

Abstract
Aptamers are short oligonucleotides isolated in vitro from randomized libraries that can bind to specific molecules with high affinity, and offer a number of advantages relative to antibodies as biorecognition elements in biosensors. However, it remains difficult and labor-intensive to develop aptamer-based sensors for small-molecule detection. Here, we provide a detailed review of the challenges and advances in the isolation and characterization of small-molecule-binding DNA aptamers and their utilization in sensors. First, we discuss in vitro methodologies for the isolation of aptamers, and provide strategies and guidance on selecting the appropriate strategy for generating aptamers with optimal binding properties for a given application. We next examine techniques for robust characterizing aptamer-target binding and structure. Afterwards, we discuss various small-molecule sensing platforms based on both original or engineered aptamers, and their strengths and limitations for detection applications. Finally, we conclude with a general workflow to develop aptamer-based small-molecule sensors for real-world applications.
Keywords: Aptamer, Aptamer characterization, Small molecule, Biosensor, SELEX
Graphical Abstract
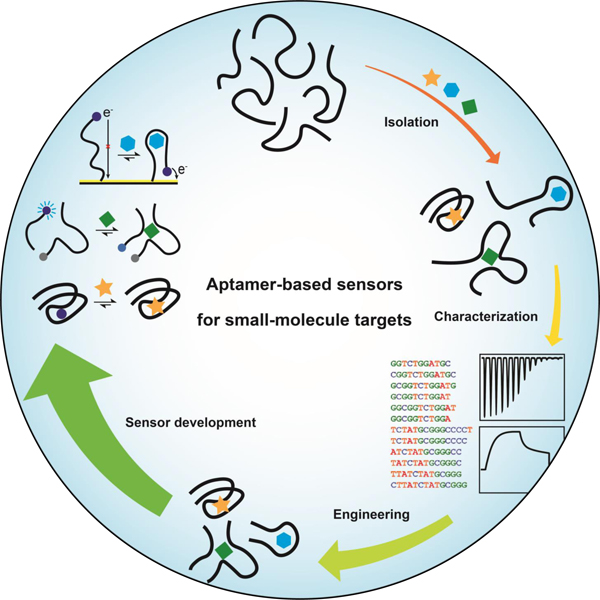
Aptamers are oligonucleotide-based recognition elements isolated from random libraries that have several favorable attributes for biosensing. This review comprehensively examines state-of-the-art methods and advances in the isolation and characterization of small-molecule-binding aptamers and their use in various biosensors. Factors limiting aptamer-based sensors and potential solutions to these issues are also discussed.
1. Introduction
1.1. Small-molecule biosensing
Sensitive and accurate detection of small-molecule targets is critical for diverse applications including environmental monitoring (e.g., toxins, heavy metals), food safety (e.g., antibiotics and additives), law enforcement (e.g., illicit drugs), and medical diagnostics (e.g., metabolites, neurotransmitters).[1–3] Methods based on chromatography and mass spectroscopy enable accurate and quantitative detection, but are largely restricted to lab settings and require sophisticated, expensive instrumentation and trained personnel. Biosensors have emerged as an alternative for simple and portable detection applications, ranging from on-site drug tests and personal glucose meters[4] to point-of-care disease biomarker screens[5]. The widespread proliferation of biosensors can be attributed to the fact that they can couple high sensitivity and specificity with ease of use, cost-effectiveness, and rapid turnaround times.[6]
In general, biosensors consist of three components: a bioreceptor, a transducer, and a signal readout system. Bioreceptors are macromolecules such as enzymes, antibodies, and oligonucleotides that can specifically recognize an analyte of interest with high affinity and specificity.[6] The transducer converts bioreceptor-analyte binding events into a measurable signal readout based on changes in optical,[7] electrochemical,[8] or electrical output[9]. Many biosensors employ enzymes that serve as both a bioreceptor and a transducer; the most well-known enzyme-based biosensor is the personal glucose meter, which utilizes glucose oxidase to generate an electrochemical signal in the presence of glucose.[10] Enzyme-based sensors are highly sensitive, specific, respond rapidly, and allow for quantitative target detection directly in complex biological media such as cell lysate and blood.[11] However, such sensing platforms are not generalizable, and only a limited number of analytes can serve as substrates for the small amount of currently available enzymes suitable for detection applications. In addition, enzymes are temperature-sensitive and have a short shelf life. Another common type of biosensor is the immunoassay, which employs antibodies as recognition elements.[12] Antibodies are proteins-based bioaffinity elements that can bind to specific analytes with high affinity,[13] enabling highly sensitive analyte detection in complex biosamples when used as bioreceptors.[14] Immunoassays have been successfully adapted onto paper substrates to fabricate lateral-flow devices,[15] a sensitive and low-cost assay that is currently the most popular biosensor format. However, antibodies have several disadvantages that limit their utility for biosensing.[16,17] Generating and characterizing new antibodies is time-consuming and laborious—the whole process of antibody generation, which includes target preparation, immunization of animals, antibody purification, and antibody characterization, is lengthy and labor intensive and can take half a year or more.[13] Additionally, the in vivo nature of the antibody generation process allows little control over their binding properties. For example, several antibodies currently used for small-molecule detection have been shown to non-specifically bind structurally similar non-target interferents.[18,19] Moreover, it is very challenging to generate antibodies that recognize entire families of small-molecule targets, such as illicit drugs, antibiotics, or pesticides.[20,21] In the case of emerging threats such as new designer drugs, for example, minor modifications to a drug’s core structure can greatly impair binding with existing detection antibodies.[22] High production costs, batch-to-batch variation, and low shelf stability also make antibodies less than ideal for on-site biosensing.[16,23]
1.2. History of small-molecule-binding aptamers
Aptamers are short oligonucleotides typically ranging from 20 to 80 nucleotides in length isolated in vitro from randomized libraries to bind specific molecules with high affinity.[24,25] Ligand recognition by aptamers is based on intermolecular forces such as electrostatic interactions, hydrogen bonding, π-π stacking, and van der Waals forces.[26] The isolation of aptamers was first described in 1990 by two independent research groups. Tuerk and Gold termed the process of isolating such molecules as SELEX (Systematic Evolution of Ligands by Exponential Enrichment),[24] and Ellington and Szostak coined the term ‘aptamer,’[25] from the Greek word aptus, which means “to fit”. The first few small-molecule-binding aptamers were isolated by Szostak group in the early 1990s.[25,27–30] Early targets included biomolecules such as amino acids, cofactors, and nucleotides. In parallel, in vitro selection experiments emerged as a means of studying binding interactions between nucleic acid motifs and ligands such as aminoglycoside antibiotics[31,32] and chloramphenicol[33]. The concept of using aptamers as bioreagents for small-molecule sensing was first explored in the mid-1990s.[34] However, the exploration of small-molecule-binding aptamers as sensing elements did not gain momentum until the early 2000s. Many of the aptamer-based optical and electrochemical sensing platforms used today were established by the mid-2000s.[35–37]
1.3. The advantage of aptamers in biosensing
To date, hundreds of aptamers have been isolated for a great number of small-molecule targets.[17] The number of publications related to the use of aptamer-based sensors for small-molecule detection has grown exponentially. Aptamers offer several exceptional advantages as bioreceptors for sensing compared with antibodies (Table 1).[23,38] For example, since small molecules have low immunogenicity, they generally need to be subjected to a challenging process of conjugation to carrier proteins prior to immunization in order to elicit an effective antibody response.[39] In contrast, the in vitro nature of SELEX allows for the direct isolation of aptamers for virtually any small molecule, including those that are non-immunogenic or toxic. Additionally, experimental conditions can be customized for the intended application, such as the working buffer ionic strength, pH, and even solvent identity. Furthermore, aptamer target-binding affinity and specificity can be closely controlled by employing different selection strategies and manipulating the selection conditions during the SELEX process. In contrast, the in vivo antibody generation process precludes such control. Moreover, aptamer isolation typically takes only a few weeks. This is critical for emerging and rapidly evolving targets such as designer drugs and pathogens, where the lengthy antibody-development process—which can take more than a year—cannot keep pace.[40–42] Aptamers are also far less prone to degradation and denaturation under harsh conditions (e.g., high temperature) relative to antibodies, and therefore have much longer shelf-lives. Aptamers can also be rapidly chemically synthesized, which makes them more economical to produce with lower batch-to-batch variation relative to antibody generation. The sequence engineering of aptamers is also straightforward, enabling introduction of diverse sensing functionalities. Indeed, various chemistries are commercially available to modify aptamers with different fluorescent,[43] electrochemical,[44] or enzymatic[45] tags for signal reporting purposes.
Table 1.
| Features | Enzyme | Antibody | Aptamer |
|---|---|---|---|
| Target | Limited to specific substrates | Limited for small molecules and toxic compounds | No limitation |
| Development Time | N/A | 6–18 months | 2–6 weeks |
| Production cost | High | Very high | Low |
| Batch-to-batch variation | High | High | Low |
| Binding profile | Uncontrollable | Uncontrollable | Highly tuneable |
| Chemical Stability | Low | Low | High |
1.4. Aptamer-based sensors for small-molecule detection
Aptamers have been incorporated into a variety of sensing platforms for the fluorescent, colorimetric, and electrochemical detection of small-molecule targets. Most aptamer-based assays operate on a system wherein aptamer-target binding induces a major conformational change in the aptamer (e.g., folding, assembly, or strand-displacement), which in turn transduces the binding event into a measurable signal. Stojanovic et al. pioneered the first aptamer-based fluorescence sensors targeting small molecules. They designed aptamer constructs such as split-aptamer fragments[43] and self-folding structure-switching aptamers[35] labeled with fluorophore-quencher pairs, where target binding causes a relative spatial reorientation of the pair, resulting in a change in fluorescence. Although these assays were rapid, they required chemically-labeled aptamers and specialized instrumentation for detection, and achieved only micromolar detection limits in buffer. Liu and Lu developed a colorimetric sensing platform for small-molecule detection based on DNA-modified gold nanoparticle networks cross-linked with aptamers,[36] where target binding to the aptamers mediates the disassembly of the nanoparticle network, triggering a blue-to-red color change that can be observed with the naked eye. However, this sensor platform has yet to be used successfully for detection in biological samples. Plaxco group developed the electrochemical aptamer-based (E-AB) sensing platform and a suite of such sensors for the rapid detection of various small molecules in complex samples such as serum,[37] soil,[46] foodstuffs,[47] and whole blood.[48] E-AB sensors consist of aptamers modified with a redox reporter such as methylene blue, and a thiol group for coupling to the surface of a gold electrode at their terminal ends.[49] Aptamer-target binding induces a conformational change in the aptamer that repositions the redox label relative to the electrode surface, which can be transduced into an electrochemical signal that is proportional to the concentration of target. E-AB sensors exhibit high selectivity and rapidity, and have recently been employed for real-time small molecule detection in live animals.[50] For reviews on aptamer-based small-molecule sensors, readers should refer to the following articles.[51–54]
However, even though more than 200 small-molecule binding aptamers have been reported[17] and thousands of aptamer-based small molecule sensors have been developed to date, only a few of them have the necessary sensitivity and specificity for real-world analytical applications. This is due to a variety of issues, including the low affinity and specificity of many small-molecule-binding aptamers, challenges in aptamer characterization, and the difficulty of engineering functionalized aptamers for sensor development. In this review, we will discuss the challenges and advances in aptamer isolation, characterization, and engineering for small molecule sensing. Finally, we will discuss some innovative strategies for enhancing the affinity and specificity of aptamers, performing robust high-throughput characterization, and developing universal sensing platforms based on non-engineered aptamers for detecting small-molecule targets in a rapid, cost-effective manner. We will primarily focus on DNA aptamers, as most aptamer sensors and the most-cited small-molecule-binding aptamers at present are DNA-based.[17] In addition, despite the greater conformational flexibility and structural complexity achieved by RNA, DNA and RNA aptamers generally have similar target-binding affinities,[55] while DNA has the additional advantages of high chemical stability and greater ease of handling.[17]
2. Challenges and advances in aptamer isolation
2.1. The SELEX procedure
SELEX is a multi-round process (Figure 1),[25] where each round entails incubating the target of interest with an oligonucleotide library (DNA or RNA), separating binding strands from non-binders, amplifying the binders via PCR, and then generating a single-stranded pool from these double-stranded amplicons for the next round of selection. This process is repeated until the pool is primarily populated with sequences that bind to the target. Thereafter, the sequences of the aptamers are identified using DNA sequencing technologies such as Sanger sequencing [25] or high-throughput sequencing (HTS).[56]
Figure 1.
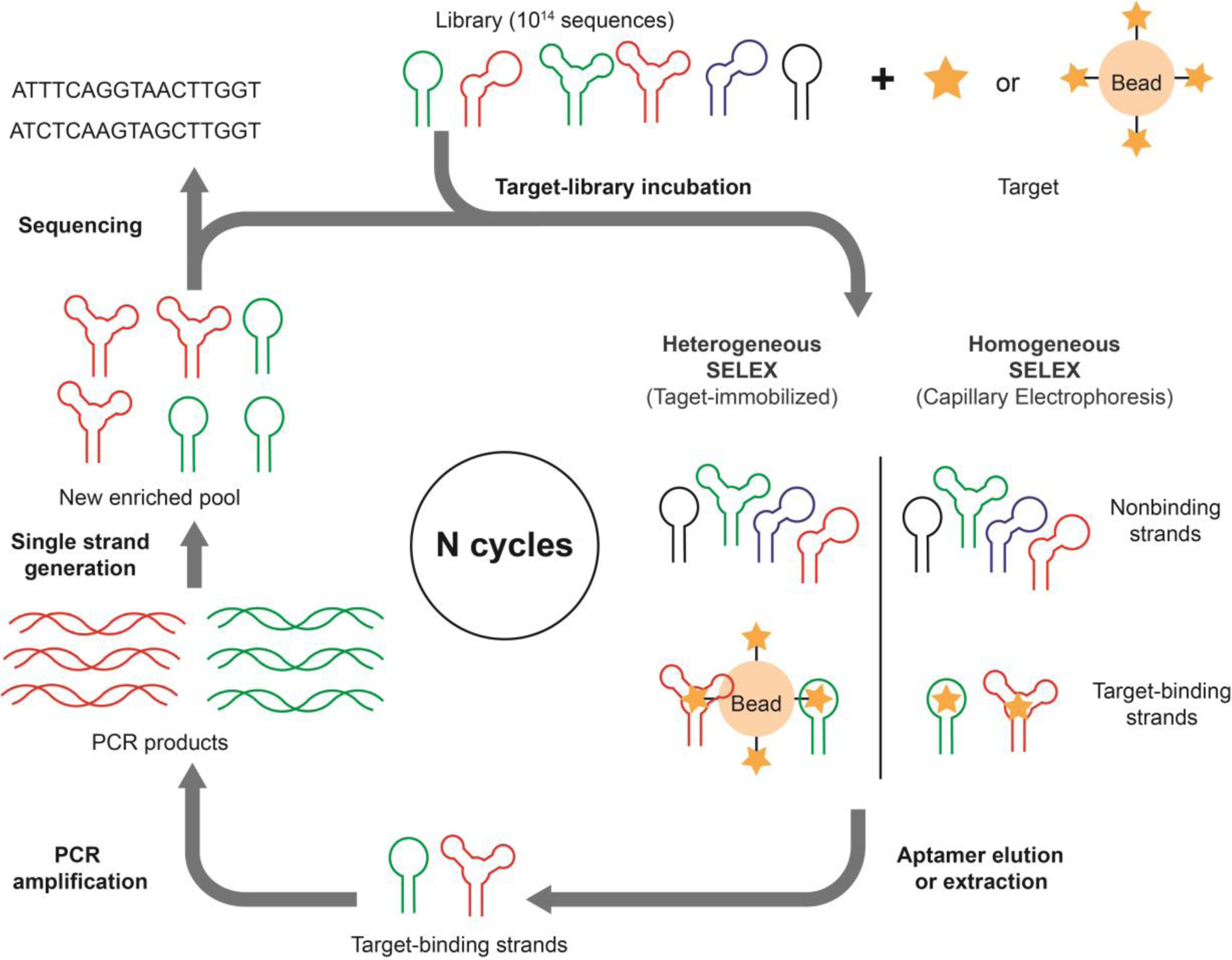
The general process of SELEX entails multiple cycles of target incubation with a nucleic acid library, target-binding strand separation, PCR amplification, and single-stranded DNA generation to regenerate a new library for another iteration of this process.
2.2. Small molecules are challenging targets for aptamer isolation
Isolating aptamers that bind small-molecule targets with high affinity and specificity is particularly challenging, mainly due to the properties of these molecules (Table 2).
Table 2.
Differences in the physiochemical properties between small molecules and proteins.
| Properties | Small molecules | Proteins |
|---|---|---|
| Epitopes available for target binding | Few | Many |
| Moieties available for conjugation | Limited, conjugation masks the few functional groups that are present in these targets. Requires specialized chemistry for conjugation due to high chemical diversity. | Abundant, conjugation has little impact on aptamer interaction. Targets share generic features that enable conjugation via standard methods |
| Structural complexity | Low, resulting in fewer potential aptamers | High, resulting in a greater number of aptamers |
| Molecular weight | < 1kDa | > 10 kDa |
| Solubility | Varies widely | Varies. Usually water soluble. |
| Accessibility | Can be readily procured commercially or synthesized | Expression and purification of certain proteins can be challenging |
Compared to larger targets like proteins, small molecules have less accessible surface area, fewer chemical moieties, and lower structural complexity. This limits the number and strength of binding interactions between small molecules and aptamers, resulting in lower aptamer-target binding affinities and sacrifices in aptamer specificity. Additionally, the aptamer isolation process typically requires conjugation of the small-molecule target to a solid surface, such as microbeads, to facilitate the separation of target-binding aptamers from non-binding strands in the library. Proteins can be easily conjugated to surfaces through standard chemistries such as EDC-NHS chemistry[57] which usually leaves most of the protein surface available for binding. However, the conjugation of small molecules comes with several challenges and complications. First, small molecules are more diverse in terms of the spectrum of functional groups that they can potentially contain compared to proteins, requiring specialized conjugation chemistries or even extensive chemical synthesis to achieve target immobilization. On the other hand, any one of these small-molecule targets generally comprises just a small number of functional groups in total, limiting their amenability for conjugation and impeding interactions between the aptamer and the immobilized target. Indeed, the attachment of linkers or moieties for conjugation can greatly change the physiochemical properties of the small-molecule target, and it has been reported that aptamers isolated against a conjugated small-molecule target exhibit greatly reduced or no affinity for the free target relative to the conjugated target.[58] The low molecular weight and abundance of charged moieties on small molecules also makes it challenging to achieve direct separation of target-aptamer complexes from free aptamers via certain solution-phase techniques such as capillary electrophoresis (CE)-SELEX, which can be highly efficient when used for protein targets.[59]
There is no absolute set of rules that can be used to determine whether an aptamer can be isolated for a particular small-molecule target. However, the physicochemical properties of the target can be used to roughly predict the degree of difficulty in isolating aptamers with high affinity. Generally, targets with higher molecular weights are better targets, because they have more moieties that the aptamer can bind to. Molecules with fewer rotatable bonds are also better targets because they have lower entropic binding penalties.[60] This includes targets with aromatic moieties, which also have the added benefit of being able to achieve π-π stacking with DNA bases. Since nucleic acid aptamers are negatively charged, there is a higher likelihood of obtaining high affinity binders for positively- versus negatively-charged targets. Targets with extremely high or low water solubility are also challenging, and may require additional measures for successful aptamer isolation. For instance, the highly hydrophobic small molecule tetrahydrocannabinol has been described as a difficult target. To overcome this problem, the Mayer group utilized a base-modified aptamer library containing benzyl-modified deoxyuridine bases.[61] Very hydrophilic targets (e.g., carbohydrates) have also proven difficult. Stojanovic and coworkers remedied this problem by isolating aptamers against these targets bound to organometallic receptors, effectively increasing the number of epitopes available for aptamer binding.[62]
2.3. Library design
The libraries used for SELEX comprise pools of single-stranded oligonucleotides with randomized sequences and secondary structures. In this review, we will focus on DNA libraries, because a majority of small molecule sensors use DNA aptamers. Oligonucleotide libraries containing base modifications have been used to isolate aptamers. However, although it has been well-established for protein targets,[63] it remains unclear whether base-modified aptamers consistently improve the binding capabilities of small-molecule-binding aptamers. For example, Imaizumi et al. reported that a base-modified library containing (E)-5-(2-(N-(2-(N6-adeninyl)ethyl))carbamylvinyl)-uracil yielded aptamers with better affinity and specificity for the small-molecule target camptothecin compared to natural DNA aptamers.[64] However, aptamers isolated from libraries containing amino functional groups[65,66] did not exhibit meaningfully improved affinity for ATP relative to unmodified RNA[29] and DNA[67] aptamers. More in-depth head-to-head comparisons will be required to draw definitive conclusions about the benefits of base-modified nucleotides for isolating small-molecule-binding aptamers.
DNA libraries are chemically produced using solid-phase synthesis.[68] Each library strand typically features a randomized region that serves as the putative target-binding domain, which is flanked by consensus sequences that enable binding to PCR primers (Figure 2A). The number of nucleotides (N) in the random region determines the number of total possible unique sequences (4N) in the library, and the length of the random region can range from as few as 8 to as many as 200 nucleotides.[55] Longer random regions have more sequence diversity, which in principle allows for the formation of more complicated motifs that may facilitate aptamer-target binding affinity and specificity. However, not all possible sequences can be practically represented in a single SELEX experiment.[69] Libraries with short random regions have certain advantages. First, the selection of aptamers is generally more rapid, because there is a greater number of copies of each sequence in the initial library. Second, since the randomized domains are smaller, it is easier to identify the target-binding domain, which makes subsequent sequence engineering processes for introducing signal reporting functionalities more facile and economical. Given that small-molecule targets are relatively miniscule and have few epitopes for binding, libraries with short random regions may be sufficient for aptamer isolation. For example, Yang et al. demonstrated the isolation of several aptamers for steroids using a library with only eight randomized nucleotides.[70] In later work, however, this group found that performing selection for the same steroid targets with libraries containing 20–30 random nucleotides yielded aptamers with at least 10-fold improved affinity and greater specificity.[71] It is therefore more beneficial to use libraries with greater randomness to ensure high aptamer quality. Since the shortest binding sequences of most small-molecule binding aptamers (e.g. for cocaine,[43] ATP,[67], kanamycin[72], synthetic cathinones,[73] serotonin,[74] and dopamine[74]) are usually 30–40 nucleotides in length, we believe libraries with ~30 randomized nucleotides should be sufficient to isolate small-molecule-binding aptamers for biosensing purposes.
Figure 2.
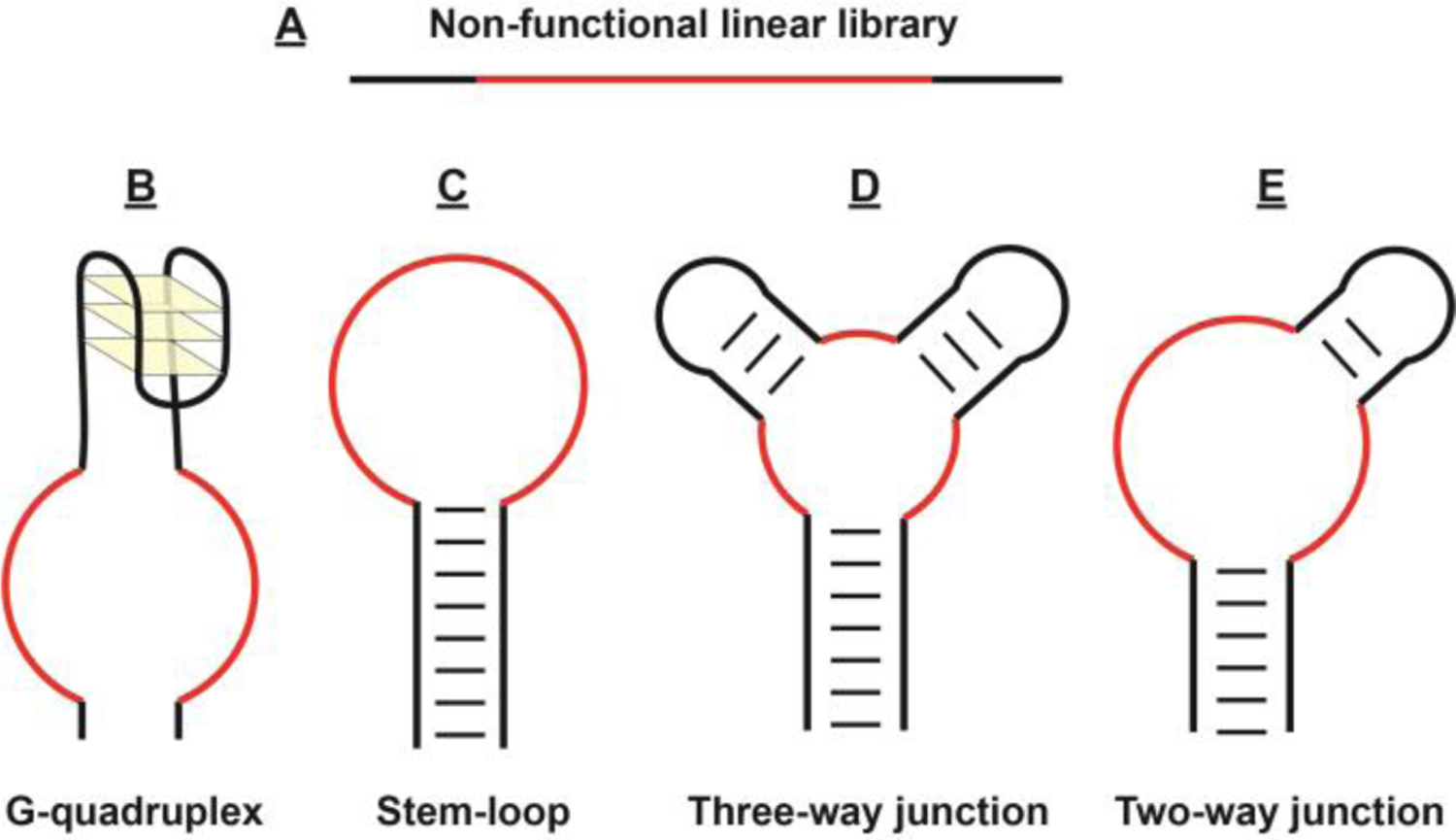
SELEX library designs including (A) unstructured linear libraries as well as functionalized (B) G-quadruplex, (C) stem-loop, (D) three-way junction, and (E) two-way junction structured libraries. Black- and red-colored regions indicate conserved and randomized regions, respectively.
The design of the secondary structure of the library members is also an important consideration. For example, one can incorporate G-quadruplexes (Figure 2B),[75] stem-loops (Figure 2C),[62] or two-way[71] (Figure 2D) or three-way (Figure 2E) junctions[70,76] into a library in order to isolate aptamers with pre-defined structures, binding domains, or sensing functionalities. G-quadruplex-structured libraries can yield aptamers with reporting functionalities, such that target binding triggers the formation of a G-quadruplex that act as a label-free signal reporter when paired with G-quadruplex-binding dyes.[75] Aptamers derived from stem-loop structured libraries can be directly incorporated into strand-displacement fluorescence sensors by labeling the aptamer and a complementary strand with a fluorophore-quencher pair.[62] Two- or three-way junction-structured libraries can be used to generate splittable aptamers that can be directly incorporated into a variety of sensing platforms with minimal post-engineering requirements.[76,77] These sensing platforms will be discussed in detail in Section 3.
2.4. Types of SELEX techniques
SELEX approaches primarily differ in how target-binders are partitioned from non-binding sequences. In general, these can be grouped into two broad categories: heterogeneous and homogeneous. Heterogeneous SELEX methods require immobilization of either the target or oligonucleotide library onto a solid matrix, while in homogeneous SELEX, both the target and library are free in solution.
2.4.1. Heterogenous SELEX
Heterogeneous, bead-based SELEX methods have been widely utilized to isolate hundreds of aptamers for small-molecule targets such as pharmaceuticals,[78] steroids,[70] nucleotides,[67,79,80] and antibiotics,[31,81–85] with nanomolar to micromolar binding affinities after 10–30 rounds of selection.[86] These methods require immobilization of either the target of interest or the library onto a solid substrate for partitioning. For target-immobilized SELEX (Figure 3A), a small-molecule target is conjugated to a solid-phase carrier (e.g., magnetic microbeads) directly or via a linker. When the target-conjugated carrier is incubated with the library, strands binding to the target strongly adhere to the solid phase and are retained, while non- or weakly target-binding strands are removed by washing with buffer. The stringency of the selection can be controlled by eluting binding strands with varying concentrations of free target or using buffers with high elution capacity. Since the target is covalently conjugated to the beads, highly stringent approaches such as multiple washing steps,[87] high temperature,[88] or volume dilution[89] can be used to isolate high-affinity aptamers within fewer rounds. However, target-immobilized SELEX has several technical challenges.[90,91] As mentioned before, the conjugation of small-molecule targets to solid substrates is challenging, and may yield aptamers that have reduced binding affinity for the free target. The background adsorption of library strands to the bead surface or the linker also leads to the enrichment of non-specific binders, which prolongs the SELEX process or can even lead to failure of the selection procedure. Given these limitations, library-immobilized SELEX (also known as capture-SELEX) may offer a more appropriate approach for many small-molecule selections (Figure 3B).[92] This method utilizes short, bead-conjugated complementary DNA (cDNA) sequences that hybridize to a specific region of the library strands to immobilize them onto the surface of beads. Upon addition of the target, non-binding sequences remain attached to the bead, while target-binding strands undergo a conformational change that causes them to separate from the cDNA and releases them into solution, so that they can be collected for further enrichment. Since the small-molecule targets are free in solution, non-specific enrichment of sequences that bind the carrier or linker can be avoided. However, the hybridization between the library and cDNA strand is vulnerable to spontaneous dissociation during the target elution step,[93] which makes the separation efficiency of this method low and increases the number of rounds needed to complete SELEX. Notably, both of these heterogeneous SELEX approaches restrict the ability of bead-bound target or library molecules to interact with their binding partner due to steric hindrance and/or functional group masking (in the case of target-immobilized SELEX), reducing the likelihood of successful aptamer isolation.
Figure 3.
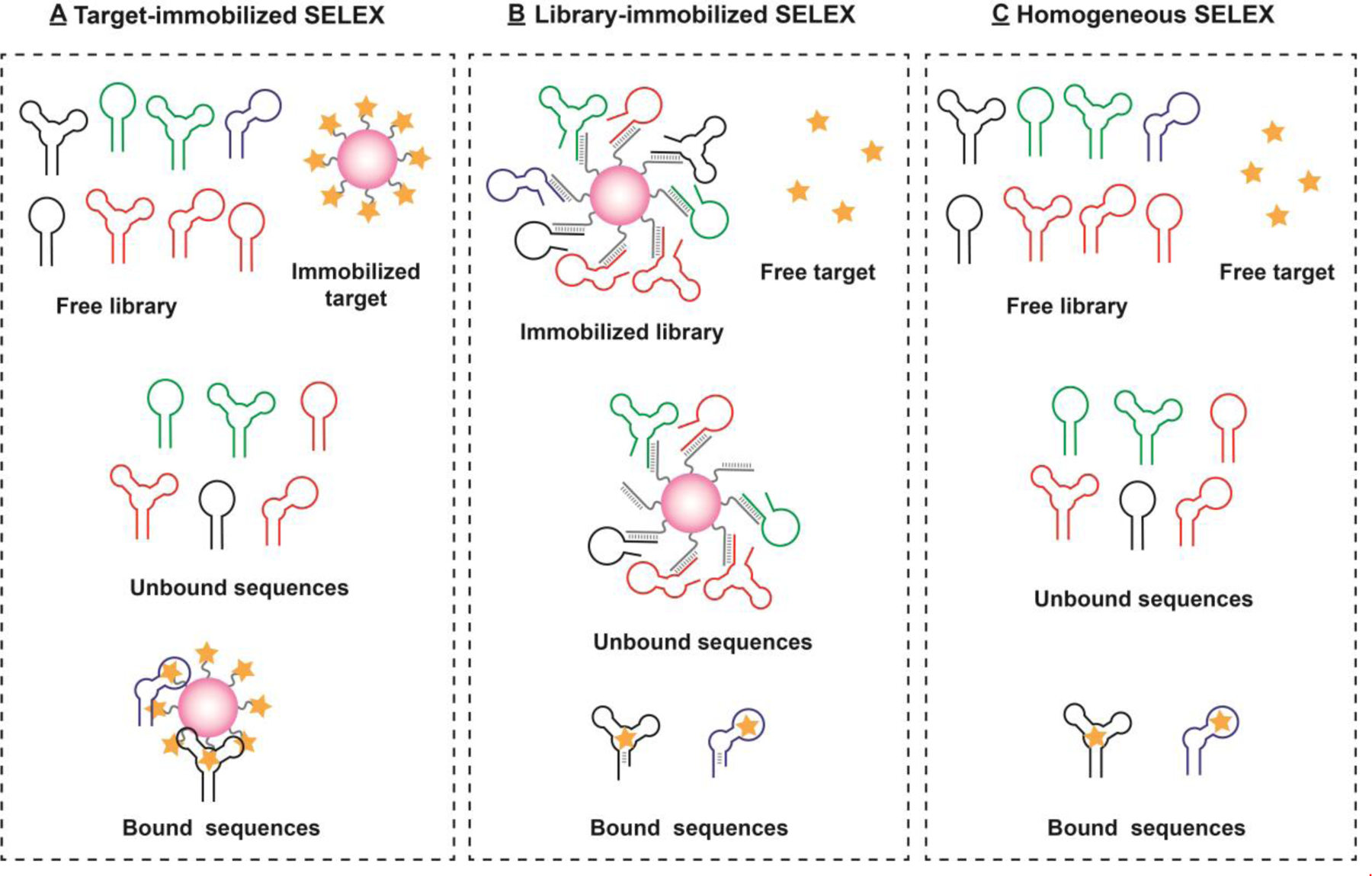
Common strategies to separate aptamers from non-binding strands in oligonucleotide libraries including (A) target-immobilized SELEX, (B) library-immobilized SELEX, and (C) homogeneous SELEX. The red sphere represents a solid substrate like a microbead.
2.4.2. Homogenous SELEX
Homogeneous SELEX techniques have higher separation efficiency, allowing for the isolation of high-affinity aptamers within just a few rounds of selection. Here, target-binders and non-binders are separated in solution, without any library or target immobilization (Figure 3C). Although homogeneous SELEX methods are less applicable for small-molecule targets compared to proteins, a few successful cases have been reported. The most well-known homogenous SELEX platform is CE-SELEX, which exploits the differential mobility between unbound and target-bound library molecules to separate them based on their charge and size under an external electric field.[59] This technique has been successfully used to isolate several aptamers for protein targets.[94,95] However, CE-SELEX offers limited utility for small-molecule targets because the mobility of small-molecule target-aptamer complexes is very similar to that of unbound oligonucleotides, resulting in poor separation resolution.[96] Alternatively, graphene oxide has been used to remove non-target-bound molecules during the separation step, as this material binds more strongly to single-stranded DNA than to target-bound folded DNA structures.[97] This technique has produced DNA aptamers for several small-molecule targets, with KD ranging from 1–2,000 nM after 5–10 rounds.[98–101] Another homogenous technique, sol-gel SELEX, utilizes a microfluidic device that incorporates a silicon chip spotted with droplets of sol-gels containing the target. This enables the entrapped molecules to remain in their native conformation and eliminates the need for target immobilization as in traditional bead-based SELEX.[102] Library-target incubation, partitioning of binding and non-binding sequences, and thermal elution of the binders are all performed within the microfluidic device. Using a sol-gel, Bae et al. isolated a DNA aptamer binding to xanthine with a binding affinity of 4.2 µM after seven rounds of selection.[103] The main challenge of this technique is to select optimal sol-gels that have nanoscale compartments that can retain the small-molecule target, allow DNA strands to freely move through microscale pores, and trap the aptamer when it binds to the target.
2.5. Selection strategies for aptamer isolation
2.5.1. Selection strategies for controlling the binding affinity of aptamers
One of the major advantages of SELEX relative to in vivo antibody production methods is that the selection conditions can be precisely controlled throughout the whole process to selectively enrich aptamers with desired binding affinities. For example, by using low-stringency conditions, aptamers with relatively low binding affinity can be retained and enriched, while highly stringent conditions can be used to select for aptamers with the highest target-binding affinity.[104–106] The most common means of modulating selection stringency include altering the concentration of target or library, target-library incubation time, incubation temperature, or buffer ionic strength.[55,104,105] Although it makes logical sense to employ high-stringency selection conditions from the beginning of SELEX as a means for maximizing aptamer affinity, this also increases the risk of losing target-binding sequences that are present only at low copy-numbers in earlier rounds. Therefore, relatively lower stringency is typically employed in earlier rounds to retain all possible target-binding sequences, with stringency gradually increased in later rounds to enrich for high-affinity aptamers after some amplification has occurred.[107,108] Some mathematical models have been established to guide selection stringency during the SELEX process, but in most cases adjustments in stringency between rounds are made empirically.[108]
2.5.2. Selection strategies to modulate aptamer specificity
One important disadvantage of target-immobilized SELEX is that it can enrich for aptamers that bind non-specifically to the beads themselves. Negative-SELEX was developed to overcome this problem (Figure 4A).[27] Specifically, the library is first incubated with non-modified beads to adsorb bead-binding strands, which are then discarded. The remaining library strands are then incubated with target-immobilized beads for positive selection against the target. Similarly, counter-SELEX can be used to remove sequences from the library that bind to non-target interferent compounds (or ‘counter-targets’), thereby ensuring that only highly-specific isolated aptamers are isolated (Figure 4A).[34] In this strategy, the library is incubated with interferents, interferent-bound strands are separated and discarded from the library, and the ‘cleaned’ pool is used for positive selection. The interferents can be applied to the pool either individually or as a mixture. Generally, low concentrations of counter-targets are used in the first few rounds of SELEX, but these are then greatly increased in later rounds. The counter-SELEX protocol can be revised from round to round to further fine-tune the specificity of the final aptamers. For example, using a very stringent counter-SELEX regime, Polisky et al. isolated an aptamer that has 10,000-fold greater affinity for theophylline relative to caffeine, a molecule that differs from the target by only a methyl group.[34]
Figure 4.
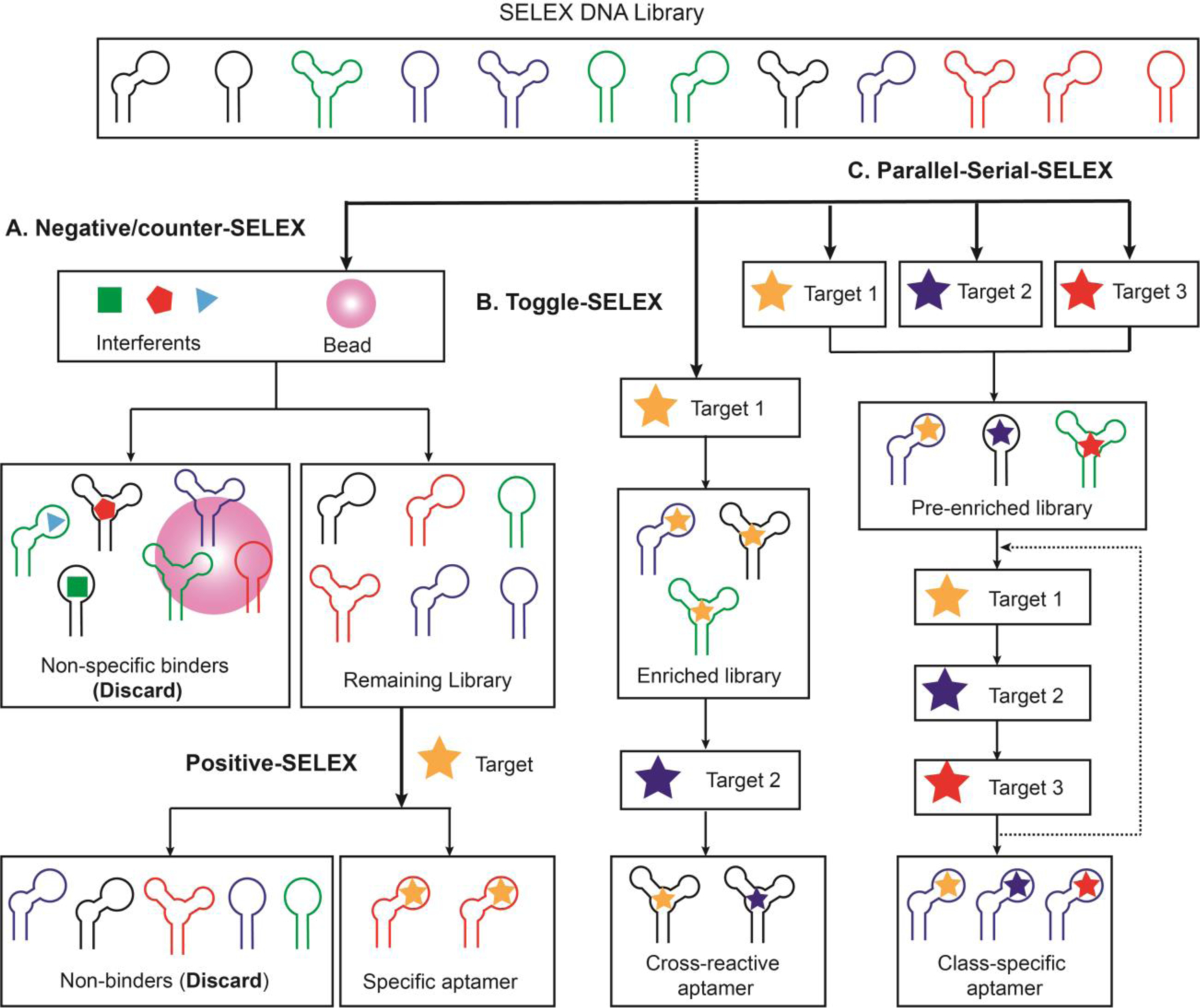
Common SELEX strategies for modulating aptamer specificity. Working principle of (A) negative/counter-SELEX, (B) Toggle-SELEX, and (C) parallel-serial SELEX.
Alternatively, the SELEX process can be designed to isolate aptamers with high cross-reactivity for a set of structurally-related target compounds—for example, highly similar designer drugs that may differ only by one functional group. Sullenger and coworkers developed a toggle-SELEX strategy (Figure 4B) to isolate aptamers that cross-reacted to both human and porcine thrombin.[109] The initial library was incubated with a mixture of both targets to enrich all potential aptamers that can bind either target, after which human and porcine thrombin were used as alternating selection targets every round. Consequently, only library strands that bound to epitopes present on both targets were enriched, while aptamers that specifically bound to only one were removed. The final aptamer bound to both human and porcine thrombin, with a KD of 2.8 and 0.1 nM, respectively. In comparison, an aptamer isolated using only porcine thrombin demonstrated >10,000-fold higher specificity for this target relative to human thrombin.[109] Toggle-SELEX has proven to be a powerful method of isolating cross-reactive aptamers for protein and cell targets,[110–112] but it has proven challenging to isolate cross-reactive aptamers for small-molecule targets using this approach[81,84,85] due to their small size and lack of binding epitopes. For example, Derbyshire et al. performed toggle-SELEX with four different pairs of targets to isolate cross-reactive aptamers to aminoglycoside antibiotics. However, among the 80 aptamer candidates identified in the four sets of toggle-SELEX screens, only one demonstrated cross-reactivity to all eight aminoglycoside targets.[81] As a solution, we have recently developed a ‘parallel-and-serial’ SELEX strategy (Figure 4C) to isolate aptamers that specifically recognize families of targets that share the same core structure.[73] This strategy entails performing selection against various members of a target family in parallel, followed by combining of the resulting pools and challenging with each target one-by-one. This selection strategy was further supplemented with a counter-SELEX procedure to remove sequences that bind to structurally-similar non-target molecules. Using this approach, we isolated a class-specific aptamer that binds to more than 12 members of the synthetic cathinone drug family with nanomolar affinities, but not to 17 structurally similar/dissimilar interferents.
2.6. Monitoring SELEX and identification of aptamer candidates
2.6.1. Characterization of pool affinity and specificity during SELEX.
The SELEX process is monitored to determine if aptamers with the desired binding properties are being enriched in pools. This is most commonly done by quantifying the library molecules collected upon target addition. For example, during target-immobilized SELEX, the library molecules captured on the target-immobilized solid support are typically eluted into solution using a combination of heat, urea, and EDTA.[61,113,114] Alternatively, high concentrations of free target molecule can be added,[80,115,116] preferentially eluting library strands that retain strong affinity for the free target. Quantification can be achieved by using libraries that have been previously tagged with molecules such as fluorescein[114,117] or 32P[61] or by performing PCR with chemically-labeled primers. In rare cases where the fluorescent properties of a target change upon binding to the library, fluorescence enhancement can be used to monitor enrichment of the pool.[118] Alternatively, one can perform gel electrophoresis of the unlabeled library with a DNA-binding dye[119]. Library-immobilized SELEX entails similar approaches, but since the library molecules that bind to the target are already being released into solution, an elution step is not necessary. The collected library strands can again be quantified by tagging with fluorescein[120] or 32P,[121] with fluorescence enhancement as needed[122], or by performing gel electrophoresis with a DNA-binding dye.[108]
2.6.2. Sequence techniques to identify aptamers from SELEX
The SELEX process can be considered ‘completed’ once the pools have clear target-binding affinity and specificity, and the pool affinity ceases to change over subsequent selection rounds. The most common approach to identify aptamer sequences after the completion of SELEX is by cloning of the aptamer pool and Sanger sequencing. This can provide as many as 50–100 sequences,[123] and the most abundant sequences are typically chosen for further characterization. HTS technologies now enable simultaneous identification of millions of sequences from individual selection pools, and this has opened new avenues for monitoring the SELEX process and the identification of aptamers.
Compared to Sanger sequencing, HTS has higher sequence coverage, allowing us to measure the frequency of sequences over the entire SELEX process,[123,124] offering the ability to distinguish truly enriched sequences from background[56,125] and identify aptamer candidates with specific binding properties.[119,126] The ability to characterize aptamers based on enrichment-fold is a critical benefit of HTS, and this is sometimes a more appropriate metric for choosing aptamer candidates than read-count, which can be affected by factors such as PCR amplification bias. For example, Cho et al. performed three rounds of microfluidic SELEX and sequenced each round of SELEX via HTS.[56] They discovered that the sequence with the highest affinity for the target in the final round did not have the highest copy number, but rather the greatest enrichment-fold between rounds of selection. Valenzano et al.[127] also used enrichment to identify highly-specific tyramine-binding aptamers from HTS data, in a process that involved counter-selection against the structurally-similar molecules histamine and tryptamine. They found that the most abundant sequence prior to the initiation of counter-selection was completely removed after counter-selection, indicating poor specificity, whereas sequences specific to the target were greatly enriched. The best aptamer they identified had a sub-micromolar KD, with lower binding affinity for the counter-targets. HTS was also used to monitor changes in the population of certain sequences during SELEX, which is an indicator of specificity or combined with counter-SELEX to isolate aptamers with specific binding profiles. Jauset-Rubio et al.[119] used HTS to identify aptamers that are highly specific or cross-reactive to the steroid hormones estradiol, progesterone, and testosterone. They first pre-enriched a library pool using estradiol, and then performed a single round of selection against the three individual compounds in parallel. They identified specific aptamers for each target and cross-reactive aptamers by sequencing the pre-enriched pool and the three parallel pools and then analyzing the change in frequency of each sequence in all pools. Sequences enriched in all pools were found to be cross-reactive, while sequences enriched only in a single pool were specific. With the development of user-friendly software (Table 3) it is possible for researchers to analyze HTS data without programming expertise.
Table 3.
Software for analyzing HTS data.
| Software | Platform | Counting | Clustering | Notes | Ref. |
|---|---|---|---|---|---|
| FASTAptamer | Command line | Yes | Based on sequence | Easy-to-use software - only requires perl | [134] |
| APTANI | Command line | Yes | Based on secondary structure | APTANI2 update includes GUI for structure analysis | [135] |
| MEME | Command line | Yes | Based on secondary structure | Web server is available for small data sets | [136] |
| MEMERIS | Command line | Yes | Based on secondary structure | Updated MEME that includes secondary structure analysis | [137] |
| AptaSUITE | Graphical user interface | Yes | Based on secondary structure | Includes AptaPLEX, AptaMUT, AptaSIM, AptaCLUSTER, and AptaTRACE | [138] |
| Galaxy Project | Web server | Yes | N/A | Does not include clustering capability | [139] |
| SMART-Aptamer | Command line | Yes | Based on secondary structure | Includes QGRS mapper for G-quadruplex prediction | [140] |
It is common that for multiple sequences that bind a common target to share a consensus motif. Monitoring the evolution of aptamer families via HTS is an effective strategy for identifying such consensus sequences. Aptamer families are identified via clustering, in which similar sequences from HTS datasets are classified by consensus sequence, secondary structure, or shared motifs. The simplest and most commonly used clustering approach ranks sequence similarity based on the Levenshtein distance,[128] which is the minimum number of nucleotide insertions, deletions, or mutations necessary to change one sequence into the seed sequence of a cluster. For example, Spiga et al.[125] monitored every other round of a SELEX screen against tobramycin using HTS and used this approach to cluster these sequences and identify families with 95% base conservation. Clusters from later rounds featured many family members (>250 sequences), indicating high enrichment of particular aptamer sequences, whereas clusters from early rounds did not exceed >25 sequences.[125] Importantly, consensus sequences from different clusters were identified as early as the second round of SELEX through HTS, indicating that enrichment of aptamer families begins during the earliest stages of selection and can be followed throughout the selection. To improve the determination of binding motifs, newer algorithms such as MEMERIS, APTANI, and AptaSUITE (AptaMOTIF[129]) (Table 3) can make use of secondary structure information. SMART-Aptamer includes QGRS mapper[130] alongside mfold[131], which enables the prediction of secondary and tertiary motifs such as G-quadruplexes. This in-depth clustering analysis alongside enrichment monitoring can elucidate an aptamer family’s secondary structure, and is especially powerful when combined with second-generation aptamer maturation techniques such as doped-SELEX,[132,133] which in turn allows for more expansive exploration of aptamer sequence space. A review on the application of HTS to aptamer selection has been published Quang et al.[123]
The use of HTS in this area has historically been limited by the high cost of instrumentation and reagents, the need for high computational power, and the lack of user-friendly software for data analysis. Today, however, these roadblocks have been largely overcome, and HTS has become as affordable as Sanger sequencing. Improvements in computing hardware have made it possible to analyze HTS data with just a personal computer, and software is publicly available for the analysis of aptamer pools,[123] as well as the identification of target-binding domains, scaffold regions, and even secondary structural motifs.[134,138] However, there are still areas that are lacking. The use of HTS may expand beyond sequence compositions of final pools into characterization of the thermodynamic properties of aptamer candidates as well. This could enable the extraction of binding affinity information using HTS data to eliminate the need for binding affinity testing for thousands of potential aptamer candidates. This concept was demonstrated by Lambert et al.[141], who used RNA bind-n-seq to determine the binding affinity of an RNA-binding protein for an RNA library containing several potential motifs recognized by the protein. They incubated their library with various concentrations of protein-immobilized beads and sequenced the captured RNAs. By constructing binding curves for each of the RNA motifs sequenced, they obtained binding affinities for each individual motif that correlated well with the gold-standard technique SPR.[141] Given this example, we anticipate that similar assays can be constructed for small-molecule-binding aptamers to expedite characterization of aptamer candidate binding affinities. We generally foresee that the use of HTS in the aptamer development process will become much more widespread moving forward.
3. Challenges and advances in aptamer characterization
Depending on the sequencing method applied, the SELEX process can provide anywhere from fewer than ten to thousands of aptamer candidate sequences. Once these sequences have been obtained, it is important to characterize their binding affinity to the target as well as specificity against interferents to determine if they are suitable for analytical applications. Characterization methods can be differentiated based on the information they can provide as well as their complexity and level of throughput. Here, we will first analyze methods based on specialized instrumentation and then focus on simpler ‘competition’-based assays that are more amenable for screening purposes. A summary of the advantages and limitations of common characterization techniques can be found in Supporting Information, Table S1.
3.1. Determination of aptamer binding affinity using specialized instrumentation
Several standard methodologies for characterizing protein-based receptor interactions have been adapted to study aptamer-small-molecule binding. These methods require specialized instrumentation, but can provide in-depth information on thermodynamic facets of binding such as affinity, enthalpy and entropy of binding, and kinetic parameters such as on- and off-rate constants.
3.1.1. Isothermal titration calorimetry (ITC)
ITC can provide detailed information on aptamer KD within a range of nM to µM, as well as ligand-binding stoichiometry, enthalpy, and entropy. The earliest use of ITC for studying small-molecule-aptamer interactions dates to the beginning of the millennium.[142] Since then, this method has been routinely used[99,143–145] including by our group[76,122] for such purposes. In a typical ITC experiment, a small-molecule ligand is loaded into the syringe and titrated to the aptamer loaded in the isothermal cell through a series of microinjections, with both molecules dissolved in the same buffer. Heat absorbed or released by binding events during each injection is measured by the calorimeter. The titration is performed until the aptamer is saturated, which is indicated by a minimal heat changes upon injection of the titrant. The heat values are integrated with respect to time, and the resulting data is plotted against the molar ratio of the ligand to the aptamer. This isotherm can be fit with a binding polynomial to obtain aptamer-ligand binding stoichiometry, KD values, and other thermodynamic constants. Advantageously, these experiments can be performed at a variety of temperatures (2–80 °C) with flexible choice of buffer pH and composition, including customization of salt concentration and the inclusion of organic solvents. Meanwhile, multiple binding parameters can be obtained with just a single experiment without any need for aptamer labeling, engineering, or immobilization. ITC data is generally analyzed using software that are provided with the instrument or open-source software.[146] New data analysis tools have enabled the extraction of more information from ITC experiments. For example, Affinimeter has ITC software that can not only determine normal thermodynamic metrics, but also kinetic parameters such as kon and koff.[147] This software and the SEDPHAT software developed by the National Institutes of Health can also be used to perform global fitting analysis of multiple ITC datasets, which allows for more accurate determination of binding mechanisms and parameters.[148] ITC results are most accurate when the concentration of the aptamer is 1–100-fold greater than the KD. For small-molecule-binding aptamers, which typically have KDs of 1–100 μM, high concentrations of aptamer (10–100 μM) and even higher concentrations of ligand (100 μM–1 mM or more) are usually required. This is not only costly, but also creates solubility issues, especially for hydrophobic small molecules. Organic solvents can partially solve this problem, but these may alter the binding properties of the aptamer. ITC experiments with sub-optimal concentrations of aptamer may still provide accurate KD, but lead to erroneous binding stoichiometries. The low binding enthalpy of small molecules also limits the characterization of aptamers with high binding affinity (KD < 1 nM), since the optimal aptamer and ligand concentration needed to produce a suitable binding isotherm does not generate an adequate heat change that can be accurately detected by the calorimeter. This challenge can be overcome by using a competition-based method.[149] In addition, the cost of ITC instruments is generally above $100K, which makes the method quite expensive for occasional users. The large quantity of reagents required for the experiment and the low-throughput nature of the method also make ITC unsuitable for intensive profiling of aptamer binding properties. A comprehensive review on the use of ITC for studying small-molecule aptamer interactions has been published by the Johnson group.[150]
3.1.2. Surface plasmon resonance (SPR)
SPR is another standard method for determining the binding parameters of aptamers.[151] One advantage of SPR is its capability to not only characterize binding affinity and stoichiometry, but also kinetic metrics such as kon and koff.[152] This method measures changes in the intensity of light reflected by a thin gold film at various angles after molecules adsorb onto the metal surface. Either the aptamer or the target must be immobilized onto the surface of the gold sensor chip through thiol-gold, carboxy-amino, or epoxy chemistry. The binding partner is then flowed over the chip surface, and aptamer-ligand binding results in a change in resonance angle due to alteration of refractive index at the metal surface. Once the resonance angle ceases to change (which is due to the saturation of binding sites), the non-immobilized binding partner is washed away with buffer, which returns the refractive index to its initial value. The resulting resonance angle time-plot can be used to determine kon and koff, which can in turn be used to calculate KD. More accurate measurements of binding affinity can be achieved by performing SPR experiments with different concentrations of the non-immobilized binding partner and plotting resonance angle at equilibrium against the concentration of the binding partner and fitting the isotherm with a binding polynomial. However, the characterization of small-molecule-binding aptamers using SPR is challenging. Typically, the aptamer is immobilized onto the SPR chip, and the binding of small molecules to the aptamer typically results in small changes in refractive index that are difficult to confidently measure. The target can be immobilized onto the chip, so that a larger change in SPR occurs upon aptamer-ligand binding,[78] but this approach has several disadvantages. First, not all small molecules have functional groups amenable for covalent attachment to the sensor surface. Second, immobilization of small-molecule targets can impede aptamer binding, as discussed above. In addition, the binding parameters obtained through this strategy do not necessarily equal those of the aptamer and ligand interacting in solution. Recently, Chang et al. developed a general SPR-based aptamer characterization method that avoids the challenges associated with aptamer/target immobilization while achieving high sensitivity.[153] They immobilized the aptamer with a poly-A tail via a complementary poly-T sequence tethered to the sensor chip surface to avoid steric hindrance and mitigate the negative effects of immobilization. To achieve more sensitive detection of binding, they used a high-density carboxymethyl CM5 sensor chip, which could support higher aptamer surface density than previous chips. Therefore, more small molecules could adhere to the surface, generating a larger signal. Using this strategy, the binding kinetics and affinity of a variety of small-molecule-binding aptamers with sub-micromolar to micromolar KDs could be determined, which could not be achieved with conventional SPR immobilization methods.
3.1.3. Microscale thermophoresis (MST)
MST is a relatively new technique that has become popular for characterizing the KD of aptamers for small molecules.[154–156] This method exploits the fact that bound aptamers or ligands have different – typically lower – diffusion rates in solution than their free counterparts, and these can be measured using thermophoresis. To perform the assay, the aptamer needs a fluorescent label, or else the small molecule must be fluorescent so that they can be spatially tracked. Specific instrumentation has been developed for MST; typically, 12 to 16 samples containing a fixed concentration of target but a varying concentration of aptamer are added to microcapillary tubes. The fluorescence of a specific region of each capillary tube is continuously monitored, and after the target and aptamer have equilibrated, the monitored region is rapidly heated with an infrared laser. This causes fluorescent molecules in the heated region to diffuse away, resulting in a reduction of fluorescence. The extent of this fluorescence depletion is related to the extent of aptamer-target binding. A binding isotherm can be created by plotting the ratio of fluorescence before and after heating versus the concentration of aptamer, and the curve is fitted with a binding polynomial to determine KD.[157] Baaske et al. first demonstrated the utility of MST to study the binding of a well-studied ATP binding aptamer.[154] They found that the aptamer bound to ATP as well as AMP with similar micromolar affinities. Rangel et al. recently reported the use of MST to determine the binding affinity of ochratoxin A aptamer.[158] One key advantage of MST is that KD can be determined in complex samples, as demonstrated by Rangel et al. in their study of the ochratoxin A-binding aptamer in human serum.
3.2. Determination of aptamer binding affinity using conventional instrumentation
3.2.1. Methods based on aptamer-target binding
3.2.1.1. Characterization based on binding-induced changes in target fluorescence
For certain targets, one can study aptamer affinity and specificity by measuring differences in the fluorescence of a small-molecule ligand when it is bound to an aptamer compared to when it is free in solution. A binding isotherm can be generated by recording the changes in fluorescence of the small molecule as a function of the concentration of the aptamer. Most commonly, aptamers for dyes have been studied using this method.[159,160] However, for a small subset of non-dye small molecules, their fluorescence emission intensity can change upon aptamer binding.[161] For example, Shoara et al. obtained an aptamer’s affinity for cocaine and quinine based on the reduction in fluorescence upon aptamer binding.[162] Idili et al. evaluated the binding affinity of an aptamer for irinotecan by measuring the fluorescence of the target in the presence of various concentrations of aptamer and fitting the curve to a Langmuir binding model.[163] Samokhvalov et al. demonstrated that the inherent fluorescence of ochratoxin A could be exploited to determine its affinity to an aptamer.[164] Although this method is label-free and very simple to perform, its generality is very limited because most instances of a ligand binding to an aptamer have no influence on the ligand’s optical properties.
3.2.1.2. Fluorescence polarization
Fluorescence polarization is another method for measuring aptamer-target binding affinity in solution.[165] It is based on the principle that receptor-ligand binding reduces the degrees of freedom of either binding partner, which can be detected by measuring the polarization of fluorescent light emitted by one of the species. This method is particularly useful when the target is fluorescent.[166] For example, Kobayashi et al. were able to study the affinity of porphyrin-binding aptamers using fluorescence polarization, because porphyrins themselves are fluorescent.[167] If the target is not fluorescent, either it or the aptamer need to be labeled with a fluorophore to enable analysis.[168] However, labeling of a small molecule is challenging, as mentioned before, and may alter its binding properties. On the other hand, a fluorescently labeled aptamer may not exhibit a detectable change in degrees of freedom upon target binding. As a result, this method cannot be generally applied.[169]
3.2.2. Methods based on competition
3.2.2.1. Bead-based binding assays
These affinity chromatography techniques serve as the basis of a wide array of aptamer affinity characterization methods.[25,29,61,67,170,171] Specifically, the ligand or aptamer is immobilized onto a solid substrate, such as microbeads, and then incubated with varying concentrations of its binding partner and washed extensively to remove unbound strands. The non-immobilized binding partner retained on the column is then eluted using high temperatures, chaotropic agents, or the free form of the immobilized partner. A binding isotherm can be made by plotting the amount of bound partner as a function of the total amount of binding partner added to the substrate. In their inaugural work with aptamers, Ellington and Szostak used a small-molecule target-immobilized column to characterize the binding affinity and specificity of their aptamers.[25] This method is simple and rapid, but it is limited by the requirement for target/aptamer immobilization, which can be problematic for the reasons discussed above. We have developed a bead-based gel-elution assay for characterizing aptamer affinity and specificity that is compatible with aptamers isolated through library-immobilized SELEX.[76] The aptamer is immobilized on streptavidin-coated agarose beads via a biotinylated complementary strand. Aptamer-ligand binding releases the aptamer from the beads, and the released aptamers are then quantified. Although we have primarily applied this technique for studying pools of aptamers, we believe it can also be applied to characterize the binding properties of individual aptamers.
3.2.2.2. Gold nanoparticle (AuNP)-based assays
Assays utilizing AuNPs provide a simple and rapid means for screening the binding of aptamers to specific molecules. The binding of a ligand to an aptamer can be converted into a colorimetric signal using unmodified AuNPs.[172] In the absence of ligand, DNA/RNA aptamers adsorb strongly onto AuNPs, which prevents them from aggregating, and the resulting solution appears red. Ligand binding to the aptamer causes it to dissociate from the AuNP surface, which destabilizes the particles and causes them to aggregate, which turns the solution blue. Derbyshire et al. used an AuNP-based assay to examine the cross-reactivity of three aminoglycoside-binding aptamers to eight different aminoglycosides.[81] DeRosa et al. likewise reported the use of AuNP assays to assess the binding capabilities of seven different small-molecule-binding aptamers.[169] Indeed, AuNPs can be used to rapidly screen a relatively large number of aptamer-ligand pairs. The main drawback of AuNP-based assays is that they are prone to erroneous results. This is because AuNP aggregation can be non-specifically triggered by several factors that are unrelated to aptamer-target binding such as the properties of the target, buffer composition, and structure of the aptamer.[173] Therefore, a great deal of caution should be practiced when interpreting the results of AuNP-based assays.
3.2.2.3. Strand-displacement assays
Strand-displacement assays are a reliable way to measure the target-binding affinity and specificity of small-molecule-binding aptamers. This method is based on the target-induced displacement of a complementary DNA (cDNA) strand that is hybridized with a portion of the aptamer. To determine the aptamer KD for the ligand, one must determine both the affinity between aptamer and cDNA (KD1) and the ligand and the aptamer-cDNA complex (KD2). KD1 is determined by titrating various concentrations of cDNA against the aptamer, while KD2 is determined by titrating varying concentrations of ligand against a fixed concentration of aptamer-cDNA complexes and measuring the extent of strand displacement. The KD is equal to the ratio of KD1/KD2. This general paradigm was first established by Easley and Hu, who determined the complexation state of an aptamer using microchip electrophoresis.[174] Stojanovic et al. introduced a more accessible fluorescence-based variant of this assay, in which they labeled the aptamer and cDNA with fluorophore-quencher pairs.[62] Strand-displacement assays have several advantages, including simplicity, high accuracy, low reagent requirements, and the ability to provide binding parameters in solution.[74] However, the successful performance of this method requires some trial-and-error to select suitable cDNA sequences and the inclusion of controls to account for the effect that ligands may have on the optical properties of the fluorophore (e.g. fluorescence enhancement/quenching) and quencher.
3.2.2.4. Dye-displacement assays
Dye-displacement assays represent an alternative approach for screening aptamer-ligand binding affinity and specificity. In the absence of target, some aptamers display the ability to bind certain dyes either in or near their target-binding domain. In the presence of target, the dye is displaced from the aptamer, which results in a change in the optical properties of the dye. Stojanovic first demonstrated this concept using a cocaine-binding aptamer with the cyanine dye diethylthiotricarbocyanine (Cy7).[175] We recently used this assay to profile the cross-reactivity of aptamers to a panel of as many as 29 different small-molecule compounds.[176] McKeague et al. have utilized the dye SYBR Green I to determine the binding affinity of a variety of small-molecule-binding aptamers, achieving similar levels of precision as SPR and fluorescence polarization methods.[156,169] The dye-displacement assay is simple to perform and does not require any aptamer engineering or prior knowledge of the target-binding domain. However, not all aptamers can bind to and release dyes in a ligand-binding dependent manner.[177]
3.3. Approaches for determining aptamer structure
It is now well established that DNA and RNA aptamers can fold into a myriad of complex architectures, including junctions, bulges, pseudoknots, G-quadruplexes, and triple-stem structures.[178] Determining aptamer structure is another crucial aspect of the characterization process that can provide useful information for engineering aptamer affinity, specificity, and the introduction of additional functionalities useful for sensing purposes. The secondary structure of an aptamer can be elucidated based on its sequence by using readily available software to identify potential double-stranded stems and single-stranded loops in the aptamer. Computer-assisted three-dimensional modeling can also provide certain predictions on aptamer tertiary structure. However, accurately determining higher-order aptamer structure requires more advanced techniques and instrumentation, which may be inaccessible to some groups due to their cost and complexity.
3.3.1. Computational methods
Software available online including mfold[179] and NUPACK[180], can be used to determine the secondary structure of aptamers. These programs use the nearest-neighbor model[181] to calculate the free energy of oligonucleotide structures based on Watson-Crick base pairing. NUPACK provides the structure of the lowest free energy folded oligonucleotide, while mfold can provide multiple putative structures with low folding energies. The temperature and concentration of Na+ and Mg2+ can be controlled in these models to determine structure of aptamer in varying buffer conditions. These programs can elucidate possible double- and single-stranded regions in an aptamer, which is useful for sequence engineering when performing truncation, splitting, or designing new aptamer constructs. They can also predict the structure and free energy of aptamers complexed with complementary oligonucleotide probes or sets of split aptamer fragments. However, they cannot predict the formation of more complicated structures, such as G-quadruplexes. Therefore, users of these tools should take into consideration that an aptamer’s actual secondary structure could be vastly different and that its three-dimensional structure is typically much more nuanced and complex.
While the secondary structures of aptamers can be predicted quite readily based on nucleotide sequence, tertiary structure prediction is far more challenging. Recently, nucleic acid modeling techniques such as coarse-grained modeling have enabled the prediction of tertiary structures either ab initio or from secondary structures determined by software like Mfold.[182–185] However, these algorithms use force fields that are only applicable for RNA structures. To model DNA in this framework, one must first convert DNA sequences into RNA by changing thymine to uracil and then input this RNA sequence into the modeling software. The resulting three-dimensional structure can then be manually converted to DNA by altering the sugar backbone and uracil base, followed by structural refinements via molecular dynamics simulations.[186] With recent updates to the MacroMolecule Builder (MMB) software,[187,188] three-dimensional models from DNA sequences can be directly generated. For example, Eisold and Labudde used MMB to generate three-dimensional structures of a DNA aptamer that binds to estradiol. They were able to perform molecular dynamics simulations to study the binding interactions between estradiol and the aptamer.[189] An obvious benefit of coarse-grained modeling is that it is easier to perform than traditional structure determination techniques like X-ray crystallography and NMR. However, it is difficult to ascertain whether the structures and information gathered from these coarse-grained models are true and accurate. In addition, because coarse-grained modeling typically provides several possible three-dimensional models, other techniques such as motif analysis from HTS SELEX datasets are needed to select the correct structure.
3.3.2. Circular dichroism (CD)
CD spectroscopy is a method that can provide structural information about aptamers based on their ability to absorb circularly polarized light. Aptamers contain a variety of chromophores that absorb UV light in the wavelength range of 200–300 nm. Different nucleic acid structures have differing CD signatures.[190] For example, the CD spectra of double stranded B-form DNA consists of a ‘positive’ peak between 260–280 nm and a ‘negative’ peak at ~245 nm. The CD spectrum of an aptamer can be compared to reference spectra to elucidate its possible secondary or tertiary structure, and Kypr et al. have provided detailed descriptions of the CD spectra of various nucleic acid structures.[191] Since the CD spectrum of an aptamer is sensitive to its structure, CD spectroscopy can also be used to identify conformational changes upon target binding. For example, Plaxco et al. were able to determine that their truncated aminoglycoside-binding aptamer could undergo target-induced structure-switching based on differences in CD spectra upon the addition of the target.[72] CD spectroscopy can be challenging when the small-molecule ligand being studied has high UV absorption. Although the contribution of signal by the ligand can be subtracted, it generally results in high levels of noise that distort the spectrum of the aptamer. CD spectroscopy also has low resolution, and it is difficult to deconvolute overlapping peaks in the spectra. In addition, many factors can affect CD spectra, such as aptamer sequence, ionic strength, pH, and temperature.[190] These may lead to misinterpretation of spectra and, consequently, false conclusions. For example, an ATP aptamer produces a CD spectra suggesting the presence of a G-quadruplex,[192] but NMR experiments have shown that the aptamer does not adopt such a structure.[193]
3.3.3. Nuclear magnetic resonance (NMR) spectroscopy
NMR spectroscopy is the most common technique for determining the tertiary structure of aptamers in aqueous solution. It is based on the interaction between radio-frequency electromagnetic energy with the nuclei of atoms within the aptamer as well as the target in a strong magnetic field. Two-dimensional NMR experiments, such as nuclear Overhauser effect spectroscopy (NOESY), can reveal the distance between specific atoms in the aptamer. The basic approach for aptamer structural determination is to gather structural constraints (i.e. distances between atoms and angle restraints) by NMR spectroscopy, and then input this information into molecular dynamics software, which generates a three-dimensional arrangement of the atoms in the aptamer. A variety of NMR experiments need to be performed to gather enough structural constraints. These include homonuclear 1H, 1H-NOESY, ¹H-¹H correlation spectroscopy (COSY), and total correlation spectroscopy (TOCSY), and heteronuclear 1H,13C-NOESY, 1H,15N-NOESY, and 13C,15N-NOESY.[194] NMR experiments require a relatively large amount of highly pure aptamer (sub- to single digit millimolar concentrations in a volume of hundreds of microliters) due to the generally low sensitivity of the technique. It is preferable to study aptamers that are as short as possible, as the more atoms the aptamer contains, the more signals need to be discerned and analyzed and the more costly the oligonucleotide. The Johnson group has used NMR spectroscopy to understand the interaction between a cocaine-binding aptamer and its target.[143] They were able to acquire the secondary structure of this aptamer, precisely pinpointing the target-binding domain, and determined which portions of the aptamer undergo conformational changes upon binding. Recently, Xu et al. were able to improve the affinity of an ochratoxin A-binding aptamer based on secondary structure determined through NMR.[195] By engineering unstable scaffold regions around the target-binding domain, they were able to improve the affinity of the aptamer by an order of magnitude. Although performing NMR experiments is straightforward, analysis of the resulting data to determine tertiary structure is difficult. Only a handful of small-molecule-binding aptamers and their complexes with ligands have been determined with NMR.[196] For example, the three-dimensional structure of the ATP aptamer bound to AMP was determined using homonuclear and heteronuclear NMR.[193] These findings provided a greater understanding of the specific nucleobases involved in binding and the binding mechanism of the aptamer, which has been used as a basis to engineer the aptamer for sensing purposes[197] and to alter aptamer binding affinity and specificity through rational mutations.[155,198]
3.3.4. X-ray crystallography
X-ray crystallography can be used to determine the three-dimensional structure of aptamers by exposing a crystalline sample of the aptamer to X-rays and detecting the spatial distribution and intensity of the diffracted rays. The resulting diffraction pattern is analyzed to determine the spatial arrangement of the atoms that make up the aptamer. Compared to NMR, X-ray crystallography can provide higher-resolution aptamer structures, and the data analysis process is more straightforward. Also, there are no restrictions on the length of the aptamer. However, the process of generating aptamers in crystalline form is challenging, laborious, and requires a large quantity of aptamer. In addition, the crystal structure of an aptamer may differ from its structure in solution. Sussman et al. used X-ray crystallography to determine the structure of an aptamer that binds to vitamin B12.[199] They determined that the aptamer has a complex architecture containing both duplexes and triplexes that form a binding site for interaction with the target. Ferré-D’Amaré et al. used X-ray crystallography to determine the structure of the fluorogenic aptamer, Spinach.[200] They were able to determine that the aptamer has a G-quadruplex region that binds and restrains the fluorescent dye ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one), thereby increasing its quantum yield.
4. Challenges and advances in developing aptamer-based small-molecule sensors
After the completion of SELEX and characterization of the resulting aptamers, sensing functionalities are engineered into the aptamer to transduce target-binding into a measurable signal. Most such sensors employ structure-switching aptamers, which undergo a major conformational change upon target binding. Recent advances in SELEX technology have made it possible to incorporate sensing functionality into the library design, which allows for the direct incorporation of the resulting aptamers into sensors. Several sensing platforms have also been developed that operate without the need for structure-switching, which allows for direct use of original aptamers in sensors without any post-selection engineering processes.
4.1. Sensors based on conformation-changing aptamers
Standard SELEX protocols typically use target binding as the sole selection force.[92] The isolated aptamers are usually fully folded in their unbound state, such that target binding does not induce any meaningful conformational change. It is therefore necessary to introduce structure-switching functionality into aptamers through post-SELEX engineering. In a typical aptamer, the target-binding domain cannot be altered during the engineering process, in order to avoid impairment of binding. However, this domain is typically flanked by scaffold regions that are amenable for sequencing engineering, because they do not directly interact with the target but rather assist in stabilizing the target-binding domain.[143,155,201] Therefore, it is important to identify the target-binding domain prior to beginning post-SELEX engineering. This can be achieved by truncating the aptamer, based on stem-loop structures identified by software such as mfold[179] or NUPACK[180]. However, this strategy requires extensive trial-and-error, which makes it costly, time-consuming, and laborious. After identifying the target-binding domain, structure-switching functionality can then be introduced via truncation,[35] splitting,[43] or utilization of a complementary strand.[202] The general underlying principle of these approaches is that when scaffold regions surrounding the binding domain are disrupted, the aptamer unfolds in the absence of target. However, the destabilized aptamer may still retain the capacity for target recognition and binding, allowing it to refold into its original secondary structure upon binding the target.
4.1.1. Sensors based on truncated aptamers
Truncating aptamers is the most intuitive way of introducing structure-switching functionality (Figure 5A). Aptamers usually have double-stranded stems that serve as a scaffold region. Shortening these stems reduces the melting temperature of the aptamer, shifting the equilibrium from a double- to single-stranded structure in the unbound state. However, the truncated aptamer can still bind to its target to form a folded aptamer-target complex. For example, the well-characterized cocaine-binding aptamer MNS-4.1 (KD = 5 μM) is structurally stable and forms a three-way-junction binding domain even in its unbound state.[35] Stojanovic et al. truncated one of the three stems to destabilize the aptamer, and attached a fluorophore-quencher pair to the termini of the aptamer for signal reporting.[35] The truncated aptamer is partially unfolded in the absence of target, and the fluorophore is largely separate from the quencher, resulting in strong fluorescence. The aptamer refolds into a three-way-junction structure upon binding cocaine, bringing the quencher into close proximity to the fluorophore and resulting in great reduction of fluorescence. This sensor demonstrated a limit of detection of 10 μM cocaine.[35] Truncated aptamers can be generally applied to other sensing platforms. For example, the same truncated cocaine aptamer has also been modified with the electroactive tag methylene blue for electrochemical detection of cocaine in the most challenging sample matrix, whole blood.[203] To date, many structure-switching aptamers have been engineered by truncation for folding-based sensor development.[204] However, engineering structure-switching functionality into aptamers via truncation is laborious, and relies greatly on trial and error. After the scaffold regions and binding domain of an aptamer have been identified, multiple truncated aptamers with different portions of the scaffold regions removed are synthesized and tested to determine if they have structure-switching functionality, with the most optimal aptamer used for downstream sensor development.[205–207] For example, Neves et al. identified the target-binding domain and secondary structure requirements of a cocaine-binding aptamer by interrogating 24 truncated or mutated aptamers using ITC,[208] followed by NMR analysis of the parent aptamer and two variants to characterize the optimized structure-switching aptamer.[143] To expedite the aptamer engineering process, we recently developed a one-step nuclease-directed truncation method to generate structure-switching small-molecule-binding aptamers.[209] This method is based on the enzyme Exonuclease III (Exo III), which completely degrades aptamers in the absence of their target. We found that Exo III can be strongly inhibited a few nucleotides prior to the target-binding domain when the aptamer is bound to the target. The resulting digestion product not only retained substantial target-binding affinity, but also exhibited structure-switching functionality, which enabled the rapid development of folding-based sensors. But since small-molecule-binding aptamers usually have relatively high (∼μM) KD,[86] a substantial scaffold region is still required to stabilize the aptamer’s structure and maintain target-binding affinity, which promotes problematic background folding. For example, the optimized truncated structure-switching cocaine-binding aptamer remains partially folded in the absence of target, resulting in high background that greatly limited sensor sensitivity.[35,37] However, further truncation of this aptamer resulted in complete loss of target affinity.[208]
Figure 5.
Different sensing strategies to develop aptamer-based sensors from a fully-folded aptamer through a variety of strategies, including (A) aptamer truncation, (B) splitting, (C) introducing a competitive strand, (D) use of a dye probe, and (E) exonuclease digestion.
4.1.2. Sensors based on split aptamers
An alternative way to program aptamers to undergo target-induced conformational change is to split the aptamer into two[43] or three[210] fragments. These fragments are separated in the absence of target but can reassemble into a complex in the presence of target (Figure 5B).[211] Splitting is typically performed at non-binding loop regions of an aptamer, and therefore requires knowledge of the aptamer’s target-binding domain and scaffold regions. Recently, Heemstra and coworkers reported a general approach for engineering split aptamers from three-way-junction-structured small-molecule-binding DNA aptamers.[212] Their approach entailed removing a single loop region of the aptamer and truncating the number of base pairs in the remaining stem regions to further destabilize the aptamer.[212] Although this method is straightforward for three-way-junction aptamers, it still requires trial and error as with truncation methods. Moreover, there is no general method for splitting aptamers with other secondary structures. Splitting is a more aggressive approach of destabilizing aptamers relative to truncation, which allows split aptamers to achieve lower background signals at the cost of notably reduced target affinity.[43] Nevertheless, sensors based on split aptamers typically achieve lower limits of detection than truncated aptamers. For example, fluorescence and E-AB sensors respectively developed by Stojanovic et al.[35] and Baker et al.[37] based on a truncated cocaine aptamer had 10-fold poorer sensitivity than sensing platforms using split-cocaine aptamers.[197] Alternatively, Zhang et al. reported that unmodified AuNPs were stabilized by adsorption of aptamer fragments in the absence of cocaine.[213] In the presence of cocaine, the two fragments assembled into a stable target-aptamer complex. The AuNPs, which were now destabilized, formed aggregates in solution, producing a red-to-blue color change. Under optimal conditions, they were able to detect 2 µM cocaine via naked eye. Recently, Zhu et al. utilized two split aptamer fragments to develop a sandwich aptamer lateral-flow assay for detecting ATP.[214] They conjugated one thiol-modified aptamer fragment onto AuNPs, and immobilized the other biotin-labeled fragment onto a nitrocellulose membrane through biotin-streptavidin interaction. In the presence of ATP, the AuNP-labeled and surface-immobilized fragments reassembled, resulting in the accumulation of AuNP-labeled fragments, producing a visible red line on the test zone. Under optimal experimental conditions, this test achieved a limit of detection of 0.5 µM ATP in buffer. The Zeng group used a similar sandwich concept to develop lateral-flow strips that contain “OR” and “AND” logic gates based on the assembly of split aptamer fragments for simultaneous detection of thrombin and ATP.[215] This flexible platform offers the possibility of multiplex detection from a single sample. Several comprehensive reviews on aptamer-based lateral-flow assays have been recently published.[216]
To further improve sensor sensitivity, Nie et al. developed an amplified assay based on an enzyme-linked split aptamer to perform colorimetric cocaine detection.[45] One of the fragments was conjugated to a plastic surface; in the presence of cocaine, this fragment formed a complex with another fragment, which was modified with biotin in order to bind streptavidin-linked horseradish peroxidase for signal amplification. However, the assay’s requirement for multiple washes caused dissociation of a subset of the assembled tripartite complexes, resulting in a limit of detection of just 2.8 μM in buffer and 50 μM in 10% serum. To prevent such dissociation, Sharma et al. incorporated a proximity ligation strategy into their split aptamer sensor.[217] Specifically, cocaine binding facilitated the assembly of azide- and cyclooctyne-modified split aptamer fragments, bringing these two chemical groups into close proximity to form covalent bonds via copper-catalyzed click chemistry. This greatly improved the sensor’s detection performance, with detection limits of 0.1 μM in buffer and 1 μM in 50% serum. Although signal amplification can improve the sensitivity of split-aptamer-based sensors, further improvements remained limited to the poor binding affinity of split aptamers. To overcome this limitation, we incorporated cooperative-binding functionality into split-aptamer constructs to achieve both low background and high target affinity.[218] This new cooperative binding split aptamer (CBSA) construct contains two tandem target-binding domains, wherein the first target-binding event stabilizes the structure of the split aptamer and facilitates subsequent target-binding at the second binding site. Compared with split aptamers with a single target-binding domain, CBSAs exhibit higher target affinity and far more responsive target-induced aptamer assembly, enabling sensitive target detection. For example, we developed a CBSA-based fluorescence assay that detected cocaine within 15 min with a limit of detection of 50 nM in both buffer and 10% saliva, surpassing the performance of previous split-aptamer-based sensors.[218] Notably, CBSAs can be easily developed from any three-way-junction-structured aptamer, and readily incorporated into various sensor platforms for sensitive small molecule detection.[77,219]
4.1.3. Sensors based on target-induced strand-displacement
Unlike truncated and split aptamers, the strand-displacement strategy allows for the generation of structure-switching aptamers without sequence engineering (Figure 5C).[202] This strategy employs a short cDNA strand that is complementary to part of the aptamer. In the absence of target, the cDNA hybridizes with the aptamer and disrupts its folded structure. The aptamer undergoes a conformational change upon target binding and folds into its original structure, inducing displacement of the cDNA. This target-induced structure-switching event can be readily transduced into an optical[202] or electrochemical[44] signal. For example, Nutiu and Li attached a fluorophore to an ATP-binding aptamer and a quencher onto a cDNA competitor. In the absence of the target, the hybridization of aptamer and the cDNA brought the fluorophore into proximity with the quencher, yielding low fluorescence. Upon addition of the target, the competitive strand was displaced, resulting in a strong fluorescent signal.[202] Although no sequence engineering is necessary, foreknowledge of the aptamer binding domain is required to identify competitor sequences that can be displaced upon target binding. To bypass this trial-and-error process, Nutiu and Li developed a method for directly isolating aptamers with accompanying competitive strands that can be directly used for sensing, greatly accelerating the sensor development process.[92] Recently, Munzar et al. designed an array-based method to optimize cDNA length for aptamer-based strand-displacement sensors.[220] Their array contained thousands of spots, each containing a cluster of unique covalently-attached DNA of differing lengths and sequence that are complementary to various regions of an aptamer. The aptamer, labeled with a fluorophore, was attached to the array surface via hybridization with the immobilized cDNA strands. Target binding caused the aptamer to dissociate, which resulted in a reduction of fluorescence. Based on fluorescence changes at specific locations on the array, the regions of the aptamer responsible for target binding could be identified. This assay could also provide information about the binding mechanism of aptamers (i.e. induced fit or conformational selection) and the cDNA sequences that were most suitable for sensor development.
4.2. Sensors that do not require target-induced conformational change
Recently, several sensing strategies have been developed for the detection of small molecules without the need for structure-switching aptamers. We have recently written a detailed review on these strategies,[53] and will only discuss them briefly here. The dye-displacement sensing platform is among the first aptamer-based detection schemes that do not require structure-switching. These assays[73,76,175,177,201,221–223] rely on small-molecule dyes that can bind to the target-binding domain of an aptamer in its non-target-bound state (Figure 5D). Target binding displaces the dye from the binding domain, resulting in a change in the absorbance or fluorescence of the dye. These assays are label-free and do not require any chemical modification or sequence engineering. Since such aptamers typically bind to the dye and small-molecule target with similar affinities dye-displacement assays can achieve limits of detection that are 10-fold lower than the KD of the aptamer being used.[73,76,177,201,221] However, this strategy is not generalizable, because there is a limited number of dye molecules that can bind to aptamers with varying sequences and structure and undergo target-specific displacement. Nuclease-based methods have been previously developed based on structure-switching aptamers for small-molecule detection.[224,225] We have recently develop a label-free assay that utilizes exonucleases to detect small-molecule targets with no aptamer engineering requirements. In the absence of target, aptamers are completely digested by exonucleases.188 However, upon target binding, digestion by the enzymes is strongly inhibited (Figure 5E). The remaining oligonucleotides can be stained using the DNA-binding dye SYBR Gold to quantify target concentrations in a label-free manner. Using this approach, we were able to detect cocaine in 10% saliva and dehydroisoandrosterone 3-sulfate in 50% urine with limits of detection of 0.1 and 0.5 µM, respectively. Importantly, this sensing platform can be generally applied to different aptamers with various secondary structures, including three-way-junctions and stem-loops.[226] More recently, receptor-modified field-effect transistors (FETs) have gained attention in the biosensing field as an alternative means of detecting small molecules. FET is ultrasensitive to the change in its surface charge.[227] Recent work by Nakatsuka et al. demonstrated that small-molecule binding to fully-folded aptamers modified onto FETs can induce a change in surface charge via aptamer ternary structure rearrangements. Using this strategy, they demonstrated femtomolar-level detection of both charged and non-charged small molecules in phosphate buffered saline and artificial cerebrospinal fluid.[74] However, the generalizability of this strategy for other small-molecule-binding aptamers needs to be assessed in the future.
4.3. Strengths and limitations of aptamer-based sensors and utility for real applications.
All analytical techniques have their strengths and limitations, including aptamer-based sensors. Two aspects need to be considered: the sensing mechanism, which happens at the level of aptamer-ligand recognition, and the signal reporting mechanism, which transduces binding events into signals that can be measured. Table S2 in the supporting information lists sensing and signal reporting mechanisms commonly used in aptamer-based sensors. This allows certain generalizations to be made about what types of sensors could be useful for a given analytical problem. For example, for non-expert users performing on-site analysis, sensors that provide results that can readily be interpreted with the naked eye might be ideal, such as colorimetric signal reporters based on organic dyes or gold nanoparticles. For applications that require target detection in complex samples, such as medical diagnostics, a high-specificity quantitative detection method with minimal sample prep requirements would be preferable. Here, the ideal sensing platform may be a reagentless E-AB sensor based on a truncated structure-switching aptamer, which operates via the aptamer folding mechanism. However, this would require aptamer engineering, which makes sensor development less straightforward than systems that utilize dyes or nanoparticles. As a final example, for applications that require analysis of multiple samples, which is common in analytical laboratories, fluorescence-based assays are ideal because of their high sensitivity. A combination of the strand-displacement, aptamer assembly (e.g., CBSAs) or exonuclease digestion sensing mechanisms with high-throughput analysis utilizing multi-well microplates would enable simple and specific analysis of many samples with the assistance of a robotic liquid-handing system.
5. Summary and outlook
To fabricate aptamer-based sensors that are suitable for small molecule detection in real samples, extensive preparation and forethought is necessary. The first step is to understand the application. Considerations here include the concentration of the analyte, the identity and composition of the relevant sample matrix, the temperature used for the measurements, the presence of potential interferents in the sample, and the required turnaround time. These factors will have an important effect in terms of choosing the most suitable sensing platform from the various strategies described above. The second step is to isolate appropriate aptamers for the sensing application. Through the SELEX process, aptamers must be isolated that not only bind to the specified target with high affinity, but must also have sensing functionalities that can be directly used for downstream sensor development. The design of the library is critical here, as a good library design allows for the introduction of functionality during SELEX, greatly reducing or even eliminating the need for subsequent engineering. For example, a three-way-junction-structured library can be used to generate aptamers for dye-displacement or split-aptamer-based assays. Aptamers isolated from stem-loop structured libraries can be directly used in exonuclease inhibition assays, or truncated to adopt structure-switching functionality. Library-immobilized SELEX methods have also been developed to generate aptamers that can be directly incorporated in strand-displacement assays—notably, this SELEX approach has no limitations in terms of the design of the library. The buffer in which aptamer isolation will be performed is also an important consideration. The composition of the selection buffer should ideally mimic that of the real sample matrix in terms of factors such as salt concentration, pH, and temperature. This ensures that the isolated aptamer will function as intended when used for sensing in real samples. During the selection process, aptamers with a particular level of affinity can be attained by fine tuning selection stringency. To obtain highly specific aptamers, counter-SELEX can be performed to remove sequences in the oligonucleotide library that bind potential structurally similar interferents in the sample matrices. Alternatively, toggle and parallel-and-serial SELEX strategies can be used to isolate aptamers with broad cross-reactivity. These different selection strategies can be employed separately or together during SELEX to isolate aptamers with customized binding properties and sensing functionalities.
As SELEX is traditionally a labor-intensive process, there have been efforts to automate SELEX with robotic liquid-handling systems.[228–232] These automated approaches reduce the time required for SELEX from weeks to days, and enable simultaneous aptamer isolation against multiple targets. More recently, microfluidic devices have been developed that integrate strand separation and strand amplification – the major steps of SELEX – on a single chip. For example, Kim et al. recently performed SELEX using a microfluidic device, and was able to isolate micromolar affinity aptamers for bisboronic-acid-bound glucose within three rounds of SELEX. The whole selection process took 10 hours.[233] Although most automation schemata have been applied for the isolation of aptamers that bind to protein and cell targets,[234] there are no factors that necessarily limit the application of these approaches to isolate small-molecule-binding aptamers, with some modifications to existing designs and protocols.
During SELEX, it is important to periodically characterize the affinity and specificity of the enriched pools. This allows one to make modifications to the selection protocol if necessary, which aids in enriching aptamers with the desired set of binding properties. For example, the gel elution has enabled the rapid evaluation of pool target-binding affinity and cross-reactivity. In recent years, HTS has become a powerful tool for aptamer candidate analysis and monitoring the enrichment of sequences during the SELEX process, thanks to rapidly falling costs and the availability of software for analyzing the large quantity of outputted data. The millions of sequences obtained using this method allow for the more efficient identification of aptamer candidates with the desired set of properties, such as high affinity and specificity, broad cross-reactivity, or different motifs and secondary structures. If a single aptamer cannot fulfill the needs of a given application, a combination of multiple aptamers with different functionalities and structures can be used to achieve finer tuning of a sensor’s limits of detection, dynamic range, or cross-reactivity. For example, we recently demonstrated the successful manipulation of aptamer-based sensors to achieve broad detection of a designer drug family by employing a mixture of two aptamers with differing ligand specificity.[176]
Once aptamer candidates are identified via HTS, they must be robustly characterized, and we suggest that affinity and specificity towards the target and all potential interferents should first be screened in a high-throughput manner. Methods such as ITC and SPR can determine quantitative binding thermodynamics and kinetics, but are ill-suited for extensively screening aptamer specificity due to low throughput. We have recently developed exonuclease-based assays based on the inhibition of aptamer digestion by enzymes upon ligand binding to characterize the binding between multiple aptamer-ligand pairs simultaneously in a single-pot reaction (in submission). Based on our preliminary success in characterizing a variety of aptamers with diverse structure and many ligands with wide-ranging physicochemical properties, we believe that this assay can be readily up-scaled to characterize aptamer binding profile in a high-throughput, automated fashion. After narrowing down the aptamer candidates, precise quantification of target-binding affinity of aptamers is recommended. In most SELEX work to date, the affinity of newly-isolated aptamers is characterized with only one method. If possible, we recommend for future aptamer selections that at least two well-accepted methods of characterization to determine binding affinity, ideally in the intended sample matrix.
After an aptamer has been thoroughly characterized, it can be used to fabricate a sensor either directly or after sequence engineering to introduce sensing functionality. As discussed above, several engineering strategies have been developed to adopt structure-switching functionality into aptamers. For example, we have recently developed a generalizable Exo III-based assay for the single-step introduction of structure-switching functionality into small-molecule-binding aptamers with various secondary structures.[209] Importantly, the library used in this work was designed to offer a compatible structure for this Exo III-based assay (e.g., stem-containing aptamers), eliminating the need for aptamer engineering. We have also recently incorporated Exo I, a single-strand DNA exonuclease, into this truncation strategy to remove single-stranded scaffold regions alongside the double-stranded areas removed by Exo III, enabling more efficient generation of minimized structure-switching aptamers.[176]
Despite the wide acclaim gathered by aptamers, they have not become as widely used as antibodies for analytical purposes due to several reasons. First, it is important to note that the field of aptamer-based sensor development did not actually begin until the early 2000s. Second, most aptamers isolated to date have insufficient affinity and/or specificity for real-world applications. This is mainly because aptamer isolation efforts have largely taken place at academic labs for research purposes, and only recently has this focus shifted from discovery to use in commercially viable real-world applications. Third, the process of isolating high-quality aptamers largely remains laborious, costly, and inherently trial-and-error. By integrating rational library design, optimal selection conditions, automated selection protocols, high-throughput sequencing and characterization methods, as well as rapid post-SELEX engineering techniques, we believe that the process of developing sensitive and robust aptamer-based sensors for small molecule detection with the capability for use in real applications can be dramatically accelerated, reducing the time required for this whole process from years to a matter of weeks. Therefore, we foresee more aptamer-based small molecule sensors that can be used broadly and commercialized for real-world applications.
Supplementary Material
Acknowledgements
This work was supported by National Institute of Justice, Office of Justice Programs, U.S. Department of Justice Awards (2015-R2-CX-0034), the National Institutes of Health - National Institute on Drug Abuse (R21DA045334-01A1), and the National Science Foundation (1905143).
Biographies

Haixiang Yu received his B.S. degree in Biotechnology from the China Pharmaceutical University in 2013. He then pursued a doctoral studies of Biochemistry major at Florida International University, where he conducted research with Dr. Yi Xiao in the areas of small-molecule binding aptamer isolation, engineering, and sensor development. After receiving his Ph.D. in 2019, he joined the Sullenger group at Duke University to develop therapeutic aptamers.
Obtin Alkhamis received his B.A. degree in Chemistry from Florida International University in 2017. He joined the laboratory of Dr. Yi Xiao in 2018 as an FIU Presidential Research Fellow. He has worked on the characterization of affinity and specificity of aptamers and is now developing new methods to isolate and characterize small-molecule-binding aptamers.

Juan Canoura received his B.S. degree in Chemistry from Florida International University in 2016 were he conducted research on aptamer-enzyme interactions and their use in aptamer-based sensors. He continued his doctoral studies as a McNair and National Institute of Justice graduate fellow at Florida International University with Dr. Yi Xiao, during which he worked on aptamer isolation and engineering using exonuclease-based techniques.

Yingzhu Liu received her B.S. degree in Chemical Engineering from Jingchu University of Technology in 2012. Then, she received her master’s degree in Chemical Engineering from Wuhan University of Science and Technology in 2015. In 2016, she pursued her doctoral degree at Florida International University in Chemistry under the supervision of Dr. Yi Xiao. She is currently working on aptamer isolation for small molecule detection and the development of electrochemical aptamer-based sensors.

Yi Xiao received her PhD in Chemistry from Nanjing University in 2000 and completed her postdoctoral training with Dr. Itamar Willner at The Hebrew University of Jerusalem and Dr. Kevin Plaxco and Dr. Alan Heeger at the University of California at Santa Barbara. In 2007, Dr. Xiao worked in Dr. Tom Soh’s lab at UCSB as a research faculty and devised microfluidic SELEX techniques for the rapid isolation of aptamers. She joined FIU as an Assistant Professor in 2011 and was tenured in 2017. Prof. Xiao’s current research direction encompasses the entirety of the aptamer and sensor development process, from aptamer isolation, to characterization, to sensor development, with the end goal of making aptamer-based sensors for real-world applications.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- [1].Peltomaa R, Glahn-Martínez B, Benito-Peña E, Moreno-Bondi MC, Sensors 2018, 18, 4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Patel S, Nanda R, Sahoo S, Mohapatra E, Biochem. Res. Int 2016, Article ID: 3130469. [DOI] [PMC free article] [PubMed]
- [3].Nguyen VT, Kwon YS, Gu MB, Curr. Opin. Biotechnol 2017, 45, 15–23. [DOI] [PubMed] [Google Scholar]
- [4].Lee TM-H, Sensors 2008, 8, 5535–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Metkar SK, Girigoswami K, Biocatal. Agric. Biotechnol 2019, 17, 271–283. [Google Scholar]
- [6].Bhalla N, Jolly P, Formisano N, Estrela P, Essays Biochem 2016, 60, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Howes PD, Chandrawati R, Stevens MM, Science 2014, 346, 1247390. [DOI] [PubMed] [Google Scholar]
- [8].Ronkainen NJ, Halsall HB, Heineman WR, Chem. Soc. Rev 2010, 39, 1747–1763. [DOI] [PubMed] [Google Scholar]
- [9].Luo X, Davis JJ, Chem. Soc. Rev 2013, 42, 5944–5962. [DOI] [PubMed] [Google Scholar]
- [10].Zhang L, Gu C, Ma H, Zhu L, Wen J, Xu H, Liu H, Li L, Anal. Bioanal. Chem 2019, 411, 21–36. [DOI] [PubMed] [Google Scholar]
- [11].Ispas CR, Crivat G, Andreescu S, Anal. Lett 2012, 45, 168–186. [Google Scholar]
- [12].Darwish IA, Int. J. Biomed. Sci 2006, 2, 217–235. [PMC free article] [PubMed] [Google Scholar]
- [13].Liu JKH, Ann. Med. Surg 2014, 3, 113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Koczula KM, Gallotta A, Essays Biochem 2016, 60, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Posthuma-Trumpie GA, Korf J, van Amerongen A, Anal. Bioanal. Chem 2009, 393, 569–582. [DOI] [PubMed] [Google Scholar]
- [16].Jayasena SD, Clin. Chem 1999, 45, 1628–1650. [PubMed] [Google Scholar]
- [17].Dunn MR, Jimenez RM, Chaput JC, Nat. Rev. Chem 2017, 1, 0076. [Google Scholar]
- [18].Pradidarcheep W, Labruyère WT, Dabhoiwala NF, Lamers WH, J. Histochem. Cytochem 2008, 56, 1099–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Baden LR, Horowitz G, Jacoby H, Eliopoulos GM, J. Am. Med. Assoc 2001, 286, 3115–3119. [DOI] [PubMed] [Google Scholar]
- [20].Spinks CA, Trends Food Sci. Technol 2000, 11, 210–217. [Google Scholar]
- [21].Zhang H, Wang S, Immunol J. Methods 2009, 350, 1–13. [DOI] [PubMed] [Google Scholar]
- [22].Ellefsen KN, Anizan S, Castaneto MS, Desrosiers NA, Martin TM, Klette KL, Huestis MA, Drug Test. Anal 2014, 6, 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou J, Rossi J, Nat. Rev. Drug Discov 2017, 16, 181–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tuerk C, Gold L, Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- [25].Ellington AD, Szostak JW, Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- [26].Hermann T, Patel DJ, Science 2000, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- [27].Ellington AD, Szostak JW, Nature 1992, 355, 850–852. [DOI] [PubMed] [Google Scholar]
- [28].Famulok M, Szostak JW, J. Am. Chem. Soc 1992, 114, 3990–3991. [Google Scholar]
- [29].Sassanfar M, Szostak JW, Nature 1993, 364, 550–553. [DOI] [PubMed] [Google Scholar]
- [30].Lorsch JR, Szostak JW, Biochemistry 1994, 33, 973–982. [DOI] [PubMed] [Google Scholar]
- [31].Wang Y, Rando RR, Chem. Biol 1995, 2, 281–290. [DOI] [PubMed] [Google Scholar]
- [32].Wallis MG, von Ahsen U, Schroeder R, Famulok M, Chem. Biol 1995, 2, 543–52. [DOI] [PubMed] [Google Scholar]
- [33].Burke DH, Hoffman DC, Brown A, Hansen M, Pardi A, Gold L, Chem. Biol 1997, 4, 833–843. [DOI] [PubMed] [Google Scholar]
- [34].Jenison RD, Gill SC, Pardi A, Polisky B, Science 1994, 263, 1425–1429. [DOI] [PubMed] [Google Scholar]
- [35].Stojanovic MN, de Prada P, Landry DW, J. Am. Chem. Soc 2001, 123, 4928–4931. [DOI] [PubMed] [Google Scholar]
- [36].Liu J, Lu Y, Angew. Chem., Int. Ed 2006, 45, 90–94. [Google Scholar]
- [37].Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW, J. Am. Chem. Soc 2006, 128, 3138–3139. [DOI] [PubMed] [Google Scholar]
- [38].Toh SY, Citartan M, Gopinath SCB, Tang T-H, Biosens. Bioelectron 2015, 64, 392–403. [DOI] [PubMed] [Google Scholar]
- [39].Harlow E, Lane D, Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York, 1988. [Google Scholar]
- [40].Rosenbaum CD, Carreiro SP, Babu KM, J. Med. Toxicol 2012, 8, 15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhuo Z, Yu Y, Wang M, Li J, Zhang Z, Liu J, Wu X, Lu A, Zhang G, Zhang B, Int. J. Mol. Sci 2017, 18, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ilgu M, Fazlioglu R, Ozturk M, Ozsurekci Y, Nilsen-Hamilton M, in Recent Adv. Anal. Chem (Eds.: Ince M, Ince OK), IntechOpen, London, 2019. [Google Scholar]
- [43].Stojanovic MN, de Prada P, Landry DW, J. Am. Chem. Soc 2000, 122, 11547–11548. [DOI] [PubMed] [Google Scholar]
- [44].Xiao Y, Lubin AA, Heeger AJ, Plaxco KW, Angew. Chem., Int. Ed 2005, 44, 5456–5459. [DOI] [PubMed] [Google Scholar]
- [45].Nie J, Deng Y, Deng QP, Zhang DW, Zhou YL, Zhang XX, Talanta 2013, 106, 309–314. [DOI] [PubMed] [Google Scholar]
- [46].Xiao Y, Rowe AA, Plaxco KW, J. Am. Chem. Soc 2007, 129, 262–263. [DOI] [PubMed] [Google Scholar]
- [47].Li H, Somerson J, Xia F, Plaxco KW, Anal. Chem 2018, 90, 10641–10645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Swensen JS, Xiao Y, Ferguson BS, Lubin AA, Lai RY, Heeger AJ, Plaxco KW, Soh HT, J. Am. Chem. Soc 2009, 131, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Xiao Y, Lai RY, Plaxco KW, Nat. Protoc 2007, 2, 2875–2880. [DOI] [PubMed] [Google Scholar]
- [50].Ferguson BS, Hoggarth DA, Maliniak D, Ploense K, White RJ, Woodward N, Hsieh K, Bonham AJ, Eisenstein M, Kippin TE, Plaxco KW, Soh HT, Sci. Transl. Med 2013, 5, 213ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dong Y, Zhang T, Lin X, Feng J, Luo F, Gao H, Wu Y, Deng R, He Q, Microchim. Acta 2020, 187, 179. [DOI] [PubMed] [Google Scholar]
- [52].Gao T, Luo Y, Li W, Cao Y, Pei R, Analyst 2020, 145, 701–718. [DOI] [PubMed] [Google Scholar]
- [53].Alkhamis O, Canoura J, Yu H, Liu Y, Xiao Y, TrAC - Trends Anal. Chem 2019, 121, 115699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu J, Cao Z, Lu Y, Chem. Rev 2009, 109, 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].McKeague M, McConnell EM, Cruz-Toledo J, Bernard ED, Pach A, Mastronardi E, Zhang X, Beking M, Francis T, Giamberardino A, Cabecinha A, Ruscito A, Aranda-Rodriguez R,Dumontier M, DeRosa MC, J. Mol. Evol 2015, 81, 150–161. [DOI] [PubMed] [Google Scholar]
- [56].Cho M, Xiao Y, Nie J, Stewart R, Csordas AT, Oh SS, Thomson JA, Soh HT, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 15373–15378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hermanson GT, Bioconjugate Techniques, Academic Press, San Diego, 2008. [Google Scholar]
- [58].Ohsawa K, Kasamatsu T, Nagashima J, Hanawa K, Kuwahara M, Ozaki H, Sawai H, Anal. Sci 2008, 24, 167–172. [DOI] [PubMed] [Google Scholar]
- [59].Mendonsa SD, Bowser MT, J. Am. Chem. Soc 2004, 126, 20–21. [DOI] [PubMed] [Google Scholar]
- [60].Carothers JM, Goler JA, Kapoor Y, Lara L, Keasling JD, Nucleic Acids Res 2010, 38, 2736–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rosenthal M, Pfeiffer F, Mayer G, Angew. Chem., Int. Ed 2019, 58, 10752–10755. [DOI] [PubMed] [Google Scholar]
- [62].Yang K-A, Barbu M, Halim M, Pallavi P, Kim B, Kolpashchikov DM, Pecic S, Taylor S, Worgall TS, Stojanovic MN, Nat. Chem 2014, 6, 1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, et al. , PLoS One 2010, 5, e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Imaizumi Y, Kasahara Y, Fujita H, Kitadume S, Ozaki H, Endoh T, Kuwahara M, Sugimoto N, J. Am. Chem. Soc 2013, 135, 9412–9419. [DOI] [PubMed] [Google Scholar]
- [65].Battersby TR, Ang DN, Burgstaller P, Jurczyk SC, Bowser MT, Buchanan DD, Kennedy RT, Benner SA, J. Am. Chem. Soc 1999, 121, 9781–9789. [DOI] [PubMed] [Google Scholar]
- [66].Vaish NK, Larralde R, Fraley AW, Szostak JW, McLaughlin LW, Biochemistry 2003, 42, 8842–8851. [DOI] [PubMed] [Google Scholar]
- [67].Huizenga DE, Szostak JW, Biochemistry 1995, 34, 656–665. [DOI] [PubMed] [Google Scholar]
- [68].Caruthers MH, Barone AD, Beaucage SL, Dodds DR, Fisher EF, McBride LJ, Matteucci M, Stabinsky Z, Tang JY, Methods Enzymol 1987, 154, 287–313. [DOI] [PubMed] [Google Scholar]
- [69].Pobanz K, Lupták A, Methods 2016, 106, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yang KA, Pei RJ, Stefanovic D, Stojanovic MN, J. Am. Chem. Soc 2012, 134, 1642–1647. [DOI] [PubMed] [Google Scholar]
- [71].Yang KA, Chun H, Zhang Y, Pecic S, Nakatsuka N, Andrews AM, Worgall TS, Stojanovic MN, ACS Chem. Biol 2017, 12, 3103–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Rowe AA, Miller EA, Plaxco KW, Anal. Chem 2010, 82, 7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Yang W, Yu H, Alkhamis O, Liu Y, Canoura J, Fu F, Xiao Y, Nucleic Acids Res 2019, 47, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Nakatsuka N, Yang K-A, Abendroth JM, Cheung KM, Xu X, Yang H, Zhao C, Zhu B, Rim YS, Yang Y, Weiss PS, Stojanovic MN, Andrews AM, Science 2018, 362, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Oh SS, Plakos K, Xiao Y, Eisenstein M, Soh HT, ACS Nano 2013, 7, 9675–9683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Yu H, Yang W, Alkhamis O, Canoura J, Yang K-A, Xiao Y, Nucleic Acids Res 2018, 46, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Luo Y, Yu H, Alkhamis O, Liu Y, Lou X, Yu B, Xiao Y, Anal. Chem 2019, 91, 7199–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Win MN, Klein JS, Smolke CD, Nucleic Acids Res 2006, 34, 5670–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kiga D, Futamura Y, Sakamoto K, Yokoyama S, Nucleic Acids Res 1998, 26, 1755–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Barbu M, Stojanovic MN, ChemBioChem 2012, 13, 658–660. [DOI] [PubMed] [Google Scholar]
- [81].Derbyshire N, White SJ, Bunka DHJ, Song L, Stead S, Tarbin J, Sharman M, Zhou D, Stockley PG, Anal. Chem 2012, 84, 6595–6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stoltenburg R, Nikolaus N, Strehlitz B, Anal J. Methods Chem 2012, 415697. [DOI] [PMC free article] [PubMed]
- [83].Schürer H, Stembera K, Knoll D, Mayer G, Blind M, Förster HH, Famulok M, Welzel P, Hahn U, Bioorg. Med. Chem 2001, 9, 2557–2563. [DOI] [PubMed] [Google Scholar]
- [84].Niazi JH, Lee SJ, Gu MB, Bioorg. Med. Chem 2008, 16, 7245–7253. [DOI] [PubMed] [Google Scholar]
- [85].Reinemann C, Freiin von Fritsch U, Rudolph S, Strehlitz B, Biosens. Bioelectron 2016, 77, 1039–1047. [DOI] [PubMed] [Google Scholar]
- [86].McKeague M, DeRosa MC, J. Nucleic Acids 2012, 2012, 748913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lou XH, Qian JR, Xiao Y, Viel L, Gerdon AE, Lagally ET, Atzberger P, Tarasow TM, Heeger AJ, Soh HT, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 2989–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Geiger A, Burgstaller P, von der Eltz H, Roeder A, Famulok M, Nucleic Acids Res 1996, 24, 1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Oh SS, Ahmad KM, Cho M, Kim S, Xiao Y, Soh HT, Anal. Chem 2011, 83, 6883–6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ozer A, Pagano JM, Lis JT, Mol. Ther. Nucleic Acids 2014, 3, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Klug SJ, Famulok M, Mol. Biol. Rep 1994, 20, 97–107. [DOI] [PubMed] [Google Scholar]
- [92].Nutiu R, Li Y, Angew. Chem., Int. Ed 2005, 44, 1061–1065. [DOI] [PubMed] [Google Scholar]
- [93].Spill F, Weinstein ZB, Shemirani AI, Ho N, Desai D, Zaman MH, Proc. Natl. Acad. Sci 2016, 113, 12076–12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Mosing RK, Mendonsa SD, Bowser MT, Anal. Chem 2005, 77, 6107–6112. [DOI] [PubMed] [Google Scholar]
- [95].Mendonsa SD, Bowser MT, Anal. Chem 2004, 76, 5387–5392. [DOI] [PubMed] [Google Scholar]
- [96].Yang J, Bowser MT, Anal. Chem 2013, 85, 1525–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Park JW, Tatavarty R, Kim DW, Jung HT, Gu MB, Chem. Commun 2012, 48, 2071–2073. [DOI] [PubMed] [Google Scholar]
- [98].Gu H, Duan N, Wu S, Hao L, Xia Y, Ma X, Wang Z, Sci. Rep 2016, 6, 21665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Nguyen V-T, Kwon YS, Kim JH, Gu MB, Chem. Commun 2014, 50, 10513–10516. [DOI] [PubMed] [Google Scholar]
- [100].Chen X, Huang Y, Duan N, Wu S, Xia Y, Ma X, Zhu C, Jiang Y, Wang Z, Agric J. Food Chem 2014, 62, 10368–10374. [DOI] [PubMed] [Google Scholar]
- [101].Xing L, Zhang Y, Yang J, Biochem. Biophys. Res. Commun 2019, 514, 134–139. [DOI] [PubMed] [Google Scholar]
- [102].Park SM, Ahn JY, Jo M, Lee D, Lis JT, Craighead HG, Kim S, Lab Chip 2009, 9, 1206–1212. [DOI] [PubMed] [Google Scholar]
- [103].Bae H, Ren S, Kang J, Kim M, Jiang Y, Jin MM, Min IM, Kim S, Nucleic Acid Ther 2013, 23, 443–449. [DOI] [PubMed] [Google Scholar]
- [104].Levine HA, Nilsen-Hamilton M, Comput. Biol. Chem 2007, 31, 11–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Irvine D, Tuerk C, Gold L, J. Mol. Biol 1991, 222, 739–761. [DOI] [PubMed] [Google Scholar]
- [106].Wang J, Rudzinski JF, Gong Q, Soh HT, Atzberger PJ, PLoS One 2012, 7, e43940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Ruscito A, DeRosa MC, Front. Chem 2016, 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Yang K-A, Pei R, Stojanovic MN, Methods 2016, 106, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].White R, Rusconi C, Scardino E, Wolberg A, Lawson J, Hoffman M, Sullenger B, Mol. Ther 2001, 4, 567–573. [DOI] [PubMed] [Google Scholar]
- [110].Bianchini M, Radrizzani M, Brocardo MG, Reyes GB, Solveyra CG, Santa-Coloma TA, Immunol J. Methods 2001, 252, 191–197. [DOI] [PubMed] [Google Scholar]
- [111].Song MY, Nguyen D, Hong SW, Kim BC, Sci. Rep 2017, 7, 43641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Gray BP, Kelly L, Ahrens DP, Barry AP, Kratschmer C, Levy M, Sullenger BA, Proc. Natl. Acad. Sci. U. S. A 2018, 115, 4761–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Jiménez GC, Eissa S, Ng A, Alhadrami H, Zourob M, Siaj M, Anal. Chem 2015, 87, 1075–1082. [DOI] [PubMed] [Google Scholar]
- [114].Mei H, Bing T, Yang X, Qi C, Chang T, Liu X, Cao Z, Shangguan D, Anal. Chem 2012, 84, 7323–7329. [DOI] [PubMed] [Google Scholar]
- [115].Oguro A, Yanagida A, Fujieda Y, Amano R, Otsu M, Sakamoto T, Kawai G, Matsufuji S, J. Biochem 2017, 161, 197–206. [DOI] [PubMed] [Google Scholar]
- [116].Sharma S, Zajac M, Krishnan Y, ChemBioChem 2020, 21, 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Eissa S, Zourob M, Anal. Chem 2017, 89, 3138–3145. [DOI] [PubMed] [Google Scholar]
- [118].Lee J, Lee KH, Jeon J, Dragulescu-Andrasi A, Xiao F, Rao J, ACS Chem. Biol 2010, 5, 1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jauset-Rubio M, Botero ML, Skouridou V, Aktas GB, Svobodova M, Bashammakh AS, El-Shahawi MS, Alyoubi AO, O’Sullivan CK, ACS Omega 2019, 4, 20188–20196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Paniel N, Istamboulié G, Triki A, Lozano C, Barthelmebs L, Noguer T, Talanta 2017, 162, 232–240. [DOI] [PubMed] [Google Scholar]
- [121].Gu C, Lan T, Shi H, Lu Y, Anal. Chem 2015, 87, 7676–7682. [DOI] [PubMed] [Google Scholar]
- [122].Li W, Luo Y, Gao T, Yang L, Wang J, Pei R, J. Mol. Evol 2019, 87, 231–239. [DOI] [PubMed] [Google Scholar]
- [123].Quang NN, Perret G, Ducongé F, Pharmaceuticals 2016, 9, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Schütze T, Wilhelm B, Greiner N, Braun H, Peter F, Mörl M, Erdmann VA, Lehrach H, Konthur Z, Menger M, Arndt PF, Glökler J, PLoS One 2011, 6, e29604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Spiga FM, Maietta P, Guiducci C, ACS Comb. Sci 2015, 17, 326–333. [DOI] [PubMed] [Google Scholar]
- [126].Dupont DM, Larsen N, Jensen JK, Andreasen PA, Kjems J, Nucleic Acids Res 2015, 43, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Valenzano S, De Girolamo A, DeRosa MC, McKeague M, Schena R, Catucci L, Pascale M, ACS Comb. Sci 2016, 18, 302–313. [DOI] [PubMed] [Google Scholar]
- [128].Levenshtein VI, Sov. Phys. Dokl 1966, 10, 707–710. [Google Scholar]
- [129].Hoinka J, Zotenko E, Friedman A, Sauna ZE, Przytycka TM, Bioinformatics 2012, 28, i215–i223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Kikin O, D’Antonio L, Bagga PS, Nucleic Acids Res 2006, 34, W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Zuker M, Mathews DH, Turner DH, in RNA Biochem. Biotechnol (Eds.: Barciszewski J, Clark BFC), Springer: Dordecht, 1999, pp. 11–43. [Google Scholar]
- [132].Bartel DP, Zapp ML, Green MR, Szostak JW, Cell 1991, 67, 529–536. [DOI] [PubMed] [Google Scholar]
- [133].Famulok M, J. Am. Chem. Soc 1994, 116, 1698–1706. [Google Scholar]
- [134].Alam KK, Chang JL, Burke DH, Mol. Ther. - Nucleic Acids 2015, 4, e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Caroli J, Taccioli C, De La Fuente A, Serafini P, Bicciato S, Bioinformatics 2016, 32, 161–164. [DOI] [PubMed] [Google Scholar]
- [136].Bailey TL, Elkan C, Proc Int Conf Intell Syst Mol Biol 1994, 2, 28–36. [PubMed] [Google Scholar]
- [137].Hiller M, Pudimat R, Busch A, Backofen R, Nucleic Acids Res 2006, 34, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Hoinka J, Backofen R, Przytycka TM, Mol. Ther. - Nucleic Acids 2018, 11, 515–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D, Nucleic Acids Res 2018, 46, W537–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Song J, Zheng Y, Huang M, Wu L, Wang W, Zhu Z, Song Y, Yang C, Anal. Chem 2020, 92, 3307–3314. [DOI] [PubMed] [Google Scholar]
- [141].Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB, Mol. Cell 2014, 54, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Cowan JA, Ohyama T, Wang D, Natarajan K, Nucleic Acids Res 2000, 28, 2935–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Neves MAD, Reinstein O, Johnson PE, Biochemistry 2010, 49, 8478–8487. [DOI] [PubMed] [Google Scholar]
- [144].Zhang Z, Oni O, Liu J, Nucleic Acids Res 2017, 45, 7593–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Vogel M, Suess B, in Nucleic Acid Aptamers (Ed.: Mayer G), Humana Press, New York, 2016, pp. 113–125. [Google Scholar]
- [146].Duvvuri H, Wheeler LC, Harms MJ, Biochemistry 2018, 57, 2578–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Piñeiro Á, Muñoz E, Sabín J, Costas M, Bastos M, Velázquez-Campoy A, Garrido PF, Dumas P, Ennifar E, García-Río L, Rial J, Pérez D, Fraga P, Rodríguez A, Cotelo C, Anal. Biochem 2019, 577, 117–134. [DOI] [PubMed] [Google Scholar]
- [148].Zhao H, Piszczek G, Schuck P, Methods 2015, 76, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Krainer G, Keller S, Methods 2015, 76, 116–123. [DOI] [PubMed] [Google Scholar]
- [150].Slavkovic S, Johnson PE, Aptamers 2018, 2, 45–51. [Google Scholar]
- [151].Chang AL, McKeague M, Smolke CD, Methods Enzymol 2014, 549, 451–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hendrix M, Priestley ES, Joyce GF, Wong CH, J. Am. Chem. Soc 1997, 119, 3641–3648. [DOI] [PubMed] [Google Scholar]
- [153].Chang AL, McKeague M, Liang JC, Smolke CD, Anal. Chem 2014, 86, 3273–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Baaske P, Wienken CJ, Reineck P, Duhr S, Braun D, Angew. Chem., Int. Ed 2010, 49, 2238–2241. [DOI] [PubMed] [Google Scholar]
- [155].Biniuri Y, Albada B, Willner I, J. Phys. Chem. B 2018, 122, 9102–9109. [DOI] [PubMed] [Google Scholar]
- [156].McKeague M, Velu R, De Girolamo A, Valenzano S, Pascale M, Smith M, Derosa MC, Toxins 2016, 8, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Entzian C, Schubert T, Methods 2016, 97, 27–34. [DOI] [PubMed] [Google Scholar]
- [158].Rangel AE, Chen Z, Ayele TM, Heemstra JM, Nucleic Acids Res 2018, 46, 8057–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Paige JS, Wu KY, Jaffrey SR, Science 2011, 333, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Tan X, Constantin TP, Sloane KL, Waggoner AS, Bruchez MP, Armitage BA, J. Am. Chem. Soc 2017, 139, 9001–9009. [DOI] [PubMed] [Google Scholar]
- [161].Lauhon CT, Szostak JW, J. Am. Chem. Soc 1995, 117, 1246–1257. [DOI] [PubMed] [Google Scholar]
- [162].Shoara AA, Slavkovic S, Donaldson LW, Johnson PE, Can. J. Chem 2017, 95, 1253–1260. [Google Scholar]
- [163].Idili A, Arroyo-Currás N, Ploense KL, Csordas AT, Kuwahara M, Kippin TE, Plaxco KW, Chem. Sci 2019, 10, 8164–8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Samokhvalov AV, Safenkova IV, Zherdev AV, Dzantiev BB, Biochem. Biophys. Res. Commun 2018, 505, 536–541. [DOI] [PubMed] [Google Scholar]
- [165].Perez-Gonzalez C, Lafontaine DA, Penedo JC, Front. Chem 2016, 4, Article 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Holeman LA, Robinson SL, Szostak JW, Wilson C, Fold. Des 1998, 3, 423–431. [DOI] [PubMed] [Google Scholar]
- [167].Okazawa A, Maeda H, Fukusaki E, Katakura Y, Kobayashi A, Bioorganic Med. Chem. Lett 2000, 10, 2653–2656. [DOI] [PubMed] [Google Scholar]
- [168].Wang Y, Killian J, Hamasaki K, Rando RR, Biochemistry 1996, 35, 12338–12346. [DOI] [PubMed] [Google Scholar]
- [169].McKeague M, De Girolamo A, Valenzano S, Pascale M, Ruscito A, Velu R, Frost NR, Hill K, Smith M, McConnell EM, DeRosa MC, Anal. Chem 2015, 87, 8608–8612. [DOI] [PubMed] [Google Scholar]
- [170].Tao J, Frankel AD, Biochemistry 1996, 35, 2229–2238. [DOI] [PubMed] [Google Scholar]
- [171].McKeague M, Bradley CR, de Girolamo A, Visconti A, David Miller J, de Rosa MC, Int. J. Mol. Sci 2010, 11, 4864–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Wang L, Li J, Song S, Li D, Fan C, Phys J. D. Appl. Phys 2009, 42, 203001. [Google Scholar]
- [173].Zong C, Liu J, Anal. Chem 2019, 91, 10887–10893. [DOI] [PubMed] [Google Scholar]
- [174].Hu J, Easley CJ, Analyst 2011, 136, 3461–3468. [DOI] [PubMed] [Google Scholar]
- [175].Stojanovic MN, Landry DW, J. Am. Chem. Soc 2002, 124, 9678–9679. [DOI] [PubMed] [Google Scholar]
- [176].Liu Y, Yu H, Alkhamis O, Moliver J, Xiao Y, Anal. Chem 2020, 92, 5041–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Pei R, Stojanovic MN, Anal. Bioanal. Chem 2008, 390, 1093–1099. [DOI] [PubMed] [Google Scholar]
- [178].Cai S, Yan J, Xiong H, Liu Y, Peng D, Liu Z, Analyst 2018, 143, 5317–5338. [DOI] [PubMed] [Google Scholar]
- [179].Zuker M, Nucleic Acids Res 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, Pierce NA, J. Comput. Chem 2011, 32, 170–173. [DOI] [PubMed] [Google Scholar]
- [181].SantaLucia J, Proc. Natl. Acad. Sci. U. S. A 1998, 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Parisien M, Major F, Nature 2008, 452, 51–55. [DOI] [PubMed] [Google Scholar]
- [183].Popenda M, Szachniuk M, Antczak M, Purzycka KJ, Lukasiak P, Bartol N, Blazewicz J, Adamiak RW, Nucleic Acids Res 2012, 40, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Frellsen J, Moltke I, Thiim M, Mardia KV, Ferkinghoff-Borg J, Hamelryck T, PLoS Comput. Biol 2009, 5, e1000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Jossinet F, Ludwig TE, Westhof E, Bioinformatics 2010, 26, 2057–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Jeddi I, Saiz L, Sci. Rep 2017, 7, 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Flores SC, Altman RB, RNA 2010, 16, 1769–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Dourado DFAR, Flores SC, Proteins Struct. Funct. Bioinforma 2014, 82, 2681–2690. [DOI] [PubMed] [Google Scholar]
- [189].Eisold A, Labudde D, Molecules 2018, 23, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Johnson WC, in Circ. Dichroism Conform. Anal. Biomol (Ed.: Fasman GD), Springer, 1996, pp. 433–468. [Google Scholar]
- [191].Kypr J, Kejnovska I, Renciuk D, Vorlickova M, Nucleic Acids Res 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Miyachi Y, Ogino C, Kondo A, Nucleosides, Nucleotides and Nucleic Acids 2014, 33, 31–39. [DOI] [PubMed] [Google Scholar]
- [193].Lin CH, Patel DJ, Chem. Biol 1997, 4, 817–832. [DOI] [PubMed] [Google Scholar]
- [194].Spring-Connell AM, Evich M, Germann MW, Curr. Protoc. Nucleic Acid Chem 2018, 72, 7.28.1–7.28.39. [DOI] [PubMed] [Google Scholar]
- [195].Xu G, Zhao J, Liu N, Yang M, Zhao Q, Li C, Liu M, Nucleic Acids Res 2019, 47, 5963–5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Sakamoto T, Aptamers 2017, 1, 13–18. [Google Scholar]
- [197].Zuo X, Xiao Y, Plaxco KW, J. Am. Chem. Soc 2009, 131, 6944–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Li Y, Liu B, Huang Z, Liu J, Chem. Sci 2020, 11, 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Sussman D, Nix J, Wilson C, Nat. Struct. Biol 2000, 7, 53–57. [DOI] [PubMed] [Google Scholar]
- [200].Warner KD, Chen MC, Song W, Strack RL, Thorn A, Jaffrey SR, Ferré-D’Amaré AR, Nat. Struct. Mol. Biol 2014, 21, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Roncancio D, Yu H, Xu X, Wu S, Liu R, Debord J, Lou X, Xiao Y, Anal. Chem 2014, 86, 11100–11106. [DOI] [PubMed] [Google Scholar]
- [202].Nutiu R, Li YF, J. Am. Chem. Soc 2003, 125, 4771–4778. [DOI] [PubMed] [Google Scholar]
- [203].Arroyo-Currás N, Somerson J, Vieira PA, Ploense KL, Kippin TE, Plaxco KW, Proc. Natl. Acad. Sci 2017, 114, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Han K, Liang Z, Zhou N, Sensors 2010, 10, 4541–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Gao S, Zheng X, Jiao B, Wang L, Anal. Bioanal. Chem 2016, 408, 4567–4573. [DOI] [PubMed] [Google Scholar]
- [206].Fischer NO, Tok JB-H, Tarasow TM, PLoS One 2008, 3, e2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Shangguan D, Tang Z, Mallikaratchy P, Xiao Z, Tan W, ChemBioChem 2007, 8, 603–606. [DOI] [PubMed] [Google Scholar]
- [208].Neves MAD, Reinstein O, Saad M, Johnson PE, Biophys. Chem 2010, 153, 9–16. [DOI] [PubMed] [Google Scholar]
- [209].Wang Z, Yu H, Canoura J, Liu Y, Alkhamis O, Fu F, Xiao Y, Nucleic Acids Res 2018, 46, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Zou R, Lou X, Ou H, Zhang Y, Wang W, Yuan M, Guan M, Luo Z, Liu Y, RSC Adv 2012, 2, 4636–4638. [Google Scholar]
- [211].Chen A, Yan M, Yang S, TrAC Trends Anal. Chem 2016, 80, 581–593. [Google Scholar]
- [212].Kent AD, Spiropulos NG, Heemstra JM, Anal. Chem 2013, 85, 9916–9923. [DOI] [PubMed] [Google Scholar]
- [213].Zhang J, Wang L, Pan D, Song S, Boey FYC, Zhang H, Fan C, Small 2008, 4, 1196–1200. [DOI] [PubMed] [Google Scholar]
- [214].Zhu C, Zhao Y, Yan M, Huang Y, Yan J, Bai W, Chen A, Anal. Bioanal. Chem 2016, 408, 4151–4158. [DOI] [PubMed] [Google Scholar]
- [215].Chen J, Fang Z, Lie P, Zeng L, Anal. Chem 2012, 84, 6321–6325. [DOI] [PubMed] [Google Scholar]
- [216].Jauset-Rubio M, El-Shahawi MS, Bashammakh AS, Alyoubi AO, O′Sullivan CK, TrAC - Trends Anal. Chem 2017, 97, 385–398. [Google Scholar]
- [217].Sharma AK, Heemstra JM, J. Am. Chem. Soc 2011, 133, 12426–12429. [DOI] [PubMed] [Google Scholar]
- [218].Yu H, Canoura J, Guntupalli B, Lou X, Xiao Y, Chem. Sci 2017, 8, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Yu H, Canoura J, Guntupalli B, Alkhamis O, Xiao Y, Anal. Chem 2018, 90, 1748–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Munzar JD, Ng A, Juncker D, Nat. Commun 2018, 9, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Ji DY, Wang HQ, Ge J, Zhang L, Li JJ, Bai DM, Chen J, Li ZH, Anal. Biochem 2017, 526, 22–28. [DOI] [PubMed] [Google Scholar]
- [222].Wang J, Jiang Y, Zhou C, Fang X, Anal. Chem 2005, 77, 3542–3546. [DOI] [PubMed] [Google Scholar]
- [223].Pei R, Rothman J, Xie Y, Stojanovic MN, Nucleic Acids Res 2009, 37, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Zheng D, Zou R, Lou X, Anal. Chem 2012, 84, 3554–3560. [DOI] [PubMed] [Google Scholar]
- [225].Lu C-H, Li J, Lin M-H, Wang Y-W, Yang H-H, Chen X, Chen G-N, Angew. Chem., Int. Ed 2010, 49, 8454–8457. [DOI] [PubMed] [Google Scholar]
- [226].Canoura J, Wang Z, Yu H, Alkhamis O, Fu F, Xiao Y, J. Am. Chem. Soc 2018, 140, 9961–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Syu Y-C, Hsu W-E, Lin C-T, ECS J Solid State Sci. Technol 2018, 7, Q3196–Q3207. [Google Scholar]
- [228].Cox JC, Ellington AD, Bioorganic Med. Chem 2001, 9, 2525–2531. [DOI] [PubMed] [Google Scholar]
- [229].Cox JC, Nucleic Acids Res 2002, 30, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Eulberg D, Buchner K, Maasch C, Klussmann S, Nucleic Acids Res 2005, 33, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Breuers S, Bryant LL, Legen T, Mayer G, Methods 2019, 161, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Ohuchi SP, Ohtsu T, Nakamura Y, Biochimie 2006, 88, 897–904. [DOI] [PubMed] [Google Scholar]
- [233].Kim J, Olsen TR, Zhu J, Hilton JP, Yang KA, Pei R, Stojanovic MN, Lin Q, Sci. Rep 2016, 6, 26139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Liu W-T, Lee W-B, Tsai Y-C, Chuang Y-J, Hsu K-F, Lee G-B, Biomicrofluidics 2019, 13, 014114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.