

Abstract
Background/Aims:
The US Food and Drug Administration (FDA) outlines clinical studies as postmarketing requirements and commitments to be fulfilled following FDA approval of new drugs and biologics (“therapeutics”). Regulators have increasingly emphasized lifecycle evaluation of approved therapeutics, and postmarketing studies are intended to advance our understanding of therapeutic safety and efficacy. However, little is known about the indications that clinical studies outlined in postmarketing requirements and commitments investigate, including whether they are intended to generate evidence for the approved or other clinical indications. Therefore, we characterized FDA postmarketing requirements and commitments for new therapeutics approved from 2009 to 2018.
Methods:
We conducted a cross-sectional study of all novel therapeutics, including small molecule drugs and biologics, receiving original FDA approval from 2009 to 2018, using approval letters accessed through the Drug@FDA database. Outcomes included the number and characteristics of FDA postmarketing requirements and commitments for new therapeutics at original approval, including types of studies outlined, indications to be investigated, and clinical evidence to be generated.
Results:
From 2009 to 2018, FDA approved 343 new therapeutics with 1978 postmarketing requirements and commitments. Overall, 750 (37.9%) postmarketing requirements and commitments outlined clinical studies. For 71 of 343 (20.7%) therapeutics, no postmarketing requirements nor commitments for clinical studies were outlined, while at least 1 was outlined for 272 (79.3%; median 2 (IQR, 1–4)). Among these 272 therapeutics, the number of postmarketing requirements and commitments for clinical studies per therapeutic did not change from 2009 (median 2 (IQR, 1–4)) to 2018 (median 2 (IQR, 1–3)). Among the 750 postmarketing requirements and commitments for clinical studies, 448 (59.7%) outlined new prospective cohort studies, registries, or clinical trials, while the remainder outlined retrospective studies, secondary analyses, or completion of ongoing studies. Although 455 (60.7%) clinical studies investigated only original approved therapeutic indications, 123 (16.4%) enrolled from an expansion of the approved disease population and 61 (8.1%) investigated diseases unrelated to approved indications.
Conclusions:
Most therapeutics are approved by FDA with at least 1 postmarketing requirement or commitment for a clinical study, which outline investigations of safety or efficacy for both approved and unapproved indications. The median number of 2 clinical studies outlined has remained relatively constant over the last decade. Given increasing emphasis by FDA on faster approval and lifecycle evaluation of therapeutics, these findings suggest that more postmarketing requirements and commitments may be necessary to address gaps in the clinical evidence available for therapeutics at approval.
Keywords: The US Food and Drug Administration, pharmaceutical regulation, postmarketing requirements, postmarketing commitments, clinical trials, clinical evidence
Introduction
To receive regulatory approval by the US Food and Drug Administration (FDA), new small molecule drugs and biologics (“therapeutics”) are generally required to be supported by two or more well-controlled studies demonstrating safety and efficacy.1,2 However, FDA has increasingly emphasized postmarket evidence generation in support of lifecycle evaluation of therapeutics.3 Furthermore, use of FDA’s expedited review programs,4 intended to speed the entry of therapeutics to market,5,6 has contributed to more therapeutics being approved on the basis of fewer pivotal trials,7 and trials using surrogate markers as primary endpoints.8 To generate evidence not available at therapeutic approval, FDA has the authority, under four statutes (the FDA Amendments Act, the Pediatric Research Equity Act, Accelerated Approval, and the Animal Efficacy Rule),9 to require sponsors to fulfill postmarketing requirements for clinical studies intended to generate safety data, confirm clinical benefit, or clarify the optimal use of therapeutics.10 FDA also collaborates with sponsors on voluntary postmarketing commitments to generate clinical evidence in support of ongoing therapeutic evaluation.10
While postmarketing requirements and commitments represent an increasingly important source of safety and efficacy evidence for approved therapeutic indications, numerous analyses have noted shortcomings in their use.11–13 For instance, therapeutics are often approved without postmarketing requirements or commitments outlining new sources of clinical evidence, such as prospective cohort studies, registries, or clinical trials.14 In such cases, sponsors rarely conduct clinical studies of original approved indications, instead investigating therapeutic uses for unapproved diseases (i.e. off-label) or expanded patient populations.14 These studies may support regulatory submissions for supplemental indication approvals, but can also promote off-label use of therapeutics without regulatory oversight of new clinical investigations.15–17 Previous analyses of new therapeutics approved from 2009 to 2012 found that postmarketing requirements and commitments outlined at approval rarely require new prospective cohort studies, registries, or clinical trials, even though these are important sources of clinical evidence for understanding therapeutic efficacy and safety.18,19 Furthermore, new clinical studies from postmarketing requirements and commitments are inconsistently completed and disseminated,18–21 despite generous timelines.22
Given the opportunity for postmarketing requirements and commitments to promote the generation of clinical evidence for therapeutics, particularly with respect to safety and efficacy for original approved indications, it is important to understand the studies FDA has outlined in postmarketing requirements and commitments, the indications they investigate, and whether they are intended to generate safety and/or efficacy data. Therefore, we characterized postmarketing requirements and commitments outlined for therapeutics receiving original FDA approval from 2009 to 2018, an interval notable for FDA’s increasing emphasis on lifecycle evaluation and expanding use of expedited review programs,3,7 including the introduction of new programs.4 We assessed the utilization of each postmarketing requirement or commitment authority, the types of studies outlined, and the indications for which postmarketing requirements and commitments are anticipated to generate evidence.
Methods
Study design and sample
Three authors (J.J.S., A.D.Z., J.D.W.) used the publicly available Drugs@FDA database to identify all therapeutics that received original FDA approval from 1 January 2009 to 31 December 2018.23 We excluded generic drugs, reformulations and new combinations of previously approved therapeutics, and non-therapeutic agents (e.g., contrast agents), using previous methodology.8 Approval data were identified from FDA novel drugs summaries and related publications,24–26 including the type of application (New Drug Application vs. Biologic License Application), whether the therapeutic received any expedited review program designations (priority review, accelerated approval, fast track, and/or breakthrough therapy), and whether the therapeutic received orphan drug designation.
Using original FDA approval letters and drug labels, we abstracted the original FDA-approved indication(s) for each therapeutic. To define original approved indications, we recorded indicated disease(s) (e.g., inflammatory bowel disease, non-small cell lung cancer), disease characteristics (e.g., moderate-to-severe, metastatic), and treatment characteristics (e.g., second-line therapy, component of multi-drug regimen). Contraindications and other information relevant to therapeutic use was also collected. Indications were classified according to the World Health Organization Anatomic Therapeutic Classification system,27 collapsed into 7 categories (Table 1).
Table 1.
Characteristics of 343 new therapeutics receiving original Food and Drug Administration approval, 2009–2018.
| Therapeutic characteristic | Number of therapeutics | No. (%) | P values | |
|---|---|---|---|---|
| No clinical PMRs or PMCs at approvala | At least one clinical PMR or PMC at approval | |||
| Total | 343 | 71 | 272 | - |
| Year of approval | ||||
| 2009 | 26 (7.6) | 5 (7.0) | 21 (7.7) | .99 |
| 2010 | 21 (6.1) | 4 (5.6) | 17 (6.3) | |
| 2011 | 28 (8.2) | 6 (8.5) | 22 (8.1) | |
| 2012 | 36 (10.5) | 8 (11.3) | 28 (10.3) | |
| 2013 | 24 (7.0) | 6 (8.5) | 18 (6.6) | |
| 2014 | 39 (11.4) | 9 (12.7) | 30 (11.0) | |
| 2015 | 45 (13.1) | 8 (11.3) | 37 (13.6) | |
| 2016 | 20 (5.8) | 3 (4.2) | 17 (6.3) | |
| 2017 | 45 (13.1) | 11 (15.5) | 34 (12.5) | |
| 2018 | 59 (17.2) | 11 (15.5) | 48 (17.6) | |
| Class | ||||
| Drug | 258 (75.2) | 59 (83.1) | 199 (73.2) | .09 |
| Biologic | 85 (24.8) | 12 (16.9) | 73 (26.8) | |
| Therapeutic area | ||||
| Autoimmune, musculoskeletal, and dermatology | 39 (11.4) | 6 (8.5) | 33 (12.1) | < .001 |
| Cancer and hematology | 97 (28.3) | 16 (22.5) | 81 (29.8) | |
| Cardiovascular and diabetes | 40 (11.7) | 10 (14.1) | 30 (11.0) | |
| Gastrointestinal and metabolism | 31 (9.0) | 3 (4.2) | 28 (10.3) | |
| Infectious disease | 55 (16.0) | 5 (7.0) | 50 (18.4) | |
| Neurology and psychiatry | 34 (9.9) | 7 (9.9) | 27 (9.9) | |
| Other | 47 (13.7) | 24 (33.8) | 23 (8.5) | |
| Priority review | ||||
| Yes | 185 (53.9) | 33 (46.5) | 152 (55.9) | .18 |
| No | 158 (46.1) | 38 (53.5) | 120 (44.1) | |
| Fast track | ||||
| Yes | 134 (39.1) | 26 (36.6) | 108 (39.7) | .68 |
| No | 209 (60.9) | 45 (63.4) | 164 (60.3) | |
| Accelerated approval | ||||
| Yes | 41 (12.0) | 0 (0) | 41 (15.1) | < .001 |
| No | 302 (88.0) | 71 (100.0) | 231 (84.9) | |
| Breakthrough therapy | ||||
| Yes | 59 (17.2) | 10 (14.1) | 49 (18.0) | .49 |
| Nob | 284 (82.8) | 61 (85.9) | 223 (82.0) | |
| Orphan drug designation | ||||
| Yes | 142 (41.4) | 33 (46.5) | 109 (40.1) | .35 |
| No | 201 (58.6) | 38 (53.5) | 163 (59.9) | |
PMRs: postmarketing requirements; PMCs: postmarketing commitments.
Includes therapeutics approved with no postmarketing requirements or commitments and therapeutics approved with non-clinical postmarketing requirements and/or commitments only.
Includes 88 (25.7%) therapeutics approved prior to origination of the Breakthrough Therapy designation on 9 July 2012.
Identifying postmarketing requirements and commitments
Building upon data collected for therapeutics receiving original FDA approval from 1 January 2009 to 31 December 2012,18,19 we used original FDA approval letters to identify all postmarketing requirements and commitments outlined at the time of original approval for each therapeutic and the regulatory authority under which each was issued (eTable 1-Supplemental Material). Postmarketing requirements and commitments were categorized based on the type of study (e.g., new prospective, ongoing prospective, animal) described in approval letters, using previous methodology.18,19 Any postmarketing requirement or commitment describing a study with safety and/or efficacy outcomes (i.e., a clinical study) was considered a “clinical postmarketing requirement or commitment”; all others (e.g., animal or drug-drug interaction studies), were considered non-clinical (eBox-Supplemental Material). Clinical study categories included: new prospective cohort studies, registries, or clinical trials (“new prospective clinical studies”); completion of ongoing prospective clinical studies; new retrospective observational studies; or new analyses or follow-up for any clinical studies. Evidence generated by clinical postmarketing requirements and commitments was characterized as safety, efficacy, or both.
Characterizing indications of postmarketing requirements and commitments
We classified the indications for clinical postmarketing requirements and commitments by comparing descriptions in approval letters of proposed postmarket studies to original FDA-approved therapeutic indications (Table 2). For each clinical postmarketing requirement and commitment, we abstracted the disease to be investigated, as well as patient and treatment characteristics, in a manner similar to the abstraction of original approved indications. Postmarketing requirements and commitments were then classified as generating evidence for the original approved disease (“original indication,” i.e., the investigated indication matched the disease, patient, and treatment characteristics described for the original approved indication), expanded disease populations beyond the scope of the original indication (“modified indication,” e.g., use in treatment-naïve patients when originally approved as second-line therapy for that disease), or new diseases not included in an original indication (“new indication”, e.g., chronic obstructive pulmonary disease when originally approved for the treatment of asthma). Postmarketing requirements and commitments outlining studies for original approved diseases were also classified as enrolling from the entirety of the indicated population (“general population”) or from a demographic (e.g., pediatric) or clinical (e.g., patients with the original approved disease and comorbid chronic kidney disease) subgroup of the indicated population (eAppendix – Supplemental Material). Pediatric studies for original approved diseases were considered to investigate a demographic subgroup of the indicated population, as these studies, when issued under the Pediatric Research Equity Act, are considered a deferred component of the original application for that indication.28
Table 2.
Sample classifications of indications for studies outlined in clinical postmarketing requirements and commitments.
| Therapeutic | Original FDA approved indication | PMR/PMC: authority;a description23 | PMR/PMC study population | PMR/PMC indication classificationb |
|---|---|---|---|---|
| Lurasidone | Schizophrenia | PMC: 506B; “To evaluate the longer-term, i.e. maintenance, efficacy of lurasidone in the treatment of adults with schizophrenia…” | Adult patients with schizophrenia |
Original indication, general population |
| Simeprevir | Chronic hepatitis C infection, as a component of a combination antiviral treatment regimen | PMC: 506B; “Submit the final report and datasets for trial… [in] Subjects who are Co-Infected with Human Immunodeficiency Virus Type 1 (HIV-1)” | Chronic hepatitis C infection, with HIV-1 coinfection | Original indication, clinical subgroup |
| Linagliptin | Type 2 diabetes mellitus | PMR: PREA; “…evaluate efficacy, safety, and pharmacokinetics of linagliptin for the treatment of type 2 diabetes mellitus in pediatric patients ages 10 to 16 years…” | Pediatric patients with type 2 diabetes mellitus | Original indication, demographic subgroup |
| Cabazitaxel | Hormone-refractory metastatic prostate cancer, in combination with prednisone, for patients previously treated with a docetaxel-containing treatment regimen | PMR: FDAAA; “Conduct a Phase 3 randomized controlled trial in patients with hormone-refractory metastatic prostate cancer…with prednisone as first-line therapy.” | First-line therapy with prednisone for (i.e., previously untreated) patients with hormone-refractory metastatic prostate cancer | Modified indication |
| IncobotulinumtoxinA | Cervical dystonia and blepharospasm | PMC: 506B; “Randomized, double-blind, adequate and well controlled, multiple fixed-dose, parallel group clinical trial… [in] adults with lower extremity spasticity.” | Adult patients with lower extremity spasticity | New indication |
PMR: postmarketing requirement; PMC: postmarketing commitment; FDAAA: Food and Drug Administration Amendments Act; PREA: Pediatric Research Equity Act.
“506B” refers to postmarketing commitments subject to reporting requirements under section 506B of the Federal Food, Drug, and Cosmetic Act.
Additional example postmarketing requirement and commitment classifications are included in eAppendix 1.
Clinical postmarketing requirements and commitments were abstracted using a prespecified algorithm, using descriptions from FDA approval letters supplemented as necessary by corresponding study registrations on ClinicalTrials.gov to abstract study design, indication, and safety and/or efficacy endpoints. Postmarketing requirements and commitments generating evidence for both original and modified or new indications were classified as “modified” or “new indication,” as applicable. Postmarketing requirements and commitments were abstracted by one author (J.J.S.), with uncertainties resolved via consensus amongst all authors. Another author (J.D.W.) validated abstractions using a 20% random sample of postmarketing requirements and commitments, with disagreements resolved via consensus amongst two authors (J.J.S. and J.D.W.; agreement rate, 98%). Analyses were conducted from 29 July 2019 to 23 March 2020.
Statistical analysis
We used descriptive statistics to summarize new therapeutics receiving FDA approval from 2009 to 2018 and to characterize postmarketing requirements and commitments by FDA, including characteristics such as issuing authority, study type, investigated indications, and evidence generated. We used Fisher’s exact and Kruskal-Wallis tests to evaluate associations between therapeutic characteristics and the issuance of postmarketing requirements and commitments. All statistical tests were 2-sided, significance was set at 0.05, and analyses were performed using R (version 3.5.1).
Ethical review and reporting guideline
This study was conducted using publicly available, nonclinical data and did not require institutional review board approval. It adheres to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline for cross-sectional studies.
Results
Characteristics of new therapeutics
From 2009 to 2018, FDA approved a total of 356 new therapeutics for 388 original indications. After excluding ineligible diagnostic agents, there were 343 (96.3%) therapeutics approved for 375 original indications included in our analyses (Table 1; eTable 2). Among the 343 therapeutics, 258 (75.2%) were small molecule drugs and 85 (24.8%) were biologics; orphan designation was granted for 142 (41.4%). A total of 185 (53.9%) therapeutics underwent priority review, 134 (39.1%) received fast track designation, 41 (12.0%) received accelerated approval designation, and 59 (17.2%) received breakthrough therapy designation. The most frequently represented therapeutic area was cancer or hematologic disease (97, 28.3%).
A total of 311 (90.7%) therapeutics were approved with at least 1 postmarketing requirement or commitment. For 272 (79.3%) therapeutics, at least 1 clinical postmarketing requirement or commitment was outlined, while none were outlined for 71 (20.7%) therapeutics. All 41 therapeutics granted accelerated approval designation were approved with at least 1 clinical postmarketing requirement or commitment. Therapeutics approved with at least 1 clinical postmarketing requirement or commitment were more likely to have been granted accelerated approval designation when compared with therapeutics approved without clinical postmarketing requirements or commitments (P < .001), and there were differences by therapeutic area (P < .001). No other expedited review program designation, nor orphan drug designation, was associated with the issuance of a clinical postmarketing requirement or commitment at original approval.
Postmarketing requirements and commitments outlined for new therapeutics
We identified a total of 1978 postmarketing requirements and commitments for therapeutics receiving original FDA approval from 2009 to 2018, including 1123 (56.8%) Postmarketing requirements and 855 (43.2%) postmarketing commitments. There were 1228 (62.1%) postmarketing requirements and commitments outlining non-clinical studies and 750 (37.9%) outlining clinical studies (eTable 3). Among the 750 clinical studies, four-fifths (600/750, 80.0%) were outlined in postmarketing requirements and one-fifth (150/750, 20.0%) in postmarketing commitments. Most clinical postmarketing requirements and commitments (448/750, 59.7%) outlined new prospective clinical studies (i.e., prospective cohort studies (48/750, 6.4%), registries (45/750, 6.0%), or clinical trials (355/750, 47.3%)); 125 of 750 (16.7%) outlined the completion or submission of results from ongoing prospective clinical studies. Three-quarters of postmarketing requirements issued under the Pediatric Research Equity Act (191/257, 74.3%) and one-half issued under accelerated approval (31/62, 50.0%) outlined new prospective clinical studies. Only 71 (71/448, 15.8%) postmarketing requirements and commitments for new prospective clinical studies were outlined for cancer and hematology therapeutics, despite those products representing over one-quarter of new therapeutic approvals (eTable 4).
The median number of postmarketing requirements and commitments for new therapeutics overall was 5 (interquartile range (IQR), 2–8), and the median number of clinical postmarketing requirements and commitments was 2 (IQR, 1–3). There was a non-significant decrease in the median number of clinical postmarketing requirements and commitments for new therapeutics overall from 2009 (2, IQR, 1–2) to 2018 (1, IQR, 1–3) (Figure 1, P = .54). Among therapeutics approved with at least 1 clinical postmarketing requirement or commitment, the median number of clinical postmarketing requirements and commitments was 2 for most years from 2009 (2, IQR,1–4) to 2018 (2, IQR, 1–3).
Figure 1. Postmarketing requirements and commitments outlined for therapeutics receiving original Food and Drug Administration approval, 2009–2018.
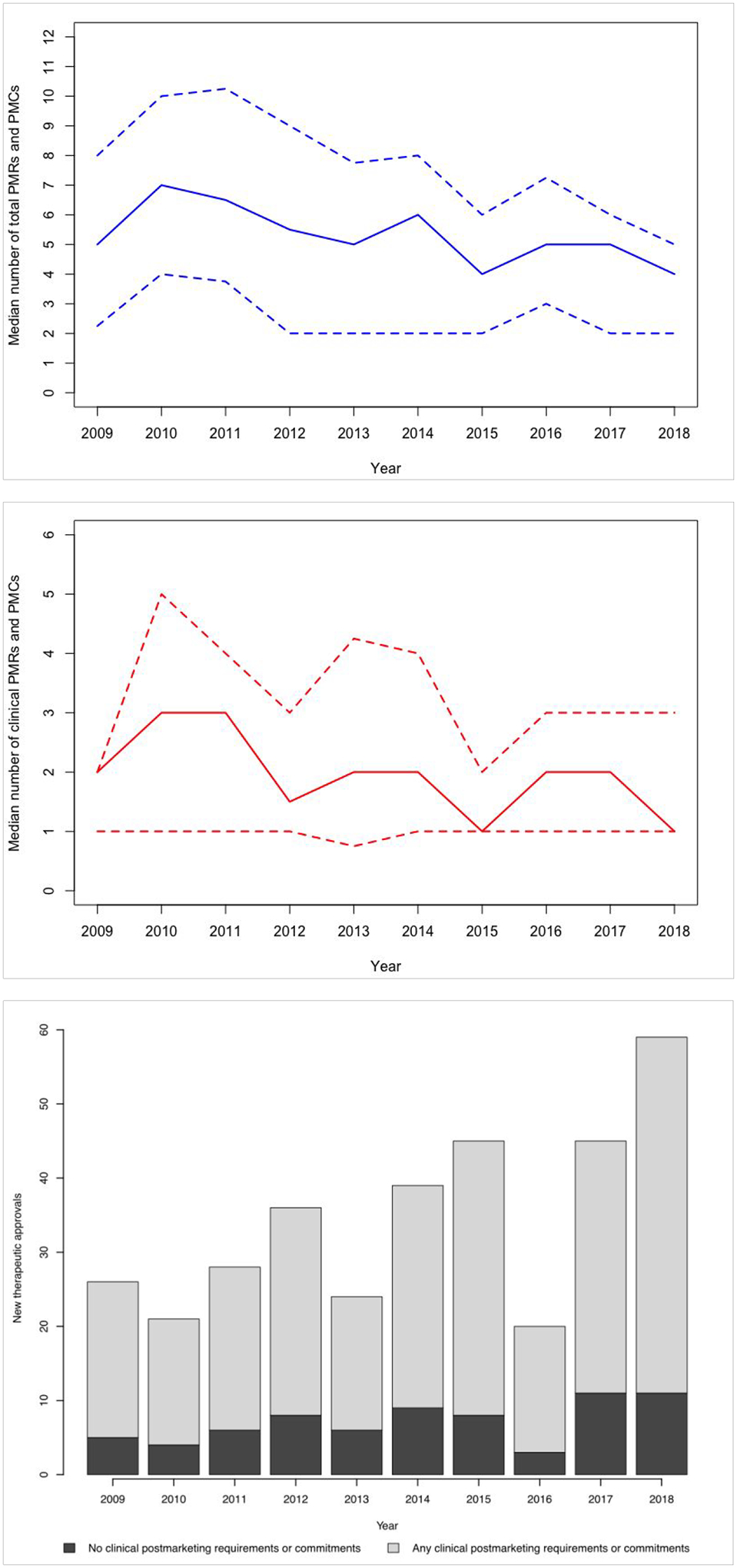
(a) Median (solid line; interquartile range between dashed lines) total number of postmarketing requirements and commitments outlined for new therapeutics, 2009–2018. (b) Median (solid line; interquartile range between dashed lines) number of clinical postmarketing requirements and commitments outlined for new therapeutics, 2009–2018. (c) Number of new therapeutics approved by US Food and Drug Administration, 2009–2018, with and without clinical postmarketing requirements and commitments.
Characteristics of clinical studies outlined in postmarketing requirements and commitments
Most clinical postmarketing requirements and commitments outlined safety endpoints to be evaluated, either with (314/750, 41.9%) or without (330/750, 44.0%) additional efficacy endpoints (Table 3). Efficacy endpoints were outlined in 420 of 750 (56.0%) clinical postmarketing requirements and commitments. Among 330 clinical postmarketing requirements and commitments specifying only safety endpoints, over three-fourths were issued under the FDA Amendments Act (258/330, 78.2%). However, out of the 331 postmarketing requirements issued under this authority, one-fifth included an efficacy endpoint (73/331, 22.1%; n=8 with only efficacy endpoints and n=65 with both efficacy and safety endpoints).
Table 3.
Characteristics of clinical postmarketing requirements and commitments for new therapeutics receiving original Food and Drug Administration approval, 2009–2018.
| PMR or PMC authority, No. (%) | Total clinical PMRs or PMCsb | PMR/PMC study indication | Study design | Study objective | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Original indication | Modified indication | New indication | Uncleard | Trial | Observational | Safety | Efficacy | Safety and Efficacy | ||||
| General population | Subgroupc | |||||||||||
| Total | 750 | 150 (20.0) | 305 (40.7) | 123 (16.4) | 61 (8.1) | 111 (14.8) | 576 (76.8) | 174 (23.2) | 330 (44.0) | 106 (14.1) | 314 (41.9) | |
| PMR | FDAAA | 331 | 94 (28.4) | 61 (18.4) | 48 (14.5) | 23 (6.9) | 105 (31.7) | 173 (52.3) | 158 (47.7) | 258 (77.9) | 8 (2.4) | 65 (19.6) |
| PREA | 207 | 0 (0) | 166 (80.2) | 19 (9.2) | 18 (8.7) | 4 (1.9) | 207 (100) | 0 (0) | 64 (30.9) | 3 (1.4) | 140 (67.6) | |
| AA | 59 | 19 (32.2) | 10 (16.9) | 24 (40.7) | 6 (10.2) | 0 (0) | 58 (98.3) | 1 (1.7) | 1 (1.7) | 34 (57.6) | 24 (40.7) | |
| AER | 3 | 3 (100.0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (100.0) | 0 (0) | 0 (0) | 3 (100.0) | |
| PMC | 506Ba | 141 | 34 (24.1) | 66 (46.8) | 29 (20.6) | 10 (7.1) | 2 (1.4) | 129 (91.5) | 12 (8.5) | 7 (5.0) | 58 (41.1) | 76 (53.9) |
| Non-506Ba | 9 | 0 (0) | 2 (22.2) | 3 (33.3) | 4 (44.4) | 0 (0) | 9 (100.0) | 0 (0) | 0 (0) | 3 (33.3) | 6 (66.7) | |
PMR: postmarketing requirement; PMC: postmarketing commitment; FDAAA: Food and Drug Administration Amendments Act; PREA: Pediatric Research Equity Act; AA: Accelerated Approval; AER: Animal Efficacy Rule.
“506B” refers to postmarketing commitments subject to reporting requirements under section 506B of the Federal Food, Drug, and Cosmetic Act. Postmarketing commitments not subject to this rule are denoted as “Non-506B.”
“Clinical PMRs or PMCs” include those outlining new prospective cohort studies, registries, or clinical trials; completion or submission of results from ongoing prospective clinical studies; new retrospective observational studies; or new analysis, follow-up, or flexible analyses of clinical studies.
Postmarketing requirements and commitments generating evidence on subgroups of the original indication may evaluate demographic subgroups, clinical subgroups, or both.
“Unclear” indications were those for which a defined disease population could not be determined from postmarketing requirement or commitment description and, if identified, corresponding study registration on ClinicalTrials.gov.
The majority of the 750 clinical postmarketing requirements and commitments were intended to generate evidence for original approved indications, either for the general disease population (150/750, 20.0%) or for a clinical and/or demographic subgroup (305/750, 40.7%) (Table 2). However, nearly one-quarter (184/750, 24.5%) of clinical postmarketing requirements and commitments described studies of unapproved indications, including 123 (123/184, 66.8%) evaluating modified indications expanding therapeutic uses within original disease populations (Table 4). An additional 61 (61/184, 33.2%) clinical postmarketing requirements and commitments evaluated therapeutic uses for unapproved diseases unrelated to original indications, including approximately one-third (21/61, 34.4%) generating preliminary safety and/or efficacy data in patients with nonspecific diagnoses such as “solid tumors” or “bacterial infections.” For all years from 2009 to 2018, at least one-half of therapeutics were approved with a postmarketing requirement or commitment describing a study of their original approved indication. In every year of the study period, a greater proportion of therapeutics with at least one expedited review program designation, as compared to therapeutics with no designations, were approved with a postmarketing requirement or commitment evaluating a modified indication. Postmarketing requirements or commitments evaluating a new indication were more often outlined for therapeutics with at least one expedited review program designation for all but two years from 2009 to 2018 (eFigure 1). Clinical postmarketing requirements and commitments for modified or new indications most often outlined new prospective clinical studies (103/184, 56.0%), of which nearly one-quarter (24/103, 23.3%) investigated therapeutic uses unrelated to original indications. For clinical postmarketing requirements issued under the accelerated approval authority, 30 of 59 (50.8%) outlined clinical studies generating evidence on modified or new therapeutic indications (Table 3).
Table 4.
Clinical postmarketing requirements and commitments investigating modified or new therapeutic indications.
| Study type, No. (%) | Clinical PMRs and PMCs investigating modified or new indications | Clinical PMRs and PMCs investigating modified indications | Clinical PMRs and PMCs investigating new indicationsa | ||
|---|---|---|---|---|---|
| Total | New therapeutic useb | Clinical safety and efficacy datab | |||
| Total | 184 | 123 (66.8) | 61 (33.2) | 40 (65.6) | 21 (34.4) |
| New prospective cohort study, registry, or clinical trial | 103 | 65 (63.1) | 38 (36.9) | 24 (63.2) | 14 (36.8) |
| Complete or submit results from prospective cohort study, registry, or clinical trial | 41 | 30 (73.2) | 11 (26.8) | 6 (54.5) | 5 (45.5) |
| New retrospective observational study | 2 | 1 (50.0) | 1 (50.0) | 1 (100.0) | 0 (0) |
| New analysis or follow-up for cohort study, registry, or clinical trial, or “flexible” requirements | 38 | 27 (71.1) | 11 (28.9) | 9 (81.8) | 2 (18.2) |
PMR: postmarketing requirement; PMC: postmarketing commitment.
Clinical postmarketing requirements and commitments were considered to investigate a new indication when the outlined study was to be conducted in a disease population not related to the original approved indication. This included studies of unapproved therapeutic uses in new disease populations as well as studies intended to generate basic clinical data in a nonspecific disease population (e.g., patients with “solid tumors” or “bacterial infections”).
Values in parentheses reflect percentages of clinical postmarketing requirements and commitments investigating new therapeutic indications.
Discussion
Among 1978 postmarketing requirements and commitments outlined for 343 therapeutics originally approved by FDA from 2009 to 2018, we found variation in the number, design, and characteristics of clinical studies they described. Just under 40% of all postmarketing requirements and commitments outlined clinical studies, and even fewer described new prospective cohort studies, registries, or clinical trials to be conducted in the postmarket period. Although the majority of new therapeutics were approved with at least 1 clinical postmarketing requirement or commitment, the median number of clinical postmarketing requirements and/or postmarketing commitments outlined for those therapeutics was 2, and was relatively consistent from 2009 to 2018. Clinical studies outlined in postmarketing requirements and commitments were frequently intended to generate safety and efficacy evidence for approved indications, but nearly one-quarter of clinical postmarketing requirements and commitments described studies with the potential to generate evidence on therapeutic uses not encompassed by their original approved indications. These findings suggest that a greater number of postmarketing requirements and commitments outlining clinical studies of approved indications may be needed to address evidentiary gaps and inform clinical decision making.
Despite FDA’s increasing emphasis on lifecycle evaluation,3 we did not identify a change in the number of clinical postmarketing requirements and commitments outlined for new therapeutics over the last decade, with fewer than one-quarter of postmarketing requirements and commitments outlining new prospective studies such as clinical trials. Between 2009 and 2018, the median number of clinical postmarketing requirements and commitments outlined at therapeutic approval remained 2 for therapeutics approved with at least 1 clinical postmarketing requirement or commitment and decreased non-significantly from 2 to 1 for therapeutics overall. This occurred in the context of therapeutic approvals increasingly being based on fewer pivotal trials,7 often using surrogate endpoints.29 Use of FDA’s expedited review programs is increasing,4 including the breakthrough therapy designation implemented in 2012, which supports the approval of new therapeutics considered promising on the basis of preliminary clinical evidence.6 These programs reduce the amount of clinical evidence available for new therapeutics at approval,30 suggesting an expanded role for postmarketing requirements and commitments in outlining clinical studies designed to address evidentiary gaps for approved indications. However, the minority of postmarketing requirements and commitments outlined in the previous decade are for new prospective clinical studies, which are most likely to inform clinicians’ use of new therapeutics. With the exception of accelerated approval, therapeutics with FDA expedited review program designations were not more likely to be approved with a clinical postmarketing requirement or commitment than those with no designations. Together, these findings suggest that clinical postmarketing requirements and commitments may not fully compensate for decreasing numbers of premarket clinical trials for new therapeutics. Expanded use of postmarketing requirements and commitments to outline prospective clinical studies may represent the most effective approach to supplement decreasing numbers of premarket clinical trials and generate postmarket evidence to inform clinical decision making.
We found that clinical postmarketing requirements and commitments frequently were expected to address both safety and efficacy endpoints, possibly reflecting FDA’s vision for postmarketing requirements and commitments as flexible responses to clinical questions arising at the time of or following approval.31 However, nearly one-quarter of clinical postmarketing requirements and commitments focused on modified or unapproved therapeutic indications, including one-half of confirmatory postmarketing requirements for therapeutics receiving accelerated approval designation. This rate is comparable to that observed for postmarket clinical studies overall, which are primarily sponsored by industry,32 frequently evaluate new therapeutic uses,33,34 and may play a role in promoting medication use after approval.35 This has also been observed for therapeutics receiving accelerated approval designation, which are integrated into clinical practice, including new applications, without confirmation of clinical benefit for original indications.36 New prospective clinical studies investigating therapeutic uses not included in their original approved indications may reflect interest in generating evidence for anticipated off-label uses.37 For example, ofatumumab originally received FDA accelerated approval for the treatment of chronic lymphocytic leukemia refractory to alemtuzumab and fludarabine. However, a confirmatory postmarketing requirement outlined under the accelerated approval authority required completion of a clinical trial of ofatumumab in previously untreated patients with chronic lymphocytic leukemia, potentially reflecting FDA expectation of this use by clinicians despite the absence of confirmatory evidence of efficacy. For deferiprone, indicated for the treatment of transfusional iron overload in patients with thalassemia syndromes, a postmarketing requirement outlined under the accelerated approval authority investigated use in patients with sickle cell disease, for which current pharmacologic management is limited.38 While postmarketing requirements and commitments investigating novel indications may address clinician needs for the treatment of more severe or earlier stage disease, or even distinct diseases, they may not generate evidence to inform management of original approved indications.
Accelerating therapeutic approvals represent a tradeoff between the benefit to patients of faster access to novel therapies and the need for comprehensive evidence demonstrating safety and efficacy.39 Following therapeutic approval, adverse event reports, electronic health records, and insurance claims allow FDA to monitor therapeutic use and identify safety signals requiring communication to the public or regulatory action.40,41 However, there are shortcomings to real world data sources.42,43 A report by the U.S. Office of the Inspector General noted that postmarketing requirements frequently result in labeling changes and other actions by FDA to support therapeutic safety, suggesting their importance for generating evidence of value to clinical practice.44 Postmarketing requirements and commitments enable FDA to target evidentiary shortcomings for new therapeutics. For example, section 505(o) of the FDA Amendments Act empowers FDA to require sponsors to conduct clinical trials or develop patient registries, such as therapeutic exposure during pregnancy, to assess known or suspected risks of therapeutic use and inform clinical decision making. Postmarketing commitments represent voluntary studies designed in collaboration with sponsors and can include both clinical and non-clinical investigations. Opportunities exist to use postmarketing requirements and commitments in coordination with real world evidence to refine assessments of therapeutic safety and efficacy and support FDA’s transition to lifecycle evaluation, such as through the conduct of pragmatic clinical trials. FDA has an opportunity to outline a greater number of new prospective clinical studies in postmarketing requirements and commitments for new therapeutics, advancing our understanding of their optimal uses as they are integrated into clinical practice. While postmarketing requirements and commitments can be used to investigate expanded or new therapeutic uses, these should not replace studies generating clinical evidence for original indications.
Limitations
Our study has several limitations. First, we abstracted postmarketing requirements and commitments based on descriptions in original approval letters, which are sometimes too brief to comprehensively characterize study indications, endpoints, or design elements such as enrollment or trial duration.18 Although we used study registrations on ClinicalTrials.gov to supplement postmarketing requirement and commitment descriptions, these were nearly always, but not universally, available. Because we focused on postmarketing requirements and commitments describing studies generating safety or efficacy evidence, it is possible that some of those categorized as non-clinical may have been intended to inform clinical practice, such as a requirement for a pharmacokinetic study with the potential to support therapeutic dosage modifications. However, the brief descriptions available for many postmarketing requirements and commitments prevent further characterization of non-clinical studies.18,19 Second, we characterized indications based on publicly available data, and therefore may not have captured FDA’s objectives for some postmarketing requirements and commitments. It is possible that postmarketing requirements and commitments outlining clinical studies enrolling from broad populations are intended primarily to support lifecycle evaluation of original approved therapeutic indications. However, in the absence of additional information from FDA, postmarketing requirement and commitment descriptions represent the best public resource for characterizing these postmarket studies, which may also generate clinical evidence supporting new uses of therapeutics. Third, postmarketing requirements and commitments were abstracted by one author. However, an independent second author validated a 20% random sample of clinical postmarketing requirements and commitments. The agreement rate between authors was 98%, with minor disagreements resolved via consensus. Fourth, we did not evaluate the completion of or reporting of results from clinical postmarketing requirements and commitments, which previous studies have suggested takes place for only approximately 50% of clinical postmarketing requirements and commitments.18–20 Further analyses may provide additional information about how often postmarket clinical evidence becomes available to clinicians and patients. Lastly, we limited our analyses to postmarketing requirements and commitments from 10 years of new therapeutic approvals. Although postmarketing studies were outlined prior to 2008, the term “postmarketing study commitments” referred to both required and agreed-upon studies, making it difficult to differentiate postmarketing requirements from postmarketing commitments.10 Our sample represents, to our knowledge, the largest analysis of postmarketing requirements and commitments since terminology was standardized.
Conclusions
Among 343 therapeutics that received original FDA approval from 2009 to 2018, most were approved with at least 1 clinical postmarketing requirement or commitment. However, the median number of studies investigating therapeutic safety and/or efficacy was 2 per approval, fewer than one-quarter outlined new prospective clinical studies, and studies investigated both approved and unapproved indications. Given FDA’s commitment to expedited approval and increasing emphasis of lifecycle therapeutic evaluation, a greater number of postmarketing requirements and commitments outlining clinical studies of approved indications may be needed to address evidentiary gaps and inform clinical decision making.
Supplementary Material
Acknowledgments
Funding/support and role of the sponsor
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: J.D.W. is supported by the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health and under award K01AA028258. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest disclosure
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: In the past 36 months, J.D.W. received research support through the Collaboration for Research Integrity and Transparency from the Laura and John Arnold Foundation and through the Center for Excellence in Regulatory Science and Innovation (CERSI) at Yale University and the Mayo Clinic (grant no. U01FD005938). J.S.R. is a former Associate Editor of JAMA Internal Medicine, a current Research Editor at BMJ and received research support through Yale from Johnson and Johnson to develop methods of clinical trial data sharing, from Medtronic, Inc. and the Food and Drug Administration (FDA) to develop methods for postmarket surveillance of medical devices (grant no. U01FD004585), from the Centers of Medicare and Medicaid Services (CMS) to develop and maintain performance measures that are used for public reporting, from the FDA to establish a Center for Excellence in Regulatory Science and Innovation (CERSI) at Yale University and the Mayo Clinic (grant no. U01FD005938), from the Blue Cross Blue Shield Association to better under- stand medical technology evaluation, from the Agency for Healthcare Research and Quality (grant no. R01HS022882), and from the Laura and John Arnold Foundation. S.S.D. received support as a Scholar in the Yale University/Mayo Clinic FDA CERSI, from the Greenwall Foundation, from the National Institutes of Health/National Heart, Lung, and Blood Institute (grant no. K12HL138046), from Arnold Ventures, from the US Food and Drug Administration, and from the National Evaluation System for health Technology Coordinating Center (NESTcc).
Footnotes
Data sharing: Data will be shared on osf.io upon publication.
References
- 1.Ross JS, Kesselheim AS. FDA Policy and Cardiovascular Medicine. Circulation 2015; 132: 1136–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.United States Food and Drug Administration (FDA). Guidance for Industry: Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products, https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072008.pdf (1998, accessed 29 June 2020).
- 3.Psaty BM, Meslin EM, Breckenridge A. A Lifecycle Approach to the Evaluation of FDA Approval Methods and Regulatory Actions: Opportunities Provided by a New IOM Report. JAMA 2012; 307: 2491–2492. [DOI] [PubMed] [Google Scholar]
- 4.Kesselheim AS, Wang B, Franklin JM, et al. Trends in utilization of FDA expedited drug development and approval programs, 1987–2014: cohort study. BMJ; 351:h4633. DOI: 10.1136/bmj.h4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liberti L, Bujar M, Breckenridge A, et al. FDA Facilitated Regulatory Pathways: Visualizing Their Characteristics, Development, and Authorization Timelines. Front Pharmacol; 8:161. DOI: 10.3389/fphar.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.United States Food and Drug Administration (FDA). Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics, https://www.fda.gov/media/86377/download (2014, accessed 29 June 2020).
- 7.Zhang AD, Puthumana J, Downing NS, et al. Assessment of Clinical Trials Supporting US Food and Drug Administration Approval of Novel Therapeutic Agents, 1995–2017. JAMA Netw Open 2020; 3: e203284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Downing NS, Aminawung JA, Shah ND, et al. Clinical Trial Evidence Supporting FDA Approval of Novel Therapeutic Agents, 2005–2012. JAMA 2014; 311: 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.United States Food and Drug Administration (FDA). Postmarketing Requirements and Commitments: Introduction, https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/post-marketingphaseivcommitments/ (accessed 29 June 2020).
- 10.United States Food and Drug Administration (FDA). SOPP 8415: Procedures for Developing Postmarketing Requirements and Commitments, https://www.fda.gov/media/90591/download (2019, accessed 29 June 2020).
- 11.Wallach JD, Ross JS, Naci H. The US Food and Drug Administration’s expedited approval programs: Evidentiary standards, regulatory trade-offs, and potential improvements. Clin Trials 2018; 15: 219–229. [DOI] [PubMed] [Google Scholar]
- 12.Wallach JD, Ross JS, Naci H. The US Food and Drug Administration’s expedited approval programs: Addressing premarket flexibility with enhanced postmarket evidence generation. Clin Trials 2018; 15: 243–246. [DOI] [PubMed] [Google Scholar]
- 13.Herder M Pharmaceutical Drugs of Uncertain Value, Lifecycle Regulation at the US Food and Drug Administration, and Institutional Incumbency. Milbank Q 2019; 97: 820–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skydel JJ, Luxkaranayagam AT, Dhruva SS, et al. Analysis of Postapproval Clinical Trials of Therapeutics Approved by the US Food and Drug Administration Without Clinical Postmarketing Requirements or Commitments. JAMA Netw Open 2019; 2: e193410–e193410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berndt ER, Cockburn IM, Grépin KA. The impact of incremental innovation in biopharmaceuticals: drug utilisation in original and supplemental indications. PharmacoEconomics 2006; 24 Suppl 2: 69–86. [DOI] [PubMed] [Google Scholar]
- 16.DiMasi JA. Innovating by developing new uses of already-approved drugs: trends in the marketing approval of supplemental indications. Clin Ther 2013; 35: 808–818. [DOI] [PubMed] [Google Scholar]
- 17.Federico CA, Wang T, Doussau A, et al. Assessment of Pregabalin Postapproval Trials and the Suggestion of Efficacy for New Indications: A Systematic Review. JAMA Intern Med 2019; 179: 90–97. [DOI] [PubMed] [Google Scholar]
- 18.Wallach JD, Egilman AC, Dhruva SS, et al. Postmarket studies required by the US Food and Drug Administration for new drugs and biologics approved between 2009 and 2012: cross sectional analysis. BMJ 2018; 361: k2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wallach JD, Luxkaranayagam AT, Dhruva SS, et al. Postmarketing commitments for novel drugs and biologics approved by the US Food and Drug Administration: a cross-sectional analysis. BMC Med 2019; 17: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woloshin S, Schwartz LM, White B, et al. The Fate of FDA Postapproval Studies. N Engl J Med 2017; 377: 1114–1117. [DOI] [PubMed] [Google Scholar]
- 21.Fain K, Daubresse M, Alexander GC. The Food and Drug Administration Amendments Act and Postmarketing Commitments. JAMA 2013; 310: 202–204. [DOI] [PubMed] [Google Scholar]
- 22.Wallach JD, Egilman AC, Ross JS, et al. Timeliness of Postmarket Studies for New Pharmaceuticals Approved Between 2009 and 2012: a Cross-Sectional Analysis. J Gen Intern Med 2019; 34(4):492–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.United States Food and Drug Administration (FDA). Drugs@FDA: FDA Approved Drug Products. Drugs@FDA, https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed 29 June 2020).
- 24.United States Food and Drug Administration (FDA). New Drugs at FDA: CDER’s New Molecular Entities and New Therapeutic Biological Products. FDA, http://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (2020, accessed 29 June 2020).
- 25.Hughes B 2009 FDA drug approvals. Nat Rev Drug Discov 2010; 9: 89–92. [DOI] [PubMed] [Google Scholar]
- 26.Mullard A 2010 FDA drug approvals. Nat Rev Drug Discov 2011; 10: 82–85. [DOI] [PubMed] [Google Scholar]
- 27.World Health Organization (WHO) Collaborating Center for Drug Statistics Methodology. WHOCC - ATC/DDD Index, https://www.whocc.no/atc_ddd_index/ (2019, accessed 29 June 2020).
- 28.United States Food and Drug Administration (FDA). Guidance for Industry: How to Comply with the Pediatric Research Equity Act, https://www.fda.gov/media/72274/download (2005, accessed 5 February 2021).
- 29.Pease AM, Krumholz HM, Downing NS, et al. Postapproval studies of drugs initially approved by the FDA on the basis of limited evidence: systematic review. BMJ 2017; 357: j1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puthumana J, Wallach JD, Ross JS. Clinical Trial Evidence Supporting FDA Approval of Drugs Granted Breakthrough Therapy Designation. JAMA 2018; 320: 301–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.United States Food and Drug Administration (FDA). Postmarketing Studies and Clinical Trials—Implementation of Section 505(o)(3) of the Federal Food, Drug, and Cosmetic Act Guidance for Industry, http://www.fda.gov/regulatory-information/search-fda-guidance-documents/postmarketing-studies-and-clinical-trials-implementation-section-505o3-federal-food-drug-and-0 (2019, accessed 29 June 2020).
- 32.Zeitoun J-D, Ross JS, Atal I, et al. Factors Associated With Postmarketing Research for Approved Indications for Novel Medicines Approved by Both the FDA and EMA Between 2005 and 2010: A Multivariable Analysis. Clin Pharmacol Ther 2018; 104: 1000–1007. [DOI] [PubMed] [Google Scholar]
- 33.Zeitoun J-D, Ross JS, Atal I, et al. Postmarketing studies for novel drugs approved by both the FDA and EMA between 2005 and 2010: a cross-sectional study. BMJ Open 2017; 7: e018587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeitoun J-D, Baron G, Vivot A, et al. Post-marketing research and its outcome for novel anticancer agents approved by both the FDA and EMA between 2005 and 2010: A cross-sectional study. Int J Cancer 2018; 142: 414–423. [DOI] [PubMed] [Google Scholar]
- 35.Koch C, Schleeff J, Techen F, et al. Impact of physicians’ participation in non-interventional postmarketing studies on their prescription habits: A retrospective 2-armed cohort study in Germany. PLOS Med 2020; 17: e1003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naci H, Wouters OJ, Gupta R, et al. Timing and Characteristics of Cumulative Evidence Available on Novel Therapeutic Agents Receiving Food and Drug Administration Accelerated Approval. Milbank Q 2017; 95: 261–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dal Pan GJ. Monitoring the Safety of Medicines Used Off-Label. Clin Pharmacol Ther 2012; 91: 787–795. [DOI] [PubMed] [Google Scholar]
- 38.Ware RE, de Montalembert M, Tshilolo L, et al. Sickle cell disease. The Lancet 2017; 390: 311–323. [DOI] [PubMed] [Google Scholar]
- 39.Jena AB, Zhang J, Lakdawalla DN. The Trade-off Between Speed and Safety in Drug Approvals. JAMA Oncol 2017; 3: 1465–1466. [DOI] [PubMed] [Google Scholar]
- 40.United States Food and Drug Administration (FDA). Best Practices in Drug and Biological Product Postmarket Safety Surveillance for FDA Staff, https://www.fda.gov/media/130216/download (2019, accessed 29 June 2020).
- 41.Robb MA, Racoosin JA, Sherman RE, et al. The US Food and Drug Administration’s Sentinel Initiative: Expanding the horizons of medical product safety. Pharmacoepidemiol Drug Saf 2012; 21: 9–11. [DOI] [PubMed] [Google Scholar]
- 42.Moore TJ, Furberg CD. Electronic Health Data for Postmarket Surveillance: A Vision Not Realized. Drug Saf 2015; 38: 601–610. [DOI] [PubMed] [Google Scholar]
- 43.Moses C, Celi LA, Marshall J. Pharmacovigilance: An Active Surveillance System to Proactively Identify Risks for Adverse Events. Popul Health Manag 2013; 16: 147–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.United States Department of Health and Human Services Office of Inspector General. FDA is Issuing More Postmarketing Requirements, but Challenges with Oversight Persist Report, https://oig.hhs.gov/oei/reports/oei-01-14-00390.asp (2016, accessed 29 June 2020).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.