

Abstract
Brain-enriched microRNA-338 (miR-338) is known to play a central role in brain mitochondrial function, however the role of miR-338 in stroke injury remains unknown. This study investigated the role of miR-338 in injury from transient focal cerebral ischemia in mice, and in cell survival and mitochondrial function after in vitro ischemia in astrocyte and neuronal cultures. Pre-treatment of mice with intracerebroventricular injection of miR-338 antagomir 24h prior to 1h of middle cerebral artery occlusion (MCAO) significantly reduced infarct size and improved neurological score at both 24h and 7d after injury. Levels of the miR-338 target cytochrome-c oxidase subunit 4I1 (COX4I1), which plays an essential role in maintaining brain mitochondrial ATP production, were increased in miR-338 antagomir-treated mice. Mouse primary astrocyte cell cultures subjected to glucose deprivation exhibited increased cell survival when pre-treated with miR-338 inhibitor, and greater cell death with miR-338 mimic. Decreased miR-338 levels were associated with increased ATP production, augmented cytochrome c oxidative (CcO) activity and preservation of COX4I1. In vitro protection with miR-338 inhibitor was blocked by concurrent knockdown of COX4I1 with small interfering RNA. Parallel studies in mouse neuronal N2a cultures resulted in preserved ATP content and CcO activity with miR-338 inhibition, indicating a shared miR-338-dependent response to ischemic stress between brain cell types. These results suggest that miR-338 inhibition and/or COX4I1-targeted therapies may be novel clinical strategies to protect against stroke injury via preservation of mitochondrial function in multiple cell types.
Keywords: stroke, TMRE, oxidative phosphorylation, ATP, ROS, glia, miRNA
INTRODUCTION
Stroke remains the second leading cause of death worldwide and the most frequent source of permanent disability in adults worldwide [1]. It is responsible for nearly 5.5 million deaths every year, and 44 million disability-adjusted life-years lost, and projected to increase to 61 million by 2020 [2]. Despite hundreds of promising preclinical trials in animal studies focusing on neuronal gene targets, treatment options remain limited to thrombolysis within a short therapeutic window [3]. The failure to develop any effective therapies aimed at traditional neuronal gene targets suggests that focusing on alternative cellular targets is warranted. Astrocytes are specialized glial cells that coordinate neuronal function in health and disease [4]. As the most abundant cell type in the brain it is not surprising that new therapies that account for astrocytes have been emerging as a promising avenue for an alternative stroke treatment [5].
Mitochondria are central to ischemic cell death, by regulating oxidative stress, ATP production and intracellular Ca2+ handling [6], and mitochondrial dysfunction strongly contributes to ischemia/reperfusion injury [7]. Cytochrome c oxidase IV (COX IV) plays an essential role in the assembly of the cytochrome c oxidase complex (CcO; complex IV), controlling ATP production and mitochondrial biogenesis [8]. Ischemia promotes inhibition of COX IV protein synthesis and mitochondrial complex IV activity [9, 10], while post-ischemic reperfusion and recovery is associated with increased COX IV levels [10]. However, the relevant molecular mechanisms that coordinate COX IV expression and activity in the setting of cerebral ischemia remain poorly described.
MicroRNAs (miRNAs) are small (19–22 nucleotide) non-coding RNAs that regulate gene expression primarily at the post-transcriptional level by inhibiting the translation of target coding genes, and growing evidence supports a central role for microRNAs (miRNAs) in the molecular response to cerebral ischemia [11]. Modulation of brain-enriched miR-338 has been shown to alter mitochondrial oxygen consumption, ATP generation and the production of reactive oxygen species (ROS) in the axon [12]. Levels of miR-338 were upregulated more than 2-fold after 20 min of global ischemia followed by 30 min reperfusion in rat hippocampus [13], and observed to be increased in the cerebral spinal fluid (CSF) of ischemic stroke patients in the subacute stage [14]. MiR-338 has been utilized as a highly sensitive biomarker of mitochondrial toxicity [15], and alteration of miR-338 levels was shown to modulate the expression of COX IV and the subunit of mitochondrial ATP synthase ATP5G1 [16], and markedly affect neuronal ROS levels and axonal growth [16, 17]. However, the role of miR-338 and potential gene targets in the evolution of stroke injury has not been previously investigated. Therefore, in the present study we tested the hypothesis that elevated miR-338 contributes to stroke injury in vivo, and in astrocyte and neuronal mitochondrial dysfunction in vitro, by targeting COX4I1, the brain enriched isoform of COX IV.
MATERIALS AND METHODS
All experimental protocols using animals were approved by the Stanford University Animal Care and Use Committee and conducted in accordance with the NIH guide for the care and use of laboratory animals.
Intracerebroventricular (ICV) Pre-treatment
Adult male C57/B6 mice (age 8–10 wks, 25–30 g, Jackson Lab, Bar Harbor, ME, USA) were randomly assigned to either intracerebroventricular (ICV) pre-treatment with miR-338 antagomir, or mismatch-control 24h prior to 1h middle cerebral artery occlusion (MCAO). A power analysis using the power procedure in SAS 9.3 to achieve 80% power at the significance level α =0.05) with a predicted effect size of 25% identified cohorts of n=8 mice per treatment group were required. Mice were anesthetized with 2% isoflurane by facemask and placed in a stereotaxic frame. A 26-gauge brain infusion cannula was placed stereotactically into the left lateral ventricle (bregma: − 0.58 mm; dorsoventral: 2.1 mm; lateral: 1.2 mm) as previously described [18, 19]. 30 pmol miR-338 antagomir (in 2μL), mimic or mismatch-control (Life Technologies, Carlsbad, CA) was mixed with cationic lipid DOTAP (4μl; 6μl total volume; Roche Diagnostics, Indianapolis, IN) and infused over 20min.
Transient Focal Cerebral Ischemia
Adult male CB57/B6 mice (25–30 g from Jackson Lab) were anesthetized with 2% isoflurane in O2 by facemask and focal cerebral ischemia was produced by 1h of MCAO with a silicone-coated 6–0 monofilament (Doccol Co, Redlands, CA, USA) followed by reperfusion as described previously [20]. Rectal temperature was maintained at 37±0.5°C controlled by a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA, USA). Temperature and respiratory rate were monitored continuously. Mice with no evidence of acute neurological deficit or with evidence of hemorrhage were excluded from analysis. A total of 132 mice were subjected to sham or MCAO surgery, 9 were excluded from analysis (5 had evidence of hemorrhage and 4 had no evidence of neurological deficit acutely). No significant differences were observed in rates of death or exclusion between treatment groups.
Determination of Infarct Volume and Neurological Status
Cerebral infarction volume was determined at 24h or 7d reperfusion with 2,3,5 triphenyltetrazolium chloride (TTC, Sigma, T8877, Louis, MO) or Cresyl violet (EMD-Millipore Chemicals, Hayward, CA) after transcardiac perfusion with saline then fixation with 4% paraformaldehyde [21]. Infarct volume was quantified by a blinded observer assessing four 50μm coronal sections/brain and corrected for edema using Adobe Photoshop CS3 as described previously [22]. Neurological status was assessed after 24h and 7d of reperfusion by a blinded observer using a 4 point neurologic deficit score [18, 21, 23]. Neurological deficit was scored as follows: 0 - no observable neurological deficits, 1 - failure to extend right forepaw, 2 - circling to the right, 3 - falling to the right, 4 - cannot walk spontaneously.
Cell Cultures
Primary astrocyte cultures were prepared from postnatal (days 1–3) Swiss Webster mice (Charles River Laboratories, Wilmington, MA) as described previously [24]. Isolated astrocytes were seeded on 24-well plates in plating medium consisting of Eagle’s Minimal Essential Medium (Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum and 10% equine serum (Hyclone, Logan, UT), 21 mM (final concentration) glucose, and 10 ng/mL epidermal growth factor. Cultures were maintained at 37°C in a 5% CO2 incubator. N2a cell cultures were grown in high-glucose DMEM (Invitrogen, Carlsbad) supplemented with 8% fetal bovine serum (Hyclone) and antibiotics (50 U/mL penicillin + 50 μg/mL streptomycin; Invitrogen) in a humidified atmosphere containing 5% CO2 at 37 °C as we have previously reported [18]. For all experiments 3–4 independent cultures were tested as replicates within each experiment, and the experiment was repeated 3–4 times using cells obtained from different dissections.
In vitro Transfection Protocols
Primary astrocyte and N2a cultures were transfected with mismatch control, miR-338 mimic or inhibitor (Dharmacon Inc., Lafayette, CO, USA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol on day-in-vitro (DIV) 10 and 5, respectively. Expression of miR-338 was confirmed by RT-qPCR, as described below. Silencing of COX4I1 was carried out using Accell SMARTpool siRNAs (Dharmacon Inc.), each of which contains four small interfering RNAs (siRNAs) according to the manufacturer’s instructions:
5′-UGAGAAAGUUCAGUUGUAC-3′
5′-CCCUCAUACUUUCGAUCGU-3′
5′-CGCUCGUUCUGAUUUGGGA -3′
5′-CAUUUCUACUUCGGUGUGC-3′
Primary astrocyte cultures were transfected with 1μM siRNA and subsequently cultured in Accell delivery mix (Dharmacon Inc.). The efficiency of the knockdown was assessed by quantification of COX4I1 protein expression evaluated 72h after transfection by immunoblot. Neuronal cultures were transfected
Injury Paradigms and assessment of cell death
Glucose deprivation (GD) injury was selected as an ischemia-like stress for astrocyte cultures as it reliably induces mitochondrial dysfunction with increased ROS production [25], and was performed as we have described previously [7]. Briefly cells were washed twice with medium lacking glucose separated by a 15min equilibration period. Because vulnerability of primary astrocyte cultures to ischemic injury increases with age [26] an extended duration (72h) of GD was necessary to induce an adequate level of cell death in these relatively younger (DIV 10–12) cultures. As neuronal cultures are more resistant to GD injury, N2a injury was induced via serum-deprivation plus exposure to 500 μM H2O2 for 24h as we have previously performed (Stary). Assessment of cell viability and cell counting were performed after staining with Hoechst 33342 (5 μM, Sigma Chemicals) and propidium iodide (PI, 5 μM, Sigma Chemicals). PI stains dead cells while Hoechst is a cell-permeant nucleic acid stain that labels nuclei of both live and dead cells. PI-positive (dead) cells were manually counted by a blinded investigator; numbers of Hoechst-positive cells were calculated using an automated macro (Image J, v1.49b, National Institutes of Health, USA). PI-positive and Hoechst-positive cells were counted in 3 microscopic fields per well at 200X magnification using an automated LumaScope™ 720 (Etaluma; Carlsbad, CA, USA). The number of PI-positive cells was expressed as a percent of the total number of cells. In parallel, viability of primary astrocytes was quantified by measuring the concentration of intracellular lactate dehydrogenase released from dead cells into the media [27]. The results were expressed as the percentage of lactate dehydrogenase release compared with lactate dehydrogenase release after freeze-thaw to kill all cells.
Live-cell fluorescent imaging
Living primary astrocyte cultures were imaged using an automated LumaScope™ 720 (Etaluma) with a 20X fluor objective. Cells were maintained at 37°C and 5% CO2 in an atmospherically controlled imaging chamber (Ibidi GmbH, Martinsried, Germany) for imaging of live-cell intracellular kinetics. For assessment of mitochondrial membrane potential (MMP), astrocytes were incubated for 30min with tetramethylrhodamine ethylester (TMRE, 50nM, ThermoFisher Scientific) at 37°C, and the same concentration of TMRE was maintained in all bathing solutions throughout the experiments. Oxygen radical production was monitored with hydroethidine (HEt, ThermoFisher Scientific, Waltham, MA, USA). Cultures were incubated in the dark with 5μM HEt in BSS5.5 (30min, 37°C), and the same concentration of HEt was maintained in the bath throughout each experiment. Mean intensity of fluorescence was quantified by Image-J v1.49b software (NIH). Changes in fluorescence were normalized to the basal fluorescence for each cell at the start of the experiment.
Measurement of Mitochondrial Function
Cytochrome c oxidase (CcO) activity was determined using Cytochrome Oxidase Activity Colorimetric Assay Kit (BioVision, Milpitas, CA) according to the manufacturer’s instructions. Cells were harvested 6h after exposure to glucose-free medium or normal medium. Absorbance was measured at 550 nm every 30 sec for 300 sec. Cellular ATP concentrations were measured using the CellTiter-Glo luminescent ATP assay kit, based on the luciferase/luciferin reaction, from Promega (Madison, WI, USA) according to the manufacturer’s instructions. After 6h of GD, cells were lysed in the same plate with the reagent included in the assay kit for 10min. Then the mixtures were transferred into opaque-walled 96-well plates, and luminescence was assessed using a multimode microplate reader (Spark 10M, Tecan Group LTD, Mannedorf, Switzerland).
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated with TRIzol® (ThermoFisher Scientific, Waltham, MA). Reverse transcription was performed as previously described [20] using the TaqMan MicroRNA Reverse Transcription Kit for miR-338 and total RNA (Applied Biosystems, Foster City, CA). Predesigned primer/probes for PCR were obtained from ThermoFisher Scientific for mmu-miRNA-338–3p (#02252), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, #4331182) mRNA, COX4I1 mRNA and U6 small nuclear RNA (U6, #01973). PCR reactions were conducted as previously described [20] using the TaqMan® Assay Kit (Applied Biosystems). Measurements for miR-338 were normalized to U6 (ΔCt), those for COX4I1 were normalized to GAPDH. Comparisons were calculated as the inverse log of the ΔΔCT from controls [28].
Immunoblotting
Immunoblotting was performed as previously described [22]. Briefly, equal amounts (50μg) of protein were loaded and separated on a 4–12% polyacrylamide gel (Invitrogen), then electrotransferred to an Immobilon polyvinylidene fluoride membrane (EMD Millipore Corp. Burlington, MA, USA). Membranes were blocked and incubated overnight with primary antibody against COX4I1 (1:1000, PA5–29992, ThermoFisher Scientific) or COX4I2 (1:1000, PA5–49919, ThermoFisher Scientific), and β-actin (1:1000, 926–42,210, LI-COR Bioscience, Lincoln, NE, USA), washed and then incubated with 1:15,000 anti-rabbit antibody (926–32,221, LI-COR Bioscience). Immunoreactive bands were visualized using the LI-COR Odyssey™ infrared imaging system according to the manufacturer’s protocol. Densitometric analysis of bands was performed using Image-J software (v1.49b NIH).
Statistics
Statistical difference was determined using one-way ANOVA with Tukey’s post-hoc comparison for experiments with >2 groups at a single time point, a 2-way ANOVA with repeated measures for comparison of groups with multiple measurements over time (TMRE and HEt fluorescence), or student’s t-test for comparison of two groups at a single time point. In all tests, P<0.05 was considered significant. Data reported are means ± SD.
RESULTS
Brain levels of miR-338 were significantly increased 24h after MCAO, (Fig. 1A), before returning to baseline levels by 3d of reperfusion. A significant rise in brain levels of COX4I1 protein observed at 3h after MCAO was abolished coincident with the rise in miR-338 (Fig. 1A). A dose-response curve was performed for miR-338 inhibitor to identify the concentration to be used. Brain levels of miR-338 was assessed 24h after ICV injection of 3, 6, or 10 pmol/g miR-338 antagomir, which each resulted in a significant decrease (Fig. 1B). ICV injection of antagomir (10 pmol/g) 24h prior to MCAO significantly decreased levels of miR-338 by 73% with an associated increase in post-MCAO COX4I1 expression. (Fig. 1C).
Figure 1.
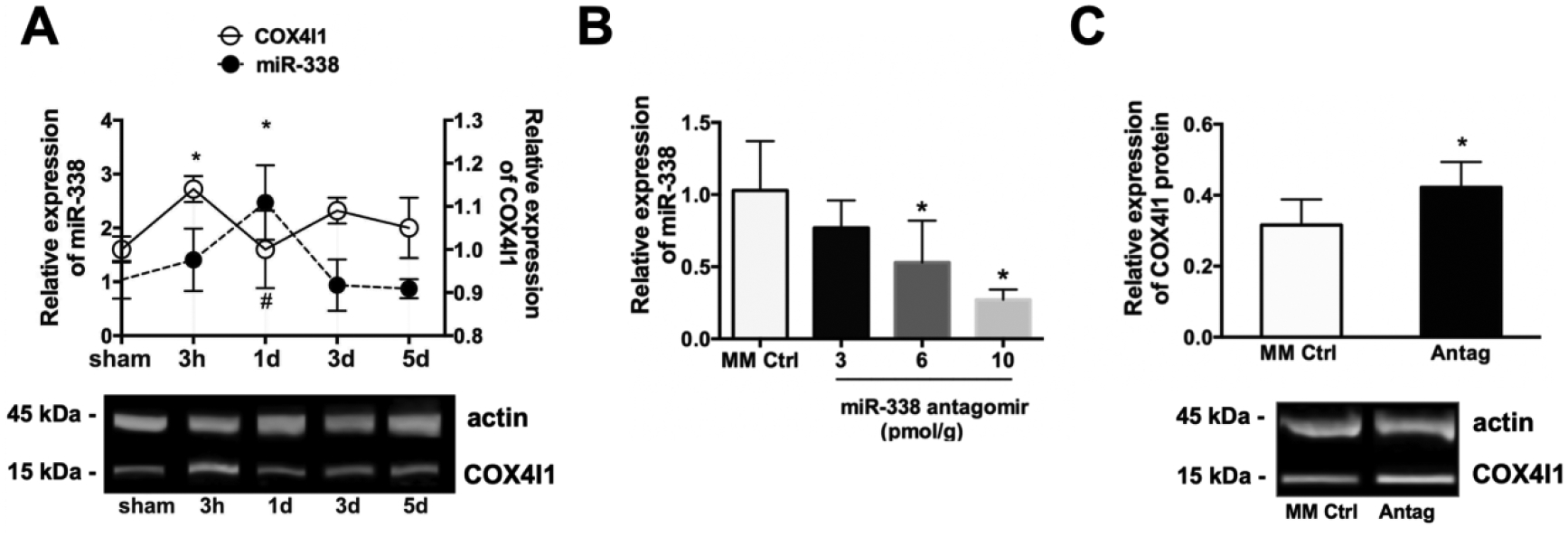
(A) Levels of miR-338 and COX4I1 protein in mouse brain subjected to 1 h MCAO (mean±SD, n=8 animals per treatment group; *P<0.05 vs. 0h). (B) Examples of immunoblots for COX4I1 and actin following MCAO. (C) Dose-response curve of miR-338 expression in brain with increasing concentration of miR-338 antag; *P<0.05 vs. mismatch control (MM Ctrl, n=4 each group; mean±SD, *P<0.05 vs. MM Ctrl). (A) Post-injury COX4I1 levels in the brain of mice pre-treated with ICV injection of 10 pmol miR-338 antagomir (Antag) or MM Ctrl (mean±SD, n=4 each group; *P<0.05 vs. MM Ctrl).
Stroke injury assessed by TTC staining (Fig. 2A) or Cresyl violet staining (Fig. 2B) was significantly (p<0.05) decreased in miR-338 antagomir pre-treated relative to MM-control at both 24h after reperfusion (29.43±7.0% versus 41.15±13.4%, respectively, Fig. 2C) and 7d after reperfusion (11.54±4.5% versus 17.68±4.6%, respectively, Fig. 2D). Concurrently, miR-338 antagomir pre-treatment resulted in significantly (P<0.05) improved post-stroke neurologic score at both 24h (Fig. 2E) and 7d (Fig. 2E) of reperfusion. Substantially decreased stroke injury and improved neurological score at 7d relative to 24h reperfusion is consistent with prior observations by others [29].
Figure 2. Effect of miR-338 inhibition on transient focal ceberal ischemia.
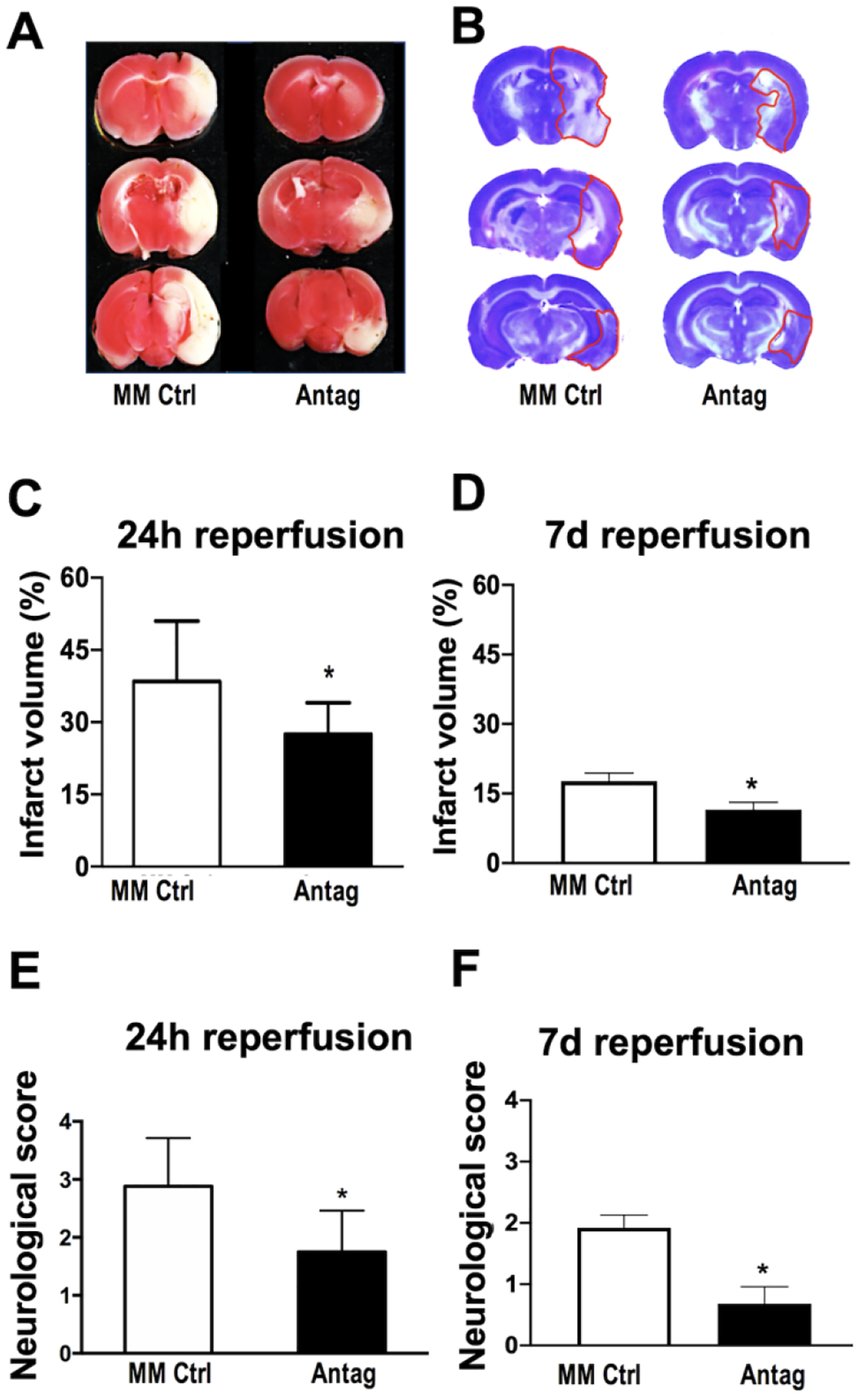
Representative TTC-stained (A) and Cresyl violet stained (B) coronal sections of brains to assess injury (lighter areas) 24h (A) and 7d (B) after 1h transient middle cerebral artery occlusion (MCAO) in mice pre-treated with either miR-338 antagomir (Antag) or mismatch control (MM Ctrl). (C,D) Quantification of infarct volume 24h after MCAO (C) and 7d after MCAO (D) in mice pre-treated with either Antag or MM Ctrl. (E,F) Neurologic deficit score 24h after MCAO (E) and 7d after MCAO (F) in mice pre-treated with either Antag or MM Ctrl. (mean±SD, n=8 animals per treatment group; *P<0.05 vs. MM Ctrl). TTC = 2,3,5-Triphenyltetrazolium chloride.
To investigate the effect of altered miR-338 levels on astrocyte cell survival, primary astrocyte cultures were transfected with miR-338 mimic, inhibitor, or MM-control 24h prior to exposure to glucose-free or normal medium. Glucose deprivation (GD, 6h) alone increased levels of miR-338 greater than 2.5 times compared to MM-control, an effect attenuated by miR-338 inhibitor (Fig. 3A). Pre-treatment with miR-338 mimic increased miR-338 levels more than 1800-fold (Fig. 3A). Increased levels of miR-338 with mimic did not result in increased cell death from GD, however pre-treatment with miR-338 inhibitor provided protection when assessed by LDH assay (Fig. 3B), and qualitatively verified with PI/Hoechst staining (Fig. 3C).
Figure 3. Effect of miR-338 inhibition on astrocyte survival following in vitro ischemic injury.
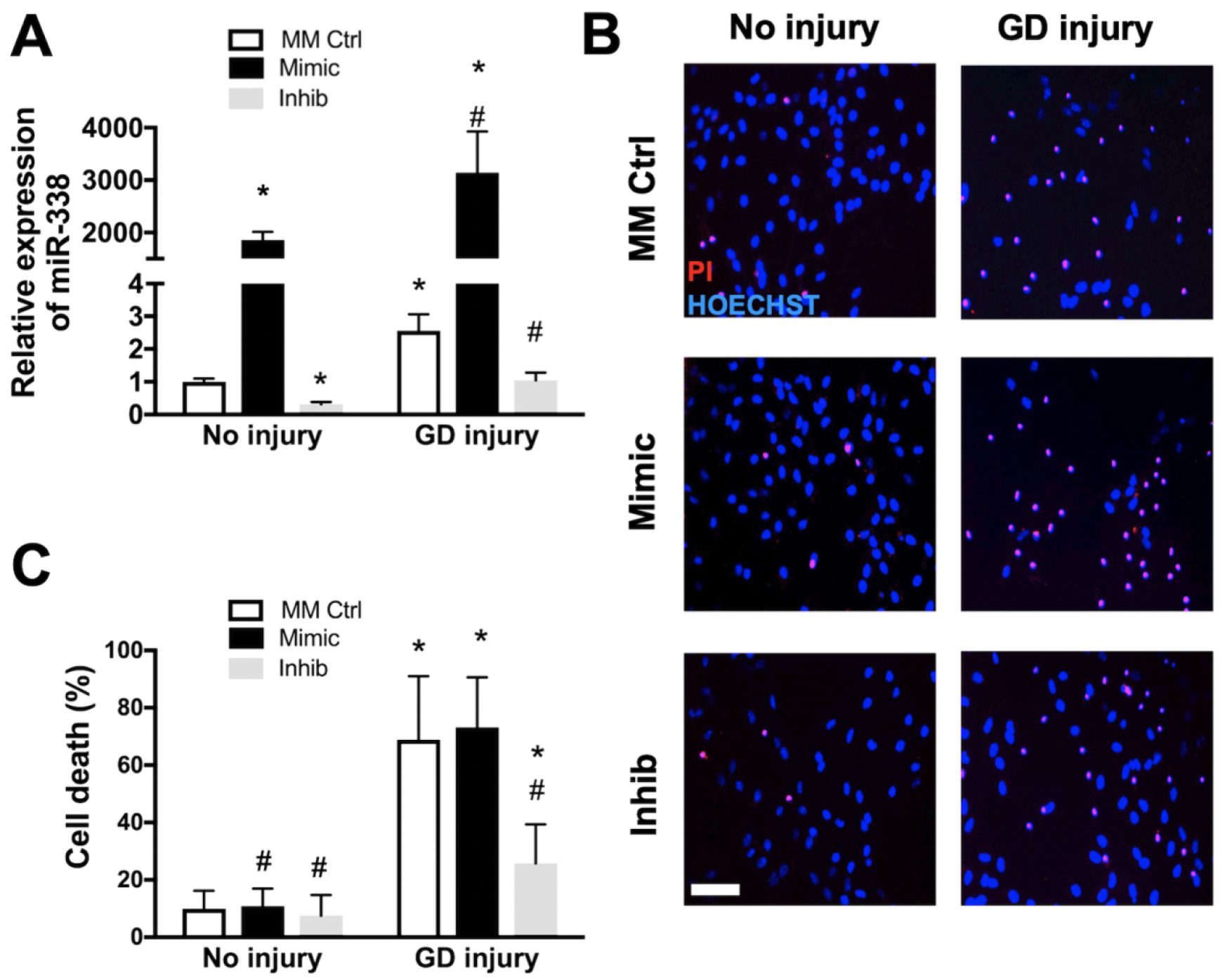
(A) MiR-338 expression in primary astrocyte cultures 72h after 6h glucose deprivation (GD) injury. (B) Astrocytes pre-treated with mismatch control (MM Ctrl), miR-338 mimic (Mimic) or miR-338 inhibitor (Inhib) stained with propidium iodide (red, dead cells) and Hoechst (blue, all cell nuclei) after 72h GD. (C) Quantification of cell death in astrocytes pre-treated with MM Ctrl, mimic or inhib with or without GD injury assessed by lactate dehydrogenase release. All graphs mean±SD; each graph represents data pooled from at least 5 experiments with n=3–4 per treatment group *P<0.05 vs. No injury + MM Ctrl, #P<0.05 vs. GD injury + MM Ctrl. Scale bar = 25μm.
Pre-treatment of astrocyte cultures with COX4I1 siRNA resulted in a significant (P<0.05) decrease in COX4I1 protein levels (Fig. 4A). However co-treatment with COX4I1 siRNA and miR-338 inhibitor notably significantly blocked the protective effect of miR-338 inhibition (Figs. 4B,C). These results demonstrate that the protective effect of miR-338 against in vitro ischemic injury in these astrocytes cultures occurs, at least in part, via augmentation of COX4I1 protein expression.
Figure 4. Effect of COX4I1 knockdown in astrocytes following in vitro ischemic injury.
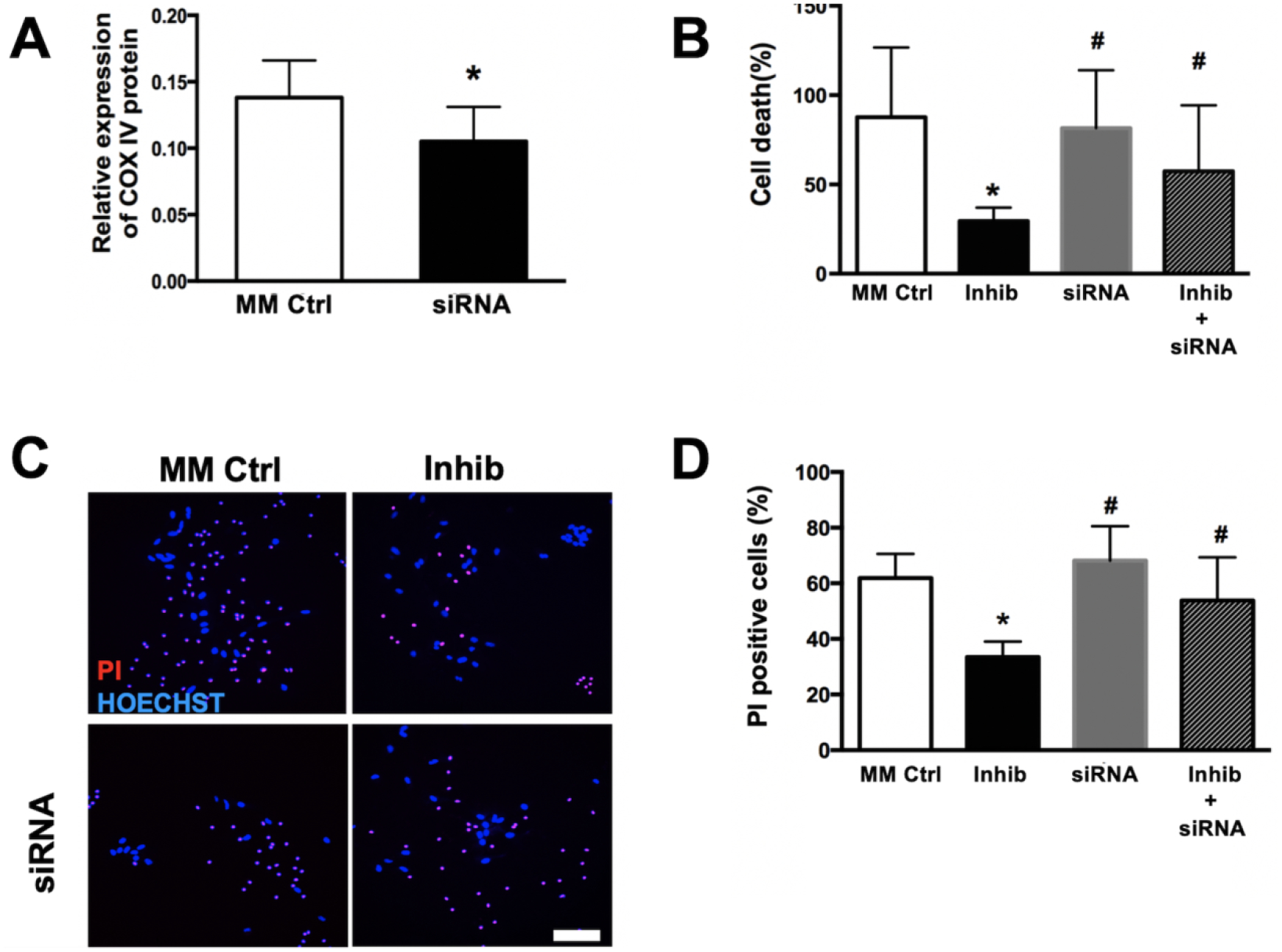
(A) COX4I1 protein expression after 24h of 6h glucose deprivatoin (GD) in primary astrocyte cultures, with and without pre-treatment with COX4I1 small interfering RNA (siRNA). (B) Quantification of cell death following 72h after 6h GD in astrocytes pre-treated with mismatch control (MM Ctrl), miR-338 inhibitor (Inhib) and COX4I1 siRNA +/− Inhib co-transfection. Representative pictographs (C) and quantification (D) of cell death with Hoechst (blue, all nuclei) and propidium iodide (red, dead cells) 72h after 6h GD is astrocytes pre-treated with MM Ctrl, Inhib and COX4I1 siRNA +/− Inhib co-transfection. All graphs mean±SD; each graph represents data pooled from at least 4 experiments with n=3–4 per treatment group; *P<0.05 vs. Ctrl, #P<0.05 vs. Inhib. Scale bar = 25μm.
Disruption in astrocyte mitochondrial function was determined qualitatively with the potentiometric fluorescent dyes TMRE (MMT) and HEt (oxidative stress), respectively. After 2h of GD, a significant decrease in TMRE fluorescence intensity was observed in all treatment conditions (Figs. 5A,B). Pre-treatment with miR-338 mimic trended towards a greater reduction in TMRE fluorescence (P=0.091), while pre-treatment with miR-338 inhibitor trended towards attenuation in TMRE fluorescence (P=0.094). After 2h of GD HEt fluorescence intensity was significantly (P>0.05) increased in all groups, with no differences between treatment groups. (Fig. 5C). 24h after 6hr GD resulted in a significant (P<0.05) decrease in astrocyte ATP levels in both MM-control and mir-338 mimic groups, however pre-treatment with miR-338 inhibitor attenuated the decline (Fig 5D). CcO activity decreased significantly 24h after 6h GD injury, an effect significantly (P<0.05) enhanced by pre-treatment with miR-338 mimic and attenuated by inhibitor (Fig. 5E). COX4I1 mRNA expression was significantly (P<0.05) reduced 24h after 6h GD injury, an effect attenuated by miR-338 inhibitor pretreatment (Fig. 5F).
Figure 5. Effect of miR-338 on astrocyte mitochondrial function following in vitro ischemic injury.
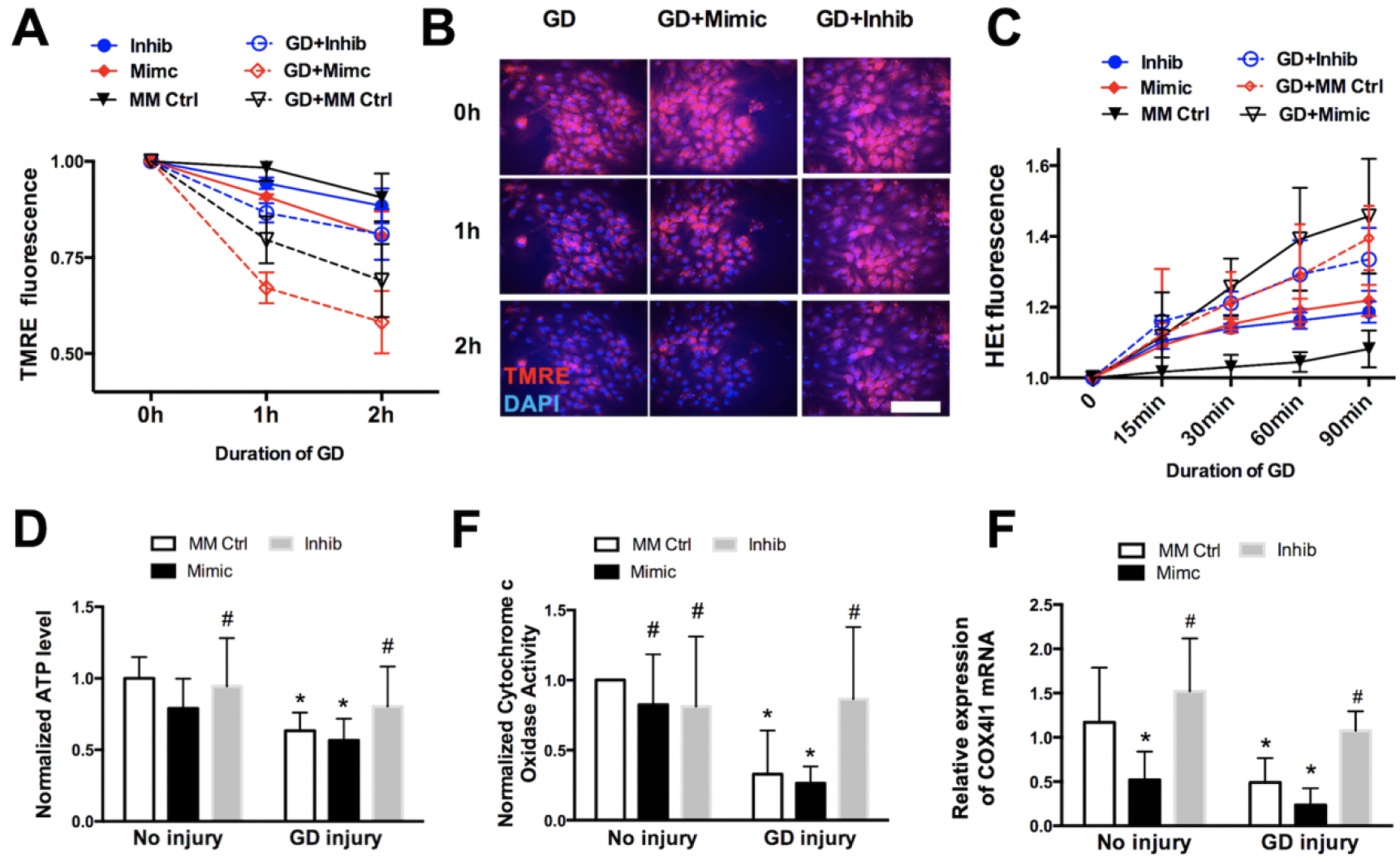
(A) Assessment of mitochondrial membrane potential with TMRE fluorescence in primary astrocyte cultures with and without glucose deprivation (GD) injury, pre-treated with either mismatch control (MM Ctrl), miR-338 mimic (Mimic) or miR-338 injibitor (Inhib). (B) Representative fluorescence micrographs of TMRE (red) fluorescence in astrocytes pre-treated with MM Ctrl, Mimic or Inhib 2h after GD or wash control. (C) Assessment of oxidative streass with hydroethedine (HEt) fluorescence in astrocytes pre-treated with MM Ctrl, Mimic or Inhib and subjected to GD or wash control. (D) ATP production, and (E) activity of cytochrome c oxidase in astrocyte cultures pre-treated with MM Ctrl, Mimic or Inhib 24h after 6h after GD or wash control. (F) COX4I1 levels in transfected astrocytes 24h after 6h GD or wash control. All graphs mean±SD; each graph represents data pooled from at least 4 experiments with n=3–4 per treatment group; *P<0.05 vs. No inury + MM Ctrl; #P<0.05 vs. GD injury + MM Ctrl. TMRE: tetramethylrhodamine ethylester. Scale bar = 25μm.
In neuronal N2a cultures, mitochondrial function was determined before and after 24h of SD+H2O2 injury, with either miR-338 mimic, inhibitor or MM-control pre-treatment. Similar to astrocyte cultures simulated ischemia in neuronal cultures resulted in a significant (P<0.05) decrease in astrocyte ATP levels in both MM-control and mir-338 mimic groups, however pre-treatment with miR-338 inhibitor attenuated the decline (Fig 6A). CcO activity in N2a cells also paralleled astrocyte cultures with a significant decrease after SD+H2O2 injury, that was significantly (P<0.05) worsened by pre-treatment with miR-338 mimic and attenuated by miR-338 inhibitor (Fig. 6B)
Figure 6. Effect of miR-338 on mitochondrial function in N2a cell cultures following in vitro ischemic injury.
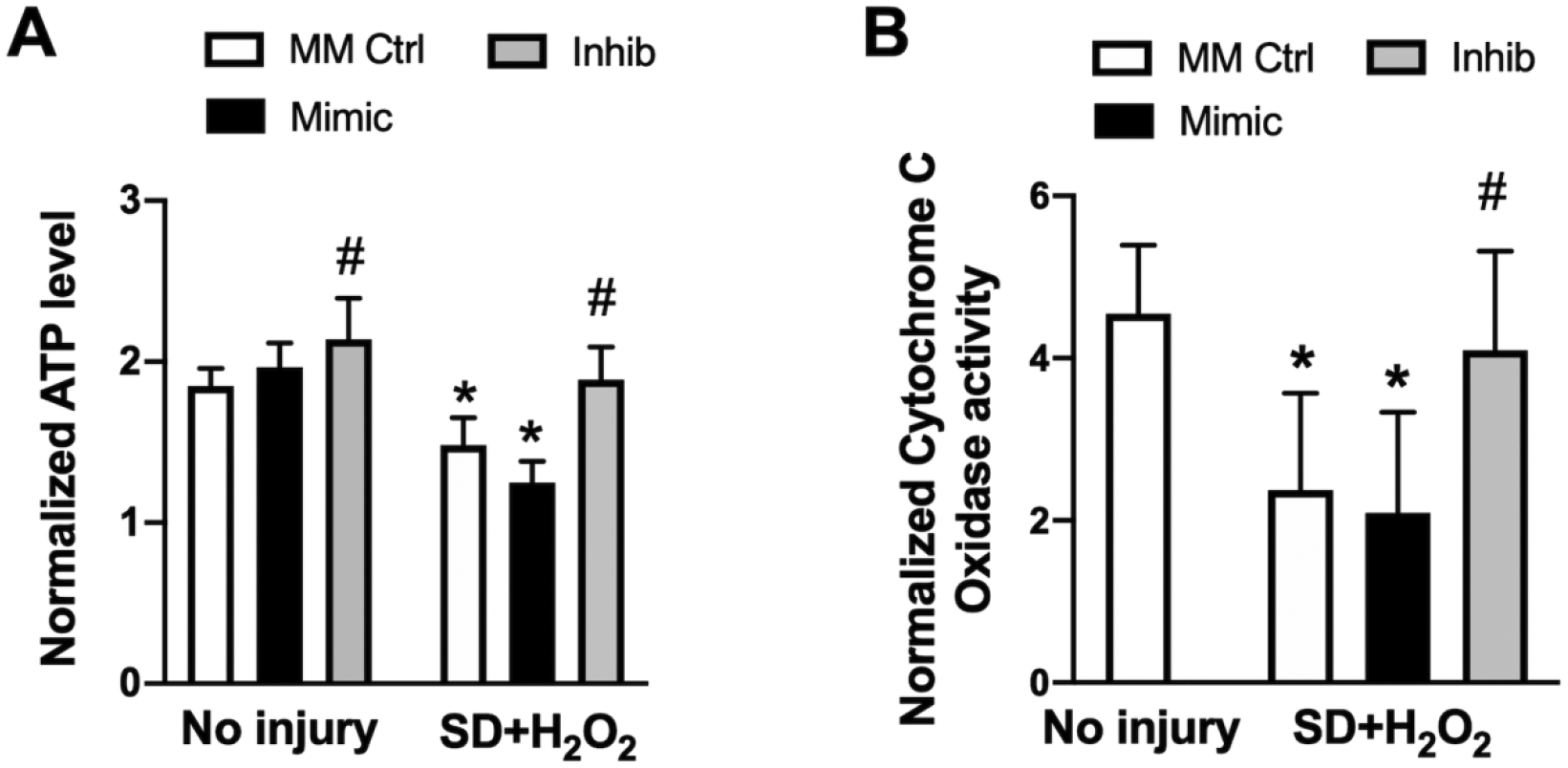
(A) ATP levels, and (B) activity of cytochrome c oxidase in mouse neuroblastoma (N2a) cell cultures pre-treated with mismatch control (MM Ctrl), miR-338 mimic (Mimic) or miR-338 inhibitor (Inhib) after 24h serum deprivation (SD) plus 500μM H2O2 or wash control. All graphs mean±SD; each graph represents data pooled from at least 4 experiments with n=3–4 per treatment group; *P<0.05 vs. No inury + MM Ctrl; #P<0.05 vs. GD injury + MM Ctrl.
DISCUSSION
In the present study, we demonstrated that preventing the early increase in miR-338 in the brain following MCAO is protective, evidenced by reduced infarction volume and improved neurological status. One miR-338 target that is increased following injury is COX4I1. Deficiency in COX IV reduces CcO activity and the rate of mitochondrial respiration and increases the accumulation of cellular and mitochondrial ROS [30]. Levels of COX IV have been shown to be increased by post-stroke exercise, correlating with significant improvements in behavioral scores and cerebral infarct volume [31]. Notably, it has been shown that astrocyte mitochondrial damage contributes to neurodegeneration [32]. Consistent with these observations, miR-338 inhibition preserved mitochondrial function in both astrocyte and neuronal cultures after in vitro ischemia. Based on these observations we propose a model (Fig. 7) whereby elevations in brain levels of miR-338 that occur in the acute phase after transient cerebral ischemia induce disruptions in mitochondrial oxidative phosphorylation via suppression of COX4I1, resulting in augmented ROS production, decreased ATP availability, and ultimately leading to brain cell death and accompanying neurologic impairment.
Figure 7.
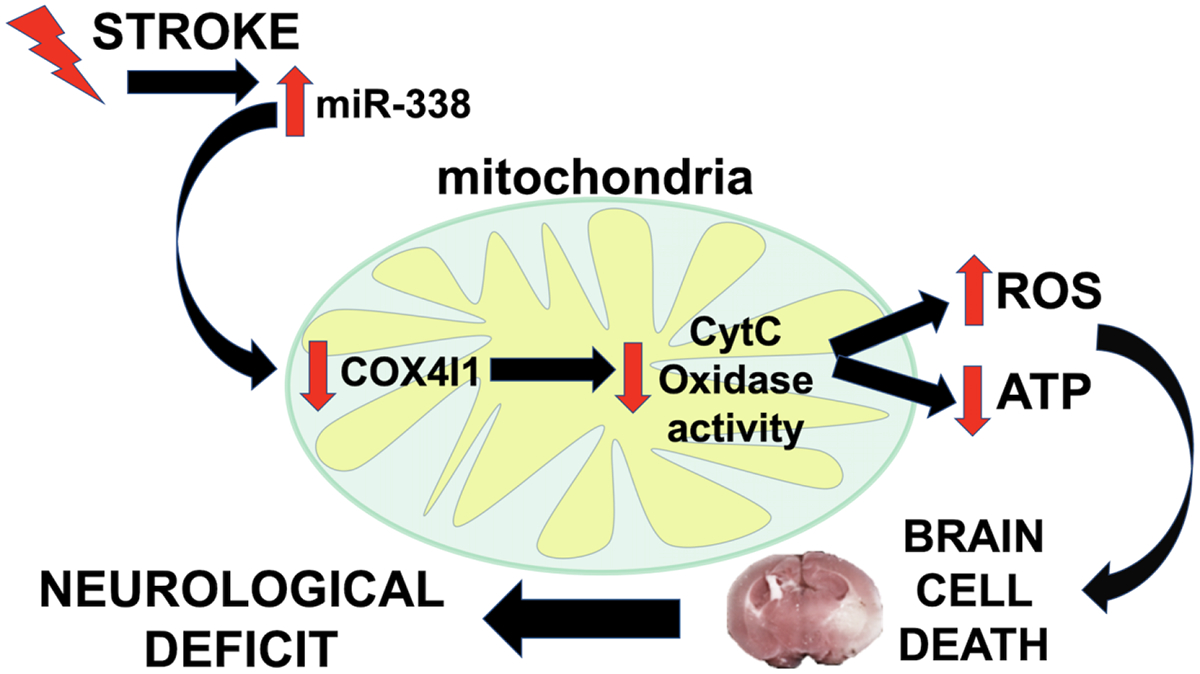
Schematic illustrating miR-338 role in early mitochondrial dysfunction and downstream neurological impairment after stroke.
Evidence provided by Vargas et al. [33] suggests that mitochondrial-localized pre-miR-338 may serve as a reservoir of miRNAs to regulate subcellular translation of nuclear-encoded mitochondrial mRNAs. COX IV, a target of miR-338 [17], is nuclear-encoded and also a precursor protein, and as such is translated in the cell body and imported into mitochondria using a mitochondrial-targeting sequence. There are two isoforms of COX4I: COX4I1 and COX4I2. COX4I2 is specific to lung and trachea [34] and was not changed in our MCAO model. In silico analysis (TargetScan v7.1) and prior reports [17] identify COX4I1 and not COX4I2 as a target of miR-338, consistent with the results of the present study. COX4I1 protein levels changed inversely relative to miR-338 levels in brains after MCAO, and in cultures. COX IV is a conserved protein whose levels are generally stable [35]. Unlike the protein, COX IV mRNA levels are more prone to regulation. Zhang et al. [36] demonstrated that the half-life of COX IV mRNA was increased from 50 min to 84 min in primary neurons following depolarizing KCl treatment that also increased CcO activity. Their findings suggested that COX IV could be regulated at both the synthetic and the degradative levels. Although COX4I1 mRNA levels in primary astrocytes remained unchanged with altered miR-338 levels, protein levels decreased, suggesting translational silencing of COX4I1 by miR-338, a known effect of miRNAs. Moreover, the protective effect of miR-338 inhibition on GD-induced cell death in astrocyte cultures was blocked by COX4I1 knockdown with siRNA, suggesting that the protective effect of miR-338 inhibition occurred at least in part by preventing targeted silencing of COX4I1 translation.
MiR-338 has also been shown to have additional targets relevant to mitochondrial function, including ATP5G1, parvalbumin, and MAP3K2, and has therefore been proposed as a general biomarker of mitochondrial toxicity [15]. It is possible that alternative mitochondria-related targets of miR-338 contributed to the observed protection conveyed by miR-338 inhibition. Numerous studies have demonstrated that mitochondrial dysfunction plays a key role in the pathophysiology of neurological diseases, including stroke [37]. Ischemic neuronal injury occurs secondary to glutamate excitotoxicity, calcium overload, and generation of ROS [38], and mitochondria are critical to many of these processes. Astrocyte mitochondrial homeostasis is essential for neuroglial protective mechanisms as inhibition of astrocytic mitochondrial function induces neuronal dysfunction and cell death [32]. In the normal physiologic state astrocytes provide metabolic support to neurons in the form of ATP and substrate for glycolysis [4]. Therefore, the observed protection with miR-338 inhibition in vivo could be due to a combination of direct protection of both astrocytes and neurons, as well as indirect neuronal support by preserving astrocytic mitochondrial homeostasis.
Notably, in the present study we observed that while GD injury induced MMP depolarization, ROS accumulation and decreased ATP levels in astrocytes, miR-338 inhibition attenuated MMP depolarization and ATP depletion, but not increased ROS levels. One explanation for this observation is that astrocytes are known to regulate the status of major redox coupling systems in the brain [39] through their high content of antioxidant compounds and enzymes. Therefore, astrocytes are well equipped to protect themselves when exposed to excess ROS [40], an effect that translates to improved capacity to protect neurons from injury. However, parallel studies in neuronal cultures verified a shared protective mechanism with miR-338 inhibitor, whereby ATP content and CcO activity were preserved with in vitro ischemia. Further studies investigating the role of miR-338 in neuroglial interactions are therefore warranted. Interestingly, Sharma et al.[41] reported an increase in COX IV expression in primary neuronal cultures from males but not female cultures after in vitro ischemic injury, suggesting that sex differences in the miR-338 response to cerebral ischemia may also be relevant.
CONCLUSIONS
In the present study we observed that in vivo ICV pre-treatment with miR-338 antagomir reduced infarct volume and improved neurological status following transient focal cerebral ischemia at both 24h and 7d after injury, which was associated with preserved COX4I1 levels. In parallel, in vitro experiments in primary astrocyte and neuronal N2a cultures pre-treated with miR-338 inhibitor demonstrated preservation of mitochondrial function after in vitro ischemia. Knockdown of the miR-338 target COX4I1 confirmed that miR-338 regulates ischemic injury via targeting of COX4I1, at least in part. These results imply that miR-338/COX4I1-mediated strategies could protect against focal cerebral ischemic injury in a manner independent of cell-type. Future investigations should therefore individually test alternative miR-338 gene targets and/or pharmacologic manipulation of COX4I1 activity as promising therapeutic treatment strategies for stroke, including testing in clinically relevant aged and female cohorts.
HIGHLIGHTS.
Pre-treatment with miR-338 antagomir reduced stroke severity and improved neurobehavior 24h and 7d after injury.
Silencing of miR-338 in vivo and in vitro augmented COX4I1 protein levels and improved cell survival in astrocytes subjected to simulated ischemia, an effect blocked by COX4I1 siRNA.
MiR-338 inhibition preserved ATP levels and cytochrome c oxidase activity in both astrocyte and neuronal cultures subjected to in vitro ischemia.
FUNDING
This study was funded by NIH grants NS084396 and NS080177 to RGG, American Heart Association Grant 18POST33990395 to BBG, and American Heart Association grants FTF-19970029 and NIH NS107445 to CMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no financial disclosures.
DISCLOSURES
None
REFERENCES
- [1].Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010, Lancet, 380 (2012) 2197–2223. [DOI] [PubMed] [Google Scholar]
- [2].Mukherjee D, Patil CG, Epidemiology and the global burden of stroke, World Neurosurg, 76 (2011) S85–90. [DOI] [PubMed] [Google Scholar]
- [3].Adams HP Jr., del Zoppo G, Alberts MJ, Bhatt DL, Brass L, Furlan A, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists, Stroke, 38 (2007) 1655–1711. [DOI] [PubMed] [Google Scholar]
- [4].Clarke LE, Barres BA, Emerging roles of astrocytes in neural circuit development, Nat Rev Neurosci, 14 (2013) 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu LJ, Ouyang YB, Xiong X, Stary CM, Giffard RG, Post-stroke treatment with miR-181 antagomir reduces injury and improves long-term behavioral recovery in mice after focal cerebral ischemia, Exp Neurol, 264 (2015) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B, Quiroga C, et al. Mitochondria, myocardial remodeling, and cardiovascular disease, Current hypertension reports, 14 (2012) 532–539. [DOI] [PubMed] [Google Scholar]
- [7].Stary CM, Sun X, Ouyang Y, Li L, Giffard RG, miR-29a differentially regulates cell survival in astrocytes from cornu ammonis 1 and dentate gyrus by targeting VDAC1, Mitochondrion, 30 (2016) 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li Y, Park JS, Deng JH, Bai Y, Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex, J Bioenerg Biomembr, 38 (2006) 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vijayasarathy C, Damle S, Prabu SK, Otto CM, Avadhani NG, Adaptive changes in the expression of nuclear and mitochondrial encoded subunits of cytochrome c oxidase and the catalytic activity during hypoxia, Eur J Biochem, 270 (2003) 871–879. [DOI] [PubMed] [Google Scholar]
- [10].Racay P, Tatarkova Z, Drgova A, Kaplan P, Dobrota D, Ischemia-reperfusion induces inhibition of mitochondrial protein synthesis and cytochrome c oxidase activity in rat hippocampus, Physiol Res, 58 (2009) 127–138. [DOI] [PubMed] [Google Scholar]
- [11].Ouyang YB, Stary CM, Yang GY, Giffard R, microRNAs: innovative targets for cerebral ischemia and stroke, Current drug targets, 14 (2013) 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aschrafi A, Kar AN, Natera-Naranjo O, Macgibeny MA, Gioio AE, Kaplan BB, MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery, Cellular and molecular life sciences : CMLS, DOI 10.1007/s00018-012-1064-8(2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M, MicroRNAs expression and function in cerebral ischemia reperfusion injury, J Mol Neurosci, 53 (2014) 242–250. [DOI] [PubMed] [Google Scholar]
- [14].Peng G, Yuan Y, Wu S, He F, Hu Y, Luo B, MicroRNA let-7e Is a Potential Circulating Biomarker of Acute Stage Ischemic Stroke, Transl Stroke Res, 6 (2015) 437–445. [DOI] [PubMed] [Google Scholar]
- [15].Baumgart BR, Gray KL, Woicke J, Bunch RT, Sanderson TP, Van Vleet TR, MicroRNA as biomarkers of mitochondrial toxicity, Toxicol Appl Pharmacol, 312 (2016) 26–33. [DOI] [PubMed] [Google Scholar]
- [16].Aschrafi A, Kar AN, Natera-Naranjo O, MacGibeny MA, Gioio AE, Kaplan BB, MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery, Cell Mol Life Sci, 69 (2012) 4017–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB, MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons, The Journal of neuroscience : the official journal of the Society for Neuroscience, 28 (2008) 12581–12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stary CM, Xu L, Sun X, Ouyang YB, White RE, Leong J, Li J, Xiong X, Giffard RG, MicroRNA-200c contributes to injury from transient focal cerebral ischemia by targeting Reelin, Stroke, 46 (2015) 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG, Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia, Stroke; a journal of cerebral circulation, 42 (2011) 2026–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ouyang YB, Lu Y, Yue S, Xu LJ, Xiong XX, White RE, Sun X, Giffard RG, miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo, Neurobiol Dis, 45 (2012) 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Stary CM, Xu L, Li L, Sun X, Ouyang YB, Xiong X, Zhao J, Giffard RG, Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-alpha, Mol Cell Neurosci, 82 (2017) 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG, Postischemic brain injury is attenuated in mice lacking the beta2-adrenergic receptor, Anesthesia and analgesia, 108 (2009) 280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Garcia JH, Wagner S, Liu KF, Hu XJ, Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation, Stroke, 26 (1995) 627–634; discussion 635. [DOI] [PubMed] [Google Scholar]
- [24].Ouyang YB, Xu LJ, Sun YJ, Giffard RG, Overexpression of inducible heat shock protein 70 and its mutants in astrocytes is associated with maintenance of mitochondrial physiology during glucose deprivation stress, Cell stress & chaperones, 11 (2006) 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Papadopoulos MC, Koumenis IL, Dugan LL, Giffard RG, Vulnerability to glucose deprivation injury correlates with glutathione levels in astrocytes, Brain Res, 748 (1997) 151–156. [DOI] [PubMed] [Google Scholar]
- [26].Papadopoulos MC, Koumenis IL, Yuan TY, Giffard RG, Increasing vulnerability of astrocytes to oxidative injury with age despite constant antioxidant defenses, Neuroscience, 82 (1998) 915–925. [DOI] [PubMed] [Google Scholar]
- [27].Xu L, Koumenis IL, Tilly JL, Giffard RG, Overexpression of bcl-xL protects astrocytes from glucose deprivation and is associated with higher glutathione, ferritin, and iron levels, Anesthesiology, 91 (1999) 1036–1046. [DOI] [PubMed] [Google Scholar]
- [28].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods, 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [29].Rewell SS, Churilov L, Sidon TK, Aleksoska E, Cox SF, Macleod MR, Howells DW, Evolution of ischemic damage and behavioural deficit over 6 months after MCAo in the rat: Selecting the optimal outcomes and statistical power for multi-centre preclinical trials, PLoS One, 12 (2017) e0171688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Oliva CR, Markert T, Gillespie GY, Griguer CE, Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas, Oncotarget, 6 (2015) 4330–4344.25726526 [Google Scholar]
- [31].Zhang Q, Wu Y, Zhang P, Sha H, Jia J, Hu Y, Zhu J, Exercise induces mitochondrial biogenesis after brain ischemia in rats, Neuroscience, 205 (2012) 10–17. [DOI] [PubMed] [Google Scholar]
- 32.Voloboueva LA, Suh SW, Swanson RA, Giffard RG, Inhibition of mitochondrial function in astrocytes: implications for neuroprotection, J Neurochem, 102 (2007) 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vargas JN, Kar AN, Kowalak JA, Gale JR, Aschrafi A, Chen CY, Gioio AE, Kaplan BB, Axonal localization and mitochondrial association of precursor microRNA 338, Cellular and molecular life sciences : CMLS, DOI 10.1007/s00018-016-2270-6(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huttemann M, Kadenbach B, Grossman LI, Mammalian subunit IV isoforms of cytochrome c oxidase, Gene, 267 (2001) 111–123. [DOI] [PubMed] [Google Scholar]
- 35.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S, The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A, Science, 272 (1996) 1136–1144. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C, Wong-Riley MT, Synthesis and degradation of cytochrome oxidase subunit mRNAs in neurons: differential bigenomic regulation by neuronal activity, Journal of neuroscience research, 60 (2000) 338–344. [DOI] [PubMed] [Google Scholar]
- [37].Ouyang YB, Giffard RG, MicroRNAs affect BCL-2 family proteins in the setting of cerebral ischemia, Neurochem Int, 77 (2014) 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lo EH, Dalkara T, Moskowitz MA, Mechanisms, challenges and opportunities in stroke, Nature reviews. Neuroscience, 4 (2003) 399–415. [DOI] [PubMed] [Google Scholar]
- [39].Schreiner B, Romanelli E, Liberski P, Ingold-Heppner B, Sobottka-Brillout B, et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS, Cell Rep, 12 (2015) 1377–1384. [DOI] [PubMed] [Google Scholar]
- [40].Dringen R, Pfeiffer B, Hamprecht B, Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione, The Journal of neuroscience : the official journal of the Society for Neuroscience, 19 (1999) 562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sharma J, Johnston MV, Hossain MA, Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation, BMC Neurosci, 15 (2014) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]