

Abstract
Showdomycin is a C-nucleoside bearing an electrophilic maleimide base. Herein, the biosynthetic pathway of showdomycin is presented. The initial stages of the pathway involve non-ribosomal peptide synthetase (NRPS) mediated assembly of a 2-amino-1H-pyrrole-5-carboxylic acid intermediate. This intermediate is prone to air oxidation whereupon it undergoes oxidative decarboxylation to yield an imine of maleimide, which in turn yields the maleimide upon acidification. It is also shown that this pyrrole intermediate serves as the substrate for the C-glycosidase SdmA in the pathway. After coupling with ribose 5-phosphate, the resulting C-nucleoside undergoes a similar sequence of oxidation, decarboxylation and deamination to afford showdomcyin after exposure to air. These results suggest that showdomycin could be an artifact due to aerobic isolation; however, the autoxidation may also serve to convert an otherwise inert product of the biosynthetic pathway to an electrophilic C-nucleotide thereby endowing showdomycin with its observed bioactivities.
Keywords: C-nucleoside, maleimide, biosynthesis, C-glycosidation, autoxidation
Graphical Abstract
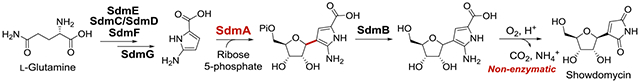
C-nucleoside biosynthesis: The biosynthetic pathway of the C-nucleoside showdomycin was reconstituted in vitro. Key steps include cyclization of l-glutamine to yield an oxygen-labile pyrrole intermediate that serves as the substrate for C-glycosidation catalyzed by SdmA. Autoxidative maturation of the pyrrole nucleobase to the maleimide moiety of showdomycin can then proceed non-enzymatically.
Introduction
Showdomycin (1) along with formycin A (2), pyrazofurin (3), pseudouridine (4), alnumycin C1 (5) and minimycin (6) are C-nucleosides with structures characterized by a C−C ribosyl linkage between ribofuranose and a heterocyclic base (Figure 1). [1–4] Showdomycin (1) isolated from Streptomyces showdoensis [5, 6] is known to inhibit DNA and RNA polymerases isolated from Escherichia coli leading to antibacterial and anti-tumor activities. [7–10] It was also found to inactivate the nucleoside uptake system, and this inhibition can be reversed by nucleoside and thiol compounds such as cysteine.[11,12] It was therefore suggested that the bioactivity of showdomycin is a consequence of its maleimide moiety, which likely acts as a Michael acceptor and thus an alkylating agent when it undergoes nucleophilic addition by a sulfhydryl group on its inhibitory targets.[11,12] On the basis of its reactivity towards thiols, showdomycin and its synthetic derivatives have also been designed as probes for the labeling and identification of clinically important enzymes in pathogenic bacteria.[13]
Figure 1.
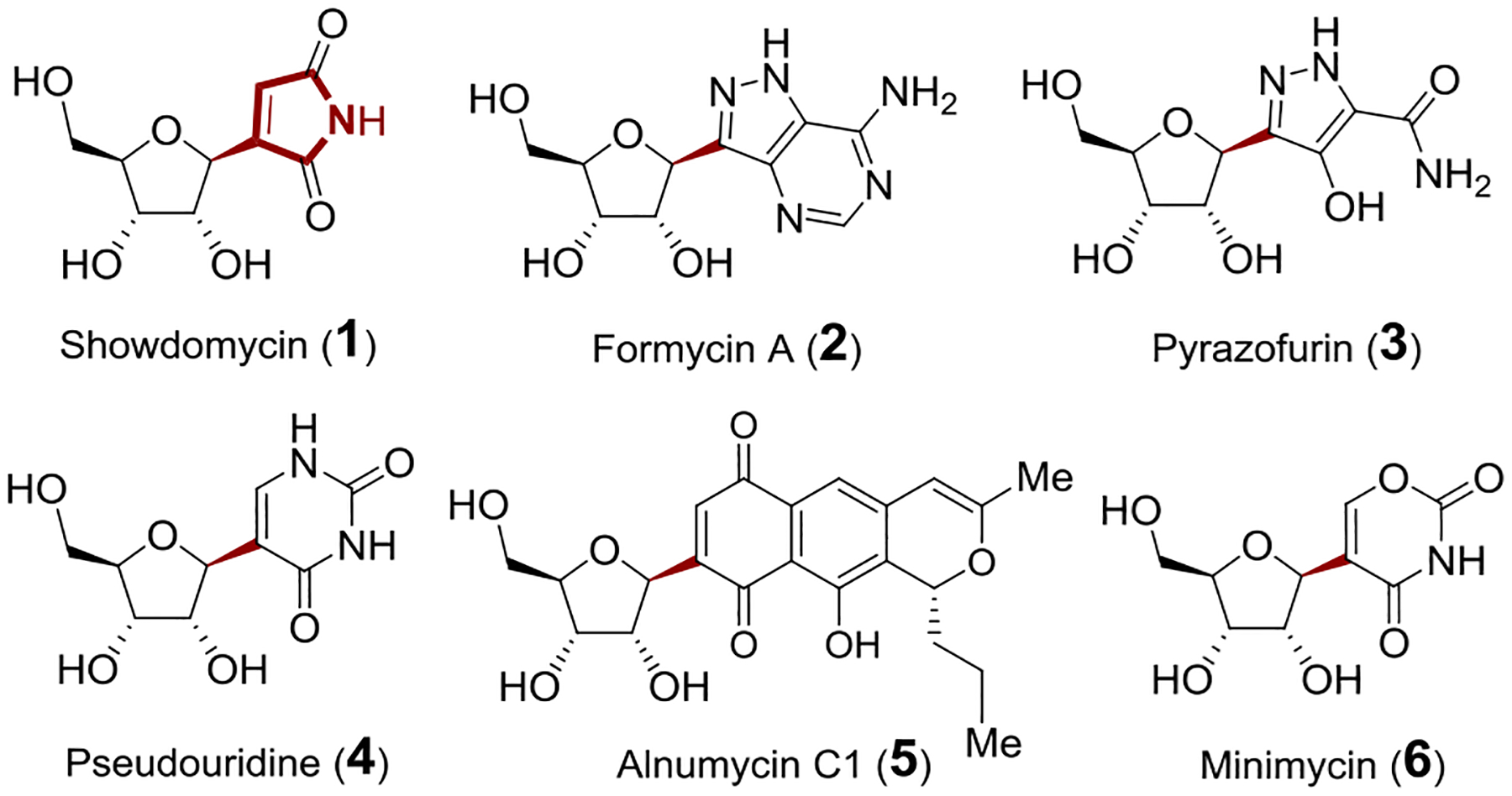
C-Ribosylnucleoside antibiotics.
How the C-nucleosides are assembled in nature has been a subject of current interest. Recently, the C-glycosylation reactions in the formycin A (2) and pyrazofurin (3) biosynthetic pathways were shown to be catalyzed by ForT and PyfQ, respectively.[14–16] Both utilize phosphoribosyl pyrophosphate (PRPP) as the ribose donor, and the reactions proceed via electrophilic aromatic substitution.[14,16] In contrast to ForT and PyfQ, however, early studies of pseudouridine (4) biosynthesis showed that pseudouridine monophosphate synthase (YeiN) catalyzes C-glycosidic bond formation and cleavage between ribose 5-phosphate (14, R5P) and uracil through a ribose ring-opening mechanism.[17,18] Likewise, AlnA in alnumycin C1 (5) biosynthesis exhibits moderate homology to YeiN and appears to catalyze the attachment of R5P to the polyketide aglycone via a mechanism involving an enediolate intermediate.[19,20] Finally, the gene cluster for showdomycin biosynthesis (sdm) was recently identified in S. showdoensis by screening for homologs of the C-glycosidase AlnA.[21] The corresponding C-glycosidase in showdomycin pathway has thus been hypothesized to be the gene product SdmA, and the observation that deletion of the sdmA gene prevents showdomycin biosynthesis in vivo is consistent with this hypothesis.[21] Nevertheless, this gene assignment has not been tested in vitro such that the biological assembly of showdomycin remains to be definitively established.
Results and Discussion
To address these unresolved questions, the sdm gene cluster was analyzed, and a cassette consisting of sdmC, D, E, F and G was found to be homologous to five genes in the kosinostatin (8) biosynthetic gene cluster (kstB1, B2, B3, B4 and B6, respectively) (Figures 2A and 2B).[22] Showdomycin (1) and kosinostatin (8) are both characterized by a moiety having a pyrrolidine skeleton (see ring F in 8), and it was therefore hypothesized that construction of this moiety (e.g., 7/12 in Figure 2C) is catalyzed by the gene products of the two homologous cassettes shared by each gene cluster. This hypothesis also implies that the early steps of showdomycin biosynthesis involve nonribosomal peptide synthesis, because sdmC and sdmD (kstB1 and kstB2) are annotated as encoding nonribosomal peptide synthetases (NRPS). SdmG, which is annotated as a transglutaminase-like protein, may then catalyze hydrolysis of the thioester linkage to release the free acid 13 as the substrate for the C-glycosidase SdmA. Subsequent tailoring steps including dephosphorylation, deamination, oxidation and decarboxylation would then lead to maturation of the nucleobase in showdomycin (1).
Figure 2.
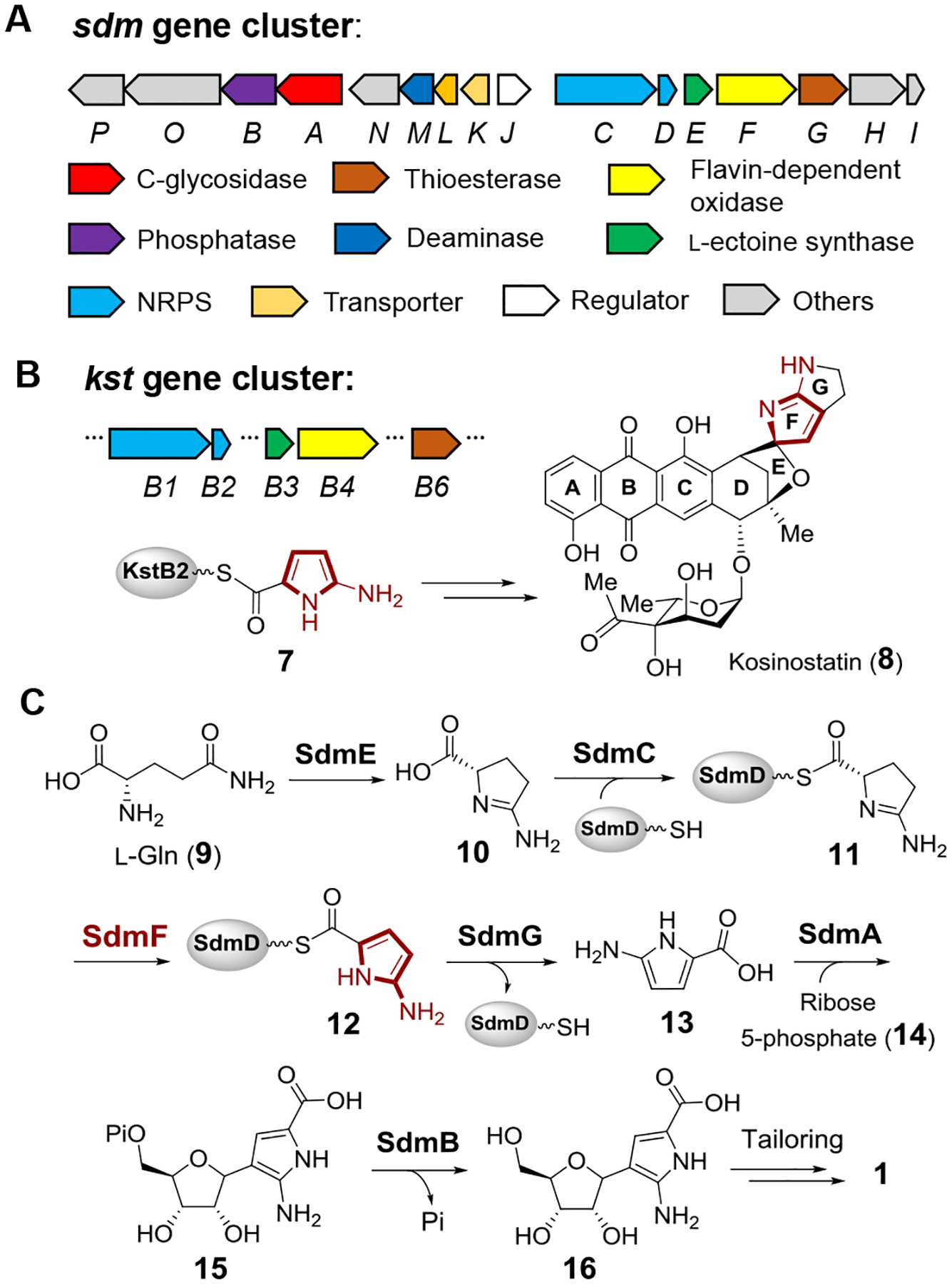
(A) Biosynthetic gene cluster of showdomycin in S. showdoensis. (B) Partial gene cluster of kosinostatin (8). The F ring was proposed to be biosynthesized from precursor 7.[22] (C) Proposed biosynthetic pathway of showdomycin (1).
A possible biosynthetic pathway for showdomycin biosynthesis can thus be envisaged in which a protein-tethered 2-aminopyrrole species 12 (7 in the kosinostatin pathway) is generated via the actions of SdmE/C/D/F and serves as a key intermediate (see Figure 2C). The proposed pathway is likely initiated with the cyclization of l-glutamine (9) to 10 catalyzed by SdmE, a homolog of l-ectoine synthase (EctC). Canonical EctC is responsible for the conversion of 17 to l-ectoine (18, see Figure 3A)[23] and can also catalyze the condensation of 9 to 10 as a side reaction.[24] Ant24 is a homolog of SdmE that is similarly believed to catalyze the same reaction (i.e., 9 → 10, see Figure 3B) during the biosynthesis of anthelvencin A (19) according to gene deletion and chemical complementation assays by feeding the mutant with 10.[25] Indeed, when 10 μM purified SdmE (Figure S1) was incubated with 5 mM l-glutamine in tris(hydroxymethyl)amino-methane (Tris) buffer (pH 8.0) and the reaction followed by 1H NMR, new signals were observed consistent with a synthetically prepared standard of 10 (see Figure 3C) in support of this hypothesis. This observation is also consistent with the report that none of the 20 proteinogenic amino acids can be uploaded onto SdmD by SdmC.[21] Thus, the conversion of l-glutamine (9) to 10 appears to be necessary to provide the proper substrate form recognized by the SdmD/SdmC NRPS system.
Figure 3.
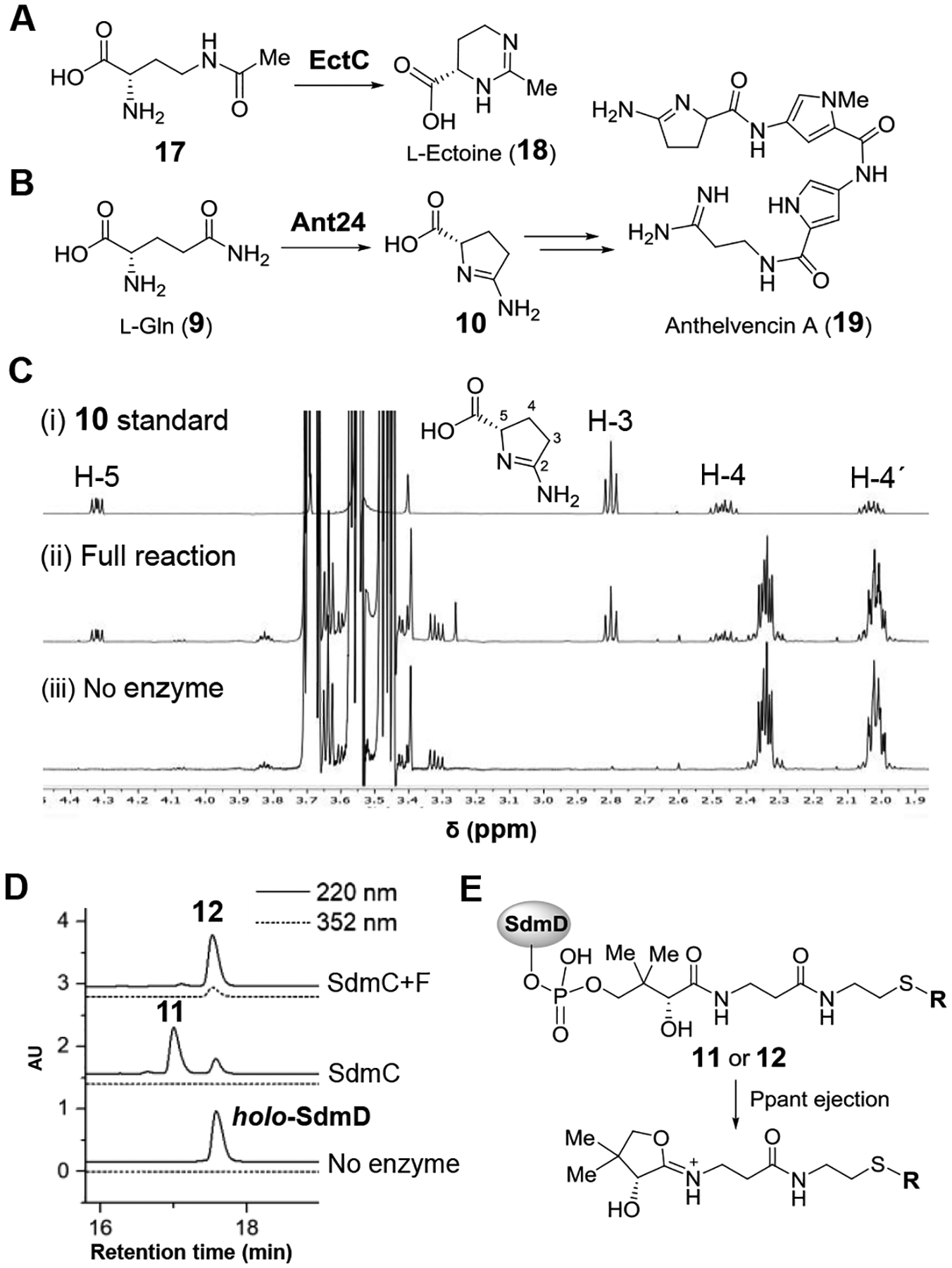
(A) EctC-catalyzed condensation of 17 to l-ectoine (18). (B) Biosynthesis of anthelvencin A (19) involving cyclization of l-Gln (9) to 10 by Ant24. (C) Monitoring product formation in the SdmE catalyzed reaction using 1H NMR spectroscopy. (D) HPLC analysis of the SdmC and SdmF catalyzed reactions. holo-SdmD coelutes with 12. (E) Ejection of Ppant-containing ions by MS fragmentation.[26]
To test this hypothesis, 50 μM apo-SdmD was incubated with 1 mM 10 in the presence of 2 μM SdmC, 2 μM phosphopantetheinyl transferase (Sfp), 1 mM ATP, 75 μM coenzyme A, 10 mM MgCl2 and 5 mM tris-(2-carboxyethyl)phosphine (TCEP) in 100 mM Tris buffer (pH 8.0) for 2 h. After quenching the reaction with acetonitrile, HPLC analysis of the supernatant revealed the presence of a new peptidyl carrier protein species (Figure 3D). To track any change at the acylation carrier site of its phosphopantetheinyl (Ppant) arm, ESI-MS was employed to detect the Ppant ion fragment ejected from the intact protein (see Figure 3E and supporting information). Indeed, an MS signal was observed for an ion fragment having the mass expected for Ppant modified with 11 (Figure S2). This implies that showdomycin biosynthesis begins with formation of the modified SdmD peptide-carrier-protein following cyclization of l-glutamine (9 → 10 → 11).
Formation of a 2-aminopyrrole from the 2-aminopyrroline intermediate 11 requires an oxidation reaction that may be catalyzed by SdmF, which is annotated as a flavin-dependent oxidase. When 1 μM SdmF was included in the above assay under aerobic condition, a new species was observed by MS with a molecular weight two mass units less than 11 (Figures 3D and S2). Purification of the SdmD/SdmC/SdmF coupled reaction mixture by gel filtration led to isolation of the modified SdmD, which exhibited a UV-Vis absorption maximum at 352 nm at pH 8.0 (Figure S3). A similar spectrum with a maximum absorbance at 350 nm (pH 8.0) was also observed with a synthesized N-acetylcysteamine (S-NAc) analog of 12 (Figure S3), suggesting that the dehydrogenated product formed in the presence of SdmF is the proposed intermediate 12, which contains a 2-aminopyrrole moiety conjugated to a thioester. Furthermore, when SdmF was incubated with 10 in the absence of SdmC and SdmD, no substrate consumption was observed by 1H-NMR spectroscopy (Figure S4). These observations imply that SdmF catalyzes the dehydrogenation of 11 to 12 in a reaction that takes place only after loading of 10 onto the peptide-carrier-protein SdmD.
Incubation of the purified 12 with SdmG resulted in the decrease of the absorbance at 352 nm and an increase in absorbance at 288 nm with an isosbestic point at 310 nm (Figure 4A). Therefore, it was hypothesized that SdmG catalyzes the subsequent hydrolysis of 12 to generate the free 2-amino-1H-pyrrole-5-carboxylic acid 13. However, LC-MS analysis failed to detect 13 following deproteinization of the reaction mixture via ultrafiltration, and the absorbance at 288 nm was also found to diminish over time (Figure 4B). One possible explanation for the results with SdmG and intermediate 12 is that 13 is unstable under aerobic conditions, because 2-aminopyrroles are prone to autoxidation.[27–29] Moreover, the pyrrole-2-carboxylate intermediates identified in the biosynthesis of violacein[30] and rebeccamycin[31,32] have also been shown to react with oxygen resulting in oxidative decarboxylation to the corresponding oxo-products (Figure S5). These early reports suggested that 13 may undergo a similar autoxidation and subsequent decarboxylation under aerobic conditions to yield 2-iminopyrrolone 21 and maleimide 22 following SdmG catalyzed hydrolysis of 12 (Figures 4B and 4C). Given that 21 and 22 are potential Michael acceptors and holo-SdmD would also be released in the SdmG reaction, the free thiol of the phosphopantetheinyl arm of holo-SdmD may in turn react with 21 or 22 to form adducts such as 23 or 24 (Figure 4D).
Figure 4.
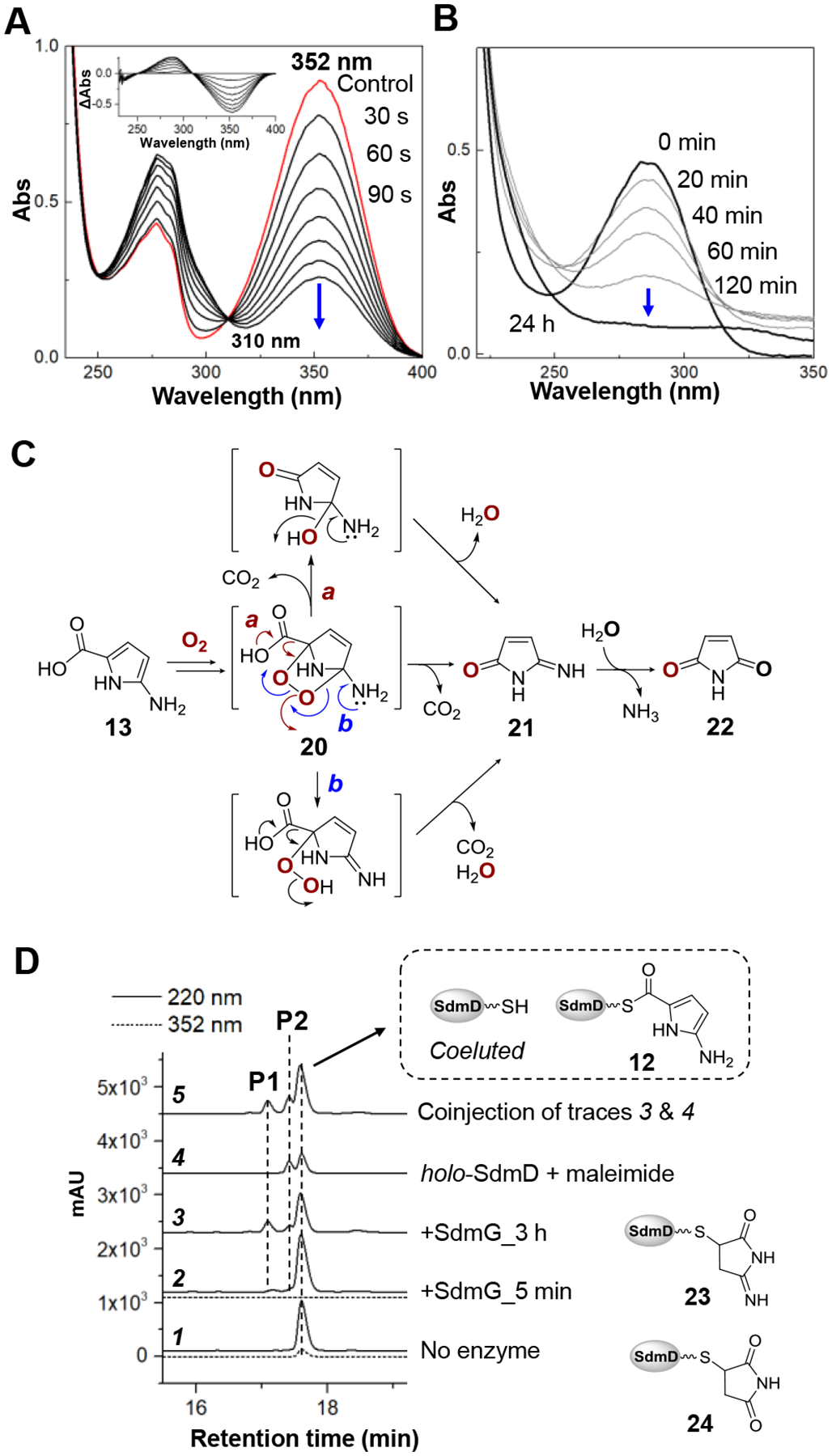
(A) Time-course of the SdmG-catalyzed reaction monitored by UV-Vis spectroscopy. The final enzyme and substrate concentrations were 50 nM and 30 μM, respectively. The inset shows difference spectra calculated by subtracting the spectrum of a control with no SdmG from each time-point. (B) Decomposition of the SdmG product determined by the diminishing absorbance at 288 nm of the reaction filtrate over 24 h. (C) The proposed autoxidation of 13. Decomposition of the endoperoxide intermediate 20 to 21 and 22 may proceed via either direct decarboxylation (route a) or O-O bond cleavage triggered by imine formation (route b). (D) HPLC analysis of the proteinaceous products from the aerobic SdmG reaction. The elution was monitored at 352 nm (dash line) to determine the consumption of substrate 12.
In order to test these hypotheses, the SdmG assay was again performed and analyzed by LC-MS to investigate the formation of modified peptidyl carrier proteins. HPLC analysis revealed the complete disappearance of substrate 12 within 5 min as indicated by the absence of any signal at 352 nm (Figure 4D trace 2). Meanwhile, gradual formation of two new peaks P1 and P2 over 3 h were found as the major SdmD-bound species in the SdmG reaction (Figure 4D). MS analysis of the Ppant ejection ions of P1 and P2 were consistent with the proposed structures of 23 and 24 (Figure S6). Furthermore, a standard of 24 prepared by reacting maleimide with holo-SdmD was observed to coelute with P2 produced in the SdmG catalyzed reaction (Figure 4D trace 5). Finally, when 15N2-l-glutamine was used in the SdmE/SdmC/SdmD/SdmF/SdmG reaction, an increase of two mass units of the Ppant ejection ion during LC-MS analysis of P1 was observed consistent with the assigned structure 23 (Figure S7). Therefore, the observed consumption of substrate 12 and formation of 23 and 24 agrees with the proposed reaction sequence in which 13 is slowly oxidized and covalently trapped by holo-SdmD.
To further investigate the mechanism by which 13 undergoes oxidative decarboxylation, 50 μM 12 was incubated with 1 μM SdmG in Tris buffer (pH 8.0) under an 18O2 atmosphere for 3 h. A mass increase of 2 Da was observed with both 23 and 24 indicating incorporation of a single 18O center from molecular oxygen to each (Figures 5A and 5B). In contrast, neither adduct contained 18O when the incubation was conducted with natural abundance O2 and 18O-enriched water. The absence of 18O incorporation from solvent in 24 suggested that 24 might be derived from the hydrolysis of 23 during HPLC separation and/or LC-MS analysis rather than during the assay. Indeed, 23 was found to be stable at pH 8.0 (SdmG assay conditions) but could be converted to 24 at pH 2.0 (HPLC and LC-MS conditions) as shown in Figures 5C and 5D. These results agree with the proposed transformation of 13 to 21. In a separate experiment where the SdmG reaction was carried out anaerobically and the reaction filtrate was promptly analyzed by LC-MS, a signal consistent with 13 was observed directly (calc. m/z for C5H7N2O2+ [M+H]+ 127.0508; found 127.0503) along with the in-source decarboxylated fragment (calc. m/z for C4H7N2+ [M−CO2+H]+ 83.0609; found 83.0604) (Figure S8).
Figure 5.
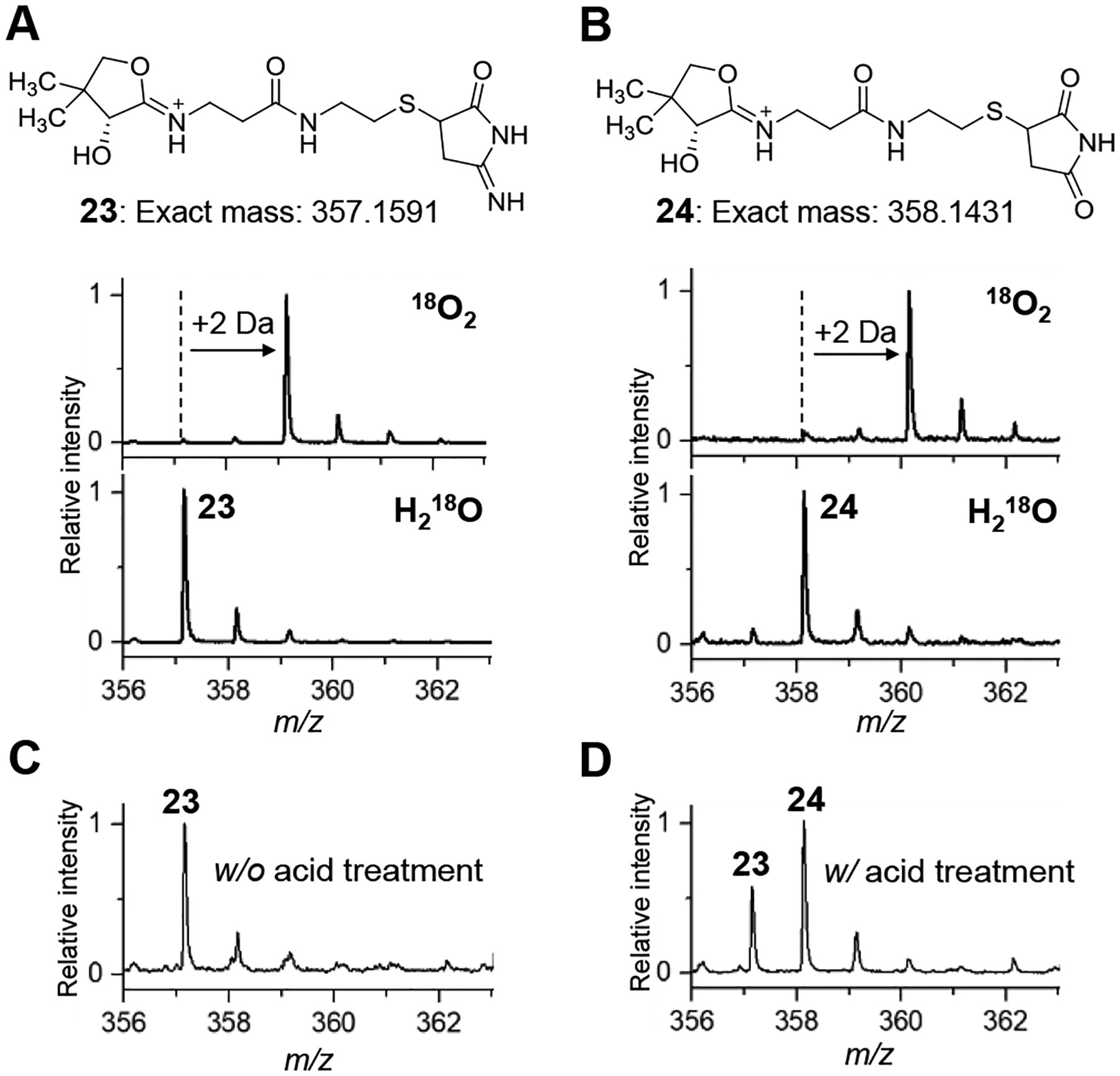
Ppant ejection mass spectra of adducts (A) 23 and (B) 24 in the presence of 18O2 + natural abundance H2O (upper trace) or natural abundance O2 + H218O (lower trace). (C) Ppant ejection mass spectra of SdmG reaction mixture without and (D) with excess 0.1% TFA pre-treatment.
The demonstrated reactivity of 13 toward O2 suggested that it is electron-rich and may thus serve as a nucleophile in the formation of the C-ribosyl linkage catalyzed by SdmA, which is annotated as a C-glycosidase in the sdm cluster. To test this hypothesis, the in vitro characterization of SdmA using ribose 5-phosphate (R5P, 14) as the electrophile was conducted under anaerobic conditions to prevent autoxidation of 13. First, 13 was freshly prepared by incubating 12 with SdmG and then removing the enzymes by ultrafiltration. When 10 μM SdmA was added to the resulting solution containing 13 (ca. 80 μM) along with 0.8 mM R5P (14) and 5 mM MgCl2, a new species was detected by HPLC with a UV-vis absorption spectrum nearly identical to that of 13 but with a different HPLC retention time (Figures 6B and S9). HRMS analysis of this new product revealed a chemical composition of C10H16N2O9P, which is consistent with the structure of 15 (calc. 339.0593, found 339.0599). The in-source decarboxylated fragment was also observed by MS (calc. m/z for C9H16N2O7P [M-CO2]+ 295.0695, found 295.0710) (Figure S10), indicating the presence of a labile carboxyl group and supporting the assignment of the SdmA product as 15. However, similar results would also be expected for a product containing a C4-C1’ linkage (i.e., 27) rather than a C3-C1’ linkage as shown in 15 (Figure 6A). While the isolation and direct characterization of 15 failed due to its instability, the regiochemistry of adduct formation was determined by using the [4-2H]-isotopolog of 13 prepared enzymatically from [3,3-2H2]-l-glutamine. LC-MS analysis of the corresponding SdmA product showed retention of one deuterium (see Figure S11), indicating 15 rather than 27 is the enzymatic product of SdmA. This regioselectivity is also consistent with the increased nucleophilicity of C3 of 13 due to the amino substituent at the C2 position. Moreover, if SdmB, which is annotated as a phosphatase, was present in the reaction, the correct mass of the dephosphorylated product 16 was detected (calc. m/z for C10H15N2O6 [M+H]+ 259.0930, found 259.0923) (Figure S10). Interestingly, unlike the cases of ForT and PyfQ, PRPP is not a substrate for SdmA (Figure 6B). It was also found that maleimide (22) could not be processed by SdmA (Figure S12). Thus, glycosidation of 13 to 15 most likely occurs prior to oxidative decarboxylation of 13.
Figure 6.
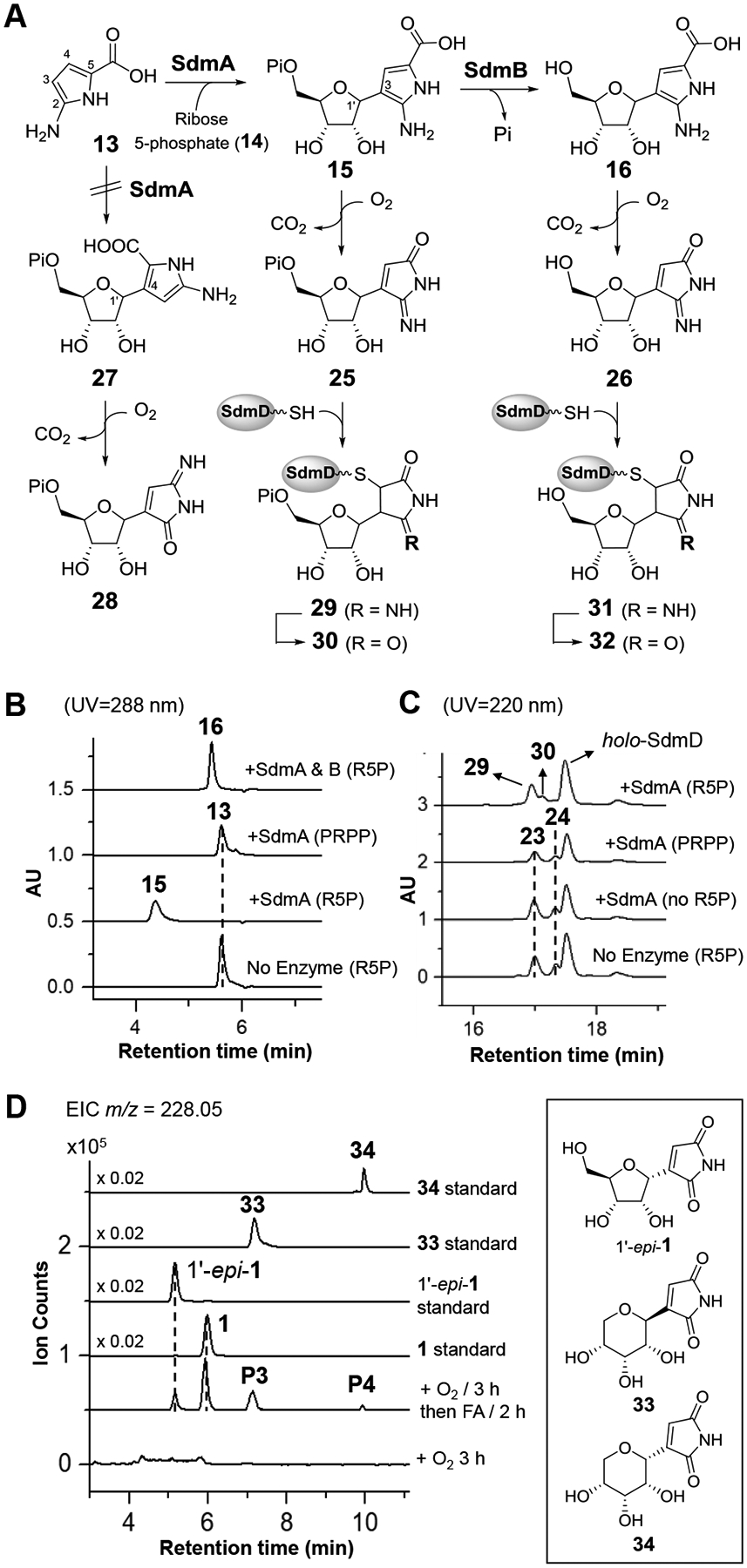
(A) Products generated in the reactions catalyzed by SdmA and SdmB. The nascent enzyme products 15 and 16 were susceptible to oxidative decarboxylation and covalent modification by holo-SdmD. (B) HPLC analysis of anaerobic incubations of SdmA and SdmB. (C) HPLC analysis of aerobic incubations of 12 and SdmA coupled with SdmG. (D) LC-MS analysis of autoxidation of 16. Extracted ion chromatogram (EIC) traces corresponding to [M - H]− signals from 1 and 1’-epi-1 are shown. The unknown P3 and P4 have the same exact mass as showdomycin and have the same retention time with standards of pyranose isomer of showdomycin (33 and 34, respectively). Proposed mechanism for the formation of 1’-epi-1, 33 and 34 is shown in Figure S15.
Given that the C-glycosylation products 15 and 16 carry the same pyrrole moiety as 13, they may also undergo autoxidation in air to generate 29 and 31, respectively. To trap and characterize the anticipated oxidative decarboxylation products, a coupled assay of 12 and 14 with SdmG/SdmA was conducted under aerobic conditions. When 50 μM 12 was incubated with 0.2 μM SdmG, 10 μM SdmA in the presence of 0.5 mM ribose 5-phosphate (14), 5 mM MgCl2 in Tris buffer (pH 8.0), LC-MS analysis revealed the presence of two new SdmD-bound adducts 29 and 30 accompanied by a small amount of 23 and 24 due to autoxidation of the intermediate 13 (Figures 6C and S13). Adducts 29 and 30 were not detected when R5P was replaced by PRPP, which is consistent with the aforementioned results of the anaerobic assays. Moreover, when SdmB was included in the coupled reaction assay, products with the correct mass for 26 as well as showdomycin (1) covalently linked to holo-SdmD (i.e., adducts 31 and 32, respectively, Figure S14) could be detected. To directly test if 16 can be converted to showdomycin via autoxidation, the SdmA/SdmB coupled reaction filtrate from anaerobic incubation was exposed to air for 3 h during which time the correct mass of 26 was detected (calc. m/z for C9H11N2O5 [M-H]− 227.0668, found 227.0675). When this mixture was acidified with excess 0.1% formic acid (FA) to pH ~3 for 2 h, LC-MS analysis revealed the presence of both showdomycin (1) and the C1’ epimer of showdomycin (1’-epi-1) (Figure 6D). Two new peaks (P3 and P4) which have the same exact mass as 1 and 1’-epi-1 were also noted. These species were assigned to be pyranose isomers of showdomycin (33 and 34) based on their coelution with the chemically prepared standards. These observations are consistent with the proposed pathway in Figure 2C whereby 16 is a key intermediate en route to the observed natural product showdomycin (1).
Conclusion
The biosynthetic pathway for showdomycin as encoded by the sdm gene cluster has now been reconstituted in vitro. The biosynthetic precursors to showdomycin are l-glutamine (9) and ribose 5-phosphate (14) with no apparent dependence on PRPP in contrast to the pyrazole C-nucleosides formycin A (2) and pyrazofurin (3).[14–16] Assembly is initiated by SdmE catalyzed cyclization of l-glutamine to yield a 2-amino-1-pyrroline-5-carboxylate intermediate that is subsequently loaded onto the phosphopantetheine arm of the peptidyl carrier protein SdmD (9 → 10 → 11). Oxidation of the resulting thioester-linked pyrroline is then catalyzed by the flavoenzyme SdmF (11 → 12) before SdmG mediated hydrolysis to yield the free 2-amino-1H-pyrrole-5-carboxylate species 13 as the key intermediate. While it is unclear why an NRPS system (SdmC and SdmD) participates in producing 13, it nevertheless is necessary as SdmF is unable to catalyze the direct oxidation of 10. In contrast, kosinostatin biosynthesis, which involves homologs of the showdomycin SdmE, SdmC, SdmD, SdmF and SdmG enzymes, has been proposed to require further NRPS catalyzed modifications in addition to oxidation.[22] Thus, while showdomycin and kosinostatin biosynthetic gene clusters may share a common evolutionary ancestor, further development may have led to the observed specificity of SdmF despite other NRPS functions having been lost.
The key step in the showdomycin biosynthetic pathway is regioselective formation of the C–C linkage between C3 of 13 and C1′ of ribose 5-phosphate (14) to yield 15, which is unstable in the presence of molecular oxygen. This reaction is catalyzed by SdmA. Given the high sequence homology between SdmA and the pseudouridine monophosphate C-glycosidase YeiN (42% identity, 59% similarity) as well as their dependence on ribose 5-phosphate but not PRPP, an analogous C-glycosidation mechanism can be envisioned as depicted in Figure 7.[17,18] Subsequent dephosphorylation catalyzed by SdmB yields the showdomycin precursor 16 that is similarly unstable towards autoxidation. Although showdomycin and 1’-epi-showdomycin were both detected as autoxidation products of 16, this result cannot be referred to the stereochemistry of 16 or 15 without more direct evidence. This is because C-nucleosides are known to be susceptible to epimerization at C1’ under acidic conditions.[33–36]
Figure 7.
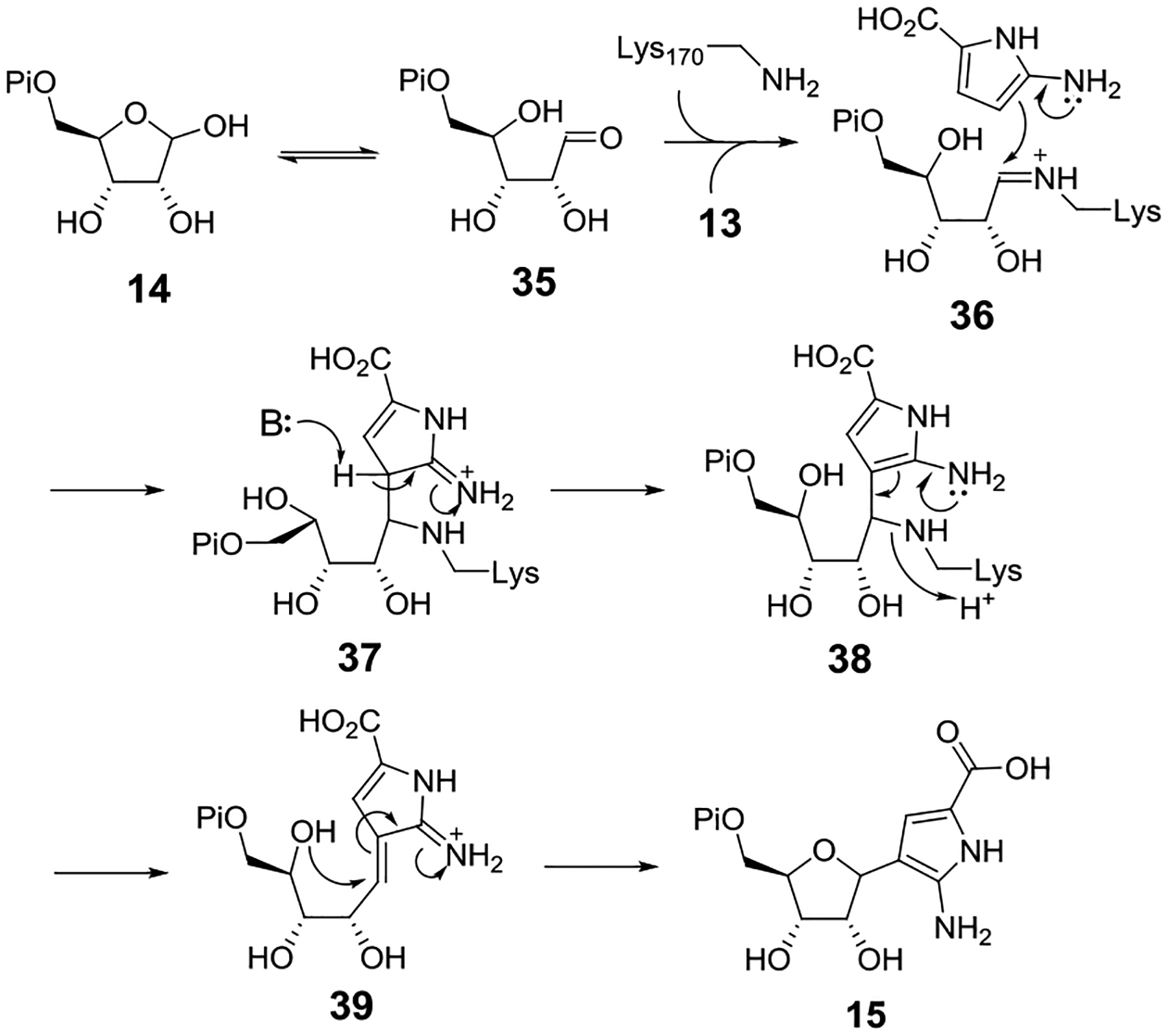
Proposed mechanism of glycosidic bond formation catalyzed by SdmA. Based on sequence alignment with YeiN[17], Lys170 in SdmA is proposed to form a Schiff base with the ring-opened ribose 5-phosphate precursor (35) prior to the nucleophilic addition of 13.
The final tailoring steps that convert 16 to showdomycin are expected to include oxidation, decarboxylation and deamination reactions. However, the observed instability of compound 16 indicates that the final steps of showdomycin formation can proceed via a series of nonenzymatic transformations to afford 1, 1’-epi-1 and the pyranose isomers of showdomycin (33 and 34). Hence, the C-nucleoside 16 may actually be the true product of the showdomycin biosynthetic pathway with showdomycin itself being an artifact due to isolation under aerobic and acidic conditions. Similar examples of misidentified biosynthetic products due to oxidation, decomposition or further modifications upon isolation have also been reported in other natural product systems, making such a possibility not without precedent.[37,38] Alternatively, the nonenzymatic conversion of 16 to showdomycin may indeed represent the final stages of the biosynthetic pathway, given that 16 is not expected to exhibit the same biological activity typically ascribed to the more electrophilic showdomycin. While these hypotheses remain to be further explored, the present results offer a full description of showdomycin biosynthesis as well as its peculiar features within the context of C-nucleoside biochemistry. Importantly, the in vitro reconstitution of showdomycin biosynthesis unveils the double-edged effect of autoxidation as it can deplete the pyrrole intermediate prior to C-glycosidation but also appears to be necessary for the putative maturation of the biologically active natural product.
Supplementary Material
Acknowledgements
We thank Dr. Mark Ruszczycky for his critical review and help of preparing the manuscript. This work was supported by grants from the National Institutes of Health (GM040541, GM035906) and the Welch Foundation (F-1511).
References
- [1].Suhadolnik RJ in Nucleoside Antibiotics; Wiley-Interscience: New York, 1970. [Google Scholar]
- [2].Buchanan JG, Fortschr. Chem. Org. Naturst 1983, 44, 243–299. [DOI] [PubMed] [Google Scholar]
- [3].Stambaský J, Hocek M, Kocovský P, Chem. Rev 2009, 109, 6729–6764. [DOI] [PubMed] [Google Scholar]
- [4].De Clercq E, J. Med. Chem 2016, 59, 2301–2311. [DOI] [PubMed] [Google Scholar]
- [5].Nishimura H, Mayama M, Komatsu Y, Kato H, Shimaoka N, Tanaka Y, J. Antibiot. (Tokyo) 1964, 17, 148–155. [PubMed] [Google Scholar]
- [6].Darnall KR, Townsend LB, Robins RK, Proc. Natl. Acad. Sci. U. S. A 1967, 57, 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Maryanka D, Johnston IR, FEBS Lett. 1970, 7, 125–128. [DOI] [PubMed] [Google Scholar]
- [8].Roy-Burman P, Recent Results Cancer Res. 1970, 25, 70–82. [Google Scholar]
- [9].Matsuura S, Shiratori O, Katagiri K, J. Antibiot. (Tokyo) 1964, 17, 234–237. [PubMed] [Google Scholar]
- [10].Roy-Burman S, Roy-Burman P, Visser DW, Cancer Res. 1968, 28, 1605–1610. [PubMed] [Google Scholar]
- [11].Nishimura H, Komatsu Y, J. Antibiot. (Tokyo) 1968, 21, 250–254. [DOI] [PubMed] [Google Scholar]
- [12].Uehara YI, Fisher JM, Rabinowicz M, Biochem. Pharmacol 1980, 29, 2199–2204. [DOI] [PubMed] [Google Scholar]
- [13].Böttcher T, Sieber SA, J. Am. Chem. Soc 2010, 132, 6964–6972. [DOI] [PubMed] [Google Scholar]
- [14].Ren D, Wang S-A, Ko Y, Geng Y, Ogasawara Y, Liu H.-w., Angew. Chem. Int. Ed. Engl 2019, 58, 16512–16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhang M, Zhang P, Xu G, Zhou W, Gao Y, Gong R, Cai YS, Cong H, Deng Z, Price NPJ, Mao X, Chen W, Appl. Environ. Microbiol 2020, 86, e01971–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gao S, Radadiya A, Li W, Liu H, Zhu W, de Crécy-Lagard V, Richards NGJ, Naismith JH, Chem. Commun. (Camb.) 2020, 56, 7617–7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huang S, Mahanta N, Begley TP, Ealick SE, Biochemistry 2012, 51, 9245–9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pfeiffer M, Nidetzky B, Nat. Commun 2020, 11, 6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oja T, Klika KD, Appassamy L, Sinkkonen J, Mäntsälä P, Niemi J, Metsä-Ketelä M, Proc. Natl. Acad. Sci. U. S. A 2012, 109, 6024–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oja T, Niiranen L, Sandalova T, Mäntsälä P, Schneider G, Metsä-Ketelä M, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Palmu K, Rosenqvist P, Thapa K, Ilina Y, Siitonen V, Baral B, Mäkinen J, Belogurov G, Virta P, Niemi J, Metsä-Ketelä M, ACS Chem. Biol 2017, 12, 1472–1477. [DOI] [PubMed] [Google Scholar]
- [22].Ma H-M, Zhou Q, Tang Y-M, Zhang Z, Chen Y-S, He H-Y, Pan H-X, Tang M-C, Gao J-F, Zhao S-Y, Igarashi Y, Tang G-L, Chem. Biol 2013, 20, 796–805. [DOI] [PubMed] [Google Scholar]
- [23].Peters P, Galinski EA, Trüper HG, FEMS Microbiol. Lett 1990, 71, 157–162. [Google Scholar]
- [24].Witt EM, Davies NW, Galinski EA, Appl. Microbiol. Biotechnol 2011, 91, 113–122. [DOI] [PubMed] [Google Scholar]
- [25].Aubry C, Clerici P, Gerbaud C, Micouin L, Pernodet JL, Lautru S, ACS Chem. Biol 2020, 15, 945–951. [DOI] [PubMed] [Google Scholar]
- [26].Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL, Biochemistry 2006, 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].De Rosa M, Issac RP, Houghton G, Tetrahedron Lett. 1995, 36, 9261–9264. [Google Scholar]
- [28].De Rosa M, Issac RP, Marquez M, Orozco M, Luque FJ, Timken MD, J. Chem. Soc., Perkin Trans. 2 1999, 7, 1433–1438. [Google Scholar]
- [29].Pichon-Santander C, Shankar R, Scott AI, Tetrahedron Lett. 1997, 38, 1293–1296. [Google Scholar]
- [30].Shinoda K, Hasegawa T, Sato H, Shinozaki M, Kuramoto H, Takamiya Y, Sato T, Nikaidou N, Watanabe T, Hoshino T, Chem. Commun 2007, 40, 4140–4142. [DOI] [PubMed] [Google Scholar]
- [31].Howard-Jones AR, Walsh CT, J. Am. Chem. Soc 2007, 129, 11016–11017. [DOI] [PubMed] [Google Scholar]
- [32].Ryan KS, Howard-Jones AR, Hamill MJ, Elliott SJ, Walsh CT, Drennan CL, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 15311–15316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cohn WE, J. Biol. Chem 1960, 235, 1488–1498. [PubMed] [Google Scholar]
- [34].Sharpiro R, Chambers RW, J. Am. Chem. Soc 1961, 83, 3920–3921. [Google Scholar]
- [35].Chambers RW, Kurkov V, Sharpiro R, Biochemistry 1963, 2, 1192–1203. [DOI] [PubMed] [Google Scholar]
- [36].Jiang YL, Stivers JT, Tetrahedron Lett. 2003, 44, 85–88. [Google Scholar]
- [37].Huang C, Yang C, Zhang W, Zhang L, De BC, Zhu Y, Jiang X, Fang C, Zhang Q, Yuan C-S, Liu H.-w., Zhang C.-s., Nat. Commun 2018, 9, 2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Capon RJ, Nat. Prod. Rep 2020, 37, 55–79. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.