

Abstract
Infectious diseases present a major threat to public health globally. Pathogens can acquire resistance to anti-infectious agents via several means including transporter-mediated efflux. Typically, multidrug transporters feature spacious, dynamic, and chemically malleable binding sites to aid in the recognition and transport of chemically diverse substrates across cell membranes. Here, we discuss recent structural investigations of multidrug transporters involved in resistance to infectious diseases that belong to the ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the drug/metabolite transporter (DMT) superfamily, the multidrug and toxic compound extrusion (MATE) family, the small multidrug resistance (SMR) family, and the resistance-nodulation-division (RND) superfamily. These structural insights provide invaluable information for understanding and combatting multidrug resistance.
Keywords: membrane transporters, multidrug resistance, structural biology, infectious disease, ABC transporters, MFS transporters, DMT, SMR transporters, MATE transporters, RND transporters, tripartite complexes
Graphical Abstract
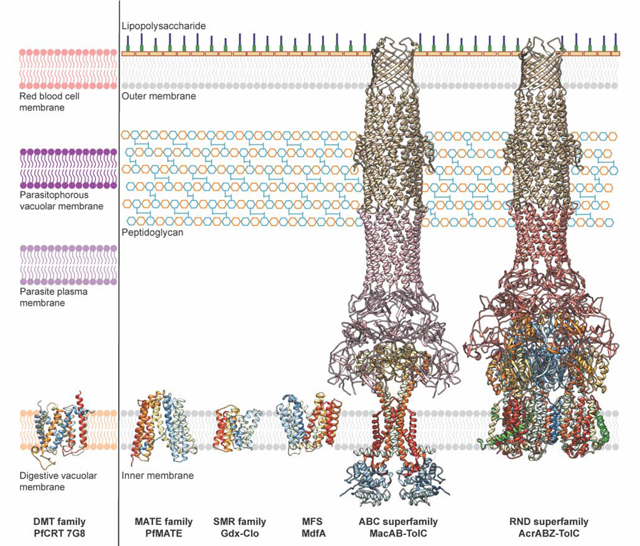
Representative multidrug transporter structures and their cellular locations. Representative structures of multidrug transporters from the DMT, MATE, SMR, MFS, ABC, and RND (super)families. Left: PfCRT (PDB ID: 6UKJ) is expressed in the digestive vacuolar membrane of the malaria-causing parasite Plasmodium falciparum, where it mediates efflux of 4-aminoquinolines from their site of action. Right: Multidrug transporters including PfMATE (ID: 6FHZ), Gdx-Clo (PDB ID: 6WK5), MdfA (PDB ID: 6GV1), MacB (PDB ID: 5NIK), and AcrB (PDB ID: 5NG5) are shown in the inner membrane of Gram-negative bacteria where they mediate efflux of a broad range of cytotoxic compounds (see Table 1). MacB and AcrB form tripartite complexes with TolC via the PAPs MacA and AcrA, respectively, to mediate efflux across the periplasm and outer membrane. Additionally, the AcrAB-TolC tripartite complex interacts with the inner membrane peptide AcrZ (green). Inner membrane proteins are shown in ribbon representation and colored in rainbow from the N- (blue) to the C- (red) terminus. MacA, AcrA, and TolC are colored in pink, salmon, and gold, respectively. Figure inspired by Dijun et al., 2018 [14].
Introduction
Drug resistance poses an increasingly serious threat to public health with devastating consequences for the treatment of many pathologies, including cancer and infectious diseases. In the latter case, this resistance is a mechanism that pathogenic microorganisms such as fungi, parasites, and bacteria have developed to mitigate the effects of cytotoxic drugs, and ultimately promote their survival. While sources of resistance are various and complex [1], the ability of bacteria to export cytotoxic drugs through efflux transporters is a major underlying cause. Exposure to drugs often triggers cellular pathways that alter the expression of transporter genes, ultimately providing increased opportunity for drug efflux. Multidrug transporters typically have broad substrate profiles and thus provide an export route for many different, often chemically unrelated cytotoxic compounds. There are several families of multidrug efflux transporters expressed by common pathogens including: the ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the drug/metabolite transporter (DMT) superfamily, the multidrug and toxic compound extrusion (MATE) family, the small multidrug resistance (SMR) family, the resistance-nodulation-division (RND) superfamily, the proteobacterial antimicrobial compound efflux (PACE) family, and the p-aminobenzoyl-glutamate transporter (AbgT) family [2–5]. With the exception of primary-active ABC transporters that utilize energy from ATP hydrolysis to catalyze drug export, these transporters are typically ‘secondary-active’ that harness energy from electrochemical gradients to power drug efflux.
Over the past two decades, technological advances in membrane protein structural biology have allowed for many structures of multidrug transporters to be determined using either X-ray protein crystallography, nuclear magnetic resonance (NMR), single-particle cryogenic-electron microscopy (cryo-EM), and very recently cryo-electron tomography (cryo-ET) [6]. These structures provide unprecedented insight into the molecular mechanisms by which transporters recognize and export broad spectrums of structurally distinct substrates from pathogenic invaders. Furthermore, these structures can provide guidance for the development of inhibitors that block multidrug transporters, and anti-infectious agents that are chemically similar enough to maintain efficacy, but distinct enough to prevent efflux.
Here, we discuss recent structural investigations of multidrug transporters involved in resistance to infectious diseases that belong to the ABC, MFS, MATE, DMT, SMR, and RND transporter (super)families (Table 1). We focus on the molecular bases of substrate recognition and transport, as well as the locations of known resistance-causing mutations, and how these findings translate into a mechanistic understanding of multidrug resistance.
Table 1.
Structures of multidrug transporters involved in resistance to infectious diseases
| Family | Protein | Drugs Resistance Profile | Organism | PDB | References |
|---|---|---|---|---|---|
| ABC transporter | MacB | Macrolides, bacitracin, colistin, enterotoxin STII, protoporphyrin | Acinetobacter baumannii | 5WS4 | [164] |
| Aggregatibacter actinomycetemcomi tans | 5LIL | [161] | |||
| Escherichia coli | 5NIL | [102] | |||
| Streptococcus pneumoniae | 5XU1 | [166] | |||
| Sav1866 | Hoechst 33342, verapamil, tetraphenylphosphonium (TPP+) | Staphylococcus aureus | 2HYD | [130] | |
| 2ONJ | [167] | ||||
| TM287/288 | Hoechst 33342, daunomycin | Thermotoga maritima | 3QF4 | PMID: 22447242; 25030449; 31113958 | |
| TmrAB | Hoechst 33342 | Thermus thermophilus | 5MKK | [168] | |
| 6RAN | [137] | ||||
| DMT | PfCRT 7G8 | 4-aminoquinolines (chloroquine, piperaquine, amodiaquine) | Plasmodium falciparum | 6UKJ | [33] |
| YddG | Aromatic amino acid, paraquat | Starkeya novella | 5I20 | [32] | |
| MATE - NorM | NorM | Norfloxacin, ciprofloxacin, doxorubicin and acriflavine, fluoroquinolone, TPP+, ethidium bromide, rhodamine 6G | Vibrio cholerae | 3MKT | [88] |
| Neisseria gonorrhoeae | 4HUK | [90] | |||
| 5C6P | [93] | ||||
| MATE - DinF | PfMATE | Br-norfloxacin, fluoroquinolone, ethidium bromide | Pyrococcus furiosus | 3VVN | [91] |
| 6FHZ | [94] | ||||
| DinF-BH | Rhodamine 6G, ethidium, TPP+, fluoroquinolone | Bacillus halodurans | 4LZ6 | [92] | |
| 5C6N | [93] | ||||
| ClbM | Colibactin | Escherichia coli | 4Z3N | [96] | |
| VcmN | Br-norfloxacin | Vibrio cholerae | 6IDP | [95] | |
| MFS | EmrD | Meta-chloro carbonylcyanide phenylhydrazone (CCCP), tetrachlorosalicylanilide (TSA) | Escherichia coli | 2GFP | [20] |
| LmrP | Lincosamides (clindamycin), macrolides (azithromycin, clarithromycin, erythromycin and roxithromycin), streptogramins (dalfopristin and RP 59500), tetracyclines | Lactococcus lactis | 6T1Z | [21] | |
| MdfA | Chloramphenicol, erythromycin, thiamphenicol, ciprofloxacin, TPP+, ethidium bromide, Rifampin, Dequalinium | Escherichia coli | 4ZP2 | [18] | |
| 6OOQ | [169] | ||||
| 6VS0 | [170] | ||||
| 6GV1 | [25] | ||||
| 6EUQ | [171] | ||||
| SotB | Toxic sugars or sugar metabolites | Escherichia coli | 6KKI | [22] | |
| YajR | N/A | Escherichia coli | 3WDO | [19] | |
| RND | AcrB | Acriflavine, crystal violet, ethidium bromide, rhodamine 6G, penicillin, cephalosporins, fluoroquinolones, macrolides, chloramphenicol, tetracyclines, novobiocin, fusidic acid, oxazolidinones, rifampicin | Escherichia coli | 2DHH | [110] |
| 1IWG | [106] | ||||
| 1OY9 | [172] | ||||
| 1OYD | |||||
| 4ZIT | [173] | ||||
| 2I6W | [174] | ||||
| 6BAJ | [175] | ||||
| 5YIL | [176] | ||||
| 3AOA | [109] | ||||
| 2HQG | [177] | ||||
| 2HQF | |||||
| 2HQD | |||||
| 2HQC | |||||
| 1T9U | [178] | ||||
| 1T9T | |||||
| 1T9Y | |||||
| 1T9X | |||||
| 1T9W | |||||
| 1T9V | |||||
| 4DX5 | [108] | ||||
| 4DX6 | |||||
| 4DX7 | |||||
| 4U95 | [111] | ||||
| 4U96 | |||||
| 4U8V | |||||
| 4U8Y | |||||
| 2W1B | [179] | ||||
| 2HRT | [117] | ||||
| 2J8S | [180] | ||||
| 2RDD | [181] | ||||
| 3W9H | [114] | ||||
| 4K7Q | [115] | ||||
| 3NOG | [182] | ||||
| 3D9B | [183] | ||||
| 4CDI | [99] | ||||
| 5O66 | [113] | ||||
| 6SGS | [105] | ||||
| 5JMN | [184] | ||||
| 6Q4N | [185] | ||||
| Salmonella enterica | 6Z12 | [186] | |||
| AdeB | β-lactams, tetracyclines, fluoroquinolones, aminoglycosides, chloramphenicol, trimethoprim, cefepime, novobiocin, tigecycline, colistin | Acinetobacter baumannii | 6OWS | [187] | |
| CmeB | Fluoroquinolone and macrolide (florfenicol) | Campylobacter jejuni | 4MT4 | [188] | |
| MexB | β-lactams, chloramphenicol, tetracycline, nalidixic acid, ciprofloxacin, streptonigrin | Pseudomonas aeruginosa | 3W9I | [114] | |
| 2V50 | [189] | ||||
| 6T7S | [190] | ||||
| 6IIA | [191] | ||||
| 6IOL | [192] | ||||
| MtrD | β-lactams, macrolides, host-derived antimicrobials (cationic antimicrobial peptides and bile salts), azithromycin, ceftriaxone, erythromycin, ampicillin | Neisseria gonorrhoeae | 6VKT | [193] | |
| 4MT1 | [194] | ||||
| SMR | EmrE | Aminoglycosides, quaternary cation compounds, TPP+, ethidium, methyl viologen, acriflavine, dequalinium | Escherichia coli | 3B5D | [59] |
| 7JK8 | [77] | ||||
| Gdx-Clo | Hydrophobic guanidinyl compounds, sulfonamides, β-lactams, aminoglycosides | Clostridiales bacterium | 6WK5 | [51] | |
The major facilitator superfamily of transporters
MFS proteins constitute one of the largest and most diverse families of membrane transporters, and are found across all domains of life as either uniporters, symporters, or antiporters [7]. MFS transporters typically contain twelve transmembrane (TM) helices arranged into two pseudosymmetric six-helix bundles termed the N- and C- domains, which are linked by a large cytoplasmic loop. The majority of structural data available supports a “rocker-switch” mechanism of transport in which the N- and C- domains undergo rigid-body conformational changes relative to each other to transition between inward- and outward-facing states. These two states provide a centrally bound substrate with alternating access to either the cytosolic or extra-cytosolic solutions, respectively [8, 9]. MFS transporters generally have a narrow substrate-specificity profile, but some sub-families, for example the drug-proton antiporter 1 (DHA1) and 2 (DHA2) families, mediate the efflux of a broad range of cytotoxic substrates and thus confer multidrug resistance [10]. In Gram-negative bacteria, some DHA transporters form tripartite complexes. For example, EmrB is expressed in the inner membrane and forms a complex with the periplasmic adaptor protein EmrA and the outer membrane channel TolC to transport drugs across the periplasm into the extracellular space [11–13]. Other DHA transporters, such as MdfA, function as simple monomeric exporters and have been extensively characterized [14, 15].
Overexpression of MdfA has been observed in Escherichia coli (E. coli) strains isolated from patients presenting multidrug resistance, and MdfA orthologs are expressed by several pathogenic bacterial species [16]. MdfA couples the efflux of various electroneutral and monovalent cationic drugs to the influx of a single proton [15, 17]. The first structure of MdfA was determined in 2015 and captured the transport deficient mutant Q131R in an inward-facing conformation with the antibiotic chloramphenicol bound centrally at the interface of the N- and C-domains [18]. Within this binding site, chloramphenicol directly interacts with N33 and D34 from TM1 via hydrogen bonding, and is also surrounded by hydrophobic residues including Y30, M58, L62, A150, L151, L235, L236, and I239 (Fig. 1a). Notably, this region is broadly conserved across the DHA family [19–22], and other cationic substrates and inhibitors have been observed binding to MdfA in a similar manner [23]. D34 has also been demonstrated to serve as a proton binding site (alongside E26) and is thus a key residue in coupling substrate export to proton import [24]. Subsequent structural studies of MdfA have captured the transporter in an outward-facing conformation (stabilized by forming a complex with an Fab antibody fragment), suggesting that MdfA mediates substrate transport using a classic MFS rocker-switch mechanism with an additional twisting of TM5 [25]. In this transport model (Fig. 1b), MdfA begins in an inward-facing conformation with D34 protonated. Substrate loading induces D34 deprotonation, freeing this residue to directly interact with the substrate via hydrogen bonding. This in turn triggers transition to an outward-facing conformation where substrate is released to the other side of the membrane and E26 is protonated. This protonation prompts a return to the inward-facing state where the proton is then transferred from E26 to D34, priming the protein for the next transport event [18].
Figure 1 |. MFS Transporters MdfA and LmrP mediate multidrug efflux.
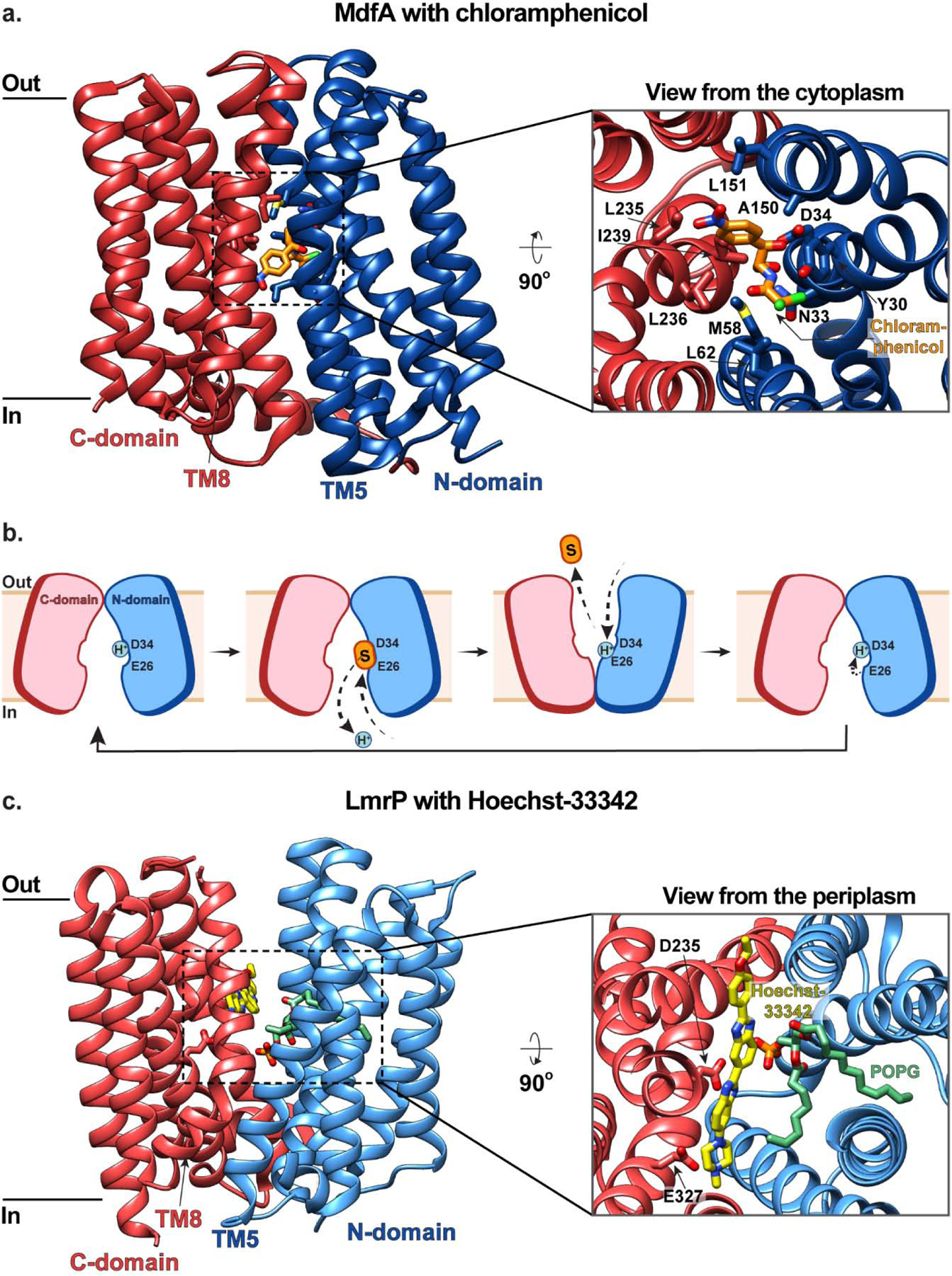
(a) The inward-facing structure of MdfA in the plane of the membrane (left) and as viewed from the cytoplasm (right inset) with chloramphenicol (orange) bound (PDB ID: 4ZOW). The N- and C- domains are colored in blue and red, respectively, and residues coordinating substrate are shown in stick representation. (b) Schematic representations of MdfA-mediated multidrug efflux. MdfA begins in an inward-facing conformation with D34 protonated. Substrate (orange) loading induces D34 deprotonation which triggers transition to an outward-facing conformation where substrate is released and E26 is protonated. This protonation prompts a return to the inward-facing state where the proton is transferred from E26 to D34, priming the protein for the next transport event. (c) The outward-facing structure of LmrP in the plane of the membrane (left) and as viewed from the periplasm (right inset). with Hoechst 33342 (yellow) and a structural POPG molecule (green) bound (PDB ID: 6T1Z). LmrP is represented the same as MdfA in panel (a).
While similar rocker-switch mechanisms have been proposed for various DHA family members including YajR [19], EmrD [20], LmrP [21], and SotB [22], recent DEER spectroscopy and biochemical cysteine cross-linking analyses of MdfA in nanodiscs have suggested that the transporter in fact does not function via a canonical rocker-switch mechanism [26]. This modified mechanism involves recruitment of hydrophobic drugs through a lateral gate that opens between TM5 and TM8 to the inner leaflet of the membrane, and hydrophilic drugs from the cytosol. Once substrate is bound in the central cavity, the lateral gate and opening to the cytosol close, and subtle rearrangements of TM helices allow for release of substrate into the extra-cytosolic space [26]. Importantly, these rearrangements are more subtle than those observed in the Fab-stabilized outward-facing structure, and it has been suggested that this adapted rocker-switch mechanism may contribute to MdfA’s broad substrate specificity profile [25]. Interestingly, MdfA can interact with neutral and positively-charged substrates simultaneously, for example chloramphenicol and tetraphenylphosphonium (TPP+), with the presence of one affecting the affinity for the other [27]. This raises the concept of distinct but interacting binding sites, which is intriguing when considering the conundrum of multidrug recognition. Furthermore, it has been proposed that DHA transporters are more dynamic than substrate-specific MFS transporters, which assists them in binding and transporting a variety of chemically and structurally distinct substrates [28].
Recently, the structure of the MdfA relative LmrP was determined and revealed the presence of a structural POPG lipid within the central cavity, where it stabilizes substrate binding [21] (Fig. 1c). It has been proposed that this embedded lipid has greater conformational plasticity than LmrP itself, and thus may provide a malleable hydrophobic environment to accommodate structurally diverse substrates [21]. Whilst this hypothesis is intriguing, it warrants further investigation given that it has not been observed previously.
The drug/metabolite transporter superfamily
The DMT superfamily represents a large group of topologically and functionally diverse transporters expressed by eukaryotes, bacteria, and archaea, that mediate efflux of a wide range of substrates, including sugars, metabolites, and toxins [3]. Several members of the DMT superfamily have been implicated in mediating multidrug resistance in infectious disease by actively extruding cytotoxic compounds from their active sites within pathogens [29, 30]. DMTs are believed to have evolved from SMR transporters – which are dimers of monomers that contain four TMs – first by gaining an extra TM helix to result in five-TM helical bacterial/archaeal transporter (BAT), and then via gene duplication to form the ten-TM helical DMT [3, 29, 31].
The structure of YddG – a bacterial DMT transporter that mediates efflux of various metabolites and exogenous toxic compounds – was the first DMT structure solved, and captured the transporter in an outward-facing conformation [32] (Fig. 2a). This structure revealed that YddG consists of ten TM helices arranged into two five-helix bundles with an inverted topology (Fig. 2a, b). Eight of these TMs (TMs1–4,6–9) form four two-helix hairpins that are arranged alternately to surround a central cavity which faces the periplasm. In this structure, a monoolein molecule (used for crystallization in lipidic cubic phase) was bound within the central cavity where it was coordinated by several conserved residues (W17, Y78, W101 and W163). This observation led to the hypothesis that this cavity may serve as a substrate-binding site. Indeed, this central cavity also features several other hydrophobic (Y78, Y82, and Y99) and hydrophilic (H79, S244, and S251) residues – several of which are critical for substrate binding/transport – and thus likely provides accommodation for a broad range of chemically diverse substrates (Fig. 2a). While this structure of YddG captured the transporter in an outward-facing state, an inward-facing model has been generated from this structure and confirmed by crosslinking and evolutionary covariation analysis. Together, these two conformational states indicate that YddG utilizes a unique alternating-access mechanism whereby bending and straightening of two-helix hairpins – TM3-TM4 and TM8-TM9 – induces tilting and upright motions of TM6 and TM1, respectively, which provides substrate with alternating access to the periplasm and cytosol to mediate substrate transport [32] (Fig. 2c).
Figure 2 |. Structures and proposed mechanism of the DMT transporters YddG and PfCRT.
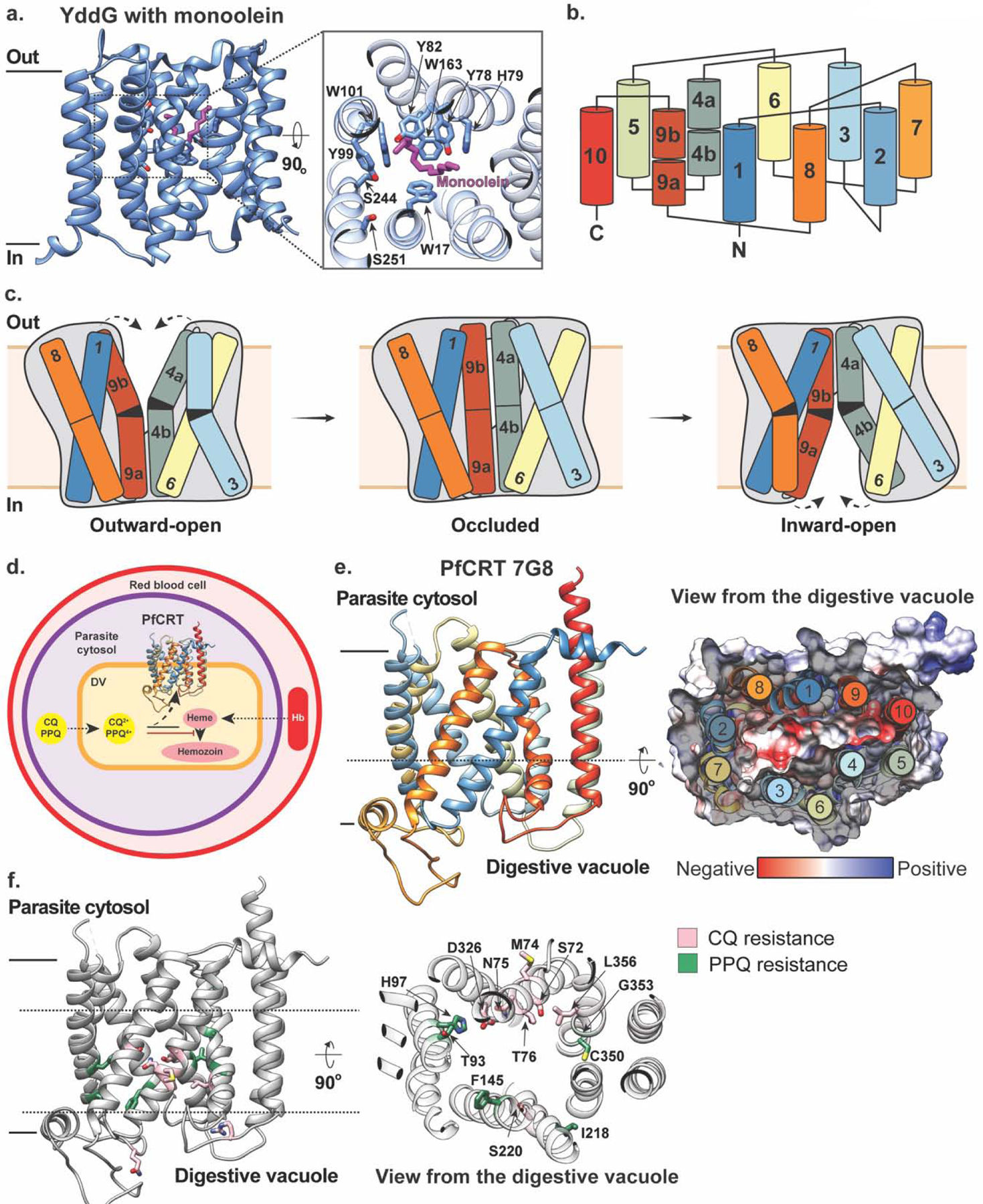
(a) The outward-facing structure of YddG in the plane of the membrane (left) and as viewed from the periplasm (right inset) with monoolein (magenta) bound (PDB ID: 5I20). Residues interacting with the monoolein molecule are shown in stick representation. (b) Topology diagram of YddG colored blue to red from the N- to C-terminus. (c) Schematic representations of a proposed transport mechanism of YddG. Bending and straightening of two-helix hairpins – TM3-TM4 and TM8-TM9 – induces tilting and upright motions of TM6 and TM1, respectively, which provides substrate with alternating access to the periplasm and cytosol to mediate substrate transport. The molecular envelopes are indicated by grey silhouette. Figure adapted from [32] (d) PfCRT is expressed in the membrane of digestive vacuole (DV) of the malaria-causing parasite Plasmodium falciparum, where host hemoglobin (Hb) is degraded into toxic heme. Mutated version of PfCRT in drug-resistant strains of Plasmodium falciparum mediates efflux of 4-aminoquinolines, such as chloroquine (CQ) and piperaquine (PPQ), from their site of action (PDB ID: 6UKJ). (e) Left panel: Structure of PfCRT in an inward-facing conformation on the digestive vacuolar membrane with TM helices colored in rainbow. Right panel: A central slice through the structure in surface representation of the electrostatic potential of the central cavity as viewed from the digestive vacuole with negatively and positively charged residues colored in red and blue, respectively, and the arrangement of TM helices shown and colored as in the left panel. (f) Left panel: The structure of PfCRT with mutations implicated in chloroquine and piperaquine resistance colored in pink and green, respectively, and shown in stick representation. Right panel: a view of these residues from the digestive vacuole.
Recently, the structure of the DMT transporter Plasmodium falciparum chloroquine resistance transporter (PfCRT), which is involved in resistance to antimalarials (Fig. 2d), was solved using cryo-EM [33] (Fig. 2e). Malaria infections occur when female Anopheles mosquitos carrying parasites such as Plasmodium falciparum feed on blood from a human host and consequently release the parasites into the bloodstream. For many decades, malaria was treated using chloroquine (CQ), which effectively eliminated disease-causing parasitic infections such as Plasmodium falciparum, until CQ resistance arose in the 1950s, independently in South America and Southeast Asia, later spreading to Africa [34]. Furthermore, strains of Plasmodium falciparum from Southeast Asia that confer resistance to the current first-line drug piperaquine (PPQ) have recently been identified [35, 36]. These highly virulent Plasmodium falciparum strains acquired resistance to these 4-aminoquinoline antimalarials via distinct sets of point mutations in PfCRT which is expressed in the parasite’s acidic digestive vacuolar membrane [30, 37] (Fig. 2d). Within the digestive vacuole, hemoglobin from human red blood cells is degraded into peptides and amino acids that are essential for parasite protein synthesis [38]. Currently available 4-aminoquinoline drugs inhibit the parasite’s hemoglobin degradation pathway to elicit their antimalarial effect [39, 40] (Fig. 2d). While the precise identification of its endogenous substrate remains elusive [41, 42], mutated versions of PfCRT in drug-resistant strains of Plasmodium falciparum enable efflux of CQ and PPQ from the digestive vacuole, thus removing them from their site of action (Fig. 2d). Notably, wild-type PfCRT can bind but not transport CQ and PPQ [33], and while a K76T substitution exists in all resistant isoforms (regardless of geographical origin) and is considered the hallmark of resistance, it mediates drug efflux in combination with other geographic-specific mutations [43, 44].
The 7G8 isoform of PfCRT harbors five mutations – C72S, K76T, A220S, N326D and I356L – which enable it to transport CQ but not PPQ, and hence confer resistance to the former but not the latter [33]. The structure of this isoform captures the transporter in an inward-open conformation and revealed that PfCRT 7G8 is a monomer consisting of ten TM helices and two juxtamembrane helices – one on either side of the membrane. The ten TM helices are arranged as five two-helix hairpins with an inverted topology, with TM1-TM4 and TM6-TM9 forming two sides of a large, negatively-charged cavity (Fig. 2e). Mutations that enable CQ and PPQ transport across different resistant isoforms primarily line this central cavity, consistent with the hypothesis that this region constitutes the binding site for positively-charged CQ and PPQ [33] (Fig. 2f). This work provides molecular insight into how specific mutations can alter this transporter’s interactions with CQ and PPQ. For example, newly emerging PPQ-resistant strains of Plasmodium falciparum in Asia and South America feature PfCRT isoforms with point mutations F145I and C350R, respectively [35, 36, 45]. These residues are located in helices lining the central cavity, and when these mutations were individually introduced into PfCRT 7G8, they both reversed the substrate profile of the 7G8 isoform, enabling transport of PPQ (PPQ-resistant) while diminishing efflux of CQ (CQ-sensitive) [33]. Molecular dynamics (MD) simulations of these mutant constructs showed that the introduction of F145I causes a rearrangement of TM helices lining the cavity. On the other hand, introduction of C350R diminishes the overall electronegativity of the cavity, which could explain the reduced binding affinity for positively-charged PPQ and the resulting transport-positive phenotype of this variant [33]. Together, these observations demonstrate that point mutations in PfCRT isoforms mediate resistance to various antimalarials by altering binding affinities and promoting conformational changes required for transport [46]. Importantly, these findings provide a powerful tool to not only understand but also predict how and which emerging PfCRT variants might evolve to mediate multidrug resistance.
Small multidrug resistant transporter family
SMR transporters are a family within the DMT superfamily that are expressed broadly across bacteria and archaea [3, 47]. As the name suggests, they are indeed small – containing just four TM helices – and they function as either homo- or hetero-dimer proton-coupled antiporters to export quaternary ammonium compounds and polyaromatic cations [48–51]. These transporters are associated with resistance to clinically relevant pharmaceuticals used to treat pathogenic infections such as those caused by Mycobacterium tuberculosis [52] and Acinetobacter baumannii [53]. Within the SMR transporter family there are two major subtypes – the Gdx cluster and the Qac cluster – that share ~40% sequence identity. The Gdx subfamily has only recently been characterized, whereas the Qac cluster has been extensively studied over many years [14, 51, 54]. Members of the Qac cluster transport structurally diverse quaternary ammonium cations (e.g., ethidium, TPP+, methyl viologen), and include the prototypical SMR member, EmrE, from E. coli. Although EmrE has not been demonstrated to be directly involved in resistance towards antibiotics used clinically to treat E. coli infections, resistance can be easily developed in vitro by mutation of just a few residues [55, 56]. This makes EmrE an elegant model transporter to elucidate the mechanism of SMR mediated drug efflux.
The first insights into the structure of EmrE were obtained from low resolution 2D (~7.5 Å resolution in the plane and ~16 Å resolution perpendicular to the membrane) [57] and 3D cryo-EM maps of the transporter (~7 Å resolution) [58]. These structures demonstrated that EmrE is a dimer comprised of two antiparallel monomers (designated ‘A’ and ‘B’) with no obvious two-fold symmetry, and a binding site for the high-affinity substrate TPP+ at the dimer interface. This was highly controversial at the time given that an antiparallel topology had never been observed for any integral membrane protein. While the limited resolutions of these structures prevented the monomers from being unambiguously delineated or for TM helices to be assigned in a sequence-specific manner, a subsequent 3.8 Å crystal structure of EmrE allowed the Cα backbone to be resolved and confirmed an antiparallel topology [59]. This structure also revealed that each EmrE monomer is made up of four TM helices, the first three form the substrate and proton binding sites, and the fourth maintains interactions at the dimer interface (Fig. 3a). Alongside structural investigations of EmrE, a plethora of biochemical and biophysical studies have established that asymmetric antiparallel EmrE dimers transport using an alternating-access mechanism in which the substrate/proton binding site, comprising two conserved glutamate residues (E14A and E14B), alternates between an outward-facing state for proton binding and an inward-facing state for drug binding [49, 56, 60–74] (Fig. 3b). Given the antiparallel nature of EmrE, the inward- and outward-facing conformations are structurally identical and differ only in orientation within the membrane.
Figure 3 |. The SMR transporters ErmE and Gdx-Clo utilize an antiparallel topology to mediate multidrug efflux.
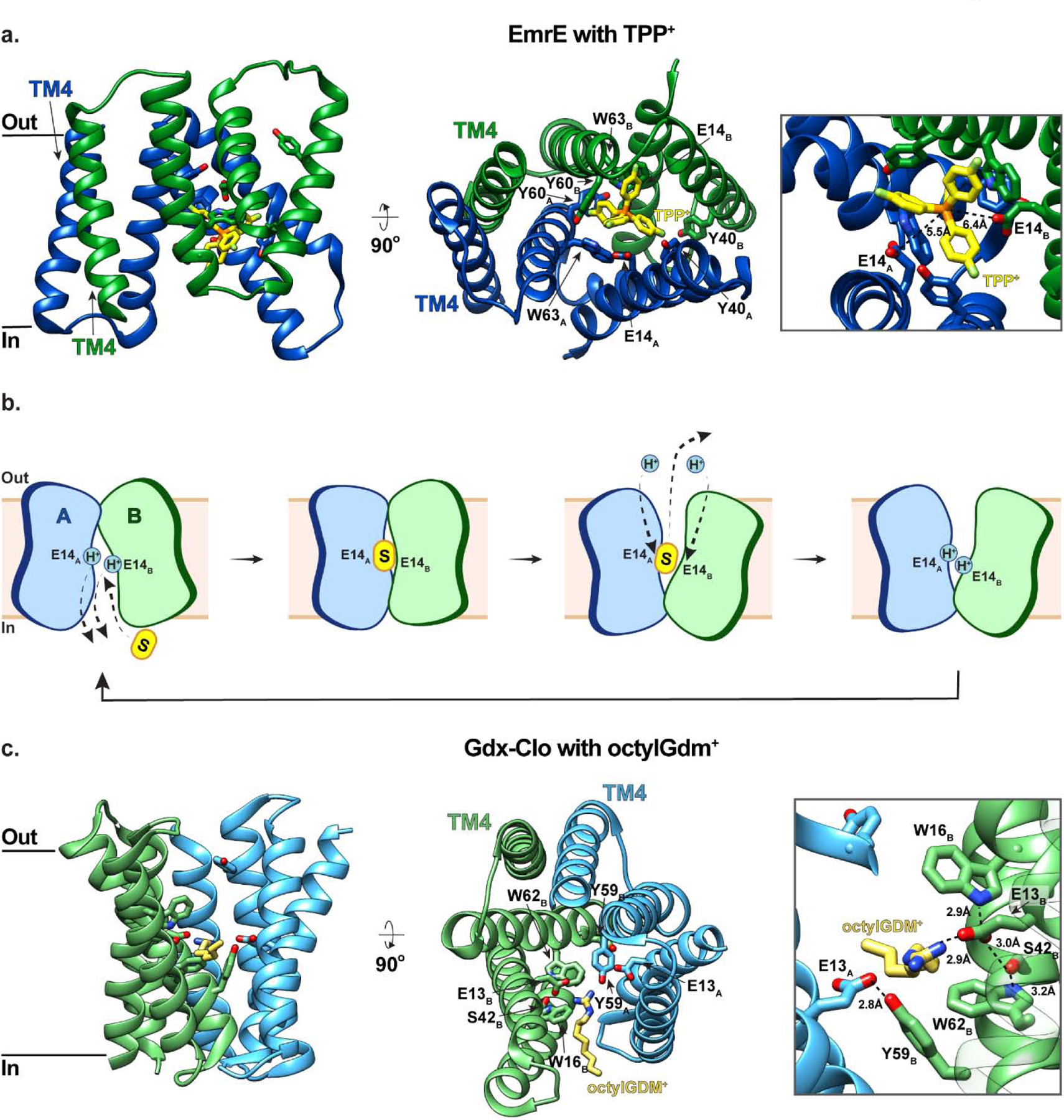
(a) Side and top-down views of EmrE with fluorinated TPP+ bound (yellow; PDB ID: 7JK8). Monomers A and B of EmrE are shown in blue and green, respectively, and residues coordinating substrate are shown in stick representation. The right panel shows a close-up of the TPP+ binding site. (b) Schematic representations of EmrE-mediated multidrug efflux. EmrE dimers transport using an alternating-access mechanism in which the substrate/proton binding site, comprising E14A and E14B, alternates between an outward-facing state for proton binding and an inward-facing state for drug binding. The antiparallel nature of EmrE, means that the inward- and outward-facing conformations are structurally identical, differing only in orientation within the membrane. (c) Side and top-down views of Gdx-Clo with octylGDM+ bound (yellow; PDB ID: 6WK9). The right panel shows a close-up of the octylGDM+ binding site. Gdx-Clo is coloured the same as EmrE in panel (a).
While these studies provided great insight into the mechanism of SMR mediated transport, it is only recently that high-resolution structures of SMRs have been reported and the substrate-binding sites characterized at an atomic level. This is not entirely surprising given that these transporters are small, highly dynamic [56, 60–67], and lack sizable soluble domains required to obtain well-diffracting crystals or align particles for cryo-EM studies. In 2020, Kermani et al. used monobodies as crystallization chaperones to determine the first high-resolution structures of an SMR transporter [51]. These structures capture Gdx-Clo, a member of the Gdx cluster, in an apo state and in complex with the substrates phenylGdm+ and octylGdm+ to 3.5 Å, 2.5 Å and 2.3 Å resolution, respectively (Fig. 3c). This work confirmed that Gdx transporters also have a dual topology architecture and revealed that an opening between TM2A and TM2B provides access to the central substrate binding site from the membrane. It has been proposed that this opening to the membrane is likely exploited by hydrophobic substrates to gain access to the binding site [51]. Within the substrate binding site, the substrate’s guanidinyl group is coordinated by E13B (Fig. 3c). This glutamate is stabilized by a conserved hydrogen-bonding network consisting of W62B, S42B, and W16B, which has previously been demonstrated to be important for substrate transport [75, 76]. While E13A is also in close proximity to the substrate’s guanidinyl group, its sidechain interacts with Y59B, thus preventing it from forming a hydrogen bond with this group. Notably, Y59 is completely conserved among SMR transporters and undergoes a large conformational rearrangement upon transition between the inward- and outward-facing states [51]. Given this, it has been proposed that Y59B and the substrate’s guanidinium group compete for E13A, and that displacement of Y59B by the guanidinyl group initiates conformational rearrangements required for alternating access. Kermani et al. also demonstrated that both Gdx-Clo and EmrE transport a broad, somewhat overlapping range of hydrophobic-substituted cations despite belonging to separate SMR subfamilies [51]. More specifically, Gdx-Clo preferentially transports Gdm+ and guanidinyl compounds with single hydrophobic substitutions compared to doubly substituted guanidinyl compounds and tetramethylGdm+, indicating that while some bulk is tolerated for Gdx-Clo-mediated transport, there is an upper limit. In contrast, EmrE cannot transport Gdm+, but preferentially transports more hydrophobic and bulky substrates.
Just a few months after these structures of Gdx-Clo, Shcherbakov et al. reported the first high-resolution structure of EmrE, determined with fluorinated TPP+ bound, by magic-angle-spinning solid-state NMR spectroscopy [77] (Fig. 3a). Shcherbakov et al. took advantage of the EmrE mutant S64V, which maintains a high affinity for TPP+ but slows turnover [78], thus making it more amenable to structural studies. This structure confirmed a dual topology architecture and placed the substrate in a similar location compared to that observed in previous low-resolution structures and MD models [59, 79], but the higher resolution structure allowed for better definition of the substrate’s orientation and position. In this structure, TPP+ is surrounded by E14, Y40, Y60, and W63 from both the A and B monomers (Fig. 3a). Among these, Y40 is the furthest away from TPP+, consistent with biochemical data suggesting that this residue regulates substrate specificity, whereas the others are essential for binding/transport [80]. Notably, TPP+ is bound asymmetrically within this binding site, with more stabilization provided by E14A than E14B (Fig. 3a). Overall, this binding pocket is loosely packed and spacious with most inter-residue distances exceeding that of hydrogen-bond length, and protein–substrate distances ranging from ~4.6–10 Å. This loose, multivalent, spacious binding site is consistent with the diverse substrate profile of EmrE and provides accommodation for the observed dynamic substrate reorientation.
Unexpectedly, recent studies have revealed that in addition to functioning as a H+-coupled antiporter, EmrE may also function as a H+-coupled symporter or uncoupled uniporter [61, 81]. Given the negative-inside membrane potential in bacteria, both symport and uniport raise the possibility for concentrative uptake of toxic substrates rather than export, adding complexity to the SMR story and warranting further investigation and consideration.
The multidrug and toxic compound extrusion family
MATE transporters are expressed by both prokaryotes and eukaryotes and function as secondary active antiporters that couple the export of cationic substrates to the influx of H+ or Na+ along their electrochemical gradients [82–84]. Within the MATE transporter family, there are three major subtypes: NorM, DinF, and eukaryotic MATE (eMATE) [82, 83]. Bacterial MATE transporters typically belong to the NorM and DinF subfamilies, whereas the archaeal and eukaryotic MATE transporters belong to the DinF and eMATE subfamilies, respectively [85]. In bacteria, MATE transporters mediate the efflux of various cationic drugs including ethidium bromide, TPP+, berberine, acriflavine, and norfloxacin, thus conferring resistance to these compounds (Table 1). Changes in expression of MATE transporters can cause drug resistance. For example, overexpression of MepA from Staphylococcus aureus results in resistance to tigecycline, an antibiotic used to treat methicillin- and vancomycin-resistant Staphylococcus aureus infections [86, 87].
To date, there are more than 20 structures of MATE transporters (Table 1). These have provided invaluable molecular insights into MATE-mediated substrate recognition and transport [85]. MATE transporters typically contain twelve TM helices arranged into two pseudosymmetric six-helix bundles (TM1–6 and TM7–12) named the N- and C-lobes. While this architecture is reminiscent of MFS transporters, the arrangement of helices within the MATE and MFS N- and C- lobes/domains is distinct, and thus they have unique topologies [83, 88] (Fig. 4a). The first MATE transporter structure was determined by X-ray crystallography in 2010 and captured NorM from Vibrio cholerae (NorM-VC) in an outward-facing conformation [88]. This structure revealed that E255 and D371 contribute to a negatively-charged pocket within a V-shaped cavity in the C-lobe which binds cations. This observation is consistent with previous analysis of NorM from Vibrio parahaemolyticus (NorM-VP) which demonstrated that mutagenesis of three conserved acidic residues D32, E51 and D367 (corresponding to D36, E255 and D371 in NorM-VC) abolish transport activity [89]. Subsequent outward-facing structures of NorM from Neisseria gonorrhea (NorM-NG) in complex with monobodies as crystallization chaperones revealed a multidrug-binding site at the interface of the N- and C-lobes near the membrane-periplasm interface. Within this site, D41, S61, Q284, S288, D355, and D356 directly coordinate the substrates TPP+, rhodamine-6G, and ethidium via hydrogen- and ionic-bonds, while A57 and F265 stabilize bound substrate via hydrophobic interactions [90] (Fig. 4b). Interestingly, this binding site is unlike that of other multidrug transporters, which typically overlap with the proton binding site and employ hydrophobic residues to accommodate binding of multiple substrates [90]. While this is typically accepted as the NorM-NG substrate binding site across the field, it is noteworthy that there seem to be several discrepancies (including Ramachandran analysis) in these substrates-bound structures, and their electron density maps, that require further examination and clarification.
Figure 4 |. MATE transporters NorM-NG and PfMATE belong to two major MATE subfamilies and mediate multidrug efflux.
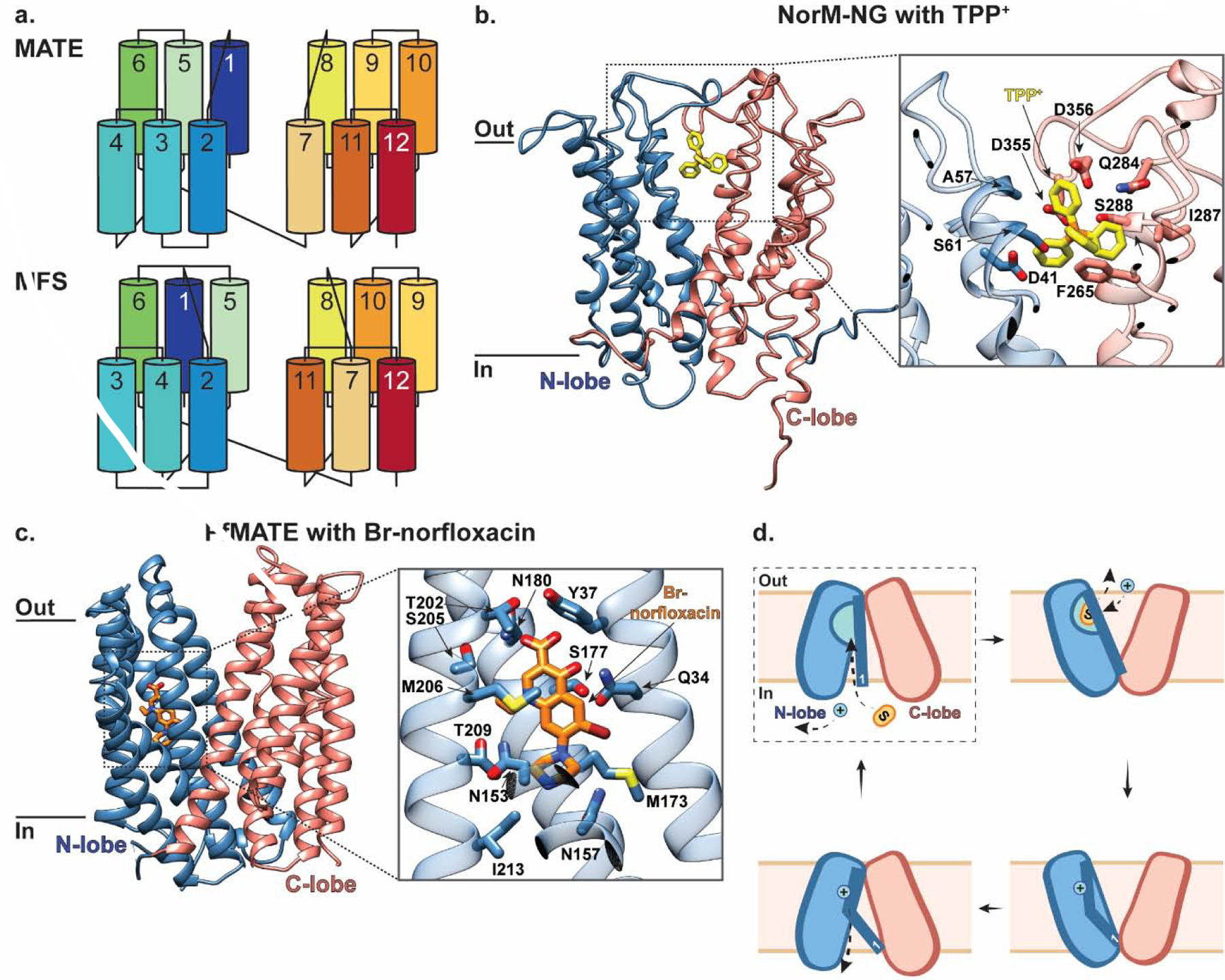
(a) Topology diagram comparing MATE and MFS transporters colored blue to red from the N- to C-terminus, adapted from [85] (b) The outward-facing structure of Norm-NG in the plane of the membrane. Inset shows a close-up of residues interacting with TPP+ (yellow; PDB ID: 4HUK). The N- and C- lobes are colored in blue and salmon, respectively, and the residues coordinating substrate are shown in stick representation. (c) The outward-facing structure of PfMATE, part of the DinF subfamily, in the plane of the membrane. Inset shows a close-up of residues interacting with Br-norflaxacin (orange; PDB ID: 3VVP). PfMATE is colored the same as NorM-NG in panel (b), and the residues coordinating substrate are shown in stick representation. (d) Schematic representation of PfMATE-mediated multidrug efflux. PfMATE switches between inward- and outward-facing conformations to provide an N-lobe localized binding pocket with alternating access to the cytosolic and extra-cytosolic solutions, respectively. In the inward-facing state, TM1 is largely kinked with D41 protonated (dashed box indicates this state is not yet structurally characterized). Deprotonation of D41 in this inward-facing state has been proposed to straighten TM1, which allows space for substrate to bind within the N-lobe cavity. This substrate binding induces a conformational switch to the outward-facing state with TM1 still straight. Here, D41 is re-protonated which causes TM1 to kink again and the N-lobe cavity to collapse, thus triggering substrate release and subsequent transition back to an inward-facing conformation.
Several structures of DinF subfamily members have been determined [91–96], including the H+-coupled MATE transporter from Pyrococcus furiosus (PfMATE) in both outward-facing apo and substrate-bound conformations [91]. The substrate-bound structure of PfMATE revealed that despite sharing a similar architecture with NorM-VC, it has a unique substrate binding site located within the N-lobe [91]. In the outward-facing substrate-bound conformation, a norfloxacin-derivative compound (Br-NRF) directly interacts with Q34 from TM1, N157 from TM4, and N180 from TM5 via hydrogen-bond interactions, and is also surrounded by residues including Y37, N153, M173, S177, T202, S205, M206, T209 and I213 (Fig. 4c). Interestingly, the amino acids of PfMATE corresponding to the cation binding site of NorM-VC are not conserved, raising the possibility that the cation binding site in PfMATE may also be unique. In combination with functional analyses, these structures demonstrate that protonation of D41, a highly conserved residue among prokaryotic MATEs, induces TM1 bending at P26, which in turn collapses the N-lobe cavity to facilitate substrate release into the extra-cytosolic region [91]. Recently, the first structure of a MATE transporter in an inward-open conformation was determined from crystals of PfMATE grown by vapor diffusion in the presence of native lipids extracted from Pyrococcus furiosus [94]. This structure displays rigid body rearrangement of TM2–6 in the N-lobe and TM8–12 in the C-lobe compared to that observed in the outward-facing state to form an inverted central cavity open to the cytosolic space. This rearrangement is facilitated by conformational flexibility of TM1 – largely kinked in this state – and TM7, which appear to be only loosely associated with the N- and C-lobes, respectively. This study, together with previously reported outward-facing structures [91], indicate that MATE transporters utilize an alternating-access mechanism that can be summarized as follows [94] (Fig. 4d). The transporter begins in the inward-facing state, where TM1 is largely kinked and D41 is protonated. Deprotonation of D41 in this state has been proposed to straighten TM1, which in turn creates space for the substrate to bind within the N-lobe cavity. Substrate binding then induces a conformational switch to the outward-facing state with TM1 maintained straight. Here, D41 is re-protonated, causing TM1 to kink and the N-lobe cavity to collapse, triggering substrate release. The protonated transporter then reorients to an inward-facing conformation and the cycle repeats. While no inward-facing substrate-bound or occluded states have been structurally characterized to date, this current model has substantially furthered our understanding of MATE-mediated multidrug transport.
Resistance-nodulation-cell-division transporter superfamily
Unlike Gram-positive bacteria, which are enclosed by a single cell membrane and peptidoglycan layer, Gram-negative bacteria are surrounded by two membrane layers between which lies the periplasm [97]. The efflux of drugs from Gram-negative bacteria therefore involves two steps. The first step requires transport across the inner membrane by various single-component transporters including ABC, MFS, MATE, and SMR (super)families described in other sections. The second step involves transport from either the outer leaflet of the inner membrane, or the periplasmic space itself, across the outer membrane and into the extracellular space. This step is mediated by large multicomponent pumps called tripartite complexes [98] (Fig. 5a). Tripartite complexes typically comprise an RND transporter in the inner membrane that is connected to an outer membrane channel (OMC) via a periplasmic adaptor protein (PAP) [98–100]. Some transporter families other than RND contain members that can also interact with PAPs and OMCs to form tripartite complexes, such as the ABC transporter MacB and the MFS transporter EmrB, but unlike RND transporters, these other families also contain members that function as single-component inner membrane transport systems [11–13, 101, 102]. The fact that numerous transporters fulfil the first step of this process, while tripartite complexes are solely responsible for the latter, illustrates the central role they play in the efflux machinery of Gram-negative bacteria. Accordingly, RND transporters have very broad substrate profiles that correspond to the conglomerate of substrates transported by various single component multidrug exporters [103]. This makes them one of the most complex and intriguing secondary transporters known to date from both structural and mechanistic perspectives.
Figure 5 |. RND-containing tripartite complexes mediate drug efflux across the outer membrane of Gram-negative bacteria.
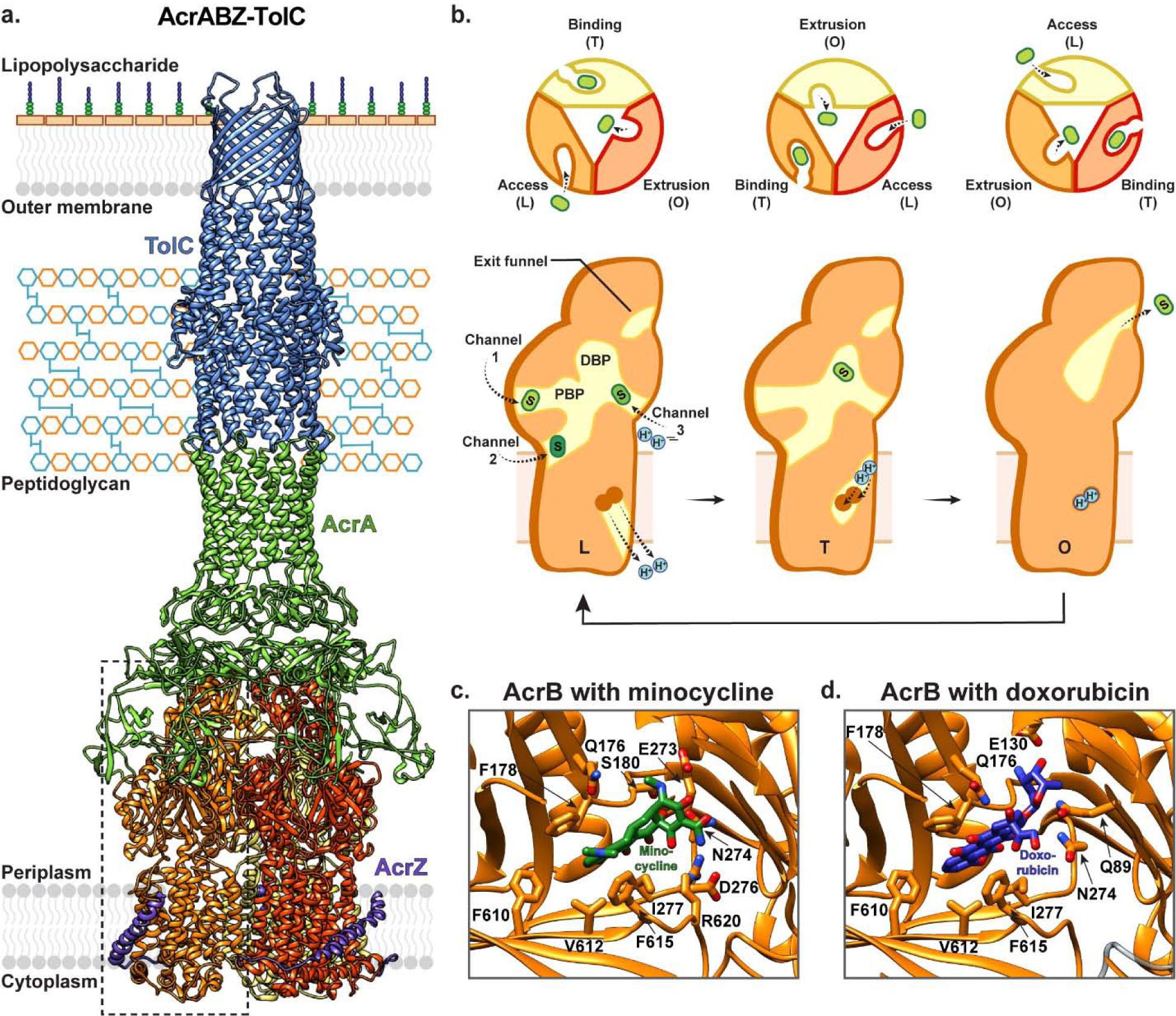
(a) AcrB is a trimeric transporter expressed in the inner membrane of Gram-negative bacteria that forms a tripartite complex with AcrA (green) and TolC (blue). Additionally, this AcrAB-TolC tripartite complex interacts with the regulatory inner membrane peptide AcrZ (purple) (PDB ID: 5NG5). The three protomers of AcrB are coloured in red, orange and yellow. (b) RND transporters such as AcrB mediate drug efflux from the periplasm, or outer leaflet of the inner membrane. In presence of substrate, each monomer within the AcrB trimer cycles between three structural states: the access/loose (L) state, the extrusion/open (O) state, and the binding/tight (T) state, with high cooperativity. Initially, the monomer enters the L state, where substrate can access the proximal binding pocket (PBP). Next, substrate enters and binds to the universal distal binding pocket (DBP) in the T state. The transporter enters the O state where the periplasmic entry pathway is closed, and a funnel connected to the AcrA-TolC channel is opened, through which substrate is released. In the L state, alternate pathways (channels 1–3) provide substrates with access to the DBP, and different drugs (indicated in different shades of green) prefer different access channels. Changes between these states are driven by proton movement across the inner membrane, with proton loading and release in the L state, transfer in the T to O transition, and binding in the O state. The distal binding pocket of the RND transporter AcrB with (c) Minocycline (green; PDB ID: 4DX5) and (d) Doxorubicin (purple; PDB ID: 4DX7) bound.
AcrB from E. coli is one of the most well-characterized RND transporters. This inner membrane protein forms a tripartite complex with the PAP AcrA and the OMC TolC, with a stoichiometry of 3:6:3 [98–100]. Additionally, AcrB has a 49 amino acid peptide partner – AcrZ – which takes the form of a single alpha helix and makes extensive contacts with the AcrB TM domain to enhance export of certain antibiotics including tetracycline, puromycin, and chloramphenicol [99, 104, 105] (Fig. 5a). Several structures of AcrB in different conformations have been determined under different conditions [100, 105–115] (Table 1). These structures have revealed that AcrB is a homotrimer, with each protomer comprising twelve TM helices and two highly structured periplasmic domains. The periplasmic domains account for approximately 60% of molecular weight of each protomer and project ~70 Å away from the inner membrane. Unlike most secondary active transporters, RND transporters, including AcrB, do not appear to feature a transmembrane translocation pathway that transports drugs across the inner membrane. Instead, the TM domains contain salt-bridged titratable residues – including D407 and D408 in TM4, and K940 in TM10 – that provide a pathway for proton movement from the periplasm to the cytoplasm, which serves as a driving force for transport [111, 116]. This proton influx involves conformational changes within the TM domain that are in turn transmitted to the periplasmic domains. Here, substrate is sequestered from either the outer leaflet of the inner membrane or the periplasm itself. Each monomer’s periplasmic domain contains four sub-domains that together constitute the two main drug binding cavities which are referred to as the proximal and distal pockets, respectively [106, 110, 117]. The most distant region of the periplasmic domain from the inner membrane features a funnel that is connected to TolC via AcrA.
While the specific details of this transport mechanism are yet to be fully elucidated, a general model in which AcrB functions as a highly cooperative homotrimer has been established [14, 100] (Fig. 5b). This model involves each protomer functionally rotating between three different states that provide substrate with alternating access to the distal binding site from either the periplasm or the AcrA-TolC channel. In the apo conformation, the trimer is symmetric [106], however upon addition of substrate, each protomer adopts a distinct conformation — namely the ‘loose’ (L), ‘tight’ (T), and ‘open’ (O) states — that represent the conformations visited by each protomer throughout the transport cycle [110, 117]. The transport cycle begins with a monomer in the L state, where substrate can access the proximal binding site. Following this, a pathway to the distal binding site opens, allowing substrate to move in deeper and bind at this distal site in the T state. Next, the transporter enters the O state, in which the periplasmic entry pathway is closed, and a funnel connected to the AcrA-TolC channel is opened. Substrate then moves through this funnel and the AcrA-TolC channel for release into the extracellular solution, and the monomer reverts to the initial L state, where it is again primed for substrate binding.
The substrate profile of AcrB and other RND transporters is incredibly diverse and encompasses hydrophobic, amphipathic, cationic, neutral, and anionic molecules [14, 100, 116, 118, 119]. Remarkably, the distal pocket is believed to be a universal binding site for all known AcrB substrates, and several mechanisms to support poly-specificity have been proposed [100]. The distal binding site predominantly comprises hydrophobic residues including F136, V139, F178, I227, P326, Y327, F610, V612, F615, and F628, with a cluster of polar/charged residues at one end including Q176, S180, E273, N274 (Fig. 5c, d). This binding site is larger than that typically seen for multidrug resistant transporters from other families, and different ligands bind preferentially to different areas. For example, doxorubicin binds predominantly in the end of the pocket that lacks polar/charged residues (Fig. 5d), whereas minocycline binds more deeply within the pocket where it interacts with the polar/charged residues [108] (Fig. 5c). While substrate-bound structures of AcrB show binding of individual drugs to this pocket, it has been demonstrated that this binding site can accommodate multiple substrates concomitantly [120], and that water molecules can stabilize both substrate and inhibitor binding to this site, properties that are both compatible with the poly-specificity of AcrB [108, 112, 121]. Additionally, it has been suggested that substrates may gain access to the distal binding pocket via other channels located at the membrane–periplasm interface. One channel provides entry from the periplasm through the periplasmic domain (channel 1), a second provides entry from the outer leaflet of the inner membrane through a groove between TM7, TM8, and TM9 (channel 2), and a third bypasses the proximal binding site to provide entry via a central periplasmic cavity formed at the interface of the three protomers (channel 3) (Fig. 5b). Interestingly, it has been suggested that different substrates may prefer distinct channels to access the distal binding site, and that this may contribute to substrate poly-specificity (see [14, 100, 116, 122] for more in-depth review).
Recently, the AcrABZ-TolC complex has been determined both in vitro via single-particle cryo-EM [113, 123] (Fig. 5a) and in situ using cryo-ET and sub-tomogram averaging [124]. These structures reveal that in the absence of substrate, the TolC channel is closed, and that substrate binding induces large scale movements in AcrB (as described above) which initiate opening of the AcrA-TolC channel. In the presence of substrate, the three AcrB protomers within the trimer adopt either an L-T-O, L-T-T, or L-L-T conformation, and interestingly, the AcrB-AcrA and TolC-AcrA interfaces do not significantly differ between the different substrate-bound conformations. This repacking of AcrA is critical to maintain all interprotein interfaces such that no leakages to the periplasm can occur. Furthermore, in situ sub-tomogram averages suggest that the AcrAB complex likely forms first, and then associates with TolC, and that the components are localized by the peptidoglycan layer [124].
ATP-binding cassette transporter superfamily
ABC transporters are found across all kingdoms of life. These primary active transporters utilize ATP binding and hydrolysis to drive the translocation of chemically diverse substrates such as amino acids, lipids, sugars, peptides, metabolites, and toxins across cell membranes [125]. Bacterial ABC transporters such as LmrA, LmrCD, HorA, BmrA, PatAB, EfrAB, OmrA, VcaM, and MacAB have been demonstrated to export antibiotics from both Gram-positive and Gram-negative bacteria and thus contribute to antibiotic resistance [126–128].
Over the past two decades, a growing number of ABC transporter structures in distinct conformational states have provided molecular details of their architecture, substrate recognition, and the mechanism by which they function [129–146] (Fig. 6a, b and Table 1). ABC transporters are typically made up of four core domains including two transmembrane domains (TMDs) and two highly conserved cytoplasmic nucleotide-binding domains (NBDs). The NBDs bind and hydrolyze ATP, in turn inducing conformational changes in the TMDs that mediate substrate transport [147, 148] (Fig. 6c). In bacteria and archaea, ABC transporters exist as either homo- or hetero-dimers of ‘half-transporters’, each containing one TMD and one NBD, while eukaryotic ABC transporters typically consist of the four domains assembled into a single polypeptide chain [149].
Figure 6 |. Representative ABC transporter structures and their general transport mechanism of mediating drug resistance.
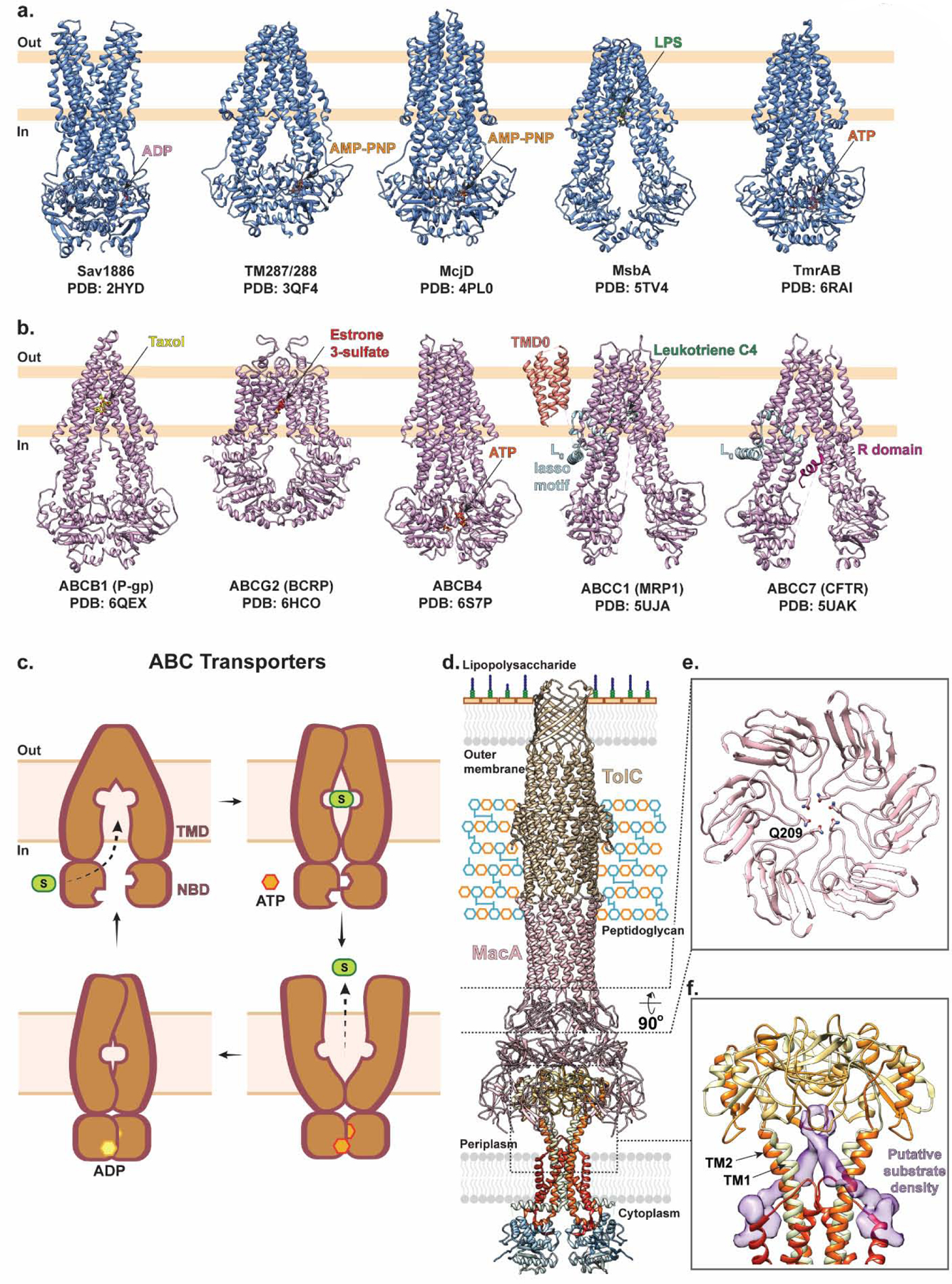
(a) Representative structures of bacterial ABC exporter including Sav1886 with ADP bound (pink; PDB ID: 2HYD), TM287/288 with AMP-PNP bound (orange; PDB ID: 3QF4), McjD with AMP-PNP bound (orange; PDB ID: 4PL0), MsbA with LPS bound (green; PDB ID: 5TV4), and TmrAB with ATP bound (dark orange; PDB ID: 6RAI). (b) Representative structures of mammalian ABC exporters including human multidrug resistance efflux pump P-glycoprotein (P-gp; ABCB1) with Taxol bound (yellow; PDB ID: 6QEX), human multidrug transporter breast cancer resistance protein (BCRP; ABCG2) with Estrone 3-sulfate bound (red; PDB ID: 6HCO), human lipid exporter ABCB4 with ATP bound (dark orange; PDB ID: 6S7P), bovine multidrug resistance-associated protein 1 (MRP1; ABCC1) with Leukotriene C4 bound (green; PDB ID: 5UJA), and human cystic fibrosis transmembrane conductance regulator anion channel (CFTR; ABCC7) with regulatory (R) domain (purple; PDB ID: 5UAK). (c) ABC exporters alternate between outward- and inward-facing states as follows. The transporter begins in an apo inward-facing state with the two cytosolic nucleotide binding domains (NBDs) separated, providing space for the substrates to access the central binding site at the interface of the transmembrane domains (TMDs). Once substrate binds, the NBDs come closer in proximity and the TMD interface closes to occlude the substrate. ATP then binds and induces NBD dimerization which in turn triggers the TMDs to adopt an outward-facing state. Here, substrate is released, ATP is hydrolyzed, and the transporter returns to the inward-facing state. (d) MacB forms a tripartite complex with PAP Mac A (pink) and OMC TolC (gold) in a 2:6:3 stoichiometric ratio for drug efflux across the periplasm and outer membrane of gram-negative bacteria. MacB is shown in ribbon representation and colored in rainbow from the N- (blue) to C- (red) terminus (PDB ID: 5NIL). (e) A central slice through the structure of MacA viewed from the bottom with Q209 shown in stick representation, forming a ring to prevent backflow of substrate into the periplasm. (f) Close-up of the MacB dimer within the MacAB-TolC tripartite complex (PDB ID: 5NIL) with unassigned, elongated, non-protein density shown as a purple surface between the periplasmic extensions of TM1 and TM2.
The first structure of a bacterial multidrug ABC exporter was determined in 2006. This structure captured Sav1886, which mediates the efflux of verapamil, tetraphenylphosphochloride, and Hoechst 33342 from Staphylococcus aureus, in an outward-facing conformation [130] (Fig. 6a). In this state, the two NBDs are in close contact, and the two TMDs form a large central cavity open to the outer leaflet of the membrane and the extracytosolic space. Subsequently, several structures of MsbA – which flips lipid A and lipopolysaccharide across the inner membrane of Gram-negative bacteria into the periplasm – and McjD – which exports the antibacterial peptide MccJ25 to establish self-immunity – have been determined in a range of different conformations including the nucleotide-free inward-facing open, nucleotide-free occluded, and nucleotide-bound outward-facing conformations [141, 150, 151] (Fig. 6a). While these are not of the same category per se, it is believed that bacterial multidrug ABC transporters likely transport via a similar mechanism to expel cytotoxic compounds from the cytoplasm.
The structures of mammalian multidrug resistance ABC transporters – such as P-glycoprotein (P-gp; ABCB1), breast cancer resistance protein (BCRP; ABCG2), and the multidrug resistance-associated protein 1 (MRP1; ABCC1) – have also been determined and provided substantial structural insights into the mechanistic basis of poly-specificity and transport in the context of multidrug resistance (Fig. 6b). For example, the structure of human multidrug transporter ABCG2 reveals a deep, slit-like hydrophobic cavity which enables binding of structurally diverse molecules including hematoporphyrin, mitoxantrone, as well as inhibitors such as gefitinib and the fumitremorgin C-derived molecule Ko143 [144–146]. The structures of ABCG2 in a substrate bound (pre-translocation) state, and an ATP bound (post-translocation) state resemble that of other ABC exporters, suggesting that ABC multidrug transporters utilize a broadly conserved alternating-access model to efflux drugs [144–146, 149].
These structural studies, in concert with spectroscopic, biophysical, and functional experiments, have established an alternating-access model for ABC mediated export [142, 152–156] (Fig. 6c). In this model, the transporter begins in an apo inward-facing state where the two cytosolic NBDs are separated, allowing substrates to access their binding site at the interface of the TMD. Once substrate binds, the NBDs come closer in proximity and induce a closure of the TMD interface resulting in a substrate-bound occluded state. ATP then binds and induces NBD dimerization which in turn triggers a rearrangement of the TMDs such that they adapt an outward-facing state. Here, the transporter has reduced affinity for substrate, favoring its release into the extra-cytosolic space. Finally, ATP is hydrolyzed, which triggers a conformational switch back to the inward-facing state, primed for the next transport event [134, 135] (see [149] and [157] for more in-depth review). Interestingly, it has been proposed that some bacterial ABC transporters such as PatAB from Streptococcus pneumoniae – which confers resistance to fluoroquinolone – favor GTP over ATP as an energy source [158].
As described previously, in Gram-negative bacteria some ABC transporters can interact with PAPs and OMCs to form tripartite complexes [11, 101, 102, 122, 128, 159]. For example, in E. coli, the inner membrane ABC transporter MacB forms a complex with the PAP MacA and the OMC TolC (Fig. 6d). This MacAB-TolC tripartite complex mediates the efflux of macrolide antibiotics [160] and has also been suggested to confer resistance to cyclic peptide-like antibiotics such as colistin and bacitracin [161]. Additionally, this complex mediates the secretion of small endogenous peptides including a heme-precursor protoporphyrin and a heat-stable polypeptide virulence factor enterotoxin STII [162, 163].
Advances in cryo-EM have recently enabled the structure of the MacAB-TolC tripartite complex to be determined [102]. This complex assembles with a 2:6:3 stoichiometry of MacB:MacA:TolC and presents a noncanonical ABC transporter fold of the homodimeric MacB, in which each protomer contains the TMD and cytosolic NBD as well as an atypical and extensive periplasmic domain. The TMD of each protomer comprises four TM helices, among which TM1 and TM2 extend into the periplasm to form a large domain. This periplasmic domain forms contacts with MacA via a conserved glutamine in each MacA monomer, which together form a glutamine ring that appears to act as a seal to prevent backflow of substrate into the periplasm (Fig. 6e). The overall architecture of MacB within this complex is consistent with crystal structures of MacB homologues obtained from two independent groups [161, 164] and previous biophysical experiments [165]. Remarkably, the MacAB-TolC cryo-EM structure reveals that the MacB dimer interface in the membrane is tightly packed, leaving no space for substrate binding despite the two cytosolic NBDs being physically separated. Instead, each MacB monomer within the dimer features an opening between the periplasmic extensions of TM1 and TM2, where unassigned density has been observed that may be attributed to an endogenous substrate (Fig. 6f). Based on this observation, and the mechanism of transport for other tripartite complexes such as AcrABZ-TolC (see above), it has been hypothesized that periplasmic substrates may access this central binding site via the opening between the periplasmic regions of TM1 and TM2, after which they are shuttled into the AcrA-TolC channel for release to the extracellular space.
Conclusions and Perspective
Rapidly evolving resistance to current drugs in clinical use and the subsequent spread of multidrug resistant strains of pathogenic organisms are major setbacks in combating infectious diseases and represent an increasingly serious public health crisis. This review reveals that while multidrug transporters may vary in fold, size, oligomerization, and coupling mechanism, they typically feature large substrate binding sites that comprise both hydrophobic and charged residues. This common feature allows them to interact with broad spectrums of substrates which can accommodate different regions of the binding sites via distinct interactions unique to their chemical identity. Additionally, other molecules such as lipids and waters, have recently been observed within the drug binding sites of certain multidrug transporters where they provide an added layer of malleability for protein-drug interactions. This is, in our opinion, a fascinating phenomenon and we are intrigued to learn if it is perhaps a common feature across multidrug transporters.
Finally, it is becoming ever more critical to understand molecular mechanisms of resistance and identify novel targets for anti-infectious agents. Technological advances, persistence of structural biologists, and importantly, corroboration with biochemical, spectroscopic, and biophysical experiments have and will continue to expand our knowledge of the molecular bases of multidrug resistance. Progress in understanding this complex, multifactorial problem has been tremendous. It is now becoming imperative that we improve how we leverage these structural insights and translate them into powerful tools to aid the development of novel, urgently required therapeutics to combat multidrug resistance and improve our treatment options for infectious diseases.
Highlights.
Transporter-mediated export is a major cause of resistance to anti-infectious agents.
Advances in structural biology have allowed for many structures of multidrug transporters to be determined.
Transporters can acquire point mutations to enable efflux of anti-infectious agents and promote survival.
Substrate binding sites are spacious, dynamic, and chemically malleable to facilitate multidrug transport.
Structural insights provide invaluable information for understanding and combatting multidrug resistance.
Acknowledgments
This work was supported by NIH grants (R35 GM132120 and R01 AI147628 to F.M.). R.J.C. was supported by the Simons Society of Fellows (Award Number: 578646).
Abbreviations:
- ABC superfamily
ATP-binding cassette superfamily
- MFS
major facilitator superfamily
- DMT superfamily
drug/metabolite transporter superfamily
- MATE family
multidrug and toxic compound extrusion family
- SMR family
small multidrug resistance protein family
- RND superfamily
resistance-nodulation-division family
- PACE family
proteobacterial antimicrobial compound efflux family
- AbgT family
p-aminobenzoyl-glutamate transporter family
- NMR
nuclear magnetic resonance
- cryo-EM
cryogenic-electron microscopy
- cryo-ET
cryo-electron tomography
- TM
transmembrane
- DHA
drug-proton antiporter family
- TPP+
tetraphenylphosphonium
- CQ
chloroquine
- PPQ
piperaquine
- PfCRT
Plasmodium falciparum chloroquine resistance transporter
- OMC
outer membrane channel
- PAP
periplasmic adaptor protein
- TMD
transmembrane domain
- NBD
nucleotide-binding domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Bolhuis H, van Veen HW, Poolman B, Driessen AJ, Konings WN. Mechanisms of multidrug transporters. FEMS Microbiol Rev. 1997;21:55–84. [DOI] [PubMed] [Google Scholar]
- [2].Putman M, Van Veen HW, Degener JE, Konings WN. Antibiotic resistance: era of the multidrug pump. Mol Microbiol. 2000;36:772–3. [DOI] [PubMed] [Google Scholar]
- [3].Jack DL, Yang NM, Saier MH Jr. The drug/metabolite transporter superfamily. Eur J Biochem. 2001;268:3620–39. [DOI] [PubMed] [Google Scholar]
- [4].Delmar JA, Yu EW. The AbgT family: A novel class of antimetabolite transporters. Protein Sci. 2016;25:322–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hassan KA, Liu Q, Elbourne LDH, Ahmad I, Sharples D, Naidu V, et al. Pacing across the membrane: the novel PACE family of efflux pumps is widespread in Gram-negative pathogens. Res Microbiol. 2018;169:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Choy BC, Cater RJ, Mancia F, Pryor EE, Jr. A 10-year meta-analysis of membrane protein structural biology: Detergents, membrane mimetics, and structure determination techniques. Biochim Biophys Acta Biomembr. 2020;1863:183533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yan N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem Sci. 2013;38:151–9. [DOI] [PubMed] [Google Scholar]
- [8].Yan N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu Rev Biophys. 2015;44:257–83. [DOI] [PubMed] [Google Scholar]
- [9].Quistgaard EM, Low C, Guettou F, Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol. 2016;17:123–32. [DOI] [PubMed] [Google Scholar]
- [10].Sá-Correia I, dos Santos SC, Teixeira MC, Cabrito TR, Mira NP. Drug: H+ antiporters in chemical stress response in yeast. Trends in microbiology. 2009;17:22–31. [DOI] [PubMed] [Google Scholar]
- [11].Tanabe M, Szakonyi G, Brown KA, Henderson PJ, Nield J, Byrne B. The multidrug resistance efflux complex, EmrAB from Escherichia coli forms a dimer in vitro. Biochemical and biophysical research communications. 2009;380:338–42. [DOI] [PubMed] [Google Scholar]
- [12].Lewis K. Translocases: a bacterial tunnel for drugs and proteins. Curr Biol. 2000;10:R678–81. [DOI] [PubMed] [Google Scholar]
- [13].Yousefian N, Ornik-Cha A, Poussard S, Decossas M, Berbon M, Daury L, et al. Structural characterization of the EmrAB-TolC efflux complex from E. coli. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2021;1863:183488. [DOI] [PubMed] [Google Scholar]
- [14].Du D, Wang-Kan X, Neuberger A, van Veen HW, Pos KM, Piddock LJV, et al. Multidrug efflux pumps: structure, function and regulation. Nature Reviews Microbiology. 2018;16:523–39. [DOI] [PubMed] [Google Scholar]
- [15].Fluman N, Bibi E. Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2009;1794:738–47. [DOI] [PubMed] [Google Scholar]
- [16].Wang D, Hu E, Chen J, Tao X, Gutierrez K, Qi Y. Characterization of novel ybjG and dacC variants in Escherichia coli. J Med Microbiol. 2013;62:1728–34. [DOI] [PubMed] [Google Scholar]
- [17].Edgar R, Bibi E. MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J Bacteriol. 1997;179:2274–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Heng J, Zhao Y, Liu M, Liu Y, Fan J, Wang X, et al. Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 2015;25:1060–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jiang D, Zhao Y, Wang X, Fan J, Heng J, Liu X, et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc Natl Acad Sci U S A. 2013;110:14664–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312:741–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Debruycker V, Hutchin A, Masureel M, Ficici E, Martens C, Legrand P, et al. An embedded lipid in the multidrug transporter LmrP suggests a mechanism for polyspecificity. Nat Struct Mol Biol. 2020;27:829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Xiao Q, Sun B, Zhou Y, Wang C, Guo L, He J, et al. Visualizing the nonlinear changes of a drug-proton antiporter from inward-open to occluded state. Biochem Biophys Res Commun. 2021;534:272–8. [DOI] [PubMed] [Google Scholar]
- [23].Liu M, Heng J, Gao Y, Wang X. Crystal structures of MdfA complexed with acetylcholine and inhibitor reserpine. Biophys Rep. 2016;2:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fluman N, Ryan CM, Whitelegge JP, Bibi E. Dissection of mechanistic principles of a secondary multidrug efflux protein. Molecular cell. 2012;47:777–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nagarathinam K, Nakada-Nakura Y, Parthier C, Terada T, Juge N, Jaenecke F, et al. Outward open conformation of a Major Facilitator Superfamily multidrug/H(+) antiporter provides insights into switching mechanism. Nat Commun. 2018;9:4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yardeni EH, Mishra S, Stein RA, Bibi E, McHaourab HS. The Multidrug Transporter MdfA Deviates from the Canonical Model of Alternating Access of MFS Transporters. J Mol Biol. 2020;432:5665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lewinson O, Bibi E. Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry. 2001;40:12612–8. [DOI] [PubMed] [Google Scholar]
- [28].Yardeni EH, Zomot E, Bibi E. The fascinating but mysterious mechanistic aspects of multidrug transport by MdfA from Escherichia coli. Research in microbiology. 2018;169:455–60. [DOI] [PubMed] [Google Scholar]
- [29].Vastermark A, Almen MS, Simmen MW, Fredriksson R, Schioth HB. Functional specialization in nucleotide sugar transporters occurred through differentiation of the gene cluster EamA (DUF6) before the radiation of Viridiplantae. BMC Evol Biol. 2011;11:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Martin RE, Kirk K. The malaria parasite’s chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol Biol Evol. 2004;21:1938–49. [DOI] [PubMed] [Google Scholar]
- [31].Lolkema JS, Dobrowolski A, Slotboom DJ. Evolution of antiparallel two-domain membrane proteins: tracing multiple gene duplication events in the DUF606 family. J Mol Biol. 2008;378:596–606. [DOI] [PubMed] [Google Scholar]
- [32].Tsuchiya H, Doki S, Takemoto M, Ikuta T, Higuchi T, Fukui K, et al. Structural basis for amino acid export by DMT superfamily transporter YddG. Nature. 2016;534:417–20. [DOI] [PubMed] [Google Scholar]
- [33].Kim J, Tan YZ, Wicht KJ, Erramilli SK, Dhingra SK, Okombo J, et al. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature. 2019;576:315–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19:156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Agrawal S, Moser KA, Morton L, Cummings MP, Parihar A, Dwivedi A, et al. Association of a Novel Mutation in the Plasmodium falciparum Chloroquine Resistance Transporter With Decreased Piperaquine Sensitivity. J Infect Dis. 2017;216:468–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ross LS, Dhingra SK, Mok S, Yeo T, Wicht KJ, Kumpornsin K, et al. Emerging Southeast Asian PfCRT mutations confer Plasmodium falciparum resistance to the first-line antimalarial piperaquine. Nat Commun. 2018;9:3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu J, Istvan ES, Gluzman IY, Gross J, Goldberg DE. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proceedings of the National Academy of Sciences. 2006;103:8840–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sullivan DJ. Quinolines block every step of malaria heme crystal growth. Proceedings of the National Academy of Sciences. 2017;114:7483–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dhingra SK, Redhi D, Combrinck JM, Yeo T, Okombo J, Henrich PP, et al. A variant PfCRT isoform can contribute to Plasmodium falciparum resistance to the first-line partner drug piperaquine. MBio. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasitol. 2012;28:504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shafik SH, Cobbold SA, Barkat K, Richards SN, Lancaster NS, Llinas M, et al. The natural function of the malaria parasite’s chloroquine resistance transporter. Nat Commun. 2020;11:3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lakshmanan V, Bray PG, Verdier-Pinard D, Johnson DJ, Horrocks P, Muhle RA, et al. A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. The EMBO journal. 2005;24:2294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gabryszewski SJ, Modchang C, Musset L, Chookajorn T, Fidock DA. Combinatorial genetic modeling of pfcrt-mediated drug resistance evolution in Plasmodium falciparum. Molecular biology and evolution. 2016;33:1554–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pelleau S, Moss EL, Dhingra SK, Volney B, Casteras J, Gabryszewski SJ, et al. Adaptive evolution of malaria parasites in French Guiana: Reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci U S A. 2015;112:11672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wicht KJ, Mok S, Fidock DA. Molecular Mechanisms of Drug Resistance in Plasmodium falciparum Malaria. Annu Rev Microbiol. 2020;74:431–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bay DC, Turner RJ. Diversity and evolution of the small multidrug resistance protein family. BMC Evol Biol. 2009;9:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bay DC, Rommens KL, Turner RJ. Small multidrug resistance proteins: a multidrug transporter family that continues to grow. Biochim Biophys Acta. 2008;1778:1814–38. [DOI] [PubMed] [Google Scholar]
- [49].Korkhov VM, Tate CG. An emerging consensus for the structure of EmrE. Acta Crystallogr D Biol Crystallogr. 2009;65:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dutta S, Morrison EA, Henzler-Wildman KA. EmrE dimerization depends on membrane environment. Biochim Biophys Acta. 2014;1838:1817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kermani AA, Macdonald CB, Burata OE, Ben Koff B, Koide A, Denbaum E, et al. The structural basis of promiscuity in small multidrug resistance transporters. Nat Commun. 2020;11:6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Gupta AK, Katoch VM, Chauhan DS, Sharma R, Singh M, Venkatesan K, et al. Microarray analysis of efflux pump genes in multidrug-resistant Mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb Drug Resist. 2010;16:21–8. [DOI] [PubMed] [Google Scholar]
- [53].Srinivasan VB, Rajamohan G, Gebreyes WA. Role of AbeS, a novel efflux pump of the SMR family of transporters, in resistance to antimicrobial agents in Acinetobacter baumannii. Antimicrob Agents Chemother. 2009;53:5312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kermani AA, Macdonald CB, Gundepudi R, Stockbridge RB. Guanidinium export is the primal function of SMR family transporters. Proc Natl Acad Sci U S A. 2018;115:3060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Brill S, Falk OS, Schuldiner S. Transforming a drug/H+ antiporter into a polyamine importer by a single mutation. Proc Natl Acad Sci U S A. 2012;109:16894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Leninger M, Sae Her A, Traaseth NJ. Inducing conformational preference of the membrane protein transporter EmrE through conservative mutations. Elife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tate CG, Kunji ER, Lebendiker M, Schuldiner S. The projection structure of EmrE, a proton-linked multidrug transporter from Escherichia coli, at 7 A resolution. EMBO J. 2001;20:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. Three-dimensional structure of the bacterial multidrug transporter EmrE shows it is an asymmetric homodimer. EMBO J. 2003;22:6175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. X-ray structure of EmrE supports dual topology model. Proc Natl Acad Sci U S A. 2007;104:18999–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Dutta S, Morrison EA, Henzler-Wildman KA. Blocking dynamics of the SMR transporter EmrE impairs efflux activity. Biophys J. 2014;107:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Robinson AE, Thomas NE, Morrison EA, Balthazor BM, Henzler-Wildman KA. New free-exchange model of EmrE transport. Proc Natl Acad Sci U S A. 2017;114:E10083–E91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gayen A, Leninger M, Traaseth NJ. Protonation of a glutamate residue modulates the dynamics of the drug transporter EmrE. Nat Chem Biol. 2016;12:141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tate CG, Ubarretxena-Belandia I, Baldwin JM. Conformational changes in the multidrug transporter EmrE associated with substrate binding. J Mol Biol. 2003;332:229–42. [DOI] [PubMed] [Google Scholar]
- [64].Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, et al. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature. 2011;481:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Dastvan R, Fischer AW, Mishra S, Meiler J, McHaourab HS. Protonation-dependent conformational dynamics of the multidrug transporter EmrE. Proc Natl Acad Sci U S A. 2016;113:1220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gayen A, Banigan JR, Traaseth NJ. Ligand-induced conformational changes of the multidrug resistance transporter EmrE probed by oriented solid-state NMR spectroscopy. Angew Chem Int Ed Engl. 2013;52:10321–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Amadi ST, Koteiche HA, Mishra S, McHaourab HS. Structure, dynamics, and substrate-induced conformational changes of the multidrug transporter EmrE in liposomes. J Biol Chem. 2010;285:26710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Korkhov VM, Tate CG. Electron crystallography reveals plasticity within the drug binding site of the small multidrug transporter EmrE. J Mol Biol. 2008;377:1094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Morrison EA, Henzler-Wildman KA. Transported substrate determines exchange rate in the multidrug resistance transporter EmrE. J Biol Chem. 2014;289:6825–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Thomas NE, Wu C, Morrison EA, Robinson AE, Werner JP, Henzler-Wildman KA. The C terminus of the bacterial multidrug transporter EmrE couples drug binding to proton release. J Biol Chem. 2018;293:19137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Morrison EA, Robinson AE, Liu Y, Henzler-Wildman KA. Asymmetric protonation of EmrE. J Gen Physiol. 2015;146:445–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cho MK, Gayen A, Banigan JR, Leninger M, Traaseth NJ. Intrinsic conformational plasticity of native EmrE provides a pathway for multidrug resistance. J Am Chem Soc. 2014;136:8072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lloris-Garcera P, Slusky JS, Seppala S, Priess M, Schafer LV, von Heijne G. In vivo trp scanning of the small multidrug resistance protein EmrE confirms 3D structure models’. J Mol Biol. 2013;425:4642–51. [DOI] [PubMed] [Google Scholar]
- [74].Rapp M, Granseth E, Seppala S, von Heijne G. Identification and evolution of dual-topology membrane proteins. Nat Struct Mol Biol. 2006;13:112–6. [DOI] [PubMed] [Google Scholar]
- [75].Brill S, Sade-Falk O, Elbaz-Alon Y, Schuldiner S. Specificity determinants in small multidrug transporters. Journal of molecular biology. 2015;427:468–77. [DOI] [PubMed] [Google Scholar]
- [76].Elbaz Y, Tayer N, Steinfels E, Steiner-Mordoch S, Schuldiner S. Substrate-induced tryptophan fluorescence changes in EmrE, the smallest ion-coupled multidrug transporter. Biochemistry. 2005;44:7369–77. [DOI] [PubMed] [Google Scholar]
- [77].Shcherbakov AA, Hisao G, Mandala VS, Thomas NE, Soltani M, Salter EA, et al. Structure and dynamics of the drug-bound bacterial transporter EmrE in lipid bilayers. Nat Commun. 2021;12:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wu C, Wynne SA, Thomas NE, Uhlemann EM, Tate CG, Henzler-Wildman KA. Identification of an Alternating-Access Dynamics Mutant of EmrE with Impaired Transport. J Mol Biol. 2019;431:2777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ovchinnikov V, Stone TA, Deber CM, Karplus M. Structure of the EmrE multidrug transporter and its use for inhibitor peptide design. Proceedings of the National Academy of Sciences. 2018;115:E7932–E41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Schuldiner S. EmrE, a model for studying evolution and mechanism of ion-coupled transporters. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2009;1794:748–62. [DOI] [PubMed] [Google Scholar]
- [81].Hussey GA, Thomas NE, Henzler-Wildman KA. Highly coupled transport can be achieved in free-exchange transport models. J Gen Physiol. 2020;152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Omote H, Hiasa M, Matsumoto T, Otsuka M, Moriyama Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol Sci. 2006;27:587–93. [DOI] [PubMed] [Google Scholar]
- [83].Brown MH, Paulsen IT, Skurray RA. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol. 1999;31:394–5. [DOI] [PubMed] [Google Scholar]
- [84].Morita Y, Kataoka A, Shiota S, Mizushima T, Tsuchiya T. NorM of vibrio parahaemolyticus is an Na(+)-driven multidrug efflux pump. J Bacteriol. 2000;182:6694–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kusakizako T, Miyauchi H, Ishitani R, Nureki O. Structural biology of the multidrug and toxic compound extrusion superfamily transporters. Biochim Biophys Acta Biomembr. 2020;1862:183154. [DOI] [PubMed] [Google Scholar]
- [86].McAleese F, Petersen P, Ruzin A, Dunman PM, Murphy E, Projan SJ, et al. A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob Agents Chemother. 2005;49:1865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kaatz GW, McAleese F, Seo SM. Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob Agents Chemother. 2005;49:1857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].He X, Szewczyk P, Karyakin A, Evin M, Hong WX, Zhang Q, et al. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010;467:991–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Otsuka M, Yasuda M, Morita Y, Otsuka C, Tsuchiya T, Omote H, et al. Identification of essential amino acid residues of the NorM Na+/multidrug antiporter in Vibrio parahaemolyticus. J Bacteriol. 2005;187:1552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lu M, Symersky J, Radchenko M, Koide A, Guo Y, Nie R, et al. Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc Natl Acad Sci U S A. 2013;110:2099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Tanaka Y, Hipolito CJ, Maturana AD, Ito K, Kuroda T, Higuchi T, et al. Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature. 2013;496:247–51. [DOI] [PubMed] [Google Scholar]
- [92].Lu M, Radchenko M, Symersky J, Nie R, Guo Y. Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat Struct Mol Biol. 2013;20:1310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Radchenko M, Symersky J, Nie R, Lu M. Structural basis for the blockade of MATE multidrug efflux pumps. Nat Commun. 2015;6:7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zakrzewska S, Mehdipour AR, Malviya VN, Nonaka T, Koepke J, Muenke C, et al. Inward-facing conformation of a multidrug resistance MATE family transporter. Proc Natl Acad Sci U S A. 2019;116:12275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Kusakizako T, Claxton DP, Tanaka Y, Maturana AD, Kuroda T, Ishitani R, et al. Structural Basis of H(+)-Dependent Conformational Change in a Bacterial MATE Transporter. Structure. 2019;27:293–301 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Mousa JJ, Yang Y, Tomkovich S, Shima A, Newsome RC, Tripathi P, et al. MATE transport of the E. coli-derived genotoxin colibactin. Nat Microbiol. 2016;1:15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harbor perspectives in biology. 2010;2:a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Du D, van Veen HW, Luisi BF. Assembly and operation of bacterial tripartite multidrug efflux pumps. Trends in microbiology. 2015;23:311–9. [DOI] [PubMed] [Google Scholar]
- [99].Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, et al. Structure of the AcrAB–TolC multidrug efflux pump. Nature. 2014;509:512–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Neuberger A, Du D, Luisi BF. Structure and mechanism of bacterial tripartite efflux pumps. Research in microbiology. 2018;169:401–13. [DOI] [PubMed] [Google Scholar]
- [101].Lee M, Kim H-L, Song S, Joo M, Lee S, Kim D, et al. The α-barrel tip region of Escherichia coli TolC homologs of Vibrio vulnificus interacts with the MacA protein to form the functional macrolide-specific efflux pump MacAB-TolC. Journal of Microbiology. 2013;51:154–9. [DOI] [PubMed] [Google Scholar]
- [102].Fitzpatrick AWP, Llabres S, Neuberger A, Blaza JN, Bai XC, Okada U, et al. Structure of the MacAB-TolC ABC-type tripartite multidrug efflux pump. Nat Microbiol. 2017;2:17070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Nikaido H, Zgurskaya HI. AcrAB and related multidrug efflux pumps of Escherichia coli. Journal of molecular microbiology and biotechnology. 2001;3:215–8. [PubMed] [Google Scholar]
- [104].Hobbs EC, Yin X, Paul BJ, Astarita JL, Storz G. Conserved small protein associates with the multidrug efflux pump AcrB and differentially affects antibiotic resistance. Proceedings of the National Academy of Sciences. 2012;109:16696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Du D, Neuberger A, Orr MW, Newman CE, Hsu P-C, Samsudin F, et al. Interactions of a bacterial RND transporter with a transmembrane small protein in a lipid environment. Structure. 2020;28:625–34. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419:587–93. [DOI] [PubMed] [Google Scholar]
- [107].He X, Szewczyk P, Karyakin A, Evin M, Hong W-X, Zhang Q, et al. Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature. 2010;467:991–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Eicher T, Cha H-j, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proceedings of the National Academy of Sciences. 2012;109:5687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature. 2011;480:565–9. [DOI] [PubMed] [Google Scholar]
- [110].Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–9. [DOI] [PubMed] [Google Scholar]
- [111].Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstätter L, Verrey F, et al. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife. 2014;3:e03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Sjuts H, Vargiu AV, Kwasny SM, Nguyen ST, Kim H-S, Ding X, et al. Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proceedings of the National Academy of Sciences. 2016;113:3509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, et al. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. elife. 2017;6:e24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, et al. Structural basis for the inhibition of bacterial multidrug exporters. Nature. 2013;500:102–6. [DOI] [PubMed] [Google Scholar]
- [115].Hung L-W, Kim H-B, Murakami S, Gupta G, Kim C-Y, Terwilliger TC. Crystal structure of AcrB complexed with linezolid at 3.5 Å resolution. Journal of structural and functional genomics. 2013;14:71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zwama M, Yamaguchi A. Molecular mechanisms of AcrB-mediated multidrug export. Research in microbiology. 2018;169:372–83. [DOI] [PubMed] [Google Scholar]
- [117].Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313:1295–8. [DOI] [PubMed] [Google Scholar]
- [118].Takatsuka Y, Chen C, Nikaido H. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proceedings of the National Academy of Sciences. 2010;107:6559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Edward WY, Aires JR, Nikaido H. AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. Journal of bacteriology. 2003;185:5657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kinana AD, Vargiu AV, Nikaido H. Some ligands enhance the efflux of other ligands by the Escherichia coli multidrug pump AcrB. Biochemistry. 2013;52:8342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Vargiu AV, Ramaswamy VK, Malvacio I, Malloci G, Kleinekathöfer U, Ruggerone P. Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump. Biochimica et Biophysica Acta (BBA)-General Subjects. 2018;1862:836–45. [DOI] [PubMed] [Google Scholar]
- [122].Murakami S, Okada U, van Veen HW. Tripartite transporters as mechanotransmitters in periplasmic alternating-access mechanisms. FEBS Lett. 2020. [DOI] [PubMed] [Google Scholar]
- [123].Daury L, Orange F, Taveau J-C, Verchère A, Monlezun L, Gounou C, et al. Tripartite assembly of RND multidrug efflux pumps. Nature communications. 2016;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Shi X, Chen M, Yu Z, Bell JM, Wang H, Forrester I, et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nature Communications. 2019;10:2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67–113. [DOI] [PubMed] [Google Scholar]
- [126].El-Awady R, Saleh E, Hashim A, Soliman N, Dallah A, Elrasheed A, et al. The Role of Eukaryotic and Prokaryotic ABC Transporter Family in Failure of Chemotherapy. Front Pharmacol. 2016;7:535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Davidson AL, Dassa E, Orelle C, Chen J. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol Mol Biol Rev. 2008;72:317–64, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Lubelski J, Konings WN, Driessen AJ. Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiol Mol Biol Rev. 2007;71:463–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Locher KP, Lee AT, Rees DC. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 2002;296:1091–8. [DOI] [PubMed] [Google Scholar]
- [130].Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–5. [DOI] [PubMed] [Google Scholar]
- [131].Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Esser L, Zhou F, Pluchino KM, Shiloach J, Ma J, Tang WK, et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J Biol Chem. 2017;292:446–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Jin MS, Oldham ML, Zhang Q, Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Alam A, Kowal J, Broude E, Roninson I, Locher KP. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science. 2019;363:753–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Kim Y, Chen J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science. 2018;359:915–9. [DOI] [PubMed] [Google Scholar]
- [136].Nosol K, Romane K, Irobalieva RN, Alam A, Kowal J, Fujita N, et al. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc Natl Acad Sci U S A. 2020;117:26245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Hofmann S, Januliene D, Mehdipour AR, Thomas C, Stefan E, Bruchert S, et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature. 2019;571:580–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Liu F, Zhang Z, Csanady L, Gadsby DC, Chen J. Molecular Structure of the Human CFTR Ion Channel. Cell. 2017;169:85–95 e8. [DOI] [PubMed] [Google Scholar]
- [139].Zhang Z, Liu F, Chen J. Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell. 2017;170:483–91 e8. [DOI] [PubMed] [Google Scholar]
- [140].Liu F, Zhang Z, Levit A, Levring J, Touhara KK, Shoichet BK, et al. Structural identification of a hotspot on CFTR for potentiation. Science. 2019;364:1184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Ward A, Reyes CL, Yu J, Roth CB, Chang G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc Natl Acad Sci U S A. 2007;104:19005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Mi W, Li Y, Yoon SH, Ernst RK, Walz T, Liao M. Structural basis of MsbA-mediated lipopolysaccharide transport. Nature. 2017;549:233–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Padayatti PS, Lee SC, Stanfield RL, Wen PC, Tajkhorshid E, Wilson IA, et al. Structural Insights into the Lipid A Transport Pathway in MsbA. Structure. 2019;27:1114–23 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Taylor NMI, Manolaridis I, Jackson SM, Kowal J, Stahlberg H, Locher KP. Structure of the human multidrug transporter ABCG2. Nature. 2017;546:504–9. [DOI] [PubMed] [Google Scholar]
- [145].Manolaridis I, Jackson SM, Taylor NMI, Kowal J, Stahlberg H, Locher KP. Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states. Nature. 2018;563:426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Jackson SM, Manolaridis I, Kowal J, Zechner M, Taylor NMI, Bause M, et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat Struct Mol Biol. 2018;25:333–40. [DOI] [PubMed] [Google Scholar]
- [147].Locher KP. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol. 2016;23:487–93. [DOI] [PubMed] [Google Scholar]
- [148].Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10:218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Thomas C, Tampe R. Structural and Mechanistic Principles of ABC Transporters. Annu Rev Biochem. 2020;89:605–36. [DOI] [PubMed] [Google Scholar]
- [150].Choudhury HG, Tong Z, Mathavan I, Li Y, Iwata S, Zirah S, et al. Structure of an antibacterial peptide ATP-binding cassette transporter in a novel outward occluded state. Proc Natl Acad Sci U S A. 2014;111:9145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Bountra K, Hagelueken G, Choudhury HG, Corradi V, El Omari K, Wagner A, et al. Structural basis for antibacterial peptide self-immunity by the bacterial ABC transporter McjD. EMBO J. 2017;36:3062–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Doshi R, van Veen HW. Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J Biol Chem. 2013;288:21638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Padayatti PS, Lee SC, Stanfield RL, Wen P-C, Tajkhorshid E, Wilson IA, et al. Structural insights into the lipid A transport pathway in MsbA. Structure. 2019;27:1114–23. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Romano M, Fusco G, Choudhury HG, Mehmood S, Robinson CV, Zirah S, et al. Structural Basis for Natural Product Selection and Export by Bacterial ABC Transporters. ACS Chem Biol. 2018;13:1598–609. [DOI] [PubMed] [Google Scholar]
- [155].Zou P, Bortolus M, McHaourab HS. Conformational cycle of the ABC transporter MsbA in liposomes: detailed analysis using double electron-electron resonance spectroscopy. J Mol Biol. 2009;393:586–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Zoghbi ME, Cooper RS, Altenberg GA. The Lipid Bilayer Modulates the Structure and Function of an ATP-binding Cassette Exporter. J Biol Chem. 2016;291:4453–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Srikant S, Gaudet R. Mechanics and pharmacology of substrate selection and transport by eukaryotic ABC exporters. Nature structural & molecular biology. 2019;26:792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Orelle C, Durmort C, Mathieu K, Duchene B, Aros S, Fenaille F, et al. A multidrug ABC transporter with a taste for GTP. Sci Rep. 2018;8:2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Greene NP, Kaplan E, Crow A, Koronakis V. Antibiotic Resistance Mediated by the MacB ABC Transporter Family: A Structural and Functional Perspective. Front Microbiol. 2018;9:950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Kobayashi N, Nishino K, Yamaguchi A. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 2001;183:5639–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Crow A, Greene NP, Kaplan E, Koronakis V. Structure and mechanotransmission mechanism of the MacB ABC transporter superfamily. Proc Natl Acad Sci U S A. 2017;114:12572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Turlin E, Heuck G, Simoes Brandao MI, Szili N, Mellin JR, Lange N, et al. Protoporphyrin (PPIX) efflux by the MacAB-TolC pump in Escherichia coli. Microbiologyopen. 2014;3:849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Yamanaka H, Kobayashi H, Takahashi E, Okamoto K. MacAB is involved in the secretion of Escherichia coli heat-stable enterotoxin II. J Bacteriol. 2008;190:7693–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Okada U, Yamashita E, Neuberger A, Morimoto M, van Veen HW, Murakami S. Crystal structure of tripartite-type ABC transporter MacB from Acinetobacter baumannii. Nat Commun. 2017;8:1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Lin HT, Bavro VN, Barrera NP, Frankish HM, Velamakanni S, van Veen HW, et al. MacB ABC transporter is a dimer whose ATPase activity and macrolide-binding capacity are regulated by the membrane fusion protein MacA. J Biol Chem. 2009;284:1145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Yang HB, Hou WT, Cheng MT, Jiang YL, Chen Y, Zhou CZ. Structure of a MacAB-like efflux pump from Streptococcus pneumoniae. Nat Commun. 2018;9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Dawson RJ, Locher KP. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS letters. 2007;581:935–8. [DOI] [PubMed] [Google Scholar]
- [168].Noll A, Thomas C, Herbring V, Zollmann T, Barth K, Mehdipour AR, et al. Crystal structure and mechanistic basis of a functional homolog of the antigen transporter TAP. Proc Natl Acad Sci U S A. 2017;114:E438–E47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Wu HH, Symersky J, Lu M. Structure of an engineered multidrug transporter MdfA reveals the molecular basis for substrate recognition. Commun Biol. 2019;2:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Wu HH, Symersky J, Lu M. Structure and mechanism of a redesigned multidrug transporter from the Major Facilitator Superfamily. Sci Rep. 2020;10:3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Zomot E, Yardeni EH, Vargiu AV, Tam HK, Malloci G, Ramaswamy VK, et al. A New Critical Conformational Determinant of Multidrug Efflux by an MFS Transporter. J Mol Biol. 2018;430:1368–85. [DOI] [PubMed] [Google Scholar]
- [172].Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE, Jr. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science. 2003;300:976–80. [DOI] [PubMed] [Google Scholar]
- [173].Ababou A, Koronakis V. Structures of Gate Loop Variants of the AcrB Drug Efflux Pump Bound by Erythromycin Substrate. PLoS One. 2016;11:e0159154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, et al. Crystal structure of the multidrug efflux transporter AcrB at 3.1A resolution reveals the N-terminal region with conserved amino acids. J Struct Biol. 2007;158:494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Qiu W, Fu Z, Xu GG, Grassucci RA, Zhang Y, Frank J, et al. Structure and activity of lipid bilayer within a membrane-protein transporter. Proc Natl Acad Sci U S A. 2018;115:12985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Zwama M, Hayashi K, Sakurai K, Nakashima R, Kitagawa K, Nishino K, et al. Hoisting- Loop in Bacterial Multidrug Exporter AcrB Is a Highly Flexible Hinge That Enables the Large Motion of the Subdomains. Front Microbiol. 2017;8:2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, et al. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol. 2006;188:7290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187:6804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Drew D, Klepsch MM, Newstead S, Flaig R, De Gier JW, Iwata S, et al. The structure of the efflux pump AcrB in complex with bile acid. Mol Membr Biol. 2008;25:677–82. [DOI] [PubMed] [Google Scholar]
- [180].Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grutter MG. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol. 2007;5:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Tornroth-Horsefield S, Gourdon P, Horsefield R, Brive L, Yamamoto N, Mori H, et al. Crystal structure of AcrB in complex with a single transmembrane subunit reveals another twist. Structure. 2007;15:1663–73. [DOI] [PubMed] [Google Scholar]
- [182].Monroe N, Sennhauser G, Seeger MA, Briand C, Grutter MG. Designed ankyrin repeat protein binders for the crystallization of AcrB: plasticity of the dominant interface. J Struct Biol. 2011;174:269–81. [DOI] [PubMed] [Google Scholar]
- [183].Veesler D, Blangy S, Cambillau C, Sciara G. There is a baby in the bath water: AcrB contamination is a major problem in membrane-protein crystallization. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Oswald C, Tam HK, Pos KM. Transport of lipophilic carboxylates is mediated by transmembrane helix 2 in multidrug transporter AcrB. Nat Commun. 2016;7:13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Tam HK, Malviya VN, Foong WE, Herrmann A, Malloci G, Ruggerone P, et al. Binding and Transport of Carboxylated Drugs by the Multidrug Transporter AcrB. J Mol Biol. 2020;432:861–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Johnson RM, Fais C, Parmar M, Cheruvara H, Marshall RL, Hesketh SJ, et al. Cryo-EM Structure and Molecular Dynamics Analysis of the Fluoroquinolone Resistant Mutant of the AcrB Transporter from Salmonella. Microorganisms. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Su CC, Morgan CE, Kambakam S, Rajavel M, Scott H, Huang W, et al. Cryo-Electron Microscopy Structure of an Acinetobacter baumannii Multidrug Efflux Pump. mBio. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Su CC, Yin L, Kumar N, Dai L, Radhakrishnan A, Bolla JR, et al. Structures and transport dynamics of a Campylobacter jejuni multidrug efflux pump. Nat Commun. 2017;8:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Sennhauser G, Bukowska MA, Briand C, Grutter MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol. 2009;389:134–45. [DOI] [PubMed] [Google Scholar]
- [190].Glavier M, Puvanendran D, Salvador D, Decossas M, Phan G, Garnier C, et al. Antibiotic export by MexB multidrug efflux transporter is allosterically controlled by a MexA-OprM chaperone-like complex. Nat Commun. 2020;11:4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Sakurai K, Yamasaki S, Nakao K, Nishino K, Yamaguchi A, Nakashima R. Crystal structures of multidrug efflux pump MexB bound with high-molecular-mass compounds. Sci Rep. 2019;9:4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Tsutsumi K, Yonehara R, Ishizaka-Ikeda E, Miyazaki N, Maeda S, Iwasaki K, et al. Structures of the wild-type MexAB-OprM tripartite pump reveal its complex formation and drug efflux mechanism. Nat Commun. 2019;10:1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Lyu M, Moseng MA, Reimche JL, Holley CL, Dhulipala V, Su CC, et al. Cryo-EM Structures of a Gonococcal Multidrug Efflux Pump Illuminate a Mechanism of Drug Recognition and Resistance. mBio. 2020;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lei HT, Chou TH, Su CC, Bolla JR, Kumar N, Radhakrishnan A, et al. Crystal structure of the open state of the Neisseria gonorrhoeae MtrE outer membrane channel. PLoS One. 2014;9:e97475. [DOI] [PMC free article] [PubMed] [Google Scholar]