

Abstract
Solid-state photodecarbonylation is an attractive but underutilized methodology to forge hindered C−C bonds in complex molecules. This study discloses the use of this reaction to assemble the vicinal quaternary stereocenter motif present in bis(cyclotryptamine) alkaloids. Our strategy was enabled by experimental and computational investigations of the role of substrate conformation on the success or failure of the solid-state photodecarbonylation reaction. This informed a crystal engineering strategy to optimize the key step of the total synthesis. Ultimately, this endeavor culminated in the successful synthesis of the bis(cyclotryptamine) alkaloid “psychotriadine,” which features the elusive piperidinoindoline framework. Psychotriadine, a previously unknown compound, was identified in the extracts of the flower Psychotria colorata, suggesting it is a naturally occurring metabolite.
Graphical Abstract

INTRODUCTION
Photochemistry has become an increasingly powerful tool in modern organic synthesis.1–4 One particularly interesting class of photochemical transformations with great potential involves reactions conducted in the crystalline solid state. Such transformations are attractive because of the opportunity to control various selectivities (stereo-, regio-, and chemo-), potential for scalable green chemistry, and ability to form strained or congested frameworks.5–7
Our laboratory has been particularly interested in photodecarbonylation reactions that occur in the crystalline solid state, and we have demonstrated their potential for the stereospecific assembly of vicinal quaternary stereocenters (Figure 1a).5,8–13 The introduction of vicinal quaternary stereocenters has remained a long-standing challenge in organic synthesis.14–16 An attractive approach to this motif, 1, is via direct coupling of two prochiral aliphatic radicals 2.17 In turn, radicals 2 can be generated from the photodecarbonylation of hexasubstituted ketone 3. While in solution, the coupling of prochiral radicals can lead to complex product mixtures, the corresponding process in the crystalline solid state can result in high-yielding stereospecific recombination.5 Despite promising investigations into fundamental reactivity, this solid-state approach has seen little use in natural product synthesis10,11 and has not been used to construct complex alkaloids. Moreover, the understanding of how substrate conformations relate to the success or failure of solid-state photodecarbonylation reactions has remained underexplored.
Figure 1.
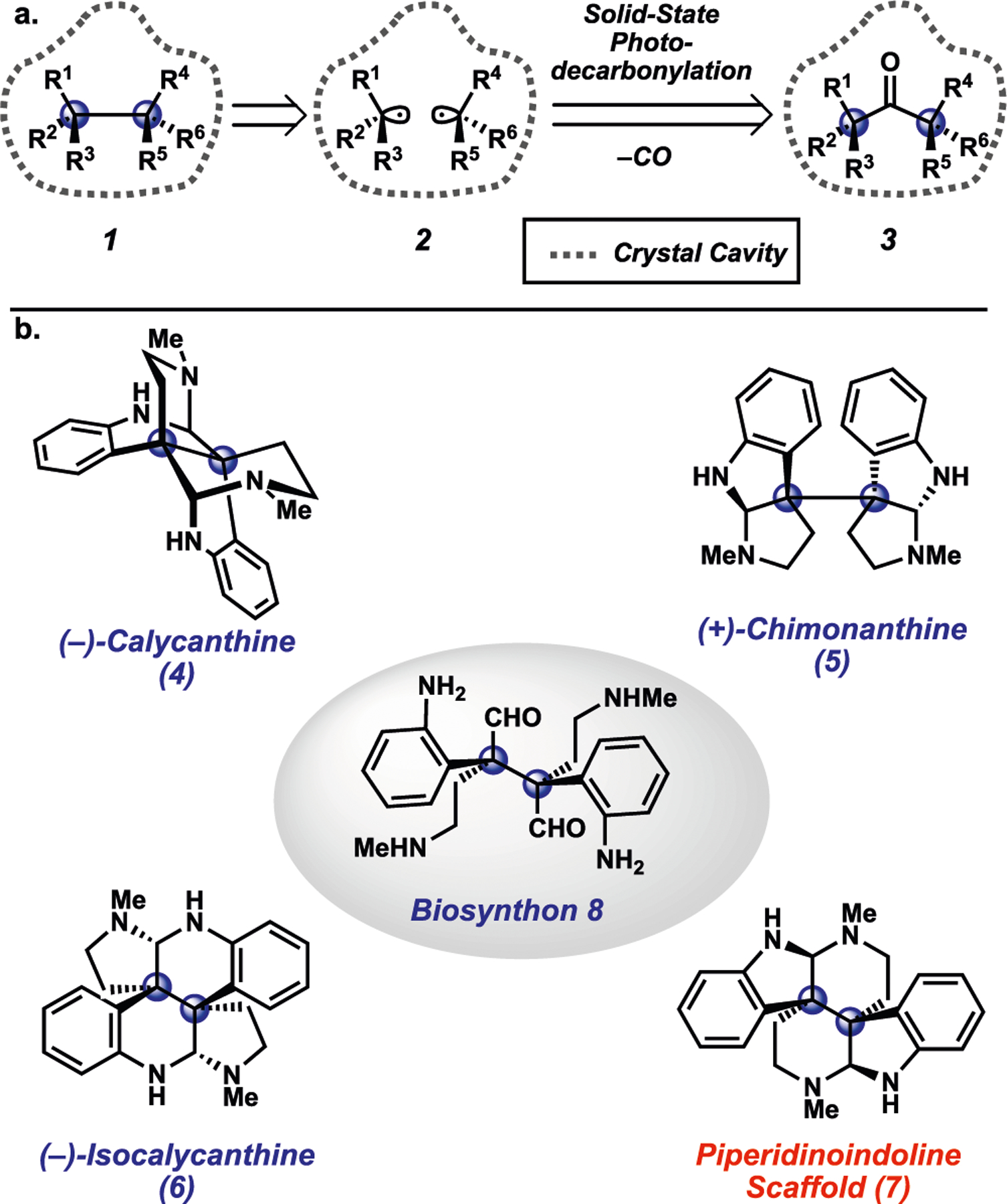
Solid-state photodecarbonylation to introduce vicinal quaternary stereocenters and overview of select bis(cyclotryptamine) alkaloids.
With the aforementioned motivations in mind, we sought to evaluate solid-state photodecarbonylation chemistry in the context of the bis(cyclotryptamine) alkaloids (Figure 1b). This class of natural products, arising from common biosynthon 8, features vicinal quaternary stereocenters and has been popular among synthetic chemists for many decades.17–22 In addition, this family of small molecules possesses a rich history, stemming from the isolation report of calycanthine (4) in 1888 and the subsequent discovery of related23 natural products (e.g., 5 and 6), representing a total of four unique constitutional isomers.24 Interestingly, a fifth piperidinoindoline isomer 7 was proposed by Robinson in 1954 but has never been isolated from a natural source.25–27 Movassaghi and coworkers have synthesized a highly substituted derivative of 7 in the context of communesin alkaloid total synthesis studies.17 Given the widespread interest in these compounds and their complex structures bearing vicinal quaternary stereocenters, we deemed the bis(cyclotryptamine) alkaloids an ideal arena to test the solid-state photodecarbonylation methodology.
Our laboratory recently described the successful application of solid-state photodecarbonylation chemistry in the total synthesis of a bis(cyclotryptamine) alkaloid.28 Here, we disclose our full investigation of the key photodecarbonylation step, including the evaluation of 11 different crystalline substrates. By analysis of substrate conformation via X-ray structures and computational modeling, we provide the physical organic underpinnings that explain the success or failure of the key photodecarbonylation step. These efforts demonstrate that the transformation can be optimized by manipulating the crystalline conformation of the necessary hexasubstituted ketone substrate. Ultimately, our crystalline conformation-based approach enabled the formation of the challenging vicinal quaternary stereocenters present in biosynthon 8. These efforts culminated in the total synthesis of a natural product that had previously been overlooked in plant isolations and features the final, elusive piperidinoindoline scaffold of the bis(cyclotryptamine) alkaloids.
RESULTS AND DISCUSSION
Retrosynthetic Analysis.
Our retrosynthetic analysis of common biosynthon 8 is depicted in Figure 2. It was envisioned that 8, or a dehydrated synthetic congener thereof, would arise from late-stage bis(amination) and reduction of intermediate 9. In turn, 9, bearing the requisite vicinal quaternary stereocenters, would be accessed via a key solid-state photodecarbonylation of hexasubstituted ketone 11 via the intermediacy of radical pair 10. Although both radical centers would be stereochemically labile in intermediate 10, it was anticipated that the rigidity of the crystalline lattice would affect the transfer of stereochemical information from 11 to 9. Finally, 11 would be constructed rapidly by the convergent coupling of two equivalents of lithium enolate 12 with an equivalent of phosgene (13).
Figure 2.
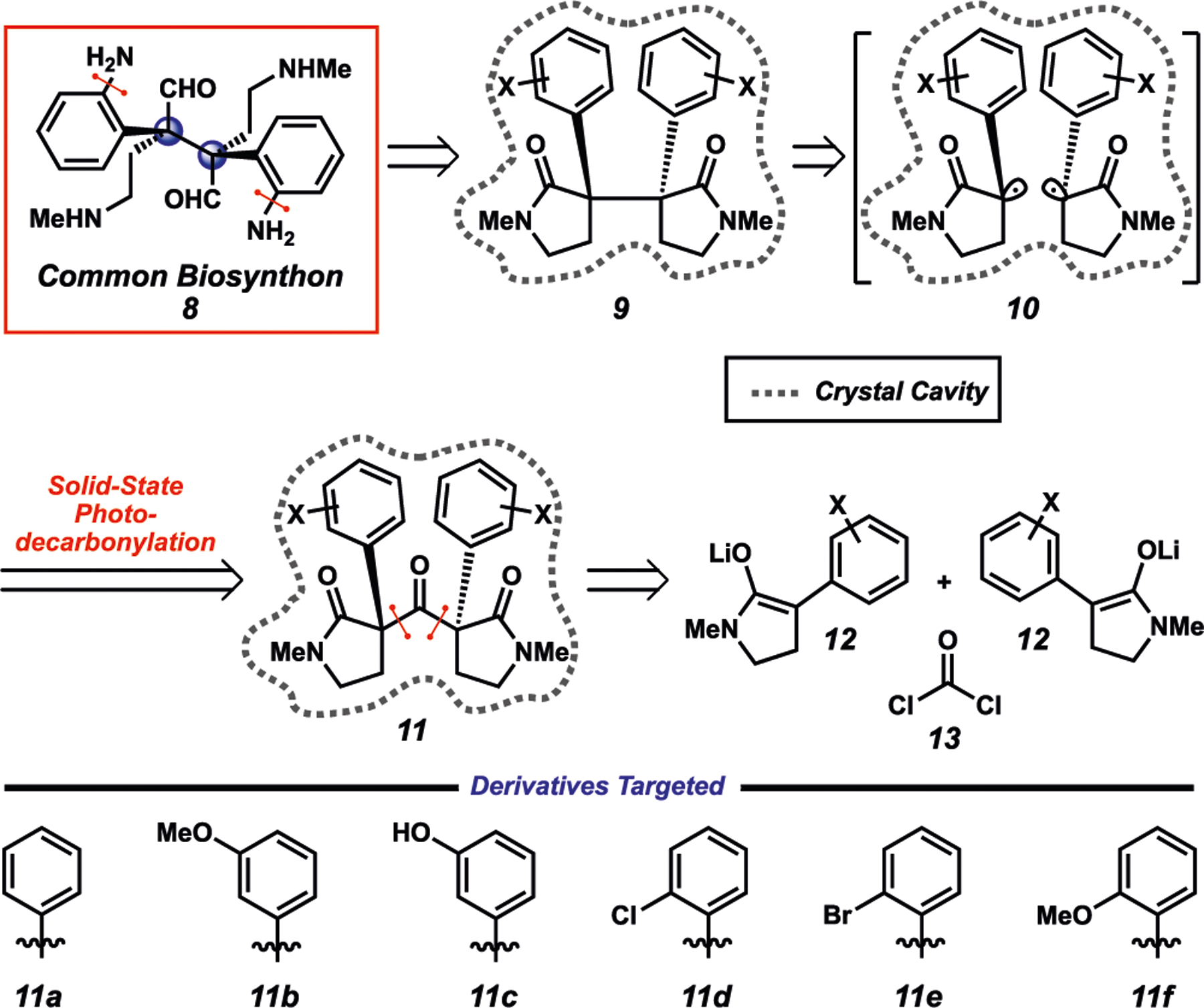
Retrosynthetic analysis of biosynthon 8.
At the outset of this research pursuit, it was anticipated that a major challenge in our synthetic strategy would be the preparation of 9 bearing amines on the ortho position of the aromatic rings. Introduction of protected amines at this position early in the synthesis would be ideal, but the steric crowding imparted by ortho substituents in close proximity to the quaternary centers would make the assembly of the corresponding ketone challenging.29 Additionally, it was envisioned that the increased steric bulk resulting from these substituents may also hinder C−C bond formation from radical pair 10. To address these concerns, two classes of ketone substrates, ortho C−H (11a−c) and ortho C−X (11d−f) (Figure 2), that would be synthetically accessible from a common route were targeted. It was hypothesized that the photodecarbonylation of ketones 11a−c would occur selectively and the resulting products 9a−c could be functionalized by directed ortho C−H metalation/amination. It was predicted that ketones 11d−f could be competent substrates for photodecarbonylation to access 9d−f, and that the ortho-Cl, -Br, and -OMe substituents could function as synthetic handles for a metal-catalyzed amination.30
Synthesis and Photodecarbonylation of Crystalline Ketones.
Photodecarbonylation substrates 11a−c were prepared in a straightforward manner from readily available diesters 14 (Scheme 1).31 Malonic esters 14 were subjected to a two-step alkylation/reductive cyclization sequence to furnish pyrrolidinones 15 in good yield. N-methylation of 15 under basic conditions provided 16 in 60–87% yield. In turn, N-methylpyrrolidinones 16 were cleanly converted to 17 via a one-pot protocol involving ester saponification and subsequent acid-mediated decarboxylation. Finally, treatment of lactams 17 with LiHMDS provided the corresponding enolates, which were coupled with phosgene (13) to deliver ketones 11a,b in 49–64% yield and with high diastereoselectivity. While 11a was a high-melting crystalline solid, 11b was a viscous oil at room temperature (and therefore not a candidate for a solid-state photodecarbonylation reaction).32 However, after efficiently cleaving the aryl-methyl ethers of 11b using BBr3, we were delighted to find that phenolic ketone 11c was a crystalline solid.
Scheme 1.

Synthesis of Ketone Substrates 11a−c
The preparation of ketones 11d−f is shown in Scheme 2.31 The beginning of this synthetic sequence mirrored that for ketones 11a−c. A two-step annulation of malonic esters 14 and subsequent N-methylation cleanly furnished pyrrolidinones 16. Saponification of the ester in 16, followed by decarboxylation, provided enolate precursors 17 in 79–90% yield. Unfortunately, we observed that the reaction of 17d−f with phosgene (13) did not provide ketone 11 and instead resulted in substantial nonspecific decomposition (not shown). To circumvent this issue, we constructed acid chloride 18 in a two-step sequence from pyrrolidinone 16. This electrophile was then efficiently coupled with the lithium enolate derived from 17 to provide ketones 11d−f in good to excellent yields and with high diastereoselectivity.
Scheme 2.

Synthesis of Ketone Substrates 11d−f
Once we had prepared ketones 11a−f, we next turned our attention to evaluating their reactivities in the key solid-state photodecarbonylation reaction that forges the vicinal quaternary stereocenters present in the bis(cyclotryptamine) alkaloids (Figure 3).33 In exploring the photodecarbonylation of these, a wide range of reactivity was observed. Substrates 11a and 11c were irradiated in the solid-state and proceeded smoothly to provide 9a and 9c in 59% and 35% yield, respectively.34 Although identifiable side products were not observed from the photodecarbonylation of 11c,35 the reaction of 11a also gave rise to products 17a and 19a (Figure 3, top), presumably via a competing radical pair disproportionation process.36 Unfortunately, ortho-substituted ketones 11d−f performed poorly in the desired C−C bond formation.37 Solid-state irradiation of ketones 11d and 11e resulted in 9% and 8% yields of 9d and 9e, respectively, while irradiation of ketone 11f gave no identifiable recombination product 9f.38 As was the case for ketone 11a, the photodecarbonylation of ketones 11d−f displayed a competitive disproportionation reaction pathway that was the major reaction outcome for these substrates.39
Figure 3.

Photodecarbonylation of ketones 11 (yields resulting from decarbonylation of substrates 11c−f determined using qNMR).
Interestingly, we noted a correlation between the relative selectivity of radical pair 10 undergoing recombination over disproportionation to the size of the ortho-substituent (quantified by the A-value).40 Ketones 11a and 11c bearing only a hydrogen atom (A-value = 0) at the ortho position underwent recombination to give 9a and 9c, respectively, as the major products. Ketones 11d−f, bearing ortho-substituents, displayed lower yields of 9d−f and showed a higher incidence of disproportionation. Intrigued by these results, we sought to understand the structural factors that guide the outcome of the solid-state photodecarbonylation reaction.
On the basis of close inspection of substrate X-ray crystal structures, we offer the analysis shown in Figure 4 to explain the sensitivity of the photodecarbonylation reaction to aromatic substituents. We postulate that radical pair 10 is prevented from forming a C−C bond due to steric clash between the ortho-substituent on one radical fragment and the aromatic ring on the other (Figure 4a). The proposed repulsive interactions can further be observed in the X-ray crystal structures of the substrates (Figure 4b and c). For ketones 11d−f, steric congestion is observed in the interaction between the ortho-substituent on one-half of the molecule and the aryl ring on the other (interaction depicted with a red arrow). Notably, such interactions are not present in ketones without ortho functionality (i.e., 11a and 11c).41 Since the radical centers in 10 must move closer to one another by approximately 1.2 Å to generate a C−C bond, these close interactions in the substrate are likely to become strongly repulsive at the transition state. It is important to note that reactions that take place in the crystalline solid state are governed by the topochemical principle and favor reaction pathways with minimal atomic motion.42 As such, the conformational features observed in the X-ray crystal structures of the substrate are likely to also be present in the reactive intermediates and transition-state structures.
Figure 4.

Crystalline conformation dictates radical−radical recombination trajectory.
Given the efficiency of the solid-state photodecarbonylation of 11a to give 9a, we pursued a C−H functionalization approach to install the requisite ortho-C−N bonds in 9a (Scheme 3).43–45 We hypothesized that the pyrrolidinone oxygen atoms may function as convenient directing groups for either transition metal-catalyzed ortho functionalization or directed lithiation.46 Unfortunately, despite extensive experimentation, we were unable to functionalize the arene and most attempts either returned starting material or resulted in substantial nonspecific decomposition.47 Interestingly, we found that the use of alkyllithium bases resulted in deprotonation of the N-methyl groups on 9a to give the corresponding primary carbanion (not shown).48 In order to circumvent this issue, we sought to construct a derivative of 9a with cleavable protecting groups on the nitrogen. This would allow for the introduction of N-substituents that would not undergo competitive activation.
Scheme 3.

Attempted Directed Ortho-Metalation/Functionalization
In pursuit of a derivative of 9a with cleavable N-substituents, ketone precursor 20 (Figure 5) was prepared.49 To our surprise, inspection of the X-ray crystal structure of 20 revealed a different ketone conformation compared to our previous observations, leading us to suspend our efforts toward C−H activation and instead focus on manipulating the conformation of the ketone substrate to optimize the solid-state photodecarbonylation reaction (Figure 5). As noted previously, it is likely that the solid-state conformations of ketones 11d−f prevent successful decarbonylative C−C bond formation due to a growing steric clash in radical pair 10. On the basis of the striking change in crystalline conformation between 11a and 20 (Figure 5a), we hypothesized that the conformation of ortho-functionalized ketone 21 could also be altered by modifying the N-substituents. An alternative conformation of 21 could lead to hypothetical radical pair 22 that does not feature a steric clash that prevents the formation of 23. Therefore, we sought to explore whether conformational manipulation could be used to give a ketone that both includes ortho- aryl halides or pseudohalides for late-stage metal-catalyzed amination and the ability to undergo efficient solid-state photodecarbonylation to forge the desired vicinal quaternary stereocenters.
Figure 5.

Conformational change could enhance chemoselectivity of the photodecarbonylation reaction.
In order to prepare ketones 21 bearing modifiable N-substituents, we conducted the synthetic sequence depicted in Scheme 4.50 First, ortho-chloro and ortho-bromo pyrrolidinones 15 were cleanly converted to para-methoxybenzyl (PMB) protected substrates 24 under basic conditions. At this point, the synthesis diverged. Esters 24 were saponified and then thermally decarboxylated to provide 25 in 79–90% yield. Esters 24 were also converted to acid chlorides 26 by a two-step saponification/dehydrochlorination protocol. Pyrrolidinones 25 were then deprotonated with LiHMDS and the resultant enolates were coupled with acid chlorides 26 to furnish PMB-protected amides 27. As was observed in the construction of 11, formation of 27 occurred with exquisite diastereoselectivity, presumably due to a highly ordered transition state mediated by Li+ chelation.49 In efforts to cleave the PMB protecting groups, ketones 27 were then treated with ceric ammonium nitrate (CAN), which unexpectedly furnished a mixture of imide products 28 and 29. Interestingly, ketones 28 and 29 were both high-melting point crystalline solids. Given this observation, we then sought to test their efficiency in the solid-state photodecarbonylation reaction.
Scheme 4.

Synthesis of Ketones 28 and 29
The results of the solid-state photodecarbonylation of symmetric chloride-containing ketone 28d are depicted in Figure 6a. We were pleased to find that initial efforts toward the conversion of 28d to 30d proceeded smoothly and with retention of stereochemistry, as verified by single-crystal X-ray diffraction. Furthermore, a competing disproportionation reaction pathway was not observed in the photodecarbonylation of 28d. This result validated our hypothesis that exchanging the N-substituents could alter the chemoselectivity of the photodecarbonylation reaction, presumably due to the new conformation of 28d relative to ketone 11d. Furthermore, the efficiency of the reaction demonstrated that solid-state photodecarbonylation is a competent transformation to forge the vicinal quaternary stereocenter motif present in the bis(cyclotryptamine) alkaloids.
Figure 6.

Solid-state photochemistry of ketones 28d and 28e (the R-groups on imides 28d and 28e were removed from the X-ray renderings for clarity).
Despite initial success, it was found that the solid-state photodecarbonylation reaction was difficult to reproduce with new batches of 28d (Figure 6a). While exposure of 28d to ultraviolet light initially led to full consumption of starting material within 24–48 h, similar irradiation of new samples of 28d only resulted in recovered starting material. After verifying the robustness of the light source and ruling out the possibility of impurities being present in the substrate, we realized that a new, more stable polymorph of 28d, termed “unreactive polymorph,” had formed. Single-crystal X-ray diffraction revealed that this new polymorph adopts a different conformation than the “reactive polymorph” (see X-ray crystal structures in Figure 6a). Unfortunately, once the unreactive polymorph was present, it was exceedingly difficult to prepare the reactive polymorph, despite extensive screening of crystallization conditions. These results suggest that the reactive polymorph was likely formed kinetically, while the unreactive polymorph was thermodynamically favored.51,52 Trace quantities of the more stable and unreactive polymorph can act as seed crystals in subsequent recrystallization attempts, thereby preventing further preparation of the reactive polymorph.53
Due to the poor reaction reproducibility stemming from the conformational polymorphism of 28d, we turned our attention to evaluating the ortho-bromo substrate 28e (Figure 6b). While two conformationally distinct polymorphs were observed for 28d, we only identified a single polymorph of 28e. The conformation of 28e observed in the X-ray structure was almost identical to that of the 28d unreactive polymorph. As was expected, based on the similar conformation, 28e was also unreactive under solid-state irradiation.
The dramatically different behavior of 28d (reactive polymorph) relative to 28d (unreactive polymorph) and 28e is likely controlled by substrate conformation in the crystalline solid state (Figure 6c). Solid-state photodecarbonylation requires stabilization of the breaking C−C sigma bonds by neighboring π-systems. The extent of these hyperconjugative interactions in substrate 28d (both reactive and unreactive polymorphs) and 28e can be correlated to the dihedral angle between the breaking C−C σ bond and the nearest C−C bond of the aromatic π-system. A dihedral angle of 90° is ideal, allowing for maximum orbital overlap (see bonds highlighted in blue). Alternatively, if the dihedral angle is 0°, the C−C σ-bond and π-system will be orthogonal, resulting in no electronic stabilization (see bonds highlighted in red). In considering substrate 28d (unreactive polymorph) and 28e, the relevant dihedral angles are 82° and 18° in the former and 85° and 20° in the latter. The smaller of the two dihedral angles for each substrate, 18° and 20°, presumably leads to negligible orbital overlap and failed bond homolysis. The relevant dihedral angles in 28d (reactive polymorph) are 64° and 68°, which we surmise provide sufficient orbital overlap to facilitate decarbonylation.
Having explored the reactivity of symmetric ketones 28 in solid-state photochemistry, we next investigated nonsym-metrical ketones 29 (Figure 7). To our delight, both 29d and 29e underwent productive photodecarbonylation. Upon exposure to UV light and subsequent pyrrolidinone deprotection, 29d and 29e efficiently provided 31d and 31e, respectively. Unlike substrate 28d, the solid-state transformations of 29d and 29e were reproducible even when performed on >300 mg scale.54 These results were consistent with our understanding of the role of solid-state conformation and photochemical lability of the ketone carbonyl. The relevant dihedral angles between breaking C−C bonds and the nearest C−C bond of the aromatic π-system (see bonds highlighted in blue in the microelectron diffraction (microED) structures) are 66° and 68° for 29d, and 70° and 72° for 29e.55,56 These values, which were similar to those seen in the reactive polymorph of 28d, presumably provide enough hyperconjugative stabilization to the breaking C−C bonds to facilitate decarbonylation. Fortuitously, we have only observed reactive polymorphs of 29d and 29e.
Figure 7.

Solid-state photochemistry of ketones 29d and 29e (the R-groups on imides 29d and 29e were removed from the X-ray renderings for clarity). Crystal structures obtained by both X-ray diffraction and microcrystal electron diffraction (microED).
Computational Analysis of C−C Bond Formation.
In order to better understand the role of substrate conformation in reaction selectivity, we conducted a computational study of the radical pairs 10a and 10c−f (Figures 8 and 9). At the outset, we recognized that effective computational simulation of the crystalline lattice would be critical in achieving meaningful results. While gas-phase and simple solvation models would be unlikely to accurately simulate the crystalline environment, full density functional theory (DFT) treatment of the crystalline lattice would be untenable due to prohibitive computational cost. Therefore, the ONIOM57 approach within the hybrid QM/MM58 method was employed, in order to estimate the intermolecular interactions within the crystal lattice environment.59 The central part consisted of a single radical pair computed with the unrestricted open-shell dispersion-corrected ωB97X-D/6–311G(d,p) level of theory.60 The surrounding shell of molecules, extracted from the experimentally determined crystal structure, was computed with the electronically embedded UFF force field.61
Figure 8.

QM/MM hybrid approach to model radical−radical recombination in the crystalline solid state. “Ball-and-stick” representation was used for the atoms treated by the DFT method (ωB97X-D/6–311G(d,p) level of theory) and a “wireframe” representation for the atoms in the low-level layer (UFF).
Figure 9.

Computational investigation of radical pair 10.
Using this methodology, we investigated the recombination barriers for radical pairs 10 in the solid state (Figure 9). Radical pairs 10 were generated computationally by removing the carbonyls from ketones 11 and then performing an energy minimization calculation. Interestingly, the success of the ground-state optimization for radical fragments was critically dependent on embedding them in their respective crystal cavities. In the gas phase, the radical pair would spontaneously recombine to form the C−C bond. However, when conformationally restrained by the crystal cavity, the radical pair was an energetic minimum and existed as a discrete species. Transition states for the recombination reaction were then identified for each of the radical pairs in their respective lattices. Radical pairs 10a and 10c, which lack ortho substitution, displayed the lowest barriers of 4.8 and 1.7 kcal/mol, respectively. Recombination barriers for ortho-functionalized radical pairs 10d−f were uniformly higher, with values between 6.1 and 6.8 kcal/mol. Given the successful calculation of transition states for C−C bond formation from radical pairs 10, we were additionally interested in probing the competing disproportionation process. The results of these calculations suggest that hydrogen atom tunneling may be responsible for the observed disproportionation (for further discussion, see SI).
After determining the recombination barriers for radical pairs 10, we turned our attention to radical pair 22d (arising from decarbonylation of ketone 28d) (Figure 10). For this calculation, we used the coordinates derived from the reactive polymorph of 28d. While radical pairs 10 all displayed small but non-negligible barriers to C−C bond formation, the corresponding process for radical pair 22d was barrierless. In fact, all attempts at energy minimization of 22d resulted in radical recombination to form the C−C bond. This result was consistent with experimental results described previously, which demonstrated that photodecarbonylation of ketone 28d (reactive polymorph) led to efficient C−C bond formation (Figure 6a).
Figure 10.
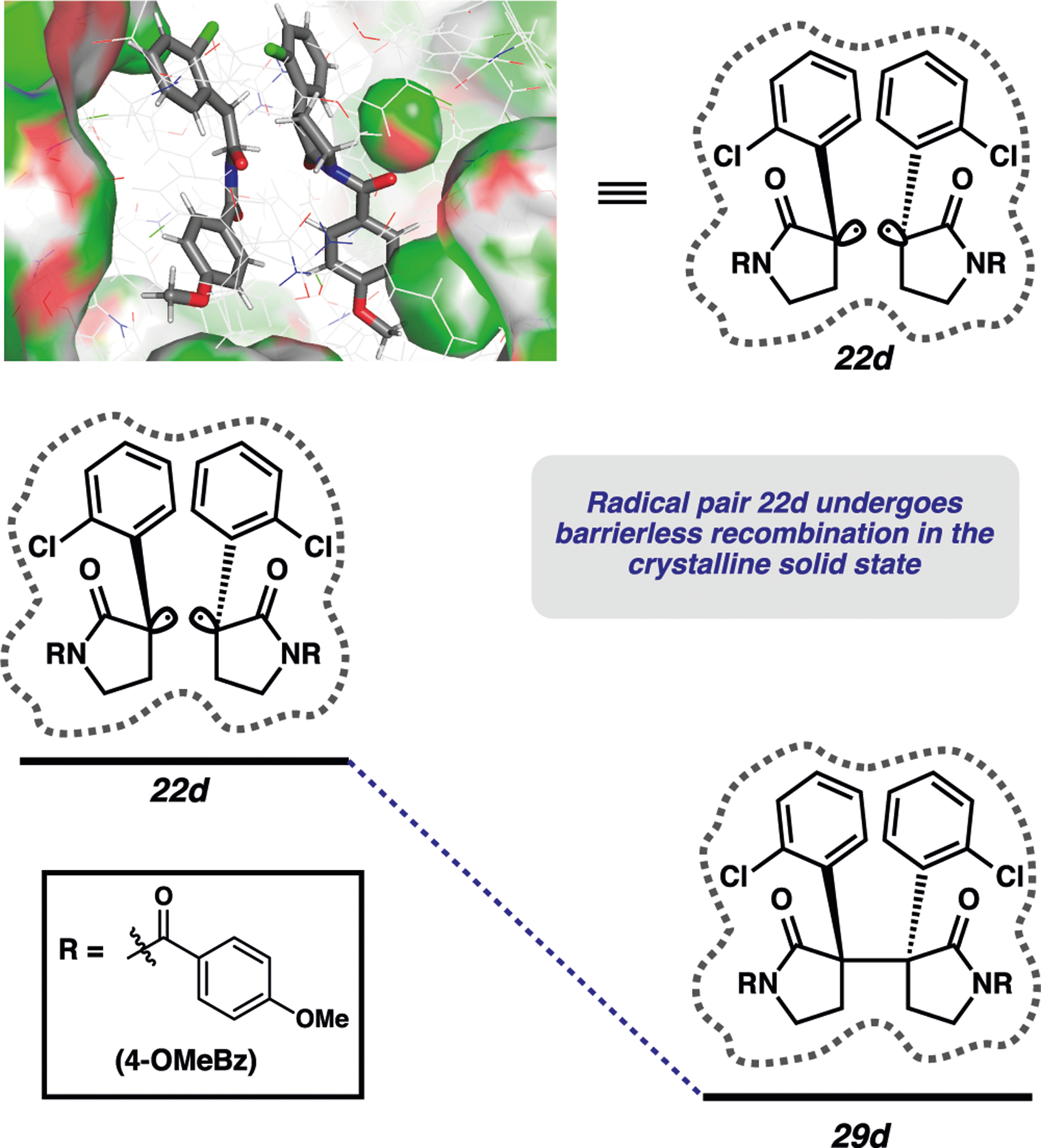
Computational investigation of radical pair 22d.
On the basis of the computational results depicted in Figures 9 and 10 and the experimental results previously discussed, we were able to draw a number of conclusions about the key photodecarbonylation reaction. Conformational restriction can sometimes lead to activation barriers for recombination. In the photodecarbonylation reaction of ketones 11a and 11c−f, the computationally generated radical pairs 10a and 10c−f were not energetic minima unless they were confined by the crystalline lattice. This is consistent with our hypothesis that in the solid state, unfavorable ground-state substrate conformations can lead to repulsive interactions in the course of the reaction, realized by the presence of an activation barrier. Comparatively, the reactive polymorph of ketone 28d likely undergoes a barrierless recombination due to a favorable ground state conformation. This is reflected both by the barrierless recombination observed in computational analysis and by the high-yielding formation of photoproduct 30d. Additionally, the magnitude of the barrier to radical recombination for radical pairs derived from ketones 11 is related to the presence of an ortho-substituent. The computational results clearly show a higher activation energy for recombination when an ortho-substituent is present (6.1–6.8 kcal/mol) and a diminished barrier for radical pairs that lack ortho substitution (1.7–4.8 kcal/mol).62
Summary of the Optimization and Mechanistic Analysis of the Solid-State Photodecarbonylation Reaction.
The optimization and analysis en route to the bis(cyclotryptamine) alkaloids gave insight into the role of substrate conformation on the success or failure of the solid-state photodecarbonylation reaction. It was found that the performance of the transformation was highly sensitive to remote substituent effects. While the reaction was low-yielding for N-methyl ketones 11d−f, the solid-state photodecarbonylation reaction of N-acyl ketones 28d and 29d,e gave up to 61% yield of the desired vicinal quaternary stereocenter-containing product. We hypothesize that changes to the solid-state conformation of the substrate underpin these dramatic substituent effects on photodecarbonylation. This hypothesis was further supported computationally through study of the C−C bond forming events for radical pairs 10a, 10c−f, and 22d. Additionally, a key stereoelectronic relationship between the conformation of the substrate and the photolability of the ketone carbonyl was identified. This led to dramatically different reactivity of two polymorphic forms of 28d, the failed reaction of 28e, and efficient decarbonylative C−C bond formation from 29d,e. Ultimately, this optimization employed crystal engineering of the solid-state conformation to improve the key step of our synthetic endeavor. To our knowledge, this is the first use of a crystal engineering-based optimization in a natural product total synthesis campaign.
Total Synthesis of a Bis(Cyclotryptamine) Alkaloid with the Piperidinoindoline Framework.
Having optimized the solid-state photochemical methodology to assemble the daunting vicinal quaternary stereocenter motif, we were poised to elaborate 31 to access members of the bis(cyclotryptamine) alkaloid family. We first focused our efforts on synthetic elaboration of 31d (Scheme 5). It was found that treatment of 31d with sodium hydride and iodomethane furnished N-methylamide 9d in 85% yield. Next, we attempted to install the requisite nitrogen substituents on the aromatic rings through a metal-catalyzed double amination reaction. Although robust methods for amination of hindered aryl chlorides have been reported,63 attempts at amination of 9d were uniformly unsuccessful. We surmise that the dense steric encumbrance of the vicinal quaternary stereocenters and the potential for the amide carbonyls in 9d to chelate catalysts in a bidentate fashion is responsible for the poor reactivity.
Scheme 5.
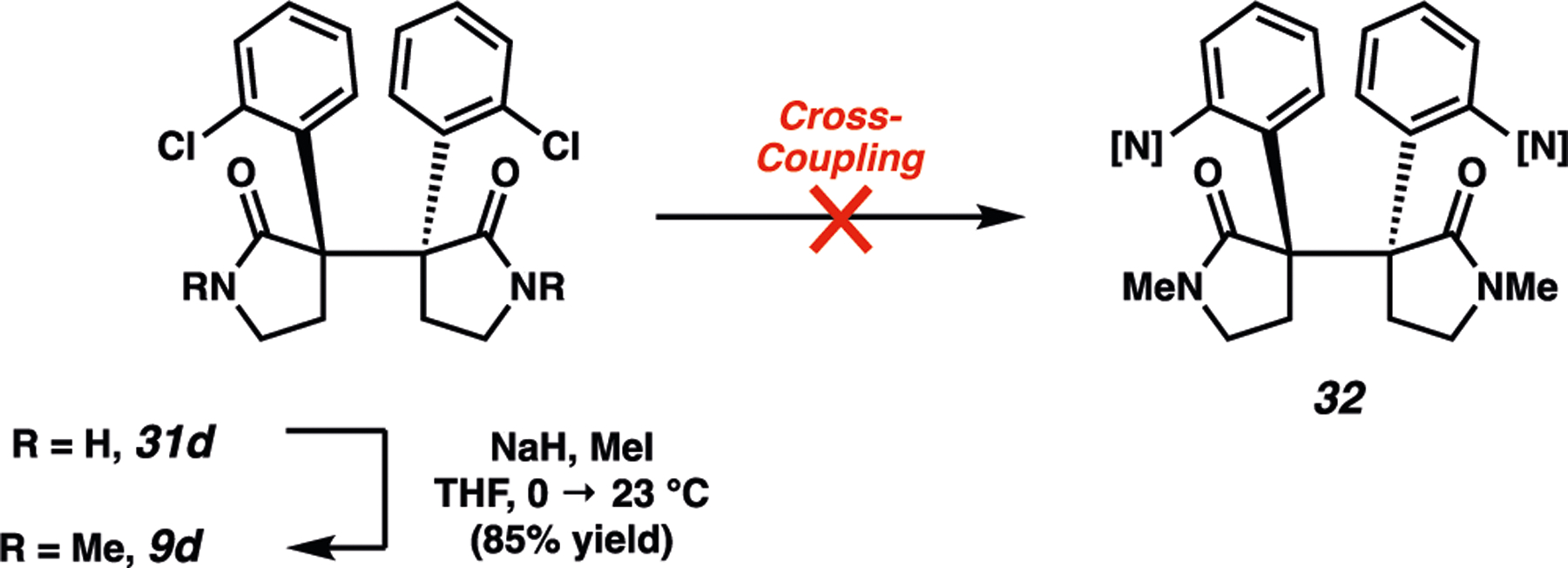
Failed Cross-Coupling Attempts Using Bis(Arylchloride) 9d
Unabated by the difficulty of functionalizing the C−Cl bond of 9d, we turned our efforts to the potentially more reactive ortho-bromo variant, 9e (Scheme 6). Efficient N-methylation of 31e was realized through a base-mediated alkylation to provide 9e. After subjecting 9e to a variety of conditions to effect amination of the aryl bromides, we found that a modification of Ma’s copper-catalyzed azidation conditions yielded the desired double azidation product, 33.64 We then subjected 33 to LiAlH4 to enact a global reduction in an effort to access an equivalent of biosynthon 8, and thereby the bis(cyclotryptamine) framework. To our surprise, under strongly reducing conditions, 33 underwent a skeletal rearrangement to provide piperidinoindoline 36.65 While there are several mechanistic possibilities to explain this reaction, one plausible pathway involves double azide reduction to give 34, followed by Lewis acid-mediated transamidation to generate bis(oxindole) 35. Finally, reductive cyclocondensation of 35 resulted in formation of piperidinoindoline 36. Although single-crystal X-ray diffraction ultimately confirmed the structure, we were initially unsure if the correct structural assignment of 36 was the piperidinoindoline depicted. As shown in Scheme 7, it was plausible that intermediate 34 could have undergone double cyclodehydration to provide dehydrobhesine (37), followed by monoamidine reduction to give bhesine (38).66
Scheme 6.

Cross-Coupling and Assembly of “Dihydropsychotriadine” 36
Scheme 7.

Possible Competing Pathway to Give 37 or 38a
aNot observed.
Before we ultimately obtained the single-crystal X-ray structure of 36, we sought to confirm the structural identity of 36 by oxidizing the aminal to the corresponding amidine (Scheme 8). To accomplish this transformation, 36 was converted to bis(amidine) 39 under Ley−Griffith oxidation conditions.67 Initially surmising that we may have generated dehydrobhesine (37), we compared our synthetic material to an authentic sample of 37 extracted from the leaves of the Psychotria colorata flower.68 Upon NMR spectroscopic analysis, it was observed that the natural sample contained a 7:1 mixture of 37 and a second, unknown isomeric alkaloid. Surprisingly, the spectrum of our synthetic material 39 matched that of the unknown alkaloid present in the natural material. On the basis of this analysis and the unambiguous crystallographic characterization of 36, we propose that our synthetic material bears the depicted piperidinoindoline scaffold. Furthermore, its presence in the extracts of Psychotria colorata suggests it is a naturally occurring alkaloid that we have termed “psychotriadine.”
Scheme 8.
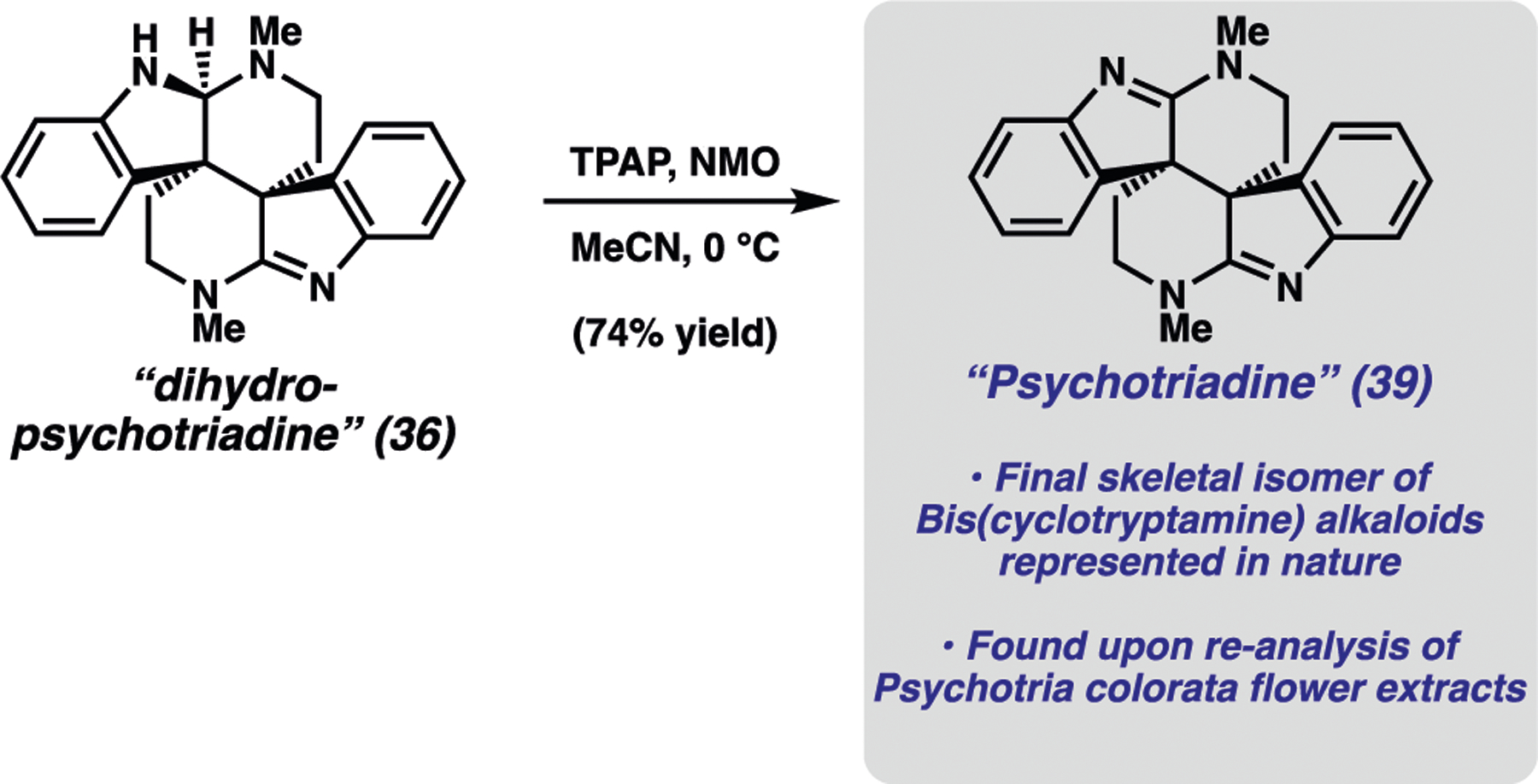
Total Synthesis of “Psychotriadine” 39
CONCLUSIONS
We completed the first total synthesis of a naturally occurring bis(cyclotryptamine) alkaloid featuring the elusive piperidinoindoline scaffold and confirmed its natural occurrence in the Psychotria colorata flower. These studies demonstrate that all five of the structurally distinct bis(cyclotryptamine) ring systems originally proposed by R. B. Woodward and Robert Robinson are, in fact, seen in natural products. Essential to the success of this endeavor was the use of the solid-state photodecarbonylation reaction to stereoselectively forge the daunting vicinal quaternary stereocenter motif and a fortuitous reductive rearrangement of bis(azide) 33 to give piperidinoindoline 36.
Psychotriadine (39) marks the most complex, synthetically challenging natural product accessed by solid-state photochemistry to date. In deploying the key photodecarbonylation reaction, two primary hurdles were faced: poor chemoselectivity for recombination and a lack of substrate reactivity. Both of these challenges were overcome by manipulation of the substrate conformation. To our knowledge, this is the first use of crystal engineering to optimize a reaction in a total synthesis endeavor.69 While these efforts showcase the power of crystal engineering as a tool to optimize solid-state reactions, the unpredictability of crystallization remains a major challenge. As is reflected in this work, optimization of the crystalline conformation of a substrate currently relies on trial-and-error based experimentation. Although the use of easily modifiable motifs expedites this process, de novo prediction of crystal conformation represents the ideal solution. As such, the advent of computational prediction of crystal structures would offer a rapid means of predictive control of reactivity and selectivity in solid-state organic reactions.70
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Science Foundation (CHE-1855342 for M.G.G.) and the Trueblood Family (for N.K.G.) for financial support. J.J.D. acknowledges the UCLA Graduate Division for a Dissertation Year Fellowship. We thank Professor Luisella Verotta (University of Milan, Italy) for helpful discussions and for providing authentic samples of 37 and 38. We also thank Chih-Te Zee and Jose A. Rodriguez for their generous help solving the microED structures of 29d and 29e. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c01100.
Detailed experimental procedures and compound characterization data (PDF)
Accession Codes
CCDC 2006460–2006463 and 2006465–2006469 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac. uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c01100
The authors declare no competing financial interest.
Contributor Information
Jordan J. Dotson, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
Ieva Liepuoniute, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States;.
J. Logan Bachman, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Vince M. Hipwell, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
Saeed I. Khan, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
K. N. Houk, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States;.
Neil K. Garg, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States;.
Miguel A. Garcia-Garibay, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States;.
REFERENCES
- (1).For other reviews of photochemical reactions being used in organic synthesis, also see references 2−4.; Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kärkäs MD; Porco JA; Stephenson CRJ Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev 2016, 116, 9683–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bach T; Hahn J Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew. Chem., Int. Ed 2011, 50, 1000–1045. [DOI] [PubMed] [Google Scholar]
- (4).Nicholls TP; Leonori D; Bissember AC Applications of Visible Light Photoredox Catalysis to the Synthesis of Natural Products and Related Compounds. Nat. Prod. Rep 2016, 33, 1248–1254. [DOI] [PubMed] [Google Scholar]
- (5).For other reviews of photochemical reactions conducted in the solid state, also see references 6 and 7.; Dotson JJ; Perez-Estrada S; Garcia-Garibay MA Taming Radical Pairs in Nanocrystalline Ketones: Photochemical Synthesis of Compounds with Vicinal Stereogenic All-Carbon Quaternary Centers. J. Am. Chem. Soc 2018, 140, 8359–8371. [DOI] [PubMed] [Google Scholar]
- (6).Ramamurthy V; Venkatesan K Photochemical Reactions of Organic Crystals. Chem. Rev 1987, 87, 433–481. [Google Scholar]
- (7).Ramamurthy V; Sivaguru J Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition. Chem. Rev 2016, 116, 9914–9993. [DOI] [PubMed] [Google Scholar]
- (8).Hernandez-Linares MG; Guerrero-Luna G; Perez-Estrada S; Ellison M; Ortin M-M; Garcia-Garibay MA Large-Scale Green Chemical Synthesis of Adjacent Quaternary Chiral Centers by Continuous Flow Photodecarbonylation of Aqueous Suspensions of Nanocrystalline Ketones. J. Am. Chem. Soc 2015, 137, 1679–168. [DOI] [PubMed] [Google Scholar]
- (9).Mortko CJ; Garcia-Garibay MA Green Chemistry Strategies Using Crystal-to-Crystal Photoreactions: Stereoselective Synthesis and Decarbonylation of trans-α,α’-Dialkenoylcyclohexanones. J. Am. Chem. Soc 2005, 127, 7994–7995. [DOI] [PubMed] [Google Scholar]
- (10).Ng D; Yang Z; Garcia-Garibay MA Total Synthesis of (±)-Herbertenolide by Stereospecific Formation of Vicinal Quaternary Centers in a Crystalline Ketone. Org. Lett 2004, 6, 645–647. [DOI] [PubMed] [Google Scholar]
- (11).Natarajan A; Ng D; Yang Z; Garcia-Garibay MA Parallel Synthesis of (+)- and (−)-)-Cuparenone by Radical Combination in Crystalline Solids. Angew. Chem., Int. Ed 2007, 46, 6485–6487. [DOI] [PubMed] [Google Scholar]
- (12).Yang Z; Photodecarbonylation of crystalline ketones and its application to the synthesis of tochuinyl acetate, Ph.D. Thesis. University of California, Los Angeles, 2003. Available from ProQuest Dissertations & Theses Global database. (Accession Order No. AAT 3133088). [Google Scholar]
- (13).Chang TY; Dotson JJ; Garcia-Garibay MA Org. Lett 2020, 22, 8855–8859. [DOI] [PubMed] [Google Scholar]
- (14).Peterson EA; Overman LE Contiguous Stereogenic Quaternary Carbons: A Daunting Challenge in Natural Products Synthesis. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 11943–11948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Long R; Huang J; Gong J; Yang Z Direct Construction of Vicinal All-Carbon Quaternary Stereocenters in Natural Product Synthesis. Nat. Prod. Rep 2015, 32, 1584–1601. [DOI] [PubMed] [Google Scholar]
- (16).Liu Y; Han S; Liu W; Stoltz BM Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res 2015, 48, 740–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pompeo MM; Cheah JH; Movassaghi M Total Synthesis and Anti-Cancer Activity of All Known Communesin Alkaloids and Related Derivatives. J. Am. Chem. Soc 2019, 141, 14411–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For other reviews on the isolation and synthesis of bis(cyclotryptamine) alkaloids, also see references 18–21.; May JA; Stoltz B The Structural and Synthetic Implications of the Biosynthesis of the Calycanthaceous Alkaloids, the Communesins, and Nomofungin. Tetrahedron 2006, 62, 5262–5271. [Google Scholar]
- (19).Schmidt MA; Movassaghi M New Strategies for the Synthesis of Hexahydropyrroloindole Alkaloids Inspired by Bio-synthetic Hypotheses. Synlett 2008, 2008, 313–324. [Google Scholar]
- (20).Steven A; Overman LE Total Synthesis of Complex Cyclotryptamine Alkaloids: Stereocontrolled Construction of Quaternary Carbon Stereocenters. Angew. Chem., Int. Ed 2007, 46, 5488–5508. [DOI] [PubMed] [Google Scholar]
- (21).Trost BM; Osipov M Recent Advances on the Total Synthesis of Communesin Alkaloids and Perophoramidine. Chem. -Eur. J 2015, 21, 16318–16343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xu J-B; Cheng K-J Studies on the Alkaloids of the Calycanthaceae and their Syntheses. Molecules 2015, 20, 6715–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).The fifth scaffold, not depicted, has only been observed in a higher oxidation state and possesses opposite relative stereochemistry compared to 4–7. For this scaffold, see; Verbitski SM; Mayne CL; Davis RA; Concepcion GP; Ireland CM Isolation, Structure Determination, and Biological Activity of a Novel Alkaloid, Perophoramidine, from the Philippine As-cidian Perophoranamei. J. Org. Chem 2002, 67, 7124–7126. [DOI] [PubMed] [Google Scholar]
- (24).Eccles RG Calycanthine. Druggists’ Circular and Chemical Gazette 1888, 32, 65. [Google Scholar]
- (25).Robinson R; Teuber HJ Reactions with Nitrosodisulfonate. IV. Calycanthine and Calycanthidine. Chem. Ind. (London) 1954, 783–784. [Google Scholar]
- (26).Manske RH The Alkaloids of Calycanthacaea. Alkaloids 1965, 8, 581–589. [Google Scholar]
- (27).While this structure has never been isolated from a natural source, a structure with this ring system (albeit the meso isomer) was reported synthetically. For further details, see the following reference.; Hall ES; McCapra F; Scott AI Biogenetic-Type Synthesis of the Calycanthaceous Alkaloids. Tetrahedron 1967, 23, 4131–4141. [DOI] [PubMed] [Google Scholar]
- (28).Dotson JJ; Bachman JL; Garcia-Garibay MA; Garg NK Discovery and Total Synthesis of a Bis(Cyclotryptamine) Alkaloid Bearing the Elusive Piperidinoindoline Scaffold. J. Am. Chem. Soc 2020, 142, 11685–11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).When a ketone bearing an ortho-amine was synthesized, spontaneous condensation of the amine onto the ketone to form the corresponding spirocyclic aminal was observed.
- (30).Although direct amination of the methoxy-substituted arene may be challenging, 9f could also be converted to the corresponding aryl triflate, which should be more reactive toward cross-coupling.
- (31).The common synthetic route used to access of ketones 11 was adapted from studies previously reported by our group. For details see the following reference:; Resendiz MJE; Natarajan A; Garcia-Garibay MA Diastereoselective Synthesis and Spin-Dependent Photodecarbonylation of Di(3-Phenyl-2-Pyrrolidinon-3-yl)Ketones: Synthesis of Nonadjacent and Adjacent Stereogenic Quaternary Centers. Chem. Commun 2008, 193–195. [DOI] [PubMed] [Google Scholar]
- (32).Dotson JJ; Garg NK; Garcia-Garibay MA Evaluation of the Photodecarbonylation of Crystalline Ketones for the Installation of Reverse Prenyl Groups on the Pyrrolidinoindoline Scaffold. Tetrahedron [Online early access]. DOI: 10.1016/j.tet.2020.131181. Published Online: Apr 10, 2020. https://www.sciencedirect.com/science/article/abs/pii/S0040402020303161?via%3Dihub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).It should be noted that all of the ketones that are disclosed in this study crystallized as racemic mixtures and their photodecarbonylation resulted in racemic products. For discussion of the solid-state photodecarbonylation being used to prepare enantioenriched products, see reference 2.
- (34).Note that starting material (11a) remained after irradiation. The yield based on remaining starting material is 54% (9a).
- (35).The loss of mass balance was attributed to unidentified, insoluble decomposition products.
- (36).The yield of disproportionation was determined based on the yield of 17a.
- (37).It was noted that a significant amount of mass was lost in the photodecarbonylation reactions that were low yielding (reactions of ketones 11d, 11e, and 11f). In these cases, we observed a significant quantity of insoluble solid material following irradiation. We were unable to identify the chemical identity of this unknown solid but believe it to be the primary source of lost mass balance.
- (38).Note that starting material (11d) remained after irradiation. The yield based on remaining starting material is 17% (9d) and 24% (17d).
- (39).Even substrates that exclusively undergo disproportionation upon solution-phase irradiation show full suppression of the disproportionation pathway in the crystalline solid state. For more details, see; Choe T; Khan SI; Garcia-Garibay MA Combination vs. Disproportionation in Dialkyl Biradicals. Selectivity Reversal in a Crystalline Solid. Photochem. Photobiol. Sci 2006, 5, 449–451. [DOI] [PubMed] [Google Scholar]
- (40).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006; pp 104. [Google Scholar]
- (41).Consistent with our experimental results, this repulsive interaction results in diminished recombination yields for ketones with larger ortho moieties.
- (42).The topochemical principle states that reactions in the crystalline solid state will favor pathways that feature a minimum amount of atomic or molecular movement. For further discussion of the topochemical principle, see reference 2c and the following book:; Schmidt GMJ; et al. in Solid State Photochemistry (Ed.: Ginsburg D) Verlag Chemie: Weinheim, 1976; pp, 1–280. [Google Scholar]
- (43).We were also interested in a directed C-H amination of 9c; however, we recognized that the meta-hydroxy moieties would need to be reductively cleaved to provide the bis(pyrrolidinoindoline) alkaloids. Functionalization of 9a would lead to a more ideal synthesis and was therefore prioritized in these studies. For functionalization of C(sp2)-H bonds ortho to an amide directing group, also see references 44,45.; Qiu F-C; Yang W-C; Chang Y-Z; Guan B-T Palladium-Catalyzed ortho-Halogenation of Tertiary Benzamides. Asian J. Org. Chem 2017, 6, 1361–136. [Google Scholar]
- (44).Wykypiel W; Lohmann J-J; Seebach D Lithiation in W-Position to the N-Atom of Triphenylacetamides from Cyclic Secondary Amines. Rearrangements of Metalated Triphenylacetamides by 1,3-Shift of Carbamoyl Groups. Helv. Chim. Acta 1981, 64, 1337–1346. [Google Scholar]
- (45).Yeung CS; Zhao X; Borduas N; Dong VM Pd-Catalyzed ortho-Arylation of Phenylacetamides, Benzamides, and Anilides with Simple Arenes Using Sodium Persulfate. Chem. Sci 2010, 1, 331–336. [Google Scholar]
- (46).For a review of directed aromatic lithiation of tertiary amides, see:; Beak P; Snieckus V Directed Lithiation of Aromatic Tertiary Amides: An Evolving Synthetic Methodology for Polysubstituted Aromatics. Acc. Chem. Res 1982, 15, 306–312. [Google Scholar]
- (47).We did not investigate electrophilic or radical mediated amination of 9a, because such transformations would likely suffer from issues of poor site-selectivity (i.e., ortho vs para functionalization). This could conceivably be mitigated with removable blocking groups installed at the 4-position of the phenyl rings, however, such strategies were not pursued.
- (48).Formation of the carbanion was determined by quenching with D2O followed by NMR spectroscopic analysis.
- (49).Compound 20 was easily prepared in one step from a ketone previously reported by our group. For details, see reference 31.
- (50).Since we anticipated that aryl halides would be ideal latent electrophiles for metal-catalyzed amination, we pursued the synthesis of chloro- and bromoketones 27d and 27e.
- (51).The occurrence of a new, more stable polymorph that precludes formation of the desired polymorph is well-known and is often referred to as the “disappearing polymorph problem.” For further discussion of this topic, also see reference 52; Cruz-Cabeza AJ; Bernstein J Conformational Polymorphism. Chem. Rev 2014, 114, 2170–2191. [DOI] [PubMed] [Google Scholar]
- (52).Snider DA; Addicks W; Owens W Polymorphism in Generic Drug Product Development. Adv. Drug Delivery Rev 2004, 56, 391–395. [DOI] [PubMed] [Google Scholar]
- (53).While highly irreproducible, several successful crystallizations to provide the reactive polymorph were accomplished when they were conducted in a different lab room, using new glassware, and new lab coat to avoid contamination of the unreactive polymorph. In these instances, small quantities of the reactive polymorph were used to seed crystallization.
- (54).Larger scale transformations were not attempted. However, we surmise that the inherent scalability of suspension photochemistry would permit increased scale up without loss of reaction performance. See the following references for the use of crystalline suspensions to scale up the solid-state photodecarbonylation reaction. (For examples, see reference 8 and; Veerman; Resendiz MJE; Garcia-Garibay MA Large-Scale Photochemical Reactions of Nanocrystalline Suspensions: A Promising Green Chemistry Method. Org. Lett 2006, 8, 2615–2617. [DOI] [PubMed] [Google Scholar]
- (55).The structures of 29d and 29e were elucidated using both single-crystal X-ray diffraction as well as microED. Both methods resulted in nearly identical solutions for both structures. For a side-by-side comparison, see Figure S1 in the Supporting Information. For further discussion of microED-based structural determination, also see reference 56; Rodriguez JA; Eisenberg DS; Gonen T Taking the measure of MicroED. Curr. Opin. Struct. Biol 2017, 46, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jones CG; Martynowycz MW; Hattne J; Fulton TJ; Stoltz BM; Rodriguez JA; Nelson HM; Gonen T The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci 2018, 4, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Dapprich S; Komáromi I; Byun KS; Morokuma K; Frisch MJ A New ONIOM Implementation in Gaussian98. Part I. The Calculation of Energies, Gradients, Vibrational Frequencies and Electric Field Derivatives. J. Mol. Struct.: THEOCHEM 1999, 461, 1–21. [Google Scholar]
- (58).Vreven T; Byun KS; Komáromi I; Dapprich S; Montgomery JA; Morokuma K; Frisch MJ Combining Quantum Mechanics Methods with Molecular Mechanics Methods in ONIOM. J. Chem. Theory Comput 2006, 2, 815–826. [DOI] [PubMed] [Google Scholar]
- (59).This type of QM/MM strategy to model a reaction in the crystalline solid state was reported in the following reference:; Keating AE; Shin SH; Houk KN; Garcia-Garibay MA Combining Quantum Mechanical Reaction Pathways with Force Field Lattice Interactions to Model a Solid-State Phototransformation. J. Am. Chem. Soc 1997, 119, 1474–1475. [Google Scholar]
- (60).Chai JD; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. [DOI] [PubMed] [Google Scholar]
- (61).We favor the employment of a molecular mechanics method to describe the low-level layer (over semiempirical methods or quantum chemical calculations with very small basis sets) since the MM force field by definition includes a description of dispersion effects between all atoms in the MM region and between QM and MM regions. Indeed, this hybrid approach has been shown to be accurate in similar computational studies of solid-state transformations of stilbene where crystalline environment calculations were included in the low-level layer (see reference 59).
- (62).We do not consider the ordering of barriers for 10d-f to be meaningful as they fall within a 0.7 kcal/mol range and are therefore within computational error of one another.
- (63).For a review of catalytic amination of aryl halides, see; Heravi MM; Kheilkordi Z; Zadsirjan V; Heydari M; Malmir M Buchwald-Hartwig Reaction: An Overview. J. Organomet. Chem 2018, 861, 17–104. [Google Scholar]
- (64).Unfortunately, despite extensive efforts, 33 could not be fully purified and was, therefore, carried forward directly in the subsequent step.
- (65).Note that the double azidation reaction to convert 9e to 33 was challenging to enact and resulted in < 35% yield (based on 1H NMR analysis of the semipure material). As such, the reductive cyclization to convert 33 into 36 likely took place in > 75% yield.
- (66).The observed 1H and 13C NMR and spectra of 36 were not identical to the published spectra of bhesine (38). We hypothesized that we may have either produced an aminal isomer of 38 or subtle changes in solvent acidity may have shifted the 1H and 13C resonances. For the first isolation of 38, see; Balayer A; Sévenet T; Schaller H; Haudi AHA; Chiaroni A; Riche C; Païs M Dihydroquinoline-type Alkaloids from Bhesa Paniculata, Celastraceae. Nat. Prod. Lett 1993, 2, 61–67. [Google Scholar]
- (67).Oxidation conditions adapted from:; Higuchi K; Sato Y; Tsuchimochi M; Sugiura K; Hatori M; Kawasaki T First Total Synthesis of Hinckdentine A. Org. Lett 2009, 11, 197–199. [DOI] [PubMed] [Google Scholar]
- (68).Verotta L; Pilati T; Tató M; Elisabetsky E; Amaor TA; Nunes DS Pyrrolidinoindoline Alkaloids from Psychotria colorata. J. Nat. Prod 1998, 61, 392–396. [DOI] [PubMed] [Google Scholar]
- (69).We anticipate that the rapid structural determination enabled by microED technology will streamline the use of crystal engineering-based reaction optimization in future studies. Only two structures (29d and 29e) were obtained using microED in this report because the requisite technological capabilities were not available to us until the end of this study.
- (70).Computational prediction of crystal structures has been recognized as an unsolved problem in the field of computational chemistry. For more discussion, see the following review:; Houk KN; Liu F Holy Grails for Computational Organic Chemistry and Biochemistry. Acc. Chem. Res 2017, 50, 539–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.