

SUMMARY
Chronic myeloid leukemia (CML) is propagated by leukemia stem cells (LSCs) that are not eradicated by tyrosine kinase inhibitor (TKI) treatment and persist as a source of disease recurrence. Bone marrow (BM) mesenchymal niches play an essential role in hematopoietic stem cell (HSC) and LSC maintenance. Using a murine CML model, we examine leukemia-induced alterations in mesenchymal cell populations. We show that 6C3+ stromal progenitors expand in CML BM and exhibit increased LSC but reduced HSC supportive capacity. Tumor necrosis factor alpha (TNF-α) signaling mediates expansion and higher expression of CXCL1 in CML BM 6C3+ cells and higher expression of the CXCL1 receptor CXCR2 in LSCs. CXCL1 enhances LSC proliferation and self-renewal, whereas CXCR2 inhibition reduces LSC growth and enhances LSC targeting in combination with tyrosine kinase inhibitors (TKIs). We find that TNF-α-mediated alterations in CML BM stromal niches enhance support of LSC maintenance and growth via CXCL1-CXCR2 signaling and that CXCR2 inhibition effectively depletes CML LSCs.
Graphical abstract
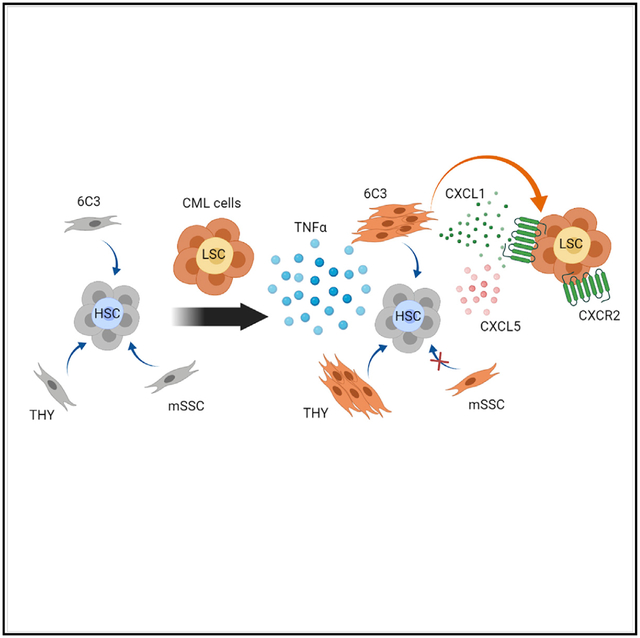
In brief
Chronic myeloid leukemia (CML) leukemia stem cells (LSCs) resist elimination by treatment. Agarwal et al. show that CML 6C3+ bone marrow stromal progenitors support LSC maintenance. TNF-α signaling mediates increased CXCL1 expression in 6C3+ cells, which support LSC proliferation and self-renewal. Targeting the CXCL1 receptor CXCR2 inhibits LSC maintenance.
INTRODUCTION
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder that arises from hematopoietic stem cell (HSC) transformation because of a chromosomal translocation between chromosomes 9 and 22 [t(9;22)], leading to development of the BCR-ABL fusion oncoprotein with abnormal tyrosine kinase activity. Leukemic stem cells (LSCs) in CML demonstrate enhanced proliferation while retaining differentiation capacity. BCR-ABL tyrosine kinase inhibitors (TKIs) have revolutionized treatment of CML and induce remission and prolong survival in the majority of individuals. However, LSCs are not eliminated in individuals receiving TKI and persist as a source of disease recurrence (Bhatia et al., 2003; Chu et al., 2011). LSC resistance has been shown to be mediated by kinase-independent mechanisms (Corbin et al., 2011; Hamilton et al., 2012; Perl and Carroll, 2011).
HSCs are regulated by cell-intrinsic mechanisms and by extrinsic signals from the bone marrow microenvironment (BMM) (Hoggatt et al., 2016). The BMM is characterized by diverse cell types (Morrison and Scadden, 2014). The HSC niche describes a specialized microenvironment that regulates HSC function in vivo. Several non-hematopoietic cell populations, including mesenchymal stem/stromal cells (MSCs) (Pinho et al., 2013), osteoblasts (Bowers et al., 2015; Calvi et al., 2003), adipocytes (Naveiras et al., 2009), sinusoidal and arteriolar endothelial cells (Kiel et al., 2005), and megakaryocytes (Bruns et al., 2014) have been implicated in niche regulation of HSCs (Birbrair and Frenette, 2016). There is considerable interest in understanding how extrinsic signals from niche cells regulate HSC maintenance, self-renewal, and differentiation (Agarwal and Bhatia, 2015).
We and others have reported that BMM factors, including CXCL12 (Agarwal et al., 2019), Wnt (Agarwal et al., 2017; Zhang et al., 2013), and interleukin-1 (IL-1) (Zhang et al., 2016), can protect CML LSCs from TKIs. Development of CML leads to altered levels of inflammatory cytokines and chemokines within the BM and to alterations in BMM function that provide CML LSCs with a growth advantage over normal HSCs (Bhatia et al., 1995; Schepers et al., 2013; Zhang et al., 2012). These observations are consistent with reports showing that BMM alterations can support leukemia development in mouse models of myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPNs), and acute myeloid leukemia (AML) (Medyouf, 2017). Diverse BMM subsets have been implicated in acute and chronic leukemia models (Arranz et al., 2014; Balderman et al., 2016; Bowers et al., 2015; Hanoun et al., 2014; Krause et al., 2013; Mead et al., 2017; Schneider et al., 2017). However, the nature of BM niches for LSCs, the influence of leukemia on niche function, and key niche mechanisms regulating LSC growth and resistance to treatment are not well understood.
MSCs in the postnatal BM generate bone, cartilage, hematopoiesis-supportive stromata, and marrow adipocytes (Bianco and Robey, 2015). A phenotypically defined hierarchy of mesenchymal stem and progenitor cells within murine BM has been delineated using single-cell analyses and lineage tracing methods and has been similarly identified for human mesenchymal cells (Chan et al., 2018). Here we used the SCL-tTABCR-ABL transgenic CML mouse model to examine how CML development affects murine BM mesenchymal subpopulations, how leukemia-induced changes in MSCs affect LSC and HSC maintenance, and specific molecular mechanisms of BM MSCLSC crosstalk.
RESULTS
Alterations in stroma-forming and bone-forming progenitors in CML BM
A hierarchy of mesenchymal stem and progenitor cells has been delineated within CD45−Ter119−Tie2−AlphaV+ cells in murine BM (Chan et al., 2015) that includes primitive mSSC (murine skeletal stem cell; Thy1−6C3−CD105−CD200+), multipotent pre-BCSP (pre-bone, cartilage, and stromal progenitor; Thy1−6C3−CD105−CD200−), and BCSP (Thy1−6C3−CD 105+) populations and oligo-lineage pro-chondrogenic progenitors (PCPs; Thy1+6C3−CD105+CD200+), bone-forming Thy1 (CD90)-expressing progenitors (THY+, Thy1+6C3−CD105+CD 200−), B cell lymphocyte stromal progenitors (BLSPs; Thy1+6C3−CD105−), 6C3 (aminopeptidase A, CD249)-expressing stromal progenitors (6C3+, Thy1−6C3+CD105+), and hepatic leukemia factor-expressing cells (HECs; Thy1−6C3+ CD105−) (Figure 1A). We examined the effect of CML development on the distribution of mesenchymal stem and progenitor cells using a well-established SCL-tTA-BCR-ABL-inducible transgenic mouse model of CML (Koschmieder et al., 2005). Tetracycline was withdrawn to allow BCR-ABL expression in HSCs and development of a CML-like myeloproliferative disorder (Figure 1B). We observed significantly increased frequencies and numbers of THY+ bone-forming progenitors and 6C3+ stromal progenitors in BM of CML mice compared with wild-type (WT) mice (Figures 1C, 1D, S1A, and S1B), but no significant change in the frequencies of other subsets (Figure S1C).
Figure 1. Increased stroma-forming and bone-forming progenitors in CML BM.
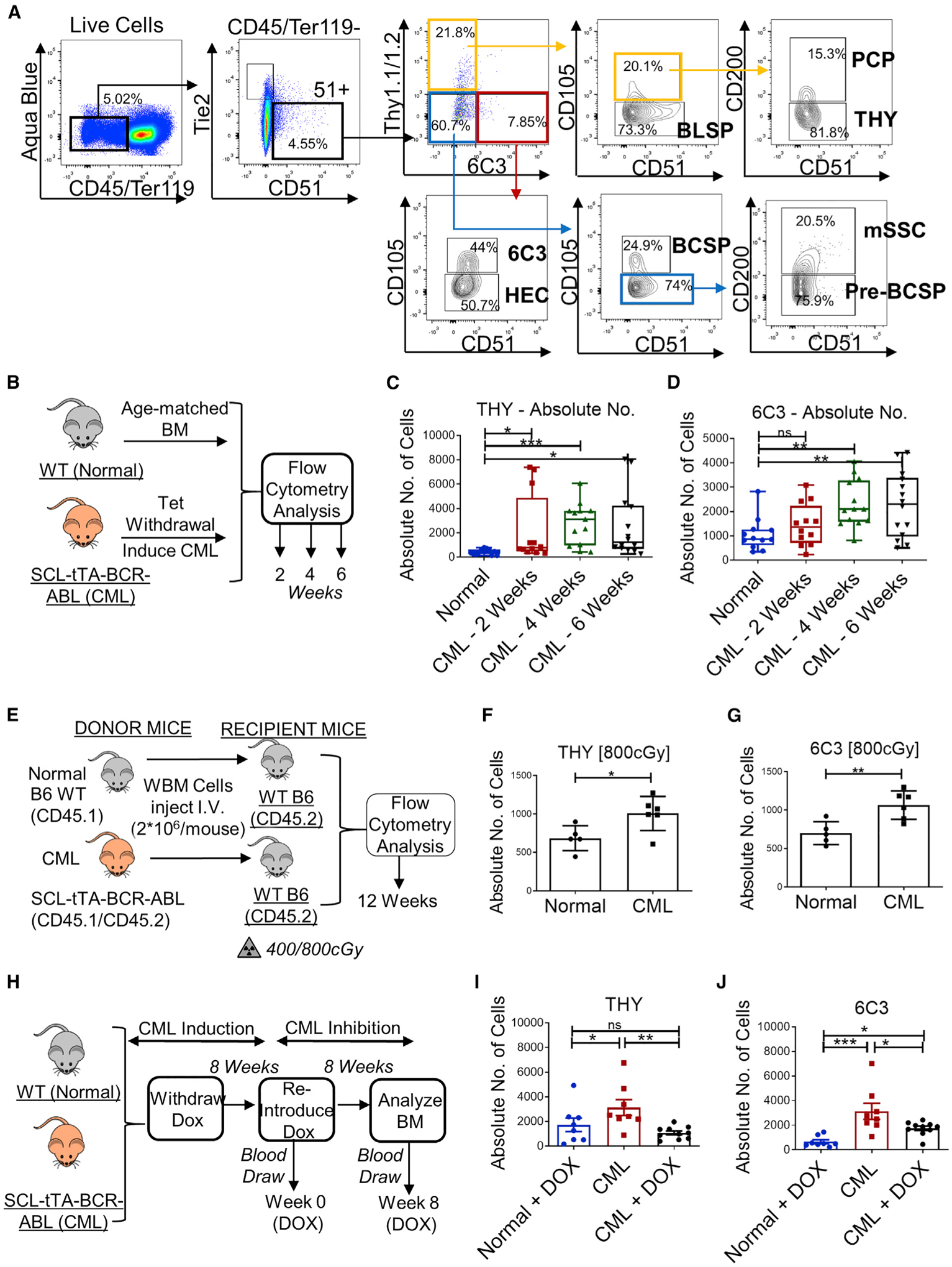
(A) Gating strategy for identification of eight distinct BM mesenchymal subpopulations: mSSCs (murine skeletal stem cells), pre-BCSPs (pre-bone, cartilage, and stromal progenitors), BCSPs, pro-chondrogenic progenitors (PCPs), THY+1+ progenitors (THY+), B cell lymphocyte stromal progenitors (BLSPs), 6C3+ stromal progenitors (6C3+), and hepatic leukemia factor-expressing cells (HECs). Percentages are of parent cells. Results are representative of multiple replicates.
(B) Experimental schema. BCR-ABL expression was induced in SCL-tTA-BCR-ABL CML mice by doxycycline (Tet) withdrawal. BM cells obtained from CML mice were analyzed for mesenchymal subpopulations 2, 4, and 6 weeks after leukemia induction (n = 12–14 mice/group) and compared with aged-matched wild-type (WT) normal mice.
(C and D) Absolute numbers of THY+ cells (C) and 6C3+ cells (D).
(E) Experimental schema. Whole BM (WBM) cells from WT normal mice and CML mice (2 × 106 cells/mouse; n = 4–8 mice/group) were transplanted into 6- to 10-week-old littermates irradiated at 400 cGy or 800 cGy, and BM mesenchymal subpopulations were analyzed after 12 weeks.
(F and G) Absolute numbers of THY+ (F) and 6C3+ cells (G) from recipient mice irradiated at 800cGy.
(H) Experimental schema. BCR-ABL expression was induced in a cohort of SCL-tTA-BCR-ABL mice by doxycycline withdrawal for 16 weeks (CML). A second cohort of SCL-tTA-BCR-ABL mice was withdrawn from doxycycline to induce CML for 8 weeks, and then doxycycline was re-introduced for 8 weeks to inhibit CML (CML + DOX) (n = 8–10 mice/group).
(I and J) Absolute number of THY+ (I) and 6C3+ cells (J).
Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Mesenchymal stem/progenitor cells identified using different cell surface receptors or genetic reporters can show considerable overlap (Pinho and Frenette, 2019). In this context, 6C3 and THY cells are present within the Nestin+ population in the BM (Chan et al., 2013) and are uniformly LeprR+ because nearly all CD51+ BM stromal cells are LepR+ and vice versa (Yue et al., 2016). Mesenchymal stem/progenitor cells expressing PDGFRα (Pα) and Sca-1 surface markers (Pα+Sca-1+; PαS cells) (Morikawa et al., 2009) showed maximum representation within THY+ cells (Figures S1D and S1E). Schepers et al. (2013) showed that MSCs defined as CD51+Sca1+ and osteoblasts defined as CD51+ are increased in CML mice. The stromal subsets studied here were first gated on CD51 and then additional markers. Thus, all subsets were CD51+, and more than 60% of the THY population was CD51+Sca1+ (Figures S1D and S1E). Lineage tracing of mesenchymal progenitors, osteoprogenitors, and osteoblasts using Prrx1-Cre (Prx1-Cre), Sp7-Cre (Osx1-Cre), and bone gamma carboxyglutamate protein (BGLAP)-Cre (Ocn-Cre) strains crossed with Rosa26CAG-tdTomato mice (Agarwal et al., 2019) showed maximum labeling of mSSCs and 6C3+ cells in Prx1-Cre lines, consistent with being mesenchymal stem/progenitor cells (Greenbaum et al., 2013), whereas maximum labeling of THY+ cells was seen in Osx1-Cre lines (Figure S1F), consistent with being osteoprogenitors (Mizoguchi et al., 2014). Finally, we evaluated the overlap of gene expression between 6C3+ cells and diverse populations identified using single-cell analysis (data not shown) (Baccin et al., 2020; Baryawno et al., 2019; Tikhonova et al., 2019; Wolock et al., 2019). These analyses showed overlap of 6C3+ cells with LepR+ perivascular MSCs with pre-adipocytic features.
As described for BM MSCs from individuals with CML (Jootar et al., 2006; Wöhrer et al., 2007), mesenchymal stem and progenitor cells in the CML mouse model did not express the BCR-ABL gene (Figure S1G). Transplantation of CML cells into sub-lethally (4 Gy) or lethally irradiated (8 Gy) WT mice to induce leukemia resulted in similar increases in the frequency and number of THY+ and 6C3+ cells (Figures 1E–1G, S1H, and S1I), suggesting that the observed changes in mesenchymal cells in CML BM were mediated by leukemic cells. In mice in which leukemia had been induced, doxycycline administration to suppress BCR-ABL expression and restore normal hematopoiesis (Figures 1H and S1J) did not affect mSSC progenitors (Figures S1K and S1L), completely reversed the increase in THY+ progenitors (Figures 1J and S1M), but only partially reversed the increase in 6C3+ progenitors, which remained significantly higher than in normal BM (Figures 1K and S1N). These results indicate that several leukemia-induced alterations in specific mesenchymal stem/progenitors may persist long term.
Analysis of the cell cycle status of mesenchymal stem and progenitor subsets by Ki67 staining of stromal cells freshly isolated from normal and CML mice showed increased frequencies of Ki-67+ cycling 6C3 and THY cells in CML mice (Figures 2A–2C). Subsets from normal and CML mice sorted by fluorescence-activated cell sorting (FACS) were cultured in vitro for 2–3 weeks (Figure 2D). Normal and CML mSSCs had a similar colony-forming unit-fibroblast (CFU-F) capacity (Figure S2A), but CML 6C3+ and THY+ cells generated increased colonies compared with their normal counterparts (Figures 2E and 2F). THY+ cells from CML BM showed an increased osteogenic differentiation capacity (Figures 2G and S2B) and reduced adipogenic capacity compared with normal THY+ cells (Figures 2H and S2C). On the other hand, CML 6C3+ cells showed reduced osteogenic potential (Figures 2I and S2D) and an increased adipogenic differentiation (Figure 2I) capacity compared with normal 6C3+ cells (Figures 2J and S2E).
Figure 2. Altered cycling and differentiation of CML 6C3+ and THY+ progenitors.
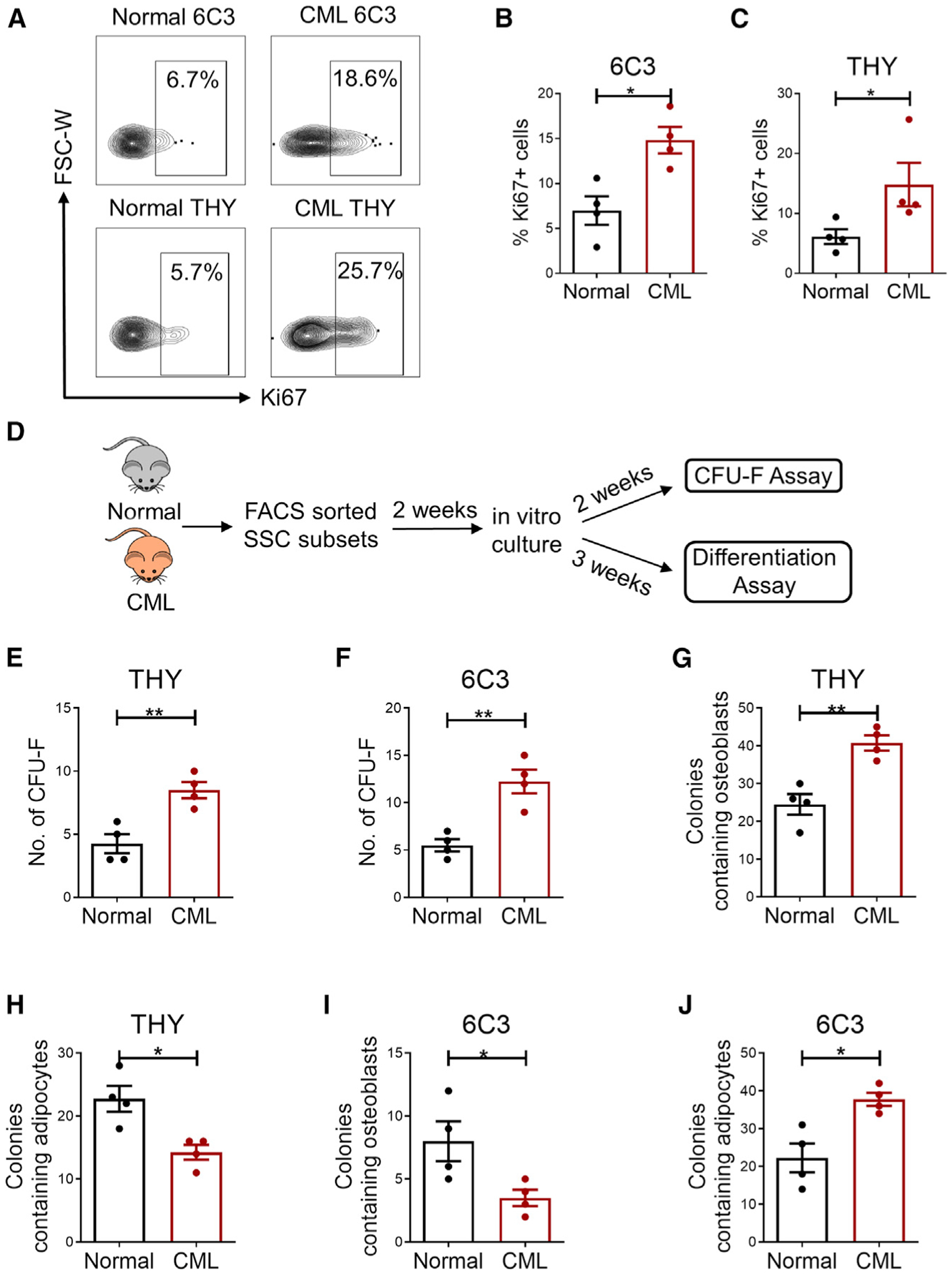
(A) Representative flow cytometry results of freshly isolated normal and CML subsets analyzed for Ki-67 staining.
(B and C) Frequency of Ki-67+ cells in normal and CML THY+ (B) and 6C3+ populations (C) (n = 4 replicates/group). Percentages are of parent cells.
(D) Experimental schema. FACS-sorted BM subsets from primary CML and aged-matched normal mice were cultured in vitro and analyzed for CFU-F and differentiation potential.
(E and F) CFU-F numbers generated from normal and CML THY+ (E) and 6C3+ cells (F) (n = 4 replicates/group).
(G and H) Osteoblast-containing (G) and adipocyte-containing (H) colonies from THY+ cells using alizarin red staining and oil red O staining, respectively.
(I and J) Osteoblast-containing (I) and adipocyte-containing (J) colonies from 6C3+ cells using alizarin red staining and oil red O staining, respectively.
n = 4 independent experiments. Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CML stromal progenitors demonstrate enhanced LSC and reduced HSC support
THY+ and 6C3+ progenitors are reported to exist in close contact with HSCs and have HSC-supporting activity (Chan et al., 2013). We investigated the ability of CML mesenchymal stem/progenitor cells to support HSC and LSC maintenance. FACS-selected long-term HSCs (LTHSCs) from normal mice (hereafter referred to as HSCs) and LTHSCs from CML mice (hereafter referred to as LSCs) were cocultured with FACS-selected mSSC, 6C3+, and THY+ cells from normal or CML mice for 3 days, followed by transplantation into lethally irradiated WT mice (Figure 3A). Long-term donor chimerism of normal HSCs was increased by coculture with normal mSSC, THY+, and 6C3+ cells compared with culture in the absence of stromata and to a greater extent than with coculture with CML mSSC THY+ or 6C3+ cells, which did not increase chimerism compared with no-stroma controls (Figure 3B). In contrast, LSC donor chimerism was not enhanced by normal or CML mSSCs but was enhanced by CML and normal Thy and 6C3 cells compared with no-stroma controls. LSC chimerism was enhanced to a greater extent by co-culture with CML compared with normal 6C3+ cells and to a lesser extent by coculture with CML compared with normal THY+ cells (Figure 3C). We confirmed multilineage engraftment of CML cells in peripheral blood (PB) under all conditions, with a significant increase in the percentage of myeloid cells and reduction in the percentage of lymphoid cells following culture of CML LSCs or CML 6C3+ cells compared with no-stroma controls (Figure 3D). We also observed increased BM chimerism of CML cells cultured with CML compared with normal 6C3+ cells and reduced BM chimerism of CML cells cultured with CML compared with normal THY+ cells (Figure 3E). These results indicate that CML BM mSSC, THY+, and 6C3+ cells have a reduced capacity to support HSC engraftment, whereas CML 6C3+ cells have an increased capacity to support LSC engraftment compared with normal 6C3+ cells.
Figure 3. CML 6C3+ stromal progenitors demonstrate enhanced LSC and reduced normal HSC support.
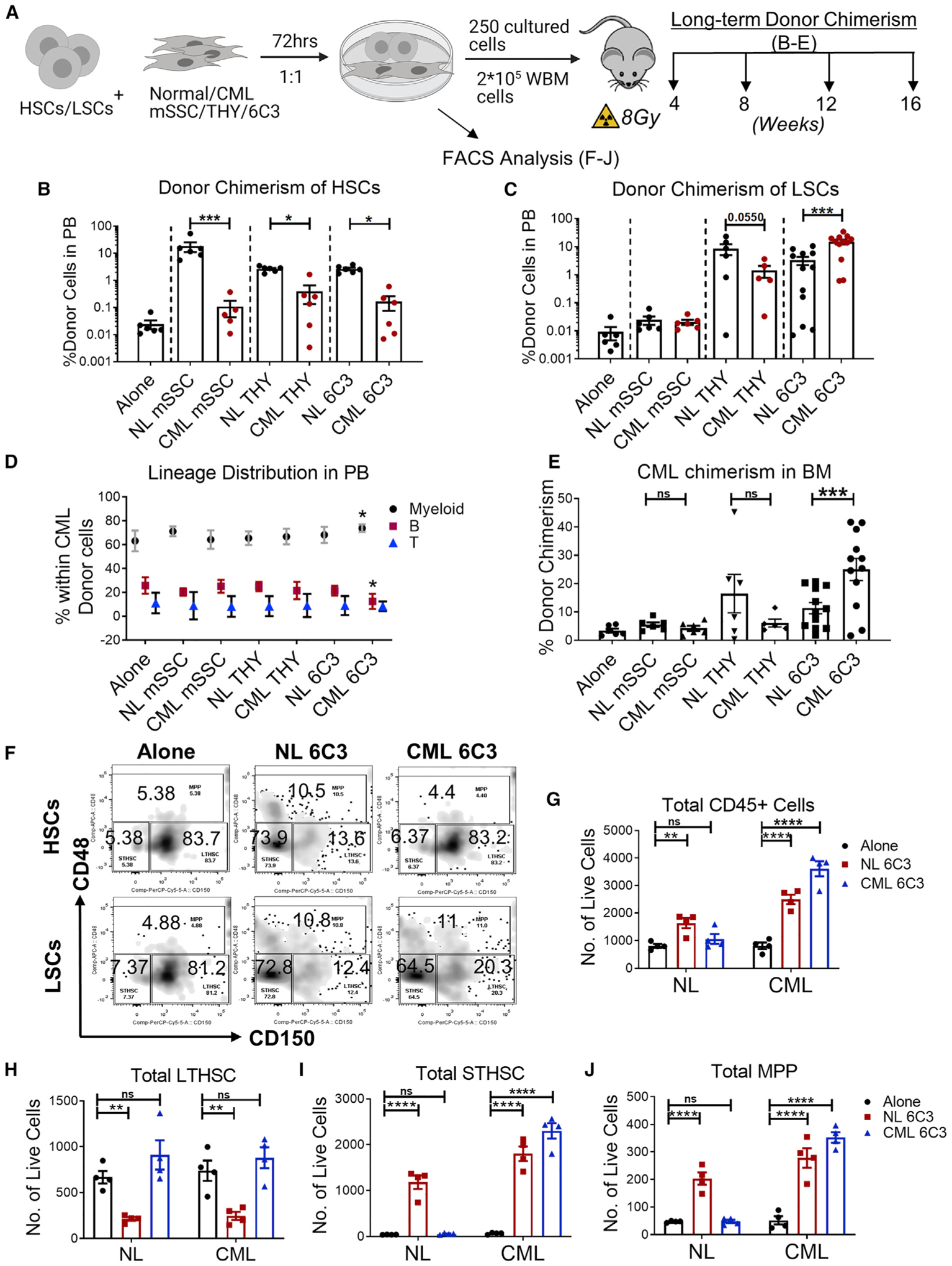
(A) Experimental schema. HSCs and LSCs were cultured with FACS-sorted BM subsets from normal (CD45.2+) and CML (CD45.1/45.2+) mice for 72 h, transplanted into irradiated WT recipients (CD45.2+), and followed for donor chimerism for 16 weeks after transplantation (n = 5–12 biological replicates per group).
(B and C) Long-term donor chimerism at 16 weeks in the PB of mice transplanted with HSCs (B) and LSCs co-cultured with normal or CML mesenchymal subsets (C).
(D and E) Multilineage donor chimerism in PB (D) and overall donor chimerism in BM (E) at 16 weeks of mice transplanted with LSCs co-cultured with normal or CML mesenchymal subsets. HSCs and LSCs were cultured for 72 h without mesenchymal cells (alone) or co-cultured with normal (NL) or CML 6C3+ cells purified from normal or CML mice at a 1:1 ratio, and cell numbers and immunophenotype (CD45, CD150, and CD48) were analyzed.
(F–J) Representative flow cytometry results (F) and absolute number of CD45+ cells (G), LTHSCs (CD150+CD48−) (H), STHSCs (CD150−CD48−) (I), and MPPs (CD48+) (J).
Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We further characterized the effects of 6C3+ cell culture on in vitro maintenance and differentiation of purified LTHSCs from normal (HSCs) and CML mice (LSCs). Normal CD45+ cell production was increased after 3 days coculture of HSCs with normal but not CML 6C3+ cells, whereas CML CD45+ cell production was increased after coculture of LSCs with CML and normal 6C3+ cells (Figure 3F). Normal short-term HSC (STHSC) and multipotent progenitor (MPP) production was increased after coculture of HSCs with normal 6C3+ cells, whereas HSCs were depleted. In contrast, normal STHSC and MPP generation was not increased, and HSC numbers were maintained after coculture of HSCs with CML 6C3+ cells. Finally, CML STHSC and MPP production was increased after coculture of LSCs with CML and normal 6C3+ cells, whereas LSCs were depleted after coculture with normal 6C3+ cells but maintained after coculture with CML 6C3 cells (Figures 3G–3J). These observations suggest that CML 6C3+ cells maintain LSC self-renewal while increasing STHSC and MPP production but reduce the ability of HSCs to generate mature progeny. The observations that similar numbers of CML and normal LTHSCs are maintained after culture of CML LSCs on CML 6C3 cells (Figure 3G) but that significantly increased donor cell chimerism is seen in mice receiving LSCs cultured on CML 6C3+ cells (Figure 3C) compared with mice receiving HSCs cultured on CML 6C3+ cells (Figure 3B) indicate that CML 6C3+ cells differentially affect the ability of LSCs versus HSCs to engraft and regenerate mature progeny.
Altered inflammatory gene expression in CML stromal progenitor cells
We compared gene expression in HSCs and LSCs and CML and normal BM 6C3+, THY+, and mSSC cells using RNA sequencing (RNA-seq) (Figure 4A). We found 298 differentially expressed genes (fold change ≥ 2, q < 0.05) in LSCs compared with HSCs (Figure 4B), 5,992 genes in CML compared with normal 6C3+ cells (Figure 4C), 6,637 genes in CML compared with normal THY+ cells (Figure 4D), and 4,341 genes in CML compared with normal mSSC cells (Figure 4E). Using gene set enrichment analysis (GSEA) analysis (Figure S3A), we found that gene sets related to tumor necrosis factor alpha (TNF-α) signaling and inflammation (Figure 4F), adipogenesis, and extracellular matrix interactions were enriched in CML compared with normal 6C3+ cells (Figures S3A and S3B) and that gene sets related to mitochondrial metabolism, MYC signaling, RNA and protein metabolism, and the cell cycle were enriched in CML compared with normal THY+ cells (Figure S3C). Gene sets related to inflammation, adipogenesis, and extracellular matrix interactions were enriched in normal compared with CML THY+ cells (Figures 4G and S3A). Consistent with in vitro differentiation assays, expression of osteogenic differentiation-related genes was increased in CML THY+ cells (Figure S3D) and of adipogenic differentiation-related genes in CML 6C3+ cells (Figure S3E).
Figure 4. Altered gene expression in CML compared with normal mesenchymal stem and progenitor cell subsets.
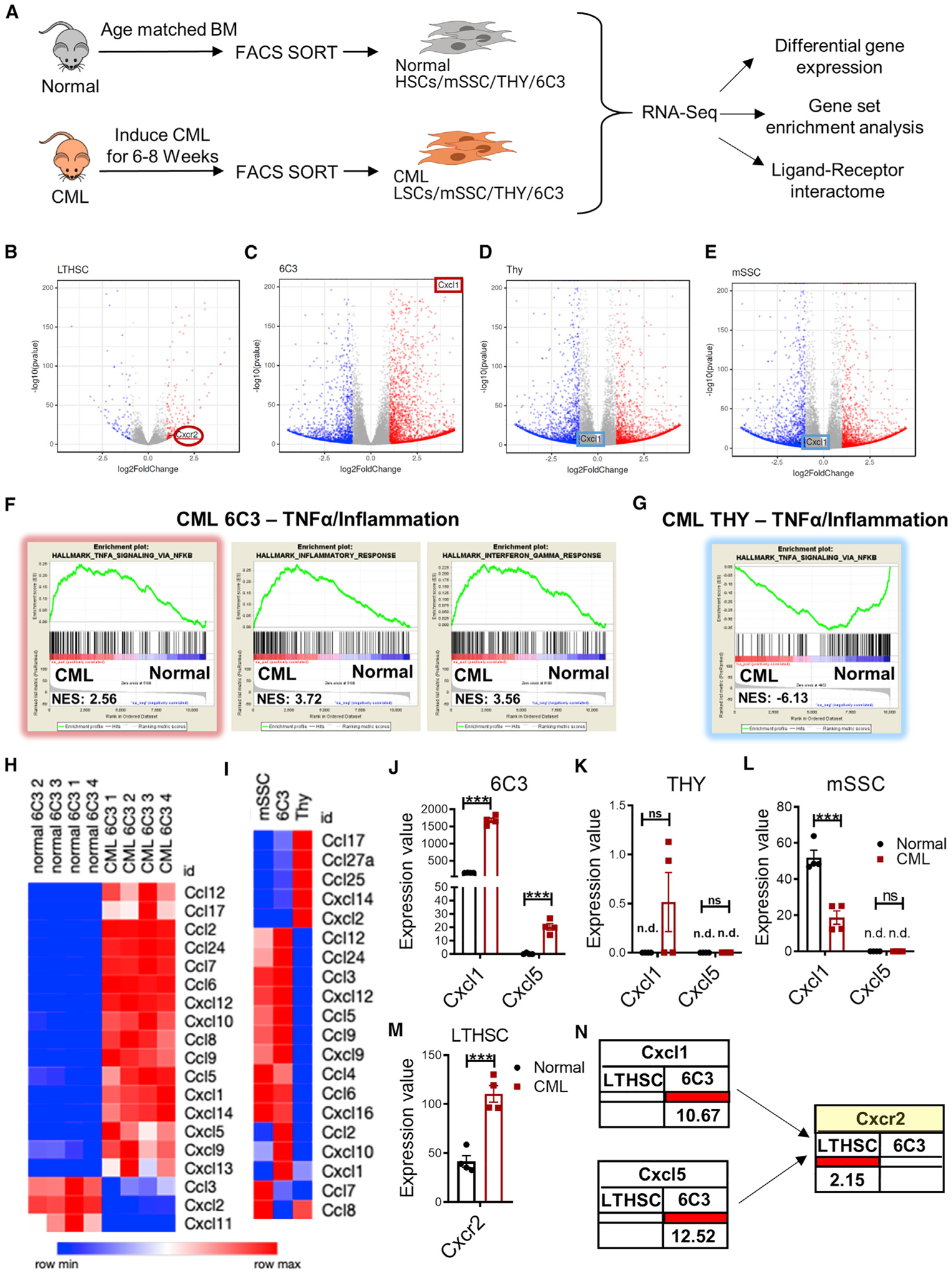
(A) Experimental schema. RNA-seq analysis was performed on FACS-sorted BM MSC subsets from normal and CML mice (8 weeks after leukemia induction) (4 biological replicates per group) and analyzed for differential gene expression, gene set enrichment analysis (GSEA), and ligand-receptor interactions.
(B–E) Volcano plot showing differentially expressed genes in FACS-sorted HSCs and LSCs (B), normal and CML 6C3+ cells (C), normal and CML THY+ cells (D), and normal and CML mSSC cells (E) (fold change ≥ 2, q < 0.05).
(F and G) GSEA of differentially expressed genes showed that CML 6C3+ cells exhibited significantly increased expression of multiple gene sets related to inflammatory response (F), including TNF-α-related genes (highlighted in red), and CML THY+ cells exhibited decreased expression of TNF-α-related genes (highlighted in blue) (G), with normalized enrichment score (NES) shown.
(H) Heatmap of cytokines and chemokines differentially expressed among normal and CML 6C3+ cells.
(I) Heatmap of cytokines and chemokines differentially expressed among CML mSSC, CML THY+ and CML 6C3+ cells.
(J–M) Pooled data from RNA-seq showing expression of CXCL1 and CXCL5 in normal and CML 6C3+ cells (J), THY+ cells (K), mSSC cells (L), and CXCR2 expression in HSCs and LSCs (M).
(N) Ligand-receptor interaction and fold change of interacting partners between CML LTHSC and CML 6C3 cells compared with normal LTHSC and normal 6C3 cells.
n.d., not determined. Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We observed altered expression of several cytokines and chemokines in CML 6C3+ cells compared with normal 6C3+ cells (Figure 4H) and other CML subsets (Figure 4I). To determine their potential significance, we analyzed how gene alterations might affect ligand-receptor interactions between LSCs and mesenchymal cells in CML BM compared with HSCs and mesenchymal cells in normal BM using an approach reported previously (Choi et al., 2017). We compiled a list of 635 pairs of ligand and receptor interactions between 182 ligands and 205 receptor genes using three public databases and mapped differentially expressed genes in CML and normal mesenchymal cells and in LSCs and HSCs to this ligand-receptor list to generate interaction maps (Table S1). This analysis revealed that expression of CXCL1 and CXCL5 was increased in CML 6C3+ cells but not THY+ or mSSC cells (Figures 4J–4L) and that expression of CXCR2, the receptor for CXCL1 and CXCL5, was increased in LSCs compared with HSCs (Figure 4M). Expression of two other CXCR2 ligands, CXCL2 and CXCL7, was reduced in CML 6C3+ cells. Therefore, CXCL1 and CXCL5 were specifically up-regulated in CML 6C3+ cells and CXCR2 in LSCs (Figure 4N). These results supported further investigation of the role of TNF-α signaling in altering 6C3+ cells and of CXCL1/5-CXCR2 interactions in LSC regulation in CML BM.
TNF-α signaling contributes to stromal progenitor alterations in CML BM
The finding of enrichment of TNF-α-related gene sets in CML compared with normal 6C3+ cells together with the knowledge that TNF-α expression is increased in CML BM led us to hypothesize that TNF-α signaling contributes to increased numbers and altered function of 6C3+ cells in CML BM. We evaluated the effect of in vivo recombinant TNF-α administration and TNF-α blockade with anti-TNF-α antibodies on mesenchymal progenitor cells in normal and CML BM (Figure 5A). TNF-α administration significantly expanded 6C3+ cells in BM of normal mice (Figure 5B), whereas administration of anti-TNF-α antibodies significantly reduced 6C3+ cells in BM of CML mice (Figure 5C). TNF-α administration also reduced THY+ cells in normal BM(Figure 5D), consistent with reports that TNF-α signaling inhibits growth of osteogenic progenitors (Osta et al., 2014). However, TNF-α administration did not alter THY+ cells in CML BM (Figure 5E), consistent with down-regulation of TNF-α signaling-related genesets in CML THY+ cells. TNF-α treatment had no significant effect on hematopoietic parameters in normal mice, as reflected by blood counts, mature lineage cells, and stem and progenitor cells. However, anti-TNF-α antibody administration reduced myeloid cells in the blood and the MPP population without major effects on other parameters (Figures S5A–S5G). In CML mice, TNF-α treatment further enhanced blood counts, with increases in myeloid cells and reduction of B cells. Anti-TNF-α antibody administration reduced overall BM cellularity, but stem/progenitor populations remained unaffected (Figures S5H–S5N). The goal of these stimulation and blocking experiments was to determine effect of TNF-α on stromal populations.However,because TNF-α has varied and complex effects on diverse hematopoietic versus microenvironmental populations, it is not possible to conclude to what extent the observed effects on hematopoiesis are related to TNF-α effects on mesenchymal cells versus other BM populations.
Figure 5. TNF-α signaling contributes to expansion and altered gene expression in 6C3+ stromal progenitors in CML BM.
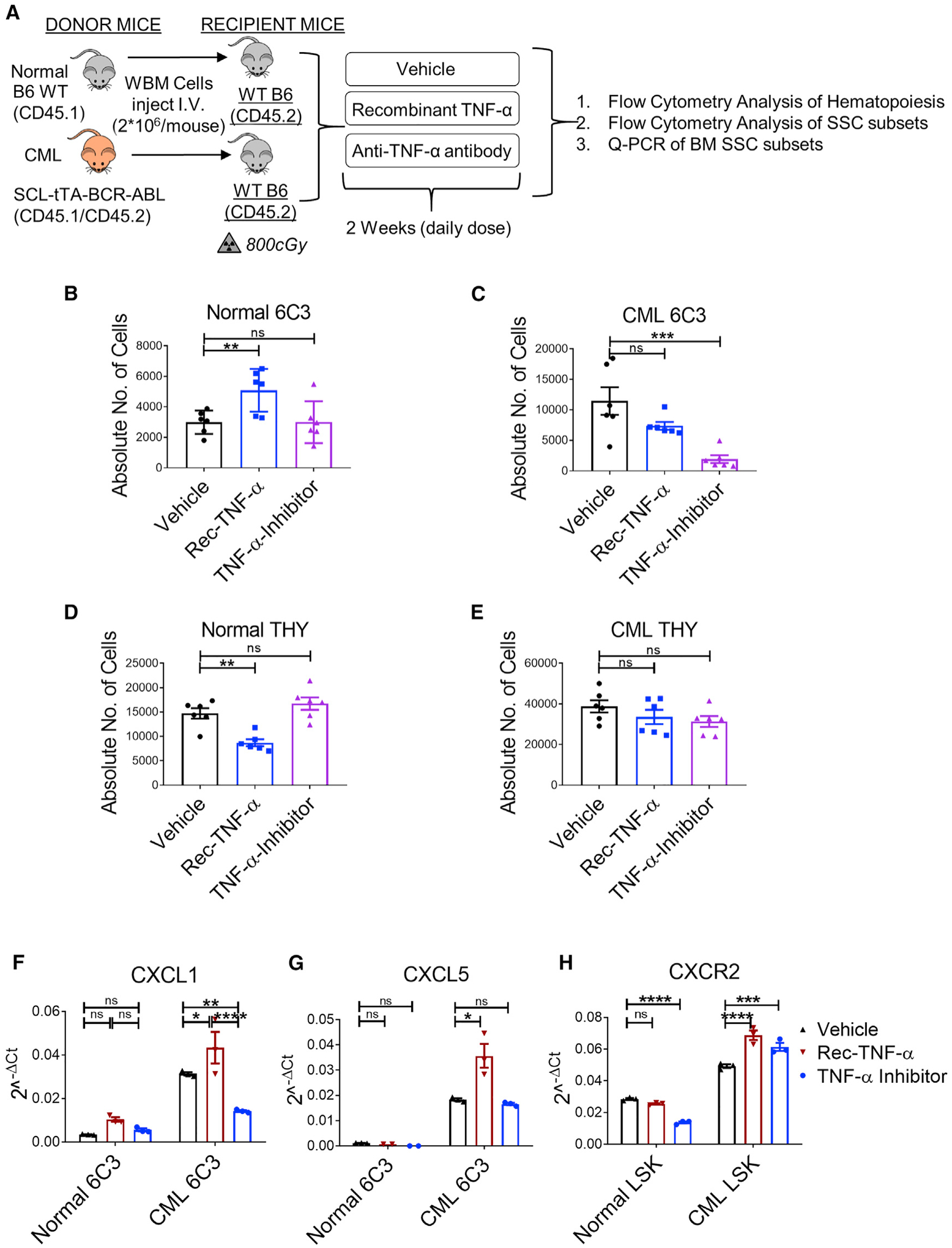
(A) Experimental schema. BCR-ABL expression was induced in SCL-tTA-BCR-ABL (CD45.1/45.2+) mice (CML) by tetracycline withdrawal for 8 weeks. BM cells from induced CML mice and aged-matched WT mice (normal) were injected into 6- to 10-week-old WT mice irradiated at 8 Gy (n = 5–6 mice/group). 8 weeks after transplantation, normal and CML mice were treated with Veh or recombinant TNF-α (0.5 μg/mouse; rec-TNF-α) or anti-TNF-α antibody (10 mg/kg, TNF-α inhibitor) once daily intraperitoneally (i.p.) for 2 weeks, after which mice were euthanized and PB and BM were analyzed.
(B and C) Absolute number of 6C3+ cells in the BM of mice engrafted with normal cells (B) and CML cells (C).
(D and E) Absolute number of THY+ cells in the BM of mice engrafted with normal cells (D) and CML cells (E).
(F and G) Relative expression of CXCL1 (F) and CXCL5 mRNA (G) determined by qPCR in normal and CML 6C3+ cells upon the indicated treatment regimens (3 biological replicates per group).
(H) Relative expression of CXCR2 determined by qPCR in normal and CML LSK stem/progenitor cells upon the indicated treatment regimens.
Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
qPCR analysis confirmed increased CXCL1 and CXCL5 expression in CML compared with normal 6C3+ cells. TNF-α treatment further increased CXCL1 (Figure 5F) and CXCL5 expression in CML 6C3+ cells (Figure 5G), and anti-TNF-α treatment significantly reduced CXCL1 but not CXCL5 expression in CML 6C3+ cells. TNF-α and anti-TNF-α treatment did not significantly affect CXCL1 or CXCL5 expression in normal BM, indicating that CML 6C3+ cells are specifically sensitive to TNF-α-induced changes in chemokine expression. These results support a role of TNF-α signaling in altered 6C3+ cell frequency and chemokine expression in CML BM.
qPCR analysis showed higher levels of CXCR2 expression in CML compared with normal hematopoietic stem/progenitors (Lin−Sca-1+c-Kit+ [LSK]). TNF-α administration enhanced CXCR2 expression in LSK from CML but not normal mice. Treatment with anti-TNF-α antibody reduced CXCR2 expression in LSK in normal but not CML mice, which instead demonstrated increased CXCR2 expression (Figure 5H). These results support a role of TNF-α in regulating CXCR2 expression in LSCs but indicate that regulation is complex and additional factors may also contribute.
CXCL1 and CXCL5 signaling through CXCR2 enhances growth of leukemic stem and progenitor cells
Murine CXCL1, CXCL5, and CXCL2/3 share the ability to signal through CXCR2 (Griffith et al., 2014). CXCR2 is expressed on neutrophils, mast cells, macrophages, and endothelial and epithelial cells and can negatively regulate myeloid progenitor cell proliferation via IL-8 (Broxmeyer et al., 1996). We used flow cytometry to confirm enhanced cell surface CXCR2 expression on CML compared with normal hematopoietic cells (Figure S6A), including CD45+ cells (Figure S6B), stem/progenitor cells (LSK) (Figure S6C), and LTHSCs (Figure S6D). To determine whether lower surface CXCR2 expression in freshly isolated LSCs is related to exogenous CXCL1 signaling, we evaluated whether CXCR2 expression is increased upon removing LSCs from a CXCL1-rich environment by measuring CXCR2 expression after culturing LSCs in the absence or presence of CXCL1 for 24–72 h. Freshly isolated LSCs were studied as controls. CML cells cultured without CXCL1 showed significantly higher CXCR2 expression compared with freshly isolated CML cells. Culture with CXCL1 led to significantly lower CXCR2 expression, consistent with this being a CXCL1-mediated effect (Figures 6A–6C). However, there was no difference in CXCR2 expression among freshly isolated normal LTHSCs or LTHSCs cultured with or without CXCL1, suggesting lack of active signaling.
Figure 6. CXCL1 and CXCL5 signaling through CXCR2 enhances growth of leukemic stem and progenitor cells.
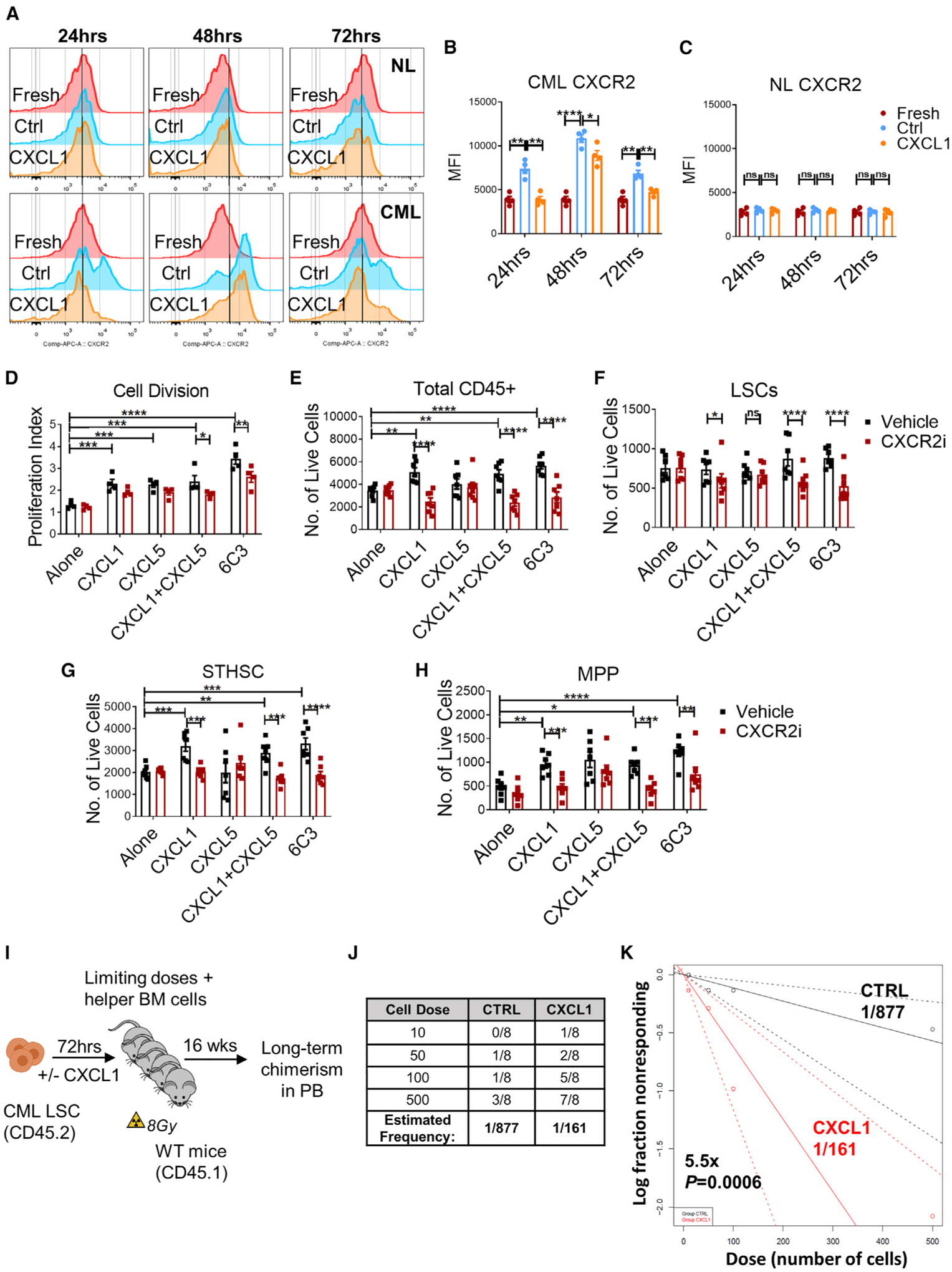
(A) Representative flow plots showing CXCR2 expression in purified HSCs from normal (NL) and LSC CML mice without in vitro culture (fresh) or cultured in the presence or absence of CXCL1 (250 ng/mL), measured at the indicated time points.
(B and C) Mean fluorescence intensity (MFI) of CXCR2 on CML LSCs (B) and normal (NL) HSCs (C) (n = 4 independent samples/group).
(D) FACS-purified CFSEmax LSCs were cultured in the presence or absence of CML 6C3+ cells, CXCL1 (250 ng/mL), CXCL5 (250 ng/mL), CXCL1 and CXCL5, or Veh with or without CXCR2i (10 μM) for 72 h (2,000 cells/condition), and their proliferation and cell numbers were analyzed.
(E–H) Absolute number of total CD45+ cells (E), LSCs (F), STHSCs (G), and MPPs (H).
(I) Experimental schema of limiting dilution competitive repopulating units (CRU) assay. FACS-purified LSCs cultured alone (CTRL) or with CXCL1 (250 ng/mL) for 72 h were transplanted in limiting doses along with helper BM cells (2.5 × 105 cells/mouse) into recipient mice irradiated at 8 cGy and followed for long-term donor chimerism in the PB for 16 weeks.
(J) Table representing the number of recipients that were positive for long-term reconstitution from the limiting dilution CRU assay (n = 8 recipients/cohort per cell dose, n = 4 donors/cohort).
(K) Line graph displaying estimates of LSC frequency in the indicated groups, with dashed lines representing 95% confidence intervals. Stem cell frequency and significance were determined using Extreme Limiting Dilution Analysis (ELDA) software.
Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To determine the response of CML LSCs to CXCL1 and CXCL5 signaling through CXCR2, we studied the effect of exposure to CXCL1, CXCL5, the combination of CXCL1 and CXCL5, and 6C3+ cells (Figure 6D) with or without the CXCR2 inhibitor (CXCR2i) SB225002 (White et al., 1998), on FACS-sorted LSCs. CML LSC cell division, as measured by carboxy-fluorescein diacetate succinimidyl ester (CFSE) labeling, was increased with each exposure but was reduced significantly in the presence of the CXCR2i (Figures 6D and S6E). Culture with CXCL1, CXCL1+CXCL5, and 6C3 cells lead to increased generation of CML STHSCs and MPPs and total number of cells. Importantly, the effects of CXCL1 and 6C3 cells were reduced significantly by addition of the CXCR2i (Figures 6E–6H). Treatment with CXCL5 had no effect on stem, progenitor, and total cell output. These results support a role of CXCL1 but not CXCL5 signaling through CXCR2 in enhancing CML LSC cycling and STHSC, MPP, and total cell production while maintaining LSC numbers. We also studied the response of HSCs to CXCL1, CXCL5, and CXCL1 plus CXCL5 on normal BM 6C3+ cells with or without the CXCR2i. HSC division was not affected by culture with CXCL1, CXCL5, or the combination. Proliferation was increased significantly in the presence of normal 6C3+ cells but was not significantly affected by the CXCR2i (Figures S6F and S6G). The total numbers of normal CD45+ cells (Figure S6H), LTHSCs (Figure S6I), STHSCs (Figure S6J), and MPPs (Figure S6K) were not changed significantly under all conditions or by addition of the CXCR2i. These results suggest that HSCs are not responsive to CXCL1 and CXCL5 stimulation and that CXCR2 signaling does not contribute to HSC support by normal BM 6C3+ cells.
We performed limiting dilution analyses to accurately quantify the effects of CXCL1 stimulation on the frequency of long-term engrafting LSCs. FACS-sorted LSCs were cultured with or without CXCL1 for 3 days and injected at limiting doses into lethally irradiated mice together with WT helper whole BM (WBM) cells. Mice were evaluated for long-term donor chimerism after 16 weeks (Figure 6I). Engraftment was scored as positive when at least 0.5% donor cells were detected in the PB (Figures 6J and 6K). The frequency of long-term repopulating LSCs was increased 5.5-fold after CXCL1 treatment, suggesting that CXCL1-CXCR2 signaling leads to expansion of functional LSCs.
CXCR2 targeting reduces LSC growth and survival alone and in combination with TKI treatment
We studied the effect of CXCR2i treatment on normal and CML hematopoiesis in vivo. BM from leukemic SCL-tTA/BCR-ABL CML mice was transplanted into irradiated congenic recipients to establish a cohort of mice with a similar time for onset of leukemia, following which mice were treated with the CXCR2i (5 mg/ kg), the second-generation BCR-ABL TKI, nilotinib (50 mg/kg), or the combination for 2 weeks (Figure 7A). CXCR2i and nilotinib treatment significantly reduced white blood cells (WBC) and neutrophils in CML mice, and the combination of CXCR2i and nilotinib resulted in significant further reduction in WBC compared with nilotinib or CXCR2i alone (Figures 7B and 7C). The combination of CXCR2i and nilotinib reduced BM cellularity compared with vehicle or nilotinib alone (Figure S7A) and markedly reduced LSC numbers compared with vehicle, nilotinib, or the CXCR2i alone (Figure 7D). Mice treated with nilotinib in combination with the CXCR2i showed significantly enhanced survival compared with mice treated with vehicle or single drugs (Figure 7E). Transplantation of FACS-purified LSCs from drug-treated mice into lethally irradiated secondary recipients showed that LSCs from nilotinib (Nil)+CXCR2i-treated mice generated significantly reduced donor chimerism and donor myeloid cells compared with LSCs from control or single drug-treated mice over 16 weeks (Figures 7F and S7B). These results indicate that in vivo treatment with a CXCR2i in combination with TKI markedly inhibits CML hematopoiesis and significantly depletes LSC with long-term repopulating capacity. Treatment with the CXCR2i and Nil alone or in combination had minimal effects on total PB counts of healthy normal mice (Figure S7C). CXCR2i treatment resulted in a small but significant increase in the frequency of neutrophils in PB (Figure S7D). No significant changes in total BM cellularity (Figure S7E) and the number of LTHSCs in BM were seen after treatment with either drug alone or in combination (Figure S7F). To further investigate the effect on HSC function, we performed secondary transplantation of FACS-purified HSCs into lethally irradiated recipient mice to evaluate their long-term repopulating capacity. Transplantation of HSCs from Nil+CXCR2i-treated mice resulted in similar donor chimerism as with HSCs from control or single drug-treated mice (Figures S7G and S7H). These results suggest that BM stromal cell-derived CXCL1-CXCR2 signaling is dispensable for HSC function, at least in the steady state.
Figure 7. CXCR2 targeting reduces LSC growth and survival, which is enhanced in combination with TKI treatment.
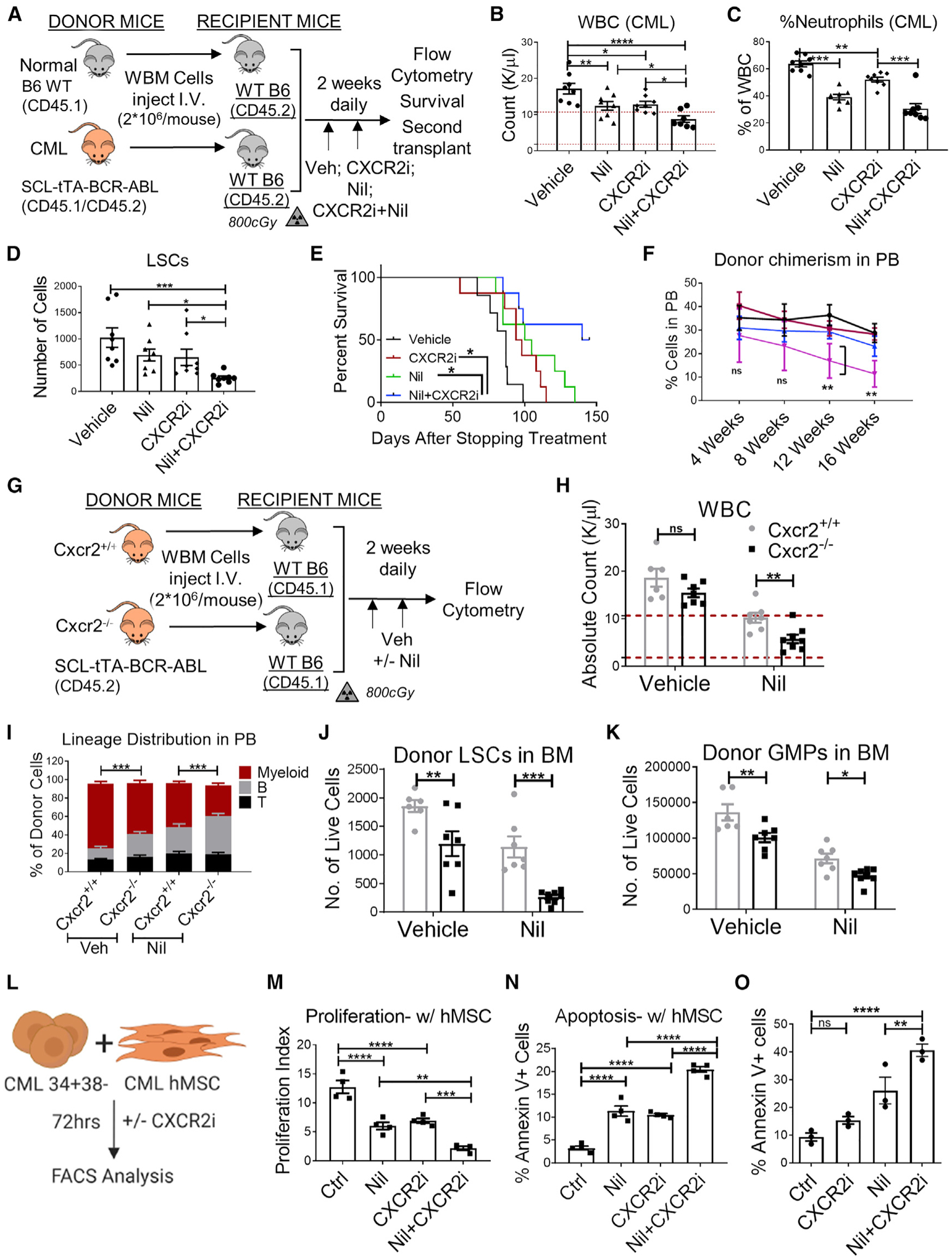
(A) Experimental schema. BCR-ABL expression was induced in SCL-tTA-BCR-ABL mice (CML) by tetracycline withdrawal. WBM cells obtained 8 weeks after leukemia induction from CML mice and from WT healthy mice (normal) were injected into WT mice irradiated at 8 Gy (n = 6–8 mice/group). Mice were treated 8 weeks after transplantation with Veh, CXCR2i (5 mg/kg), Nil (50 mg/kg), or the combination once daily by oral gavage for 2 weeks, after which they were euthanized and PB and BM were analyzed by flow cytometry.
(B–D) WBC numbers (B) and neutrophil frequency (C) in the PB and LSC numbers in (D) in the BM (2 femora + 2 tibiae) of CML mice.
(E) A cohort of mice was followed for survival after stopping treatment (n = 7–8 mice/group).
After primary transplantation of CML WBM cells from (A), LTHSCs were FACS-purified and transplanted into secondary healthy WT mice irradiated at 800 cGy.
(F) Serial blood draw was performed every 4 weeks until 16 weeks.
(G) WBM cells from Cxcr2+/+-SCL-tTA-BCR-ABL or Cxcr2−/−-SCL-tTA-BCR-ABL double-mutant mice (2 × 106 cells/mouse, n = 6–8 mice/group) were injected into WT mice irradiated at 8 Gy (n = 6–8 mice/group). Transplanted mice were maintained off doxycycline to induce BCR-ABL expression and generate leukemia. Mice were treated 8 weeks after transplantation with Veh or Nil (50 mg/kg) once daily by oral gavage for 2 weeks and euthanized, and PB and BM were analyzed by flow cytometry.
(H–K) Total WBC (H) and frequency of multi-lineage reconstitution of donor cells (I) in the PB and numbers of donor LSCs (J) and GMP (K) in the BM (2 femora + 2 tibiae).
(L) Primary human CML CD34+ cells were labeled with CFSE, and CFSEmax CD34+CD38− primitive LSCs were FACS purified and co-cultured with or without primary human BM MSC (hMSCs) with nilotinib (Nil; 1 μM), the CXCR2i (10 μM), or both or were left untreated for 72 h (n = 4 biological replicates). The proliferation index was calculated on the basis of reduction in CFSE levels.
(M and N) The effects of treatment on the proliferation index in the presence of hMSC cells (M) and the percentage of apoptosis, calculated based on Annexin V+ labeling, of CML cells in the presence of hMSCs (N) are shown.
(O) Annexin V+ labeling of murine LSCs cocultured in the presence of CML 6C3+ cells and treated with various drugs for 48 h.
Error bars represent mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
To confirm results obtained with pharmacological inhibitors, we performed genetic targeting of CXCR2 in LSCs by generating CML (SCL-tTA-BCR-ABL)-Cxcr2−/− double mutant mice. 8- to 12-week-old adult healthy WT mice (CD45.1+) were irradiated at 800 cGy and transplanted with BM cells obtained from CML-Cxcr2+/+ or CML-Cxcr2−/− mice. This approach supported assessment of the effects of CXCR2 deletion selectively in hematopoietic cells and generation of cohorts of mice with similar times of onset of leukemia. Mice were maintained off tetracycline, and PB samples were obtained 8 weeks after transplantation to confirm development of neutrophilic leukocytosis, following which mice were treated with vehicle (Veh) or Nil (50mg/kg) for 2 weeks, euthanized, and PB and BM cells were harvested for FACS analysis (Figure 7G). Consistent with results with the CXCR2i, CML-Cxcr2−/− knockout (KO) cells exhibited significantly reduced donor chimerism (Figure S7I), reduced donor myeloid (Gr-1+CD11b+), and increased donor B (B220+CD19+) cells in PB (Figures 7H and 7I) and reduced LSCs, STHSCs, and granulocyte/macrophage progenitors (GMPs) in BM compared with CML-Cxcr2+/+ mice (Figures 7J, 7K, S7J, and S7K). Treatment with Nil led to significant reductions in leukocytosis and myeloid cells, increased B cells in the PB, and drastic reductions of BM LSCs, MPPs, and GMPs in CML-Cxcr2−/− compared with CML-Cxcr2+/+ mice. Therefore, genetic targeting of CXCR2 confirms a critical role of CXCR2 signaling in CML leukemogenesis and in maintenance and TKI resistance of CML LSC.
IL-8/CXCL8 is the major CXCR2 ligand in humans but is not expressed in rodents. Murine CXCL1, CXCL5, and CXCL2/3 are homologs of human IL-8/CXCL8, CXCL5, and CXCL1/2/3. To study the effect of CXCR2i treatment on human CML LSC, CML CD34+38- stem/primitive progenitor cells were labeled with carboxyfluorescein succinimidyl ester (CFSE), cocultured with or without primary human CML BM-derived stromal cell (hMSC) monolayers, and treated with the CXCR2i, Nil, or the combination (Figure 7L). The CXCR2i reduced proliferation and enhanced apoptosis of CML CD34+CD38− cells cocultured with hMSCs (Figures 7M and 7N). The inhibitory effects of the CXCR2i on proliferation and survival were not seen in the absence of stromata (Figures S7L–S7N). Combination of the CXCR2i and Nil enhanced apoptosis and inhibited proliferation of CML CD34+CD38− cells compared with Nil alone. Finally, we also co-cultured FACS-purified murine CML LSCs and CML 6C3+ cells and treated them with the CXCR2i and Nil. As seen with the human CD34+CD38− cells, treatment with Nil in combination with the CXCR2i resulted in enhanced apoptosis of murine LSCs compared with Veh control or individual drugs (Figures 7O and S7O). These observations support the importance of CXCR2 signaling in stroma-mediated regulation of human CML LSC proliferation, survival, and TKI resistance.
DISCUSSION
The BMM is made up of diverse cell types that play an essential role in maintaining tissue homeostasis and HSC function. The critical role of the BMM in health, disease progression, and drug resistance of hematological malignancies is increasingly recognized (Arranz et al., 2014; Hanoun et al., 2014; Lim et al., 2016; Shafat et al., 2017). The current study addresses gaps in knowledge regarding the role of heterogeneous mesenchymal cells in LSC maintenance and resistance and the underlying regulatory mechanisms. We show that CML development leads to increased proliferation, clonogenic potential, and expansion of THY+ bone-forming progenitors and 6C3+ stromal progenitors. We show that, unlike their normal counterparts, CML mesenchymal populations fail to support normal HSC engraftment and regeneration of mature progeny and identify a specific role of CML 6C3+ stromal progenitors in selectively supporting CML LSC self-renewal and engraftment. Detailed characterization of 6C3+ cells indicates that they overlap with Nestin+ LepR+ Prx1−Cre+ perivascular MSCs with pre-adipocytic features. Our studies reveal an important role of TNF-α signaling in driving expansion and increased CXCL1 expression in 6C3+ stromal progenitors and of CXCL1 signaling through the CXCR2 receptor in LSC maintenance and TKI resistance. Our re sults support further evaluation of targeting the CXCL1/CXCR2 axis as a critical strategy to deplete BM niche-protected, TKI-resistant CML LSCs.
Inflammatory signaling through TNF-α has complex effects on hematopoiesis, including inducing apoptosis in differentiated hematopoietic cells while also activating stem cells to enhance regeneration and recovery. Acute TNF-α exposure has been shown to enhance nuclear factor κB (NF-κB)-dependent gene expression to promote HSC survival and myeloid differentiation (Yamashita and Passegué, 2019). Besides direct effects, inflammatory factors may also modulate MSC function to influence hematopoiesis (Raaijmakers, 2011; Takizawa and Manz, 2017). Granulocyte-derived TNF-α has been shown to promote vascular and hematopoietic regeneration in the BM (Bowers et al., 2018). TNF-α signaling promotes clonogenic capacity and disease progression in myeloid malignancies (Abegunde et al., 2018; Fleischman et al., 2011; Gallipoli et al., 2013). The current studies reveal that TNF-α also modifies BM stromal progenitors to enhance leukemic hematopoiesis. TNF-α levels are increased in CML compared with normal BM (Zhang et al., 2012). CML 6C3+ cells show increased expression of TNF-α and NF-κBB-related gene sets. Our gene expression and ligand-receptor interactome analysis reveal upregulation of CXCL1 and CXCL5 in CML 6C3+ cells and their cognate receptor CXCR2 in LSCs. We further show that TNF-α signaling contributes to increased CXCL1 expression in CML BM stromal progenitors and increased CXCR2 expression in LSCs.
IL-8/CXCL8 is the major CXCR2 ligand in humans but is not expressed in rodents (Asfaha et al., 2013). Murine CXCL1 is the homolog of human IL-8/CXCL8. CXCR2 is expressed on neutrophils, mast cells, macrophages, and endothelial and epithelial cells and mediates neutrophil mobilization and migration to sites of inflammation and the angiogenic effects of IL-8. CXCR2 expression is associated with poor outcomes in cancer. Promyelocytic leukemia (PML)-regulated expression of pro-inflammatory cytokines, including IL-6 and CXCL1, in MSCs has been shown to support leukemic cell growth (Guarnerio et al., 2018). However, the role of CXCL1 in LSC regulation in leukemic BMM has not been studied. We show here that enhanced CXCL1 expression in the CML BMM selectively expands LSCs and leukemic progenitors through CXCR2-dependent mechanisms. A CXCR2i reduced LSC proliferation and reduced leukemic stem, progenitor, and mature cells in vivo. The CXCR2i, when combined with TKI, markedly increased inhibition of leukemic cells and LSCs. The CXCR2i also reduced proliferation and enhanced apoptosis of human CML CD34+CD38− cells cocultured with human CML BM stromal cells, and the combination of a CXCR2i and Nil resulted in enhanced inhibition compared with Nil alone. Importantly, genetic targeting of CXCR2 confirmed the critical role of CXCR2 signaling in CML leukemogenesis and in maintenance and TKI resistance of CML LSCs. These studies reveal an important role for paracrine CXCL1-CXCR2 signaling in supporting growth and TKI resistance of LSCs.
Although our studies have focused on direct CXCL1 effects on LSCs, CXCL1/IL8-CXCR2 interactions may also affect leukemic hematopoiesis indirectly by effects on neutrophil migration and angiogenesis. In normal hematopoiesis, stimulation of CXCR2 signaling via CXCL1 in endothelial cells results in loss of macrophages and HSPC mobilization (Karpova et al., 2019). Similarly, BM dendritic cell regulation of BM vascular sinusoidal CXCR2 signaling and permeability plays an important role in HSPC trafficking (Zhang et al., 2019). A secreted form of DEK may also regulate mouse and human HSC function via CXCR2 signaling (Capitano et al., 2019). We have shown that CXCL12 deletion from Prx1-Cre transgene-expressing mesenchymal progenitors leads to loss of LSC quiescence and enhanced LSC sensitivity to TKI treatment (Agarwal et al., 2019). Previous studies also suggest that elevated levels of CCL3 in the BMM could contribute to CML development in the murine model (Schepers et al., 2013). The interaction between inflammatory signaling via TNF-α and IL-8/CXCL1 and the niche regulatory function of CXCL12-expressing mesenchymal progenitors and with other chemokines in LSC regulation is not known and warrants further analysis in future studies.
Our studies support an important role of inflammatory signaling through TNF-α in expanding and modifying stromal progenitors in CML BM to differentially support LSCs compared with HSCs via overexpression of CXCL1, which interacts with the CXCR2 receptor that is overexpressed on LSCs. These observations support further studies exploring targeting of CXCL1-CXCR2 interactions as a critical and effective strategy to eliminate BMM-protected, TKI-resistant LSCs. CXCR2 antagonists are being tested in clinical trials for chronic obstructive pulmonary disease (COPD), transplant rejection, and influenza (Ha et al., 2017; Kirsten et al., 2015). Our studies are also relevant to other malignancies where inflammatory signaling may play a role, including MDS and AML, where IL-8 and CXCR2 overexpression may contribute to proliferation of malignant stem/progenitor cells (Heuser et al., 2011; Schinke et al., 2015).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the lead contact, Dr. Ravi Bhatia (rbhatia@uabmc.edu).
Materials availability
All mouse lines used in this study were purchased from Jackson Laboratory. Sources and identifiers of these lines are provided in the key resources table. All unique resources generated in this study are available from the lead contact with a completed materials transfer agreement.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3 | eBioscience | Cat#13-0031-85; RRID:AB_466320 |
| Anti-mouse CD4 | eBioscience | Cat#13-0041-85; RRID:AB_466326 |
| Anti-mouse CD8 | eBioscience | Cat#13-0083-85; RRID:AB_657763 |
| Anti-mouse B220 | eBioscience | Cat#13-0452-85; RRID:AB_466450 |
| Anti-mouse CD19 | eBioscience | Cat#13-0193-85; RRID:AB_657658 |
| Anti-mouse IgM | eBioscience | Cat#13-5790-85; RRID:AB_466676 |
| Anti-mouse Gr-1 | eBioscience | Cat#13-5931-85; RRID:AB_466801 |
| Anti-mouse CD11b | eBioscience | Cat#13-0112-85; RRID:AB_466360 |
| Anti-mouse NK1.1 | eBioscience | Cat#13-5941-85; RRID:AB_466805 |
| Anti-mouse Ter119 | eBioscience | Cat#13-5921-85; RRID:AB_466798 |
| Anti-mouse CD117 | eBioscience | Cat#47-1172-82; RRID:AB_1582226 |
| Anti-mouse CD11b | eBioscience | Cat#47-0112-82; RRID:AB_1603193 |
| Anti-mouse CD16/32 | eBioscience | Cat#56-0161-82; RRID:AB_493994 |
| Anti-mouse CD45 | eBioscience | Cat#56-0451-82; RRID:AB_891454 |
| Anti-mouse B220 | eBioscience | Cat#56-0452-82; RRID:AB_891458 |
| Anti-mouse CD135 | eBioscience | Cat#15-1351-82; RRID:AB_494219 |
| Anti-mouse Sca-1 | eBioscience | Cat#12-5981-82; RRID:AB_466086 |
| Anti-mouse CD48 | eBioscience | Cat#17-0481-82; RRID:AB_469408 |
| Anti-mouse CD34 | eBioscience | Cat#50-0341-82; RRID:AB_10596826 |
| Anti-mouse CD45.1 | eBioscience | Cat#25-0453-82; RRID:AB_469629 |
| Anti-mouse CD45.2 | eBioscience | Cat#11-0454-85; RRID:AB_465062 |
| Anti-mouse CD31 | eBioscience | Cat#48-0311-82; RRID:AB_10598807 |
| Anti-mouse CD51 (Integrin alpha V) | eBioscience | Cat#13-0512-85; RRID:AB_466478 |
| Anti-mouse CD140α | eBioscience | Cat#25-1401-82; RRID:AB_2573400 |
| Anti-mouse Gr-1 | eBioscience | Cat#12-5931-83; RRID:AB_466046 |
| Anti-mouse CD19 | eBioscience | Cat#17-0193-82; RRID:AB_1659676 |
| Anti-mouse Ter119 | 3ioLegend | Cat#116220; RRID:AB_528963 |
| Anti-mouse Sca-1 | 3ioLegend | Cat#108126; RRID:AB_10645327 |
| Anti-mouse CD3 | 3ioLegend | Cat#100218; RRID:AB_1595492 |
| Anti-mouse CD150 | 3ioLegend | Cat#115922; RRID:AB_2303663 |
| Streptavidin | 3ioLegend | Cat#405229 |
| Anti-mouse Tie2 | 3ioLegend | Cat# 124006, RRID:AB_2203221 |
| Anti-mouse Thy1.1 | Thermo Fisher Scientific | Cat# 47-0900-82, RRID:AB_1272252 |
| Anti-mouse Thy1.2 | Thermo Fisher Scientific | Cat# 47-0902-82, RRID:AB_1272187 |
| Anti-mouse CD105 | BioLegend | Cat#120410; RRID:AB_1027700 |
| Anti-mouse Ly-51 (6C3/BP-1) | BioLegend | Cat#108305; RRID:AB_313362 |
| Anti-mouse CD200 | BioLegend | Cat#123810; RRID:AB_10900447 |
| Anti-mouse CD182 (CXCR2) | BioLegend | Cat#149310; RRID:AB_2566148 |
| Anti-Annexin V | BioLegend | Cat#640920; RRID:AB_2561515 |
| Anti-CD117 MicroBeads | Miltenyi Biotech | Cat# 130-091-224; RRID:AB_2753213 |
| Ki-67 Monoclonal Antibody (SolA15), PerCP-eFluor 710 | Thermo Fisher Scientific | Cat# 46-5698-82, RRID:AB_11040981 |
| Biological samples | ||
| Peripheral blood or bone marrow CML samples | University of Alabama at Birmingham Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Doxycycline Rodent Diet | Envigo | Cat#TD.09761 |
| Sulfatrim-based diet (5053/.025%Tri/.1242%Sulf ■■■ IRR) | TestDiet | Cat#5W8F |
| MEM α, nucleosides, no phenol red | Thermo Fisher Scientific | Cat# 41061029 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat#472301 |
| Nilotinib | Novartis Pharmaceuticals | N/A |
| Collagenase II | Sigma-Aldrich | Cat# C1764 |
| Dispase II | Sigma-Aldrich | Cat#4942078001 |
| DNase I | Sigma-Aldrich | Cat#D4263 |
| BSA | Sigma-Aldrich | Cat#A9418 |
| DAPI | Thermo Fisher Scientific | Cat#D1306; RRID:AB_2629482 |
| CountBright Absolute Counting Beads | Thermo Fisher Scientific | Cat# C36950 |
| Recombinant Murine IL-3 | Peprotech | Cat#213-13 |
| Recombinant Murine IL-6 | Peprotech | Cat#216-16 |
| Recombinant Murine SCF | Peprotech | Cat#250-03 |
| Recombinant Murine TPO | Peprotech | Cat#315-14 |
| Recombinant Mouse CXCL1/KC | R&D Systems | Cat#453-KC-050 |
| Recombinant Mouse CXCL5/LIX | R&D Systems | Cat#433-MC-025 |
| Methylene Blue | Millipore Sigma | Cat#M9140-25G |
| Oil Red O Solution | Lifeline Cell Technology | Cat# LL-0052 |
| Alizarin Red S (2%) | Lifeline Cell Technology | Cat# CM-0058 |
| SB225002 | Selleckchem | Cat#S7651 |
| Recombinant mouse TNFα (clone MP6-XT22) | Biolegend | Cat#718004 |
| InVivoMab anti-mouse TNFα antibody | BioXCell | Cat# BE0058, RRID:AB_1107764 |
| Critical commercial assays | ||
| RNeasy Micro Kit | QIAGEN | Cat#74004 |
| CellTrace CFSE Cell Proliferation Kit | Thermo Fisher Scientific | Cat#C34554 |
| CD34 Microbead Kit | Miltenyi Biotech | Cat#130-046-702 |
| Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat#C10633 |
| MesenCult Adipogenic Differentiation Kit (Mouse) | Stem Cell Technologies | Cat#05507 |
| MesenCult Osteogenic Stimulatory Kit (Mouse) | Stem Cell Technologies | Cat#05504 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat#FC-131-1096 |
| SMART-Seq v4 Ultra Low Input RNA Kit | Takara Bio | Cat#634891 |
| Deposited data | ||
| RNA Seq data | UAB Bioinformatics Core | GEO: GSE126547 |
| Experimental models: Organisms/strains | ||
| Mouse: SCL-tTA-BCR-ABL | Ravi Bhatia Lab | N/A |
| Mouse: B6.SJL-PtprcaPepcb/BoyCrCrl | Charles River Laboratory | Cat#564 |
| Mouse: C57BL/6NCr | Charles River Laboratory | Cat#556 |
| Mouse: Ai9 (B6.Cg-Gt (Rosa) 26Sortm9(CAG-tdTomato)Hze/J) | Jackson Laboratory | Stock No: 007909 |
| Mouse: B6.Cg-Tg(Prrx1-cre)1Cjt/J | Jackson Laboratory | Stock No: 005584 |
| Mouse: B6.Cg-Tg(Sp7-tTA,tetO-EGFP/cre) 1Amc/J | Jackson Laboratory | Stock No: 006361 |
| Mouse: B6N.FVB-Tg(BGLAP-cre)1Clem/J | Jackson Laboratory | Stock No: 19509 |
| Mouse: B6.129S2(C)-Cxcr2tm1Mwm/J | Jackson Laboratory | Stock No: 006848 |
| Mouse: Cxcr2-SCL-tTA-BCR-ABL | This paper | N/A |
| Oligonucleotides | ||
| Primers (see Table S2 for sequences) | Thermo Fisher Scientific | N/A |
| Software and algorithms | ||
| FlowJo software (version 8.5.2) | FlowJo | RRID:SCR_008520; (https://www.flowjo.com/) |
| BD FACSDiva | BD Biosciences | RRID:SCR_001456; (https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software#Overview) |
| GraphPad Prism 7 | GraphPad | RRID:SCR_002798; (https://www.graphpad.com/scientific-software/prism/) |
| ModFit LT | Verity | RRID:SCR_016106; (http://www.vsh.com/products/mflt/) |
| Extreme limiting dilution analysis (ELDA) | http://bioinf.wehi.edu.au/software/elda/ | http://bioinf.wehi.edu.au/software/elda/ |
Data and code availability
The RNA sequencing data discussed in this publication has been deposited in NCBI’s Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The GEO accession number is listed in the key resources table. This study does not report development of new code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Samples were obtained from patients with CML (aged 40–65 years) in chronic phase (CP) without prior imatinib treatment seen at the University of Alabama at Birmingham Hospital. Samples were processed for CD34+ cell selection with CliniMACS (Miltenyi Biotech, Teterow, Germany). Mononuclear cells were isolated by Ficoll-Hypaque (Sigma Diagnostics, St. Louis, MO) separation. CD34+ cells were isolated using immunomagnetic beads (Miltenyi Biotech, Auburn, CA). CD34+ CD38− cells were obtained by flow cytometry sorting. Equal numbers of male and female subjects were used. Although numbers are insufficient for statistical analysis, review of the data did not show a significant impact of sex on the results obtained. All subjects signed an informed consent form. Sample acquisition was approved by the Institutional Review Board at the University of Alabama at Birmingham Hospital, in accordance with an assurance filed with and approved by the Department of Health and Human Services, and met all requirements of the Declaration of Helsinki.
Mice
SCL-tTA-BCR-ABL mice were maintained on CD45.2+ congenic C57BL/6 background. WT C57BL/6 mice (CD45.2+; stock no. 027) were obtained from Charles River laboratories (Frederick, MD). For certain experiments, SCL-tTA-BCR-ABL mice (CD45.2+; immune competent, C57BL/6J) were crossed with B6-Ly5.1/Cr mice (CD45.1+; immune competent, C57BL/6J; Charles River, Frederick, MD) to generate mice on the CD45.1/CD45.2 C57BL/6 background. B6.129S2(C)-Cxcr2tm1Mwm/J (mIL-8Rh-, stock no. 006848) mice were obtained from Jackson Laboratory (Bar Harbor, ME), and crossed with SCL-tTA-BCR-ABL mice to generate Cxcr2−/−-SCL-tTABCR-ABL double mutant mice on CD45.2 background. Because Cxcr2−/− mice have increased susceptibility to various pathogens, they were maintained in a gnotobiotic facility. All CML mice were maintained on tetracycline by feeding them doxycycline containing food (cat no. TD.09761, Envigo, New Jersey). Unless otherwise mentioned, all experiments were performed using 6–10 weeks old mice of both sexes. All experiments involving primary CML mice were performed at 4–8 weeks after doxycycline withdrawal and confirmation of neutrophilic leukocytosis. Complete blood counts and differential counts were analyzed using Hemavet HV950FS (Drew Scientific, Miami Lakes, FL). Experimental mice were separated by sex and housed with 5 mice per cage. All the mice were drug or test naive and not involved in previous procedures. Mice of similar age were randomly divided into experimental groups. Investigators were blinded to the mice genotype during treatments or monitoring survival. All experimental mice received autoclaved water and clean rodent chow diet. Mice were subjected to 12-hour light/dark cycles, and controlled ambient room temperature (20–22°C), and air humidity (30%–70%). All mice were maintained in Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) specific-pathogen-free animal care facilities, and all procedures were carried out in accordance with federal guidelines and protocols approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham and Cincinnati Children’s Hospital Medical Center. All the various animal models utilized in this study are summarized in the Key resources table.
Primary human mesenchymal stromal cell culture (hMSC)
hMSC cultures were successfully derived from the bone marrow aspirates of patients with untreated newly diagnosed chronic-phase CML. Fresh BM mononuclear cells isolated using Ficoll-Hypaque density gradient separation (2 to 5 million cells) were cultured in α-MEM, 10% heat-inactivated fetal bovine serum, 1% penicillin–streptomycin (media and supplements from Invitrogen, Carlsbad, CA). After 72 h, non-adherent cells were removed by washing with phosphate-buffered solution and fresh media added. Over a period of 2–3 weeks, adherent, spindle-shaped cells were derived and subcultured weekly by trypsinization for ~3–4 passages. hMSC expressed the traditional CD73, CD90, CD105, and CD146 phenotypic markers when measured by flow cytometry (data not shown).
METHOD DETAILS
Analysis of apoptosis and proliferation
Apoptosis was assessed by labeling cells with Annexin V-APC (BD Biosciences PharMingen, San Diego, CA) and DAPI. Proliferation was assessed by labeling cells with 5- (and 6-) carboxy-fluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR). CML CD34+CD38-CFSEmax cells were cultured for 3 days and CFSE analyzed by flow cytometry. ModFit software (Verity, Top-sham, ME) was used to fit data, determine percentage of cells in each generation, and generate a proliferation index. The position of the parent generation was based on cell aliquots treated with 4% paraformaldehyde immediately after sorting.
Flow cytometry
Peripheral blood from mice was obtained via retro-orbital bleeding in EDTA-coated tubes. After red blood cell lysis with ACK lysis buffer, peripheral blood and BM cells were stained with appropriate antibodies as summarized in the Key resources table, and analyzed using BD Fortessa X-20 and BD LSRII flow cytometers (BD Biosciences, San Jose, CA). Mature populations were identified as follows: myeloid cells (Gr-1+Mac-1+), B cells (B220+CD19+), T cells (CD3+). For analysis of hematopoietic stem/progenitor cells, femurs and tibias were crushed gently with mortar and pestle to dissociate the BM fraction and filtered through 70-μm cell strainer (BD biosciences). Filtered cells were analyzed as follows: LSK (Lin−Sca-1+c-Kit+), long-term hematopoietic stem cells (LTHSC; Lin−Sca-1+c-Kit+Flt3-CD150+CD48−), short-term hematopoietic stem cells (STHSC; Lin−Sca-1+c-Kit+Flt3-CD150−CD48−), multipotent progenitors (MPP; Lin−Sca-1+c-Kit+Flt3-CD48+). For analysis of various stromal populations, a protocol described by Gulati et al. (2018) was followed. Briefly, after the removal of muscle and connective tissue, femurs and tibias were crushed gently with mortar and pestle to dissociate the BM fraction and filtered through 70-μm cell strainer (BD biosciences). Residual BM chips were digested with BM digestion buffer containing collagenase II (1 mg/mL; Sigma), dispase II (5 mg/mL; Sigma), DNase I (0.1 mg/mL; Sigma), BSA (1 mg/mL; Sigma) and HEPES, and gently agitated for 60 minutes at 37°C. The digested bones were then filtered through a 70 μm strainer (BD Biosciences) and the marrow cells were collected, and red blood cells were depleted with ACK lysis buffer. The BM populations were stained with antibodies mentioned in Key resources table, and analyzed by flow cytometry on BD Fortessa X-20 flow cytometer (BD Biosciences, San Jose, CA). To calculate the absolute number of various BM hematopoietic and non-hematopoietic subsets whenever required, CountBright Absolute Counting Beads (cat no. C36950; Thermo Fisher Scientific) were mixed with the cell sample (per 4 bones; 2 femurs and 2 tibia) and assayed via flow cytometry. By comparing the ratio of bead events to cell events, absolute numbers of cells in the sample were calculated.
Bone marrow transplantation
For analyzing leukemia-induced changes on BMM, 6–10 weeks old adult healthy WT mice (CD45.2+) were irradiated at 400cGy or 800cGy (split dose of 400cGy with 2–3hrs apart) and transplanted with WBM cells (2*106 cells/mouse) obtained from either age-matched normal WT mice or CML mice (8 weeks post-doxycycline withdrawal). Since the primary recipient mice develop leukemia within 8–12 weeks and are at risk of becoming sick and dying beyond this time period, mice were sacrificed at 12 weeks post-transplant to analyze the BM subsets. In co-culture experiment with BMM subsets, the long-term engraftment capacity of normal HSCs or CML LSCs was monitored in PB serially every 4 weeks until 20 weeks. For primary transplantation experiments, WBM cells were obtained from normal WT mice or CML mice and transplanted (2*106 cells/mouse) into CD45.2 recipient mice irradiated at 800 cGy. For secondary transplantation experiments, BM cells from mice receiving different treatments were pooled and 2*106 cells/ mouse (6 mice per condition) were transplanted into WT recipient mice irradiated at 800 cGy. Multi-lineage donor cell engraftment was monitored in PB serially every 4 weeks until 16 weeks. In all transplantation experiments, recipient mice were placed on sulfatrim-based food (5053/.025%Tri/.1242%Sulf ½ IRR; Cat No: 5W8F; TestDiet, Richmond, IN) for 2 weeks post-transplantation to avoid any infection/toxicity-associated with lethal irradiation.
Limiting dilution CRU assay
FACS sorted LSCs from CML mice were cultured in Stemspan serum-free enhanced medium (SFEM; Stem Cell Technologies, Vancouver, BC, Canada) supplemented with 10ng/ml murine IL-3, IL-6, SCF, TPO in the presence or absence of CXCL1 (250ng/ml) for 3 days at 37°C, 5% CO2. Cells were harvested and injected in serial dilutions into lethally irradiated recipients (CD45.1), together with 2 × 105 CD45.1+ competitor BMNC, to determine competitive repopulating units (CRU) frequency. Sixteen weeks after transplantation, the percentage of donor-derived cells was determined and CRU was calculated as described previously (Miller et al., 2008). Mice showing more than 0.5% reconstitution were considered positive. ELDA software (http://bioinf.wehi.edu.au/software/elda/; Hu and Smyth, 2009) was used to calculate LTHSC frequency and statistical significance.
Cell cycle analysis
For each biological replicate, BM from 2 mice were pooled and stained with Ki-67 Monoclonal Antibody (SolA15), PerCP-eFluor 710 (Thermo Fisher Scientific, catalog no. 46-5698-82) by following manufacturer’s instructions. Ki67+ stromal cells were then identified on a BD LSRII flow cytometer.
Fibroblastic colony forming units (CFU-F) assay
CFU-F assay was performed as described previously (Robey et al., 2014). Briefly, FACS sorted SSC subsets were seeded at a clonal density of 10,000 cells per well (1.9 cm2) of a 24-well plate (Corning, NY) in phenol red-free α-MEM (Cat no. 41061029; Thermo Fisher Scientific) plus 10% fetal bovine serum (Atlanta Biologicals, GA) and 1% penicillin/streptomycin (Invitrogen). One half of the medium was replaced after 7 days and cells were stained with methylene blue (Millipore Sigma, Burlington, MA) at day 14 and colonies were counted.
MSC differentiation culture
FACS sorted MSC subsets were first grown for 2 weeks in phenol red-free α-MEM plus 10% fetal bovine serum and 1% penicillin/streptomycin for 2 weeks with one half medium change every 3 days to reach 70% confluency. For adipocyte differentiation, MSC subsets were cultured in adipogenesis medium (MesenCult basal medium + mouse MesenCult adipogenic stimulatory supplement; Cat no. 05503; Stem Cell Technologies) for further 3 weeks by following manufacturer’s instructions. Briefly, medium was changed every 3–4 days and wells were then stained with Oil Red O solution (Cat no. LL-0052; Lifeline Cell Technology, Frederick, MD). For osteoblast differentiation, MSC subsets were cultured in osteogenic medium (MesenCult basal medium + mouse MesenCult osteogenic stimulatory kit; Cat no. 05504; Stem Cell Technologies) for further 3 weeks by following manufacturer’s instructions. Briefly, medium was changed every 3–4 days and wells were then stained with 2% Alizarin Red S (Cat no. CM-0058; Lifeline Cell Technology, Frederick, MD).
Inhibitors
Nilotinib (TKI) supplied by Novartis Pharmaceuticals, and CXCR2 inhibitor (SB225002) purchased from Selleckchem (cat no. S7651, Houston, TX) were stored in 10mM dimethylsulfoxide (DMSO) at 20°C. Recombinant mouse TNFα (animal-free, clone MP6-XT22, cat. no. 718004, BioLegend, San Diego, CA) was reconstituted in water and stored at −20°C. Undiluted TNFα neutralizing antibody (InVivoMAb anti-mouse TNFα, clone XT3.11, cat no. BE0058, BioXCell, West Lebanon, NH) was stored at 4°C, and reconstituted in InVivoPure pH 8.0 dilution buffer just before use.
In vivo drug treatment
Peripheral blood samples were obtained 8 weeks after transplantation of CML cells in WT recipient mice to confirm development of neutrophilic leukocytosis (data not shown). Mice were treated with recombinant TNFα and anti-TNFα antibody for 2 weeks daily intraperitoneally at 0.5μg/mouse and 10mg/kg, respectively. Mice were then euthanized to harvest PB, BM for Q-PCR analysis, and the enumeration of BM hematopoietic and non-hematopoietic stem/progenitor subsets by flow cytometry. In another cohort of mice, CXCR2i and Nilotinib (Nil) were dissolved in 0.5% methylcellulose/0.5% Tween 80, and mice were treated with either Vehicle (Veh) or CXCR2i (5mg/kg) or Nil (50mg/kg) or combination by oral gavage once daily for 2 weeks and euthanized to harvest PB and BM cells for Q-PCR and FACS analysis. A cohort of mice was followed for survival. In other experiment, secondary transplants were performed using purified LTHSC selected by FACS sorting from primary recipient mice that had been treated with vehicle, TKI, CXCR2i or the combination. 1000 CD45.1/2+ LTHSC were transplanted into WT CD45.2+ recipient mice irradiated at 800 cGy with 0.25*106 supporting WBM CD45.2+ cells. Multi-lineage donor cell engraftment was monitored in PB serially every 4 weeks until 16 weeks.
Primary cell co-culture
FACS sorted BM mSSC, THY+, and 6C3+ cells pooled from several either normal or CML mice were allowed to attach to gelatin-coated 96-well culture plates for 24hrs in SFEM. WBM cells were subjected to purification of c-Kit+ (CD117+) stem/progenitor cells using CD117 mouse microbeads (Miltenyi Biotech, catalog no. 130-091-224) and LTHSC from either normal or CML mice were FACS sorted and co-cultured in 1:1 ratio with BM subsets, either alone or in competition with each other, in SFEM supplemented with 10ng/ ml murine IL-3, IL-6, SCF, TPO for 3 days at 37°C, 5% CO2. Harvested suspension LTHSC were then transplanted into lethally irradiated WT mice at 800 cGy with helper WBM cells to analyze their long-term engraftment capability. For in vitro experiments, 20,000 FACS sorted CFSEmax LTHSC from normal mice (n = 8) and CML mice (n = 13) were plated in gelatin-coated 96-well plates in SFEM supplemented with 10ng/ml murine IL-3, IL-6, SCF, TPO either in the presence of sorted 6C3+ cells from normal or CML mice, or the indicated chemokines, CXCL1 (250ng/ml), CXCL5 (250ng/ml), combination with or without CXCR2i for 3 days at 37°C, 5% CO2. The division and differentiation of LTHSC were then analyzed by FACS by calculating the proliferation index of gated LTHSC, and determining total CD45+ cells, LTHSC, STHSC, and MPP subsets. In other experiments, FACS sorted LTHSC from normal and CML mice were plated in gelatin-coated 96-well plates in SFEM supplemented with 10ng/ml murine IL-3, IL-6, SCF, TPO either in the presence or absence of CXCL1 (250ng/ml) for 3 days at 37°C, 5% CO2. Expression of mouse CXCR2 was measured every day until 3 days by flow cytometry. In other experiments, FACS sorted primary human CML CD34+38-CFSEmax cells were co-cultured in the presence or absence of hMSC prepared from the BM of patients with CML in StemSpan serum-free expansion medium (Stem Cell Technologies, Vancouver, BC, Canada), supplemented with growth factors at concentrations found in stroma-conditioned medium from long-term BM cultures at 37°C (Bhatia et al., 1995) in 5% CO2, and the indicated drugs.
Quantitative reverse transcription PCR
BM cells were pooled from femurs and tibia of 2–4 mice for each sample and specific populations were FACS sorted directly into RLT lysis buffer. Total RNA was extracted using RNAeasy micro kit (QIAGEN, Valencia, CA), and cDNA was synthesized using the Superscript III First-Strand Kit (Invitrogen, Grand Island, NY). Q-PCR for all the genes were performed using TaqMan gene expression assays as summarized in Table S2.
RNA sequencing
mSSC, THY+, 6C3+, and LTHSC populations were FACS sorted from the BM (femur and tibia) of 2–4 mice for each sample replicate directly into RLT lysis buffer. RNA was prepared with four biological replicates for each group. Sequencing libraries were prepared with the SMARTer Ultra Low Input RNA Kit for Sequencing (v4, TaKaRa Clontech, catalog no. 634891) following the manufacturer’s protocol with minor modifications. Briefly, the quality of DNA-free RNA from each sample was first assessed using the Agilent 2100 Bioanalyzer (to confirm their RIN score ≥ 7), then was reverse transcribed and first-strand cDNA synthesis and subsequent amplification was carried out with 15 cycles of Q-RT-PCR using SMART (switching mechanism at 5′ end of RNA template) technology. The resulting double-stranded cDNA was purified using Agencourt AMPure XP Kit (60 mL Beckman Coulter catalog no. A63881) and validated using the Agilent 2100 Bioanalyzer and Agilent’s High Sensitivity DNA Kit (Agilent Technologies, catalog no. 5067–4626). The amplified cDNA were also quantified using Qubit dsDNA HS Assay Kit (Catalog No. Q32851; Invitrogen Life Technologies, Grand Island, NY, USA), and all samples producing ample cDNA yields (1.7–7.76ng/μl) were used for library construction. Next, DNA libraries were prepared using Nextera XT DNA Library Preparation Kit (Illumina, catalog no. FC-131–1096) from 200pg of total amplified cDNA. cDNA were tagmented with i5 and i7 barcoded adapters (Illumina), and subjected to 12 cycles of PCR to produce the final sequencing library. Sequencing was performed using the HiSeq 2500 platform with the HiSeq SBS Kit V4 (Illumina) at 50 base pairs, single end, and ~24 million reads per library following the manufacturer’s instructions (Hudsonalpha Institute for Biotechnology, Huntsville, AL, USA). Reads were mapped to the UCSC mouse transcriptome (genome build mm10) using the STAR aligner tool. An average of 77.64% of the reads mapped to the reference genome. Next, to calculate the transcript abundances and differential expression from the aligned reads, DESeq2 tool was used. 4 biological replicates for each sample are reported.
Ligand-Receptor interaction map
As previously reported (Choi et al., 2017), we constructed a ligand-receptor interaction map by compiling three public databases providing ligand-receptor binding-pair annotations: (i) a Database of Ligand-Receptor Partners (DLRP; https://dip.doe-mbi.ucla. edu/dip/DLRP.cgi) that includes 462 interactions between 176 ligands and 133 receptors; and (ii) a list of experimentally proven interactions (in vivo and/or in vitro) extracted from BioGrid v3.2 (https://thebiogrid.org/) include 64 interactions between 36 ligands and 107 receptors; and (iii) an XML file containing 242 cytokine-cytokine receptor interactions (138 ligands and 107 receptors) was downloaded from KEGG (mmu: 04062) and parsed. After deleting redundant interaction pairs, we compiled an interaction map containing 635 ligand-receptor interactions including 182 ligands and 205 receptor genes. Differentially expressed genes from the comparison of 6–10 weeks post CML induced mesenchymal subsets (CML) group to age- and sex- matched wild-type subsets (NL) and LSCs to age- and sex- matched wild-type HSCs (NL) by applying FDR q < 0.05 and fold change > 2x cutoffs, and then mapped to this ligand-receptor map. The final interaction map was automatically generated using in-house Perl script and the GraphViz graph package (http://www.graphviz.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, all results obtained from independent experiments are reported as means ± standard errors of means (SEM) of multiple replicates, and statistical analyses were performed using unpaired, nonparametric Student’s t test or 2-way analysis of variance (ANOVA), adjusting for multiple comparisons (GraphPad Prism version 7.0, La Jolla, CA). Data was examined for normality by evaluating skewness and for equivalence of variance by comparing variance between groups. Survival probabilities were estimated using Kaplan-Meier analysis and significance calculated using the log-rank (Mantel-Cox) test. p values < 0.05 were considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. The sample size was estimated based on assumptions of one-sided Type I error of 0.05 and estimates of fold change and variance from preliminary data to have > 80% power to detect differences, if present, in our groups. All data and subjects were included in the analysis. The “n” in the figures and results represents the number of samples or animals utilized in the indicated experiments and values are reported in respective Figure Legends.
Supplementary Material
Highlights.
CML bone marrow 6C3+ stromal progenitors show enhanced leukemic stem cell (LSC) support
TNF-α signaling mediates expansion and increased CXCL1 expression in 6C3+ cells
CXCL1 support LSC proliferation and self-renewal
Targeting the CXCL1 receptor CXCR2, overexpressed in CML LSCs, inhibits LSC growth
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Health (R01 CA172447 and R01 CA249794 to R.B. and R35HL135787, R01DK102759, and R01DK113639 to D.T.S), the Cincinnati Children’s Hospital Research Foundation (to D.T.S), and the Pelotonia Fellowship (to P.A.). We thank the HudsonAlpha Institute for Biotechnology Sequencing Facility, Huntsville, Alabama, for performing RNA sequencing; the UAB Comprehensive Flow Cytometry Core and Animal Resource Center for providing help with FACS and maintaining mice colonies, respectively; and Dr. David Crossman at the Heflin Center for Genomic Science Core Laboratories at University of Alabama Birmingham for analysis of the RNA-seq data. We are grateful to Maya Robinson and Amanda Mullen for procurement and processing of human samples. We thank J. Bailey and V. Summey for assistance with transplantations (Comprehensive Mouse and Cancer Core at CCHMC) and members of the Bhatia and the Starczynowski laboratories for insightful suggestions and feedback. Bio-Render was used to make selected figures.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109386.
DECLARATION OF INTERESTS
D.T.S. serves on the scientific advisory board at Kurome Therapeutics. All other authors declare no competing interests.
REFERENCES
- Abegunde SO, Buckstein R, Wells RA, and Rauh MJ (2018). An inflammatory environment containing TNFα favors Tet2-mutant clonal hematopoiesis. Exp. Hematol 59, 60–65. [DOI] [PubMed] [Google Scholar]
- Agarwal P, and Bhatia R (2015). Influence of Bone Marrow Microenvironment on Leukemic Stem Cells: Breaking Up an Intimate Relationship. Adv. Cancer Res 127, 227–252. [DOI] [PubMed] [Google Scholar]
- Agarwal P, Zhang B, Ho Y, Cook A, Li L, Mikhail FM, Wang Y, McLaughlin ME, and Bhatia R (2017). Enhanced targeting of CML stem and progenitor cells by inhibition of porcupine acyltransferase in combination with TKI. Blood 129, 1008–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P, Isringhausen S, Li H, Paterson AJ, He J, Gomariz A, Nagasawa T, Nombela-Arrieta C, and Bhatia R (2019). Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell 24, 769–784.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz L, Sánchez-Aguilera A, Martín-Pérez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntión S, Tzeng YS, Lai DM, et al. (2014). Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 512, 78–81. [DOI] [PubMed] [Google Scholar]
- Asfaha S, Dubeykovskiy AN, Tomita H, Yang X, Stokes S, Shibata W, Friedman RA, Ariyama H, Dubeykovskaya ZA, Muthupalani S, et al. (2013). Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology 144, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, Nombela-Arrieta C, Steinmetz LM, Trumpp A, and Haas S (2020). Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol 22, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderman SR, Li AJ, Hoffman CM, Frisch BJ, Goodman AN, La-Mere MW, Georger MA, Evans AG, Liesveld JL, Becker MW, and Calvi LM (2016). Targeting of the bone marrow microenvironment improves outcome in a murine model of myelodysplastic syndrome. Blood 127, 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, Kokkaliaris KD, Mercier F, Tabaka M, Hofree M, et al. (2019). A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 177, 1915–1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia R, McGlave PB, Dewald GW, Blazar BR, and Verfaillie CM (1995). Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood 85, 3636–3645. [PubMed] [Google Scholar]
- Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL, Arber DA, Slovak ML, and Forman SJ (2003). Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 101, 4701–4707. [DOI] [PubMed] [Google Scholar]
- Bianco P, and Robey PG (2015). Skeletal stem cells. Development 142, 1023–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbrair A, and Frenette PS (2016). Niche heterogeneity in the bone marrow. Ann. N Y Acad. Sci 1370, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers M, Zhang B, Ho Y, Agarwal P, Chen CC, and Bhatia R (2015). Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood 125, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers E, Slaughter A, Frenette PS, Kuick R, Pello OM, and Lucas D (2018). Granulocyte-derived TNFα promotes vascular and hematopoietic regeneration in the bone marrow. Nat. Med 24, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer HE, Cooper S, Cacalano G, Hague NL, Bailish E, and Moore MW (1996). Involvement of Interleukin (IL) 8 receptor in negative regulation of myeloid progenitor cells in vivo: evidence from mice lacking the murine IL-8 receptor homologue. J. Exp. Med 184, 1825–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheier-mann C, Schiff L, Poncz M, Bergman A, and Frenette PS (2014). Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. (2003). Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846. [DOI] [PubMed] [Google Scholar]
- Capitano ML, Mor-Vaknin N, Saha AK, Cooper S, Legendre M, Guo H, Contreras-Galindo R, Kappes F, Sartor MA, Lee CT, et al. (2019). Secreted nuclear protein DEK regulates hematopoiesis through CXCR2 signaling. J. Clin. Invest 129, 2555–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CK, Lindau P, Jiang W, Chen JY, Zhang LF, Chen CC, Seita J, Sahoo D, Kim JB, Lee A, et al. (2013). Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl. Acad. Sci. USA 110, 12643–12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, et al. (2015). Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CKF, Gulati GS, Sinha R, Tompkins JV, Lopez M, Carter AC, Ransom RC, Reinisch A, Wearda T, Murphy M, et al. (2018). Identification of the Human Skeletal Stem Cell. Cell 175, 43–56.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K, Komurov K, Fletcher JS, Jousma E, Cancelas JA, Wu J, and Ratner N (2017). An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci. Rep 7, 43315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, McDonald T, Lin A, Chakraborty S, Huang Q, Snyder DS, and Bhatia R (2011). Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood 118, 5565–5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, and Druker BJ (2011). Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Invest 121, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischman AG, Aichberger KJ, Luty SB, Bumm TG, Petersen CL, Doratotaj S, Vasudevan KB, LaTocha DH, Yang F, Press RD, et al. (2011). TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 118, 6392–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallipoli P, Pellicano F, Morrison H, Laidlaw K, Allan EK, Bhatia R, Copland M, Jørgensen HG, and Holyoake TL (2013). Autocrine TNF-α production supports CML stem and progenitor cell survival and enhances their proliferation. Blood 122, 3335–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, and Link DC (2013). CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Guarnerio J, Mendez LM, Asada N, Menon AV, Fung J, Berry K, Frenette PS, Ito K, and Pandolfi PP (2018). A non-cell-autonomous role for Pml in the maintenance of leukemia from the niche. Nat. Commun 9, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Murphy MP, Marecic O, Lopez M, Brewer RE, Koepke LS, Manjunath A, Ransom RC, Salhotra A, Weissman IL, et al. (2018). Isolation and functional assessment of mouse skeletal stem cell lineage. Nat. Protoc 13, 1294–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H, Debnath B, and Neamati N (2017). Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 7, 1543–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Müller-Tidow C, Bhatia R, Brunton VG, et al. (2012). Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood 119, 1501–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoun M, Zhang D, Mizoguchi T, Pinho S, Pierce H, Kunisaki Y, Lacombe J, Armstrong SA, Dührsen U, and Frenette PS (2014). Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell 15, 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser M, Yun H, Berg T, Yung E, Argiropoulos B, Kuchenbauer F, Park G, Hamwi I, Palmqvist L, Lai CK, et al. (2011). Cell of origin in AML: susceptibility to MN1-induced transformation is regulated by the MEIS1/AbdB-like HOX protein complex. Cancer Cell 20, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J, Kfoury Y, and Scadden DT (2016). Hematopoietic Stem Cell Niche in Health and Disease. Annu. Rev. Pathol 11, 555–581. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Jootar S, Pornprasertsud N, Petvises S, Rerkamnuaychoke B, Disthabanchong S, Pakakasama S, Ungkanont A, and Hongeng S (2006). Bone marrow derived mesenchymal stem cells from chronic myeloid leukemia t(9;22) patients are devoid of Philadelphia chromosome and support cord blood stem cell expansion. Leuk. Res 30, 1493–1498. [DOI] [PubMed] [Google Scholar]
- Karpova D, Rettig MP, Ritchey J, Cancilla D, Christ S, Gehrs L, Chen-damarai E, Evbuomwan MO, Holt M, Zhang J, et al. (2019). Targeting VLA4 integrin and CXCR2 mobilizes serially repopulating hematopoietic stem cells. J. Clin. Invest 129, 2745–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Kirsten AM, Förster K, Radeczky E, Linnhoff A, Balint B, Watz H, Wray H, Salkeld L, Cullberg M, and Larsson B (2015). The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate-to-severe COPD. Pulm. Pharmacol. Ther 31, 36–41. [DOI] [PubMed] [Google Scholar]
- Koschmieder S, Göttgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, Dayaram T, Geary K, Green AR, Tenen DG, and Huettner CS (2005). Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 105, 324–334. [DOI] [PubMed] [Google Scholar]
- Krause DS, Fulzele K, Catic A, Sun CC, Dombkowski D, Hurley MP, Lezeau S, Attar E, Wu JY, Lin HY, et al. (2013). Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med 19, 1513–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim M, Pang Y, Ma S, Hao S, Shi H, Zheng Y, Hua C, Gu X, Yang F, Yuan W, and Cheng T (2016). Altered mesenchymal niche cells impede generation of normal hematopoietic progenitor cells in leukemic bone marrow. Leukemia 30, 154–162. [DOI] [PubMed] [Google Scholar]
- Mead AJ, Neo WH, Barkas N, Matsuoka S, Giustacchini A, Facchini R, Thongjuea S, Jamieson L, Booth CAG, Fordham N, et al. (2017). Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. J. Exp. Med 214, 2005–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf H (2017). The microenvironment in human myeloid malignancies: emerging concepts and therapeutic implications. Blood 129, 1617–1626. [DOI] [PubMed] [Google Scholar]
- Miller CL, Dykstra B, and Eaves CJ (2008). Characterization of mouse hematopoietic stem and progenitor cells. Curr. Protoc. Immunol Chapter 22, Unit 22B.2. [DOI] [PubMed] [Google Scholar]
- Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, Ono N, Kronenberg HM, and Frenette PS (2014). Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev. Cell 29, 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa S, Mabuchi Y, Kubota Y, Nagai Y, Niibe K, Hiratsu E, Suzuki S, Miyauchi-Hara C, Nagoshi N, Sunabori T, et al. (2009). Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med 206, 2483–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, and Scadden DT (2014). The bone marrow niche for haematopoietic stem cells. Nature 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, and Daley GQ (2009). Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osta B, Benedetti G, and Miossec P (2014). Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front. Immunol 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A, and Carroll M (2011). BCR-ABL kinase is dead; long live the CML stem cell. J. Clin. Invest 121, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, and Frenette PS (2019). Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol 20, 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S, Lacombe J, Hanoun M, Mizoguchi T, Bruns I, Kunisaki Y, and Frenette PS (2013). PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med 210, 1351–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raaijmakers MH (2011). Niche contributions to oncogenesis: emerging concepts and implications for the hematopoietic system. Haematologica 96, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey PG, Kuznetsov SA, Riminucci M, and Bianco P (2014). Bone marrow stromal cell assays: in vitro and in vivo. Methods Mol. Biol 1130, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, and Passegué E (2013). Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, et al. (2015). IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood 125, 3144–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RK, Mullally A, Dugourd A, Peisker F, Hoogenboezem R, Van Strien PMH, Bindels EM, Heckl D, Büsche G, Fleck D, et al. (2017). Gli1+ Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 20, 785–800.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, Piddock RE, Fenech M, Zaitseva L, Abdul-Aziz A, et al. (2017). Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 129, 1320–1332. [DOI] [PubMed] [Google Scholar]
- Takizawa H, and Manz MG (2017). Impact of inflammation on early hematopoiesis and the microenvironment. Int. J. Hematol 106, 27–33. [DOI] [PubMed] [Google Scholar]
- Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, Pinho S, Akhmetzyanova I, Gao J, Witkowski M, et al. (2019). The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JR, Lee JM, Young PR, Hertzberg RP, Jurewicz AJ, Chaikin MA, Widdowson K, Foley JJ, Martin LD, Griswold DE, and Sarau HM (1998). Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J. Biol. Chem 273, 10095–10098. [DOI] [PubMed] [Google Scholar]
- Wöhrer S, Rabitsch W, Shehata M, Kondo R, Esterbauer H, Streubel B, Sillaber C, Raderer M, Jaeger U, Zielinski C, and Valent P (2007). Mesenchymal stem cells in patients with chronic myelogenous leukaemia or biphenotypic Ph+ acute leukaemia are not related to the leukaemic clone. Anticancer Res 27 (6B), 3837–3841. [PubMed] [Google Scholar]
- Wolock SL, Krishnan I, Tenen DE, Matkins V, Camacho V, Patel S, Agarwal P, Bhatia R, Tenen DG, Klein AM, and Welner RS (2019). Mapping Distinct Bone Marrow Niche Populations and Their Differentiation Paths. Cell Rep 28, 302–311.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, and Passegué E (2019). TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 25, 357–372.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, and Bhatia R (2012). Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 21, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, Shultz L, and Bhatia R (2013). Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood 121, 1824–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue R, Zhou BO, Shimada IS, Zhao Z, and Morrison SJ (2016). Leptin Receptor Promotes Adipogenesis and Reduces Osteogenesis by Regulating Mesenchymal Stromal Cells in Adult Bone Marrow. Cell Stem Cell 18, 782–796. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jørgensen HG, Lin A, Gaal K, Holyoake TL, and Bhatia R (2016). Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 128, 2671–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Supakorndej T, Krambs JR, Rao M, Abou-Ezzi G, Ye RY, Li S, Trinkaus K, and Link DC (2019). Bone marrow dendritic cells regulate hematopoietic stem/progenitor cell trafficking. J. Clin. Invest 129, 2920–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data discussed in this publication has been deposited in NCBI’s Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. The GEO accession number is listed in the key resources table. This study does not report development of new code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.