

Abstract
A bioorthogonal reaction between N,N-dialkylhydroxylamines and push-pull-activated halogenated alkynes is described. We explore the use of rehybridization effects in activating alkynes, and we show that electronic effects, when competing stereoelectronic and inductive factors are properly balanced, sufficiently activate a linear alkyne in the uncatalyzed conjugative retro-Cope elimination reaction while adequately protecting it against cellular nucleophiles. This design preserves the low steric profile of an alkyne and pairs it with a comparably unobtrusive hydroxylamine. The kinetics are on par with those of the fastest strain-promoted azide-alkyne cycloaddition reactions, the products regioselectively formed, the components sufficiently stable and easily installed, and the reaction suitable for cellular labeling.
Keywords: retro-Cope elimination, hydroamination, bioorthogonal, rehybridization effect, enamine N-oxide
Graphical Abstract

A rapid and regioselective click reaction between N,N-dialkylhydroxylamines and linear alkynes is described. This bioorthogonal reaction between two small components is enabled by the rehybridization effect of terminally halogenated alkynes and the careful balancing of alkyne π-electron density through a push-pull mechanism.
Since its introduction two decades ago,[1] the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction has proven indispensable in fields ranging from materials science to chemical biology (Figure 1A).[2–3] It is a reaction with widespread appeal for its rapid kinetics, ease of execution, and for the near universal adaptability of its reaction components. The relatively inert azide and terminal alkyne are two of the smallest functional groups available, and either component can be incorporated with ease into biological macromolecules, metabolites, and probes without imposing significant perturbations on the system under evaluation. The ability to incorporate these motifs into biological systems through unnatural amino acids using orthogonal tRNA systems and methionine auxotrophs,[4–6] through lipid and nucleotide modifications,[7–10] or through metabolic engineering[7, 11–12] have been transformational.
Figure 1.
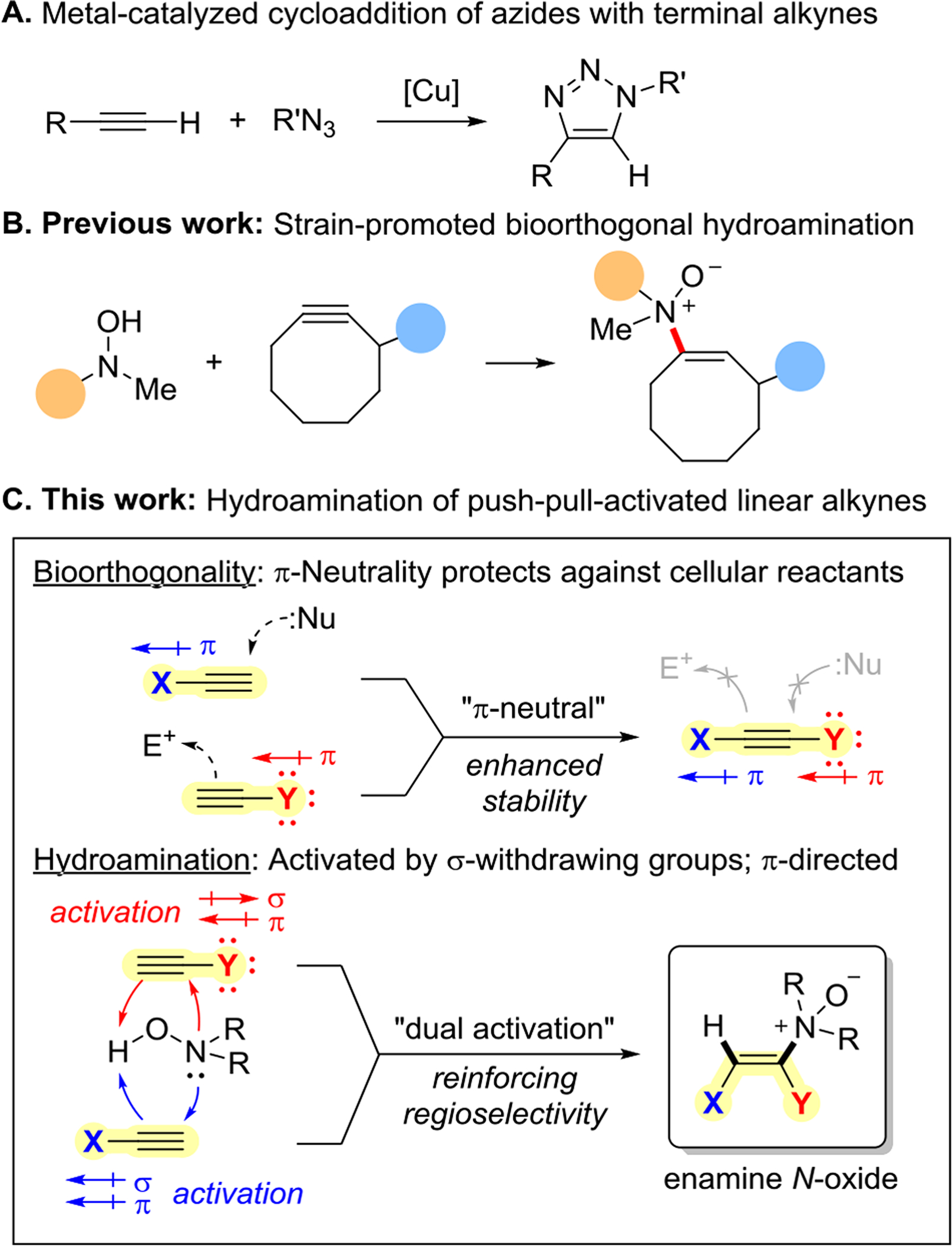
Alkyne activation. (A) Metal-catalyzed azide-alkyne cycloaddition. (B) Strain-promoted alkyne hydroamination. (C) Hydroamination of a push-pull-activated linear alkyne.
Bertozzi and co-workers have since expanded the application of the azide-alkyne cycloaddition to live cell and in vivo systems by employing strained cyclooctynes in lieu of copper catalysts.[13–14] Strain-promoted reactions are now a staple of the bioorthogonal compendium with notable examples featuring trans-cyclooctene,[15] norbornene,[16] quadricyclane,[17] and cyclopropene[18–19] in inverse-electron demand Diels-Alder cycloadditions, dipolar cycloadditions, and phosphine ligations. Importantly, cyclooctyne has undergone substantial geometric[20–24] and electronic[25–28] tuning in a bid to enhance reaction kinetics with azides,[29] tetrazines,[15, 30] sydnones,[31] diazo[32] compounds, and N,N-dialkylhydroxylamines (Figure 1B).[33]
In this work, we revisited the terminal alkyne and explored alternative modes of activation with the aim of achieving uncatalyzed reaction kinetics comparable to that of strain-promoted variants while preserving the size, regioselectivity, and bioorthogonality of this classic reaction component (Figure 1C). We first directed our attention to the alkyne’s electronics. Despite the common presumption that polarized alkenes and alkynes lack chemoselectivity in biological contexts,[29, 34–35] we reasoned it would be feasible to thread the narrow window of chemoselectivity by deconstructing the electronic effects of the alkyne’s substituents into their mesomeric and inductive components.
Positing that cellular nucleophiles are particularly attuned to π-electron density, often reacting by Michael-type addition into π-deficient alkynes, we surmised that mild π-withdrawing substituents might be tolerated if offset by compensatory π-donation from a second substituent in a push-pull manner. Moreover, such constraints on the mesomeric and stereoelectronic effects would not preclude strong σ-withdrawing effects by either or both substituents. The potential magnitude of the influence of σ-withdrawing effects sans reinforcing π-effects is apparent in the instability of 1-fluoroalkynes[36] as well as the computed acceleration of the Bergman cycloaromatization reaction per the rehybridization effect.[37] Strategic halogenation at the terminal and propargylic positions of a linear alkyne appeared promising, a search for an appropriate reaction partner notwithstanding.
We demonstrated recently that N,N-dialkylhydroxylamines and cyclooctynes form a bioorthogonal reaction pair and their union yields biologically compatible enamine N-oxide ligation products (Figure 1B). With this precedent, we reasoned that the retro-Cope elimination reaction would also prove suitable not just in our bid to miniaturize the alkyne, but to achieve an uncatalyzed bioorthogonal ligation reaction between a pair of exceptionally small reaction components, hearkening back to the CuAAC reaction. While promising in concept, few enamine N-oxide products derived by intermolecular retro-Cope elimination had been reported, and of these few, each save one was the product of a reaction with a strong Michael acceptor.[38] Alkyne π-activation appeared key in accessing these compounds and circumventing competing Cope elimination and Meisenheimer rearrangement-based degradation pathways.[39–41] Intriguingly, an outlier, ethoxyethylene, indicated that both π-donors and acceptors are actually prime for the task.[42] Agnosticism of the reaction to the dearth or surplus of π-electron density boded well for a push-pull system.
We first evaluated the impact of inductively withdrawing halogen and chalcogen substituents at the propargylic and terminal positions of the alkyne. Using reaction conditions previously optimized for product stability,[43] we found the retro-Cope elimination reaction between propargyl ether 1 and N,N-diethylhydroxylamine (2) was completed in 18 h whereas hydroamination of terminal alkyne 4 was incomplete even after 10 d. Terminal halogenation also had a similar accelerating effect as the conversion of 6-chlorohex-5-yn-1-ol (6) was nearly complete by 24 h. Most excitingly, the regioselectivities induced by the propargyl and terminal alkyne substituents appeared to be reinforcing. Hydroamination of unactivated alkyne 4 resulted in the preferential formation of the Markovnikov adduct, but this preference was overturned in favor of the anti-Markovnikov products for both propargyl ether 1 and chloroalkyne 6. These data suggested that synergy could be achieved if the propargylic and terminal substituent effects were combined.
To examine whether these predictions would be borne out experimentally, substrates 8–15 were synthesized (Figure 2A). Grignard addition of ethynylmagnesium bromide into aldehyde 16 provided propargyl alcohol 17, which was readily converted to either propargyl fluoride 9 using diethylaminosulfur trifluoride (DAST) or to propargyl difluoride 10 by sequential oxidation and difluorination with Dess-Martin periodinane and DAST, respectively. Chloroalkyne 15 was obtained by halogenation of the corresponding acetylide (Figure 2B). Difluoropropargyl ethers 11–14 were accessed by SN2’ addition of a sodium alkoxide into bromodifluoroallene 18,[44] desilylation, and acetylide halogenation (Figure 2B).
Figure 2.

Effects of terminal and propargylic modification. (A) Reactivity screen using alkynes 8–15. NMR conversion with trifluorotoluene as internal standard. (B) Synthesis of alkynes 9–15. R=PMB. PMB=p-methoxybenzyl. (C) Second-order rate constants for hydroamination of alkynes 11–15 in CD3CN at rt.
With the desired alkynes in hand, we screened their reactivity toward N,N-diethylhydroxylamine (2). Alkynes 8–15 were each incubated with 5 equivalents of hydroxylamine 2 in CD3CN at room temperature, and the reaction conversions were monitored by 1H (alkyne 8) or 19F (alkynes 9–15) NMR (Figure 2A). Robust reactivity toward N,N-dialkylhydroxylamines was observed with halogenated alkynes 13–15 while alkynes 11 and 12 exhibited moderate reactivity. Difluoroalkyne 10 did undergo partial conversion over 48 h; however, no conversion could be observed for propargyl fluoride 9 and propargyl ether 8 over the same period. Their conversion to the corresponding enamine N-oxides could only be achieved upon heating to 60 °C (see Supporting Information). The data indicated that the rate of hydroamination correlates positively with the addition of electronegative substituents to the propargylic position as well as to alkyne termini, as anticipated.
Second-order rate constants were determined for moderately reactive substrates 11 and 12 under pseudo first-order conditions using excess hydroxylamine. Rate constants for the most reactive substrates 13–15 were determined by reaction with equimolar hydroxylamine (Figure 2C). Rate accelerations of 4.1, 63, and 240-fold were achieved over the parent difluoroether 11 by addition of an iodine, bromine, or chlorine atom, respectively, to the alkyne terminus. Interestingly, removal of a propargylic oxygen from chloroalkyne 14 produced chloroalkyne 15, which was still marginally faster than bromoalkyne 13 possessing the propargylic ether. With absolute rate constants on the order of 0.1–1 M−1s−1, the rate of hydroamination of alkynes 13–15 is comparable to the fastest bioorthogonal strain-promoted azide-alkyne cycloaddition reactions.[23, 45]
We next set out to investigate the role of the rehybridization effect in driving the hydroamination of electronically modified linear alkynes. Deviation of an alkyne’s atomic orbitals from canonical hybridization schemes in accordance with Bent’s rule[46] was expected to result in ground state destabilization of haloalkynes – instability, which reaction might alleviate.[36–37, 47]
DFT calculations were performed at the M06–2X[48] level of theory for reactions between N,N-dimethylhydroxylamine (22) and model alkynes 20a–20o. We first confirmed that the computed activation energies (ΔG‡) accurately recapitulated the reactivity trends observed experimentally (Figure 3A). We then performed natural bond orbital (NBO) analysis. In ground state calculations of the alkyne component, electronegative atoms, whether at the propargylic or terminal position, reduced the s-character in the bonding orbital of the most proximal sp-hybridized carbon consistent with Bent’s rule. Notably, the effect of terminal halogenation was much more pronounced, and the s-character of C1 in the C1-X bond deviated significantly (Cl, 40%; F, 35%) from the canonical 50%. A plot of the reaction free energies against the percent s-character of C1 in the C1-X bond of terminally functionalized propynes or difluoropropargyl ethers revealed a strong positive linear relationship consistent with other reactions that exhibit rate enhancements deriving from substantial rehybridization effects (Figure 3B).[37, 47] NBO analysis also revealed that propargylic modifications had a much more muted effect on alkyne rehybridization despite its more significant impact on reducing the activation free energy. In combination with the divergent regioselectivities imparted by each modification, it is plausible that the accelerating effects of propargylic halogen substituents can be imputed more heavily on their stereoelectronic rather than inductive effects, while the opposite may be true for their terminal counterparts. Nonetheless, alkyne halogenation at either site or in combination provides an effective alternative to strain-activation in the context of the retro-Cope elimination reaction.
Figure 3.

Computational studies on the effects of alkyne halogenation. (A) s-Characters (s-char) of alkyne sp-carbons were analyzed, and activation free energies (ΔG‡) were computed for the reaction of alkynes with hydroxylamine 23. (B) Correlation of percent s-character of C1 in the C1-X bond and activation free energy for hydroamination of the alkyne with N,N-dimethylhydroxylamine.
We next evaluated the stability of alkynes 13–15 under physiologically relevant conditions (Figure 4). Alkynes 14 and 15 showed no degradation by 19F NMR over 7 d in 50% CD3CN/phosphate-buffered saline (PBS), pH 7.0 at room temperature while displaying half-lives of 14 h and 43 h, respectively, in the added presence of 2 mM glutathione. These stabilities compare favorably with contemporary bioorthogonal transformations.[49–50] Notably, bromoalkyne 13, which was less reactive toward hydroamination than alkynes 14 and 15, proved unacceptably sensitive to thiols, degrading with a half-life of <30 min under identical conditions. This observation is consistent with the hypothesis that σ-withdrawing/π-donating alkyne substituents such as halogens simultaneously promote hydroamination and attenuate conjugate addition by cellular nucleophiles. Additionally, enamine N-oxide 23 showed no observable degradation over 24 h at room temperature in aqueous solutions containing 2 mM glutathione or HEK293T cell lysate.
Figure 4.

Stability of alkynes 13–15 in 50% CD3CN/PBS in the presence or absence of glutathione (GSH, 2 mM). Stability of enamine N-oxide 23 in PBS in the presence or absence of glutathione (2 mM) or HEK293T cell lysate.
We continued to evaluate the viability of the reaction both in vitro and in cells (Figures 5A and 5B). After verifying that the reaction between chloroalkyne 14 and N,N-diethylhydroxylamine proceeds as expected under aqueous conditions and in the presence of protein (Figure S2), purified recombinant HaloTag protein[51–52] was incubated with HaloTag linker-conjugated difluoropropargyl ether 24 for 10 min at room temperature in pH 7.0 phosphate buffer to provide alkyne-conjugated protein. This protein was then treated with 200 μM TAMRA- hydroxylamine 25 and analyzed at various time points by in-gel fluorescence. Fluorophore-labeled protein was observed within 1 min, and the experiment demonstrated expected time-dependent labeling over 1 h (Figure 5C). Labeling also proved concentration-dependent across a range of concentrations up to 200 μM, as expected (Figure 5D). No labeling was observed in the absence of either the alkyne or hydroxylamine. Formation of the desired protein-fluorophore adduct upon hydroamination was verified by ESI-MS (Figure S11).
Figure 5.

In vitro and live cell labeling by bioorthogonal hydroamination. (A) HaloTag protein was conjugated to chloroalkyne 24 and modified by TAMRA-hydroxylamine 25 then visualized by in-gel fluorescence or fluorescence microscopy. (B) Structures of chloroalkyne 24 and TAMRA-hydroxylamine 25. (C) Time-dependent in-gel fluorescence analysis of hydroamination between HaloTag protein-alkyne 24 conjugate (18 μM) and hydroxylamine 25 (200 μM) for 1–60 min at rt. (D) Concentration-dependent in-gel fluorescence analysis of hydroamination between HaloTag protein-alkyne 24 conjugate (18 μM) and hydroxylamine 25 (25–200 μM) upon incubation for 1 h at rt. (E) Cell surface HaloTag-GFP expressed on HEK293T cells was labeled with TAMRA by bioorthogonal hydroamination between alkyne 24 and hydroxylamine 25. Merge is a composite of Hoechst 33342, GFP, and TAMRA channels. Scale bar = 50 μm. TAMRA = tetramethylrhodamine.
Finally, we explored live cell labeling by hydroamination (Figures 5E and S12). HEK293T cells were transiently transfected with a cell surface HaloTag-GFP construct, treated with 10 μM HaloTag linker-conjugated difluoropropargyl ether 24, washed, and incubated with 50 μM TAMRA-conjugated hydroxylamine 25 for 1 h. The cells were then fixed and visualized by confocal microscopy. TAMRA signal from cells treated with alkyne 24 and hydroxylamine 25 localized to the cell surface and co-localized with the GFP signal of transfected cells. Importantly, negative controls lacking the alkyne and/or hydroxylamine did not exhibit significant labeling. These experiments demonstrated the specificity and efficacy of the reaction in cellular ligation applications.
We have described a bioorthogonal reaction between N,N-dialkylhydroxylamines and halogenated alkynes. We explored the use of rehybridization effects in activating alkynes, and we found that electronic effects, when competing stereoelectronic and inductive factors are properly balanced, sufficiently activate a linear alkyne in the uncatalyzed conjugative retro-Cope elimination reaction while adequately protecting it against cellular nucleophiles. This design preserves the low steric profile of an alkyne and pairs it with a comparably unobtrusive hydroxylamine. The kinetics are on par with those of the fastest strain-promoted azide-alkyne cycloaddition reactions, the products regioselectively formed, the components sufficiently stable and easily installed, and the reaction suitable for cellular labeling. Furthermore, this method introduces a unique enamine N-oxide molecular linchpin, which has potential for further functional development.
Supplementary Material
Scheme 1.

Substituent effects on hydroamination.
Acknowledgements
We thank Dr. Scott Ficarro and the DFCI Blais Proteomics Center for assistance with protein mass spectrometry. This research was supported by the NIH NIEHS (1DP2ES030448) and the Claudia Adams Barr Program for Innovative Cancer Research.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB, Angew. Chem., Int. Ed 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- [2].Hein JE, Fokin VV, Chem. Soc. Rev 2010, 39, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Neumann S, Biewend M, Rana S, Binder WH, Macromol. Rapid Commun 2020, 41, 1900359. [DOI] [PubMed] [Google Scholar]
- [4].Kiick KL, Saxon E, Tirrell DA, Bertozzi CR, Proc. Natl. Acad. Sci. U.S.A 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Prescher JA, Bertozzi CR, Nat. Chem. Biol 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- [6].Plass T, Milles S, Koehler C, Schultz C, Lemke EA, Angew. Chem., Int. Ed 2011, 50, 3878–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Parker CG, Pratt MR, Cell 2020, 180, 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hang HC, Wilson JP, Charron G, Acc. Chem. Res 2011, 44, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Laguerre A, Schultz C, Curr. Opin. Cell Biol 2018, 53, 97–104. [DOI] [PubMed] [Google Scholar]
- [10].Flores J, White BM, Brea RJ, Baskin JM, Devaraj NK, Chem. Soc. Rev 2020, 49, 4602–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Agard NJ, Bertozzi CR, Acc. Chem. Res 2009, 42, 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Laughlin ST, Bertozzi CR, Proc. Natl. Acad. Sci. U.S.A 2009, 106, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sletten EM, Bertozzi CR, Acc. Chem. Res 2011, 44, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR, Science 2008, 320, 664–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Blackman ML, Royzen M, Fox JM, J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Devaraj NK, Weissleder R, Hilderbrand SA, Bioconjugate Chem 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sletten EM, Bertozzi CR, J. Am. Chem. Soc 2011, 133, 17570–17573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Patterson DM, Nazarova LA, Xie B, Kamber DN, Prescher JA, J. Am. Chem. Soc 2012, 134, 18638–18643. [DOI] [PubMed] [Google Scholar]
- [19].Row RD, Shih H-W, Alexander AT, Mehl RA, Prescher JA, J. Am. Chem. Soc 2017, 139, 7370–7375. [DOI] [PubMed] [Google Scholar]
- [20].Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL, Angew. Chem., Int. Ed 2010, 49, 9422–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ning X, Guo J, Wolfert MA, Boons G-J, Angew. Chem., Int. Ed 2008, 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mbua NE, Guo J, Wolfert MA, Steet R, Boons G-J, ChemBioChem 2011, 12, 1912–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jewett JC, Sletten EM, Bertozzi CR, J. Am. Chem. Soc 2010, 132, 3688–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].de Almeida G, Sletten EM, Nakamura H, Palaniappan KK, Bertozzi, Angew. Chem., Int. Ed 2012, 51, 2443–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR, ACS Chem. Biol 2006, 1, 644–648. [DOI] [PubMed] [Google Scholar]
- [26].Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR, Proc. Natl. Acad. Sci. U.S.A 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ni R, Mitsuda N, Kashiwagi T, Igawa K, Tomooka K, Angew. Chem., Int. Ed 2015, 54, 1190–1194. [DOI] [PubMed] [Google Scholar]
- [28].Hu Y, Roberts JM, Kilgore HR, Mat Lani AS, Raines RT, Schomaker JM, J. Am. Chem. Soc 2020, 142, 18826–18835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Agard NJ, Prescher JA, Bertozzi CR, J. Am. Chem. Soc 2004, 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- [30].Yang J, Šečkutė J, Cole CM, Devaraj NK, Angew. Chem., Int. Ed 2012, 51, 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tao H, Liu F, Zeng R, Shao Z, Zou L, Cao Y, Murphy JM, Houk KN, Liang Y, Chem. Commun. (Cambridge, U. K.) 2018, 54, 5082–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Andersen KA, Aronoff MR, McGrath NA, Raines RT, J. Am. Chem. Soc 2015, 137, 2412–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kang D, Kim J, J. Am. Chem. Soc 2021, 143, 5616–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McGrath NA, Raines RT, Chem. Sci 2012, 3, 3237–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Algar WR, Dawson P, Medintz IL, Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications, Wiley-VCH, Weinheim, 2017. [Google Scholar]
- [36].Hanamoto T, Koga Y, Kawanami T, Furuno H, Inanaga J, Angew. Chem., Int. Ed 2004, 43, 3582–3584. [DOI] [PubMed] [Google Scholar]
- [37].Alabugin IV, Manoharan M, J. Comput. Chem 2007, 28, 373–390. [DOI] [PubMed] [Google Scholar]
- [38].O’Neil IA, McConville M, Zhou K, Brooke C, Robertson CM, Berry NG, Chem. Commun. (Cambridge, U. K.) 2014, 50, 7336–7339. [DOI] [PubMed] [Google Scholar]
- [39].Castagnoli N, Cymerman Craig J, Melikian AP, Roy SK, Tetrahedron 1970, 26, 4319–4327. [Google Scholar]
- [40].Moran J, Gorelsky SI, Dimitrijevic E, Lebrun M-E, Bédard A-C, Séguin C, Beauchemin AM, J. Am. Chem. Soc 2008, 130, 17893–17906. [DOI] [PubMed] [Google Scholar]
- [41].Beauchemin AM, Org. Biomol. Chem 2013, 11, 7039–7050. [DOI] [PubMed] [Google Scholar]
- [42].Ciganek E, Read JM, Calabrese JC, J. Org. Chem 1995, 60, 5795–5802. [Google Scholar]
- [43].Kang D, Cheung ST, Wong-Rolle A, Kim J, ACS Cent. Sci 2021, 7, 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu B, Hammond GB, Angew. Chem., Int. Ed 2005, 44, 7404–7407. [DOI] [PubMed] [Google Scholar]
- [45].Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL, Chem. Commun. (Cambridge, U. K.) 2010, 46, 97–99. [DOI] [PubMed] [Google Scholar]
- [46].Bent HA, Chem. Rev 1961, 61, 275–311. [Google Scholar]
- [47].Alabugin IV, Bresch S, dos Passos Gomes G, J. Phys. Org. Chem 2015, 28, 147–162. [Google Scholar]
- [48].Zhao Y, Truhlar DG, Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- [49].Oliveira BL, Guo Z, Bernardes GJL, Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- [50].Tian Y, Lin Q, ACS Chem. Biol 2019, 14, 2489–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV, ACS Chem. Biol 2008, 3, 373–382. [DOI] [PubMed] [Google Scholar]
- [52].Murrey HE, Judkins JC, am Ende CW, Ballard TE, Fang Y, Riccardi K, Di L, Guilmette ER, Schwartz JW, Fox JM, Johnson DS, J. Am. Chem. Soc 2015, 137, 11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.