggest that the relative proportions of each modification within each cell line vary significantly, which is likely connected to gene regulation states that underlie phenotypic and species-specific differences. Our studies indicate that exogenously delivered plasmid DNA interact with histone proteins and that several epigenetic enzymes including histone deacetylases, histone demethylases, and sirtuins influence polymer-mediated transgene expression. This study also indicates novel molecular interventions, including pairwise combination treatments, that may be employed for enhancing the efficacy of non-viral gene delivery vehicles for various applications in industrial biotechnology and medicine.
EXPERIMENTAL
Small Molecule Inhibitors of Epigenetics Enzymes
A library of 89 small-molecule inhibitors of chromatin-modifying enzymes was purchased from Cayman Chemicals (item number 11076, 10 mM in DMSO). Based on a combination of published IC50 values (dose at half-maximal target inhibition) and concentration ranges typically used in the literature, all inhibitors were categorized either as “high dose candidates” (screened at 500 nM, 5 μM, and 50 μM working concentrations) or “low dose candidates” (screened at 50 nM, 500 nM, and 5 μM working concentrations). Stock solutions were prepared in DMSO at 100X the highest concentration used in the screen (5 mM for high dose candidates, 0.5 mM for low dose candidates). Table S1 (Supporting Information section) contains detailed information on the doses used for each inhibitor.
Inhibitor Screening
Approximately 16 hours prior to screening, 10,000 UMUC3 human bladder cancer or CHO-K1 cells per well were plated in 96 well plates in RPMI 1640 media (10% fetal bovine serum and penicillin (100 units/mL)-streptomycin (100 μg/mL). On the following morning, a solution of 0.375 mg/mL neomycin resorcinol diglycidyl ether (N-RDGE) [26] was prepared in 1X PBS and filtered through a 0.2 μm regenerated cellulose syringe filter; this polymer was previously identified as an effective candidate for transgene expression via combinatorial synthesis and screening in our laboratory. The pEF-Luc plasmid (Addgene, plasmid #22524) was prepared at 15 ng/μL in EB buffer (Qiagen) and used in all screening studies; the plasmid expresses luciferase protein under the control of the EF1-α promoter. Small-molecule inhibitors from the library were added corresponding to the experimental plate position in a sterile deep well 96-well block. Cell-containing plates and the deep well inhibitor-containing block were then placed inside a Biomek FXp liquid handing system (LHS). Serum-containing RPMI media was then added to the 100X concentrated inhibitors and mixed using an orbital shaker, in order to dilute them to the highest concentration tested in these studies. Old media was removed from cells and replenished with drug-containing media by the LHS. The LHS carried out 10X dilutions of the original inhibitor solution to prepare media at the two lower working inhibitor concentrations to be tested. A reservoir of polymer-pDNA complexes (polyplexes) was prepared manually in a laminar flow hood at a 25:1 (w/w) ratio and allowed to form for 20 minutes at room temperature. Polyplexes containing 75 ng DNA/well were then added to all cells (10 μL/well) by the LHS, and transfection was allowed to proceed for 48 hours in an incubator maintained at 37°C with 5% CO2.
Quantification of Transgene Expression
Cells were assayed for transgene (luciferase) expression 48 hours after transfection. Cell lysis was carried out using cell culture lysis reagent (Promega) and luminescence was detected using a Synergy 2 plate reader (Biotek, Winooski, VT). MTT viability assays (ATCC) were carried out in parallel plates, and luminescence values were normalized to viability to account for cell proliferation reduction effects on total transgene expression.
Optimization of Inhibitor Dose
Select lead inhibitors from the small-molecule inhibitor screen were chosen for dose optimization with the pEF-Luc plasmid, expressing firefly luciferase driven by the EF1α promoter as well as the pGL3 plasmid, which also expresses luciferase, but under control of the Simian Virus 40 (SV40) promoter. These transfection experiments were carried out manually (without the LHS) using the same polymer-pDNA doses and ratios as used in the screen. In addition to the N-RDGE polymer, an efficient in-house lipid-conjugated polymer (lipopolymer), paromomycin glycerol diglycidyl ether-C18 (PG-C18) [27] was used as an additional carrier in order to further investigate the observed phenomena with a second non-viral delivery vehicle. Inhibitors chosen for dose-optimization were purchased individually from Cayman Chemical in solid form and dissolved in DMSO to 500x working concentration for each dose to be tested. Doses were chosen to test at and above (where possible) and below the optimal of three doses tested in the screen, based on screening results. The luciferase assay was employed in a similar manner as the screen. Although cell viability normalization was incorporated in luciferase expression calculations for the screening experiments to ensure lead inhibitors were not missed due to viability losses, it was not included in luciferase expression results for dose-optimization experiments; these results are expressed as total luminescence, normalized to the no inhibitor (vehicle) control.
Nuclear Localization of Plasmid DNA
The N-RDGE polymer was used to deliver 200 ng pGL3 plasmid per well in a 24-well plate in the absence and presence of the indicated small molecule inhibitors. Using a protocol and primer pairs established in our previous study [17], cell nuclei were isolated and assayed using qPCR for relative presence of the pGL3 plasmid. Briefly, nuclei of cells transfected with pGL3 plasmid were isolated using the EZ Nucleosomal DNA Prep kit (Zymo #D5220) and DNA (plasmid and genomic together) were purified using the spin columns provided by the manufacturer. Using this DNA pool as the template, qPCR with four primer pairs was carried out to detect levels of pGL3 plasmid present in target cell nuclei. Primer pairs amplifying multiple regions on the plasmid were used as a safeguard; primers were designed to detect plasmid SV40 promoter (primer pair 1), the luciferase gene (primer pairs 3 and 4), or a region bridging the promoter and gene (primer pair 2).
Native Chromatin Immunoprecipitation (N-ChIP) Analysis
Cells (250,000 / well) were plated in 6-well plates, with two full plates (i.e. pool from 12 wells) used for every condition investigated. On the following day, cells were transfected with the pEF-Luc plasmid (800 ng/well) complexed with N-RDGE polymer at a 25:1 (wt/wt) ratio. At ~48 hours, cells were washed and trypsinized for detachment. The supernatant, which contained transfection media and the PBS used in the wash step, was saved to in order to ensure that all detached cells were collected for ChIP analysis. Following the nuclear extraction and micrococcal nuclease (MNase) steps outlined by the manufacturer protocol for the EZ Nucleosomal DNA Prep kit, cell nuclei were isolated and treated with MNase to yield nucleosomal particles ready for immunoprecipitation.
Chromatin preps were incubated with 5 mg/mL BSA+PBS (Sigma) washed (3x) Magna ChIP Protein A + G beads (Millipore) for three hours at 4°C with nutation to clear non-specifically interacting particles. Pre-cleared chromatin was moved to a new tube without beads. Fifty μL of each sample were set aside as input, a theoretical maximum amount of target DNA prior to pulldown, and kept at −20°C. Five μL of each antibody, anti-histone H3 (Cell Signaling #4620), anti-acetyl histone H3 K9 (Cell Signaling #9649), or rabbit IgG (Cell Signaling #2729), were added to each tube and samples were incubated for 12 hours at 4°C with nutation. Antibody-chromatin samples were incubated with 20μl of PBS+BSA washed (3x) MagnaChIP beads for three hours at 4°C with nutation. Bead-antibody-chromatin complexes were washed once with Buffer A [50 mM Tris-HCl pH 8.1 (Sigma), ethylenediaminetetracacetic acid (EDTA) (Fisher Scientific), 50 mM sodium chloride (NaCl) (Sigma)], once with Buffer B [50 mM Tris-HCl pH 8.1 (Sigma), ethylenediaminetetracacetic acid (EDTA) (Fisher Scientific), 100 mM sodium chloride (NaCl) (Sigma)] and once with Buffer C [50 mM Tris-HCl pH 8.1 (Sigma), ethylenediaminetetracacetic acid (EDTA) (Fisher Scientific), 150 mM sodium chloride (NaCl) (Sigma)]. Each wash step was followed by 10 min nutating incubations at room temperature. Bead complexes were resuspended in 100 μL of elution buffer [1% SDS, 0.1 M sodium bicarbonate (Sigma), 0.1 M NaCl]. Fifty μL of elution buffer were added to each input sample as well. IPs and inputs were incubated at room temperature for 30 min with nutation. Samples were incubated with 10 μg of RNase A (Sigma) for 30 min at 37 °C and then with 10 μg of Proteinase K (QIAGEN) for 2 h at 62 °C. DNA from IPs and inputs was purified using Genelute PCR Cleanup Kit (Sigma) and eluted in 50 μL of nuclease-free water.
Real-Time Quantitative PCR to measure ChIP-DNA
Real-time quantitative PCR (qPCR) was conducted in 15 μL reactions using the SYBR Green detection system (Roche) in a 96-well format. Each reaction contained 7.5 μL of SYBR Green Master Mix, 2 μL of ChIP DNA, and 562.5 pmol of forward and reverse primers (each). As input samples are 1/20th (50 μL / 1000 μL) of each sample, Cp values were adjusted by subtracting log2(20) from each. Percent of input DNA [28] bound was calculated as 100 × 2 [Ct input−Ct IP], where Ct IP is the Ct value for each IP sample and Ct input represents the theoretical maximum DNA bound.
Primers were designed to detect a number of positions along the pEF-Luc plasmid; these primers and their sequences are (F: forward; R: reverse):
EF1 Prom (F: AGGAGTGGGAATTGGCTC, R: CCTTCTCTAGGCACGGTTC);
EF1 Prom-2 (F: CGTGATTCTTGATCCCGAGC, R: CTCAAGCACGAGGCGAAG);
EF1 Prom-3 (F: CTGAGTGGGTGGAGACTG, R: GTCTGAGGCTTGAGAATGAACC);
Early Luc+ (F: GCAGATCATGGAAGACGCC, R: CCACCTCGATATGTGCATCTG);
Luc+ (F: gcttttacagatgcacatatcgaggtgg,
R: GTATTCAGCCCATATCGTTTCATAGCTTC);
pBR322 Ori (F: GCCACCTCTGACTTGAGC, R: CAGCAAAAGGCCAGGAACC).
The regions of the pEF-Luc plasmid amplified by these primer pairs include the promoter (EF1 Prom 1–3), a region at the 5’ end of the luciferase gene + a small proximal upstream region (Early Luc+), a portion solely present on the luciferase gene (Luc+), and the origin of bacterial plasmid replication (pBR322 Ori).
RESULTS
Small Molecule Inhibitors of Chromatin-Modifying Enzymes Enhance Polymer-Mediated Transgene Expression
Each chromatin modulator inhibitor was tested at three different concentrations, as described in the Experimental section. Relative luciferase expression results are displayed qualitatively in the heat map shown in Figure 1A, and the vehicle-control (DMSO) normalized transgene expression at the optimal dose for each inhibitor is shown in Figure 1B. The specific doses employed for each inhibitor can be found in Table S1 (Supporting Information section). Cell viability corresponding to the trials in figures 1 and 2 are shown in Figures S1 and S2 (Supporting Information section) for CHO-K1 and UMUC3 cells, respectively.
Figure 1.
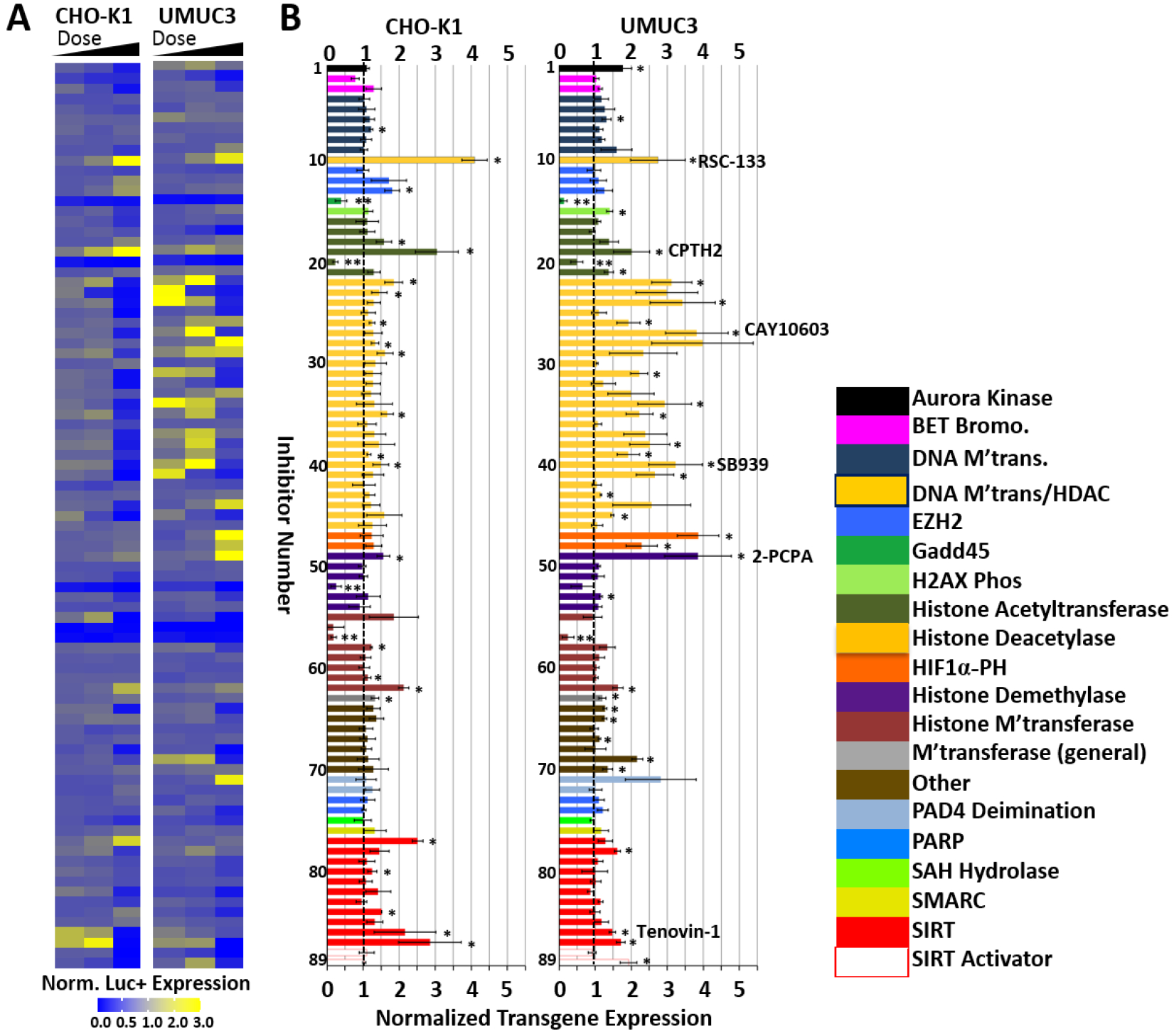
Small-molecule inhibitors of epigenetics modulators were screened to identify leads that enhanced polymer-mediated transgene expression in CHO-K1 cells and UMUC3 bladder cancer cells. A) Heat map indicating the normalized luciferase expression ranges for each of the three doses examined for each inhibitor in the screen. B) Normalized transgene expression for optimal inhibitor dose; color-coding indicates modulator family targeted by the indicated inhibitor. Vertical dashed line indicates point of no enhancement of transgene expression. Inhibitor numbers (1–89) can be referenced in Table S1, supporting information section, for each individual small molecule. Lead inhibitors discussed in more depth are listed. Polyplexes were formed by complexing the N-RDGE polymer with 75 ng EF-Luc plasmid DNA at a 25:1 (wt/wt) ratio. * = Student’s t-test p<0.05 for enhancement in transgene expression relative to DMSO vehicle (no inhibitor) + polyplex control; ** = Student’s T-test p<0.05 for reduction in transgene expression relative to DMSO vehicle (no inhibitor) + polyplex control. Chaetocin (inhibitor #56) was not considered a statistically significant reducer of transgene expression due to its high toxicity at the doses tested.
Figure 2.
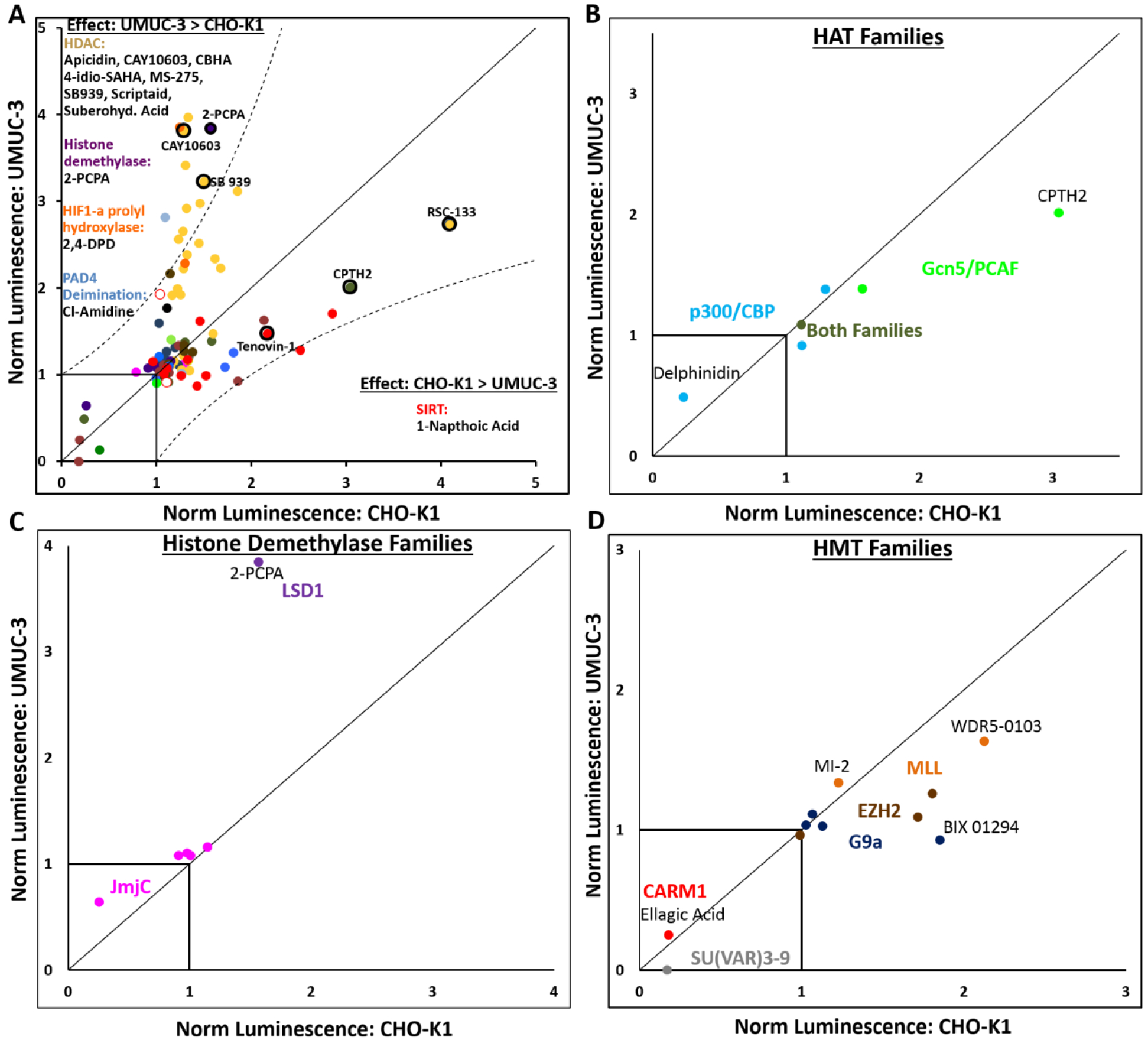
Screening results depicted on a dot plot showing normalized luciferase expression in UMUC3 cells (y-axis) and CHO-K1 cells (x-axis). A) Inhibitors falling near the y=x line have similar effect on luciferase expression in both cell lines. Those that fall outside of the y=2x and x=2y lines have 2x greater effect in UMUC3 cells than CHO-K1 cells or vice versa, respectively. Such inhibitors are listed under their family type in these regions. All inhibitors are color-coded by modulator family defined in figure 1. Data points corresponding to lead inhibitors discussed later are circled in black. B) Histone acetyltransferase inhibitors, C) histone demethylase inhibitors, and D) histone methyltransferase inhibitors depicted by their sub-families.
The screen revealed that inhibition of several epigenetics modulators resulted in enhancement of polymer-mediated transgene expression in both CHO-K1 and UMUC3 cells. In the following sections, we discuss universal and cell-line-specific effects for each family of inhibitors that affected transgene expression.
Inhibitors of Histone Deacetylase (HDACs, Classes I and II) Enhance Transgene Expression
Our screen with both, CHO-K1 and UMUC3 cells, identified several Class I/II HDAC inhibitors as enhancers of transient transgene expression (Figures 1 and 2A, yellow bars and points). As demonstrated in Figure 2A, HDAC inhibition induced more potent transgene expression enhancement in UMUC3 cells than in CHO-K1 cells (p=6.7×10−5) on average; eight HDAC inhibitors enhanced transgene expression to at least twice the extent in UMUC3 cells observed in CHO-K1 cells. The variation observed between the two cell lines could be related to differences in HDAC expression levels or alternative levels of dependence on HDAC for regulation of chromatin. The literature indicates isoform-dependence in expression levels of class I/II HDAC levels in urothelial cancer cells versus normal urothelial cell lines; HDACs 2 [29, 30], 4 [30], 5 [30], and 8 [30, 31] are transcriptionally upregulated, while HDACs 6 [30] and 9 [30] are transcriptionally downregulated; HDAC1 has been observed to be both up- and down-regulated [29, 30], and HDAC3 transcriptional differences between normal and cancer urothelial cells are not detectable [29]. To our knowledge, HDAC mRNA or protein levels are not readily available for CHO-K1 cells. Different epigenetic modulators have complementary and often synergistic effects on transcriptional regulation, making some modulators more dispensable than others. This will be discussed later in the context of combinatorial inhibitor treatments.
Entinostat (MS-275, inhibitor #34), trichostatin A (inhibitor #45), sodium butyrate (inhibitor #43), and valproic acid (inhibitor #46) are known enhancers of transgene expression. Entinostat and trichostatin A both improved transgene expression in UMUC3 cells, supporting the validity of the screen; we have previously demonstrated that Entinostat and tichostatin A enhance transgene expression in human prostate and murine bladder cancer cells [17, 18]. Valproic acid, which did not affect transgene expression in our hands, was tested well below its IC50 value due to constraints introduced by the manufacturer-provided stock concentration in the screening library.
In addition to inhibitors with known effects on transgene expression, the screen found several new class I/II HDAC inhibitors that enhance transient transgene expression. SB939 is a pan-HDAC inhibitor, inhibiting all HDAC enzymes with the exception of HDACs 6 and 7 [32]. This inhibitor was chosen for further mechanistic evaluation to represent the class I/II HDAC family, because it enhanced transgene expression in both cell lines (UMUC3 > CHO-K1) and represents the general HDAC inhibitor data from the screen well. A particularly interesting inhibitor from this group is CAY10603 (inhibitor #27), which potently and specifically inhibits HDAC6, a primarily cytoplasmic HDAC inhibitor that is known to deacetylate tubulin, affecting intracellular (cytoplasmic) trafficking. HDAC6 inhibitors have previously been found to enhance transgene expression by modifying intracellular trafficking [18, 33]. This inhibitor was also selected for further investigation. RSC-133 was chosen for further evaluation as a key lead inhibitor from the screen based on the magnitude and consistency of enhancement between the cell lines. RSC-133 has been demonstrated to inhibit both HDAC1 and DNA methyltransferase 1 (DNMT1) [34]. The magnitude of enhancement in CHO-K1 cells with RSC-133, which differs drastically from that observed by HDAC inhibition alone (Figure 1), suggests there may be a synergistic effect between DNMT and HDAC functioning that influences transgene expression from plasmids.
Inhibitors of Sirtuins (HDACs, Class III) Enhance Transgene Expression
In addition to the classical HDAC classes I, II, and IV, class III HDACs are a group of NAD+ dependent protein functionalizing enzymes known as sirtuins. All seven sirtuins (SIRT1–7) share an NAD-binding domain which facilitates their deacetylation and/or ADP ribosylation enzymatic activity; all but SIRT4 have deacetylation capability [35]. Sirtuins were identified as a potential target for transgene expression enhancement in both cell lines (Figure 1 and 2A, red bars and points). Sirtuin inhibitors are more promising for enhancing transient luciferase expression in CHO-K1 cells than in UMUC3 cells, as indicated by the majority of the sirtuin (red points) falling below the y=x line in Figure 2. Tenovin 1 and its analog Tenovin 6, identified as enhancers of transgene expression in both cell lines, are inhibitors of sirtuins SIRT1 and SIRT2. Also identified as enhancers of transgene expression in CHO-K1 cells were sirtinol, a SIRT1/2 inhibitor [36] and its derivative naphthoic acid [37], which increased transgene expression in CHO-K1 cells by double the extent of that seen in UMUC3 cells (Figure 2A). In addition, JGB1741, an inhibitor of SIRT1 and weak inhibitor of SIRT2 led to moderate transgene expression enhancement. Interestingly, piceatannol, which has been shown to activate SIRT1 expression in human monocytes [38], enhanced transgene expression in UMUC3, but not in CHO-K1 cells. Tenovin-1 was chosen for further evaluation as a promising lead from this family of inhibitors.
The effect of Histone Acetyltransferase (HAT) Inhibitors on Transgene Expression is Sub-family Dependent
HATs catalyze the addition of acetyl groups onto ε-amino groups of lysine residues present on core histones, carrying out the opposite activity of HDACs, with which they work to regulate gene transcription. Addition of these acetyl groups at nucleosomes leads to a general increase in chromatin accessibility [39], allowing interaction with necessary transcriptional machinery. While inhibition of HATs is hypothesized to decrease transgene expression, the screening revealed mixed results that point to potential dependence of the HAT sub-family involved (Figure 2B).
The screen identified CPTH2 hydrochloride as a strong enhancer of transgene expression in both cell lines (Figures 1 and 2). CPTH2 hydrochloride specifically inhibits the HAT enzyme Gcn5, which has known acetyltransferase activity at Histone H3K9 [40] and H3K14[41]. A similar protein with HAT activity, PCAF, was represented in the screening by the inhibitor CAY10669 which moderately enhanced transgene expression in CHO-K1 cells. In addition to inhibitors of the Gcn5/PCAF family of HATs, inhibitors of the CBP/p300 family of HATs were included in the screen. Anacardic acid inhibits both PCAF and p300 [42], while C646 has only been demonstrated to inhibit p300/CBP [43]; neither, however, enhanced transgene expression in either cell line. Although the inhibitor of CBP, I-CBP 112 hydrochloride, very moderately enhanced transgene expression in UMUC3 cells, delphinidin (chloride), a CBP/p300 HAT inhibitor, significantly decreased transgene expression in both cell lines, one of only three inhibitors in the library to do so. CPTH2, the lead inhibitor from the HAT family, was chosen for further mechanistic study. Taken together, inhibitors of the Gcn5/PCAF family of HAT enzymes have potential to enhance transgene expression. However, inhibitors of CBP/p300 HAT enzymes, based on the screen, were not able to enhance transgene expression.
Gcn5 HATs exhibit acetyltransferase activity on transcription and have also been shown to influence cell cycle. Specifically, cells lacking Gcn5 have demonstrated vulnerability to DNA-damaging agents and a tendency to arrest at the G2/M cell cycle transition [44], which is a particularly favorable cell cycle phase for efficient transient gene expression [45]. Additionally, Gcn5 is implicated to play a positive role in histone deposition on newly synthesized DNA following DNA replication [44]. It is possible when inhibiting Gcn5 HAT that 1) cells accumulate at the G2/M transition and / or 2) delivered plasmids are poorly bound by histones. Both of these factors can increase transgene expression in cells. Further systematic investigation with more inhibitors from each HAT sub-family would be required to elucidate the mechanism governing HAT-family dependence of transient expression efficacy.
Inhibition of the Histone Demethylase, Lysine Specific Demethylase 1 (LSD1), Enhances Transgene Expression
The removal (current section) and addition (next section) of methyl groups at a multitude of histone sites plays a vital role in regulating transcription. Lysine specific demethylase 1 (LSD1) specifically removes methyl groups from lysines H3K4 and in some cases, H3K9. Although only one inhibitor of LSD1, 2-PCPA hydrochloride, was available in the inhibitor library, it enhanced transgene expression in both cell lines (Figures 1 and 2C) and was chosen for subsequent mechanistic evaluation. In order to validate LSD1 as a target for enhancing transgene expression, a second small molecule inhibitor, OG-L002, was purchased separately and evaluated; the results, which validated LSD1’s hit in the screen, are discussed in a subsequent section. Demethylation at H3K4 is a repression mark of the histone code, so LSD1 inhibition is hypothesized to increase expression, as we have observed. LSD1, additionally, participates as part of the CoREST complex with HDAC1/2 [46], linking demethylation and deacetylation. Also, LSD1 can catalyze demethylation of H3K9, which can have either a repressive (H3K9me → H3K9) or enhancing (H3K9me2 → H3K9me) effect on transcription. Both of these lysine sites can also be trimethylated, but LSD1 is not capable of demethylating H3K4me3 or H3K9me3. Interestingly, IOX-1 (inhibitor #54) and N-Oxalylglycine (inhibitor #53), which both inhibit jumonji domain histone demethylases (JmjC) (Figure 2C), responsible for demethylating H3K9me3, demonstrated little promise in enhancing transgene expression in the screen. This indicates that the extent of methylation at one or both of these sites may play a role in the ability to enhance transgene expression.
Inhibitors of Mixed Lineage Leukemia (MLL) Methyltransferase Related Proteins and Enhancer of Zeste Homolog 2 (EZH2) Trimethyltransferase Increase Transgene Expression
Methylation of core histone tails by histone methyltransferase (HMT) enzymes plays a key role in the regulation of endogenous gene expression. WDR5 is a WD-repeat family protein that is a non-catalytic subunit of trithorax (TRX) methyltransferase complexes, including the mixed myeloid leukemia (MLL) complex, catalyzing H3K4 methylation [47]. The small-molecule inhibitor WDR5–0103 (inhibitor #62), which has been demonstrated to bind WDR5 and inhibit MLL methyltransferase activity in vitro [48], enhanced transgene expression in both cells investigated (Figures 1 and 2D). Another target that has been shown to bind with the MLL complex, menin, is inhibited by MI-2 (inhibitor #58); MI-2 demonstrated moderate enhancement in transgene expression (Figures 1 and 2D). MI-2 has been found to reduce H3K4me3 and H3K79me2 in MLL leukemia cells [49]. Interestingly, H3K4 methylation is a transcriptional activation mark, so the observation of WDR5- and menin- inhibition-induced transgene expression enhancement is contrary to the known epigenetic function of this histone mark. This suggests a possible off-target effect of MLL complexes or these particular components.
Enhancer of Zeste Homolog 2 (EZH2) is a histone trimethyltransferase that functions as the catalytic unit of the polycomb repressive complex 2 (PRC2). Within the context of PRC2, EZH2 facilitates trimethylation of histone H3, lysine 27 (H3K27me3), generally resulting in transcriptional repression [50]. The EZH2 inhibitors GSK343 (inhibitor 12) and UNC1999 (inhibitor 13) displayed moderate efficacies as enhancers of transgene expression in CHO-K1 cells (Figure 1), but they did not demonstrate this effect in UMUC3 cells (Figure 2D).
Finally, the enzyme coactivator-associated arginine methyltransferase 1 (CARM1) facilitates methylation at an arginine site (H3R17). Additionally, CARM1 methylates the p300/CBP HAT, contributing to gene activation activity of this HAT [51]. CARM1 was represented by a single inhibitor in the screen, Ellagic acid, which resulted in a drastic decline in transgene expression (Figures 1 and 2D); this behavior is consistent with the known gene activation activity of CARM1.
Other Lead Inhibitors
In addition to the aforementioned modulator families, the screen identified several interesting targets that will not be discussed in detail here. Some of these include aurora kinase, growth arrest and DNA-damage-inducible protein (Gadd45), hypoxia-inducible factor 1-alpha prolyl hydroxylase (HIF-PH), and peptidyl arginine deiminase (PAD).
The enzyme target identification covered in the previous sections highlights some key similarities between endogenous and transgene expression, as well as cases where plasmid and genomic (chromosomal) gene expression may be regulated very differently (Table 1). As summarized in the table, the screening results indicate that the activity observed for epigenetic modulators with plasmid-based expression is largely consistent with their known activity with genomic expression. The exceptions include inhibitors known to interact with and increase activity of MLL methyltransferase complexes as well Gcn5/PCAF HATs, and are shown in red in Table 1.
Table 1.
Comparison of the known effect of chromatin modifying enzyme targets on genomic gene expression and transgene expression as observed in the screen.
| Target Modulator | Enzyme Effect on Endogenous Chromatin Expression Activity | Inhibitor Effect on Transient Luciferase (Transgene) Expression |
|---|---|---|
| EZH2 | Repression[52] | Activation |
| Gadd45 | Activation [53] | Repression |
| HAT (CBP/p300) | Activation [54] | Repression |
| HDAC/Sirtuin | Repression [55] | Activation |
| LSD1 | Repression [56] | Activation |
| HAT (Gcn5/PCAF) | Activation [57] | Activation |
| WDR5/Menin-MLL* | Activation* [58] | Activation |
WDR5 and Menin are non-catalytic components of H3K4 methyltransferase complex
Dose Response Studies Optimize the Efficacy of Lead Inhibitors and Demonstrate Application with an Additional Plasmid and Delivery Vehicle
The small-molecule screen identified several inhibitors of epigenetic modulators that enhanced transgene expression in UMUC3 bladder cancer cells and/or CHO-K1 cells. However, local chromatin structure and transcriptional activity are often dependent on the promoter and the gene itself. Consequently, a subset of the lead inhibitors identified in the screen were investigated further for their abilities to enhance transgene expression using different plasmids and delivery vehicles in conjunction with inhibitor dose-optimization. In particular, the pGL3 plasmid, which is a luciferase-expressing plasmid driven by a different (SV40) promoter, was used as a second plasmid in addition to the pEF-Luc plasmid employed in the screen. Similarly, the efficacy of lead inhibitors was investigated using a second delivery vehicle not included in the screen: a lipid-conjugated polymer, paromomycin glycerol diglycidyl ether–C18, or PG-C18, which was previously developed in our laboratory [27]. Figures 3 and 4 show transgene expression using lead inhibitors and the additional plasmid and/or delivery vehicle in CHO-K1 and UMUC3 cells, respectively.
Figure 3.
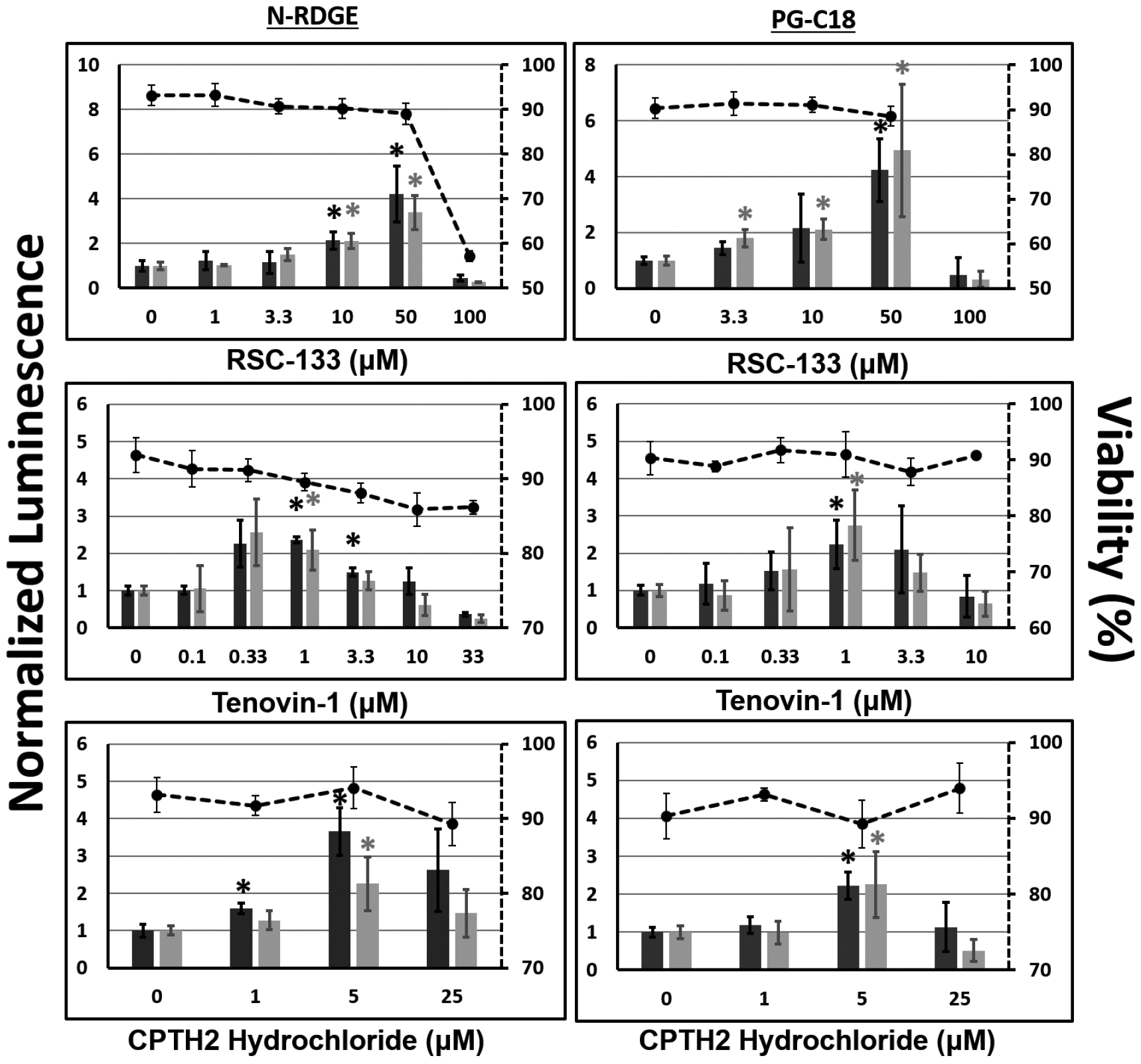
Dose optimization of lead inhibitors identified from the screen for enhancing transgene expression in CHO-K1 cells. A second plasmid, pGL3, which expresses luciferase protein under the control of the SV40 promoter was evaluated; the original screening plasmid (pEF-Luc) was also included. In addition, two polymers N-RDGE (top) and PG-C18 (bottom), were evaluated for delivering plasmid DNA. Transgene expression efficacies are reported normalized to the vehicle control (0.2% DMSO + polyplex and indicated by “0” on the x-axis, normalized to 1 on the y-axis). Cell viability, as measured using the MTT assay, are plotted on the secondary axes (dashed lines). * indicates p-value < 0.05, as determined using Student’s t-test, each condition relative to vehicle control + polyplex formed with same polymer/plasmid combination.
Figure 4.
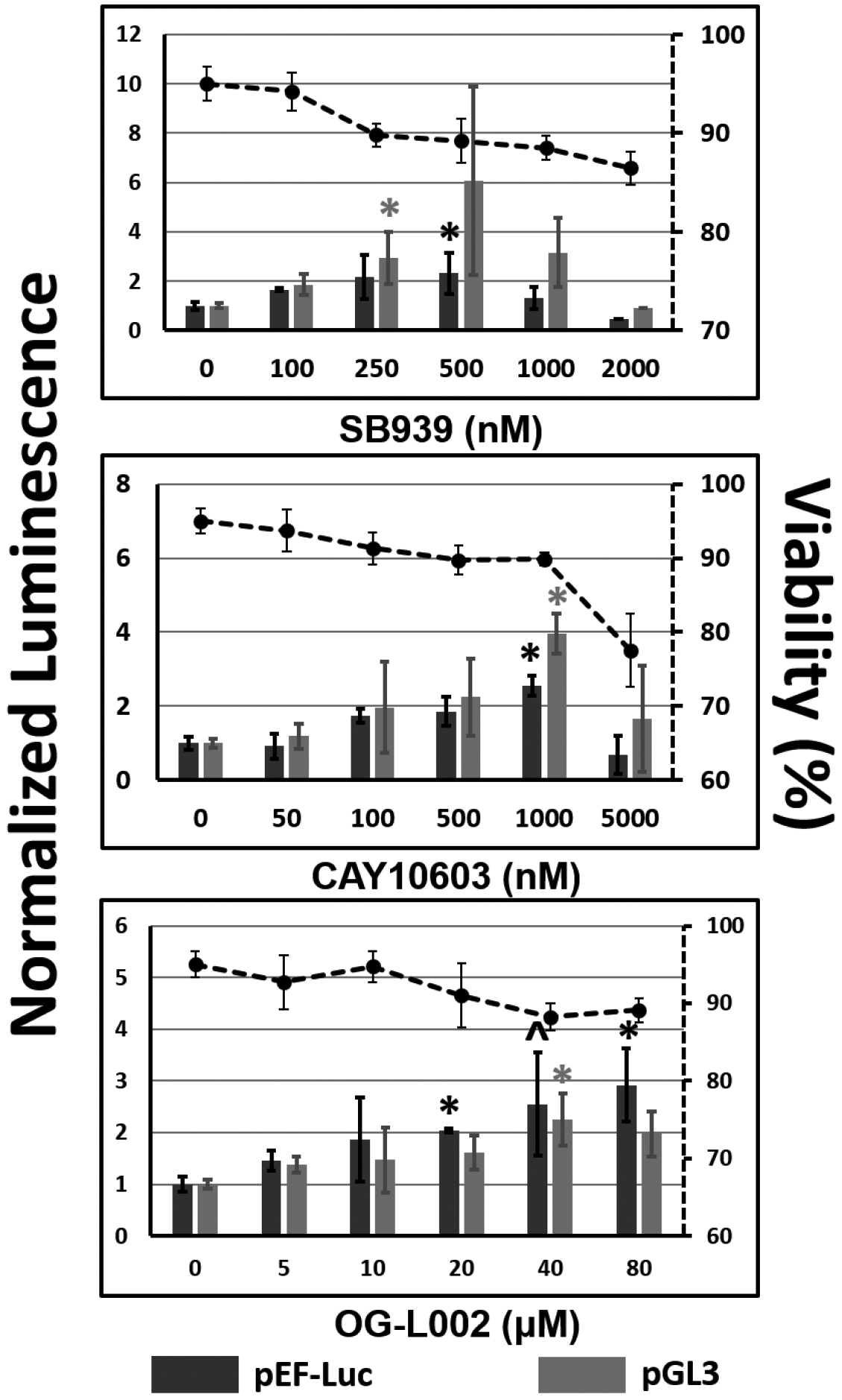
Dose optimization of lead inhibitors identified from the screen for enhancing transgene expression in UMUC3 cells. A second plasmid, pGL3, which expresses luciferase protein under the control of the SV40 promoter was evaluated; the original screening plasmid (pEF-Luc) was also included. N-RDGE was used for delivering plasmid DNA. Transgene expression efficacies are reported normalized to the vehicle control (0.2% DMSO + polyplex and indicated by “0” on x-axis, normalized to 1 on the y-axis). Cell viability, as measured using the MTT assay, is plotted on the secondary axes (dashed lines). * indicates p-value < 0.05, as determined using Student’s T-test, each condition relative to vehicle control + polyplex formed with same plasmid. ^ p=0.05
Table 2 compares our optimal inhibitor doses with published IC50 (or effective) doses. In CHO-K1 cells, RSC-133, a DNA Methyltransferase (DNMT)/HDAC inhibitor has been shown to effectively inhibit these targets at 10 μM, comparable to our findings for enhancing transgene expression. Tenovin-6 is a water-soluble analog of Tenovin-1, and has IC50 values in the tens of micromolar range, an order of magnitude above our optimal concentration for Tenovin-1 (for which published IC50 values were not available). CPTH2, a Gcn5 HAT inhibitor, has been demonstrated to effectively inhibit this target at concentrations of 50 and 800 μM, the former which is reasonably in agreement with our optimal 5–25 μM optimal range for enhancing transgene expression. For all three of these inhibitors, doses corresponding to greatest enhancement in luciferase expression are non-toxic, with cell viabilities near 90%. (Figure 3). Data for two additional inhibitors (WDR5–0103 and 2-PCPA) are shown in Figure S3 (Supporting Information section).
Table 2.
Comparison of published IC50 or effective doses with optimized concentrations determined from dose optimization studies for select inhibitors.
| Inhibitor | Optimal Concentration | Published IC50 / Effective Concentration (Target) [Reference] |
|---|---|---|
| RSC-133 | 50 μM | Effective dose: 10 μM (HDAC/DNMT) [34] |
| Tenovin-1 (Ref. values based on Tenovin-6) | 1 μM | 21, 10, 67 μM (SIRT1/2/3) [59] |
| CPTH2 | 5 μM | Effective dose of 50 [41, 60], 800 μM [60] (Gcn5) |
| SB939 | 0.25–0.5 μM | 77 nM (HDAC1) [32] |
| CAY10603 | 1 μM | 2 pM (HDAC6) [61] |
| OG-L002 | 20–80 μM | 0.2 μM (LSD1) [62] |
The optimal dose of pan-HDAC inhibitor SB939 is approximately two orders of magnitude above the published IC50 value of 77 nM in UMUC3 cells, while the cytoplasmic HDAC inhibitor was most effective in our hands many orders of magnitude above the published 2 pM IC50 value. OG-L002 (Figure 4) is an LSD1 inhibitor that was not part of the screening library. It was evaluated in these optimization studies to investigate the effect of LSD1 inhibition, because this target had only one inhibitor (2-PCPA) in the screen. The most effective concentrations for enhancing transient luciferase expression ranged from 20–80 μM, which was about two orders of magnitude above the published IC50 value. As in CHO-K1 cells, the optimal inhibitor doses for transgene expression enhancement were not highly inhibitory to UMUC-3 viability.
Although these experiments served the purpose of finding the optimal doses of the lead inhibitors for further mechanistic experimental evaluation (additional data in Figure S2, Supporting Information section), it is evident that the enhancement in transgene expression is not wholly dependent on the promoter (SV40 or EF1α) or the delivery vehicle used (N-RDGE or PG-C18 polymer).
Inhibitor Combinations Further Enhance Transgene Expression following Delivery of Plasmid DNA
Inhibition of individual chromatin modulating enzymes has clearly been demonstrated to enhance non-viral transgene expression in the sections above. We reasoned that the simultaneous inhibition of more than one epigenetics enzymes will likely result in (1) increased enhancement in transgene expression and, (2) information on mechanisms and mediators that may be independent, redundant, or synergistic. We therefore delivered plasmid DNA to CHO-K1 cells in presence of different inhibitor combinations (Figure 5). There is no detectable enhancement in transgene expression with HDAC inhibitor (SB939) treatment at the doses tested. However, the enhancement of transgene expression observed with single-agent inhibition of LSD1 using 500 μM 2-PCPA hydrochloride is amplified by inhibition of HDAC with SB939 (Figure 5, left). This indicates that inhibition of HDAC and LSD1 synergistically enhances transgene expression. Additionally, inhibition of LSD1 with 2-PCPA hydrochloride (500 μM) and inhibition of EZH2 with UNC1999 (1 μM) led to at least slightly synergistic enhancement in transgene expression in CHO-K1 cells; this dose of UNC1999 was unable to enhance transgene expression individually, but when combined with 2-PCPA, it improved the efficacy of this inhibitor. Modest synergy may be observed in the case of 500 μM 2-PCPA hydrochloride and 5 μM UNC1999; individual treatments enhance transgene expression by factors of 4.2-fold and 1.4-fold respectively, and the combination yields 7.7-fold enhancement (Figure 5, right).
Figure 5.
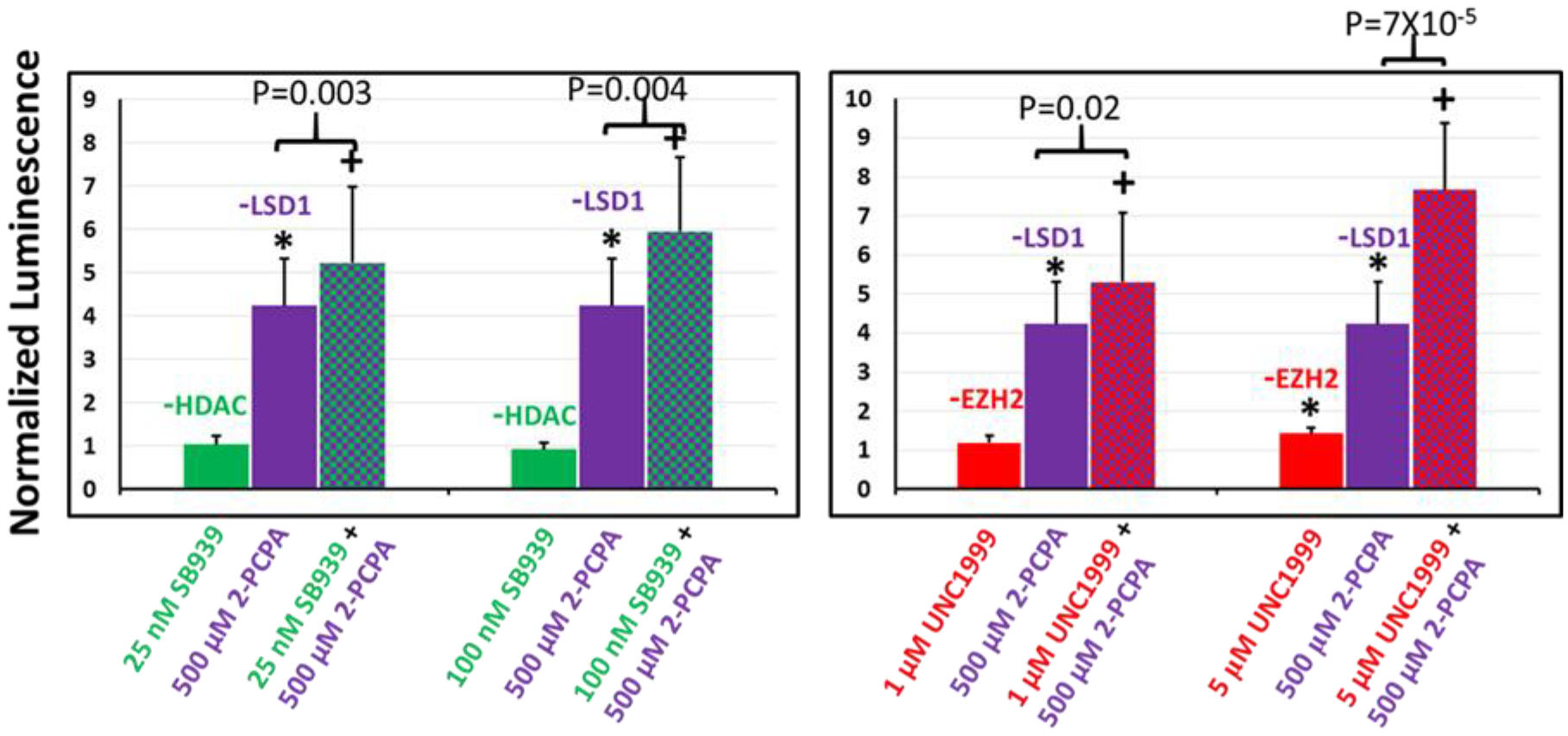
Relative transgene (luciferase) expression in CHO-K1 cells following single and dual combination treatments with epigenetics modulators targeting (left) HDAC and LSD1, and (right) EZH2 and LSD1; the N-RGDE polymer was used for delivering pDNA in these studies. Single-agent inhibitor treatments are indicated by solid bars and dual combination inhibitor treatments are indicated using checkered bars. Transgene expression in each case is normalized to that seen in presence of DMSO (carrier). * indicates p-value <0.05 relative to DMSO control treatment as determined using Student’s T-test; + indicates p-value < 0.05 relative to both individual inhibitor treatment transfections as determined using Student’s t-test. SB939, 2-PCPA hydrochloride, and UNC1999 are inhibitors of HDAC, LSD1, and EZH2 respectively. n=5 independent experiments.
The effect of inhibitor combinations was also evaluated in UMUC3 cells (Figure 6). HDAC inhibition and LSD1 inhibition, which both increase transgene expression individually, were modestly synergistic at low concentrations but additive at higher concentrations when used for simultaneously enhancing transgene expression. However, combinations involving HDAC/EZH2 and LSD1/EZH2 did not synergistically enhance transgene expression in these cells.
Figure 6.
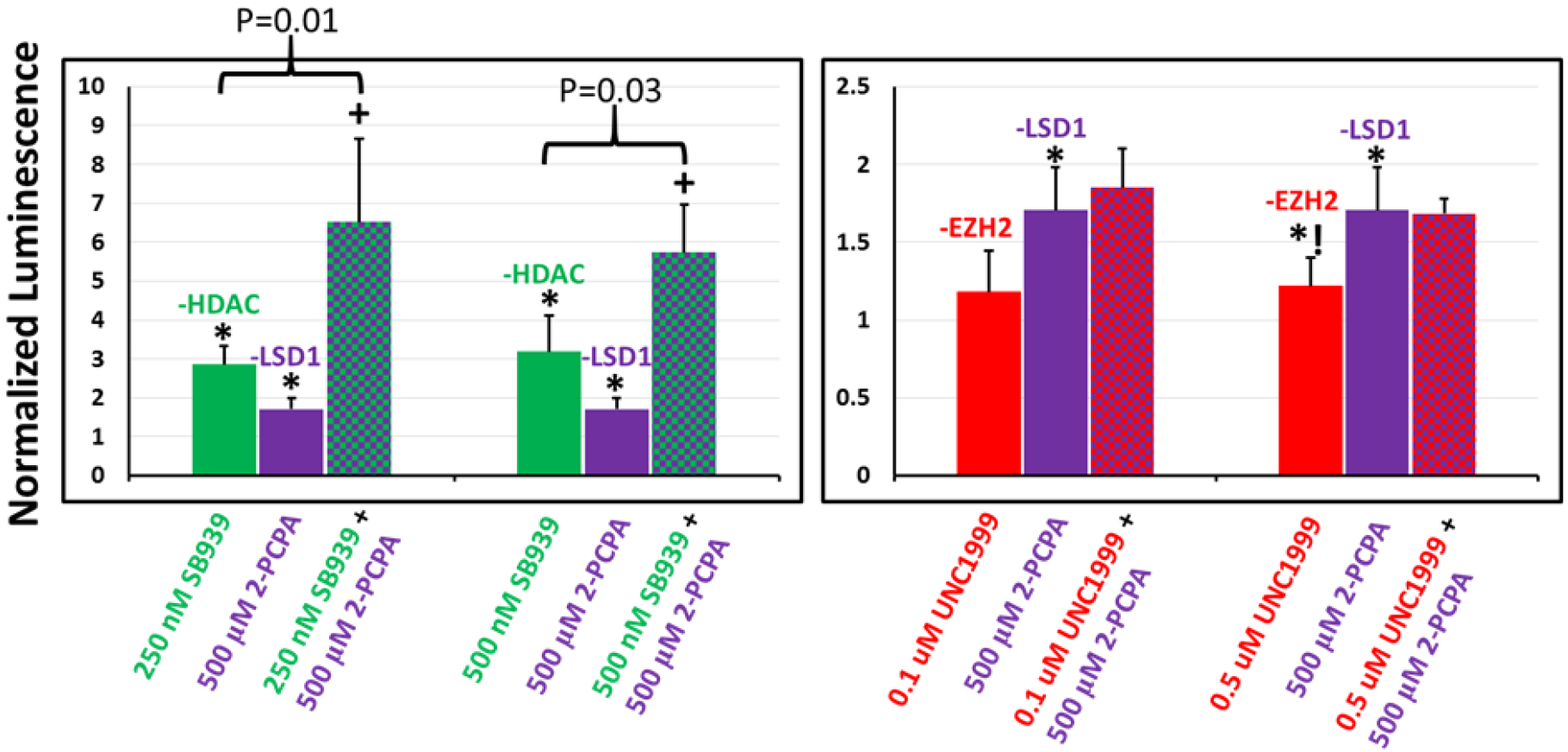
Relative transgene (luciferase) expression in UMUC3 cells following single and dual combination treatments with epigenetics modulators targeting (left) HDAC and LSD1, and (right) EZH2 and LSD1; the N-RGDE polymer was used for delivering pDNA in these studies. Single-agent inhibitor treatments are indicated by solid bars and dual combination inhibitor treatments are indicated using checkered bars. Transgene expression in each case is normalized to that seen in presence of DMSO (carrier). * indicates p-value <0.05 relative to DMSO control treatment and *! indicates p-value = 0.05 relative to DMSO control treatment as determined using Student’s T-test; + indicates p-value < 0.05 relative to both individual inhibitor treatment transfections as determined using Student’s t-test. SB939, 2-PCPA hydrochloride, and UNC1999 are inhibitors of HDAC, LSD1, and EZH2 respectively. n=5 independent experiments.
Cooperation between the aforementioned chromatin modulators in the silencing of transcription is not uncommon and, therefore, it is not surprising that synergistic effects on transgene expression are observed here. HDAC and LSD1 participate in the silencing CoREST complex, linking LSD1-mediated demethylation and HDAC-controlled deacetylation of histones. For example, LSD1/CoREST activity has been demonstrated to be inhibited by prior target histone hyperacetylation induced by HDAC inhibitor (Trichostatin A) treatment in HeLa cell isolated nucleosomes [46]. Two-way cooperation has even been observed with HDAC1 and LSD1, with inhibition of either enzyme resulting in decreased acetylation at H4K16 and methylation of H3K4, which are the respective targets of these enzymes [63]. This behavior was only observed in the pluripotent systems tested in this study, suggesting these interactions may not play the same role in vastly differing cells, such as UMUC3 and CHO-K1. LSD1 and EZH2, which showed synergistic potential in our system through dual inhibition, are also known to work together to silence chromatin as part of a long intergenic non-coding RNA (lincRNA) complex referred to as HOTAIR [64].
Inhibition of Histone Deacetylases Increases Nuclear Plasmid Levels in UMUC3 Cells
With the exception of the cytoplasmic activity of a subset of HDACs (e.g. HDAC6), most epigenetics enzymes are active primarily in the nucleus, and influence histone-DNA interactions. We therefore focused our attention to the status of the delivered plasmid in the nucleus, in order to carry out a mechanistic investigation into the enhancement of transgene expression using epigenetics modulators. Several lead inhibitors, at their optimal concentrations, were added to cells in concert with delivery of the pGL3 plasmid. Following transfection (48 h), nuclei were isolated and using primers described before [17] (Figure 7), qPCR was carried out to quantify relative amounts of plasmid inside the nuclei of transfected cells, denoted as “normalized nuclear plasmid levels”.
Figure 7.
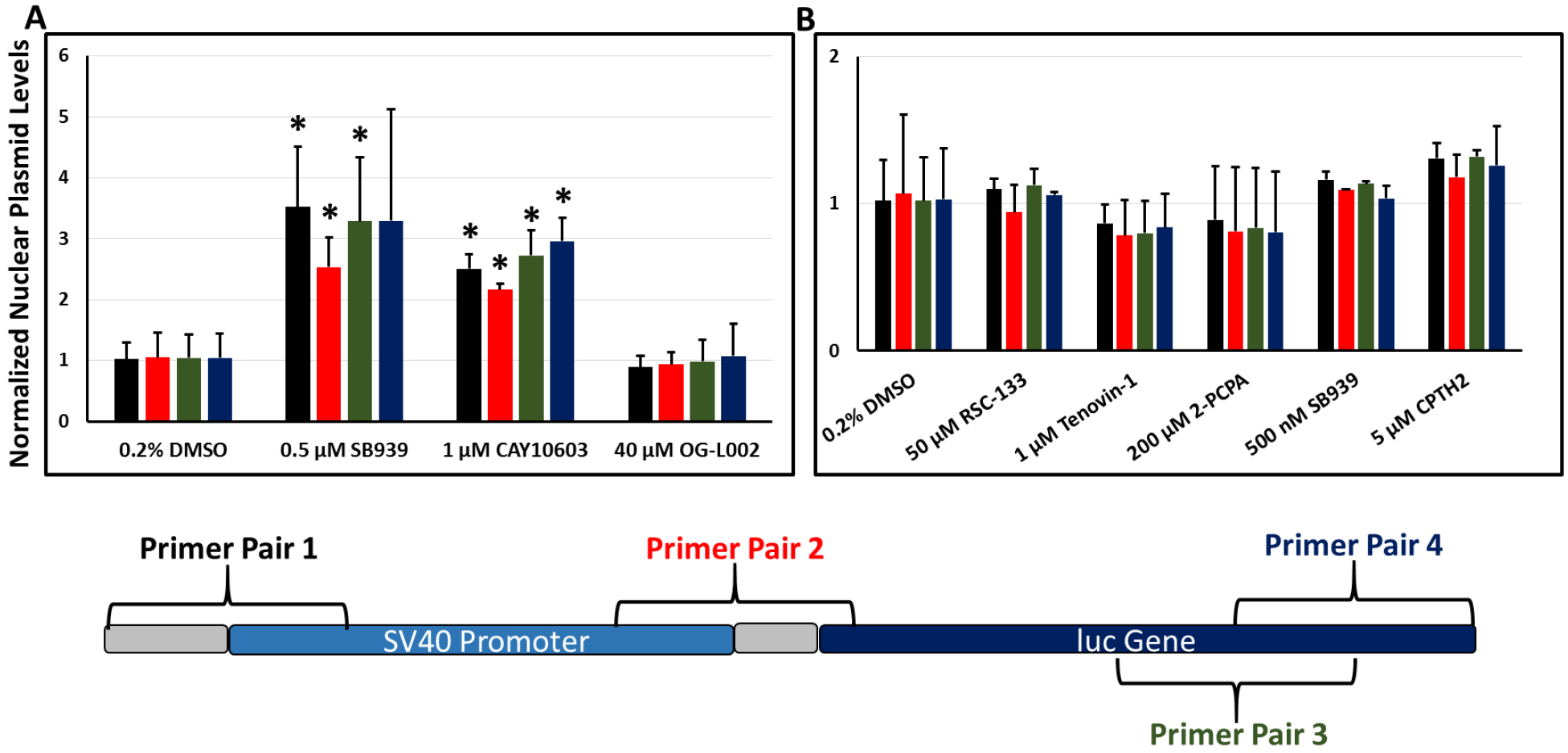
Localization of the pGL3 plasmid in the nuclear fraction of (A) UMUC3 cells (n=3 independent experiments) and (B) CHO-K1 cells (n=2 independent experiments) following delivery of the plasmid along with different epigenetics modulators. The assayed region of the pGL3 plasmid is illustrated with rough primer pair locations indicated. Bars are color-coded according to their respective primer pair label color on the plasmid map. *indicates p-value < 0.05 for plasmid presence in nuclear fraction relative to vehicle control condition. (Student’s t-test).
In UMUC3 cells (Figure 7A), the pan-HDAC inhibitor (SB939), cytoplasmic HDAC6 inhibitor (CAY10603), and LSD1 inhibitor (OG-L002) were evaluated. Although the LSD1 inhibitor did not affect plasmid presence in target nuclei, both HDAC inhibitors increased detectable intact plasmid inside nuclei, likely playing a role in the observed enhancement in transgene expression. We have previously reported increased plasmid uptake in two prostate cancer cell lines treated with the nuclear HDAC1/HDAC3 inhibitor Entinostat [17]. The increase in nuclear plasmid levels accompanying inhibition of HDAC6 is likely related to increased levels of acetylated tubulin, previously demonstrated to improve cargo transport toward the nucleus [18, 33]. Although an increase in plasmid levels in the nucleus of UMUC3 cells ostensibly explains a portion of the observed increase in transgene expression, nuclear histone modifications affecting histone/plasmid interactions also likely play a role. Unchanged levels of plasmid copy in the nucleus with the small molecule LSD1 inhibitor, OG-L002, suggests that increased transgene expression with LSD1 inhibition is governed by epigenetic mechanisms.
In CHO-K1 cells (Figure 7B), inhibitors of HDAC/DNMT (RSC-133), sirtuins (Tenovin-1), LSD1 (2-PCPA), pan-HDAC (SB939), and Gcn5 HAT (CPTH2), all of which were leads for enhancing transgene expression, did not enhance the presence of plasmid inside the nucleus. This suggests that in CHO-K1 cells, the mechanistic explanation for the observed enhancement of transgene expression lies at some point following nuclear entry, likely involving epigenetic mechanisms.
Native ChIP Detects Plasmid/Histone H3 Interactions in CHO-K1 Cells and Indicates Modulation by HDAC/LSD1 Inhibition
Histones bind chromosomal DNA in eukaryotic nuclei leading to the formation of chromatin and, in particular, organized nucleosomes. These interactions regulate DNA replication [65] and gene expression [66]. We hypothesized that, in our system, histones interact with exogenously delivered plasmid DNA in target nuclei, thus engendering similar epigenetic regulatory mechanisms by chromatin modifying enzymes on transient expression as observed with stable chromosomal DNA expression. While histone/plasmid chromatin formation in target cell nuclei has been reported before [20], in addition to demonstrating this in our system, we sought to understand the change in histone/plasmid interactions with lead inhibitors from our screen. We used chromatin immunoprecipitation (ChIP) in order to investigate physical interactions between the endogenously expressed core histone, Histone H3, and the pEF-Luc plasmid DNA in the nuclei of target CHO-K1 cells. Although cross-linking ChIP (X-ChIP) is commonly employed for studying protein-DNA interactions, excessive cross-linking can lead to the false-positive detection of protein-DNA interactions [67]. Additionally, formaldehyde treatment can reduce antibody recognition of the protein to be detected, thus reducing immunoprecipitation efficiency [67, 68]. We therefore employed the non cross-linking method, native ChIP (N-ChIP), in this study.
The first antibody used in the ChIP assay isolated histone H3, and molecules directly bound to this histone from the nuclei of transfected cells. Although other core histones H2A, H2B, and H4 play a vital role in epigenetic transcriptional regulation, particular histone H3 sites act as substrates for most of the lead epigenetic enzymes identified in the screen, as described in previous sections. Quantitative PCR (qPCR) was performed on these histone H3 pulldown contents using primers for six different regions on the pEF-Luc plasmid in order to determine if the pEF-Luc plasmid was directly interacting with histone H3 (Figure 8A). Cells were transfected in the absence of inhibitors (DMSO), in the presence of a single HDAC or LSD1 inhibitor, or with both inhibitors simultaneously. Depending on the primer pairs used for regional plasmid amplification, the plasmid-histone H3 signal produced was around 30% input in the nuclei of cells transfected with no inhibitors; in other words, 30% of the available plasmid DNA sequences present in target nuclei were isolated with immunoprecipitated Histone H3, clearly indicating specific interactions between histone H3 and pEF-Luc plasmid DNA. Interestingly, these interactions appeared to decrease when transfected cells were treated with an HDAC or LSD1 inhibitor, suggesting that HDAC and LSD1 inhibition may “loosen” histone-plasmid complexes. An additional pulldown using a general IgG antibody was used as a negative control in order to determine if the detected qPCR signal was specifically due to histone H3-plasmid interaction. Pulldowns with the IgG antibody yielded values of 0.2% input or less depending on the plasmid region, which confirmed the validity of this assay. These values can be seen in Figure S4 (Supporting Information section).
Figure 8.
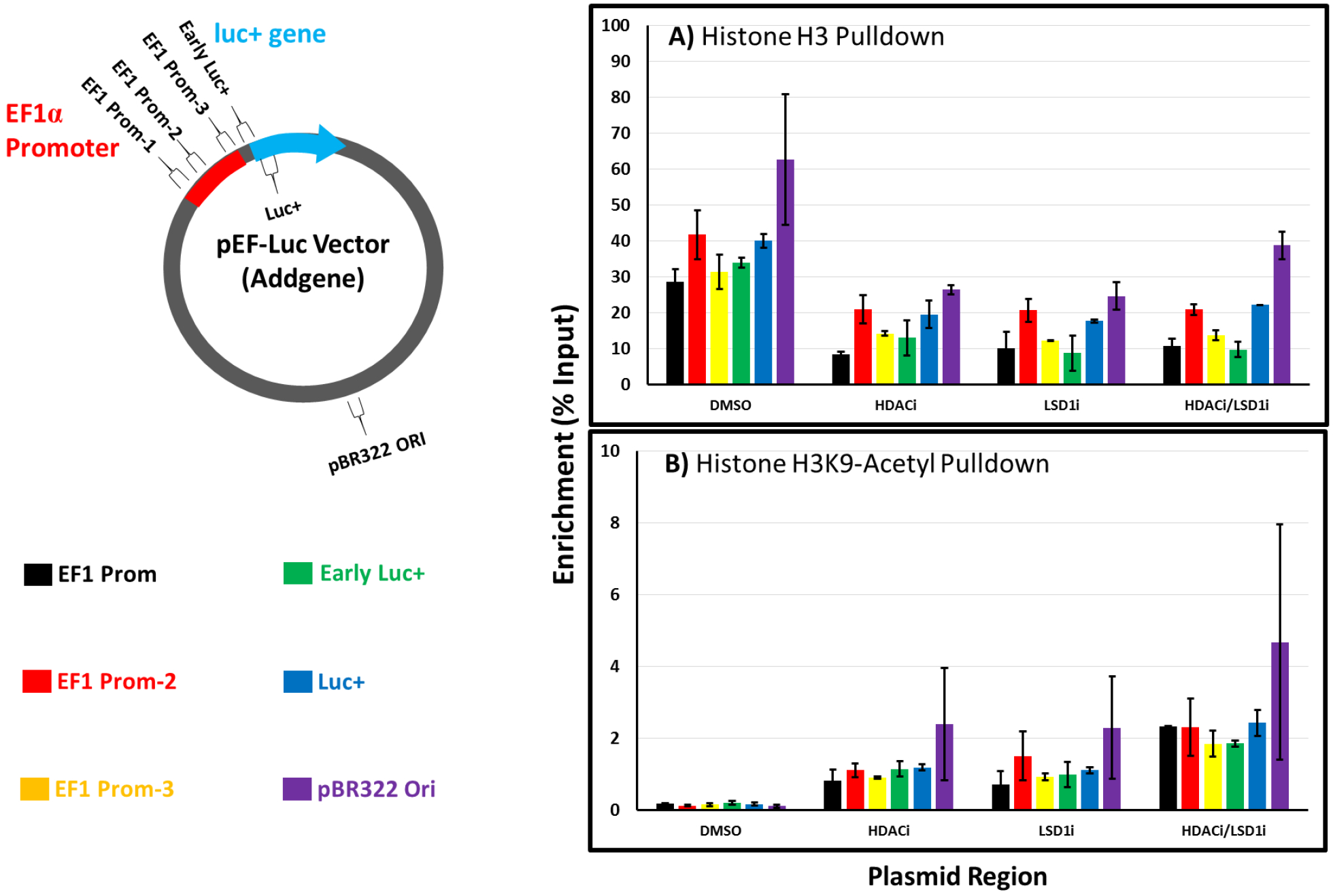
Native chromatin immunoprecipitation (ChIP) studies on histone-plasmid interactions in CHO-K1 cell nuclei. CHO-K1 cells were treated with polyplexes of N-RDGE with pEF-Luc plasmid in the absence or presence of the HDAC inhibitor SB939 and LSD1 inhibitor 2-PCPA hydrochloride, as well as the combination of the inhibitors (indicated by DMSO, HDACi and LSD1i, and HDAC1i/LSD1i, respectively). Pulldowns were carried out using A) a general Histone H3 antibody and B) an antibody detecting Histone H3 acetylated at lysine 9 (H3K9ac). All nuclei isolation and pulldowns were performed 48 hours following inhibitor treatment and polyplex delivery as in previous experiments. Error bars indicate the standard deviation of two qPCR trials on the same chromatin samples (N=1 transfection and pulldown in each case).
Additional pulldowns were performed using an antibody specific for acetylated histone H3 (specifically, acetylated at lysine 9, H3K9ac) (Figure 8B). Generally, higher gene transcription is observed when chromatin is highly acetylated [69, 70], although epigenetic regulation of gene expression is complex and local histone acetylation is not the only indicator of transcriptional activity. In the absence of inhibitors, the interaction of acetylated histone with plasmid is almost non-existent, with %input values near those observed with the IgG negative control antibody (Figure S4, Supporting Information section). HDAC inhibition with SB939 clearly increases the interaction of the pEF-Luc plasmid with histone H3 acetylated at lysine 9, even while total Histone H3-pEF-Luc plasmid interactions decrease (Figure 8A). Interestingly, LSD1 inhibition also increased H3K9ac-plasmid interactions, suggesting that LSD1 and HDAC may have an overlapping function on histone acetylation in CHO-K1 cells. As discussed above, LSD1 is one component of the CoREST complex along with HDAC [46], linking demethylation with deacetylation in one transcriptionally repressive complex. However, LSD1 inhibition with 2-PCPA hydrochloride was a much more potent inducer of transgene expression than HDAC inhibition with SB939 in CHO-K1 cells, which indicates that although histone H3-plasmid interactions are important, other factors may also play a key role, including the other three core histones which were not assayed in this study. However, it is interesting to note that simultaneously inhibiting LSD1 and HDAC results in roughly an additional doubling of H3K9ac-plasmid interactions (Figure 8B) from either individual inhibitor treatment without an observed increase in overall histone H3-plasmid interaction (Figure 8A); the exception is at the origin of replication on the plasmid backbone, where overall histone/plasmid interactions seem to increase with the inhibitor combination treatment. These results indicate that HDAC and LSD1 likely have coordinated activities in regulating histone acetylation and this likely has an effect on plasmid-histone interactions and transient luciferase expression.
DISCUSSION
As part of this work, we have identified a number of epigenetic enzymes and families that play an activating or repressive role in plasmid DNA mediated transgene expression in mammalian cells. Histone deacetylase (HDAC), lysine-specific demethylase (LSD), and enhancer of Zeste homolog 2 (EZH2) enzymes have well-established roles in repressing transcription by removing or adding functional groups to promoters of target genes. In agreement with these roles in chromosomal transcription regulation, inhibitors of these enzymes enhanced transgene expression following polymer-mediated plasmid DNA delivery in one or both cell lines. Inhibition of WDR5 (an MLL methyltransferase component) enhanced transgene expression, contrary to its role of activating target genes in the chromosome. Histone acetyltransferase (HAT) inhibition led to enhancement, reduction, or no effect on transgene expression; however, the effect on transgene expression appeared to depend on the family of HAT inhibited.
The small-molecule screen was set up with a single plasmid, pEF-Luc, in order to keep the experimental throughput to manageable levels. However, because epigenetic modifications are often dependent on the promoter driving target gene expression, we carried out additional studies in which lead inhibitors were evaluated with a second plasmid (pGL3), which was driven by a different promoter. A second polymer was also investigated for delivering pDNA, and our results indicate that the inhibitors also enhanced activity when this different gene delivery vehicle was employed. Taken together, our findings on inhibiting epigenetic enzymes for enhancing transgene expression are likely generic and are not limited to a particular polymer and / or plasmid system.
Epigenetic regulation of transcription occurs in the nucleus, and we therefore first determined if small-molecule inhibitors of epigenetic modulators influenced the amount of the delivered plasmid into the nucleus. Lead inhibitors did not result in an increase in the amount of plasmid DNA inside nuclei of CHO-K1 cells, indicating that the mechanism(s) responsible for enhanced transgene expression occurred following nuclear entry. In UMUC3 cells, however, HDAC inhibition increased the amount of plasmid DNA inside transfected cell nuclei, which may be partly responsible for the observed enhancement in transgene expression in these cells. Further, this observation was consistent with our previous study [17], which showed that treatment with Entinostat, an HDAC inhibitor, resulted in an increase of the plasmid in the nuclei of prostate cancer cells, as well as other findings which show that HDAC6-deficient cells traffic cytoplasmically microinjected plasmids more efficiently into target cell nuclei [33]. Our data indicate a currently undetermined mechanism by which HDAC inhibition enhances the delivery of plasmids to the nucleus in human cancer cells. This phenomenon was not seen for HDAC inhibition in CHO-K1 cells, which showed only a very moderate enhancement in transgene expression with HDAC inhibition in the screen. It is likely that this phenomenon is observed mainly in cancer cells which are known to possess aberrant expression of HDACs [71].
Pair-wise combinations of small molecule inhibitors indicated that simultaneous inhibition of LSD1 and HDACs resulted in significantly higher transgene expression compared to the additive effect of the single-agent treatments. This suggests synergy in the mechanisms of LSD1 and HDAC related to transgene expression. Simultaneous treatment with LSD1 and EZH2 inhibitors led to significant enhancement of transgene expression, with a modest indication of synergy. In UMUC3 cells, simultaneous inhibition of LSD1 and HDAC resulted in additive, but likely not synergistic, enhancement in transgene expression. This suggests that the mechanisms by which LSD1 and HDAC affect transgene expression in UMUC3 cells may not be linked.
Finally, ChIP experiments were carried out in an effort to determine if plasmids interact with endogenous histones following cellular delivery and if these interactions are responsive to epigenetic modulation. It was found that roughly 30% of plasmids delivered to CHO-K1 cell nuclei with an in-house gene delivery polymer interact with histone H3, and a reduction in interaction was evident with inhibition of HDAC and/or LSD1. Additionally, it was found that HDAC and LSD1 inhibition individually increased plasmid interactions with acetylated histone H3 while inhibiting both of these targets concurrently further increased this interaction. These data combined with our transgene expression results point to a coordination in the activity between LSD1 and HDAC on both plasmid-histone interactions and plasmid expression, although further experimentation is necessary to determine if these phenomena are directly related.
CONCLUSIONS
The screening experiments described here revealed several epigenetic modulators that play a role in transgene expression efficacy following polymer-mediated pDNA delivery to two very different mammalian cell lines. As indicated in Table 1, most of these enzymes demonstrated a similar effect on transient transgene expression consistent with their known activities in regulation of chromosomal transcription. The use of both individual and combinatorial enzymatic inhibitor treatments for enhancement in transgene expression is promising for a broad array of applications in biotechnology as well as in medicine, due to the involvement of epigenetic regulation in a multitude of diseases. Although some inhibitors enhanced delivery of plasmids to cell nuclei, many modulators in the current screen did not demonstrate this behavior, suggesting post-nuclear mechanisms (likely epigenetics) as the key factor governing expression from plasmid DNA. Findings from ChIP studies pointed towards interactions between the endogenous core Histone H3 and delivered plasmid DNA. Furthermore, these interactions were modulated by epigenetic enzyme inhibition, providing initial evidence for epigenetic plasmid transcriptional regulation. These mechanistic findings open the door to in-depth studies gauging spatial (histone site) and temporal interaction profiles between core histones and delivered plasmid DNA to understand how these interactions regulate transient plasmid expression in mammalian systems.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the National Science Foundation (Grant CBET 1404084) to KR and KH and the National Institute of General Medical Sciences, National Institutes of Health (Grant 1R01GM093229-01A1) to KR for funding this research. We are also thankful to Mr. Fred Pena for his excellent and generous technical help.
REFERENCES
- [1].Lund AM, Kildegaard HF, Petersen MB, Rank J, Hansen BG, Andersen MR, Mortensen UH, A versatile system for USER cloning-based assembly of expression vectors for mammalian cell engineering, PLoS One, 9 (2014) e96693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dhaliwal A, Maldonado M, Lin C, Segura T, Cellular cytoskeleton dynamics modulates non-viral gene delivery through RhoGTPases, PLoS One, 7 (2012) e35046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Munsell EV, Ross NL, Sullivan MO, Journey to the Center of the Cell: Current Nanocarrier Design Strategies Targeting Biopharmaceuticals to the Cytoplasm and Nucleus, Curr Pharm Des, 22 (2016) 1227–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reilly MJ, Larsen JD, Sullivan MO, Polyplexes Traffic through Caveolae to the Golgi and Endoplasmic Reticulum en Route to the Nucleus, Molecular pharmaceutics, 9 (2012) 1280–1290. [DOI] [PubMed] [Google Scholar]
- [5].Vaughan EE, Dean DA, Intracellular Trafficking of Plasmids during Transfection Is Mediated by Microtubules, Molecular Therapy, 13 (2006) 422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dean DA, Strong DD, Zimmer WE, Nuclear entry of nonviral vectors, Gene Ther, 12 (2005) 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lam AP, Dean DA, Progress and prospects: nuclear import of nonviral vectors, Gene Ther, 17 (2010) 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fuks F, DNA methylation and histone modifications: teaming up to silence genes, Curr Opin Genet Dev, 15 (2005) 490–495. [DOI] [PubMed] [Google Scholar]
- [9].Rasmussen TP, The epigenetics of early development: inferences from stem cells, Mol Reprod Dev, 81 (2014) 194–201. [DOI] [PubMed] [Google Scholar]
- [10].Carrer A, Wellen KE, Metabolism and epigenetics: a link cancer cells exploit, Curr Opin Biotechnol, 34 (2015) 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brunet A, Berger SL, Epigenetics of aging and aging-related disease, J Gerontol A Biol Sci Med Sci, 69 Suppl 1 (2014) S17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Johnson C, Warmoes MO, Shen X, Locasale JW, Epigenetics and cancer metabolism, Cancer Lett, 356 (2015) 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Landgrave-Gómez J, Mercado-Gómez O, Guevara-Guzmán R, Epigenetic mechanisms in neurological and neurodegenerative diseases, Front Cell Neurosci, 9 (2015) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chang Y, Ganesh T, Horton JR, Spannhoff A, Liu J, Sun A, Zhang X, Bedford MT, Shinkai Y, Snyder JP, Cheng X, Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases, J Mol Biol, 400 (2010) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jenuwein T, Allis CD, Translating the histone code, Science, 293 (2001) 1074–1080. [DOI] [PubMed] [Google Scholar]
- [16].Gorman CM, Howard BH, Reeves R, Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate, Nucleic Acids Res, 11 (1983) 7631–7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Elmer JJ, Christensen MD, Barua S, Lehrman J, Haynes KA, Rege K, The histone deacetylase inhibitor Entinostat enhances polymer-mediated transgene expression in cancer cell lines, Biotechnology and bioengineering, 113 (2016) 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barua S, Rege K, The influence of mediators of intracellular trafficking on transgene expression efficacy of polymer-plasmid DNA complexes, Biomaterials, 31 (2010) 5894–5902. [DOI] [PubMed] [Google Scholar]
- [19].Escher G, Hoang A, Georges S, Tchoua U, El-Osta A, Krozowski Z, Sviridov D, Demethylation using the epigenetic modifier, 5-azacytidine, increases the efficiency of transient transfection of macrophages, J Lipid Res, 46 (2005) 356–365. [DOI] [PubMed] [Google Scholar]
- [20].Riu E, Chen Z, Xu H, He C, Kay M, Histone Modifications are Associated with the Persistence or Silencing of Vector-mediated Transgene Expression In Vivo, Molecular Therapy, 15 (2007) 1348–1355. [DOI] [PubMed] [Google Scholar]
- [21].Nan X, Ng H, Johnson C, Laherty C, Turner B, Eisenman R, Bird A, Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex, Nature, 393 (1998) 386–389. [DOI] [PubMed] [Google Scholar]
- [22].TJIO JH, PUCK TT, Genetics of somatic mammalian cells. II. Chromosomal constitution of cells in tissue culture, J Exp Med, 108 (1958) 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Feichtinger J, Hernández I, Fischer C, Hanscho M, Auer N, Hackl M, Jadhav V, Baumann M, Krempl PM, Schmidl C, Farlik M, Schuster M, Merkel A, Sommer A, Heath S, Rico D, Bock C, Thallinger GG, Borth N, Comprehensive genome and epigenome characterization of CHO cells in response to evolutionary pressures and over time, Biotechnol Bioeng, 113 (2016) 2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, Andersen MR, Neff N, Passarelli B, Koh W, Fan HC, Wang J, Gui Y, Lee KH, Betenbaugh MJ, Quake SR, Famili I, Palsson BO, The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line, Nat Biotechnol, 29 (2011) 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Singh R, Harshman SW, Ruppert AS, Mortazavi A, Lucas DM, Thomas-Ahner JM, Clinton SK, Byrd JC, Freitas MA, Parthun MR, Proteomic profiling identifies specific histone species associated with leukemic and cancer cells, Clin Proteomics, 12 (2015) 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Potta T, Zhen Z, Grandhi T, Christensen M, Ramos J, Breneman C, Rege K, Discovery of Antibiotics-derived Polymers for Gene Delivery using Combinatorial Synthesis and Cheminformatics Modeling, Biomaterials: in press, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Miryala B, Zhen Z, Potta T, Breneman C, Rege K, Parallel Synthesis and Quantitative Structure-Activity Relationship (QSAR) Modeling of Aminoglycoside-Derived Lipopolymers for Transgene Expression, Acs Biomaterials-Science & Engineering, 1 (2015) 656–668. [DOI] [PubMed] [Google Scholar]
- [28].Lin X, Tirichine L, Bowler C, Protocol: Chromatin immunoprecipitation (ChIP) methodology to investigate histone modifications in two model diatom species, Plant Methods, 8 (2012) 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pinkerneil M, Hoffmann MJ, Deenen R, Köhrer K, Arent T, Schulz WA, Niegisch G, Inhibition of Class I Histone Deacetylases 1 and 2 Promotes Urothelial Carcinoma Cell Death by Various Mechanisms, Mol Cancer Ther, 15 (2016) 299–312. [DOI] [PubMed] [Google Scholar]
- [30].Niegisch G, Knievel J, Koch A, Hader C, Fischer U, Albers P, Schulz WA, Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers, Urol Oncol, 31 (2013) 1770–1779. [DOI] [PubMed] [Google Scholar]
- [31].Lehmann M, Hoffmann MJ, Koch A, Ulrich SM, Schulz WA, Niegisch G, Histone deacetylase 8 is deregulated in urothelial cancer but not a target for efficient treatment, J Exp Clin Cancer Res, 33 (2014) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang H, Yu N, Chen D, Lee KC, Lye PL, Chang JW, Deng W, Ng MC, Lu T, Khoo ML, Poulsen A, Sangthongpitag K, Wu X, Hu C, Goh KC, Wang X, Fang L, Goh KL, Khng HH, Goh SK, Yeo P, Liu X, Bonday Z, Wood JM, Dymock BW, Kantharaj E, Sun ET, Discovery of (2E)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1H-benzimidazol-5-yl}-N-hydroxyacrylamide (SB939), an orally active histone deacetylase inhibitor with a superior preclinical profile, J Med Chem, 54 (2011) 4694–4720. [DOI] [PubMed] [Google Scholar]
- [33].Vaughan EE, Geiger RC, Miller AM, Loh-Marley PL, Suzuki T, Miyata N, Dean DA, Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency, Mol Ther, 16 (2008) 1841–1847. [DOI] [PubMed] [Google Scholar]
- [34].Lee J, Xia Y, Son MY, Jin G, Seol B, Kim MJ, Son MJ, Do M, Lee M, Kim D, Lee K, Cho YS, A novel small molecule facilitates the reprogramming of human somatic cells into a pluripotent state and supports the maintenance of an undifferentiated state of human pluripotent stem cells, Angew Chem Int Ed Engl, 51 (2012) 12509–12513. [DOI] [PubMed] [Google Scholar]
- [35].Nakagawa T, Guarente L, Sirtuins at a glance, J Cell Sci, 124 (2011) 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL, Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening, J Biol Chem, 276 (2001) 38837–38843. [DOI] [PubMed] [Google Scholar]
- [37].Dai X, Hayashi K, Nozaki H, Cheng Y, Zhao Y, Genetic and chemical analyses of the action mechanisms of sirtinol in Arabidopsis, Proc Natl Acad Sci U S A, 102 (2005) 3129–3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kawakami S, Kinoshita Y, Maruki-Uchida H, Yanae K, Sai M, Ito T, Piceatannol and its metabolite, isorhapontigenin, induce SIRT1 expression in THP-1 human monocytic cell line, Nutrients, 6 (2014) 4794–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kuo MH, Allis CD, Roles of histone acetyltransferases and deacetylases in gene regulation, Bioessays, 20 (1998) 615–626. [DOI] [PubMed] [Google Scholar]
- [40].Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY, Ge K, Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation, EMBO J, 30 (2011) 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chimenti F, Bizzarri B, Maccioni E, Secci D, Bolasco A, Chimenti P, Fioravanti R, Granese A, Carradori S, Tosi F, Ballario P, Vernarecci S, Filetici P, A novel histone acetyltransferase inhibitor modulating Gcn5 network: cyclopentylidene-[4-(4’-chlorophenyl)thiazol-2-yl)hydrazone, J Med Chem, 52 (2009) 530–536. [DOI] [PubMed] [Google Scholar]
- [42].Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK, Small molecule modulators of histone acetyltransferase p300, J Biol Chem, 278 (2003) 19134–19140. [DOI] [PubMed] [Google Scholar]
- [43].Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA, Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor, Chem Biol, 17 (2010) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Burgess RJ, Zhang Z, Roles for Gcn5 in promoting nucleosome assembly and maintaining genome integrity, Cell Cycle, 9 (2010) 2979–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Christensen MD, Elmer JJ, Eaton S, Gonzalez-Malerva L, LaBaer J, Rege K, Kinome-level screening identifies inhibition of polo-like kinase-1 (PLK1) as a target for enhancing non-viral transgene expression, Journal of controlled release : official journal of the Controlled Release Society, 204 (2015) 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y, Regulation of LSD1 histone demethylase activity by its associated factors, Mol Cell, 19 (2005) 857–864. [DOI] [PubMed] [Google Scholar]
- [47].Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG, Regulation of MLL1 H3K4 methyltransferase activity by its core components, Nat Struct Mol Biol, 13 (2006) 713–719. [DOI] [PubMed] [Google Scholar]
- [48].Senisterra G, Wu H, Allali-Hassani A, Wasney GA, Barsyte-Lovejoy D, Dombrovski L, Dong A, Nguyen KT, Smil D, Bolshan Y, Hajian T, He H, Seitova A, Chau I, Li F, Poda G, Couture JF, Brown PJ, Al-Awar R, Schapira M, Arrowsmith CH, Vedadi M, Small-molecule inhibition of MLL activity by disruption of its interaction with WDR5, Biochem J, 449 (2013) 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Cierpicki T, Grembecka J, Targeting protein-protein interactions in hematologic malignancies: still a challenge or a great opportunity for future therapies?, Immunol Rev, 263 (2015) 279–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Margueron R, Reinberg D, The Polycomb complex PRC2 and its mark in life, Nature, 469 (2011) 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee YH, Coonrod SA, Kraus WL, Jelinek MA, Stallcup MR, Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination, Proc Natl Acad Sci U S A, 102 (2005) 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Swigut T, Wysocka J, H3K27 demethylases, at long last, Cell, 131 (2007) 29–32. [DOI] [PubMed] [Google Scholar]
- [53].Niehrs C, Schäfer A, Active DNA demethylation by Gadd45 and DNA repair, Trends Cell Biol, 22 (2012) 220–227. [DOI] [PubMed] [Google Scholar]
- [54].Sun H, Yang X, Zhu J, Lv T, Chen Y, Chen G, Zhong L, Li Y, Huang X, Huang G, Tian J, Inhibition of p300-HAT results in a reduced histone acetylation and down-regulation of gene expression in cardiac myocytes, Life Sci, 87 (2010) 707–714. [DOI] [PubMed] [Google Scholar]
- [55].Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C, HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics, Cell Res, 17 (2007) 195–211. [DOI] [PubMed] [Google Scholar]
- [56].Rudolph T, Beuch S, Reuter G, Lysine-specific histone demethylase LSD1 and the dynamic control of chromatin, Biol Chem, 394 (2013) 1019–1028. [DOI] [PubMed] [Google Scholar]
- [57].Georgakopoulos T, Thireos G, Two distinct yeast transcriptional activators require the function of the GCN5 protein to promote normal levels of transcription, EMBO J, 11 (1992) 4145–4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD, WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development, Cell, 121 (2005) 859–872. [DOI] [PubMed] [Google Scholar]
- [59].Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ, Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator, Cancer Cell, 13 (2008) 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ablack JN, Cohen M, Thillainadesan G, Fonseca GJ, Pelka P, Torchia J, Mymryk JS, Cellular GCN5 is a novel regulator of human adenovirus E1A-conserved region 3 transactivation, J Virol, 86 (2012) 8198–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD, Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6, J Med Chem, 51 (2008) 4370–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM, A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency, MBio, 4 (2013) e00558–00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T, Sun H, Lu F, Zhang H, LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16, Mol Cell Biol, 34 (2014) 158–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY, Long noncoding RNA as modular scaffold of histone modification complexes, Science, 329 (2010) 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].McNairn AJ, Gilbert DM, Epigenomic replication: linking epigenetics to DNA replication, Bioessays, 25 (2003) 647–656. [DOI] [PubMed] [Google Scholar]
- [66].Jaenisch R, Bird A, Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals, Nat Genet, 33 Suppl (2003) 245–254. [DOI] [PubMed] [Google Scholar]
- [67].Gilfillan GD, Hughes T, Sheng Y, Hjorthaug HS, Straub T, Gervin K, Harris JR, Undlien DE, Lyle R, Limitations and possibilities of low cell number ChIP-seq, BMC Genomics, 13 (2012) 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].O’Neill LP, Turner BM, Immunoprecipitation of native chromatin: NChIP, Methods, 31 (2003) 76–82. [DOI] [PubMed] [Google Scholar]
- [69].Kouzarides T, Chromatin modifications and their function, Cell, 128 (2007) 693–705. [DOI] [PubMed] [Google Scholar]
- [70].Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA, Genome-wide map of nucleosome acetylation and methylation in yeast, Cell, 122 (2005) 517–527. [DOI] [PubMed] [Google Scholar]
- [71].Li Y, Seto E, HDACs and HDAC Inhibitors in Cancer Development and Therapy, Cold Spring Harbor Perspectives in Medicine, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.