

Abstract
Dystonia is characterized by involuntary muscle contractions that cause debilitating twisting movements and postures. Although dysfunction of the basal ganglia, a brain region that mediates movement, is implicated in many forms of dystonia, the underlying mechanisms are unclear. The inherited metabolic disorder DOPA-responsive dystonia is considered a prototype for understanding basal ganglia dysfunction in dystonia because it is caused by mutations in genes necessary for the synthesis of the neurotransmitter dopamine, which mediates the activity of the basal ganglia. Therefore, to reveal abnormal striatal cellular processes and pathways implicated in dystonia, we used an unbiased proteomic approach in a knockin mouse model of DOPA-responsive dystonia, a model in which the striatum is known to play a central role in the expression of dystonia. Fifty-seven of the 1805 proteins identified were differentially regulated in DOPA-responsive dystonia mice compared to control mice. Most differentially regulated proteins were associated with gene ontology terms that implicated either mitochondrial or synaptic dysfunction whereby proteins associated with mitochondrial function were generally over-represented and proteins associated with synaptic function were largely under-represented. Remarkably, nearly 20% of the differentially regulated striatal proteins identified in our screen are associated with pathogenic variants that cause inherited disorders with dystonia as a sign in humans suggesting shared mechanisms across many different forms of dystonia.
Keywords: proteomics, dopamine, basal ganglia, mitochondria, dopamine, knockin mouse
1. Introduction
Dystonia is characterized by involuntary muscle contractions that cause debilitating twisting movements and postures [1]. Dystonia is a heterogeneous disorder that may be sporadic or inherited [2, 3] and can sometimes occur as a result of brain injury, such as the dystonia that frequently occurs in cerebral palsy patients. Although the etiologies are diverse, basal ganglia dysfunction is consistently implicated across many different forms of dystonia [4]. Lesions and structural defects of the basal ganglia and its connections are sometimes accompanied by dystonia [5–8]. Functional imaging studies have revealed abnormal metabolic activity in the basal ganglia [9–12] and dystonia improves in some patients after neurosurgical lesions or deep brain stimulation of the internal segment of the globus pallidus [13, 14]. Additionally, dopamine neurotransmission, which is integral to the normal function of the basal ganglia, is abnormal in both sporadic and inherited forms of dystonia. Mutations in genes critical for the synthesis of dopamine, including GTP-cyclohydrolase and tyrosine hydroxylase (TH) cause DOPA-responsive dystonia [DRD; 15, 16, 17]. Idiopathic forms of dystonia such as writer’s cramp and spasmodic dysphonia are also associated with abnormal striatal dopaminergic neurotransmission [18, 19]. Further, reduced striatal D2 dopamine receptor (D2R) availability is observed in both inherited and idiopathic forms of dystonia including DYT1 dystonia, blepharospasm, torticollis, writer’s cramp and laryngeal dystonia [18–23]. Despite the overwhelming evidence implicating basal ganglia dysfunction in dystonia, the precise nature of the cellular defects that give rise to dystonia are unclear.
The inherited metabolic disorder DRD is considered a prototype for understanding basal ganglia dysfunction in dystonia because the causal pathogenic variants are known to disrupt dopamine neurotransmission, which mediates striatal activity [24, 25]. DRD knockin mice carry the human DRD-causing p.381Q>K mutation in TH and exhibit the characteristic features of DRD including reduced brain dopamine concentrations and dystonic movements that improve in response to l-DOPA administration [26]. Further, it is known that striatal dopamine neurotransmission plays a central role in mediating the dystonia in DRD mice [26]. Therefore, we used an unbiased proteomic approach to analyze striatal protein expression in DRD mice to provide insight into the cellular processes and pathways underlying dystonia. Our analyses revealed 57 proteins that were differentially expressed in DRD mice compared to control mice. Remarkably, pathogenic variants of 12 of the 57 differentially regulated proteins are known to be associated with disorders in which dystonia is a prominent feature, suggesting shared mechanisms across many different forms of dystonia.
2. Methods
2.1. Mice.
Male and female adult mice homozygous for the c.1160C>A TH mutation (Thdrd/Thdrd ;DRD mice) and normal littermates (+/+) were used for all experiments. Mice were tested at 3–5 months of age when DRD mice exhibit peak dystonia [27]. The DRD mutation is coisogenic on C57BL/6J and congenic on DBA/2J. DRD and normal mice were generated as F1 hybrids of C57BL/6J +/Thdrd × DBA/2J +/Thdrd to circumvent the high perinatal lethality exhibited by inbred C57BL/6J DRD mice. Cell-type specific reporter transgenes inbred on C57BL/6J were bred onto the C57BL/6J +/Thdrd strain prior to the F1 hybrid cross to identify D1R-expressing (Drd1a-tdTomato; JAX) or D2R-expressing (Drd2-EGFP; Heintz, Rockefeller University) medium spiny neurons (MSNs); [28, 29]. Mice were maintained as described previously [26]. Briefly, from P9.5 to P16.5, the drinking water was supplemented with 1.5 mg/mL L-DOPA (Sigma-Aldrich), 0.5 mg/mL benserazide (Sigma-Aldrich), and 2.5 mg/mL ascorbic acid to facilitate feeding, growth, and normal movement. Thereafter, mice received a daily injection (s.c.) of 10 mg/kg l-DOPA, 2.5 mg/kg benserazide and 2.5 mg/mL ascorbic acid in saline. Normal (+/+) littermates received all treatments in parallel. l-DOPA supplementation was terminated >24 hours prior to euthanasia so DRD mice were dystonic. Mice were housed in standard mouse cages (2–5 mice/cage) in standard environmental conditions (72°C, 40–50% relative humidity) with a 12 hr light cycle (7am to 7 pm) and ad lib access to food and water. All procedures conformed to the NIH Guidelines for the Care and Use of Animals and were approved by the Emory University Animal Care and Use Committee.
2.2. Behavioral assessment.
Mice were habituated to the test cages (29 × 50 cm) for > 3 hrs prior to behavioral assessment. A behavioral inventory was used to identify abnormal movements including tonic flexion (forelimbs, hindlimbs, trunk, head), tonic extension (forelimbs, hindlimbs, trunk, head), clonus (forelimbs and hindlimbs), twisting (trunk, head), and tremor (forelimbs, hindlimbs, trunk, head) [30]. Testing began at 2 pm, and abnormal movements were observed and scored by an observer blinded to genotype for 30 sec at 10 min intervals for 1 hr. Abnormal movements were scored as present (1) or absent (0) for each body region during each time bin. An abnormal movement score was calculated by summing all scores.
2.3. Immunohistochemistry.
Mice were deeply anesthetized with isoflurane and perfused with 4% ice-cold, buffered paraformaldehyde (pH 7.2), incubated overnight in the paraformaldehyde perfusate at 4°C and transferred to a 30% buffered sucrose solution. Brains were sectioned (30 μm) in the coronal plane using a freezing microtome. Sections were immunostained using an anti-mCherry polyclonal antibody (1:5,000; #AB167453; Abcam) or anti-GFP polyclonal primary antibody (1:20,000; A-11122; Invitrogen). Floating sections were treated with 0.5% Triton-X in Tris-buffered saline (TBS) for 30 min and then transferred to 5% normal goat serum, 5% bovine serum albumin, 0.1% bovine gelatin, 0.05% Tween-80, and 0.01% sodium azide in TBS for 2 hrs. Sections were incubated with primary antibody for 16–24 hrs at 4°C and then for 2 hrs at 4°C with biotinylated goat anti-rabbit IgG secondary antibody (1:800, Vector Laboratories) in 5% normal goat serum in 0.1% Triton-X-100 in TBS. Sections were incubated with avidin-biotin complex (Vector Labs, Burlingame, CA) for 1 hr, and developed using 3-3’-diaminobenzidine (DAB; Sigma-Aldrich, St. Louis, MO).
2.4. Image analysis.
QuPath, an open source software for whole slide image analysis, was used to automate counts of immunoreactive cells and to define striatal regions for analysis. Striatal sections from Bregma +1.145 to 0.145, based on the Allen Coronal Mouse Brain Atlas, were quantified. Striatal regions were defined by outlining and calculating the entire area of the striatum. To consistently define dorsomedial, dorsolateral, and ventral striatum across sections and mice, the center x and y coordinate of the right and left striatum was determined using QuPath. A line between the striatal midpoints delimited the dorsal and ventral striatum and a line perpendicular to the dorsal-ventral boundary at the center x,y coordinate defined medial and lateral striatum. To determine cell counts for the dorsomedial (DM), dorsolateral (DL), and ventral striatum, the total number of positive cells from each region was divided by total area of the region to determine the number of positive cells/mm2.
2.5. Preparation of tissue for proteomic analysis.
Mice (n=4/genotype) were euthanized by cervical dislocation. Brains were rapidly removed and striata were rapidly dissected and frozen on dry ice and stored at −80° C. The protocol for tissue homogenization was adapted from a published method [31]. Samples from individual mice (~30 mg) were vortexed in 300 μL of urea lysis buffer (8 M urea, 10 mM Tris, 100 mM NaH2PO4, pH 8.5), including 3μL (100x stock) HALT(−EDTA) protease and phosphatase inhibitor cocktail (Pierce). All homogenization was performed using a Bullet Blender (Next Advance) according to the manufacturer’s protocol. Briefly, each tissue piece was added to urea lysis buffer in a 1.5 mL Rino tube (Next Advance) harboring 750 mg stainless steel beads (0.9–2 mm in diameter) and blended twice for 5 minute intervals in the cold room (4° C). Protein homogenates were transferred to 1.5 mL Eppendorf tubes and were sonicated (Sonic Dismembrator, Fisher Scientific) 3 times for 5 sec each with 15 sec intervals of rest at 30% amplitude to disrupt nucleic acids and were subsequently centrifuged at 4° C. Protein concentration was determined by the bicinchoninic acid (BCA) method, and samples were frozen in aliquots at −80 °C. Protein homogenates (100 μg) were diluted with 50 mM NH4HCO3 to a final concentration of less than 2 M urea and then were treated with 1 mM dithiothreitol (DTT) at room temperature for 30 min, followed by 5 mM iodoacetimide at room temperature for 30 min in the dark. Protein samples were digested with 1:100 (w/w) lysyl endopeptidase (Wako) at room temperature for 4 hrs and were further digested overnight with 1:50 (w/w) trypsin (Promega) at room temperature. Resulting peptides were desalted with HLB column (Waters) and were dried under vacuum.
2.6. Proteomics data acquisition.
The data acquisition by LC-MS/MS protocol was adapted from a published procedure[31] and was performed by the Integrated Proteomics Core Facility at Emory University. Derived peptides were resuspended in loading buffer (0.1% trifluoroacetic acid). Peptide mixtures (3 μL) were separated on a self-packed C18 (1.9 μm Dr. Maisch, Germany) fused silica column (25 cm × 75 μM internal diameter (ID); New Objective, Woburn, MA) by a Dionex Ultimate 3000 RSLCNano and monitored on a Fusion mass spectrometer (ThermoFisher Scientific, San Jose, CA). Elution was performed over a 125 min gradient at a rate of 300 nL/min (buffer A: 0.1% formic acid in water, buffer B: 0.1 % formic acid in acetonitrile): The gradient started with 3% buffer B and went to 7% in 5 minutes, then increased from 7% to 35% in 120 minutes, then to 99% within 10 minutes and finally staying at 99% for 10 minutes. The mass spectrometer cycle was programmed to collect at the top speed for 3 sec cycles. The MS scans (300–1500 m/z range, 200,000 AGC target, 50 ms maximum injection time) were collected at a resolution of 120,000 at m/z 200 in profile mode and the HCD MS/MS spectra (1.5 m/z isolation width, 30% collision energy, 10,000 AGC target, 35 ms maximum injection time) were detected in the ion trap. Dynamic exclusion was set to exclude previous sequenced precursor ions for 20 sec within a 10 ppm window. Precursor ions with +1, and +8 or higher charge states were excluded from sequencing.
2.7. Protein identification and quantification.
ThermoFisher generated RAW files were used for label-free quantitation (LFQ) of proteins in eight biological samples. Base peak chromatograms were inspected visually using RawMeat software. All RAW files were processed together in a single run by MaxQuant version 1.6.0.16 with default parameters unless otherwise specified (http://www.maxquant.org). Database searches were performed using the Andromeda search engine (a peptide search engine based on probabilistic scoring) with the UniProt mouse sequence database (update in 2017). MaxQuant provides a contaminants.fasta database file (a database of common laboratory contaminants) within the software that is automatically added to the list of proteins for the in-silico digestion when this feature is enabled.
Precursor mass tolerance was set to 4.5 ppm in the main search and fragment mass tolerance was set to 20 ppm. Digestion enzyme specificity was set to trypsin with a maximum of 2 missed cleavages. A minimum peptide length of 6 residues was required for identification. Up to 5 modifications per peptide were allowed; acetylation (protein N-terminal), oxidation (Met) and deamidation (NQ) were set as variable modifications, and carbamidomethylation (Cys) was set as a fixed modification. No Andromeda score threshold was set for unmodified peptides. A minimum Andromeda score of 40 was required for modified peptides. Peptide and protein false discovery rates (FDR) were both set to 1% based off a target-decoy reverse database. Proteins that shared all identified peptides were combined into a single protein group. If all identified peptides from one protein were a subset of identified peptides from another protein, these proteins were also combined into that group. Peptides that matched multiple protein groups (“razor” peptides) were assigned to the protein group with the most unique peptides.
Peaks were detected in Full MS, and a three-dimensional peak was constructed as a function of peak centroid m/z (7.5 ppm threshold) and peak area over time. Following de-isotoping, peptide intensities were determined by extracted ion chromatograms based on the peak area at the retention time with the maximum peak height. Peptide intensities were normalized to minimize overall proteome difference based on the assumption that most peptides do not change in intensity between samples. Protein LFQ intensities were calculated from the median of pairwise intensity ratios of peptides identified in two or more samples and adjusted to the cumulative intensity across samples. Quantification was performed using razor and unique peptides, including those modified by acetylation (protein N-terminal), oxidation (Met) and deamidation (NQ). A minimum peptide ratio of 1 was required for protein intensity normalization, and “Fast LFQ” was enabled.
2.8. Data analysis.
Data analysis was performed using Perseus version 1.5.0.31 (http://www.perseus-framework.org). Contaminants and protein groups identified by a single peptide were filtered from the data set. FDR was calculated as the percentage of reverse database matches out of total forward and reverse matches. Protein group LFQ intensities were log2 transformed to reduce the effect of outliers. Data were filtered by valid values. A minimum of four valid values was required in at least one group (DRD or normal). For cluster analysis and statistical comparisons between proteomes, protein groups missing LFQ values were assigned values using imputation. Missing values were assumed to be biased toward low abundance proteins that were below the MS detection limit. The missing values were replaced with random values taken from a median downshifted Gaussian distribution to simulate low abundance LFQ values. Imputation was performed separately for each sample from a distribution with a width of 0.3 and downshift of 1.8. Unsupervised hierarchical clustering was performed on Z-score normalized, log2 LFQ intensities using Euclidean distance and average linkage with k-means preprocessing (300 clusters). Log-fold changes were calculated as the difference in log2 LFQ intensity averages between experimental and control groups. Welch’s t test calculations were used in statistical tests as histograms of LFQ intensities showed that all data sets approximated normal distributions. Protein IDs/Protein names were mapped to gene symbols before further steps, The gplots package in R software (https://CRAN.R-project.org/package=gplots) was used to create the heatmap and the volcano plot was created using Graphpad Prism 9 (https://www.graphpad.com/).
2.9. Western blotting.
Striata were dissected from an independent cohort of control and DRD mice and homogenized in 50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Na deoxycholate, 1% Triton X-100, pH 8.0 plus PhosSTOP and cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche). Protein concentrations were determined using a BCA assay (Thermo Scientific). 50 μg protein/sample was denatured for 95°C for 5 min in Laemmli buffer with 2.5% ß-mercaptoethanol and separated with 4–20% Mini-PROTEAN (Bio-rad) polyacrylamide gel electrophoresis in 25 mM Tris, 192 mM glycine and 0.1% SDS running buffer. After transfer of the proteins to a nitrocellulose membrane in 25 mM Tris, 192 mM glycine and 20% methanol, the blot was blocked with Intercept Blocking Buffer (Li-cor) and subsequently incubated with primary antibodies against TOMM70a (mouse monoclonal anti-TOMM70, Santa Cruz Biotechnology #sc-390545, 1:100), TH (rabbit polyclonal anti-TH, Pel-Freez Biologicals #P40101-0, 1:1000) and β-actin (mouse monoclonal anti-β-actin, Cell Signaling Technology #3700, 1:1000) in blocking buffer overnight at 4°C. Secondary antibodies (donkey anti-mouse IRDye 680RD and goat anti-rabbit IRDye 800CW, Li-cor, 1:10,000) were incubated for 1 hr at 4°C in blocking buffer. The blot was imaged on a Li-cor odyssey (700 channel intensity of 2.0, 800 channel intensity of 5.0) and analyzed using Image Studio Lite (Version 5.2). TOMM70a and TH were normalized to β-actin.
2.9. Gene Ontology (GO) and KEGG pathway enrichment analysis of the differentially expressed proteins.
Analyses were performed using the gene list of differentially expressed proteins (p <0.05) and annotations were limited by species (mus musculus). DAVID Bioinformatic Resources v6.8 (https://david.ncifcrf.gov) [32] was used to identify KEGG pathways (Kyoto Encyclopedia of Genes and Genomes). The Gene Ontology Resource (http://geneontology.org) Panther Overrepresentation Test (released 20200407; GO database released 20200323) was used to identify enriched biological terms using the Fisher Exact test and FDR correction [33, 34].
2.10. Identification of dystonia-associated proteins.
PubMed searches were performed to determine whether proteins differentially regulated in DRD mice compared to normal mice (p < 0.05) were previously associated with dystonia. Keywords included the differentially regulated protein (protein and gene name) and “dystonia”. Studies were reviewed and only those that were clearly associated with both dystonia and the protein of interest were included. When differentially regulated proteins were components of a larger protein complex, the larger protein complex was also used as a keyword.
3. Results
3.1. Striatal anatomy
The development of the striatum is, in part, mediated by dopamine. Therefore, we first determined if anatomical features of the striatum in DRD mice differed from normal dopamine-intact mice because potential anatomical abnormalities caused by the loss of dopamine in developing DRD mice would influence the interpretation of proteomic analyses. We have previously demonstrated that TH+ midbrain nigrostriatal dopamine neurons are intact in DRD mice [27]. There was no apparent difference in the size of the striatum between normal and DRD mice (Student’s t test, p=0.43). The vast majority of neurons in the striatum are MSNs. Generally, there are two subtypes of MSNs, D1 dopamine receptor (D1R)-expressing MSNs and D2R-expressing MSNs, which each express a unique repertoire of proteins. To determine if the MSN subtype populations in DRD mice differed from normal mice, we assessed the number and distribution of D1R-expressing or D2R-expression MSNs by using reporter transgenes that express tdTomato (tdTom) in D1R-MSNs or EGFP in D2R-MSNs to identify each subtype. The density of tdTom- and EGFP-labeled cells within the striatum was not significantly different between normal and DRD mice (Figure 1). Further, the distribution of EGFP- and tdTom-positive neurons in dorsomedial, dorsolateral, and ventral striatum in DRD mice did not differ from normal mice (Figure 1). To ensure that the reporter transgenes did not spuriously alter the dystonic phenotype of DRD mice, which is known to be dependent on striatal dysfunction, we assessed abnormal movements in DRD mice carrying the fluorescent reporter transgenes. Abnormal movement scores in DRD mice carrying tdTom (5 ± 0.6, n=6), EGFP (5 ± 0.3, n=5) or both tdTom and EGFP (3.9 ± 1.2, n=7) were not significantly different from DRD mice without the transgenes (4.4 ± 0.5, n=25; Student’s t test, p>0.1 for all). These results suggest that major anatomical features of the striatum are intact in DRD mice.
Figure 1. D1R- and D2R-expressing MSNs in DRD and normal mouse striatum.

Representative micrographs of coronal sections from normal (A) and DRD (B) mouse striatum with tdTom (D1R-MSNs) and EGFP (D2R-MSNs) fluorescent proteins. C. EGFP positive cell counts (D2R-MSNs) did not differ between DRD (n=3) and normal (n=4) mice (p>0.1; Student’s t test). D. tdTom positive cell counts (D1R-MSNs) did not differ between DRD (n=3) and normal (n=3) mice (p>0.1; Student’s t test). Values represent mean ± SEM.
3.2. Proteomic analyses
The unbiased examination of the striatal proteome identified 1805 proteins. These proteins are illustrated in a volcano plot (Figure 2). TH protein expression was significantly reduced in DRD mice compared to normal mice (Figure 2) and reached a false discovery rate of q<0.05. We have previously demonstrated that Th mRNA is expressed at comparable levels in normal mice and DRD mice, but the abnormal p.Q381K TH is not efficiently transported from the midbrain to the striatum [26] likely because the mutation causes a reduction in the stability of the protein [35].
Figure 2. Volcano plot of protein expression in normal and DRD mice.

Each dot corresponds to an identified protein, including those that do not have associated gene names. The y-axis shows negative log10 transformed p-values obtained based on a two-tailed Welch’s t-tests with dotted lines representing unadjusted p-values of 0.05 and <0.01. The x-axis illustrates the log2 difference between DRD and normal mouse striatal protein expression with negative values representing proteins that are reduced in DRD mice compared to normal mice (n=4/genotype). Blue dots indicate differentially expressed proteins with unadjusted p-values < 0.05; those previously associated with dystonia are labeled with the associated gene name.
The number of differentially expressed proteins identified depends on statistical thresholds and methods. Further, it has been suggested that the false discovery rate (FDR) is too conservative for exploratory proteomics [36]. Therefore, results are presented using several different approaches including uncorrected p-values and volcano plots. We identified proteins differentially regulated in DRD compared to normal mice using the following arbitrary p-values cutoffs: p<0.001, 0.01 and 0.05. TH was significantly downregulated in DRD mice at p<0.001. Eight additional proteins were differentially regulated in the striatum of DRD compared to normal mice at p<0.01. When p<0.05 was considered, 19 additional proteins were downregulated and 29 additional proteins were upregulated in the striatum of DRD compared to normal mice. Differentially regulated proteins (p<0.05) are illustrated in Figure 3.
Figure 3. Heatmap of differentially expressed proteins in the striatum of normal and DRD mice.
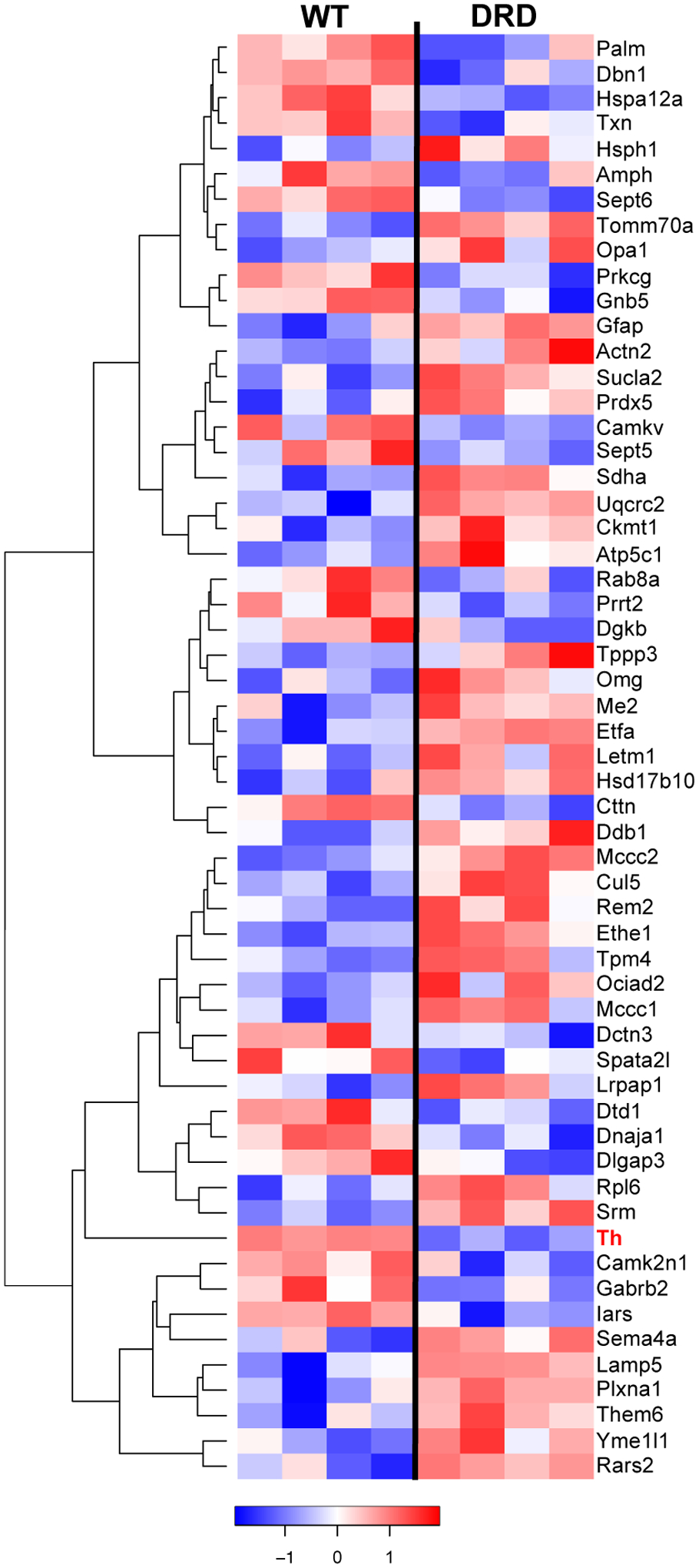
Relative levels of differentially regulated proteins (p<0.05) are presented. Rows represent proteins and columns represent samples from individual mice. Proteins are clustered based on protein expression (abundance) pattern. Unsupervised hierarchical clustering was performed on Z-score normalized, log2 LFQ intensities using Euclidean distance and average linkage. Low to high protein expression is represented by a change of color from blue to red, respectively. The color key scale bar at bottom shows z-score values for the heatmap. Each row (protein) is scaled to have mean zero and standard deviation one.
3.3. Validation of the proteomic analysis.
The results of the proteomic analysis were validated by western blot using striata obtained from an independent cohort of control and DRD mice. TH and TOMM70a (translocase of outer mitochondrial membrane 70a) were selected because 1) this is the first quantitative analysis of TH expression in the striatum of DRD mice, 2) both proteins implicated in disorders in which dystonia is a prominent feature and 3) the proteomic analysis indicated that these proteins are oppositely regulated whereby TH is underrepresented and TOMM70a is overrepresented in DRD mice compared to control mice. Western blot analysis confirmed that the striatal expression of both TH and TOMM70a was significantly different in DRD mice compared to controls (Figure 4). In fact, TH expression in DRD mice was <1% of control mice. The direction of the effect was consistent with the proteomic analysis with a significant reduction in TH (p = 0.0001) and a significant increase in TOMM70a (p = 0.014) in DRD mice compared to controls.
Figure 4. Quantification of TOMM70a and TH proteins in the striatum of normal and DRD mice.
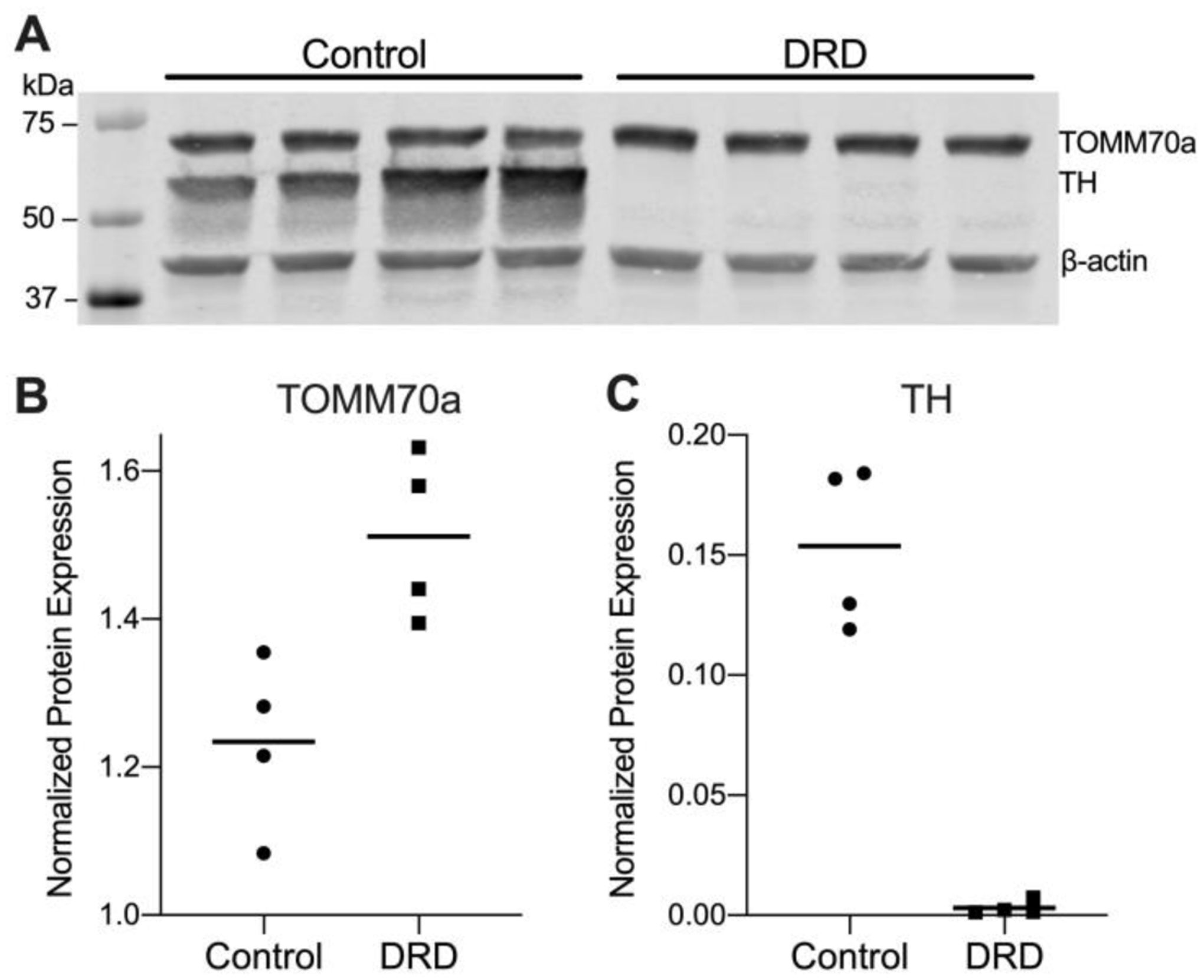
A. Western blot for TOMM70a, TH. ß-actin was used as a loading control. Each lane represents an individual mouse. The molecular weight standard is shown in the left lane. B. TOMM70a protein expression was significantly increased in DRD mice compared to control mice (Student’s two-tailed t-test, p = 0.014). C. TH protein expression was significantly reduced in DRD mice compared to control mice (Student’s two-tailed t-test, p = 0.0001). B and C illustrate means (horizontal lines) and individual values of the densitometric quantification of TOMM70a and TH which was normalized to ß-actin.
3.4. Dysregulated cellular pathways and processes in DRD mice.
Gene ontology analysis was used to identify pathways and functions that may be abnormally regulated in DRD mice. When differentially regulated proteins with p values < 0.05 were considered, several dysregulated functional groups were identified (Gene Ontology FDR p<0.005 for all). Table 1 provides a representative sample of the gene ontologies with nonspecific terms such as ‘cytoplasm’ or ‘cell projection’ or terms that included < 4 differentially expressed proteins omitted for clarity. Two prominent categories of gene ontologies were identified: mitochondrion and synapse. Proteins associated with the gene ontology term ‘mitochondrion’ accounted for ~36% of the differentially expressed proteins. Mitochondrial subcategories included both structural and functional (Reactome) terms including mitochondrial membrane or matrix and mitochondrial energetics (tricarboxylic acid cycle). All differentially-expressed proteins associated with these gene ontology terms were over-represented in DRD mice compared to normal mice. Proteins associated with the gene ontology term ‘synapse’ accounted for ~33% of the differentially expressed proteins. In contrast to the mitochondrial-associated proteins, differentially expressed proteins associated with synaptic function, including pre- and postsynapse, synaptic vesicle, axon and glutamatergic synapse, were under-represented in DRD mice compared to normal mice with the exception of Lamp5, Rpl6 and Actn2. Notably, pathways associated with many neurologic disorders including inflammatory processes and cell death were not identified.
Table 1.
Gene Ontology (GO) enrichment analysis
| GO term | Genes | P value | FDR |
|---|---|---|---|
| Mitochondrion | Sucla2|Sdha|Rars2|Mccc1|Prdx5|Txn|Hsd17b10|Ociad2|Dnaja1|Atp5c1|Opa1|Ethe1|Yme1l1|Me2|Th|Uqcrc2|Letm1|Ckmt1|Et fa|Tomm70a|Mccc2 | 1.11 E-09 | 1.10 E-06 |
| Synapse | Camk2n1|Prkcg|Lamp5|Amph|Dlgap3|Palm|Cttn|Dbn1|Rpl6|Gabrb2|Sept5|Camkv|Gnb5|Th|Actn2| Prrt2|Dgkb|Rab8a|Sept6 | 5.74 E-09 | 3.80 E-06 |
| Myelin Sheath | Omg|Sucla2|Sdha|Atp5c1|Gfap|Gnb5|Uqcrc2|Ckmt1 | 1.52 E-07 | 7.56 E-05 |
| Postsynapse | Camk2n1|Prkcg|Dlgap3|Palm|Cttn|Dbn1|Rpl6|Gabrb2|Camkv|Actn2|Prrt2|Rab8a | 6.59 E-07 | 1.63 E-04 |
| Mitochondrial Membrane | Sdha|Mccc1|Hsd17b10|Ociad2|Atp5c1|Opa1|Yme1l1|Uqcrc2|Letm1|Ckmt1|Tomm70a | 1.02 E-06 | 2.03 E-04 |
| Postsynaptic Specialization | Camk2n1|Prkcg|Dlgap3|Palm|Dbn1|Rpl6|Rab8a|Gabrb2|Actn2 | 3.17 E-06 | 4.50 E-04 |
| Axon | Prkcg|Lamp5 |Txn|Amph|Palm|Cttn|Dbn1|Sept5|Th|Prrt2|Sept6 | 3.89 E-06 | 5.15 E-04 |
| Glutamatergic Synapse | Amph|Dlgap3|Cttn|Dbn1|Camkv|Actn2|Prrt2|Dgkb|Rab8a | 1.06 E-05 | 1.05 E-03 |
| Synaptic Vesicle | Amph|Sept5|Th|Prrt2|Sept6|Rab8a | 9.60 E-05 | 7.06 E-03 |
| ATP Binding | Sucla2|Rars2|Mccc1|Prkcg|Srm|Dnaja1|Yme1l1|Camkv|Hsph1|Ckmt1|Iars|Hspa12a|Dgkb|Mccc2 | 1.98 E-05 | 7.65 E-03 |
| tRNA Binding | Hsd17b10|Dtd1|Iars|Rpl6 | 3.20 E-05 | 1.06 E-02 |
| Presynapse | Prkcg|Amph|Sept5|Gnb5|Th|Prrt2|Rab8a |Sept6 | 2.18 E-04 | 1.24 E-02 |
| Ligase Activity | Sucla2|Mccc1|Iars|Rars2|Mccc2 | 5.81 E-05 | 1.41 E-02 |
| The citric acid cycle (TCA) and respiratory electron transport | Sucla2|Sdha|Atp5c1|Opa1|Me2|Etfa | 3.78 E-05 | 2.08 E-02 |
| Dendrite | Camk2n1|Prkcg|Txn|Dlgap3|Opa1|Dbn1|Th|Rab8a | 6.94 E-04 | 3.36 E-02 |
| Cellular response to stress | Prdx5|Txn|Dnaja1|Dctn3|Hsph1|Hspa12a | 1.40 E-04 | 3.85 E-02 |
| Mitochondrial Matrix | Mccc1|Hsd17b10|Uqcrc2|Mccc2|Etfa | 9.72 E-04 | 4.02 E-02 |
Red or blue text indicates up- and downregulation, respectively, in DRD mice compared to control mice.
DAVID was used to probe for pathways within the KEGG pathway database that may be differentially regulated in DRD compared to normal mouse striatum. Eight enriched KEGG pathways were identified (Table 2), including GABAergic and endocannabinoid signaling pathways. In addition, the Parkinson’s disease pathway was identified. Both Parkinson’s disease and DRD are characterized by dopamine deficiency, a central biochemical feature of both disorders, providing additional support for the validity of this proteomic analysis.
Table 2.
Enriched KEGG pathways of differentially expressed proteins
| Pathway Description | Genes | P value | FDR q-value |
|---|---|---|---|
| Parkinson’s disease | Uqcrc2|Sept5|Sdha|Th|Atp5c1 | 0.003 | 0.259453884 |
| Valine, leucine, and isoleucine degradation | Mccc2|Hsd17b10|Mccc1 | 0.022 | 0.643639237 |
| Metabolic pathways | Uqcrc2|Sdha|Th|Atp5c1|Mccc2|Hsd17b10|Mccc1|Dgkb|SRM|Ckmt1|Sucla2 | 0.029 | 0.598691922 |
| Alzheimer’s disease | UQCRC2|SDHA|ATP5C1|HSD17B10 | 0.036 | 0.582326059 |
| GABAergic synapse | Gabrb2|Gnb5|Prkcg | 0.050 | 0.621737675 |
| Morphine addiction | Gabrb2|Gnb5|Prkcg | 0.057 | 0.599647403 |
| Retrograde endocannabinoid signaling | Gabrb2|Gnb5|Prkcg | 0.068 | 0.611250528 |
| Carbon metabolism | Me2|Sdha|Sucla2 | 0.083 | 0.640892686 |
3.5. Dysregulated proteins previously associated with disorders with dystonia as a sign.
A literature review of the 57 differentially regulated proteins (p<0.05) revealed 11 proteins in which pathogenic genetic variants are associated with disorders with dystonia as a sign (Table 3). Further, abnormal regulation of one of these proteins, succinate-coenzyme A ligase, ADP-forming, beta subunit (Sucla2), is also dysregulated in a cellular model of DYT1 dystonia [37]. Thus, nearly 20% of the differentially regulated proteins in DRD mice are implicated in other forms of dystonia.
Table 3.
Differentially regulated proteins associated with disorders with dystonia as a sign
| Protein | Gene | Disorder | p-value | Difference DRD-WT (Welch’s t-test) | References |
|---|---|---|---|---|---|
| Tyrosine hydroxylase | Th | DOPA-responsive dystonia | 0.001 | −3.72 | [16,26] |
| Translocase of outer mitochondrial membrane 70a | Tomm70a | hypotonia, hyper-reflexia, ataxia, dystonia | 0.002 | 0.75 | [38] |
| Sulfur dioxygenase | Ethe1 | ethylmalonic encephalopathy | 0.004 | 0.87 | [39] |
| Flavoprotein subunit of succinate dehydrogenase | Sdha | multisystem mitochondrial disease | 0.006 | 0.15 | [40] |
| Protein kinase C gamma | Prkcg | spinocerebellar ataxia type 14 | 0.010 | −0.31 | [41–43] |
| Succinate-coenzyme A ligase, ADP-forming, beta subunit | Sucla2 | dystonia deafness syndrome | 0.011 | 0.25 | [44, 45] |
| Proline-rich transmembrane protein 2 | Prrt2 | paroxysmal kinesigenic dyskinesias | 0.016 | −0.56 | [46, 47] |
| GABAA receptor, subunit beta 2 | Gabrb2 | cervical dystonia | 0.019 | −0.59 | [48]* |
| Glial fibrillary acidic protein | Gfap | Alexander disease | 0.030 | 0.23 | [49, 50] |
| Arginyl-tRNA synthetase 2, mitochondrial | Rars2 | pontocerebellar hypoplasia type 6 | 0.032 | 0.74 | [51] |
| OPA1, mitochondrial dynamin like GTPase | Opa1 | optic atrophy | 0.045 | 0.21 | [52] |
Indirectly associated
4. Discussion
Dystonia is generally not associated with degeneration or overt cellular pathology, suggesting that neuronal dysfunction mediates the abnormal movements. Therefore, to better understand the cellular defects underlying dystonia, we examined striatal protein expression in a mouse model of DRD because it is known that the dystonic movements in these mice are mediated by striatal dysfunction [26]. Our unbiased assessment of the DRD mouse striatal proteome revealed that of the 1805 proteins identified in the proteomic screen only 57 (~3%) were differentially regulated in DRD mice compared to normal mice. Importantly, the proteomic analysis and subsequent validation by western blotting revealed that TH protein expression in the striatum of DRD mice was reduced to <1% of control mice. This finding is consistent with our previous biochemical analyses demonstrating that TH enzyme activity is reduced to ~1% of normal in DRD mouse striatum [26].
Despite the significant deficit in TH expression that is caused by the mutation in Th, major anatomical features of the striatum, including striatal size and the distribution and number of D1R- and D2R-expressing MSNs, were intact in DRD mice. These results suggest that the proteomic analysis was not overtly confounded by gross morphological defects. That relatively few proteins were abnormally regulated is consistent with the observation that striatal anatomy is preserved in DRD mice. However, it is surprising that we did not observe more extensive protein dysregulation considering that, in adults, dopamine neurotransmission plays an important role in the strength and plasticity of glutamatergic synaptic inputs, the intrinsic excitability of MSNs and contributes to morphological changes [24, 53, 54]. Dopamine neurotransmission also plays a critical role in striatal development. Dopamine innervates the nascent striatum mid-gestation, prior to the arrival of excitatory afferents and induces the formation of immature spines through both D1Rs and D2Rs [55, 56]. Dopamine also regulates the macroscopic dendritic architecture of MSNs by stimulating neurite outgrowth in embryonic striatal neurons through activation of D1Rs but not D2Rs [57, 58], suggesting that an early-life dopamine deficit may reduce dendritic complexity. Subtle morphological changes such as spine formation and complexity would not have been detected in our gross anatomical analyses but were nonetheless reflected in the proteomic analysis.
Many of the differentially regulated proteins were associated with synaptic function, which is perhaps not surprising considering that the brain is enriched in these proteins. It is, however, striking that the majority (16/19) of the differentially expressed proteins represented in the GO ‘synapse’ pathway and related pathways were significantly under-represented in DRD mice, suggesting a general deficit in striatal neurotransmission. Specifically, the intracellular signaling protein kinase C gamma (PRKCG) and the transmitter release protein proline-rich-transmembrane-protein-2 (PRRT2) are notable because of their shared dysregulation across dystonias. PRKCG is a neuron-specific calcium-activated, phospholipid- and diacylglycerol (DAG)-dependent serine/threonine-protein kinase. Although variants in PRKCG are specifically associated with spinocerebellar ataxia type 14 [41, 42, 59], which is associated with dystonic movements, identification of PRKCG in our proteomic analysis has broader implications for dystonia in light of the role of PRKCG in the regulation of synaptic plasticity. Abnormal plasticity is observed in many different forms of dystonia in humans and in several mouse models of dystonia [60–66], suggesting that PRKCG may be a pathophysiological node. PRRT2 is a presynaptic membrane protein that interacts with SNAP-25 to mediate calcium-dependent vesicular exocytosis, particularly glutamate release [67]. Pathogenic variants in PRRT2 are associated with paroxysmal kinesigenic dyskinesia, a disorder that includes dystonic movements [46, 47, 68]. Prrt2 mRNA dysregulation is also observed in a mouse model of dystonia associated with THAP1 [69]. Because pathogenic variants of both PRKCG and PRRT2 are associated with disorders that include dystonia and are also more broadly implicated in dystonias, the encoded proteins are attractive targets for therapeutics aimed at enhancing expression and/or activity.
Approximately one-third of all differentially regulated proteins were associated with synaptic function based on the enriched GO terms. Subsets of the broad term ‘synapse’ implicated some specific functions, like ‘presynapse,’ which was expected considering that DRD is caused by a mutation in Th, which is located in nigrostriatal processes. Consistent with a presynaptic dopamine defect in DRD, the KEGG analysis identified Parkinson’s disease, which is, in part, caused by striatal dopamine deficiency that results from degeneration of nigrostriatal neurons. Enriched pathways for glutamatergic (GO) and GABAergic (KEGG) synapses were also identified. The striatum receives abundant glutamatergic innervation from both corticostriatal and thalamostriatal efferents while the vast majority of neurons in the striatum itself are GABAergic spiny projection neurons. The activity of both is mediated by dopamine neurotransmission. Thus, the enriched pathways reflect both the biochemical defect and striatal anatomy, providing additional support for the validity of the pathway analyses.
Approximately one-third of all differentially regulated proteins were associated with mitochondrial function based on the enriched GO terms. Further, fully half of the genes associated with dystonia in Table 3 play a role in mitochondrial function including Ethe1, Sdha, Sucla2, Rars2, Opa1, and Tomm70a. Several additional lines of evidence implicate mitochondrial dysfunction across different forms of dystonia. In addition to the inherited dystonias listed in Table 3, other inherited dystonias in which mitochondrial dysfunction is implicated include [70, 71] Leber’s hereditary optic neuropathy which is caused by a mutation in the mtDNA complex I gene MTND6 [72] and deafness-dystonia-optic neuropathy syndrome which is caused by a mutation in the DDP1 gene that encodes the mitochondrial translocase subunit Tim8 A [73]. Mitochondrial dysfunction is also implicated in idiopathic dystonias. In particular, a reduction in mitochondrial complex I activity was observed in patients with idiopathic focal, segmental and generalized dystonia [74, 75]. Dystonia can also be induced by accidental exposure to the mitochondrial complex II inhibitor 3-nitropropionic acid in otherwise healthy individuals [76–78]. Thus, mitochondrial dysfunction is a shared defect among inherited, acquired, and idiopathic dystonias in humans. Furthermore, mitochondrial dysregulation is also shared among mouse models, including those associated with Tor1a, Thap1, and now Th, demonstrating that mitochondrial changes can occur even when the dystonia-causing defect is apparently unrelated to mitochondrial function [37, 79]. However, it will be critical to determine whether the mitochondrial abnormalities identified here are integral to the expression of dystonia or compensatory.
Pathogenic variants of 11 of the 57 differentially regulated proteins identified in DRD mice are implicated in disorders that have dystonia, suggesting common pathological mechanisms across different forms of dystonia. Abundant evidence implicating abnormal dopaminergic neurotransmission in many different dystonias has been accumulating for decades [2]. More recently, in addition to the shared mechanisms such as mitochondrial and synaptic dysfunction mentioned previously, abnormal plasticity, developmental defects and cholinergic dysfunction have been implicated across different forms of dystonias using -omics, computational or physiological approaches in both humans and mouse models [37, 69, 79–81]. The identification of shared processes suggests that it may be possible to identify specific, druggable targets for the development of novel therapeutics that are effective in a broad range of dystonias.
Acknowledgements
We thank Alec P. Shannon for technical support.
Funding
This work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant R01 NS088528, F31 NS103363 and T32 NS007480.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
All authors state they have no conflict of interest.
References
- [1].Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, Hallett M, Jankovic J, Jinnah HA, Klein C, Lang AE, Mink JW, Teller JK, Phenomenology and classification of dystonia: a consensus update, Mov Disord 28 (2013) 863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jinnah HA, Sun YV, Dystonia genes and their biological pathways, Neurobiol Dis 129 (2019) 159–168. [DOI] [PubMed] [Google Scholar]
- [3].Balint B, Mencacci NE, Valente EM, Pisani A, Rothwell J, Jankovic J, Vidailhet M, Bhatia KP, Dystonia, Nat Rev Dis Primers 4 (2018) 25. [DOI] [PubMed] [Google Scholar]
- [4].Neychev VK, Gross RE, Lehericy S, Hess EJ, Jinnah HA, The functional neuroanatomy of dystonia, Neurobiol Dis 42 (2011) 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marsden CD, Obeso JA, Zarranz JJ, Lang AE, The anatomical basis of symptomatic hemidystonia, Brain 108 (1985) 463–483. [DOI] [PubMed] [Google Scholar]
- [6].Pettigrew LC, Jankovic J, Hemidystonia: a report of 22 patients and a review of the literature, J Neurol Neurosurg Psychiatry 48 (1985) 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Obeso JA, Gimenez-Roldan S, Clinicopathological correlation in symptomatic dystonia, Adv Neurol 50 (1988) 113–122. [PubMed] [Google Scholar]
- [8].Bhatia KP, Marsden CD, The behavioural and motor consequences of focal lesions of the basal ganglia in man, Brain 117 (1994) 859–876. [DOI] [PubMed] [Google Scholar]
- [9].Kerrison JB, Lancaster JL, Zamarripa FE, Richardson LA, Morrison JC, Holck DE, Andreason KW, Blaydon SM, Fox PT, Positron emission tomography scanning in essential blepharospasm, Am J Ophthalmol 136 (2003) 846–852. [DOI] [PubMed] [Google Scholar]
- [10].Galardi G, Perani D, Grassi F, Bressi S, Amadio S, Antoni M, Comi GC, Canal N, Fazio F, Basal ganglia and thalamo-cortical hypermetabolism in patients with spasmodic torticollis, Acta Neurol Scand 94 (1996) 172–176. [DOI] [PubMed] [Google Scholar]
- [11].Carbon M, Niethammer M, Peng S, Raymond D, Dhawan V, Chaly T, Ma Y, Bressman S, Eidelberg D, Abnormal striatal and thalamic dopamine neurotransmission: Genotype-related features of dystonia, Neurology 72 (2009) 2097–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Eidelberg D, Moeller J, Antonini R,A, Kazumata K, Nakamura T, Dhawan V, Spetsieris P, deLeon D, Bressman S, Fahn B,S, Functional brain networks in DYT1 dystonia, Ann Neurol 44 (1998) 303–312. [DOI] [PubMed] [Google Scholar]
- [13].Centen LM, Oterdoom DLM, Tijssen MAJ, Lesman-Leegte I, van Egmond ME, van Dijk JMC, Bilateral Pallidotomy for Dystonia: A Systematic Review, Mov Disord (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rodrigues FB, Duarte GS, Prescott D, Ferreira J, Costa J, Deep brain stimulation for dystonia, Cochrane Database Syst Rev 1 (2019) CD012405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, Fujita K, Nagatsu T, Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene, Nat Genet 8 (1994) 236–242. [DOI] [PubMed] [Google Scholar]
- [16].Knappskog PM, Flatmark T, Mallet J, Ludecke B, Bartholome K, Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene, Human Molecular Genetics 4 (1995) 1209–1212. [DOI] [PubMed] [Google Scholar]
- [17].van den Heuvel LP, Luiten B, Smeitink JA, de Rijk-van Andel JF, Hyland K, Steenbergen-Spanjers GC, Janssen RJ, Wevers RA, A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population, Hum Genet 102 (1998) 644–646. [DOI] [PubMed] [Google Scholar]
- [18].Berman BD, Hallett M, Herscovitch P, Simonyan K, Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp, Brain 136 (2013) 3645–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Simonyan K, Berman BD, Herscovitch P, Hallett M, Abnormal striatal dopaminergic neurotransmission during rest and task production in spasmodic dysphonia, J Neurosci 33 (2013) 14705–14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Horie C, Suzuki Y, Kiyosawa M, Mochizuki M, Wakakura M, Oda K, Ishiwata K, Ishii K, Decreased dopamine D2 receptor binding in essential blepharospasm, Acta Neurol Scand 119 (2009) 49–54. [DOI] [PubMed] [Google Scholar]
- [21].Hierholzer J, Cordes M, Schelosky L, Richter W, Keske U, Venz S, Semmler W, Poewe W, Felix R, Dopamine D2 receptor imaging with iodine-123-iodobenzamide SPECT in idiopathic rotational torticollis, J Nucl Med 35 (1994) 1921–1927. [PubMed] [Google Scholar]
- [22].Naumann M, Pirker W, Reiners K, Lange KW, Becker G, Brucke T, Imaging the pre- and postsynaptic side of striatal dopaminergic synapses in idiopathic cervical dystonia: a SPECT study using [123I] epidepride and [123I] beta-CIT, Mov Disord 13 (1998) 319–323. [DOI] [PubMed] [Google Scholar]
- [23].Asanuma K, Ma Y, Okulski J, Dhawan V, Chaly T, Carbon M, Bressman SB, Eidelberg D, Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation, Neurology 64 (2005) 347–349. [DOI] [PubMed] [Google Scholar]
- [24].Gerfen CR, Surmeier DJ, Modulation of striatal projection systems by dopamine, Annu Rev Neurosci 34 (2011) 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].DeLong MR, Wichmann T, Circuits and circuit disorders of the basal ganglia, Arch Neurol 64 (2007) 20–24. [DOI] [PubMed] [Google Scholar]
- [26].Rose SJ, Yu XY, Heinzer AK, Harrast P, Fan XL, Raike RS, Thompson VB, Pare JF, Weinshenker D, Smith Y, Jinnah HA, Hess EJ, A new knock-in mouse model of L-DOPA-responsive dystonia, Brain 138 (2015) 2987–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rose SJ, Harrast P, Donsante C, Fan X, Joers V, Tansey MG, Jinnah HA, Hess EJ, Parkinsonism without dopamine neuron degeneration in aged l-dopa-responsive dystonia knockin mice, Mov Disord 32 (2017) 1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ade KK, Wan Y, Chen M, Gloss B, Calakos N, An Improved BAC Transgenic Fluorescent Reporter Line for Sensitive and Specific Identification of Striatonigral Medium Spiny Neurons, Frontiers in systems neuroscience 5 (2011) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, Heintz N, Greengard P, Surmeier DJ, Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice, J Neurosci 32 (2012) 9124–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Raike RS, Pizoli CE, Weisz C, van den Maagdenberg AM, Jinnah HA, Hess EJ, Limited regional cerebellar dysfunction induces focal dystonia in mice, Neurobiol Dis 49 (2013) 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, Glass J, Gearing M, Thambisetty M, Troncoso JC, Geschwind DH, Lah JJ, Levey AI, A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease, Cell Syst 4 (2017) 60–72 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang da W, Sherman BT, Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat Protoc 4 (2009) 44–57. [DOI] [PubMed] [Google Scholar]
- [33].Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, Gene ontology: tool for the unification of biology. The Gene Ontology Consortium, Nat Genet 25 (2000) 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].The Gene Ontology C, The Gene Ontology Resource: 20 years and still GOing strong, Nucleic Acids Res 47 (2019) D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fossbakk A, Kleppe R, Knappskog PM, Martinez A, Haavik J, Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in dopa-responsive dystonia, Human mutation 35 (2014) 880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pascovici D, Handler DC, Wu JX, Haynes PA, Multiple testing corrections in quantitative proteomics: A useful but blunt tool, Proteomics 16 (2016) 2448–2453. [DOI] [PubMed] [Google Scholar]
- [37].Martin JN, Bair TB, Bode N, Dauer WT, Gonzalez-Alegre P, Transcriptional and proteomic profiling in a cellular model of DYT1 dystonia, Neuroscience 164 (2009) 563–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dutta D, Briere LC, Kanca O, Marcogliese PC, Walker MA, High FA, Vanderver A, Krier J, Carmichael N, Callahan C, Taft RJ, Simons C, Helman G, Network UD, Wangler MF, Yamamoto S, Sweetser DA, Bellen HJ, De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment, Hum Mol Genet 29 (2020) 1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Walsh DJ, Sills ES, Lambert DM, Gregersen N, Malone FD, Walsh AP, Novel ETHE1 mutation in a carrier couple having prior offspring affected with ethylmalonic encephalopathy: Genetic analysis, clinical management and reproductive outcome, Mol Med Rep 3 (2010) 223–226. [DOI] [PubMed] [Google Scholar]
- [40].Renkema GH, Wortmann SB, Smeets RJ, Venselaar H, Antoine M, Visser G, Ben-Omran T, van den Heuvel LP, Timmers HJ, Smeitink JA, Rodenburg RJ, SDHA mutations causing a multisystem mitochondrial disease: novel mutations and genetic overlap with hereditary tumors, Eur J Hum Genet 23 (2015) 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ganos C, Zittel S, Minnerop M, Schunke O, Heinbokel C, Gerloff C, Zuhlke C, Bauer P, Klockgether T, Munchau A, Baumer T, Clinical and neurophysiological profile of four German families with spinocerebellar ataxia type 14, Cerebellum 13 (2014) 89–96. [DOI] [PubMed] [Google Scholar]
- [42].Miura S, Nakagawara H, Kaida H, Sugita M, Noda K, Motomura K, Ohyagi Y, Ayabe M, Aizawa H, Ishibashi M, Taniwaki T, Expansion of the phenotypic spectrum of SCA14 caused by the Gly128Asp mutation in PRKCG, Clin Neurol Neurosurg 111 (2009) 211–215. [DOI] [PubMed] [Google Scholar]
- [43].Nibbeling EAR, Duarri A, Verschuuren-Bemelmans CC, Fokkens MR, Karjalainen JM, Smeets C, de Boer-Bergsma JJ, van der Vries G, Dooijes D, Bampi GB, van Diemen C, Brunt E, Ippel E, Kremer B, Vlak M, Adir N, Wijmenga C, van de Warrenburg BPC, Franke L, Sinke RJ, Verbeek DS, Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia, Brain 140 (2017) 2860–2878. [DOI] [PubMed] [Google Scholar]
- [44].Garone C, Gurgel-Giannetti J, Sanna-Cherchi S, Krishna S, Naini A, Quinzii CM, Hirano M, A Novel SUCLA2 Mutation Presenting as a Complex Childhood Movement Disorder, J Child Neurol 32 (2017) 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Maas RR, Marina AD, de Brouwer AP, Wevers RA, Rodenburg RJ, Wortmann SB, SUCLA2 Deficiency: A Deafness-Dystonia Syndrome with Distinctive Metabolic Findings (Report of a New Patient and Review of the Literature), JIMD Rep 27 (2016) 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Marano M, Motolese F, Consoli F, De Luca A, Di Lazzaro V, Paroxysmal Dyskinesias in a PRRT2 Mutation Carrier, Tremor Other Hyperkinet Mov (N Y) 8 (2018) 616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang Y, Li L, Chen W, Gan J, Liu ZG, Clinical characteristics and PRRT2 gene mutation analysis of sporadic patients with paroxysmal kinesigenic dyskinesia in China, Clin Neurol Neurosurg 159 (2017) 25–28. [DOI] [PubMed] [Google Scholar]
- [48].Berman BD, Pollard RT, Shelton E, Karki R, Smith-Jones PM, Miao Y, GABAA Receptor Availability Changes Underlie Symptoms in Isolated Cervical Dystonia, Front Neurol 9 (2018) 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Jefferson RJ, Absoud M, Jain R, Livingston JH, MS VDK, Jayawant S, Alexander disease with periventricular calcification: a novel mutation of the GFAP gene, Dev Med Child Neurol 52 (2010) 1160–1163. [DOI] [PubMed] [Google Scholar]
- [50].Machol K, Jankovic J, Vijayakumar D, Burrage LC, Jain M, Lewis RA, Fuller GN, Xu M, Penas-Prado M, Gule-Monroe MK, Rosenfeld JA, Chen R, Eng CM, Yang Y, Lee BH, Moretti PM, Undiagnosed Diseases N, Dhar SU, Atypical Alexander disease with dystonia, retinopathy, and a brain mass mimicking astrocytoma, Neurol Genet 4 (2018) e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Glamuzina E, Brown R, Hogarth K, Saunders D, Russell-Eggitt I, Pitt M, de Sousa C, Rahman S, Brown G, Grunewald S, Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2, J Inherit Metab Dis 35 (2012) 459–467. [DOI] [PubMed] [Google Scholar]
- [52].Liskova P, Ulmanova O, Tesina P, Melsova H, Diblik P, Hansikova H, Tesarova M, Votruba M, Novel OPA1 missense mutation in a family with optic atrophy and severe widespread neurological disorder, Acta Ophthalmol 91 (2013) e225–231. [DOI] [PubMed] [Google Scholar]
- [53].Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ, Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models, Nat Neurosci 9 (2006) 251–259. [DOI] [PubMed] [Google Scholar]
- [54].Villalba RM, Smith Y, Differential striatal spine pathology in Parkinson’s disease and cocaine addiction: A key role of dopamine?, Neuroscience 251 (2013) 2–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fasano C, Bourque MJ, Lapointe G, Leo D, Thibault D, Haber M, Kortleven C, Desgroseillers L, Murai KK, Trudeau LE, Dopamine facilitates dendritic spine formation by cultured striatal medium spiny neurons through both D1 and D2 dopamine receptors, Neuropharmacology 67 (2013) 432–443. [DOI] [PubMed] [Google Scholar]
- [56].Money KM, Stanwood GD, Developmental origins of brain disorders: roles for dopamine, Frontiers in cellular neuroscience 7 (2013) 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Schmidt U, Pilgrim C, Beyer C, Differentiative effects of dopamine on striatal neurons involve stimulation of the cAMP/PKA pathway, Mol Cell Neurosci 11 (1998) 9–18. [DOI] [PubMed] [Google Scholar]
- [58].Schmidt U, Beyer C, Oestreicher AB, Reisert I, Schilling K, Pilgrim C, Activation of dopaminergic D1 receptors promotes morphogenesis of developing striatal neurons, Neuroscience 74 (1996) 453–460. [DOI] [PubMed] [Google Scholar]
- [59].Nibbeling EA, Delnooz CC, de Koning TJ, Sinke RJ, Jinnah HA, Tijssen MA, Verbeek DS, Using the shared genetics of dystonia and ataxia to unravel their pathogenesis, Neurosci Biobehav Rev 75 (2017) 22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Quartarone A, Bagnato S, Rizzo V, Siebner HR, Dattola V, Scalfari A, Morgante F, Battaglia F, Romano M, Girlanda P, Abnormal associative plasticity of the human motor cortex in writer’s cramp, Brain 126 (2003) 2586–2596. [DOI] [PubMed] [Google Scholar]
- [61].Edwards MJ, Huang YZ, Mir P, Rothwell JC, Bhatia KP, Abnormalities in motor cortical plasticity differentiate manifesting and nonmanifesting DYT1 carriers, Mov Disord 21 (2006) 2181–2186. [DOI] [PubMed] [Google Scholar]
- [62].Evinger C, Benign Essential Blepharospasm is a Disorder of Neuroplasticity: Lessons From Animal Models, J Neuroophthalmol 35 (2015) 374–379. [DOI] [PubMed] [Google Scholar]
- [63].Gilbertson T, Humphries M, Steele JD, Maladaptive striatal plasticity and abnormal reward-learning in cervical dystonia, Eur J Neurosci (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A, Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine, Brain 132 (2009) 2336–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Weise D, Schramm A, Beck M, Reiners K, Classen J, Loss of topographic specificity of LTD-like plasticity is a trait marker in focal dystonia, Neurobiol Dis 42 (2011) 171–176. [DOI] [PubMed] [Google Scholar]
- [66].Meunier S, Russmann H, Shamim E, Lamy JC, Hallett M, Plasticity of cortical inhibition in dystonia is impaired after motor learning and paired-associative stimulation, Eur J Neurosci 35 (2012) 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Li M, Niu F, Zhu X, Wu X, Shen N, Peng X, Liu Y, PRRT2 Mutant Leads to Dysfunction of Glutamate Signaling, Int J Mol Sci 16 (2015) 9134–9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bruno MK, Ravina B, Garraux G, Hallett M, Ptacek L, Singleton A, Johnson J, Hanson M, Considine E, Gwinn-Hardy K, Exercise-induced dystonia as a preceding symptom of familial Parkinson’s disease, Mov Disord 19 (2004) 228–230. [DOI] [PubMed] [Google Scholar]
- [69].Frederick NM, Shah PV, Didonna A, Langley MR, Kanthasamy AG, Opal P, Loss of the dystonia gene Thap1 leads to transcriptional deficits that converge on common pathogenic pathways in dystonic syndromes, Hum Mol Genet 28 (2019) 1343–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].De Vries DD, Went LN, Bruyn GW, Scholte HR, Hofstra RM, Bolhuis PA, van Oost BA, Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia, Am J Hum Genet 58 (1996) 703–711. [PMC free article] [PubMed] [Google Scholar]
- [71].Sarzi E, Brown MD, Lebon S, Chretien D, Munnich A, Rotig A, Procaccio V, A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia, Am J Med Genet A 143A (2007) 33–41. [DOI] [PubMed] [Google Scholar]
- [72].Huoponen K, Vilkki J, Aula P, Nikoskelainen EK, Savontaus ML, A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy, Am J Hum Genet 48 (1991) 1147–1153. [PMC free article] [PubMed] [Google Scholar]
- [73].Roesch K, Curran SP, Tranebjaerg L, Koehler CM, Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex, Hum Mol Genet 11 (2002) 477–486. [DOI] [PubMed] [Google Scholar]
- [74].Benecke R, Strumper P, Weiss H, Electron transfer complex I defect in idiopathic dystonia, Ann Neurol 32 (1992) 683–686. [DOI] [PubMed] [Google Scholar]
- [75].Schapira AH, Warner T, Gash MT, Cleeter MW, Marinho CF, Cooper JM, Complex I function in familial and sporadic dystonia, Ann Neurol 41 (1997) 556–559. [DOI] [PubMed] [Google Scholar]
- [76].Ming L, Moldy sugarcane poisoning--a case report with a brief review, J Toxicol Clin Toxicol 33 (1995) 363.367. [DOI] [PubMed] [Google Scholar]
- [77].He F, Zhang S, Qian F, Zhang C, Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid), Neurology 45 (1995) 2178–2183. [DOI] [PubMed] [Google Scholar]
- [78].Peraica M, Radic B, Lucic A, Pavlovic M, Toxic effects of mycotoxins in humans, Bull World Health Organ 77 (1999) 754–766. [PMC free article] [PubMed] [Google Scholar]
- [79].Zakirova Z, Fanutza T, Bonet J, Readhead B, Zhang W, Yi Z, Beauvais G, Zwaka TP, Ozelius LJ, Blitzer RD, Gonzalez-Alegre P, Ehrlich ME, Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions, PLoS Genet 14 (2018) e1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mencacci NE, Reynolds R, Ruiz SG, Vandrovcova J, Forabosco P, Sanchez-Ferrer A, Volpato V, Consortium UKBE, C. International Parkinson’s Disease Genomics, Weale ME, Bhatia KP, Webber C, Hardy J, Botia JA, Ryten M, Dystonia genes functionally converge in specific neurons and share neurobiology with psychiatric disorders, Brain 143 (2020) 2771–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Eskow Jaunarajs KL, Scarduzio M, Ehrlich ME, McMahon LL, Standaert DG, Diverse Mechanisms Lead to Common Dysfunction of Striatal Cholinergic Interneurons in Distinct Genetic Mouse Models of Dystonia, J Neurosci 39 (2019) 7195–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]