

Abstract
Acute myeloid leukemia (AML) is characterized by the disruption of myeloid differentiation and accumulation of blast cells in the bone marrow. While AML patients respond favorably to induction chemotherapy, long-term outcomes remain poor due to a high rate of chemoresistance. Advances with targeted therapies, which can be used in combination with conventional chemotherapy, have expanded therapeutic options for patients. However, remission is often short-lived and followed by disease relapse and drug resistance. Therefore, there is a substantial need to improve treatment options by identifying novel molecular and cellular targets that regulate AML chemosensitivity. Membrane scaffolds such as the tetraspanin family of proteins often serve as signaling mediators, translating extracellular signaling cues into intracellular signaling cascades. In this review, we discuss the conventional and targeted treatment strategies for AML and review chemoresistance mechanisms with a focus on the tetraspanin family of membrane scaffold proteins.
Keywords: Acute myeloid leukemia, targeted therapy, chemoresistance, tetraspanins
1. Introduction.
AML Treatment and Outcomes.
AML is a rapid-growing cancer of the myeloid line of blood cells that is characterized by the accumulation of blast cells in the bone marrow resulting in dysfunctional hematopoiesis (Saultz & Garzon, 2016). In 2021, the American Cancer Society estimated AML to be the second most prevalent newly-diagnosed leukemia and the leading cause of leukemia-related mortality. Although the majority of cases occur in adults, AML accounts for approximately 18% of childhood leukemia diagnoses (Nasir, Giri, Nunnery, & Martin, 2017; Saultz & Garzon, 2016). Conventionally, to first reduce tumor burden and achieve remission, patients are treated with induction chemotherapy. The standard therapy regimen of cytarabine for seven consecutive days and daunorubicin on the first three days has remained the first-line treatment for AML for over four decades (Table 1). This treatment strategy has been greatly successful in disease remission, however the long-term overall survival (OS) and event-free survival for children/adolescents after induction therapy is only about 55–65% and the OS in adults is even worse, with only 20–40% of the patients gaining disease-free survival of >5 years with chemotherapy-only treatment (Nasir et al., 2017; Saultz & Garzon, 2016). The variability in patient prognosis and outcomes is largely due to the highly heterogenous nature of AML. Moreover, drug resistance is a major contributor to treatment failure in AML. Therefore, targeted therapies are being developed for AML treatment and usually given in combination with chemotherapeutics. If patients present the genetic alteration for which targeted therapies are approved, their disease prognosis is significantly improved. Some of the most common approved targeted therapies for AML include targeting CD33, aberrant Fms-Related Receptor Tyrosine Kinase 3 (FLT3) activation and isocitrate dehydrogenase-1 (IDH-1) or B-cell lymphoma 2 (BCL-2) activities (Table 1).
Table 1:
FDA approved clinical therapies for the treatment of newly diagnosed and relapsed/refractory AML. (https://www.cancer.gov/aboutcancer/treatment/drugs/leukemia#3)
| FDA Approved Treatment for AML | |||
|---|---|---|---|
| Drug(s) | Treatment Type | Newly Diagnosed Indication | Relapse or Refractory Indication |
| Cytarabine / Daunorubicin (7+3) (1973) | Cytotoxic Chemotherapy | All adult and pediatric patients | All adult and pediatric patients |
| Bone Marrow Transplantation (1977) | Allogeneic or autologous stem cell transplantation | All patients under the age of 65 years, if donor available | All patients under the age of 65 years, if donor available |
| CPX-351 (2017) | Liposomal cytarabine and daunorubicin fixed combination (5:1 molar ratio) | Newly diagnosed therapy-related AML or AML with myelodysplasia-related changes | |
| Gemtuzumab ozogamycin (2017) | Anti-CD33 antibody–drug conjugate | Adult patients with CD33-positive AML | Patients ≥ 2 years of age with refractory CD33-positive AML |
| Midostaurin (2017) | Multikinase FLT3 inhibitor | Patients with FLT3-mutated AML, in combination with standard induction chemotherapy followed by cytarabine consolidation | |
| Enasidenib (2017) | IDH2 inhibitor | IDH2- mutated newly diagnosed AML | IDH2- mutated refractory AML |
| Glasdegib (2018) | Hedgehog pathway inhibitor | Newly diagnosed AML patients ≥75 years in combination with low-dose cytarabine | |
| Venetoclax (2018) | BCL-2 inhibitor | Newly diagnosed AML patients ≥75 years in combination with azacitidine or decitabine, or low-dose cytarabine | |
| Gilteritinib (2018) | FLT3 inhibitor | Patients with relapsed or refractory FLT3-mutated AML | |
| Ivosidenib (2019) | IDH1 inhibitor | Patients ≥ 75 years old or ineligible to receive high-dose chemotherapy with IDH1- mutated AML | IDH1- mutated refractory AML |
| CC-486 (2020) | Hypomethylating agent (Oral azacitidine) | Patients who previously responded to induction chemotherapy however are unfit for high-dose chemotherapy | |
2. Targeted Therapies.
2.1. CD33 Immunotherapy.
AML has generally not been considered a prime target for immunotherapies due to reduced mutational burden compared to other cancers. However, in recent years immunotherapies for the treatment of de novo and refractory AML have been investigated, with many in clinical trials. Initial studies with unmodified monoclonal antibodies targeting CD33 displayed minimal clinical activity for the treatment of AML as single-therapy agents or in combination with chemotherapy (Sekeres et al., 2013). In contrast, a humanized anti-CD33 antibody conjugated to calicheamicin, Gemtuzumab Ozogamicin (GO), was approved for treatment of AML, although was voluntarily withdrawn in 2010 due to lack of clinical benefit. Subsequent studies altering the dosing regimen led to reapproval of GO in 2017 for newly diagnosed and relapsed AML in adults and pediatric patients (NCT00927498; NCT00091234; MyloFrance-1) (Fostvedt, Hibma, Masters, Vandendries, & Ruiz-Garcia, 2019). To reduce toxicity and side-effects like cytokine storms, GO is only recommended for use in lower or fractionated doses in combination with chemotherapy. Additionally, anti-CD33 CAR T cells have also demonstrated significant preclinical efficacy against primary AML blasts both in vitro and in humanized animal models (Kenderian et al., 2015). Unfortunately, these promising results were tempered by toxicity against nonleukemic myeloid cells and potentially HSCs, however, numerous clinical trials using anti-CD33 CAR T cells are currently underway and should be reporting results in the near future.
2.2. FLT3-Internal Tandem Duplication (ITD) Inhibitors.
The frequency and prognostic impact of FLT3 mutations have been well established in AML, which has resulted in the development of first-generation tyrosine kinase inhibitors midostaurin and sorafenib. In 2017, the Food & Drug Administration (FDA) approved midostaurin for the treatment of newly diagnosed FLT3-mutated AML in adults (NCT00651261) (Stone et al., 2017). Sorafenib also displayed strong initial response in clinical trials, however the majority of patients relapsed within 72 days of remission. This was mainly due to the emergence of D835Y and D835 resistant mutations within the FLT3 tyrosine kinase domain (Borthakur et al., 2011). Second-generation inhibitors, quizartinib and crenolanib, were created to specifically target the FLT3 kinase domain instead of a universal tyrosine kinase inhibition. These new drugs have increased selectivity and significantly reduced toxicity and off-target effects. Interestingly, crenolanib displayed ‘pan-kinase’ inhibition abilities against secondary tyrosine kinase domain mutations and was investigated in numerous clinical trials but clinical resistance remains a challenge (Smith et al., 2014). Based on the results of the ADMIRAL trial, gilteritinib, a small molecule dual inhibitor of FLT3/AXL, was recently approved by the FDA for relapse/refractory (R/R) FLT3-mutated AML, demonstrating an increased response rate and longer OS with the use of gilteritinib in comparison to salvage chemotherapy for R/R AML (NCT02421939) (Perl et al., 2019).
2.3. IDH1/IDH2 Small Molecule Inhibitors.
Approximately 20% of AML patients display gain of function mutations in IDH-1 and IDH-2 enzymes (Saultz & Garzon, 2016). Thus, targeting these enzymes for the treatment of AML has been of great interest. In 2013, Wang et al. published results from a study investigating AGI-6780, a small molecule inhibitor of the R140Q mutant IDH-2 enzyme. Using an ex vivo model of primary human AML cells, the authors showed that treatment with AGI-6780 could induce differentiation in leukemic cells, which was previously blocked (Wang et al., 2013). In 2017, after successful clinical trial (NCT01915498), the FDA granted approval of AG-221 or enasidenib as the first oral target therapy for adults with R/R AML and IDH2 mutation (Yen et al., 2017). Additionally, small molecule inhibitor, AG-120 II (Ivosidenib), demonstrated successful hematological response with median OS of 12.6 months in newly diagnosed IDH1-mutated AML, which led to the approval of Ivosidenib for newly diagnosed IDH1- mutated AML patients ≥ 75 years old and adults with R/R AML(NCT02074839) (DiNardo et al., 2018).
2.4. BCL-2 Inhibitors.
Over 30 years ago, BCL-2 was discovered at the breakpoint of the t(14; 18) in follicular lymphoma. In one of the first clinical trials targeting BCL-2 in AML, which used antisense oligonucleotide G3139, the reduction in BCL-2 expression was inconsistent and short-lived (Marcucci et al., 2003). Alternatively, preclinical studies with ABT-737 demonstrated the importance of BCL-2 inhibition in AML and efficacy of ABT-737 was established. Mechanistically, ABT-737 neutralizes BCL-2, which ultimately results in mitochondrial dysfunction, release of cytochrome c and activation of caspases that initiate cell death (Beurlet et al., 2013). However, due to the low solubility and oral bioavailability of ABT-737, further studies targeting BCL-2 focused on using ABT-199, which could induce rapid apoptosis of multiple AML cell lines with only nanomolar concentrations and significantly reduced leukemia progression in murine AML xenograft models. Of particular interest was the ability of ABT-199 to induce apoptosis in both chemotherapy-sensitive and chemorefractory primary AML cells in vitro (Pan et al., 2014). Ultimately, ABT-199, now known as venetoclax, was developed by AbbVie and approved for the treatment of R/R AML (NCT02993523; NCT03069352) and is being further investigated in combination with hypomethylating agents or low-dose cytarabine for front-line treatment of AML (Jonas & Pollyea, 2019). However, the majority of patients treated with venetoclax develop resistance, thus various combination treatment approaches are currently being investigated.
3. AML Therapy Resistance Mechanisms
Some of the most recognized mechanisms of drug resistance in AML include genetic alterations, drug resistance-related proteins, miRNAs expression, and aberrant activation of drug resistance-related signaling pathways. Gene mutations in the RAS protein resulting in its constitutive activation cause increased proliferation, metastasis, and drug resistance of AML cells (Stephen, Esposito, Bagni, & McCormick, 2014). FLT3 kinase mutations found in one-third of patients with AML can also support AML cell survival upon treatment by chemotherapeutics (Cloos et al., 2006). In addition, new mutations in FLT3 found in relapsed patients can be a consequence of epigenetics-modifying gene mutations present at initial diagnosis and these mechanisms of resistance require further investigation (Wakita et al., 2013). Additionally, signaling pathways such as PI3K/AKT, critical regulators of cellular processes like cell survival and proliferation, are also potential drug resistance mechanisms. For example, aberrant activation of the PI3K/AKT pathway was found in HL-60 cells treated with arsenic trioxide, which led to drug resistance through the JNK-p38 MAPK pathway (Roszak, Smok-Pieniazek, & Stepnik, 2017). Moreover, a broad dependence of AML cells on autocrine activation of mesenchymal-epithelial transition factor (MET) has been demonstrated, where treatment with MET kinase inhibitors resulted in resistance via the upregulation of hepatocyte growth factor signaling (Kentsis et al., 2012). Thus, drug resistance mechanisms in AML are complex and dependent on various signaling pathways.
4. AML Drug Resistance and Tetraspanins
Targeting cellular signaling modulators may provide a way of suppressing multiple potential mechanisms of drug resistance in AML. The tetraspanin family of scaffolding proteins (also called transmembrane 4 superfamily (TM4SF) proteins) are membrane-spanning proteins that are integral in the organization of various membrane proteins. Tetraspanin assembly into membrane microdomains, which function to organize plasma membrane-associated proteins, regulates signal transduction and modulates various cellular process including adhesion, migration, and the immune response (Hemler, 2005). Increasing evidence describing potential roles of tetraspanins in cancer growth, progression, metastasis, and immune evasion, has led to this family of proteins being investigated as potential therapeutic targets. With respect to AML initiation, progression and prognosis, several tetraspanins family members have been described including CD9, CD37, CD63, CD81 CD82 and Tspan3. However, at this point a limited number of tetraspanins have been described to modulate AML chemoresistance mechanisms specifically (Figure 1) For example, CD81 expression was shown to correlate with poor survival outcomes in AML, specifically in patients that express a high proportion of CD81-positive blasts (Boyer et al., 2016). Additionally, CD81 positive AML blasts were found to be 30 to 50% more resistant to conventional chemotherapeutic treatment (Gonzales et al., 2017). CD9 has also been shown to regulate leukemia development and chemotherapeutic resistance. CD9 positive AML blasts were shown to exhibit increased resistance to Ara-C chemotherapy and higher migration potential than CD9 negative cells. Mechanistically, CD9 expression was found to be regulated by alpha-2-macroglobulin (A2M) mediated signaling through the modulation of transcription factor EGR1 (Liu et al., 2021). Similarly, Tspan3 was identified to be upregulated in both Adriamycin resistant AML samples and cell lines. Here, the IncRNA potassium voltage-gated channel subfamily Q member 1 overlapping transcript 1 (KCNQ1OT1) was shown to modulate Tspan3 expression through its target, miR-193a-3p (Sun, Sun, Chen, & Xu, 2020).
Fig. 1.
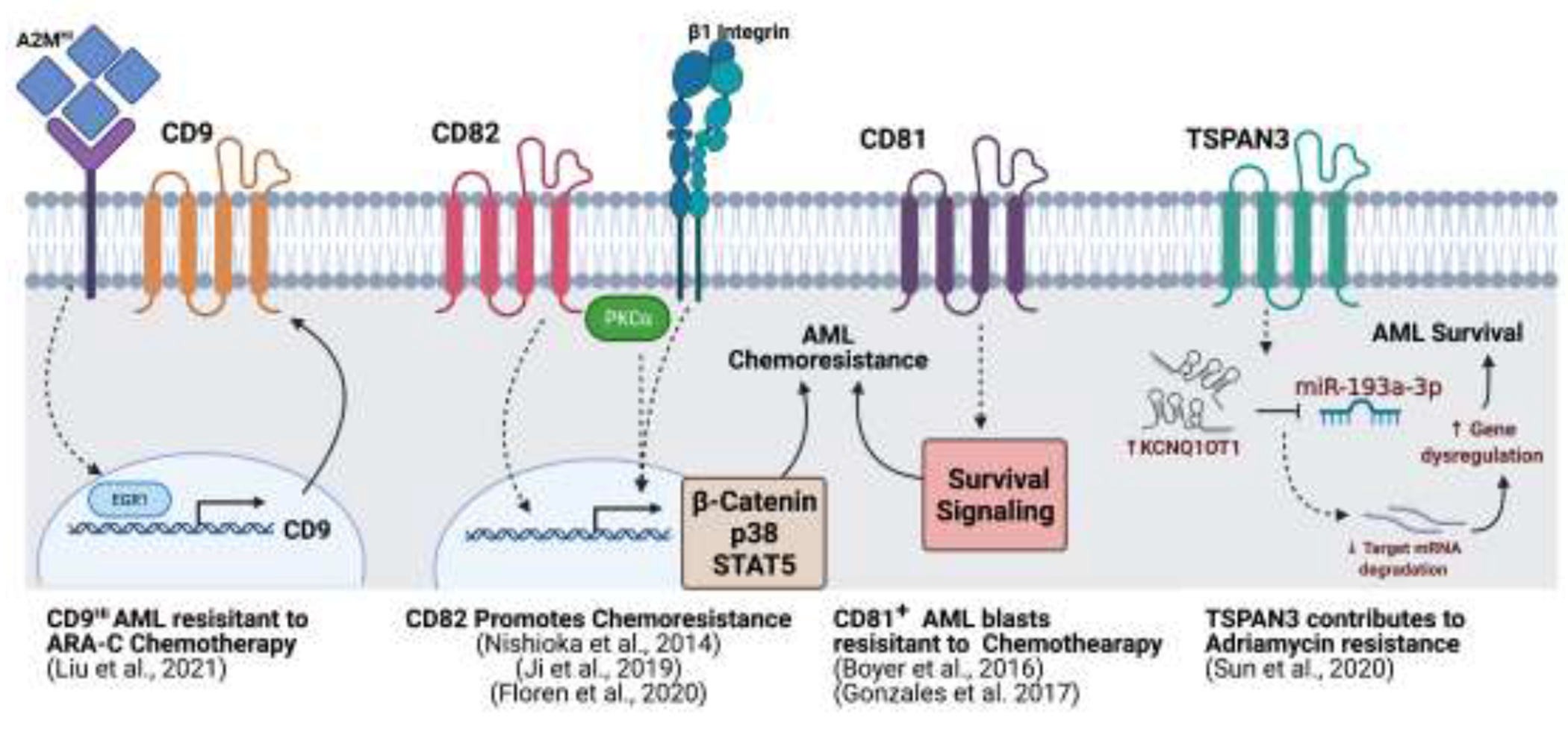
Tetraspanins regulate AML chemoresistance and survival signaling. Schematic of the AML membrane depicting different tetraspanins (CD9, CD82, CD81, TSPAN3) along with proposed signaling mechanisms involved in AML chemoresistance and survival. Created using BioRender.
More recently, our group utilized an RNA-seq approach to examine transcriptional alterations in tetraspanin family members following chemotherapy in AML and identified increased expression of the tetraspanin CD82 (Floren et al., 2020). Similarly, a previous study reported tetraspanin CD82 upregulation in chemotherapy-resistant CD34+/CD38− AML cells where CD82 positively regulated STAT5 signaling (Nishioka et al., 2014). Our analysis of pediatric and adult AML patient survival outcomes found that CD82 expression correlates with reduced OS and increased chemoresistance in vitro. Consistent with our findings, Ji et al. recently reported that CD82 regulates the proliferation and chemotherapy resistance of pediatric AML, suggesting that CD82-mediated survival is supported via the activation of the Wnt/β-catenin signaling pathway (Ji et al., 2019). In contrast, our study demonstrated the requirement of PKCα activation in CD82-mediated chemoresistance. PKCα signaling has been implicated in the regulation of Wnt/β-catenin, therefore, further studies may show potential crosstalk between these signaling pathways in promoting CD82-mediated AML survival. Collectively, our work proposes that patient stratification based on CD82 expression could potentially improve response to therapeutics targeting PKC signaling in AML.
5. Therapeutic Opportunities and Future Directions
Despite decades of clinical trials focused on improving short- and long-term responses to AML, outcomes have remained poor due to the development of chemotherapy resistance. Thus, there is an urgent need to understand the molecular mechanisms driving drug resistance in order to prevent relapse. As more resistance mechanisms are discovered, therapeutics targeting these pathways need to be further investigated in order to improve long-term patient outcomes in AML. Given the broad network of adhesion and signaling proteins that interact with tetraspanins, these membrane scaffold proteins represent a novel target to directly influence a range of biological signaling rather than targeting specific surface proteins. In fact, tetraspanins are currently being used in clinical trials for the treatment of other hematological malignancies (Beckwith, Byrd, & Muthusamy, 2015). Therefore, the ability to disrupt the membrane scaffold, which can drive multiple survival signaling pathways, may represent a novel approach to treat chemoresistant AML. In summary, relapse disease remains a significant cause of mortality for patients. Therefore, a better understanding of chemoresistant mechanisms are likely to lead to the development of novel therapeutic approaches that can offer longer-term cures for AML patients.
Key Facts.
Acute Myeloid Leukemia (AML) is a hematologic cancer caused by aberrant proliferation of the myeloid lineage of blood cells.
AML is a heterogenous disease encompassing clinically and genetically different subtypes.
AML remains the leading cause of leukemia-related mortality.
Majority of patients with AML relapse within 3 years of diagnosis.
AML has a high rate of treatment failure due to increased prevalence of therapy resistance leading to disease relapse.
Tetraspanin scaffold proteins regulate AML chemosensitivity.
Funding
This work was supported by fellowship F31CA232480 to M.F. and NHLBI investigator grant (RO1HL122483 to J.M.G.), and an American Cancer Society Research Scholar Grant (130675 to J.M.G). This work was partially supported by the UNM Spatiotemporal Modeling Center (P50GM085273) and the UNM Comprehensive Cancer Center Support Grant (P30CA118100).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
The authors declare no competing financial interests in relation to the work described.
References.
- Beckwith KA, Byrd JC, & Muthusamy N (1015). Tetraspanins as therapeutic targets in hematological malignancy: a concise review. Front Physiol, 6, 91. doi: 10.3389/fphys.2015.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurlet S, Omidvar N, Gorombei P, Krief P, Le Pogam C, Setterblad N, … Padua RA (1013). BCL-2 inhibition with ABT-737 prolongs survival in an NRAS/BCL-2 mouse model of AML by targeting primitive LSK and progenitor cells. Blood, 122(16), 2864–2876. doi: 10.1182/blood-2012-07-445635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, … Cortes JE (2011). Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica, 96(1), 62–68. doi: 10.3324/haematol.2010.030452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer T, Guihard S, Roumier C, Peyrouze P, Gonzales F, Berthon C, … Cheok M (2016). Tetraspanin CD81 is an adverse prognostic marker in acute myeloid leukemia. Oncotarget, 7(38), 62377–62385. doi: 10.18632/oncotarget.11481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloos J, Goemans BF, Hess CJ, van Oostveen JW, Waisfisz Q, Corthals S, … Kaspers GJ (2006). Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia, 20(7), 1217–1220. doi: 10.1038/sj.leu.2404246 [DOI] [PubMed] [Google Scholar]
- DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, … Kantarjian HM (2018). Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med, 378(25), 2386–2398. doi: 10.1056/NEJMoa1716984 [DOI] [PubMed] [Google Scholar]
- Floren M, Restrepo Cruz S, Termini CM, Marjon KD, Lidke KA, & Gillette JM (2020). Tetraspanin CD82 drives acute myeloid leukemia chemoresistance by modulating protein kinase C alpha and beta1 integrin activation. Oncogene, 39(19), 3910–3925. doi: 10.1038/s41388-020-1261-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fostvedt LK, Hibma JE, Masters JC, Vandendries E, & Ruiz-Garcia A (2019). Pharmacokinetic/Pharmacodynamic Modeling to Support the Re-approval of Gemtuzumab Ozogamicin. Clin Pharmacol Ther, 106(5), 1006–1017. doi: 10.1002/cpt.1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales F, Boyer T, Peyrouze P, Guihard S, Roumier C, Berthon C, … Cheok M (2017). Targeting Aberrant Expression of CD81 Impacts Cell Adhesion and Migration, Drug Resistance and Prognosis of Acute Myeloid Leukemia. Blood, 130 Supplement 1), 2675–2675. doi: 10.1182/blood.V130.Suppl_1.2675.2675 [DOI] [Google Scholar]
- Hemler ME (2005). Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol, 6(10), 801–811. doi: 10.1038/nrm1736 [DOI] [PubMed] [Google Scholar]
- Ji H, Chen L, Xing Y, Li S, Dai J, Zhao P, & Wang Y (2019). CD82 supports survival of childhood acute myeloid leukemia cells via activation of Wnt/beta-catenin signaling pathway. Pediatr Res, 85(7), 1024–1031. doi: 10.1038/s41390-019-0370-3 [DOI] [PubMed] [Google Scholar]
- Jonas BA, & Pollyea DA (2019). How we use venetoclax with hypomethylating agents for the treatment of newly diagnosed patients with acute myeloid leukemia. Leukemia, 33(12), 2795–2804. doi: 10.1038/s41375-019-0612-8 [DOI] [PubMed] [Google Scholar]
- Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, … Gill S (2015). CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia, 29(8), 1637–1647. doi: 10.1038/leu.2015.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentsis A, Reed C, Rice KL, Sanda T, Rodig SJ, Tholouli E, … Look AT (2012). Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat Med, 18(7), 1118–1122. doi: 10.1038/nm.2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang G, Zhang J, Chen X, Xu H, Heng G, … Qian C (2021). CD9, a potential leukemia stem cell marker, regulates drug resistance and leukemia development in acute myeloid leukemia. Stem Cell Res Ther, 12(1), 86. doi: 10.1186/s13287-021-02155-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcucci G, Byrd JC, Dai G, Klisovic MI, Kourlas PJ, Young DC, … Caligiuri MA (2003). Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood, 101(2), 425–432. doi: 10.1182/blood-2002-06-1899 [DOI] [PubMed] [Google Scholar]
- Nasir SS, Giri S, Nunnery S, & Martin MG (2017). Outcome of Adolescents and Young Adults Compared With Pediatric Patients With Acute Myeloid and Promyelocytic Leukemia. Clin Lymphoma Myeloma Leuk, 17(2), 126–132 e121. doi: 10.1016/j.clml.2016.09.011 [DOI] [PubMed] [Google Scholar]
- Nishioka C, Ikezoe T, Yang J, Nobumoto A, Kataoka S, Tsuda M, … Yokoyama A (2014). CD82 regulates STAT5/IL-10 and supports survival of acute myelogenous leukemia cells. Int J Cancer, 134(1), 55–64. doi: 10.1002/ijc.28348 [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, … Letai AG (2014). Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov, 4(3), 362–375. doi: 10.1158/2159-8290.CD-13-0609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, … Levis MJ (2019). Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N Engl J Med, 381(18), 1728–1740. doi: 10.1056/NEJMoa1902688 [DOI] [PubMed] [Google Scholar]
- Roszak J, Smok-Pieniazek A, & Stepnik M (2017). Transcriptomic analysis of the PI3K/Akt signaling pathway reveals the dual role of the c-Jun oncogene in cytotoxicity and the development of resistance in HL-60 leukemia cells in response to arsenic trioxide. Adv Clin Exp Med, 26(9), 1335–1342. doi: 10.17219/acem/65475 [DOI] [PubMed] [Google Scholar]
- Saultz JN, & Garzon R (2016). Acute Myeloid Leukemia: A Concise Review. J Clin Med, 5(3). doi: 10.3390/jcm5030033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekeres MA, Lancet JE, Wood BL, Grove LE, Sandalic L, Sievers EL, & Jurcic JG (2013). Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica, 98(1), 119–128. doi: 10.3324/haematol.2012.066613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Lasater EA, Lin KC, Wang Q, McCreery MQ, Stewart WK, … Shah NP (2014). Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A, 111(14), 5319–5324. doi: 10.1073/pnas.1320661111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, & McCormick F (2014). Dragging ras back in the ring. Cancer Cell, 25(3), 272–281. doi: 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, … Dohner H (2017). Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med, 377(5), 454–464. doi: 10.1056/NEJMoa1614359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Sun Y, Chen Q, & Xu Z (2020). LncRNA KCNQ1OT1 contributes to the progression and chemoresistance in acute myeloid leukemia by modulating Tspan3 through suppressing miR-193a-3p. Life Sci, 241, 117161. doi: 10.1016/j.lfs.1019.117161 [DOI] [PubMed] [Google Scholar]
- Wakita S, Yamaguchi H, Omori I, Terada K, Ueda T, Manabe E, … Inokuchi K (2013). Mutations of the epigenetics-modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia, 27(5), 1044–1052. doi: 10.1038/leu.2012.317 [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, … Yen KE (2013). Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science, 3406132), 622–626. doi: 10.1126/science.1234769 [DOI] [PubMed] [Google Scholar]
- Yen K, Travins J, Wang F, David MD, Artin E, Straley K, … Su SM (2017). AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Canter Distov, 7(5), 478–493. doi: 10.1158/2159-8290.CD-16-1034 [DOI] [PubMed] [Google Scholar]