

Abstract
Pancreatic cancer metastasis is a leading cause of cancer-related deaths, yet very little is understood regarding the underlying biology. As a result, targeted therapies to inhibit metastasis are lacking. Here, we report that the parathyroid hormone-related protein (PTHrP encoded by PTHLH) is frequently amplified as part of the KRAS amplicon in pancreatic cancer patients. PTHrP upregulation drives the growth of both primary and metastatic tumors in mice and is highly enriched in PDAC metastases. Loss of PTHrP – either genetically or pharmacologically – dramatically reduces tumor burden, eliminates metastasis, and enhances overall survival. These effects are mediated in part through a reduction in epithelial-to-mesenchymal transition, which reduces the tumor cells’ ability to initiate the metastatic cascade. Spp1, which encodes Osteopontin, is revealed to be a downstream effector of PTHrP. Our results establish a new paradigm in pancreatic cancer whereby PTHrP is a driver of disease progression and emerges as a novel therapeutic vulnerability.
Keywords: Pancreatic cancer, PTHrP, PTHLH, EMT, metastasis
INTRODUCTION
The overwhelming majority of pancreatic ductal adenocarcinoma (PDAC) patients present with metastases, and nearly all will succumb to disease within one year of diagnosis [1]. A limited understanding of the biological processes underlying metastasis has hampered the development of customized therapeutic interventions, thereby resulting in pancreatic cancer to evolve into the second leading cause of cancer-related deaths [2]. In recent years, genomic and transcriptomic characterization of PDAC has revealed that this is not a singular disease, but rather comprised of subtypes with distinct molecular aberrations and characteristics [3–5]. However, defining pathways that are activated within these subtypes to drive metastasis and implementing targeted therapies to disrupt these pathways remains elusive.
Transcriptomic profiling of PDAC has yielded its classification into two major subtypes: (1) classical/progenitor and (2) squamous/quasi-mesenchymal/basal-like, with the latter exhibiting higher tumor grade, enhanced epithelial-to-mesenchymal transition (EMT)/invasion and decreased survival [6]. The squamous subtype has been defined further by lineage-specific markers, namely TP63, PTHLH (parathyroid hormone like hormone also known as PTHrP), KRT5, and KRT6A. The transcription factor TP63 may constitute a master regulator of the other squamous lineage-specific genes [7]. Whole-genome sequencing has established that the squamous subtype is annotated by KRAS genomic amplification [8], which is significantly enriched in metastatic tumors relative to primary tumors [9]. Interestingly, co-amplification occurs in nearby genes within the KRAS amplicon, including PTHLH. Our own work, using genetically engineered mouse models of PDAC, has established that Pthlh is one of the top five most upregulated genes in a highly metastatic subset of cells that have undergone EMT [10]. We therefore hypothesized that copy number gains in the KRAS-PTHLH loci drive tumors towards a highly invasive squamous subtype that is marked by mesenchymal characteristics, potentially due to enhanced PTHLH expression and function.
PTHrP was described initially as a secreted factor produced by tumors that leads to paraneoplastic hypercalcemia [11]. It has since been demonstrated to be involved in multiple cellular processes, including regulation of intracellular calcium, proliferation/hypertrophy and differentiation [reviewed in 12, 13]. These diverse functions are likely due to the processing of the mature PTHrP protein into at least three active peptides that are thought to act independently through autocrine, paracrine, and intracrine routes [12, 13]. PTHrP isoforms are produced and secreted by malignant cells, including moieties with intact N-terminus as well as mid-region fragments. The most widely studied N-terminal peptide, PTHrP 1–34, binds to the Parathyroid Hormone Receptor 1 (PTHR1) in an autocrine/paracrine manner to activate downstream effectors such as ERK, PKA PKC, AKT, and Cyclin D1 as well as RUNX and CREB transcription factors [14–18]. Upstream drivers of Pthlh expression include members of the TGF-β superfamily that signal through the SMAD transcription factors [19, 20], the RAS family through the ETS transcription factors [21–24], and mitogenic stimuli such as EGF and IGF-1 [23–25]. Additionally, recent evidence has revealed that TP63 is a major driver of PTHLH expression in the squamous subtype of PDAC [7]. PTHrP has been described as oncogenic and fostering bone metastasis in lung, breast, head and neck, lymphoma and colon cancers [14, 18, 24, 26–29]. Paradoxically, some studies have concluded that PTHLH has a tumor suppressive role in breast cancer and skeletal metastases [30, 31], suggesting an incomplete understanding of the role that PTHrP plays during tumorigenesis. Furthermore, how PTHLH functions in vivo to modulate pancreatic cancer progression and metastasis has yet to be explored.
In the current study, we establish Pthlh as a bona fide oncogene and a novel metastasis driver in pancreatic cancer. To that end, we use genetic deletion and complementary pharmacological inhibition of PTHrP to demonstrate that its loss significantly delays primary tumor growth, reduces metastatic colonization and increases overall survival in mice. We nominate anti-PTHrP therapy as a viable candidate for the transition from preclinical models to clinical trials in PDAC.
RESULTS
PTHLH is amplified in a subset of pancreatic cancer patients
To better understand the extent to which known KRAS amplifications in PDAC also encompass PTHLH (which are separated by 2.7MB on human chromosome 12 and 2.0MB on murine chromosome 6; Supplemental Figure 1A), we first evaluated all copy number alterations for KRAS and PTHLH in PDAC patients from The Cancer Genome Atlas (TCGA). A Spearman correlation coefficient of R=0.97 (p = 8.96e−107) was observed between KRAS and PTHLH copy number (Figure 1A). Additionally, treatment of two independent murine PDAC tumor cell lines with MEK1/2 (U0126) and ERK1/2 (SCH772984) inhibitors decreased Pthlh expression (Supplemental Figure 1B–C), demonstrating that Pthlh is transcriptionally activated by RAS signaling. Further analysis of the TCGA indicated that ~25% of patients exhibited PTHLH copy number gains (Figure 1B). PTHLH was shown, in an independent patient cohort [32], to have copy number gains in nearly one-third of PDAC patients (Figure 1B). A pan-cancer analysis of the Cancer Cell Line Encyclopedia (CCLE) database showed similar PTHLH and KRAS copy number gains in other cancers (Supplemental Figure 1D). Of all CCLE cell lines, pancreatic cancers harbored the second highest PTHLH amplifications, behind only bile duct carcinomas (Figure 1C and Supplemental Figure 1E). We demonstrated that copy number gains/amplifications functionally led to increased PTHLH mRNA expression in both the TCGA and CCLE datasets (Supplemental Figure 1F–G). Additionally, we confirmed PTHLH amplification in human PDAC cell lines through fluorescence in situ hybridization (FISH), revealing multiple copies of PTHLH per cell, whereas the normal human pancreatic ductal epithelial (HPDE) cell line had the expected 2 copies per cell (Figure 1D). Importantly, Kaplan-Meier survival analysis of the TCGA-Pancreatic Adenocarcinoma (PAAD) cohort showed that patients with PTHLH copy number gains correlated with decreased survival relative to those with fewer than 2 copies (Figure 1E). Patients with >2 copies of PTHLH died 3 months earlier than those with ≤2 copies (12.1 months and 15.3 months respectively; Supplemental Figure 1H). Additionally, increased PTHLH mRNA expression in the same cohort correlated with reduced survival (Figure 1F). Finally, immunohistochemical (IHC) staining of a Tumor Microarray (TMA) of PDAC patients showed that patients with high PTHLH protein expression correlated with decreased survival (Figures 1G–H). Collectively, these data show that amplification of the KRAS locus in PDAC has concurrent PTHLH co-amplification, thereby setting the stage to pursue mechanistic studies of how PTHrP might be driving PDAC progression and/or metastasis.
Figure 1.
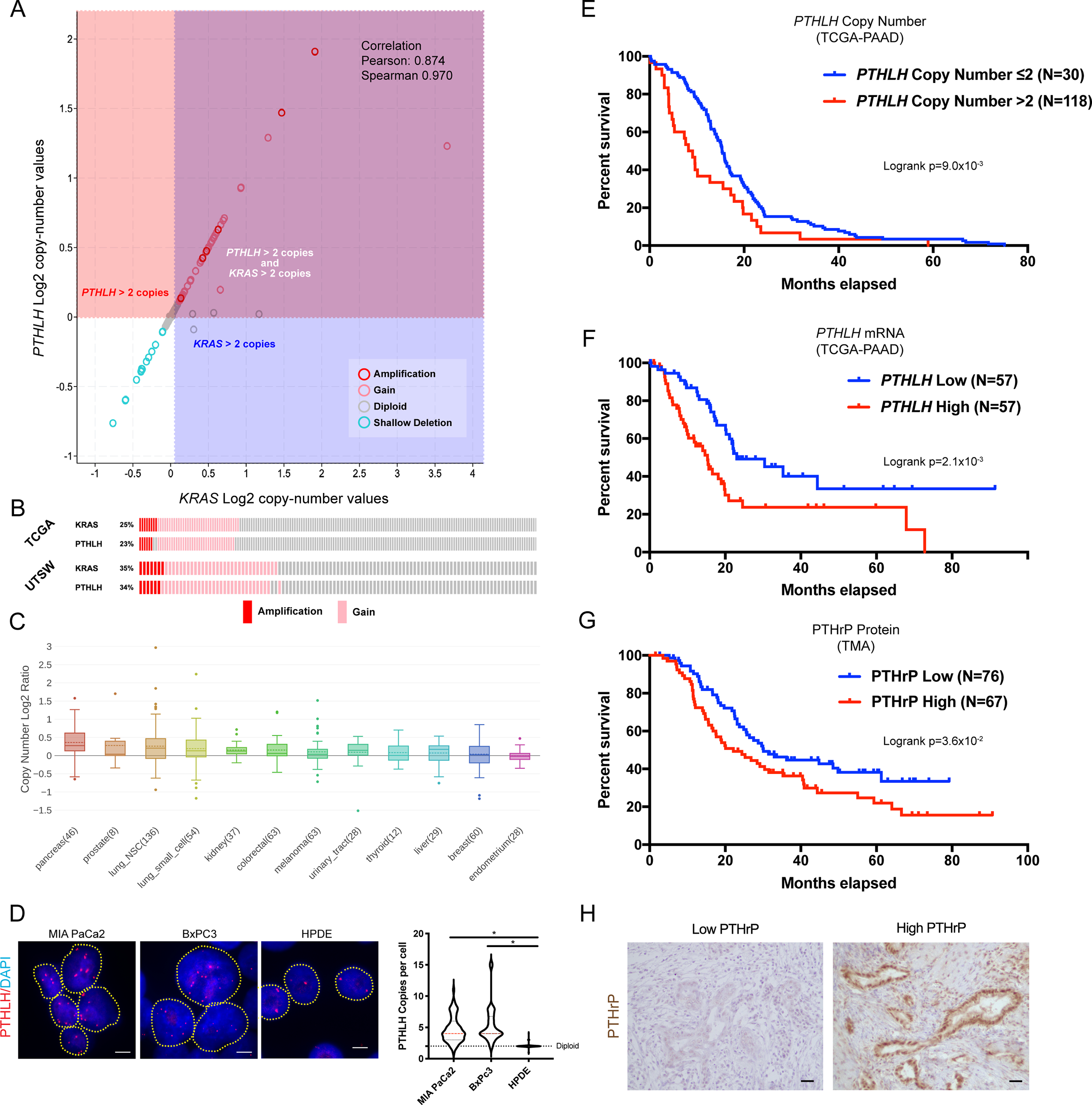
(A) PTHLH and KRAS Log2 copy-number values from TCGA-PAAD cBioportal. Each circle indicates one patient.
(B) Oncoprints of copy number gains and amplifications for PTHLH or KRAS from TCGA-PAAD or UTSW datasets.
(C) Box and whisker plot of PTHLH copy number Log2 Ratio distribution of common cancer cell lines in the CCLE database. Number of samples per cohort is indicated in parentheses.
(D) Representative FISH images of nuclei stained for PTHLH (red) and DAPI (blue) in human pancreatic cancer and normal ductal epithelial cell lines. Each red spot is one copy of PTHLH DNA. Yellow dotted lines indicate individual nuclei. Violin plots depict the number of PTHLH copies per nuclei in the indicated cell lines. Scale bars = 5μm. Statistical analysis by Mann-Whitney U test with significance indicated (*, p<0.05; N=50–100 nuclei per cell line).
(E) Kaplan-Meier overall survival plots stratified by PTHLH copy number gains (red) or PTHLH diploid (blue) status from TCGA-PAAD. Logrank p=9.0×10−3.
(F) Kaplan-Meier overall survival plots stratified by high PTHLH (red) or low PTHLH (blue) mRNA expression from TCGA-PAAD. Logrank p=2.1×10−3.
(G-H) (G) Kaplan-Meier overall survival plots stratified by high PTHrP (red) or low PTHrP (blue) protein expression from a PDAC patient TMA. Logrank p=3.6×10−2. (H) Representative IHC images of low PTHrP or high PTHrP staining in PDAC patient tissue. Scale bars = 100μm.
PTHLH is enriched in metastatic pancreatic cancer patients
PDAC samples in the TCGA are primarily non-metastatic (AJCC TMN score of M0) or metastatic burden not classified (Mx), thereby making it difficult to evaluate for any potential correlation between PTHrP expression and proclivity for metastasis. The recently launched COMPASS trial (NCT02750657), which recruited advanced stage PDAC patients for genomic analysis, has allowed a prism through which to view the genomic landscape of stage IV PDAC patients [8]. Whole genome sequencing and transcriptomic RNA-seq profiling of laser capture microdissected (LCM) tumor epithelia indicated that PTHLH is one of the top 20 genes used to classify a basal-like (analogous to squamous/quasi-mesenchymal discussed above) transcriptional signature, along with the squamous lineage genes TP63, KRT5 and KRT6A [8]. Furthermore, copy number analysis in this cohort demonstrates that PTHLH gains correlate with decreased survival (Figure 2A). Additionally, nearly one-third of patients with metastatic PDAC lesions show PTHLH copy number gains (Figure 2B). Finally, PTHLH gains were strongly enriched in the basal-like signature (48% of basal-like patients exhibited PTHLH copy number gains) relative to the classical signature (Figure 2C). These analyses show that PTHrP may be a potential driver of the invasive and highly metastatic basal subtype. To investigate this further in independent cohorts, we evaluated PTHrP mRNA expression in the publicly available dataset from Bailey et al. [3], which revealed that PTHLH mRNA was significantly enriched in the squamous subset relative to other subtypes (Figure 2D). Additionally, PTHLH expression was highest in the quasi-mesenchymal subset of the PDAC dataset established by Collisson et al. [4] (Figure 2E). PTHLH was higher in the basal subset relative to classical dataset generated by Moffitt et al. [5] (Figure 2F). Publicly available ChIP-seq of TP63 in the squamous subtype cell line BxPC3 [7] revealed that TP63 binds to the promoter of PTHLH (Supplemental Figure 2A). Thus, PTHrP is consistently highly expressed in the squamous/quasi-mesenchymal/basal-like subsets identified by others as being the most aggressive and least differentiated tumor subtype in PDAC.
Figure 2.
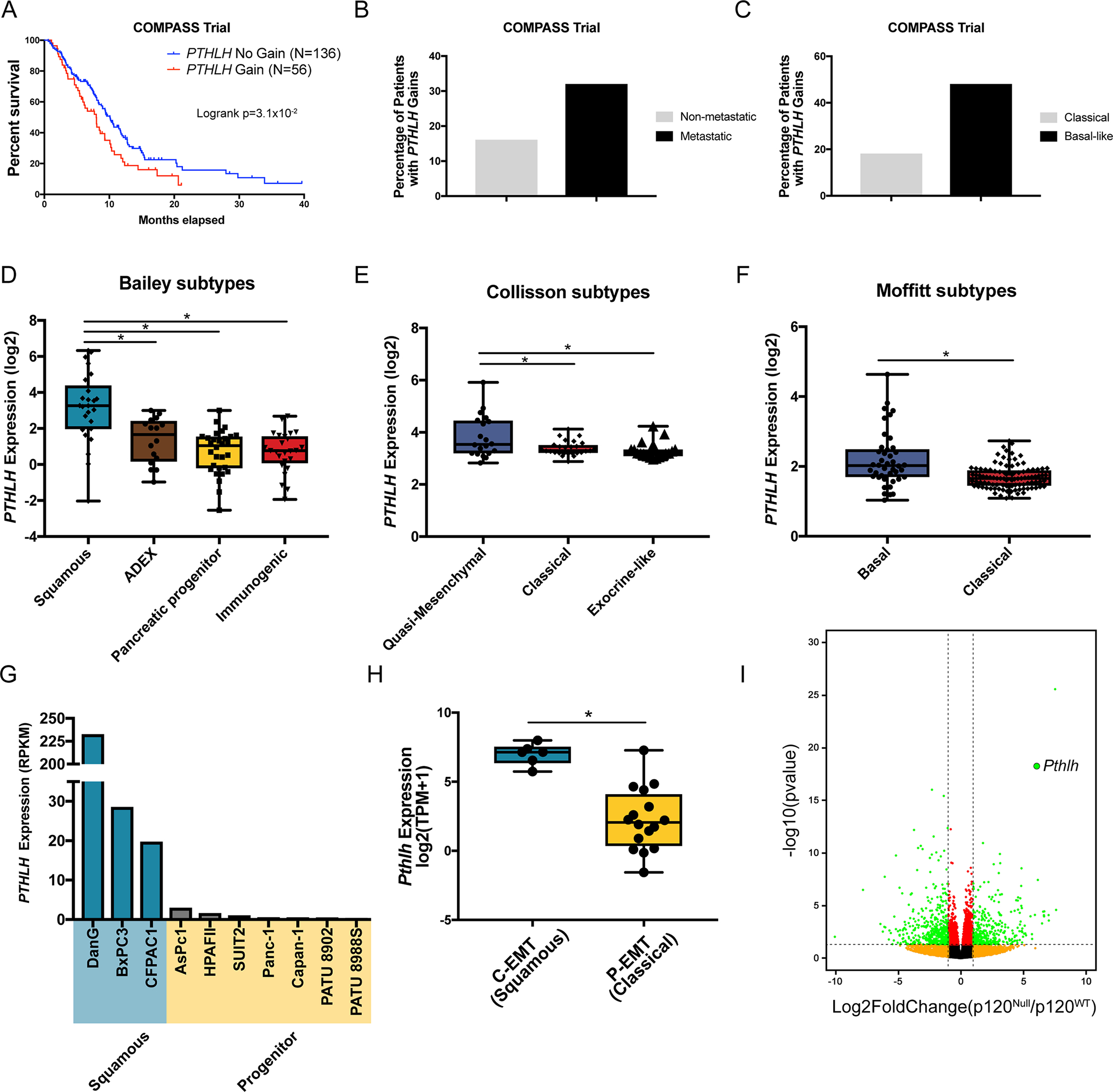
(A) Kaplan-Meier overall survival plots stratified by PTHLH copy number gains (red) or PTHLH diploid (blue) from the COMPASS trial cohort of PDAC patients. Logrank p=3.1×10−2.
(B) Percentage of patients in the COMPASS trial that have PTHLH copy number gains segregated into non-metastatic and metastatic patients.
(C) Percentage of patients in the COMPASS trial that have PTHLH copy number gains segregated into Classical and Basal-like PDAC subtypes.
(D-F) Box and whisker plots showing quantification of PTHLH mRNA expression in PDAC subtypes identified by (D) Bailey et al. [3], (E) Collisson et al. [4], and (F) Moffitt et al. [5] Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05).
(G) PTHLH mRNA expression (RPKM) in a panel of PDAC CCLE cell lines that are Squamous (blue) or Progenitor (yellow) subtypes.
(H) Box and whisker plot showing quantification of PTHLH mRNA expression in EMT subtypes identified by Aiello et al [34]. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05).
(I) Volcano plot of differentially expressed genes in p120ctn−/− versus p120ctn+/+ identified by Reichert et al [10]. Green dots passed p-value <0.05 and fold change >2 cut-offs. Pthlh is enlarged and highlighted.
Next, we found that human squamous subtype PDAC cell lines (DanG, BxPC3, and CFPACI) [7, 33] harbored >100-fold higher PTHLH mRNA expression than in PDAC cell lines of the non-squamous subtypes (Figure 2G). Analysis of RNA-seq from murine PDAC tumor cell lines classified according to an EMT subtype (complete EMT “c-EMT” versus partial EMT “p-EMT”) [34] segregates them into squamous and classical subtypes. Pthlh expression is increased significantly in squamous/c-EMT cell lines (Figure 2H). Finally, we have demonstrated previously that P120CTN loss in the pancreata of mice results in a highly aggressive and metastatic phenotype [10]. These highly mesenchymal and metastatic KPCY-p120ctn−/− tumor cells have >50-fold increased Pthlh expression relative to the KPCY-p120ctn+/+ tumor cells (Figure 2I). In fact, differential gene expression analysis of these cells indicates that Pthlh was the 2nd most up-regulated gene in the highly metastatic KPCY-p120ctn−/− cells (Figure 2I, p = 5.49e-19). Taken together, PTHrP is highly expressed in the most aggressive and metastatic subsets of human and mouse PDAC cells.
Pthlh is induced during tumorigenesis and its loss retards tumor growth
To determine the stage in which PTHrP expression is induced during PDAC progression, we evaluated PTHrP IHC of pancreatic tissues from wildtype (WT), LSL-KrasG12D; Pdx1-Cre (KC) and LSL-KrasG12D; P53R172H; Pdx1-Cre (KPC) mice. Wildtype mice exhibited very little PTHrP expression, except in islet cells, consistent with previous work showing a role in beta cell proliferation (Figure 3A) [35, 36]. Pancreatic intraepithelial neoplasia (PanIN) cells and PDAC cells were positive for PTHrP expression (Figure 3A), consistent with a role for RAS/MAPK/ETS signaling as one driver of Pthlh expression. Quantification of PTHrP staining in pancreatic tissue with PanIN lesions (KC mice; 3–4-months-old), non-metastatic primary PDACs (KPC; 3–7-months-old), and metastatic primary PDACs (KPC; 3–7-months-old) showed a trend towards increasing PTHrP levels with disease progression (Supplemental Figure 3A). Western blot analysis of lineage labeled YFP+ tumor cells isolated from KPC-Rosa26LSL-YFP (KPCY) pancreata demonstrated high PTHrP expression in the malignant epithelia relative to normal ductal cells isolated from WT (Pdx1-Cre; LSL-Rosa26YFP/YFP) mice (Figure 3B). YFP/PTHrP co-immunofluorescence of metastatic liver lesions showed that YFP+ tumor cells maintained PTHrP expression (Figure 3C). Additionally, analysis of publicly available transcriptomic datasets (GSE90824 [37] and GSE144561 [38]) showed that metastatic tumors had higher PTHrP expression relative to primary tumors (Supplemental Figure 3B), and that PTHrP expression was highest in circulating tumor cells (CTCs) from metastatic PDAC mice (Supplemental Figure 3C).
Figure 3.

(A) Representative IHC images of PTHrP staining in PanIN (from KC mice) or PDAC tumors (from KPC mice). Scale bars = 100μm.
(B) Western blots of PTHrP and ACTIN in cell lines isolated from WT pancreatic ductal epithelial cells (Pdx1-Cre; LSL-Rosa26YFP/YFP) or KPCY pancreatic cancer cells.
(C) Representative IF images of metastatic liver lesions from KPCY mice stained with YFP (green), PTHrP (red), and DAPI (blue). Scale bars = 100μm.
(D-F) (D) Representative brightfield dissection scope images of orthotopic injection tumors from KPCY-shNT, KPCY-shPthlh #1, and KPCY-shPthlh #2 cell lines 3-weeks post-implantation. (E) Quantification of pancreatic tumor burden normalized to body mass after orthotopic injection. (F) Quantification of the percentage of YFP+ metastatic tumor cells present in FACS analyzed livers in mice with implanted orthotopic primary tumors. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SD; n=5 mice/group).
Primary PDAC burden was reduced significantly following pancreatic orthotopic injection with KPCY-shPthlh knockdown cell lines (Figure 3D–E; knockdown was confirmed in Supplemental Figure 3D–E). KPCY-shPthlh showed a statistically significant, albeit modest, decrease in proliferation in vitro (Supplemental Figure 3F) in concordance with the in vivo phenotype. Fluorescence Activated Cell Sorting (FACS) analysis of livers from mice with shPthlh primary tumors showed a marked reduction in YFP+ tumor cells (Figure 3F), consistent with a reduced capacity for metastatic colonization upon Pthlh loss. To rule out the possibility that KPCY-shPthlh tumors are less metastatic due to their smaller primary tumors, we repeated the orthotopic injection of KPCY-shPthlh tumor cells and let them grow until they reached a primary tumor burden similar to shNT (5–7 weeks in shPthlh versus 3 weeks in shNT; Supplemental Figure 3G). Strikingly, these long-term KPCY-shPthlh orthotopic tumors continued to show no evidence of metastasis (Supplemental Figure 3H). Reduced primary tumor and metastatic burden were observed also upon CRISPR-mediated knockout of Pthlh in an independent KPC-derived tumor cell line (Supplemental Figure 3I–K). These loss-of-function experiments suggest a potential oncogenic role for PTHrP. To establish further the pro-proliferative function of PTHrP, we used lentiviral-mediated Pthlh overexpression plasmids in either WT murine 3D organoids (which do not express Pthlh) or KPCY tumor cells (enhancing the endogenous expression of Pthlh). RT-PCR confirmed Pthlh overexpression, which leads to enhanced proliferation in both WT 3D organoids and KPCY tumor cells (Supplemental Figure 3L–O).
Pthlh deletion reduces tumor burden, abolishes metastasis and extends survival in KPC mice
The encouraging reduction of primary and metastatic tumor burden observed in Pthlh knockdown and knockout experiments in pancreatic orthotopic transplant models suggested that PTHrP may have an oncogenic and pro-metastatic role in PDAC. In order to test this hypothesis in an autochthonous model, we bred the PthlhLoxP conditional deletion allele into KPCY mice (Figure 4A). Of note, we were unable to combine homozygous PthlhLoxP/LoxP alleles with the LSL-KrasG12D knock-in allele, likely due to the <2.5MB genomic distance that separates the endogenous Kras and Pthlh loci on mouse chromosome 6. More than 200 pups were genotyped and no LSL-KrasG12D; PthlhLoxP/LoxP animals were obtained; therefore, we proceeded with LSL-KrasG12D; P53R172H; Pdx1-Cre; LSL-Rosa26YFP/YFP; PthlhLoxP/wt (designated as KPCY-PthlhHET) mice with heterozygous Pthlh knockout for further analysis. To validate the fidelity of our model, we confirmed a reduction in PTHrP expression in the ductal epithelia of KPCY-PthlhHET PDAC tumors relative to KPCY mice (Figure 4B). We performed FACS of YFP+ tumor cells from KPCY-PthlhHET and KPCY pancreata followed by RT-PCR to confirm Pthlh deletion in vivo (Supplemental Figure 4A). In parallel experiments, we generated Pdx1-Cre; LSL-Rosa26YFP/YFP; PthlhLoxP/wt (CY-PthlhHET) animals and performed histopathological analysis of the tissues, demonstrating that the pancreas developed normally after Pthlh heterozygous deletion (Supplemental Figure 4B) with normal acinar, ductal, and endocrine compartments. Furthermore, we generated LSL-KrasG12D; Pdx1-Cre; LSL-Rosa26YFP/YFP; PthlhLoxP/wt (KCY-PthlhHET) mice and harvested pancreatic tissues at three months of age, which showed similar levels of acinar-to-ductal metaplasia (ADM) and Pancreatic Intraepithelial Neoplasia (PanIN) lesions relative to KCY control animals (Supplemental Figure 4C), indicating no difference in tumor initiation.
Figure 4.

(A) Schematic drawing showing pancreas epithelial cell-specific heterozygous deletion of Pthlh in the KPCY mouse pancreatic cancer model.
(B) Representative IHC images of PTHrP staining in pancreas from KPCY or KPCY-PthlhHET mice. Scale bars = 100μm.
(C-H) (C) Kaplan-Meier overall survival plots of KPCY or KPCY-PthlhHET mice cohorts. Logrank p=3.0×10−4. (D) Quantification of the percentage of mice with gross YFP+ metastases. (E) Quantification of endpoint pancreatic tumor mass normalized to body mass. (F) Quantification of endpoint liver mass normalized to body mass. (G) Quantification of endpoint lung mass normalized to body mass. (H) Representative brightfield and YFP images of pancreas, liver, and lungs at endpoint. Statistical analysis by Mann-Whitney U test (E) or Student’s unpaired t-test (F-G) with significance indicated (*, p<0.05; error bars indicate SD; n=41 mice and n=16 for KPCY or KPCY-PthlhHET cohorts, respectively).
We observed a dramatic increase in overall survival of KPCY-PthlhHET mice (p=3.0e−4), whose median survival was 192 days relative to 111 days in KPCY mice (Figure 4C). This survival advantage of 81-days (representing a 73% increase) is one of the strongest survival benefits observed in the aggressive KPC model to our knowledge. These results are even more impressive taking into consideration that we utilized a ~50% reduction in Pthlh expression by using KPCY-PthlhHET mice. More surprisingly, despite KPCY-PthlhHET mice being three months older, only one of the sixteen (6.25%) KPCY-PthlhHET mice showed gross metastasis by YFP analysis under a dissection microscope (Figure 4D). This is in stark contrast to the nearly 45% of KPCY mice with gross YFP+ metastases (Figure 4D). KPCY-PthlhHET mice likely lived longer due to the combined effects of having significantly decreased primary tumor burden (Figure 4E) and decreased metastases to the liver and lungs (Figure 4F–G). Representative YFP images of gross pancreata, liver, and lung demonstrate the reduced primary tumor and metastatic burden observed in KPCY-PthlhHET mice relative to controls (Figure 4H). It is possible that KPCY-PthlhHET mice may harbor fewer metastases due to their decreased primary tumor load. To address this possibility, we re-examined the metastatic burden in an independent cohort of KPCY control animals with matched primary tumor load to our KPCY-PthlhHET cohort (i.e. a restricted analysis of our historical control KPCY tumors to only include mice with tumor burden similar to that of KPCY-PthlhHET mice). This new analysis demonstrated that KPCY-PthlhHET mice continued to show reduced metastasis compared to KPCY mice with size matched primary tumor load (Supplemental Figure 4D).
Anti-PTHrP monoclonal neutralizing antibody reduces primary tumor load and metastatic outgrowth
We next sought to test the therapeutic efficacy of inhibiting PTHrP in vivo. We engrafted KPCY tumor cells by orthotopic injection into the pancreas of C57BL/6 mice, allowed them to establish for 7 days, and then began treatment with either anti-PTHrP monoclonal neutralizing antibody or control anti-IgG antibody. After three weeks of treatment, anti-PTHrP-treated tumors were significantly smaller (Figure 5A–B). Their rate of tumor growth (Figure 5C) and their endpoint masses (Figure 5D) were significantly less. None of the anti-PTHrP-treated mice showed gross YFP+ metastases, in contrast to approximately one-third of anti-IgG-treated mice with overt metastases, primarily to the liver (Figure 5E). This experiment was repeated with a second independent KPCY cell line, which showed significantly reduced tumor growth (Supplemental Figure 5A), smaller primary tumors (Supplemental Figure 5B–C), and no metastases in anti-PTHLH treated mice (Supplemental Figure 5D). To test the ability of the anti-PTHLH antibody to block metastatic outgrowth, we performed tail vein injections of KPCY cells, allowed them to engraft for 5 days, and then began anti-PTHLH treatment. After 3 weeks of growth, anti-PTHrP treated mice had reduced metastatic burden as compared to anti-IgG treated controls (Figure 5F–G). YFP staining and quantification of the percentage of lung tissue occupied by YFP+ tumor cells confirmed anti-PTHrP treatment reduced metastatic outgrowth (Figure 5G). This tail vein metastasis experiment was repeated with a second independent KPCY cell line, showing the same reduced metastatic growth (Supplemental Figure 5E–F).
Figure 5.
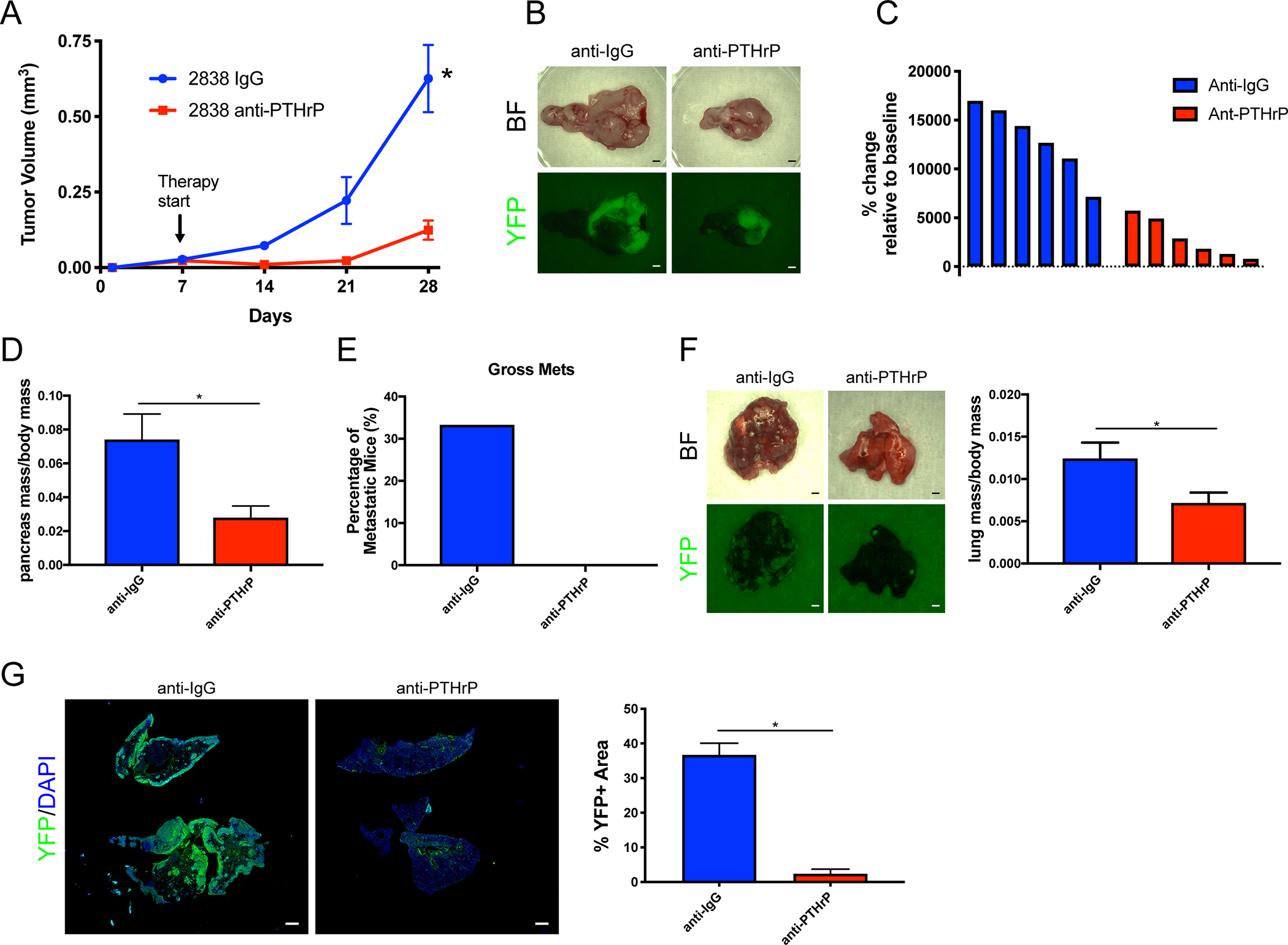
(A-E) (A) Quantification of tumor volume over time from ultrasound imaging of orthotopic implanted tumors. Arrow indicates the induction of anti-PTHrP (or anti-IgG) therapy. (B) Representative brightfield and YFP images of orthotopic pancreas tumors after treatment with the indicated therapy. (C) Waterfall plots of percent change in tumor volume relative to baseline. Each bar indicates an independent animal. (D) Quantification of pancreatic tumor mass normalized to body mass. (E) Quantification of the percentage of mice with gross YFP+ metastases. Statistical analysis by ANOVA (A) or Student’s unpaired t-test (D) with significance indicated (*, p<0.05; error bars indicate SD; n=6 mice/group).
(F-G) (F) Representative brightfield and YFP images of metastatic lung tumors 14-days post-tail vein injection and treatment with the indicated therapy beginning at day 5. Scale bars = 100μm. Quantification of lung mass normalized to body mass. (G) IF images of whole lungs after tail vein injection and treatment with the indicated therapy, stained with YFP (green) and DAPI (blue). Scale bars = 1mm. Quantification of percent YFP+ area relative to total lung area. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SD; n=6 mice/group).
Pthlh deletion or pharmacological inhibition decreases the proliferative capacity of pancreatic tumor cells
To determine if the reduction in overall tumor burden upon decreased PTHrP expression was due to a cell autonomous mechanism, we stained tissues from KPCY-PthlhHET mice with Ki-67, a marker for proliferation. We observed reduced Ki67 expression in YFP+ tumor cells from KPCY-PthlhHET mice relative to KPCY mice (Figure 6A–B). Additionally, delaminating YFP+ tumor cells in KPCY mice maintained their Ki-67 expression, which was not typical in KPCY-PthlhHET tissues (Figure 6A). PTHrP is a secreted ligand that binds to the G-protein coupled receptor PTH1R in either an autocrine or paracrine fashion, and we performed western blot analysis to determine if PDAC tumor cells express PTH1R, confirming such a status (Figure 6C). Stimulation of the receptor with recombinant PTHrP 1–34 enhanced growth (Supplemental Figure 6A). Recombinant mid-region (PTHrP 67–86) and C-terminal (PTHrP 107–111) peptides, which do not signal through PTH1R receptor, did not enhance growth (Supplemental Figure 6A), suggesting that PTHrP 1–34 signaling through PTH1R is the predominant, if not exclusive, pathway in promoting proliferation of KPCY cells. We next isolated conditioned media from shNT and shPthlh cell lines and performed media swapping experiments. To that end, shNT conditioned media (contains secreted PTHrP) rescued the reduced growth phenotype of both shPthlh clones (Supplemental Figure 6B). Conversely, treatment of shNT cells with shPthlh conditioned media (lacks secreted PTHrP) reduced growth (Supplemental Figure 6C). Finally, stimulation of shPthlh clones with PTHrP 1–34 enhanced proliferation to shNT levels (Supplemental Figure 6D). Collectively, these data support the premise that an autocrine signaling network exists whereby pancreatic tumor cells secrete PTHrP that binds to PTH1R on adjacent tumor cells, thereby stimulating growth. We cannot exclude the possibility of paracrine signaling to non-tumor cells, the topic of future investigation.
Figure 6.

(A-B) (A) Representative IF images of primary pancreatic tumors stained with YFP (green), Ki-67 (red), and DAPI (blue). White arrows indicate YFP+/Ki-67+ tumor cells in epithelial clusters; white arrowheads indicate YFP+/Ki-67+ delaminating tumor cells that have begun to metastasize. Scale bars = 100μm. (B) Quantification of YFP+/Ki-67+ proliferating tumor cells per high powered field (HPF). Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SD; n=6 mice/group).
(C) Western blots of PTH1R and ACTIN in cell lines isolated from WT pancreatic ductal epithelial cells (Pdx1-Cre; LSL-Rosa26YFP/YFP) or KPCY pancreatic cancer cells.
(D-E) (D) Cell viability assay measuring Optical Density (OD) after WST-1 cell proliferation reagent addition in a panel of murine KPCY PDAC cell lines treated with the indicated doses of anti-PTHrP monoclonal neutralizing antibody therapy for 48 hours. (E) Cell viability assay measuring OD after WST-1 cell proliferation reagent addition in 2838 and 6419 murine KPCY PDAC cell lines treated with 10μg/ml of anti-PTHrP monoclonal neutralizing antibody therapy over time. Statistical analysis by ANOVA with significance indicated (*, p<0.05; error bars indicate SEM; n=3 cultures/cell line).
(F) Cell viability assay measuring OD after WST-1 cell proliferation reagent addition in human PDAC cell lines treated with the indicated doses of anti-PTHrP therapy for 48 hours. (error bars indicate SEM; n=3 cultures/cell line).
(G-H) (G) Representative brightfield images of human pancreatic cancer patient-derived 3D organoid cultures after 48 hours of treatment. (H) Cell viability assay measuring luminescence after Cell Titer Glow 3D cell proliferation reagent addition in a panel of human pancreatic cancer patient-derived 3D organoid cultures treated with the indicated doses of anti-PTHrP therapy for 48 hours. (error bars indicate SEM; n=2 cultures/cell line).
Treatment of a panel of KPCY cell lines with an anti-PTHrP antibody showed a dose-dependent growth reduction (Figure 6D). Treatment with the same anti-PTHrP antibody over time appeared to block cellular proliferation (Figure 6E). Anti-PTHrP treatment of established human PDAC cell lines showed similar growth-reducing capabilities (Figure 6F). Finally, we harvested 3D organoid cultures from PDAC patients and treated them with anti-PTHrP antibody, which led to greatly reduced viability (Figure 6G–H), even beyond that of established cell lines. These results support the conclusion that, in part, secretion of PTHrP by tumor cells stimulates proliferation of neighboring tumor cells and provides an explanation for the reduced tumor burden upon genetic reduction or pharmacological inhibition of PTHrP in vivo.
Pthlh loss reduces EMT and metastatic competency
We next evaluated differentiation status of primary PDAC tumors from KPCY-PthlhHET and age-matched KPCY control mice, which revealed that a significant portion of KPCY-PthlhHET mice harbored only PanIN lesions and no evidence of PDAC (Figure 7A). In addition, the percentage of KPCY-PthlhHET mice that did progress to PDAC was greatly reduced relative to KPCY control mice, in this age-matched cohort (Figure 7A). Of the KPCY-PthlhHET mice that harbored PDAC, the percentage of animals with evidence of poorly-differentiated tumor cells was reduced (Figure 7B). Additionally, the percentage of the pancreata designated as poorly-differentiated in these mice was reduced greatly, with <2% of the entire pancreas being classified as poorly-differentiated in KPCY-PthlhHET mice (Figure 7C). This reduction in the poorly-differentiated status in KPCY-PthlhHET tissues is illustrated best by co-staining for YFP with ECADHERIN (ECAD). YFP+/ECAD- cells that have left their epithelial clusters and undergone EMT are abundant in KPCY tissues and absent from KPCY-PthlhHET tissues (Figure 7D). Quantification of the delaminating cells undergoing EMT demonstrates a dramatic approximately 10-fold reduction in KPCY-PthlhHET (Figure 7E).
Figure 7.
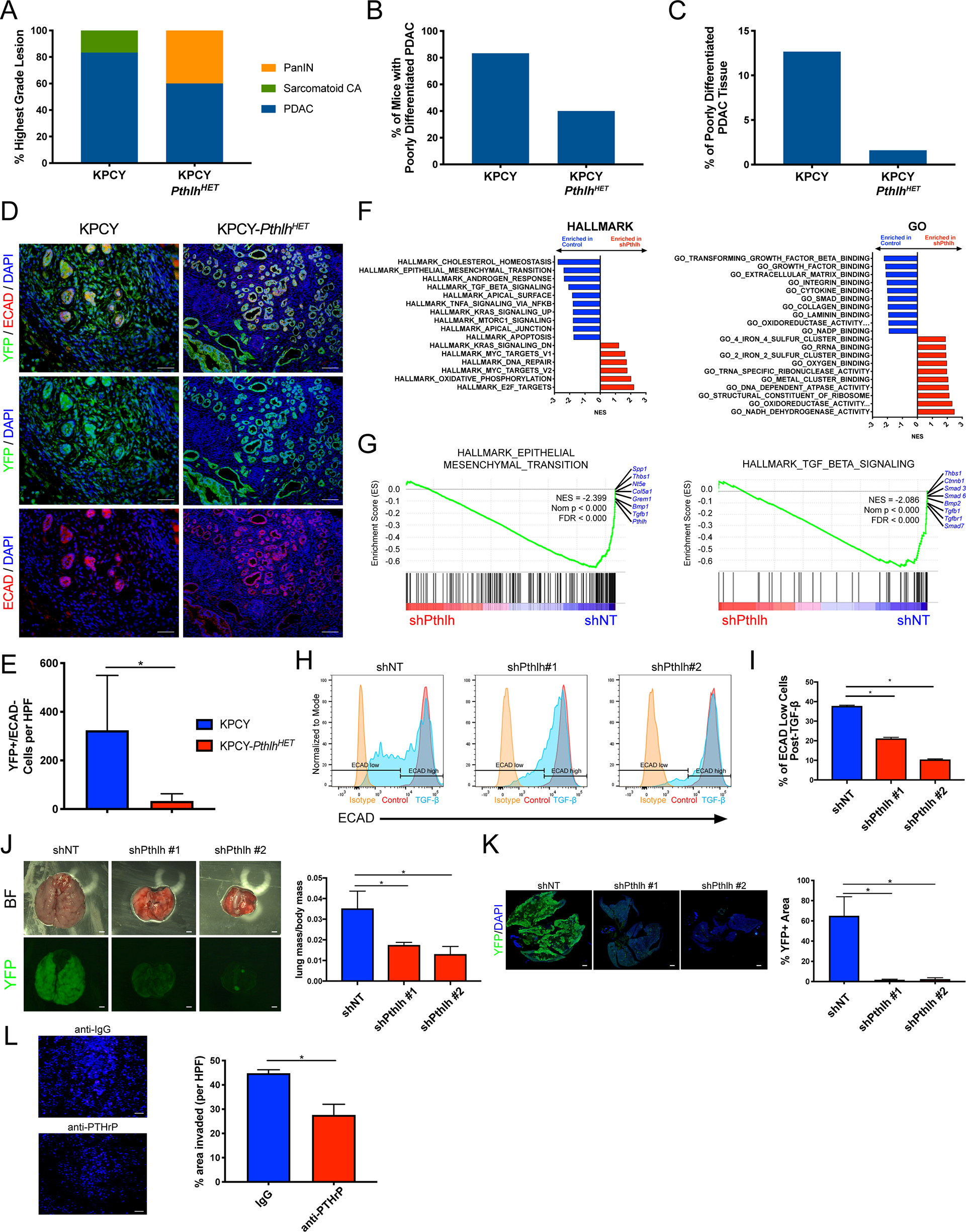
(A-C) (A) Classification of the highest-grade lesions present in 5–7-months old KPCY or KPCY-PthlhHET mice. (B) Quantification of the percentage of mice with poorly-differentiated PDAC present in 5–7-months old KPCY or KPCY-PthlhHET mice. (C) Quantification of the percentage of pancreatic tissue per mouse designated poorly-differentiated in 5–7-months old KPCY or KPCY-PthlhHET mice.
(D-E) (D) Representative IF images of primary pancreatic tumors stained with YFP (green), ECAD (red), and DAPI (blue). Scale bars = 100μm. (E) Quantification of YFP+/ECAD- delaminating tumor cells that have begun to metastasize. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SEM; n=6 mice/group).
(F-G) (F) Top gene set enrichment analyses (GSEA) plots by normalized enrichment score (NES) from the Hallmark (left) or GO (right) collections, enriched in shNT (blue) versus shPthlh (red) cells. (G) GSEA plots of the Hallmark EMT and TGF-β gene sets. The top genes lost in shPthlh and enriched in shNT cells are indicated in blue. NES, nominal p value, and FDR are indicated.
(H-I) (H) Representative ECAD flow histograms for shNT and shPthlh cells after treatment with 10 ng/ml TGF-β (or vehicle) for 48 hours. Isotype control stained cells shown in yellow, vehicle treated cells in red, and TGF-β treated in blue. (I) Quantification of the percentage of ECAD low cells post-TGF-β treatment. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SEM; n=3 cultures/group).
(J-K) (J) Representative brightfield and YFP images of metastatic lung tumors 3-weeks post-tail vein injection. Scale bars = 100μm. Quantification of lung mass normalized to body mass. (K) IF images of whole lungs after tail vein injection, stained with YFP (green) and DAPI (blue). Scale bars = 1mm. Quantification of percent YFP+ area relative to total lung area. Statistical analysis by Mann-Whitney U test (J) or Student’s unpaired t-test (K) with significance indicated (*, p<0.05; error bars indicate SD; n=5 mice/group).
(L) Representative IF images (left) and quantification (right) of DAPI-stained (blue) KPCY cells after 48 hours in a matrigel transwell cell invasion assay. Statistical analysis by Student’s unpaired t-test with significance indicated (*, p<0.05; error bars indicate SD; n=3 cultures/group).
RNA-seq and Gene Set Enrichment Analysis (GSEA) of KPCY-shPthlh tumor cells indicated that EMT and associated pathways were highly enriched in control cells relative to those without Pthlh (Figure 7F). In the latter, the top genes lost upon Pthlh knockdown include Smad3/6/7, Tgfb1, Tgfbr1/2, Bmp1/2, Bmpr2, Spp1, and Pthlh itself (Figure 7G and Supplemental Figures 7A–C). This analysis suggests that cells with reduced PTHrP are putatively unable to undergo EMT. To test this, we treated cells with TGF-β, a potent EMT inducer. KPCY cells, which express high PTHrP levels, were able to efficiently undergo EMT as measured by ECAD loss by FACS analysis (Figure 7H–I). Additionally, the downregulation of ECAD mRNA (Cdh1 expression) after TGF-β-induced EMT in KPCY cells was associated with a concomitant upregulation of Pthlh (Supplemental Figure 7D). Furthermore, stimulation with TGF-β enhanced the already high levels of secreted PTHrP present in the media (this Immunoradiometric Assay or IRMA only detects human PTHrP, thus necessitating the use of the squamous PDAC subtype cell line BxPC3; Supplemental Figure 7E). Remarkably, shPthlh cell lines were refractory to TGF-β stimulated EMT (Figure 7H–I). We additionally performed immunofluorescence for ECAD to show that control KPCY cells efficiently undergo EMT and completely lose their ECAD after TGF-β treatment, while shPthlh cells maintain their ECAD expression post-TGF-β stimulation (Supplemental Figure 7F). The reduction in ECAD in control KPCY cells coincides with a change to mesenchymal morphology, while shPthlh cells maintain their epithelial morphology after TGF-β treatment (Supplemental Figure 7F). To determine if the lack of EMT observed in shPthlh cell lines is functionally important for metastasis, we performed a tail vein metastasis assay, which revealed that cells with reduced PTHrP had a markedly reduced capacity to form lung metastases (Figure 7J–K). This was repeated in a second independent KPCY shPthlh knockdown cell line, and reduced lung metastatic burden was observed in a similar fashion (Supplemental Figure 7G–H). Treatment with anti-PTHrP antibody in a transwell migration assay, reduced the in vitro invasive capacity of KPCY tumor cells (Figure 7L).
RNA-seq analysis suggested that one downstream mechanism governing PTHrP-driven metastases may be through the known metastasis regulating gene Spp1, which was the second highest expressed transcript in shNT cells relative to shPthlh cells (Supplemental Table 1). Western blot analysis revealed a decrease in SPP1 (Osteopontin) protein levels in Pthlh knockdown cells (Supplemental Figure 7I). To determine if the reduced metastasis phenotype observed in shPthlh cells could be restored, we performed lentiviral-mediated stable overexpression of Spp1 (Spp1 OE). Western blot analysis confirmed efficient overexpression in Spp1 OE cells relative to empty vector (EV) (Supplemental Figure 7J). Tail vein injection of KPCY-shPthlh cells expressing EV or Spp1 OE partially rescued the metastatic ability in vivo (Supplemental Figure 7K–L). Additionally, Spp1 OE enhanced the in vitro migratory capacity of KPCY-shPthlh tumor cells in a transwell migration assay (Supplemental Figure 7M).
DISCUSSION
We demonstrate that a subset of human PDAC patients show PTHLH copy number gains as part of the KRAS amplicon. Increased PTHLH mRNA expression correlates strongly with the squamous/quasi-mesenchymal/basal-like subtypes of PDAC, and PTHLH copy number gains are enriched in metastatic patients. We functionally demonstrate that genetic Pthlh loss, either in genetically engineered KPCY mice or orthotopic injection models, reduces both primary and metastatic tumor burden, leading to enhanced overall survival in mice. Anti-PTHrP monoclonal antibody therapy reduced tumor cell proliferation and overall tumor burden. Finally, we propose a mechanism for reduced metastatic competency whereby tumor cells without PTHrP are unable to undergo EMT and thus cannot initiate the metastatic cascade (Supplemental Figure 8).
In the context of tumor metastasis biology, PTHrP has been studied as a potent inducer of osteolytic bone metastases through a process termed the ‘vicious cycle’. The abundance of TGF-β in the bone microenvironment stimulates local PTHrP production by tumor cells, which activates PTH1R signaling on osteoblasts, beginning a process favoring the osteoclast lineage through the RANKL/RANK axis [reviewed in 39]. This paracrine signaling causes osteoclasts to degrade bone (more permissive for metastatic growth), thereby releasing more TGF-β superfamily members, which induces the tumor cell to produce even more PTHrP [40]. In this regard, PTHrP is functioning in a manner similar to the highly related Parathyroid Hormone (PTH), a critical protein involved in bone homeostasis. This PTH-like effect on bone metastasis is fundamentally different from the cell autonomous mechanisms of metastasis we propose here, where PTHrP signaling is a necessary component of EMT within tumor cells, thereby driving invasion and dissemination to distant sites. We further demonstrate that PTHrP loss maintains cells primarily in an epithelial state, resulting in a nearly complete arrest of the macrometastatic process.
Provocatively, in KPCY-PthlhHET mice, survival was prolonged with near complete elimination of metastases in the otherwise highly aggressive KPCY autochthonous PDAC mouse model. Our work provides a foundation for the application of anti-PTHrP therapy in clinical trials of PDAC patients. Our loss-of-function experiments in complementary mouse models and human 3D organoids demonstrate that PTHrP deletion/inhibition results in less aggressive disease, with reduced primary and metastatic tumor growth. Metastatic progression is a complex multistep process consisting of invasion through the basement membrane, intravasation into adjacent vasculature, survival in circulation, extravasation into the metastatic tissue, and successful colonization at the secondary site [41]. Our results indicate that PTHrP inhibition inhibits cells from entering the metastatic cascade by hindering their ability to undergo EMT and invade. The inability of PTHrP deficient cells to undergo EMT maintains them in organized epithelial clusters, thus preventing invasion. The striking change from predominantly poorly-differentiated PDAC in KPCY mice to well-differentiated tumors in KPCY-PthlhHET mice holds promise for the ability of long-term anti-PTHrP treatment to alter the differentiation status in PDAC patients. Additionally, we show that PTHrP loss reduces cellular proliferation, which would provide a two-pronged attack for anti-PTHrP therapy – reduced invasion through inhibition of EMT and decreased proliferation in tumor cells that colonize the metastatic site.
Others have demonstrated that the metastatic cascade in pancreatic cancer is a highly choregraphed interplay between proliferation and metastatic competency, modulated by the transcription factor Runx3 [42]. PTHrP has been shown to be an upstream driver of other Runx family members, namely Runx1 and Runx2 [15, 43, 44], which may themselves induce EMT [15]. Additionally, our differential gene expression analysis indicates that expression of many RUNX3-regulated transcripts [42] are also enhanced in PTHrP-expressing cells. Most notably, RUNX3 controls the metastatic switch to promote metastasis through osteopontin (encoded by Spp1) and Col6a1 [42], which we show are both highly enriched in Pthlh intact cells relative to Pthlh knockdown cells (Supplemental Table 1). Spp1, in particular, is the second highest expressed transcript in control cells relative to Pthlh knockdown, with near complete loss in shPthlh cells. We demonstrate that PTHrP exerts its pro-metastatic effects, in part, by driving Spp1 expression, and that re-expression of Spp1 in cells lacking Pthlh increases their metastatic proclivity. Future studies will look to determine the full extent of crosstalk between PTHrP and RUNX signaling in order to find new therapeutically actionable nodes within the pathways that may be important for metastasis.
PTHrP, a marker of the squamous PDAC subtype, endows cells with the capacity for invasion and metastasis, which can be inhibited by an anti-PTHrP monoclonal antibody treatment. In a broader sense, we have uncovered a targeted therapeutic vulnerability that emerges from amplification of a “passenger” oncogene and offer a new paradigm to look at genes in close proximity to other commonly amplified oncogenes. PTHLH is co-amplified as part of the KRAS amplicon in PDAC patients. This collateral amplification (i.e. amplification of a supposed passenger gene that ends up having its own oncogenic functions) is analogous to collateral passenger deletions of genes that are lost when established tumor suppressors and their surrounding genes are deleted [45–47]. The collateral amplification confers a bona fide change in phenotype that can be attributed to the amplified “passenger” gene (PTHLH) in addition to KRAS. This genomic amplification of PTHLH does not preclude other methods of enhancing PTHrP transcription in the squamous and mesenchymal states, through factors such as TP63/ETS/SMAD. In fact, our observations indicate that the increase in PTHrP expression in PDAC is likely a complex interplay of multiple pathways. For example, it is tempting to speculate that a single PDAC patient may possess multiple means of enhancing PTHrP levels. There may be PTHLH copy number gains as part of the KRAS amplicon, the latter of which activates MAPK/ETS signaling, leading to further increased PTHrP expression. In addition, this amplicon correlates with the squamous subtype, thus activating a transcriptional program through TP63, which itself augments PTHrP expression further. On a cellular basis, individual cells undergoing EMT would have higher PTHLH expression. Collectively, our work nominates PTHrP as a novel therapeutic avenue in PDAC patients that has the potential to limit both primary and metastatic tumor growth.
MATERIALS AND METHODS
Mice
LSL-KrasG12D; Pdx1-Cre (KC) and LSL-KrasG12D; P53R172H or LoxP; Pdx1-Cre (KPCY) mice have been described previously [48]. The PthlhLoxP mice were a generous gift by Dr. Andrew Karaplis (McGill) [49]. For survival analysis, mice were palpated and examined for evidence of morbidity twice per week and sacrificed when moribund. Both male and female mice were used for survival analysis. The percentage of metastatic mice were determined based upon gross YFP positivity in the liver, lungs, and other metastatic sites. Female C57BL/6J (Stock No. 000664) or outbred athymic nude J:NU (Stock No. 007850) for tumor cell injection experiments were obtained from The Jackson Laboratory. Mice treated with anti-PTHrP monoclonal antibody (200μg; Richard Kremer laboratory, McGill University Health Center and material transfer agreement with Biochrom Pharma. Inc.) received intraperitoneal injections three times per week every 48 hours day for the indicated time. All vertebrate animals were maintained, and experiments were conducted in compliance with the NIH guidelines for animal research and approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Cell lines and culture conditions
Murine PDAC cell lines were derived from KPCY mice that were backcrossed onto the C57BL/6J strain or KPC tumors of mixed genetic background[50]. All human cell lines were originally obtained from ATCC. All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% Penicillin-Streptomycin and maintained in standard culture conditions. Cell lines were regularly tested for Mycoplasma. The following doses were used for in vitro treatment: U0126 (10 uM), SCH772983 (2 uM), recombinant PTHrP 1–34 (100nM), recombinant PTHrP 67–86 (100nM), recombinant PTHrP 107–111 (100nM), TGF-β (10 ng/mL). For conditioned media experiments, complete DMEM media (10% FBS, 1% P/S) was collected after 48 hrs. of culture and combined with fresh complete DMEM at a 1:1 ratio for subsequent treatments.
Immunohistochemistry and immunofluorescence staining
Dissected mouse pancreata were fixed in zinc-formalin, processed, embedded in paraffin, sectioned and mounted. Immunohistochemical (IHC) sections were stained using a Bond Rx autostainer (Leica). Immunofluorescent sections were deparaffinized, rehydrated and antigen retrieval performed, which was followed by overnight incubation with the specific primary antibodies at 4°C. Primary antibodies used were PTHrP (Sigma AV33885), Ki-67 (Abcam ab16667), ECAD (Takara Clone M108), and GFP (Abcam; recognizes YFP). Images were acquired using an Olympus IX71 inverted multicolor fluorescent microscope equipped with a DP71 camera, and quantification performed in ImageJ software.
Histopathological Analysis
All pathological analysis was performed by the lab of Anirban Maitra in accordance with the consensus report and recommendations for pathological analysis of genetically engineered mouse models of pancreatic exocrine cancer [51]. For tissue grading, the percentage of normal pancreas tissue, ADM, PanIN, well-differentiated carcinoma, poorly-differentiated carcinoma, sarcomoid carcinoma and necrosis was analyzed on every slide in a blinded fashion. For ADM/PanIN quantification, the size of pancreas tissue was measured and the total numbers of PanINs and ADMs were counted. For PTHrP IHC H score analysis, 0 indicates negative staining, 1 weak staining, 2 moderate staining, and 3 strong staining. H score was calculated by the following formula: [(3) x (percentage of strong staining)] + [(2) x (percentage of moderate staining)] + [(1) x (percentage of weak staining)], giving a range of 0 to 300.
Human PDAC specimens
Human 3D organoid samples were obtained from patients at the University of Pennsylvania and Columbia University under IRB approval. The PDAC tissue microarray were obtained from patients who underwent radical pancreatectomy at the Department of General Surgery, Chiba University Hospital, Japan, from June 2013 to December 2015. All patients were histologically diagnosed with primary invasive PDAC. The study protocol was approved by the Ethics Committees of Chiba University, Graduate School of Medicine and written informed consent was obtained from each patient before surgery. Formalin-fixed and paraffin-embedded samples were stained with anti-PTHrP antibody (Sigma AV33885) using standard immunohistochemistry techniques. The staining intensities of PTHrP expression was evaluated independently by two investigators and scored as follows: low expression: 0–50% tumor cells stained positive; high expression: more than 50% tumor cells stained positive.
Fluorescence in situ hybridization (FISH)
Cells were treated with Colcemin for 60 minutes prior to harvest to arrest cells at metaphase. Trypsinized and pelleted cells were re-suspended in a 75mM KCl hypotonic solution and incubated at 37°C for 20 minutes. Cells were fixed thrice in 3:1 ice cold Methanol:Acetic Acid fixative solution at −20°C for 5 minutes. After the final fixative solution wash, cell suspensions were dropped onto slides from a height of 10 cm. Slides were dehydrated through 70%/85%/100% ethanol washes and air dried. While slides dried, the probe mixture was prepared by mixing a 1:5 dilution of probe to FISH probe buffer (Empire Genomics PTHLH-CHR12–20). The probe mixture was applied to the air-dried slides and cover slips placed on top prior to denaturing slides on a slide warmer at 73°C for 2 minutes. Slides were next placed in a humified chamber and incubated overnight at 37°C. After incubation, coverslips were removed, and slides were washed in Wash Solution 1 wash buffer (Empire Genomics WS1) at 73°C for 2 minutes. Slides were transferred to Wash Solution 2 (Empire Genomics WS2) at room temperature for 2 minutes. After washes, slides were removed, cleaned, and allowed to air dry. Slides were stained with Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories H-1200–100), cover slipped and stored for imaging.
Western blot analysis
Cells were washed with ice cold PBS and lysed in RIPA buffer. Equal amounts of protein were run in reducing conditions on SDS-PAGE gels and transferred to Immoblion-FL PVDF membrane (Millipore IPFL00010). Blocking was performed in Odyssey Blocking Buffer (Li-Cor) for 1 hour at room temperature. After blocking, membranes were incubated in primary antibody diluted in Odyssey Blocking Buffer overnight at 4°C. After PBS-T washes, membranes were incubated with secondary antibody diluted in Odyssey Blocking Buffer at room temperature in the dark for 1 hour. Following PBS-T washes, membranes were imaged on a Li-Cor Odyssey Imaging System.
shRNA and CRISPR
For shRNA experiments, cells were transduced with Mission pLKO.1-puro Lentiviral particles with non-target (shNT; 5’ CCGGGATACCTAACTCAGGAAACCACTCGAGTGGTTTCCTGAGTTAGGTATCTTTTTG 3’), shPthlh #1 (5’ CCGGGATACCTAACTCAGGAAACCACTCGAGTGGTTTCCTGAGTTAGGTATCTTTTTTG 3’), or shPthlh #2 (5’ CCGGCCAATTATTCCTGTCACTGTTCTCGAGAACAGTGACAGGAATAATTGGTTTTTTG 3’) and stable colonies selected with puromycin (Sigma). For CRISPR experiments, cells were transduced with pLentiCRISPR V2 lentiviral particles with Pthlh guide RNA ( 5’ GCCCCTCCACCGAACGCCCG 3’) and stable colonies selected with puromycin (Sigma).
Lentiviral Overexpression
For Pthlh overexpression, cells were transduced with pLenti-C-Myc-DDK-P2A-Puro Lentiviral particles with empty vector (Origene #PS100092) or Pthlh ORF (Origene #MR201519L3). For Spp1 overexpression, cells were transduced with pCDH-EF1-FHC Lentiviral particles with empty vector (Addgene #64874) or Spp1 ORF cloned into the NheI-BamHI sites.
PTHrP IRMA
Conditioned media was harvested from BxPC3 cells 48- and 72-hours post-TGF-β treatment (10ng/mL) and frozen for downstream PTHrP 2-site immunoradiometric assay (IRMA) analysis. Active PTHrP IRMA (Beckman DSL8100) was performed as per the manufacturer’s instructions. Briefly, 200 μl of calibrator or sample was added to antibody-coated tubes, followed by 100 μl of Iodide-125 tracer, and gentle vortexing. Samples were incubated overnight at room temperature on a shaker and then discarded. Tubes were washed three times with deionized water and signal was measured using a PerkinElmer model 2470 automatic Gamma Counter Wizard. PTHrP concentration (pg/mL) was calculated based off of a standard curve.
Orthotopic injection
Orthotopic injection was performed as previously described [52]. Briefly, after anesthesia and sterile preparation of the abdomen, an incision was made in the upper left quadrant and the pancreas exteriorized on a sterile field. 1.0–2.5×105 cells in 100 μl sterile DMEM were injected into the tail of the pancreas via an insulin syringe. KPCY-derived tumor cells were injected into C57BL/6J mice (The Jackson Lab Stock #000664) and mixed background KPC-derived tumor cells into athymic nude J:NU mice (The Jackson Lab Stock # 002019). A cotton swab was held over the injection site to ensure that no cells leaked into the peritoneal cavity. Successful injection was verified by the appearance of a liquid bleb at the injection site. Afterwards, the pancreas was placed back into the peritoneum and the incision was closed with 4–0 coated sutures. Tumors, lungs, and livers were harvested at the indicated time points.
Tail Vein Injection Metastasis Assay
1.0–2.5×105 KPCY cells in 100 μl sterile DMEM were injected into the tail veins of C57BL/6J mice (The Jackson Lab Stock #000664) with an insulin syringe. Lungs were harvested at the indicated time points. Tail vein injected lungs were stained for YFP/DAPI, imaged on a Keyence microscope, and stitched images were generated to quantify the percentage of YFP+ area relative to the entire lung.
Flow cytometry
For ECAD flow cytometry, cells were dissociated into single cells with Hanks Enzyme Free Cell Dissociation Solution (Millipore S-004-C). Cells were stained with anti-ECAD (Bio Legend 147308) or isotype control (Bio Legend 400418) in staining solution on ice for 15 minutes. Cells were washed three times in staining solution, filtered through a 70 μM strainer, and then stained with DAPI prior to flow cytometric analysis. This assay measures only surface ECAD levels, as we did not permeabilize the cells.
For FACS of YFP+ cells, tumors were washed, minced, and digested with Collagenase Type V (2mg/ml; Sigma C9263) for 20–30 minutes at 37ºC and vortexed every 5 minutes. Digested tissue was filtered through a 70μm filter into a conical tube and complete DMEM (10% FBS, 1% P/S) was added. Cells were pelleted at 300g for 5 minutes and resuspended in cold sorting buffer (1XHBSS with 25mM HEPES, 5mM MgCl2, 17.5mM D-glucose, 1X Glutamax, 1mM Sodium Pyruvate, DNase 25 μg/mL) with DAPI. YFP+ cells were collected via FACS and RNA isolated for downstream analysis.
Cell viability assays
WST-1 Cell Proliferation Assay (Abcam ab65475) was performed per the manufacturer’s instructions. Briefly, 2.0×103-5.0×103 cells were plated on 96-well plates and grown in standard culture conditions. Media was replaced with 100μl fresh media containing 10μl WST solution, and incubated for 2 hours at 37°C. Plate was shaken and absorbance read at 420–480nm and reference wavelength of 650nm. Final absorbance was obtained by subtracting 650nm reference wavelength from the 420–480nm read.
CellTiter-Glo 3D Cell Viability Assay (Promega G9681) was performed per the manufacturer’s instructions. Briefly, a 1:1 dilution of 3D organoid culture media: Celltiter-Glu 3D reagent was added to organoid cultures and incubated for 30 minutes at 37°C. 100ul of the 1:1 mixture was added to a 96-well plate and luminescence read in triplicate.
3D organoids
Human pancreatic cancer patient-derived 3D organoid cultures were grown as previously described [53]. Matrigel embedded organoids were maintained in culture media containing A 83–01 (Tocris 2939), B27 (Thermo Fisher 17504044), hEGF (Peprotech AF-100–15), hFGF-10 (Peprotech 100–26), hGastrin (Tocris 3006), mNoggin (Peprotech 250–38), N-acetylcysteine (Sigma A9165–5G), Nicotinamide (Sigma N0636–100G), Y-27632 (Sigma Y0503–5mg), R-Spondin1 conditioned medium, and Wnt3a-conditioned medium.
Matrigel invasion assay
Transwell inserts (Corning 3464) were coated with matrigel and placed in 24 well plates above complete DMEM. Cells were seeded with serum-free DMEM on top of the matrigel layer. Cells were incubated under normal conditions for 24 hours in the presence of Hydroxyurea (HU). Transwell inserts were removed, washed three times, and fixed in 4% PFA for 15 minutes. Fixed matrigel inserts were washed three times, stained with DAPI, and imaged.
RNA-seq and GSEA
RNA samples were extracted using the Qiagen RNeasy following manufacturer’s instructions. RNA was sent out to a commercial company, Novogene, for library preparation and high-throughput sequencing using Illumina sequencers to generated paired-end results. Raw counts of gene transcripts were obtained using an alignment-independent tool, Salmon (https://combine-lab.github.io/salmon/), using standard settings. The raw count matrix was subsequently imported into R-studio (R version 3.5) and used as input file for DESeq2 analysis (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) with default settings from online software instruction for normalization and differential gene expression analysis. Salmon was used to normalize and quantitate gene expression in transcripts-per-million (tpm) through quasi-alignment. Differentially expressed genes were used as input for principal component analysis (PCA), gene set enrichment analysis (GSEA) (https://www.gsea-msigdb.org/gsea/index.jsp) and motif analysis using HOMER (http://homer.ucsd.edu/homer/ngs/index.html). Detailed Scripts and parameters used for each steps of analysis could be provided by reasonable request to the authors.
Data resources
All sequencing data has been deposited in the Gene Expression Omnibus under the series GSE154661.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Pancreatic cancer often presents with metastases, yet no strategies exist to pharmacologically inhibit this process. Herein, we establish the oncogenic and pro-metastatic roles of PTHLH, a novel amplified gene in PDAC. We demonstrate that blocking PTHrP activity reduces primary tumor growth, prevents metastasis and prolongs survival in mice.
ACKNOWLEDGEMENTS
This work was supported by American Gastroenterology Association Bern Schwartz Research Scholar Award in Pancreatic Cancer (JRP), NIH/NCI NRSA F32CA221094 (JRP), NIH/LRP (JRP), Hopper-Belmont Foundation Inspiration Award (JRP), NIH/NCI P30CA01369644, NIH/NIDDK R01 DK060694 (AKR), NIH/NCI R01 CA229803 (BZS), United States Department of Defense W81XWH-15-1-0723 (RK), Canadian Institutes of Health Research (CIHR) MOP142287, and American Cancer Society Fairfield County Comedy Against Cancer Postdoctoral Fellowship PF-19-227-01-CMS (AC). We thank the following core facilities through the Herbert Irving Comprehensive Cancer Center (Molecular Pathology, Genetically engineered mouse models, Flow Cytometry). We thank Iok In Christine Chio (Columbia) for human PDAC organoids, Karine Sellin (McGill University Health Center) for production of anti-PTHrP monoclonal antibodies, and Mary Ann Crissey for mouse colony maintenance (University of Pennsylvania).
Footnotes
RK is the inventor of the therapeutic monoclonal anti-PTHrP antibodies used in this study and authorized for use by Biochrom Pharma. RK is Founder and President of Biochrom Pharma. All other authors declare no potential conflicts of interest.
REFERENCES
- 1.Rawla P, Sunkara T, and Gaduputi V, Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol, 2019. 10(1): p. 10–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res, 2014. 74(11): p. 2913–21. [DOI] [PubMed] [Google Scholar]
- 3.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. , Genomic analyses identify molecular subtypes of pancreatic cancer. Nature, 2016. 531(7592): p. 47–52. [DOI] [PubMed] [Google Scholar]
- 4.Collisson EA, Sadanandam A, Olson P, Gibb WJ, Truitt M, Gu S, et al. , Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med, 2011. 17(4): p. 500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA, et al. , Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet, 2015. 47(10): p. 1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collisson EA, Bailey P, Chang DK, and Biankin AV, Molecular subtypes of pancreatic cancer. Nat Rev Gastroenterol Hepatol, 2019. 16(4): p. 207–220. [DOI] [PubMed] [Google Scholar]
- 7.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. , TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep, 2018. 25(7): p. 1741–1755 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan-Seng-Yue M, Kim JC, Wilson GW, Ng K, Figueroa EF, O’Kane GM, et al. , Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet, 2020. 52(2): p. 231–240. [DOI] [PubMed] [Google Scholar]
- 9.Connor AA, Denroche RE, Jang GH, Lemire M, Zhang A, Chan-Seng-Yue M, et al. , Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell, 2019. 35(2): p. 267–282 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reichert M, Bakir B, Moreira L, Pitarresi JR, Feldmann K, Simon L, et al. , Regulation of Epithelial Plasticity Determines Metastatic Organotropism in Pancreatic Cancer. Dev Cell, 2018. 45(6): p. 696–711 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Albright F, Case Records of the Massachusetts General Hospital. Case 27461. N Engl J Med, 1941. 225: p. 789–791. [Google Scholar]
- 12.Wysolmerski JJ, Parathyroid hormone-related protein: an update. J Clin Endocrinol Metab, 2012. 97(9): p. 2947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCauley LK and Martin TJ, Twenty-five years of PTHrP progress: from cancer hormone to multifunctional cytokine. J Bone Miner Res, 2012. 27(6): p. 1231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang WM, Lin YF, Su CY, Peng HY, Chang YC, Hsiao JR, et al. , Parathyroid Hormone-Like Hormone is a Poor Prognosis Marker of Head and Neck Cancer and Promotes Cell Growth via RUNX2 Regulation. Sci Rep, 2017. 7: p. 41131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He S, Xue M, Liu C, Xie F, and Bai L, Parathyroid Hormone-Like Hormone Induces Epithelial-to-Mesenchymal Transition of Intestinal Epithelial Cells by Activating the Runt-Related Transcription Factor 2. Am J Pathol, 2018. 188(6): p. 1374–1388. [DOI] [PubMed] [Google Scholar]
- 16.Tang J, Liao Y, He S, Shi J, Peng L, Xu X, et al. , Autocrine parathyroid hormone-like hormone promotes intrahepatic cholangiocarcinoma cell proliferation via increased ERK/JNK-ATF2-cyclinD1 signaling. J Transl Med, 2017. 15(1): p. 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walia MK, Ho PM, Taylor S, Ng AJ, Gupte A, Chalk AM, et al. , Activation of PTHrP-cAMP-CREB1 signaling following p53 loss is essential for osteosarcoma initiation and maintenance. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Karaplis AC, Huang DC, Siegel PM, Camirand A, Yang XF, et al. , PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J Clin Invest, 2011. 121(12): p. 4655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kakonen SM, Selander KS, Chirgwin JM, Yin JJ, Burns S, Rankin WA, et al. , Transforming growth factor-beta stimulates parathyroid hormone-related protein and osteolytic metastases via Smad and mitogen-activated protein kinase signaling pathways. J Biol Chem, 2002. 277(27): p. 24571–8. [DOI] [PubMed] [Google Scholar]
- 20.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. , TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest, 1999. 103(2): p. 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindemann RK, Ballschmieter P, Nordheim A, and Dittmer J, Transforming growth factor beta regulates parathyroid hormone-related protein expression in MDA-MB-231 breast cancer cells through a novel Smad/Ets synergism. J Biol Chem, 2001. 276(49): p. 46661–70. [DOI] [PubMed] [Google Scholar]
- 22.Richard V, Rosol TJ, and Foley J, PTHrP gene expression in cancer: do all paths lead to Ets? Crit Rev Eukaryot Gene Expr, 2005. 15(2): p. 115–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson J, Sebag M, Rhim J, Goltzman D, and Kremer R, Dysregulation of parathyroid hormone-like peptide expression and secretion in a keratinocyte model of tumor progression. Cancer Res, 1991. 51(24): p. 6521–8. [PubMed] [Google Scholar]
- 24.Kremer R, Shustik C, Tabak T, Papavasiliou V, and Goltzman D, Parathyroid-hormone-related peptide in hematologic malignancies. Am J Med, 1996. 100(4): p. 406–11. [DOI] [PubMed] [Google Scholar]
- 25.Sebag M, Henderson J, Goltzman D, and Kremer R, Regulation of parathyroid hormone-related peptide production in normal human mammary epithelial cells in vitro. Am J Physiol, 1994. 267(3 Pt 1): p. C723–30. [DOI] [PubMed] [Google Scholar]
- 26.Lv Z, Wu X, Cao W, Shen Z, Wang L, Xie F, et al. , Parathyroid hormone-related protein serves as a prognostic indicator in oral squamous cell carcinoma. J Exp Clin Cancer Res, 2014. 33: p. 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Urosevic J, Garcia-Albeniz X, Planet E, Real S, Cespedes MV, Guiu M, et al. , Colon cancer cells colonize the lung from established liver metastases through p38 MAPK signalling and PTHLH. Nat Cell Biol, 2014. 16(7): p. 685–94. [DOI] [PubMed] [Google Scholar]
- 28.Truong NU, de BEMD, Papavasiliou V, Goltzman D, and Kremer R, Parathyroid hormone-related peptide and survival of patients with cancer and hypercalcemia. Am J Med, 2003. 115(2): p. 115–21. [DOI] [PubMed] [Google Scholar]
- 29.Assaker G, Camirand A, Abdulkarim B, Omeroglu A, Deschenes J, Joseph K, et al. , PTHrP, A Biomarker for CNS Metastasis in Triple-Negative Breast Cancer and Selection for Adjuvant Chemotherapy in Node-Negative Disease. JNCI Cancer Spectr, 2020. 4(1): p. pkz063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleming NI, Trivett MK, George J, Slavin JL, Murray WK, Moseley JM, et al. , Parathyroid hormone-related protein protects against mammary tumor emergence and is associated with monocyte infiltration in ductal carcinoma in situ. Cancer Res, 2009. 69(18): p. 7473–9. [DOI] [PubMed] [Google Scholar]
- 31.Henderson M, Danks J, Moseley J, Slavin J, Harris T, McKinlay M, et al. , Parathyroid hormone-related protein production by breast cancers, improved survival, and reduced bone metastases. J Natl Cancer Inst, 2001. 93(3): p. 234–7. [DOI] [PubMed] [Google Scholar]
- 32.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. , Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun, 2015. 6: p. 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamdan FH and Johnsen SA, DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc Natl Acad Sci U S A, 2018. 115(52): p. E12343–E12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. , EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev Cell, 2018. 45(6): p. 681–695 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cebrian A, Garcia-Ocana A, Takane KK, Sipula D, Stewart AF, and Vasavada RC, Overexpression of parathyroid hormone-related protein inhibits pancreatic beta-cell death in vivo and in vitro. Diabetes, 2002. 51(10): p. 3003–13. [DOI] [PubMed] [Google Scholar]
- 36.Williams K, Abanquah D, Joshi-Gokhale S, Otero A, Lin H, Guthalu NK, et al. , Systemic and acute administration of parathyroid hormone-related peptide(1–36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia, 2011. 54(11): p. 2867–77. [DOI] [PubMed] [Google Scholar]
- 37.Chiou SH, Risca VI, Wang GX, Yang D, Gruner BM, Kathiria AS, et al. , BLIMP1 Induces Transient Metastatic Heterogeneity in Pancreatic Cancer. Cancer Discov, 2017. 7(10): p. 1184–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franses JW, Philipp J, Missios P, Bhan I, Liu A, Yashaswini C, et al. , Pancreatic circulating tumor cell profiling identifies LIN28B as a metastasis driver and drug target. Nat Commun, 2020. 11(1): p. 3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waning DL and Guise TA, Molecular mechanisms of bone metastasis and associated muscle weakness. Clin Cancer Res, 2014. 20(12): p. 3071–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soki FN, Park SI, and McCauley LK, The multifaceted actions of PTHrP in skeletal metastasis. Future Oncol, 2012. 8(7): p. 803–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambert AW, Pattabiraman DR, and Weinberg RA, Emerging Biological Principles of Metastasis. Cell, 2017. 168(4): p. 670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whittle MC, Izeradjene K, Rani PG, Feng L, Carlson MA, DelGiorno KE, et al. , RUNX3 Controls a Metastatic Switch in Pancreatic Ductal Adenocarcinoma. Cell, 2015. 161(6): p. 1345–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ardura JA, Sanz AB, Ortiz A, and Esbrit P, Parathyroid hormone-related protein protects renal tubuloepithelial cells from apoptosis by activating transcription factor Runx2. Kidney Int, 2013. 83(5): p. 825–34. [DOI] [PubMed] [Google Scholar]
- 44.Toribio RE, Brown HA, Novince CM, Marlow B, Hernon K, Lanigan LG, et al. , The midregion, nuclear localization sequence, and C terminus of PTHrP regulate skeletal development, hematopoiesis, and survival in mice. FASEB J, 2010. 24(6): p. 1947–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. , Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature, 2016. 531(7595): p. 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller FL, Aquilanti EA, and DePinho RA, Collateral Lethality: A new therapeutic strategy in oncology. Trends Cancer, 2015. 1(3): p. 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, et al. , Passenger deletions generate therapeutic vulnerabilities in cancer. Nature, 2012. 488(7411): p. 337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. , EMT and dissemination precede pancreatic tumor formation. Cell, 2012. 148(1–2): p. 349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He B, Deckelbaum RA, Miao D, Lipman ML, Pollak M, Goltzman D, et al. , Tissue-specific targeting of the pthrp gene: the generation of mice with floxed alleles. Endocrinology, 2001. 142(5): p. 2070–7. [DOI] [PubMed] [Google Scholar]
- 50.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. , Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity, 2018. 49(1): p. 178–193 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hruban RH, Adsay NV, Albores-Saavedra J, Anver MR, Biankin AV, Boivin GP, et al. , Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res, 2006. 66(1): p. 95–106. [DOI] [PubMed] [Google Scholar]
- 52.Aiello NM, Rhim AD, and Stanger BZ, Orthotopic Injection of Pancreatic Cancer Cells. Cold Spring Harb Protoc, 2016. 2016(1): p. pdb prot078360. [DOI] [PubMed] [Google Scholar]
- 53.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. , Organoid models of human and mouse ductal pancreatic cancer. Cell, 2015. 160(1–2): p. 324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.