

Abstract
Many believe that engaging in novel and mentally challenging activities promotes brain health and prevents Alzheimer’s disease in later life. However, mental stimulation may also have risks as well as benefits. As neurons release neurotransmitters, they often also release amyloid peptides and tau proteins into the extracellular space. These by-products of neural activity can aggregate into the tau tangle and amyloid plaque signatures of Alzheimer’s disease. Over time, more active brain regions accumulate more pathology. Thus, increasing brain activity can have a cost. But the neuromodulator noradrenaline, released during novel and mentally stimulating events, may have some protective effects—as well as some negative effects. Via its inhibitory and excitatory effects on neurons and microglia, noradrenaline sometimes prevents and sometimes accelerates the production and accumulation of amyloid-β and tau in various brain regions. Both α2A- and β-adrenergic receptors influence amyloid-β production and tau hyperphosphorylation. Adrenergic activity also influences clearance of amyloid-β and tau. Furthermore, some findings suggest that Alzheimer’s disease increases noradrenergic activity, at least in its early phases. Because older brains clear the by-products of synaptic activity less effectively, increased synaptic activity in the older brain risks accelerating the accumulation of Alzheimer’s pathology more than it does in the younger brain.
Keywords: Locus coeruleus, aging, Alzheimer’s disease, glutamatergic activity
1. Introduction
1.1. Associations between the locus coeruleus and Alzheimer’s disease
The locus coeruleus (LC), a small brain nucleus in the brainstem, integrates arousal-related signals and releases noradrenaline throughout much of the brain. Recent findings indicate that the integrity of the LC is associated with cognition in late life [for reviews see [1, 2, 3]. For instance, greater neuronal density in the LC at the time of death is associated with less cognitive decline in the years before death even when controlling for neuronal density in the dorsal raphe nucleus, substantia nigra and ventral tegmental area [4]. Post-mortem analyses show fewer LC cells in patients with Alzheimer’s disease compared with controls [5-8], In postmortem human brains, the LC appears to be the earliest site of Alzheimer’s related pathology [9, 10].
In vivo estimates of LC integrity have been challenging due to its small size and lack of clear structural boundaries on typical magnetic resonance images. However, due to the LC’s different magnetic properties than surrounding tissue, specialized magnetic resonance imaging (MRI) sequences show high contrast at the site of the LC compared with surrounding brainstem regions [11]. Consistent with postmortem findings of correlations between LC structure and pre-mortem cognition or disease status, measures of LC contrast are lower in patients with mild cognitive impairment and Alzheimer’s disease than in cognitively healthy adults [12-16] and among cognitively healthy adults, higher LC contrast is associated with better memory and cognition [17-20]. Furthermore, recent findings indicate that even standard T1-weighted structural MRI sequences provide some information about LC decline. In the large Alzheimer’s Disease Neuroimaging Initiative (ADNI) sample, voxel-based morphometry from standard structural scans revealed that, within the brainstem, lower volume estimates within voxels overlapping the LC predicted future diagnoses of Alzheimer’s disease [21] and were associated with lower cognition [22].
These findings indicate that a person’s LC integrity in late life relates to their likelihood of avoiding cognitive decline and Alzheimer’s disease. What is the causal relationship? Does LC degeneration accelerate cognitive deficits and neuropathology in Alzheimer’s disease, as some have argued [23]? Indeed, some have made the case that the LC is the brain substrate of cognitive reserve [3, 24, 25]. Cognitive reserve is a concept evoked to explain the phenomenon that those with more years of education tend to have a delayed onset of the behavioral and cognitive symptoms of Alzheimer’s disease [26, 27]. The LC-as-reserve theory posits that the increased LC activity evoked during cognitively stimulating activities delays onset of the behavioral and cognitive symptoms of Alzheimer’s disease in those who have had more education [3].
This perspective suggests that stimulating noradrenergic activity via arousing, engaging activities would be a helpful strategy throughout life to stave off Alzheimer’s disease [28, 29]. Indeed, cognitive training has been one of the most popular clinical trial interventions, with hundreds of randomized controlled trials published [30-36]. Researchers have also been interested in pharmacological interventions targeting noradrenergic activity. To date, clinical trials have targeted different components of the noradrenergic system, including α2-adrenergic agonists [37-39], β-adrenergic agonists and antagonists [40, 41], an α1-adrenergic antagonist [42], and a noradrenaline transport inhibitor [43]. Yet evidence that either cognitive training or noradrenergic drugs can do anything against the progression of Alzheimer’s disease in older adults is lacking. Most cognitive training studies do not include measures of Alzheimer’s pathology and most registered trials of noradrenergic drugs have not reported results, had samples too small to be interpretable, or had null findings [37].
But as reviewed in this paper, the lack of success in establishing a causal link between interventions stimulating the noradrenergic system and the progression of Alzheimer’s disease may also be explained by findings in animal models that noradrenergic activity can either speed up or slow down Alzheimer’s-related pathophysiological processes, depending on various factors, including the type of adrenergic receptors activated.
To date, it has been assumed that cognitive training should benefit older adults’ brain structure and function, or at worst have no significant impact on the brain [44-46]. But if noradrenergic activity accelerates Alzheimer’s-related pathophysiological processes in the older brain, cognitive training in older adults may have costs beyond the time and effort involved [28]. Better understanding of how behavioral interventions interact with the noradrenergic system to slow or accelerate accumulation of Alzheimer’s-related pathology in the brain may improve future intervention attempts. This review delves into the literature to address the question of when engaging noradrenergic activity via cognitively stimulating activity helps avoid Alzheimer’s pathology and when it may instead accelerate its accumulation.
1.2. The noradrenergic system’s receptor types
To understand the mechanisms of noradrenaline action, it is important to distinguish among the different noradrenergic receptor types. There are three noradrenergic receptor classes (α2, α1, and β), with α2-adrenergic receptors having the highest affinity for noradrenaline and therefore a low threshold for activation. In contrast, β-adrenergic receptors have the lowest affinity for noradrenaline and therefore a high threshold for activation [47-49].
All three types of receptors are G protein-coupled receptors that translate signals from extracellular ligands into intracellular responses by activating specific heterotrimeric G proteins [50, 51]. Each class of noradrenergic receptors typically depends on a different family of heterotrimeric G proteins; Gi/0, Gq/11, and Gs for α2, α1, and β respectively [52]. These G protein families in turn stimulate different sets of intracellular responses [50]. However, there are exceptions to these canonical couplings of receptor types to G protein families. For example, there is evidence of β2 receptors switching from Gs to Gi [53] or existing in a Gicoupled state [54].
β-adrenergic receptors are generally excitatory, stimulating the enzyme adenylate cyclase which synthesizes cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP) [55]. cAMP is a second messenger that relays information into the cell and triggers chains of biochemical events, often including activation of protein-kinase A (PKA) [56] and enhanced long-term potentiation [57, 58]. While responsible for the fight-or-flight response conveyed by catecholamines, cAMP also mediates the effects of many other neurotransmitters and hormones [59].
In contrast, α2-adrenergic receptors generally inhibit neurotransmitter release and decrease neuronal excitability [60]. Positively charged calcium ions are among the ions that can flow into a cell as it undergoes depolarization, increasing neuron excitation and neurotransmitter release [61]. The majority of α2-adrenergic receptors in the human brain are of the α2A subtype [62], which decrease calcium currents [60, 63]. Gi/0 protein activity also inhibits adenylyl cyclase, leading α2-adrenergic receptors to have opposing effects on cAMP as compared with β-adrenergic receptors [50]. α2A receptors are often found at presynaptic sites, serving as autoreceptors on noradrenergic neurons that provide feedback to diminish further noradrenaline release when activated by extracellular noradrenaline [60]. They also serve as heteroreceptors on non-adrenergic neurons and cells. In fact, in mice, most effects of α2 agonist drugs, including on pain, body temperature, sedation and lowering the required dose of anesthetics are mediated by α2 receptors on non-adrenergic neurons and cells [64]. In contrast with other brain regions, in the human and monkey prefrontal cortex α2A receptors appear to be mainly postsynaptic [65, 66].
Via Gq-protein-mediated activation of phospholipase Cβ, α1 adrenergic receptors can trigger release of calcium from intracellular stores and potentiate cAMP responses [47]. Thus, unlike α2-adrenergic receptors, they increase calcium excitability, and like β-adrenergic receptors, they mobilize cAMP. Thus, at a cellular level, α1-adrenergic receptors typically have an excitatory effect. However, α1 and β-adrenergic receptors appear to have opposing effects on long-term plasticity of synapses, with α1-adrenergic receptors facilitating long-term depression while β-adrenergic receptors facilitate long-term potentiation [47].
1.3. Benefits of environmental enrichment depend on noradrenergic activity
Education and other cognitively challenging activities involve novelty, working memory and motivated behavior which each tend to activate the LC [67]. Consistent with the link between cognitive stimulation and noradrenergic activity, research with animals shows that being housed in an enriched environment increases noradrenaline levels while not changing serotonin or dopamine levels in the olfactory bulb [68] or in parieto-tempero-occipital cortex, cerebellum or pons/medulla oblongata [69]. Presumably the enriched environment increases various aspects of arousal and thereby stimulates the LC, which in turn leads to release of noradrenaline elsewhere in the brain, as well as to release of dopamine in hippocampus [70].
Noradrenaline release in brain regions involved in cognitive processing during novel arousing situations appears to be critical for the beneficial effects of environmental enrichment, as indicated by findings that rearing rodents with diminished forebrain noradrenaline in a complex environment did not improve their maze learning, unlike their controls with intact noradrenaline neurons [71, 72].
The ability of environmental enrichment to increase neurogenesis also depends on noradrenaline. In mice exposed to a novel odor each day for 47 days, neurogenesis increased in the granular cell layer of the olfactory bulb compared with mice exposed to a stable mix of those same odors for 47 days [68]. However, the enhanced neurogenesis in the enriched odor environment was eliminated if the mice received a β-adrenergic antagonist during the 47-day period. These findings that novelty stimulates neurogenesis via β-adrenergic activity in the olfactory bulb are consistent with findings that hippocampal dentate gyrus neurogenesis depends on noradrenaline [73, 74] and that β-adrenergic receptor activity promotes dentate gyrus cell proliferation [75, 76] whereas α2-adrenergic receptor activity diminishes cell proliferation in the hippocampus [77]. As reviewed previously in Section 1.2, relatively high concentrations of noradrenaline are necessary to activate β-adrenergic receptors whereas low concentrations can activate α2-adrenergic receptors. Thus, high levels of noradrenergic activity in the hippocampus can stimulate neurogenesis. The resulting new neurons may then enhance the ability to retain new information. Thus, β-adrenergic enhanced neurogenesis is one mechanism that can explain why environmental enrichment leads to better memory.
However, increased neurogenesis is likely only one of multiple noradrenergic pathways contributing to the positive effects of environmental enrichment. Another relevant factor is that noradrenaline can promote expression of plasticity-related genes. Long-lasting adaptive changes in the brain require changes in gene transcription in cell nuclei [78]. In the brain, multiple signaling cascades transmit signals from synapses to cell nuclei. Many of these are modulated by adrenergic activity.
In particular, β-adrenergic receptors promote neuronal and synaptic plasticity during environmental enrichment [47, 79]. By initiating the cAMP/PKA/CREB pathway which promotes activity-dependent changes in gene expression [59, 80-82], β-adrenergic receptors modulate cellular processes involved in establishing and maintaining memories over the long term. Destroying locus coeruleus noradrenaline neurons in adult rats reduced expression of plasticity-related genes (e.g., CREB, Arc and BDNF) across the cerebral cortex by about 50% or to the low levels seen during sleep [83]. In particular, noradrenaline appears to be critical for CREB-induced transcription in cortical astrocytes [84].
In humans, neuroimaging demonstrates that daily immersion in cognitively challenging activities such as studying a language to become a foreign language interpreter [85], studying London street routes to become a licensed taxi driver [86], practicing a video game daily [87], or learning a new motor skill [88] leads to structural brain changes. While links between these structural changes and noradrenaline have yet to be established for humans, animal studies suggest that noradrenergic activity plays a key role in this plasticity.
1.4. Local levels of glutamate modulate noradrenergic activity
During moments of high arousal, people attend to whatever matters or stands out most right then while neglecting processing of other stimuli [89]. This increased selectivity under arousal has been linked with noradrenergic processes, in particular β-adrenergic receptors [90, 91]. But how can the LC differentially target the neural processing of stimuli depending on their current priority?
To guide attention towards high priority information, the LC must interact with cortical networks representing stimuli. Stimuli are represented in the brain in neural networks defined by their synaptic connections [92-95]. Neurons representing sensory stimuli tend to form synapses with other neurons representing the same type of stimuli [e.g., 96]. Thus, to selectively enhance the processing of a particular stimulus, the LC needs to interact differently with neural networks that currently have priority than with those that do not. To make this possible, a local signal of priority that can influence noradrenaline release at that local site seems necessary.
As the brain’s most common excitatory neurotransmitter [97, 98], glutamate is well positioned to provide a local signal of priority. Glutamate is released from synaptic vesicles in nerve terminals to the extracellular space via exocytosis [as well as via non-exocytosis mechanisms 98]. However extracellular glutamate concentrations are kept quite low (in the low micromolar range compared with the intracellular concentrations which are in the millimolar range) by glutamate transporters that import it into astrocytes and neurons [97]. While a primary role of glutamate release at synaptic terminals is to excite the post-synaptic neuron, glutamate can also leak out of the synapse [99], where it can activate extrasynaptic N-methyl d-aspartate (NMDA) receptors [100], including those located on nearby axons of LC neurons [Figure 1; 101].
Figure 1.
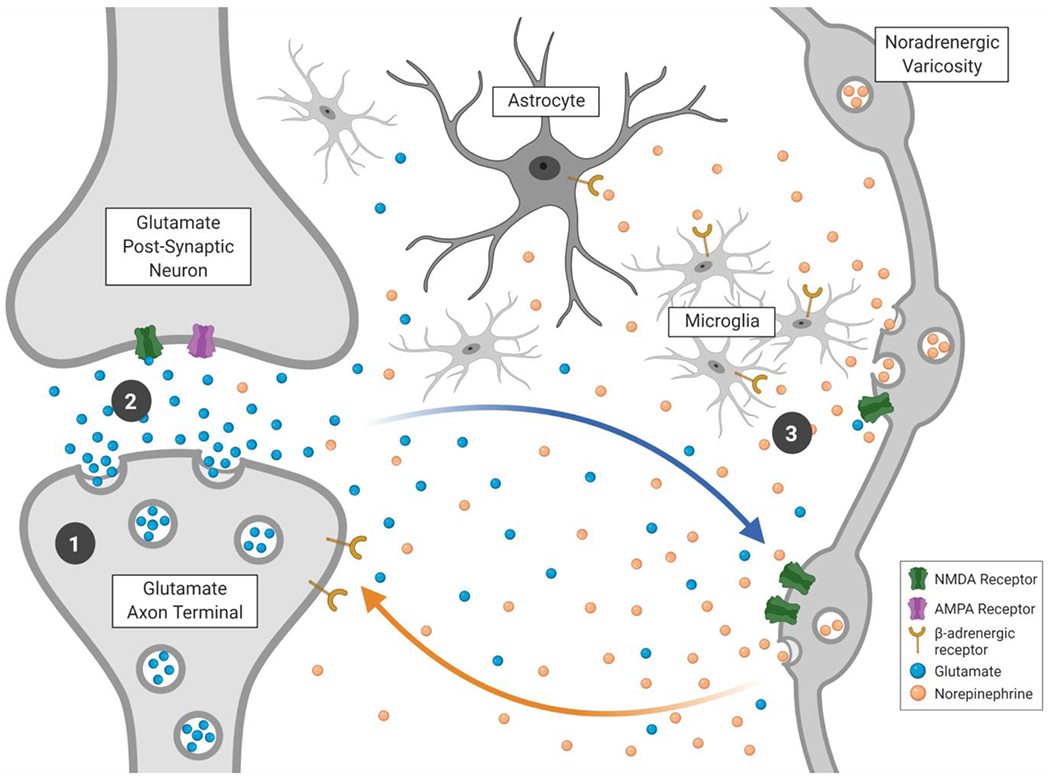
Aspects of noradrenergic hot spots that may contribute to modulating Aβ and tau release and their impact: 1) Increased vesicle trafficking due to synaptic activity is associated with increased release of Aβ and tau; 2) Increased glutamate release is associated with increased tau release; and 3) β-adrenergic microglial activation modulates toxic effects of Aβ. Figure created with BioRender.com.
The ‘glutamate amplifies noradrenergic effects” (GANE) model posits that glutamate provides a signal of where high priority processing is occurring [101]. According to GANE, when the LC is active, glutamate can modulate levels of local noradrenaline release which in turn activate excitatory β-adrenergic receptors and thereby stimulate hot spots of high synaptic activity in local regions where there is currently high glutamatergic activity (see Figure 1). In contrast, in regions without high glutamate levels, LC activity will activate lower threshold α2-adrenergic receptors. The net effect is an increase in ‘neural gain,’ in which excited neurons become even more excited and inhibited neurons even more inhibited [102, 103]. Supporting this model, prior research indicates that glutamate increases noradrenaline release via NMDA and non-NMDA receptors on LC axons, in various rodent brain regions including prefrontal cortex, hippocampus and hypothalamus [e.g., 104, 105-108]. There is also evidence for NMDA-receptor-evoked noradrenaline release in human neocortex [109-111]. As presynaptic β2-adrenergic receptors can stimulate glutamate release [112] and increase excitatory synaptic transmission [113, 114], there is the potential for a self-amplifying feedback loop.
However, an important feature of NMDA receptors is that they conduct currents only when glutamate is bound to the NMDA receptor and the neuron that receptor is on is simultaneously depolarized [115]. Thus, NMDA receptors detect the coincident activity of pre- and postsynaptic neurons. One interesting property of LC neurons is that the timing and factors that influence release of noradrenaline from the soma versus from varicosities or terminal regions differ, such that release from the soma takes longer (with latencies as long as seconds, compared with synaptic transmission in milliseconds) and requires a higher frequency of action potentials [116] and may occur via dense core vesicles that also contain neuropeptides [117]. This slow release at the LC cell body in response to high frequency action potentials activates local α2A autoreceptors, which should inhibit activity in the LC cell body and thereby decrease noradrenaline release in neocortex [118]. Thus, this provides a mechanism to shut down intense LC activity triggered by an arousal-inducing stimulus after a few seconds, and this should in turn limit the duration of glutamate-noradrenaline hot spots elsewhere in the brain.
In the GANE hot spot model, the release of noradrenaline from LC varicosities increases excitatory synaptic activity in nearby glutamatergic neurons that in turn stimulates further noradrenaline release (as illustrated in Figure 1). This noradrenergic amplification of neural gain is supported by various mechanisms at play in and around the hot spot synapses. Astrocytes likely play a key role. One of the main functions of astrocytes is to take up unused synaptically released glutamate to prevent glutamate excitotoxicity [119]. Astrocytes with immunoreactivity for β-adrenergic receptors are often located around glutamatergic synaptic junctions, thus β-adrenergic activity on these astrocytes may modulate uptake of glutamate and other amino acids [120]. β-adrenergic signaling increased astrocytic lactate release during a startle paradigm [121]. Lactate provides additional fuel to neurons when glucose levels might not be sufficient [122]. Thus, noradrenergic signaling in astrocytes provides a mechanism to support momentarily locally high levels of metabolism. Astrocytes also express α1-adrenergic receptors that modulate their Ca2+ signaling [123].
β-adrenergic signaling increases dendrite excitability, which not only promotes the spread of depolarization [124] but may promote spike timing-dependent synaptic plasticity, which is linked with associative learning [125]. α2-adrenergic autoreceptors decrease further release of noradrenaline when they sense low levels of NE, thus suppressing activity in non-hot-spot regions. In some regions of the brain, β-adrenergic receptors may also serve as autoreceptors, increasing release of noradrenaline in the presence of high levels of noradrenaline [126]. This hot-spot glutamate-noradrenaline interaction may be most prominent in sensory regions where it is important to amplify the activity associated with specific perceptual representations under arousal. In contrast, noradrenergic activity may have different effects in cortical regions involved in coordinating broad-scale activity and tracking which perceptual representations currently have the highest priority. In particular, there is evidence that noradrenergic modulatory activity plays out differently in the prefrontal cortex than elsewhere in the brain [127]. In the prefrontal cortex, α2-adrenergic postsynaptic receptors tend to enhance prefrontal function [127, 128], while there are fewer inhibitory presynaptic α2-adrenergic receptors compared with most other brain regions [66]. The opposing effects of phasic noradrenergic activity in sensory vs. control networks was seen in a recent analysis of functional MRI resting-state brain activity [129]. LC phasic bursts were associated with more within- and across-network correlated activity in and between ventral attention network, executive and default mode networks but less correlated activity in and between visual and somatomotor networks [129]. This is consistent with phasic LC activation promoting focused hot spots that are not correlated with surrounding activity in sensory regions but simultaneously promoting coordinated frontoparietal activity that helps to coordinate competition based on relative priority.
In summary, the current level of glutamatergic activity in a particular brain region has the potential to modulate local noradrenaline release when the LC is activated. Thus, during moments of high arousal, LC activity can elicit greater noradrenaline release in cortical and hippocampal regions where there are already high levels of excitatory activity, leading to hot spots of high noradrenaline. These hotspots in turn can produce β-adrenergic activation in those specific regions, leading to qualitatively different effects than activation of α2A receptors by low levels of noradrenaline.
If, as the GANE model outlines, glutamate and β-adrenergic receptor interactions can create hot spots of high synaptic activity during arousal states involving LC activation, how does this interact with the production of Alzheimer’s-related pathology? As outlined in the following sections, increased synaptic activity has been associated with increased production of both amyloid peptides and tau proteins. Does this mean that LC activity accelerates the production of amyloid and tau and increases the risk of accumulation of amyloid plaques and tau tangles in ‘hot spot’ regions where there are high levels of glutamate? Also, does the LC pathology observed in aging and/or Alzheimer’s disease change the impact of LC activity? Subsequent sections address these questions.
2. Relationship between neuronal activity and amyloid-β and tau
2.1. Synaptic activity affects production of amyloid-β
One way that noradrenaline can affect Alzheimer’s-related pathology is simply by amplifying or suppressing synaptic activity. Like the body’s other metabolic activities, brain activity produces waste that can become problematic if not disposed of (Sections 3.2 and 3.6 review how noradrenaline influences waste clearance). For instance, synaptic activity stimulates the production of amyloid-β (Aβ) peptides, which are cleavage products of amyloid precursor protein (APP). Aβ plaques are a post-mortem hallmark of Alzheimer’s disease, and although these Aβ plaques are unlikely to be the cause of the disease [130], smaller aggregate forms (oligomers) of Aβ have toxic properties, disrupting synaptic activity and memory processes [131-133]. Why does synaptic activity produce Aβ? During synaptic activity, the process of releasing synaptic vesicles containing neurotransmitters involves fusing the vesicle with the cell membrane and creating an opening in both the vesicle and the cell membrane to release the contents of the vesicle. APP covers parts of the cell membrane [134] and can end up being moved inside the cell as part of the process of vesicle recycling. Once inside the cell, APP is processed through the cell’s endosome system, which sorts through cell membrane molecules to either recycle them or dispose of them. While in the endosome, APP is sequentially cleaved by β-secretase and then γ-secretase to produce Aβ [135], which is then released into the fluid-filled interstitial spaces between brain cells [136]. Because synaptic activity increases vesicle recycling, it increases the likelihood that APP will be cleaved to form Aβ [136-138].
There is also an alternative metabolic pathway for breaking down APP that does not produce Aβ. In this alternative pathway, before it can be cleaved by β-secretase and then γ-secretase inside the neuron, APP is cleaved by α-secretase and then γ-secretase while it is still on the surface of the neuron [139]. The factors determining whether APP goes through the Aβ-promoting β-secretase or the non-Aβ-forming α-secretase pathway are not yet fully understood, but in addition to synaptic activity increasing the likelihood of the Aβ-promoting β-secretase pathway, activation of muscarinic acetylcholine receptors stimulates α-secretase and release of non-Aβ-forming APP fragments [140, 141], thus tipping the scales against the formation of amyloid plaques.
Aβ itself is not necessarily problematic. It may even have some physiologically beneficial effects. One suggestion is that it provides feedback to prevent overexcitation of neural activity where there is high synaptic activity, as elevated extracellular Aβ (at high nanomolar and low micromolar levels) locally inhibits further synaptic activity [142, 143]. However, in healthy brains, extracellular concentrations of Aβ are in the low picomolar range, far lower than the range used in studies showing Aβ-induced synaptic depression [144]. Studies investigating the effect of either endogenous Aβ or extracellular application of picomolar concentrations of Aβ have found that it enhances hippocampal long-term potentiation and memory performance [145-148]. Thus, in the healthy brain there may be some benefits to Aβ.
However, if Aβ is not cleared and instead aggregates, it can damage neurons. The cleavage of APP by β-secretase and then γ-secretase produces Aβ monomers. These singlet forms of Aβ are attracted to each other and can eventually assemble into oligomers, and strings of oligomers known as protofibrils [149]. Over time Aβ protofibrils and fibrils can aggregate into the much larger amyloid plaques that are a hallmark of Alzheimer’s disease. However, it is the smaller Aβ oligomers and protofibrils that are most toxic [149-151].
Consistent with the role of synaptic activity in the formation of Aβ, there is evidence that more active brain regions accumulate more Aβ. In positron emission tomography (PET) imaging studies, the most metabolically active regions of the brain (in particular, those comprising the brain’s ‘default mode network’) show the most uptake of Aβ markers [152, 153]. In transgenic mice expressing a mutated form of APP to mimic aspects of human Alzheimer’s disease, 30 minutes of unilateral whisker stimulation increased interstitial Aβ in the contralateral barrel cortex [154], demonstrating that an episode of high neural activity in a particular brain region can increase Aβ levels in that region. Furthermore, depriving mice of whisker sensation on one side for about a month reduced amyloid plaque growth by 78% in the contralateral hemisphere compared to the control hemisphere [154].
2.2. Neuronal activity also increases extracellular tau
Aβ is not the only potentially toxic waste product of neuronal activity. Another one is the microtubule-associated protein tau which, among its multiple roles, provides a protein scaffold on microtubules [155]. When hyperphosphorylated, the tau protein no longer binds as effectively to microtubules and more readily misfolds, creating insoluble fibrils that are the main constituent of the neurofibrillary tangles that, together with Aβ plaques, are the key pathological features of Alzheimer’s disease [10, 156].
In wild-type mice, tau is released into the extracellular space in the brain under normal conditions [157]. Increasing neuronal activity increases release of tau into the interstitial space in both in vitro and in vivo rodent models [for review see 158], and if the excited neuron contains pathological tau, its release can lead to its spread to other neurons [159]. In a transgenic mouse Alzheimer’s disease model, chemogenetic attenuation of neuronal hyperactivity reduces the spread of pathological tau [160]. Presynaptic glutamate release is sufficient to increase tau release [161], and the release apparently can be triggered by either AMPA [162] or NMDA receptors [163].
Growing evidence suggests that, as with Aβ, vesicle-trafficking stimulates the release of tau. Exosomes are small extracellular vesicles that derive from the fusion of intracellular vesicles and organelles with the cell membrane [164]. Secretion of phosphorylated tau often occurs via exosomal release [165]. Depolarization of neurons promotes release of tau-containing exosomes [166], providing additional evidence of the importance of neuronal activity in stimulating release of the building blocks of Alzheimer’s pathology. Tau secretion has been linked not just with exosomes released from neurons but also with exosomes released from microglia [167]. Microglia likely accumulate tau as part of the process of phagocytosing dying cells, debris and protein aggregates [168].
Thus, both Aβ and tau release can be stimulated by neuronal activity. Therefore, insofar as LC activity increases the gain on neural excitation/inhibition levels [101], it should also influence Aβ and tau release simply by modulating levels of synaptic activity.
2.3. Relationship between environmental enrichment and Alzheimer’s pathology
A question motivating this review is how stimulating noradrenergic activity via arousing, engaging cognitive activities will affect accumulation of Alzheimer’s pathology. The findings reviewed in Sections 2.1 and 2.2 indicate that neuronal activity stimulates release of Aβ and tau into the extracellular space. However, connecting these findings at the level of synaptic activity with broad-scale interventions such as environmental enrichment in rodents or increased years of education in humans is not straightforward. Stimulating environments will not affect activity in all brain regions in the same way. Enriched environments may also increase brain activity in a particular brain region during an initial novel phase but decrease activity in that region in the long run. For instance, as part of learning, brain regions may execute tasks more efficiently [169], and so one effect of environmental stimulation could conceivably be decreased metabolism in the long run in those brain regions initially activated by the environmental challenges.
For these reasons, perhaps it is not surprising that non-specific environmental enrichment in APP transgenic mice can either increase amyloid plaque formation [170, 171] or decrease it [172, 173]. One interesting possibility is that these contradictory findings may relate to whether the mice were placed in environmentally enriched housing immediately after weaning [172, 173] or from when they were two months old [170, 171]. Early in the life, the amyloid produced as a by-product of synaptic activity is regularly cleared out along with other waste and potentially toxic substances. In contrast, through middle-age and in late life, clearance of amyloid declines significantly in mice and rats [174, 175]. Thus, one possibility is that the neuronal activity stimulated by enriched environments leads to more amyloid accumulation later in life than the same stimulation would early in life.
In humans, random assignment to dramatically differing environments for long periods of life is not a feasible research strategy. However, we can examine whether there are correlations between factors that reflect time spent in an enriched environment (such as education levels, job complexity, or being bilingual) and development of Alzheimer’s disease. Across many studies, people with higher education or other indicators of enriched environments are less likely to have been diagnosed with Alzheimer’s disease [176, 177]. But despite this relationship between prior education and likelihood of a diagnosis, those with more education do not tend to show an advantage in terms of brain pathology. In other words, while diagnoses of Alzheimer’s disease based on cognitive performance are lower in those with more education, diagnoses based on post-mortem brain pathology show no indication that education protects against Alzheimer’s pathology. For instance, in a post-mortem sample of 2372 participants whose brain pathology met criteria for an Alzheimer’s diagnosis, there was no correlation between education and level of Alzheimer’s neuropathology [e.g., 178]. Nevertheless, in this same sample, whether they had received a clinical diagnosis before death was related to education, with those with a ante-mortem diagnosis having 2-3 years less education on average than those with no ante-mortem diagnosis. Likewise, examination of data from 835 autopsies from the United States National Alzheimer Coordinating Center found that although higher levels of education decreased the odds of cognitive impairment, this effect was not mediated by extent of pathological changes (including amyloid plaques) [179].
In a large meta-analysis employing participant level data from 55 studies (N = 8694) with individuals with normal cognition, subjective cognitive impairment or mild cognitive impairment, those with above-median education had a higher prevalence of being amyloid positive on the basis of either cerebrospinal fluid (CSF) or PET imaging than those with less education, regardless of cognitive status, age, sex or APOE-ε4 carrier status [180]. However, as this meta-analysis was not a population-based study, it is possible that highly educated people with amyloid positivity were more likely to seek out clinical care or research studies than less educated people with amyloid positivity. Indeed, a large population-based study in Minnesota (N = 942) revealed that neither years of education, degree of intellectual challenge in jobs held, nor midlife weekly cognitive activity (such as reading or crafts) were associated with PET imaging categorization as amyloid positive or negative [181]. In contrast with both of the above larger studies, one study has found that those with higher education showed less PET amyloid binding than those with lower education [182]. However, this study had a small sample (N = 30) in which education level was confounded with participant sex. Thus, consistent with post-mortem data, the best evidence from PET imaging studies indicates no relationship between education and amyloid load. Education also fails to slow the progression of cortical atrophy [183]
This disjunct between the effects of education on Alzheimer’s diagnoses versus brain pathology led to arguments that education provides some sort of reserve that allows people to keep functioning well despite the encroaching brain pathology [26, 177]. If education provides reserve capacity, it should slow cognitive decline in those who can call on that reserve when challenged by encroaching pathology. However, whether higher education attainment slows cognitive decline has been a challenging question to address due to the nonlinear nature of cognitive decline and methodological confounds that might lead to comparing those with higher and lower education at different points in their downward cognitive trajectory [184].
A recent systematic review of the literature that focused especially on studies that had longitudinal data and used appropriate statistical approaches concluded that the association between educational attainment and late-life cognitive decline is small and inconsistent, and most of the effects of education are due to those with higher education achieving a higher peak level of functioning [185]. Alzheimer’s pathology seems to cause cognitive decline at the same general rate regardless of one’s peak cognitive attainment [184]. Thus, if one starts at a higher point, it will take more pathology to bring that person down to the threshold of attaining a diagnosis based on their cognitive functioning.
In summary, the difference in environmental enrichment between completing high school versus college or graduate school fails to provide any protection from cognitive decline or Alzheimer’s disease pathology. But the rodent studies described above suggest that timing of environmental enrichment may matter, and that providing environmental enrichment early in development has the most positive effect on later resistance to Alzheimer’s pathology. Thus, perhaps environmental enrichment that occurred during childhood would have a more noticeable effect on later pathological processes.
One study estimated early-life cognitive enrichment in 813 participants with post-mortem neuropathology results. Participants were recruited from the community when they were older, and, at baseline, answered questions about their early life (about socioeconomic status, availability of cognitive resources, frequency of cognitively stimulating activities and foreign language instruction) from which an early-life cognitive enrichment score was derived [186].
This score was negatively associated with a score assessing global Alzheimer’s pathology, as well as with levels of tau and Aβ measured separately. These results provide the strongest link to date between cognitive enrichment and Alzheimer’s pathology. Comparing these findings to the lack of association between cognitive enrichment in adolescence and adulthood and Alzheimer’s pathology seen in other studies raise the possibility that childhood cognitive enrichment has more of an impact on later Alzheimer’s pathology than factors operating in late adolescence or early adulthood, such as additional years of total education attainment.
However, this study unfortunately did not compare the relationship of early and mid- or late-life cognitive enrichment. Further work is needed to directly compare the effects of environmental enrichment initiated very early in development or at a later stage on amyloid plaque formation to see if indeed the risks of stimulating amyloid plaque formation are greater when the novel mental stimulating environment is first experienced later in life.
3. Relationships between adrenergic activity and the progression of Alzheimer’s disease
3.1. β-adrenergic activation influences microglial activity
The previous sections reviewed how neuronal activity and environmental enrichment affects Alzheimer’s pathology. In the next few sections, I address how noradrenaline modulates these processes.
One important way that noradrenaline modulates the progression of Alzheimer’s disease is via its influence on microglia. Microglia are the main immune cells in the brain that dynamically survey their environment, ready to extend their processes towards disease or injury sites to clear damage [187, 188]. Microglia can either protect against Alzheimer’s disease or exacerbate the progression of the disease [189]. Postmortem analyses and Alzheimer’s mouse models indicate that in Alzheimer’s disease, microglial inflammatory activity increases while microglial clearance activity decreases [190].
Microglia express more β2-adrenergic receptors than most other cortical cell types [191, 192]. Recent advances now allow tracking of microglia dynamics in awake mice using two-photon microscopy. These in vivo methods have revealed that, during wakefulness, microglia are less active with diminished responsiveness to injury compared to during anesthesia [193, 194]. The LC is much more active during wakefulness than during sleep [195] and β2-adrenergic receptor activity decreases microglial activity during wakefulness [193, 194]. A β2-adrenergic agonist also suppresses proliferation of microglia in culture [196]. However, stress (a potent noradrenergic stimulus) activates microglia and a β-adrenergic blocker inhibits this stress-induced microglial activation [192]. Thus, effects of noradrenaline on microglial activity appear to depend on stress levels.
As would be predicted by the GANE model (see Section 1.4), levels of local neuronal activity modulate microglial β2-adrenergic receptor activity [193]. For instance, trimming a mouse’s whiskers on one side (a sensory-deprivation method that suppresses neuronal activity in the contralateral barrel cortex), led microglia in the contralateral barrel cortex to increase their process area, an effect similar to that seen under anesthesia [193]. Interestingly, whereas resting microglia primarily express β2-adrenergic receptors, they switch expression to α2A receptors under proinflammatory conditions [197], which should lower the threshold for noradrenergic influence (due to the different affinities of β and α2 receptors; see Section 1.2) and may also change its effect.
3.2. β-adrenergic activation influences Aβ accumulation and clearance
As already discussed, cortical regions with high levels of neural activity tend to produce more Aβ. An intriguing possibility is that β-adrenergic activation during high levels of neuronal activity may provide a countervailing influence that reduces the toxic influences of Aβ. Even though, as reviewed in the previous section, local β-adrenergic activation decreases microglial process surveillance, it may stimulate microglial cells to dispose of Aβ42. When isolated in a cell culture, mouse microglia cells show increased Aβ42 uptake when exposed to either noradrenaline or a β-adrenergic agonist, apparently driven by upregulation of the microglia receptor for Aβ42 [198]. One study consistent with the possibility that β-adrenergic activation helps combat the negative effects of Aβ examined the effects of environmental enrichment in wild-type mice exposed to human Aβ oligomers [79]. Mice living in an enriched environment for two months starting just two weeks after birth had higher expression of long-term potentiation (LTP) related signaling proteins in the hippocampus than did control mice. Furthermore, exposing hippocampal slices from these mice to soluble Aβ oligomers revealed the time in the enriched environment made hippocampal LTP less vulnerable to Aβ and that this protection could be up- and down-regulated by β-adrenergic agonists and antagonists, respectively. In addition, feeding normally housed mice a β-adrenergic agonist protected LTP from the toxic effects of Aβ. Exposing mice at midlife to an enriched environment also enhanced their LTP and protected it from Aβ, although it took 8 weeks to reach similar benefits as the 4-week exposure used in younger mice. Consistent with the protective effects of β-adrenergic receptors, other studies demonstrate that injection of a β-adrenergic receptor agonist before LTP induction prevents Aβ-induced LTP attenuation [199, 200]. Rats injected with human Aβ42 a week after toxin-induced degeneration of the LC showed greater inflammatory responses to Aβ42, effects that were attenuated by co-injection with noradrenaline or a specific β-adrenergic receptor agonist [201]. Furthermore, in studies with APP-transgenic mice, toxin-induced degeneration of the LC increased Aβ42 levels [202-204] and reduced the colocalization of Aβ deposits and a microglial activation marker by 70% [205]. Providing the noradrenaline-depleted mice with the noradrenaline precursor L-threo-DOPS restored microglial phagocytosis of Aβ [205]. These findings suggest that without noradrenaline, the microglia failed to take up Aβ as part of the phagocytosis process. β-adrenergic activity has been identified as the mechanism underlying the benefits of environmental enrichment on microglia exposed to Aβ [206]. In wild-type mice housed in standard cages, exposure to soluble oligomers of Aβ (isolated directly from human brains with Alzheimer’s disease) increases local microglial inflammation, an effect that is prevented by housing mice in enriched environments [207]. The positive effects of environmental enrichment can be mimicked by giving the mice in the standard cages a β-adrenergic receptor agonist or blocked by giving the environmentally enriched mice a β-adrenergic antagonist [206]. In mice genetically altered to lack β-adrenergic receptors, environmental enrichment failed to prevent microglia inflammatory responses to Aβ [206]. Also demonstrating the beneficial effects of β-adrenoceptor activity, giving amyloid precursor protein/presenilin 1 (APP/PS1) double transgenic mice (who exhibit Aβ accumulation, synaptic deficits and memory impairment at 8 months) a β-adrenergic agonist for two months starting when they were 8 months old led to decreased Aβ plaques, greater hippocampal neurogenesis, less pronounced memory deficits and increased dendritic branching and density of dendritic spines than giving them a vehicle control [208].
In contrast with the positive effects reviewed above of β-adrenergic agonists in wild-type and transgenic mice [198, 206, 208], other studies with transgenic mice show negative effects. For instance, a study using APP/PS1 double-transgenic mice found that 30 days of β-adrenergic agonists increased amyloid plaque formation [209]. Another study using Tg2576 mice which at 6 months start showing exponential increases in Aβ peptide content in the brain found that giving these mice a β-adrenergic antagonist reduced Aβ42 accumulation in the hippocampus [210]. It is not clear how to account for the different results across studies.
Data in humans also indicates that β-adrenergic activity influences the course of Alzheimer’s disease. In a study of Han Chinese, there were significant differences between a group of 109 Alzheimer’s disease patients vs. 109 controls in the Glu27Gln (rs1042714) and the Arg16Gly (rs1042713) polymorphisms of the β2-adrenergic receptor gene [211]. Both Gly16 and Glu27 polymorphisms were more likely to occur in patients with Alzheimer’s disease. It is not clear yet what mechanism leads to this association, but these polymorphisms are associated with greater vascular responsiveness to a β2-adrenergic agonist [212] and with higher low-density lipoprotein cholesterol [213], either of which might be a relevant factor in addition to potential brain effects. An observational longitudinal study of older adults with dementia found that functional decline was slower in those taking β-adrenergic antagonists for cardiovascular conditions than the other participants not taking this type of medication [214]. Likewise, a study comparing medication histories of patients newly diagnosed with dementia versus age- and sex-matched controls found that the dementia group was significantly less likely to have been continuously taking a β-adrenergic antagonist in the previous three years [215]. These findings suggest that, at least once Alzheimer’s disease is relatively advanced (as amyloid pathology is already typically evident at least a couple of years before typical diagnosis [216]), β-adrenergic receptor activity may accelerate disease progression. Currently, the human literature lacks analyses of the effects of β-adrenergic medications taken earlier in the life on later timing and progression of Alzheimer’s disease. The fact that β-adrenergic activity can have either beneficial or harmful effects in rodents suggests the effects depend on the presence of some other factor such as the level of existing pathology.
In summary, β-adrenergic receptor activity has significant effects on the accumulation of Aβ pathology, especially via its modulatory effects on microglial activity. However, whether these effects are positive or negative depends on factors that are not yet clear.
3.3. α2A-adrenergic activation influences Aβ accumulation
There is also evidence that α2A-adrenergic receptors modulate Aβ accumulation. APP/PS1 transgenic mice genetically modified to lack α2A-adrenergic receptors show reduced Aβ generation and Alzheimer’s-related pathology whereas activating the α2A-adrenergic receptor with noradrenaline (while blocking β-adrenergic receptors) enhances Aβ generation and Alzheimer’s-related pathology [217]. As reviewed in Section 1.2, α2A-adrenergic receptors are found in the presynaptic membrane of noradrenergic terminals as well as on other types of neurons. They also are widely expressed in astroglia. Thus, the findings that α2A-adrenergic receptor activity is associated with Aβ generation and Alzheimer’s-related pathology [217] could be due to a variety of possible mechanisms.
One possibility is that α2A-adrenergic receptors have an amyloid plaque promoting effect because their activation reduces APP localization to the Golgi where it could be recycled and increases APP sorting to endosomal compartments. There it is colocalized and interacts with β-secretase [217], biasing processing towards the Aβ-promoting metabolic pathway for breaking down APP. In addition to α2A-adrenergic receptors influencing APP, APP also influences α2A-adrenergic receptors, enhancing the surface retention of α2A-adrenergic receptors and their signaling intensity and duration [218]. As reviewed in a later section (4.2), Aβ oligomers also potentiate the effects of α2A-adrenergic receptors [219]. Together, these findings suggest positive feedback between α2A-adrenergic receptor activity and APP and Aβ oligomers that may accelerate Alzheimer’s pathology in brain regions where there already is some pathology — a possibility consistent with findings that taking an α2-adrenergic agonist accelerates cognitive decline in those who already are diagnosed with Alzheimer’s disease but not in healthy older adults (see Section 4.2) [219].
3.4. The locus coeruleus and pathological tau
In addition to its role in influencing the progression of Alzheimer’s disease pathology elsewhere in the brain, the LC is significantly affected itself in the course of the disease. In fact, in a large study with over 2000 post-mortem brains from age 1 to 100, it was the first site in which the pretangle stage of tau pathology was detected [9]. This pretangle stage was detected using an antibody (AT8) to tau phosphorylated on amino acids Ser202 and Thr205 [for more details see 10]. Another study using a different Ser202 antibody (CP-13) estimated that around 8% of LC neurons showed a positive antibody response in those at Braak stage 0 of Alzheimer’s disease (when there is no cortical indication of the disease, in this particular study N=13 cases with a mean age of 58) [220].
In addition to this early vulnerability of the LC to abnormal tau, linkages between adrenergic activity and tau hyperphosphorylation have been detected. Dexmedetomidine is a selective α2-adrenergic agonist that is commonly used as an anesthetic to induce a sedative state [221] because it reduces LC noradrenaline release via α2-adrenergic autoreceptor activation [222]. Dexmedetomidine induces tau hyperphosphorylation in vivo and in vitro in both transgenic human Tau mice and in non-transgenic mice, with an α2A receptor antagonist blocking this dexmedetomidine-induced tau hyperphosphorylation [223]. The hyperphosphorylation levels were significantly elevated half an hour after dexmedetomidine administration in cortex and hippocampus, with elevated rates still detectable in the hippocampus but not the cortex 6 hours afterwards.
Links with tau hyperphosphorylation have also been demonstrated with β2-adrenergicreceptors. Removing the gene encoding β2-adrenergic receptors from a mouse model overexpressing mutant human tau led to mice that lived longer and showed reduced hyperphosphorylated tau in the basal ganglia and hippocampus (but not in the cortex) than the tau model mice with β2-adrenergic receptors [224].
Interestingly, in mice, toxin-induced degeneration of the LC led to significant impairments in spatial learning and memory in P301S tau transgenic mice (who show progressive accumulation of neurofibrillary tangles) but not in wild-type mice [225], suggesting that loss of LC neurons is especially problematic within the context of tau pathology.
3.5. Interactions of Aβ and tau can be influenced by noradrenergic activity
Both human and animal research have documented that Aβ and hyperphosphorylated tau aggregates interact to accelerate Alzheimer’s disease degeneration [226, 227]. Some aspects of these interactions are mediated by noradrenergic activity. For instance, Aβ oligomers bind to an allosteric site on α2A adrenergic receptors, thereby redirecting noradrenergic signaling to glycogen synthase kinase 3β (GSK3β) activation and tau hyperphosphorylation [219]. GSK3 contributes to the abnormal phosphorylation of tau proteins in Alzheimer’s disease, as well as being involved in many other disorders [228].
3.6. Noradrenergic activity influences glymphatic clearance
The accumulation of Alzheimer’s disease pathology depends not only on the rates of production of Aβ and tau but also on how effectively these by-products of metabolic activity are cleared out of the brain. There are a number of ways that waste products are eliminated from the brain [229-231]; one of these is via the glymphatic pathway [232, 233]. Glymphatic clearance of waste occurs when CSF traveling in compartments wrapped around brain arteries exchanges with the interstitial fluid between brain cells. Eventually, the CSF exits the brain in compartments wrapped around veins, taking substances such as Aβ [234] and tau [235] that were in the interstitial space with it.
Glymphatic function is active in both naturally sleeping and anesthetized mice but suppressed during wakeful states [236, 237]. As already discussed, noradrenaline is a key regulator of arousal states, including wakefulness [67]. Indeed, it turns out that elevated noradrenaline levels help drive suppression of glymphatic function during wakefulness. Administering a cocktail of adrenergic antagonists, including β, α1, and α2-adrenergic antagonists, significantly increased the influx of cerebral spinal fluid into the cortex and the volume of the fluid-filled interstitial space between brain cells [237]. Dexmedetomidine, the commonly used anesthetic that reduces LC noradrenaline release via α2-adrenergic autoreceptor activation [222] increases glymphatic CSF influx [236, 238, 239]. One way that noradrenaline modulates glymphatic function is by lowering production of CSF at the choroid plexus [240-242]. Another potential pathway is via noradrenaline’s modulation of vasoconstriction and vasodilation [243-245]. In awake mice, slow oscillations in vasoconstriction appear to facilitate clearance of particles from interstitial fluid [246].
Thus, in summary, noradrenaline impedes glymphatic clearance both by decreasing production of CSF and by decreasing the interstitial space volume, restricting movement of interstitial fluid. This is an important effect that could accelerate the accumulation and eventual aggregation of Aβ and tau, in particular in those individuals with high levels of noradrenergic activity. As reviewed below, both age and Alzheimer’s disease have been associated with increased CSF and blood levels of noradrenaline. It is possible that disease-related increases in noradrenaline in turn lead to decreased glymphatic clearance of Aβ and tau [247].
4. Influences of Alzheimer’s disease and aging on locus coeruleus function
4.1. Locus coeruleus vulnerability in Alzheimer’s disease and other neurodegenerative diseases
Thus far, this review has focused on how noradrenergic activity affects the production and accumulation of Aβ and tau. However, we should not ignore that these effects of noradrenergic activity occur in a context in which the LC is changing with age. In particular, the LC is a key node in the age-related progression of Alzheimer’s disease pathology. As already mentioned, the LC appears to be the earliest site of pathological tau [9, 10], with post-mortem abnormally phosphorylated tau seen in the LC in most non-selected brains by age 30 [9]. In cognitively normal older adults, decreased structural volume in the LC predicts conversion to an Alzheimer’s disease diagnosis [21].
Previously, we had speculated that the high energetic demands of LC neurons contribute to their vulnerability [2]. Recent findings indicate that metabolism in the LC carries a particularly high risk due to a specific intermediate metabolite of noradrenaline [248]. This metabolite, 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL), is produced by monoamine oxidase A (MAO-A) oxidation of noradrenaline and triggers tau pathology in the LC that can spread to other brain regions [248]. DOPEGAL activates a lysosomal endopeptidase (asparagine endopeptidase) that cleaves tau at specific locations (N255 and N368) and the resulting fragments increase tau hyperphosphorylation and aggregation [249]. Postmortem LC samples from subjects with Alzheimer’s disease showed more asparagine endopeptidase along with aggregated tau and more tau cleaved at N368 than LC samples from control brains [248].
In addition to suffering from the DOPEGAL metabolic risk, the LC may also be vulnerable for a number of other reasons. Its extensive contact with central nervous system capillaries and its proximity to the fourth ventricle increases its exposure to circulating toxicants [2]. Also, viruses or other noxious substances may be transported to the LC from the gut via the vagus nerve and nucleus of the solitary tract [250]. Herpes infections may cause a particular risk, because after people recover from a herpes infection, the common herpes simplex type 1 virus lies dormant in the trigeminal ganglion, which projects to the LC [251]. Another possibility is that the narrow nature of the cerebral aqueduct connecting the third and fourth ventricles induces CSF flow (especially in those with hardened arteries that fail to absorb cardiac pulsations and so pass along greater pulsatility to upstream vessels) that alters the molecular structure of tau proteins being carried by the CSF in ways that eventually promote their aggregation [252].
Further evidence indicating the LC is especially vulnerable to pathological tau is that degeneration of the LC is a common feature of tauopathies such as progressive supranuclear palsy [253, 254]. Furthermore, the LC is a specific target of tauopathies rather than of all proteinopathies [e.g., 255].
Degeneration of the LC is also seen early in Parkinson’s disease [256]. Like other α-synucleinopathies such as dementia with Lewy bodies, Parkinson’s disease is characterized by the abnormal accumulation of aggregates of α-synuclein protein in the brain, α-synuclein promotes tau hyperphosphorylation and can trigger tauopathy [257]. Abnormal tau and α-synuclein appear to coaggregate in the locus coeruleus and elsewhere in the brain [258], which may help explain why the LC is also targeted in Parkinson’s disease and other α-synucleinopathies [259].
How do the increases in abnormal tau in the LC seen in aging and Alzheimer’s disease relate to LC function? This is still a mystery as, to date, we have only hints as to how increases in hyperphosphorylated tau affect the functioning of LC neurons [e.g., 260]. Although not documented for the LC itself, there are indications that tau hyperphosphorylation decreases excitation within the neuronal networks involved [261]. However, we have yet to have definitive information regarding even the simple question of whether LC tonic activity increases or decreases in aging. The following section reviews some of the existing relevant evidence.
4.2. Changes in noradrenergic activity in aging
Does LC activity increase or decrease with age? This is a challenging question to answer given the currently available data [for reviews see 262, 263, 264]. Postmortem measures of noradrenaline extracted from brain tissue tend to be lower in older adults than younger adults and lower in those with Alzheimer’s disease compared with cognitive normal older adults [265-268]. However, noradrenaline levels reflect production, release and re-uptake and so are not a direct measure of LC activity. Furthermore, these measures use homogenized brain tissue and so estimates of noradrenaline depend both on the number of noradrenergic neurons and their corresponding internal store of neurotransmitter as well as on how much of the neurotransmitter was circulating in the interstitial spaces between cells.
In contrast with the results from homogenized brain tissue, plasma noradrenaline concentrations are estimated to increase 10-15% per decade during adulthood [269]. Under resting conditions, noradrenaline in the bloodstream mostly comes from noradrenaline released by sympathetic nerves, with only a small proportion coming from the adrenal gland [270]. Consistent with the plasma findings, levels of noradrenaline in CSF are greater in cognitively normal older adults than in younger adults [271, 272]. Advanced Alzheimer’s disease is associated with even greater levels of noradrenaline in CSF and plasma than in cognitively normal older adults [271, 273].
Thus, despite the substantial decline in noradrenaline neurons in the LC in Alzheimer’s disease [e.g., 5], noradrenaline levels in CSF and plasma still remain high, and in many cases even higher than in normal aging or in younger adults. This opposing effect of Alzheimer’s disease on noradrenaline neurons versus on circulating levels of noradrenaline suggests that remaining noradrenaline neurons may compensate by increasing their activity levels (this could include non-LC sources of noradrenaline such as A5 and A7 cell groups and sympathetic ganglion neurons).
Research with rodents supports the possibility that intact noradrenergic neurons compensate for damaged ones by increasing their activity rates. For instance, using intraventricular administration of 6-hydroxydopamine to destroy 80-90% of noradrenergic terminals in the rat forebrain led the still-intact LC cell bodies to increase their firing rates by about four times normal levels [274]. Likewise, injection of 6-hydroxydopamine directly into mouse LC at different doses to destroy between 30-70% of the neurons led surviving LC neurons to fire more frequently and with a more irregular pattern three weeks later [275]. Compensation may also occur via increased sprouting of axonal projections. Normal-to-elevated presynaptic α2A binding site density in hippocampus and prefrontal cortex in Alzheimer’s disease patients compared with controls suggest greater numbers of axonal projections from each LC neuron into these regions in patients [276, 277].
But the human data indicating high circulating levels of noradrenaline could also result from impaired clearance processes, such as declines in noradrenaline transporter availability [278]. To address this, additional insight can be gained by examining metabolites that precede versus follow noradrenaline in its metabolic process. Studies assessing the noradrenaline metabolite methoxyhydroxyphenylglycol (MHPG) levels indicate sustained noradrenergic metabolic activity in Alzheimer’s disease patients despite a declining noradrenergic system: In studies quantifying both post-mortem brain noradrenaline and MHPG, while noradrenaline tended to be lower in patients with Alzheimer’s disease compared with controls, MHPG tended to be at least as high in the patients as in the controls [266, 279]. Other studies also show similar CSF MHPG levels between patients with Alzheimer’s disease and age-matched controls, with lower levels in younger adults [280, 281]. Of particular interest, greater CSF MHPG levels in memory clinic patients predicted a steeper decline in their memory in the next few years [282]. Alzheimer’s disease pathology in the context of low MHPG levels was not associated with significant memory decline, raising the possibility that noradrenergic hypermetabolism plays an essential role in memory declines in Alzheimer’s disease.
Another indication that, in Alzheimer’s disease, the surviving LC neurons produce more noradrenaline is that patients with Alzheimer’s disease had more tyrosine hydroxylase mRNA expression in those LC neuron cell bodies that survived the profound neuronal loss associated with the disease than in the LC cell bodies of controls [276]. The greater the neuronal loss, the more each remaining cell expressed tyrosine hydroxylase mRNA. Tyrosine hydroxylase is the rate limiting factor in catecholamine biosynthesis. The decrease seen in noradrenaline transporter density in the brain with aging [283] and with Alzheimer’s disease [284, 285] may also lead to increases in extracellular levels of noradrenaline in the brain which would be reflected in CSF levels.
A study that administered yohimbine, an α2A antagonist that increases release of noradrenaline, clonidine, an α2A agonist that decreases release of noradrenaline, or a placebo to participants revealed that CSF and blood levels of noradrenaline were modulated similarly by the adrenergic drugs in older and Alzheimer’s disease patients as in younger adults [286]. The three groups of participants also showed similar increases in dihydroxyphenylglycol (DHPG), a metabolite of noradrenaline that is an indicator of its clearance, in response to yohimbine. However, in the yohimbine condition, the older and Alzheimer’s group showed greater increases than did younger adults in CSF levels of the noradrenaline precursor dihydroxyphenylacetic acid (DOPA), an indicator of noradrenaline synthesis. These findings suggest that aging and Alzheimer’s disease increase synthesis of noradrenaline.
Alzheimer’s patients have Aβ oligomers in the LC that, based on a mouse model of increased Aβ production (APP-PSEN1), appear to contribute to its hyperexcitability [287]. Like Alzheimer’s patients (but unlike wild-type mice), APP-PSEN1 mice have LC neurons that express Aβ oligomers. Electrophysiological recordings in 2-to 4-month-old mice brain slices revealed that APP-PSEN1 LC neurons had greater firing rates than wild-type LC neurons [287]. Both GABA and glycine provide inhibitory modulation of LC [288]. APP-PSEN1 LC neurons decreased their firing significantly less in response to GABA than did wild-type LC neurons, whereas there was no significant difference in response to glycine [287]. There also were no detectable differences in levels of expression of glutamatergic synaptic markers across the two mice groups. These findings suggest that Aβ oligomers in the LC impair the activity of GABA receptors that reduce activity rates.
APP-PSEN1 mice lack tau pathology, the other main feature of Alzheimer’s disease. But recent work has created a rat model of the spread of pathological tau from the LC to cortical regions by expressing pseudohyperphosphorylated human tau in rat LC [260]. At 6 weeks post-infusion of tau/control in the LC, the rats with pathological human tau showed spread of that tau along LC axons toward forebrain targets, and by 12 weeks, it had reached the olfactory bulb and other cortical areas such as the hippocampus and piriform cortex. By 7-8 months post-infusion, the rats with pathological tau showed impaired olfactory discrimination and degeneration of LC axons in the piriform cortex. At the same time, they showed upregulation of β1-adrenoceptors in the piriform cortex. Previous studies in rats have also shown compensatory increase of cortical β-adrenoceptors after damage to noradrenergic neurons [289, 290]. Interestingly, in humans, depressed suicide victims show fewer rostral LC neurons [291] and greater prefrontal β-adrenergic receptor binding [292-294], consistent with the notion that β-adrenoceptors upregulate in response to LC degeneration. Data from Alzheimer’s patients are limited, but one post-mortem study showed increased β-adrenoreceptors in the hippocampus with a mixed profile in other brain regions (e.g., in frontal cortex β2-adrenoceptors but not β1-adrenoceptors were greater in Alzheimer’s patients than in controls) [295]. While increased cortical β-adrenoceptors may help compensate for decreased LC neurons, they may also lead to hyperexcitability, such as seen in older adults’ hippocampus [296, 297].
Along with these indications of increased noradrenergic activity and upregulation of excitatory β-adrenergic receptors, there also appears to be upregulation of α2A noradrenergic receptors in Alzheimer’s disease. Postmortem prefrontal brain samples from patients with Alzheimer’s disease show greater α2A-receptor-mediated G protein activation in response to noradrenaline even when adjusting for α2A receptor density compared with controls [219]. How could α2A receptors show a more potent response to noradrenaline in patients with Alzheimer’s disease?
G protein-coupled receptors not only have their primary binding sites for their target neurotransmitter or other ligand (known as orthosteric binding sites), but also have other (allosteric) binding sites that can modulate the effects of ligand binding at the orthosteric sites [298]. Experiments with an Alzheimer’s disease mouse model suggest that Aβ oligomers’ ability to bind to an allosteric site on α2A receptors allows this Alzheimer’s related peptide to potentiate the effects of noradrenaline binding [219].
Thus, the emerging data so far suggest a pattern of increased noradrenergic activity in Alzheimer’s disease that is accompanied by increased effectiveness of typically inhibitory α2A receptors. It is not clear yet whether one or both of these phenomena accelerate or slow down the course of the disease. However, one relevant observation comes from 267 Alzheimer’s patients who happened to be taking clonidine at some but not all clinic visits [219]. Scores on the mini-mental state examination declined more during time periods with clonidine than without clonidine, an effect not seen in normal controls. Furthermore, this decrease in cognitive function when on clonidine was more pronounced for those diagnosed with Alzheimer’s disease than those with mild cognitive impairment or who were impaired without mild cognitive impairment. As clonidine decreases LC noradrenergic activity [299], these correlational findings suggest that, at least once the Alzheimer’s disease process is fairly advanced, reducing noradrenergic activity accelerates decline.
4.3. Alzheimer’s disease is associated with an increase in extracellular glutamate
Yet another factor that might produce higher extracellular noradrenaline levels in patients with Alzheimer’s disease is the increase in extracellular glutamate that is associated with the disease [97]. In the brain, intracellular glutamate concentrations are in the millimolar range, but transporters that import glutamate into astrocytes and neurons keep the extracellular concentrations in the low micromolar range [300]. In patients with Alzheimer’s disease, post-mortem examination reveals decreased expression and activity of transporters that remove glutamate from the extracellular space [301-304]. In some studies, patients with Alzheimer’s disease have higher levels of glutamate in CSF compared with either healthy controls [305-307] or those with mild cognitive impairment [308], although a reduced level of CSF glutamate in patients with Alzheimer’s compared to controls has also been observed [309].
One reason for decreased glutamate clearance in Alzheimer’s disease may be increased levels of Aβ, as soluble Aβ peptides and oligomers reduce glutamate uptake and decrease glutamatergic transporter activity [310-312].
As outlined earlier, the GANE model posits that locally high levels of extrasynaptic glutamate can stimulate greater noradrenaline release via NMDA receptors if the LC neuron is concurrently depolarized. Thus, effects of noradrenergic activity on neuronal activity and on Aβ and tau dynamics could be amplified by Alzheimer’s-related increases in glutamate concentrations in the extrasynaptic space. Furthermore, as outlined by GANE, higher levels of local noradrenaline release can in turn lead to higher glutamate release in those regions. These interactive effects in turn may accelerate the accumulation of Alzheimer’s pathology, as in vitro work indicates that glutamate activity upregulates the tau mRNA translation involved in the transport of non-axonal tau across synapses [313] and increases the missorting of tau into dendrites [314].
5.1. Conclusions
On the one hand, researchers have posited that the LC may be the main brain mechanism underlying cognitive reserve [3, 24, 25]. On the other hand, the argument has been made that drugs that lower noradrenergic signaling should protect against a broad range of diseases, including Alzheimer’s disease [315]. This paper reviews findings suggesting that the story is not as simple as either of these opposing perspectives, as noradrenergic activity can be either beneficial or problematic when it comes to Alzheimer’s disease.
Although existing findings leave many unanswered questions, one hypothesis suggested by the existing literature is that increases in tonic LC activity due to age-related LC deterioration or stress are detrimental whereas phasic LC activity associated with cognitive stimulation may be more beneficial. However, even the phasic noradrenergic activity induced by cognitive stimulation is likely to have a higher risk-to-benefit ratio for brain health as the brain’s ability to clear out waste products associated with neural activity diminishes in aging. This set of findings suggest that it may be more challenging for cognitive training trials to have a net positive impact in older brains than in younger brains not just because of age-related decline in the processes underlying synaptic plasticity, but also because the side effects of synaptic plasticity are likely to be higher in older brains, where metabolic waste products linger longer and so can have more toxic effects.
5.2. Future directions
How does LC activity and structural integrity over the life course accelerate or slow down Alzheimer’s disease neuropathology? To address this question, in this review I had to draw mainly on research using animal models, as relevant findings in living humans are still quite limited. However, I expect that we will soon see significant advances in answering this question in living humans due to the recent and on-going methodological advances in both imaging the LC and quantifying Aβ and tau.
A first critical step along these lines would be to improve measures of LC function and structure in large longitudinal studies. Most large publicly available longitudinal imaging studies do not include any of the recently developed scan sequences that are optimal for localizing and characterizing the LC [316, 317]. We have found that, given a sufficiently large number of participants, even general-purpose magnetic resonance images identify the LC as being a brain region where decreased structural volume predicts later conversion to Alzheimer’s disease [318]. However, scan sequences specialized to detect the LC should provide more sensitive measures and should be included in longitudinal studies, especially in studies that provide measures of Alzheimer’s disease or pathology.
We also need to characterize the typical age trajectory of noradrenergic activity in the brain and how this corresponds to structural change in the LC. For instance, one hypothesis is that in aging and early Alzheimer’s disease, LC hyperactivity exacerbates Alzheimer’s pathology but that in later stages of the disease, loss of LC neurons and diminished noradrenergic activity lead to cognitive impairment and neuroinflammation [264]. As outlined in Section 4.2, we still have only scant human data regarding age differences in the levels of production and clearance of noradrenaline. Although we cannot yet measure LC neuron activity non-invasively in the human brain, we do have indirect measures such as pupil dilation [319] and the p300 event-related potential [320]. Frontoparietal attention networks provide another indirect window into LC activity and integrity [321-323]. Given the documented age differences in these measures, the degree to which resting-state LC activity is coordinated with frontoparietal attention networks [324, 325] and the variability of frontoparietal network activity at rest [326] may prove to be indicators of age differences in LC activity. Noradrenaline release may also lead to EEG α-β desynchronization [327] as well as increase stimulus-induced gamma oscillations [328]. At this point, mapping how these different measures relate to each other and to structural MRI LC contrast might help clarify which measures would be most relevant. In particular, animal work that includes functional MRI measures alongside direct measures of LC activity would be very helpful for advancing LC imaging in humans. Developing well-validated measures will be essential to guide future interventions and to characterize the trajectory of change in LC activity during aging and Alzheimer’s disease.
Indeed, the ultimate goal of this line of inquiry is to slow down the accumulation of Alzheimer’s pathology. Pharmacological, behavioral and lifestyle interventions all have some potential; however, gaining more information regarding how noradrenergic activity changes in aging and how these changes affect Aβ and tau will be essential to design the best targeted interventions.
Acknowledgements
Funding:
This work was supported by the National Institutes of Health grant number R01AG025340. I thank Christine Cho for her assistance with the figure and Martin Dahl and Lissa Riley for comments on a previous version.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
3715 McClintock Ave. University of Southern California Los Angeles, CA 90089
References
- 1.James T, et al. , Locus coeruleus in memory formation and Alzheimer’s disease. European Journal of Neuroscience, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mather M and Harley CW, The locus coeruleus: Essential for maintaining cognitive function and the aging brain. Trends in Cognitive Sciences, 2016. 20: p. 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robertson IH, A noradrenergic theory of cognitive reserve: implications for Alzheimer’s disease. Neurobiology of aging, 2013. 34(1): p. 298–308. [DOI] [PubMed] [Google Scholar]
- 4.Wilson RS, et al. , Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology, 2013. 80(13): p. 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelly SC, et al. , Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta neuropathologica communications, 2017. 5(1): p. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyness SA, Zarow C, and Chui HC, Neuron loss in key cholinergic and aminergic nuclei in Alzheimer disease: a meta-analysis. Neurobiology of aging, 2003. 24(1): p. 1–23. [DOI] [PubMed] [Google Scholar]
- 7.Ohm T, Busch C, and Bohl J, Unbiased estimation of neuronal numbers in the human nucleus coeruleus during aging. Neurobiology of aging, 1997. 18(4): p. 393–399. [DOI] [PubMed] [Google Scholar]
- 8.Theofilas P, et al. , Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimer’s & Dementia, 2017. 13(3): p. 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braak H, et al. , Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. Journal of Neuropathology & Experimental Neurology, 2011. 70(11): p. 960–969. [DOI] [PubMed] [Google Scholar]
- 10.Harley CW, et al. , The ’a, b, c’s of pretangle tau and their relation to aging and the risk of Alzheimer’s disease. Seminars in Cell and Developmental Biology, in press. [DOI] [PubMed] [Google Scholar]
- 11.Keren NI, et al. , Histologic validation of locus coeruleus MRI contrast in post-mortem tissue. NeuroImage, 2015. 113: p. 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Betts MJ, et al. , Locus coeruleus MRI contrast is reduced in Alzheimer’s disease dementia and correlates with CSF Aβ levels. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, 2019. 11: p. 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahl MJ, et al. , Locus coeruleus integrity is related to tau burden and memory loss in autosomal-dominant Alzheimer’s disease. medRxiv, 2021: p. 2020.11. 16.20232561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hou R, et al. , A case-control study of the locus coeruleus degeneration in Alzheimer’s disease. European Neuropsychopharmacology, 2021. 43: p. 153–159. [DOI] [PubMed] [Google Scholar]
- 15.Olivieri P, et al. , Early alteration of the locus coeruleus in phenotypic variants of Alzheimer’s disease. Annals of clinical and translational neurology, 2019. 6(7): p. 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takahashi J, et al. , Detection of changes in the locus coeruleus in patients with mild cognitive impairment and Alzheimer’s disease: High-resolution fast spin-echo T1-weighted imaging. Geriatrics & Gerontology International, 2015. 15(3): p. 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clewett D, et al. , Neuromelanin marks the spot: Identifying a locus coeruleus biomarker of cognitive reserve in healthy aging. Neurobiology of Aging, 2016. 37: p. 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahl MJ, et al. , Rostral locus coeruleus integrity is associated with better memory performance in older adults. Nature Human Behaviour, 2019. 3: p. 1203–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elman JA, et al. , MRI-assessed locus coeruleus integrity is heritable and associated with cognition, Alzheimer’s risk, and sleep-wake disturbance: Neuroimaging: Environmental and lifestyle factors. Alzheimer’s & Dementia, 2020. 16: p. e044862. [Google Scholar]
- 20.Liu KY, et al. , Noradrenergic-dependent functions are associated with age-related locus coeruleus signal intensity differences. Nature communications, 2020. 11(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dutt S, et al. , Brainstem volumetric integrity in preclinical and prodromal Alzheimer’s disease. Journal of Alzheimer’s Disease, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dutt S, et al. , Brainstem substructures and cognition in prodromal Alzheimer’s disease. Brain Imaging and Behavior, 2021: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matchett BJ, et al. , The mechanistic link between selective vulnerability of the locus coeruleus and neurodegeneration in Alzheimer’s disease. Acta neuropathologica, 2021: p. 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson IH, A right hemisphere role in cognitive reserve. Neurobiology of aging, 2014. 35(6): p. 1375–1385. [DOI] [PubMed] [Google Scholar]
- 25.Xu W, et al. , Cognitive reserve and Alzheimer’s disease. Molecular neurobiology, 2015. 51(1): p. 187–208. [DOI] [PubMed] [Google Scholar]
- 26.Stern Y, et al. , Influence of education and occupation on the incidence of Alzheimer’s disease. Jama, 1994. 271(13): p. 1004–1010. [PubMed] [Google Scholar]
- 27.Stern Y, et al. , Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer’s & Dementia, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mather M, How do cognitively stimulating activities affect cognition and the brain throughout life? Psychological Science in the Public Interest, 2020. 21(1): p. 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plini ERG, et al. , Examining the Role of the Noradrenergic Locus Coeruleus for Predicting Attention and Brain Maintenance in Healthy Old Age, Mild Cognitive Impairment and Alzheimer’s Disease: An MRI Structural Study On the Adni Cohort. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basak C, Qin S, and O’Connell MA, Differential effects of cognitive training modules in healthy aging and mild cognitive impairment: A comprehensive meta-analysis of randomized controlled trials. Psychology and aging, 2020. 35(2): p. 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards JD, et al. , Systematic review and meta-analyses of useful field of view cognitive training. Neuroscience & Biobehavioral Reviews, 2018. 84: p. 72–91. [DOI] [PubMed] [Google Scholar]
- 32.Kelly ME, et al. , The impact of cognitive training and mental stimulation on cognitive and everyday functioning of healthy older adults: a systematic review and meta-analysis. Ageing research reviews, 2014. 15: p. 28–43. [DOI] [PubMed] [Google Scholar]
- 33.Lampit A, Hallock H, and Valenzuela M, Computerized cognitive training in cognitively healthy older adults: a systematic review and meta-analysis of effect modifiers. PLoS medicine, 2014. 11(11): p. e1001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marusic U, Verghese J, and Mahoney JR, Cognitive-based interventions to improve mobility: a systematic review and meta-analysis. Journal of the American Medical Directors Association, 2018. 19(6): p. 484–491. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen L, Murphy K, and Andrews G, Immediate and long-term efficacy of executive functions cognitive training in older adults: A systematic review and meta-analysis. Psychological Bulletin, 2019. 145(7): p. 698–733. [DOI] [PubMed] [Google Scholar]
- 36.Tetlow AM and Edwards JD, Systematic literature review and meta-analysis of commercially available computerized cognitive training among older adults. Journal of Cognitive Enhancement, 2017. 1(4): p. 559–575. [Google Scholar]
- 37.Barcelos NM, et al. , Guanfacine treatment for prefrontal cognitive dysfunction in older participants: a randomized clinical trial. Neurobiology of aging, 2018. 70: p. 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ClinicalTrials.gov. Noradrenergic Add-on Therapy With Guanfacine. 2017. [cited 2020 Nov. 18]; Available from: https://ClinicalTrials.gov/show/NCT03116126.
- 39.ClinicalTrials.gov. Lymphatic System Health in Alzheimer’s Disease. 2019. [cited 2020 Dec. 22]; Available from: https://clinicaltrials.gov/ct2/show/.NCT04205539
- 40.ClinicalTrials.gov. Trial of Carvedilol in Alzheimer’s Disease. 2011. [cited 2020 Nov. 18]; Available from: https://clinicaltrials.gov/ct2/show/results/.NCT01354444
- 41.ClinicalTrials.gov. Improving Beta-2 Adrenergic Signaling in Alzheimer’s Disease. 2015. [cited 2020 Nov. 18]; Available from: https://clinicaltrials.gov/ct2/show/.NCT02500784
- 42.ClinicalTrials.gov. Alzheimer’s in Long-Term Care--Treatment for Agitation. 2005. [cited 2020 Dec. 22]; Available from: https://clinicaltrials.gov/ct2/show/.NCT00161473
- 43.ClinicalTrials.gov. Effects of Atomoxetine in Mild Cognitive Impairment. 2012. [cited 2020 Dec. 22]; Available from: https://clinicaltrials.gov/ct2/show/.NCT01522404
- 44.Nguyen L, Murphy K, and Andrews G, Cognitive and neural plasticity in old age: a systematic review of evidence from executive functions cognitive training. Ageing Research Reviews, 2019. 53: p. 100912. [DOI] [PubMed] [Google Scholar]
- 45.Park DC and Bischof GN, The aging mind: neuroplasticity in response to cognitive training. Dialogues in clinical neuroscience, 2013. 15(1): p. 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Balkom TD, et al. , The Effects of Cognitive Training on Brain Network Activity and Connectivity in Aging and Neurodegenerative Diseases: a Systematic Review. Neuropsychology Review, 2020: p. 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marzo A, Bai J, and Otani S, Neuroplasticity regulation by noradrenaline in mammalian brain. Current neuropharmacology, 2009. 7(4): p. 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos BP and Arnsten AFT, Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacology & Therapeutics, 2007. 113(3): p. 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salgado H, Kohr G, and Trevino M, Noradrenergic ‘one’ determines dichotomous control of cortical spike-timing-dependent plasticity. Scientific Reports, 2012. 2: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Oliveira PG, et al. , Gi/o-Protein coupled receptors in the aging brain. Frontiers in aging neuroscience, 2019. 11: p. 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosenbaum DM, Rasmussen SG, and Kobilka BK, The structure and function of G-protein-coupled receptors. Nature, 2009. 459(7245): p. 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wess J, Molecular Basis of Receptor/G-Protein-Coupling Selectivity. Pharmacology & Therapeutics, 1998. 80(3): p. 231–264. [DOI] [PubMed] [Google Scholar]
- 53.Daaka Y, Luttrell LM, and Lefkowitz RJ, Switching of the coupling of the β 2-adrenergic receptor to different G proteins by protein kinase A. Nature, 1997. 390(6655): p. 88–91. [DOI] [PubMed] [Google Scholar]
- 54.Schutsky K, et al. , Stress and glucocorticoids impair memory retrieval via β2-adrenergic, Gi/o-coupled suppression of cAMP signaling. Journal of Neuroscience, 2011. 31(40): p. 14172–14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stiles GL, Caron MG, and Lefkowitz RJ, Beta-adrenergic receptors: biochemical mechanisms of physiological regulation. Physiological reviews, 1984. 64(2): p. 661–743. [DOI] [PubMed] [Google Scholar]
- 56.Sassone-Corsi P, The cyclic AMP pathway. Cold Spring Harbor perspectives in biology, 2012. 4(12): p. a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maity S, et al. , Norepinephrine stabilizes translation-dependent, homosynaptic long-term potentiation through mechanisms requiring the cAMP sensor Epac, mTOR and MAPK. European Journal of Neuroscience, 2020. 52(7): p. 3679–3688. [DOI] [PubMed] [Google Scholar]
- 58.O’Dell TJ, et al. , β-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learning & Memory, 2015. 22(9): p. 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Musheshe N, Schmidt M, and Zaccolo M, cAMP: From long-range second messenger to nanodomain signalling. Trends in Pharmacological Sciences, 2018. 39(2): p. 209–222. [DOI] [PubMed] [Google Scholar]
- 60.Starke K, Presynaptic autoreceptors in the third decade: focus on α2-adrenoceptors. Journal of neurochemistry, 2001. 78(4): p. 685–693. [DOI] [PubMed] [Google Scholar]
- 61.Nanou E and Catterall WA, Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron, 2018. 98(3): p. 466–481. [DOI] [PubMed] [Google Scholar]
- 62.Sastre M and García-Sevilla JA, α2-Adrenoceptor Subtypes Identified by [3H] RX821002 Binding in the Human Brain: The Agonist Guanoxabenz Does Not Discriminate Different Forms of the Predominant α2a Subtype. Journal of neurochemistry, 1994. 63(3): p. 1077–1085. [DOI] [PubMed] [Google Scholar]
- 63.Li Y-W and Bayliss D, Activation of α2-adrenoceptors causes inhibition of calcium channels but does not modulate inwardly-rectifying K+ channels in caudal raphe neurons. Neuroscience, 1997. 82(3): p. 753–765. [DOI] [PubMed] [Google Scholar]
- 64.Gilsbach R, et al. , Genetic dissection of α2-adrenoceptor functions in adrenergic versus nonadrenergic cells. Molecular pharmacology, 2009. 75(5): p. 1160–1170. [DOI] [PubMed] [Google Scholar]
- 65.Arnsten A and Goldman-Rakic PS, Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science, 1985. 230(4731): p. 1273–1276. [DOI] [PubMed] [Google Scholar]
- 66.Erdozain AM, et al. , Differential α2A-and α2C-adrenoceptor protein expression in presynaptic and postsynaptic density fractions of postmortem human prefrontal cortex. Journal of Psychopharmacology, 2019. 33(2): p. 244–249. [DOI] [PubMed] [Google Scholar]
- 67.Mather M, The locus coeruleus-norepinephrine system role in cognition and how it changes with aging, in The Cognitive Neurosciences, Poeppel D, Mangun G, and Gazzaniga M, Editors. 2020, MIT Press: Cambridge, MA. p. 91–101. [Google Scholar]
- 68.Veyrac A, et al. , Novelty determines the effects of olfactory enrichment on memory and neurogenesis through noradrenergic mechanisms. Neuropsychopharmacology, 2009. 34(3): p. 786–795. [DOI] [PubMed] [Google Scholar]
- 69.Naka F, et al. , An enriched environment increases noradrenaline concentration in the mouse brain. Brain research, 2002. 924(1): p. 124–126. [DOI] [PubMed] [Google Scholar]
- 70.Duszkiewicz AJ, et al. , Novelty and dopaminergic modulation of memory persistence: a tale of two systems. Trends in Neurosciences, 2019. 42(2): p. 102–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mohammed AK, Jonsson G, and Archer T, Selective lesioning of forebrain noradrenaline neurons at birth abolishes the improved maze learning performance induced by rearing in complex environment. Brain research, 1986. 398(1): p. 6–10. [DOI] [PubMed] [Google Scholar]
- 72.Pappas BA, et al. , Forebrain norepinephrine and neurobehavioral plasticity: Neonatal 6-hydroxydopamine eliminates enriched-impoverished experience effects on maze performance. Pharmacology Biochemistry and Behavior, 1987. 27(1): p. 153–158. [DOI] [PubMed] [Google Scholar]
- 73.Coradazzi M, et al. , Selective noradrenaline depletion impairs working memory and hippocampal neurogenesis. Neurobiology of Aging, 2016. 48: p. 93–102. [DOI] [PubMed] [Google Scholar]
- 74.Kulkarni VA, Jha S, and Vaidya VA, Depletion of norepinephrine decreases the proliferation, but does not influence the survival and differentiation, of granule cell progenitors in the adult rat hippocampus. European Journal of Neuroscience, 2002. 16(10): p. 2008–2012. [DOI] [PubMed] [Google Scholar]
- 75.Bortolotto V, et al. , Salmeterol, a β2 adrenergic agonist, promotes adult hippocampal neurogenesis in a region-specific manner. Frontiers in pharmacology, 2019. 10: p. 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jhaveri DJ, et al. , Norepinephrine directly activates adult hippocampal precursors via β3-adrenergic receptors. Journal of Neuroscience, 2010. 30(7): p. 2795–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jhaveri DJ, et al. , Opposing effects of α2-and β-adrenergic receptor stimulation on quiescent neural precursor cell activity and adult hippocampal neurogenesis. PLoS One, 2014. 9(6): p. e98736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hagenston AM, Bading H, and Bas-Orth C, Functional consequences of calcium-dependent synapse-to-nucleus communication: Focus on transcription-dependent metabolic plasticity. Cold Spring Harbor perspectives in biology, 2020. 12(4): p. a035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li S, et al. , Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron, 2013. 77(5): p. 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor SS, et al. , PKA: lessons learned after twenty years. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 2013. 1834(7): p. 1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kandel ER, The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Molecular brain, 2012. 5(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kandel ER, The molecular biology of memory storage: a dialogue between genes and synapses. Science, 2001. 294(5544): p. 1030–1038. [DOI] [PubMed] [Google Scholar]
- 83.Cirelli C and Tononi G, Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. The Journal of Neuroscience, 2000. 20(24): p. 9187–9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carriba P, et al. , ATP and noradrenaline activate CREB in astrocytes via noncanonical Ca2+ and cyclic AMP independent pathways. Glia, 2012. 60(9): p. 1330–1344. [DOI] [PubMed] [Google Scholar]
- 85.Mårtensson J, et al. , Growth of language-related brain areas after foreign language learning. NeuroImage, 2012. 63(1): p. 240–244. [DOI] [PubMed] [Google Scholar]
- 86.Woollett K and Maguire EA, Acquiring “the Knowledge” of London’s layout drives structural brain changes. Current biology, 2011. 21(24): p. 2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kühn S, et al. , Playing Super Mario induces structural brain plasticity: gray matter changes resulting from training with a commercial video game. Molecular psychiatry, 2014. 19(2): p. 265–271. [DOI] [PubMed] [Google Scholar]
- 88.Wenger E, et al. , Expansion and renormalization of human brain structure during skill acquisition. Trends in cognitive sciences, 2017. 21(12): p. 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mather M and Sutherland MR, Arousal-biased competition in perception and memory. Perspectives on Psychological Science, 2011. 6: p. 114–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clewett D, et al. , Noradrenergic mechanisms of arousal’s bidirectional effects on episodic memory. Neurobiology of learning and memory, 2017. 137: p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strange BA, Hurlemann R, and Dolan RJ, An emotion-induced retrograde amnesia in humans is amygdala- and beta-adrenergic-dependent. Proceedings of the National Academy of Sciences of the United States of America, 2003. 100(23): p. 13626–13631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Abdou K, et al. , Synapse-specific representation of the identity of overlapping memory engrams. Science, 2018. 360(6394): p. 1227–1231. [DOI] [PubMed] [Google Scholar]
- 93.Blumenfeld B, et al. , Dynamics of memory representations in networks with novelty-facilitated synaptic plasticity. Neuron, 2006. 52(2): p. 383–394. [DOI] [PubMed] [Google Scholar]
- 94.O’Reilly KC, Perica MI, and Fenton AA, Synaptic plasticity/dysplasticity, process memory and item memory in rodent models of mental dysfunction. Schizophrenia research, 2019. 207: p. 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Susman L, Brenner N, and Barak O, Stable memory with unstable synapses. Nature communications, 2019. 10(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee W-CA, et al. , Anatomy and function of an excitatory network in the visual cortex. Nature, 2016. 532(7599): p. 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lewerenz J and Maher P, Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Frontiers in neuroscience, 2015. 9: p. 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou Y and Danbolt NC, Glutamate as a neurotransmitter in the healthy brain. Journal of neural transmission, 2014. 121(8): p. 799–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okubo Y and Iino M, Visualization of glutamate as a volume transmitter. The Journal of physiology, 2011. 589(3): p. 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Papouin T and Oliet SH, Organization, control and function of extrasynaptic NMDA receptors. Philosophical Transactions of the Royal Society B: Biological Sciences, 2014. 369(1654): p. 20130601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mather M, et al. , Norepinephrine ignites local hotspots of neuronal excitation: How arousal amplifies selectivity in perception and memory. Behavioral and Brain Sciences, 2016. 39: p. e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Eldar E, Cohen JD, and Niv Y, The effects of neural gain on attention and learning. Nature neuroscience, 2013. 16: p. 1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Servan-Schreiber D, Printz H, and Cohen JD, A network model of catecholamine effects: gain, signal-to-noise ratio, and behavior. Science, 1990. 249(4971): p. 892–895. [DOI] [PubMed] [Google Scholar]
- 104.Atmore KH, et al. , Differential effects of social isolation rearing on glutamate-and GABA-stimulated noradrenaline release in the rat prefrontal cortex and hippocampus. European Neuropsychopharmacology, 2020. 36: p. 111–120. [DOI] [PubMed] [Google Scholar]
- 105.Göthert M and Fink K, Stimulation of noradrenaline release in the cerebral cortex via presynaptic N-methyl-D-aspartate (NMDA) receptors and their pharmacological characterization, in Recent Advances in Neuropharmacology. 1991, Springer, p. 121–127. [DOI] [PubMed] [Google Scholar]
- 106.Pittaluga A and Raiteri M, N-methyl-D-aspartic acid (NMDA) and non-NMDA receptors regulating hippocampal norepinephrine release, I. Location on axon terminals and pharmacological characterization. Journal of Pharmacology and Experimental Therapeutics, 1992. 260(1): p. 232–237. [PubMed] [Google Scholar]
- 107.Snell LD, Yi S-J, and Johnson KM, Comparison of the effects of MK-801 and phencyclidine on catecholamine uptake and NMDA-induced norepinephrine release. European journal of pharmacology, 1988. 145(2): p. 223–226. [DOI] [PubMed] [Google Scholar]
- 108.Takahashi M, Hayashi Y, and Tanaka J, Glutamatergic modulation of noradrenaline release in the rat median preoptic area. Brain research bulletin, 2017. 130: p. 36–41. [DOI] [PubMed] [Google Scholar]
- 109.Fink K, Schultheiß R, and Göthert M, Stimulation of noradrenaline release in human cerebral cortex mediated by N-methyl-d-aspartate (NMDA) and non-NMDA receptors. British Journal of Pharmacology, 1992. 106(1): p. 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luccini E, et al. , Functional interactions between presynaptic NMDA receptors and metabotropic glutamate receptors co-expressed on rat and human noradrenergic terminals. British Journal of Pharmacology, 2007. 151(7): p. 1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pittaluga A, et al. , Activity of putative cognition enhancers in kynurenate test performed with human neocortex slices. Journal of Pharmacology and Experimental Therapeutics, 1999. 290(1): p. 423–428. [PubMed] [Google Scholar]
- 112.Egli RE, et al. , Norepinephrine modulates glutamatergic transmission in the bed nucleus of the stria terminalis. Neuropsychopharmacology, 2005. 30(4): p. 657–668. [DOI] [PubMed] [Google Scholar]
- 113.Gereau R and Conn PJ, Presynaptic enhancement of excitatory synaptic transmission by beta-adrenergic receptor activation. Journal of neurophysiology, 1994. 72(3): p. 1438–1442. [DOI] [PubMed] [Google Scholar]
- 114.Ji X-H, et al. , Pre-and postsynaptic β-adrenergic activation enhances excitatory synaptic transmission in layer V/VI pyramidal neurons of the medial prefrontal cortex of rats. Cerebral Cortex, 2008. 18(7): p. 1506–1520. [DOI] [PubMed] [Google Scholar]
- 115.Lüscher C and Malenka RC, NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspectives in Biology, 2012. 4(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang H-P, et al. , Long latency of evoked quantal transmitter release from somata of locus coeruleus neurons in rat pontine slices. Proceedings of the National Academy of Sciences, 2007. 104(4): p. 1401–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ross JA, Reyes BA, and Van Bockstaele EJ, Amyloid beta peptides, locus coeruleus-norepinephrine system and dense core vesicles. Brain research, 2019. 1702: p. 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fernández-Pastor B and Meana JJ, In vivo tonic modulation of the noradrenaline release in the rat cortex by locus coeruleus somatodendritic α2-adrenoceptors. European journal of pharmacology, 2002. 442(3): p. 225–229. [DOI] [PubMed] [Google Scholar]
- 119.Mahmoud S, et al. , Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells, 2019. 8(2): p. 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aoki C, Beta-adrenergic receptors: astrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. Journal of Neuroscience, 1992. 12(3): p. 781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zuend M, et al. , Arousal-induced cortical activity triggers lactate release from astrocytes. Nature Metabolism, 2020. 2(2): p. 179–191. [DOI] [PubMed] [Google Scholar]
- 122.Magistretti PJ. and Allaman I, Lactate in the brain: from metabolic end-product to signalling molecule. Nature Reviews Neuroscience, 2018. 19(4): p. 235. [DOI] [PubMed] [Google Scholar]
- 123.Paukert M, et al. , Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron, 2014. 82(6): p. 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoffman DA and Johnston D, Neuromodulation of dendritic action potentials. Journal of neurophysiology, 1999. 81(1): p. 408–411. [DOI] [PubMed] [Google Scholar]
- 125.Liu Y, et al. , Adrenergic gate release for spike timing-dependent synaptic potentiation. Neuron, 2017. 93(2): p. 394–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dietl H, Sinha J, and Philippu A, Presynaptic regulation of the release of catecholamines in the cat hypothalamus. Brain Research, 1981. 208(1): p. 213–218. [DOI] [PubMed] [Google Scholar]
- 127.Arnsten AF, Through the looking glass: differential noradenergic modulation of prefrontal cortical function. Neural plasticity, 2000. 7(1-2): p. 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arnsten AF, Guanfacine’s mechanism of action in treating prefrontal cortical disorders: Successful translation across species. Neurobiology of Learning and Memory, 2020. 176: p. 107327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Munn BR, et al. , The ascending arousal system shapes low-dimensional brain dynamics to mediate awareness of changes in intrinsic cognitive states. bioRxiv, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Herrup K, The case for rejecting the amyloid cascade hypothesis. Nature neuroscience, 2015. 18(6): p. 794–799. [DOI] [PubMed] [Google Scholar]
- 131.Kayed R and Lasagna-Reeves CA, Molecular mechanisms of amyloid oligomers toxicity. Journal of Alzheimer’s Disease, 2013. 33(s1): p. S67–S78. [DOI] [PubMed] [Google Scholar]
- 132.Shankar GM, et al. , Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature medicine, 2008. 14(8): p. 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee SJC, et al. , Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chemical Society Reviews, 2017. 46(2): p. 310–323. [DOI] [PubMed] [Google Scholar]
- 134.de Coninck D, et al. , Packing Density of the Amyloid Precursor Protein in the Cell Membrane. Biophysical journal, 2018. 114(5): p. 1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vassar R, et al. , β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. science, 1999. 286(5440): p. 735–741. [DOI] [PubMed] [Google Scholar]
- 136.Cirrito JR, et al. , Endocytosis is Required for Synaptic Activity-Dependent Release of Amyloid-β In Vivo. Neuron, 2008. 58(1): p. 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cirrito JR, et al. , Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron, 2005. 48(6): p. 913–922. [DOI] [PubMed] [Google Scholar]
- 138.Kamenetz F, et al. , APP Processing and Synaptic Function. Neuron, 2003. 37(6): p. 925–937. [DOI] [PubMed] [Google Scholar]
- 139.Parvathy S, et al. , Cleavage of Alzheimer’s amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry, 1999. 38(30): p. 9728–9734. [DOI] [PubMed] [Google Scholar]
- 140.Caccamo A, et al. , M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron, 2006. 49(5): p. 671–682. [DOI] [PubMed] [Google Scholar]
- 141.Nitsch RM, et al. , Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science, 1992. 258(5080): p. 304–307. [DOI] [PubMed] [Google Scholar]
- 142.Hsieh H, et al. , AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron, 2006. 52(5): p. 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walsh DM, et al. , Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature, 2002. 416(6880): p. 535–539. [DOI] [PubMed] [Google Scholar]
- 144.Lazarevic V, et al. , Physiological concentrations of amyloid beta regulate recycling of synaptic vesicles via alpha7 acetylcholine receptor and CDK5/calcineurin signaling. Frontiers in molecular neuroscience, 2017. 10: p. 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garcia-Osta A and Alberini CM, Amyloid beta mediates memory formation. Learning & memory, 2009. 16(4): p. 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gulisano W, et al. , Neuromodulatory action of picomolar extracellular Aβ42 oligomers on presynaptic and postsynaptic mechanisms underlying synaptic function and memory. Journal of Neuroscience, 2019. 39(30): p. 5986–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Puzzo D, et al. , Endogenous amyloid-β is necessary for hippocampal synaptic plasticity and memory. Annals of neurology, 2011. 69(5): p. 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Puzzo D, et al. , Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. Journal of Neuroscience, 2008. 28(53): p. 14537–14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sengupta U, Nilson AN, and Kayed R, The Role of Amyloid-β Oligomers in Toxicity Propagation, and Immunotherapy. EBioMedicine, 2016. 6: p. 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang T, et al. , Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. The Journal of Neuroscience, 2017. 37(1): p. 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li S and Selkoe DJ, A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer’s brain. Journal of Neurochemistry, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Buckner RL, et al. , Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. The Journal of Neuroscience, 2005. 25(34): p. 7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Palmqvist S, et al. , Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nature communications, 2017. 8(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bero AW, et al. , Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nature neuroscience, 2011. 14(6): p. 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morris M, et al. , The many faces of tau. Neuron, 2011. 70(3): p. 410–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lee VM, et al. , Developing therapeutic approaches to tau, selected kinases, and related neuronal protein targets. Cold Spring Harbor perspectives in medicine, 2011. 1(1): p. a006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yamada K, et al. , In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. Journal of Neuroscience, 2011. 31(37): p. 13110–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vogels T, et al. , Propagation of tau pathology: integrating insights from post mortem and in vivo studies. Biological psychiatry, 2019. [DOI] [PubMed] [Google Scholar]
- 159.Wu JW, et al. , Neuronal activity enhances tau propagation and tau pathology in vivo. Nature Neuroscience, 2016. 19: p. 1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rodriguez GA, et al. , Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Aβ and tau pathology in the hippocampus. PLoS biology, 2020. 18(8): p. e3000851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yamada K, et al. , Neuronal activity regulates extracellular tau in vivo. Journal of Experimental Medicine, 2014. 211(3): p. 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pooler AM, et al. , Physiological release of endogenous tau is stimulated by neuronal activity. EMBO reports, 2013. 14(4): p. 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schultz MK, et al. , Pharmacogenetic neuronal stimulation increases human tau pathology and trans-synaptic spread of tau to distal brain regions in mice. Neurobiology of Disease, 2018. 118: p. 161–176. [DOI] [PubMed] [Google Scholar]
- 164.Kalluri R and LeBleu VS, The biology, function, and biomedical applications of exosomes. Science, 2020. 367(6478): p. eaau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Saman S, et al. , Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. Journal of Biological Chemistry, 2012. 287(6): p. 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang Y, et al. , The release and trans-synaptic transmission of Tau via exosomes. Molecular Neurodegeneration, 2017. 12(1): p. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Asai H, et al. , Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nature neuroscience, 2015. 18(11): p. 1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hopp SC, et al. , The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. Journal of neuroinflammation, 2018. 15(1): p. 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Luft AR and Buitrago MM, Stages of motor skill learning. Molecular neurobiology, 2005. 32(3): p. 205–216. [DOI] [PubMed] [Google Scholar]
- 170.Jankowsky JL, et al. , Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. Journal of Neuropathology & Experimental Neurology, 2003. 62(12): p. 1220–1227. [DOI] [PubMed] [Google Scholar]
- 171.Jankowsky JL, et al. , Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. Journal of Neuroscience, 2005. 25(21): p. 5217–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lazarov O, et al. , Environmental enrichment reduces Aβ levels and amyloid deposition in transgenic mice. Cell, 2005. 120(5): p. 701–713. [DOI] [PubMed] [Google Scholar]
- 173.Costa DA, et al. , Enrichment improves cognition in AD mice by amyloid-related and unrelated mechanisms. Neurobiology of aging, 2007. 28(6): p. 831–844. [DOI] [PubMed] [Google Scholar]
- 174.Kress BT, et al. , Impairment of paravascular clearance pathways in the aging brain. Annals of neurology, 2014. 76(6): p. 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Silverberg GD, et al. , Amyloid efflux transporter expression at the blood-brain barrier declines in normal aging. Journal of Neuropathology & Experimental Neurology, 2010. 69(10): p. 1034–1043. [DOI] [PubMed] [Google Scholar]
- 176.Hersi M, et al. , Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology, 2017. 61: p. 143–187. [DOI] [PubMed] [Google Scholar]
- 177.Stern Y, Cognitive reserve in ageing and Alzheimer’s disease. The Lancet Neurology, 2012. 11(11): p. 1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Roe CM, et al. , Education and Alzheimer disease without dementia: support for the cognitive reserve hypothesis. Neurology, 2007. 68(3): p. 223–228. [DOI] [PubMed] [Google Scholar]
- 179.Serrano-Pozo A, et al. , Examination of the clinicopathologic continuum of Alzheimer disease in the autopsy cohort of the National Alzheimer Coordinating Center. Journal of Neuropathology & Experimental Neurology, 2013. 72(12): p. 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jansen WJ, et al. , Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. Jama, 2015. 313(19): p. 1924–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vemuri P, et al. , Evaluation of amyloid protective factors and Alzheimer disease neurodegeneration protective factors in elderly individuals. JAMA neurology, 2017. 74(6): p. 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yasuno F, et al. , Low amyloid-β deposition correlates with high education in cognitively normal older adults: a pilot study. International journal of geriatric psychiatry, 2015. 30(9): p. 919–926. [DOI] [PubMed] [Google Scholar]
- 183.Nyberg L, et al. , Educational attainment does not influence brain aging. Proceedings of the National Academy of Sciences, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Tucker-Drob EM, Cognitive aging and dementia: A life-span perspective. Annual Review of Developmental Psychology, 2019. 1: p. 177–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lövdén M, et al. , Education and cognitive functioning across the life span. Psychological Science in the Public Interest, 2020. 21(1): p. 6–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Oveisgharan S, et al. , Association of early-life cognitive enrichment with Alzheimer disease pathological changes and cognitive decline. JAMA neurology, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Davalos D, et al. , ATP mediates rapid microglial response to local brain injury in vivo. Nature neuroscience, 2005. 8(6): p. 752–758. [DOI] [PubMed] [Google Scholar]
- 188.Perry VH, Nicoll JA, and Holmes C, Microglia in neurodegenerative disease. Nature Reviews Neurology, 2010. 6(4): p. 193. [DOI] [PubMed] [Google Scholar]
- 189.Hansen DV, Hanson JE, and Sheng M, Microglia in Alzheimer’s disease. Journal of Cell Biology, 2018. 217(2): p. 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sarlus H and Heneka MT, Microglia in Alzheimer’s disease. The Journal of Clinical Investigation, 2017. 127(9): p. 3240–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.O’Donnell J, et al. , Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochemical research, 2012. 37(11): p. 2496–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sugama S, et al. , Stress-induced microglial activation occurs through β-adrenergic receptor: noradrenaline as a key neurotransmitter in microglial activation. Journal of neuroinflammation, 2019. 16(1): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Liu YU, et al. , Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nature neuroscience, 2019: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Stowell RD, et al. , Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nature neuroscience, 2019. 22(11): p. 1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Takahashi K, et al. , Locus coeruleus neuronal activity during the sleep-waking cycle in mice. Neuroscience, 2010. 169(3): p. 1115–1126. [DOI] [PubMed] [Google Scholar]
- 196.Fujita H, et al. , Adrenergic agonists suppress the proliferation of microglia through β2-adrenergic receptor. Neuroscience letters, 1998. 242(1): p. 37–40. [DOI] [PubMed] [Google Scholar]
- 197.Gyoneva S and Traynelis SF, Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. Journal of Biological Chemistry, 2013. 288(21): p. 15291–15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kong Y, et al. , Norepinephrine promotes microglia to uptake and degrade amyloid β peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. The Journal of Neuroscience, 2010. 30(35): p. 11848–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tamano H, et al. , Adrenergic β receptor activation in the basolateral amygdala, which is intracellular Zn2+−dependent, rescues amyloid β1-42-induced attenuation of dentate gyrus LTP. Neurochemistry international, 2018. 120: p. 43–48. [DOI] [PubMed] [Google Scholar]
- 200.Tamano H, et al. , Adrenergic β receptor activation reduces amyloid β1-42-mediated intracellular Zn2+ toxicity in dentate granule cells followed by rescuing impairment of dentate gyrus LTP. NeuroToxicology, 2020. [DOI] [PubMed] [Google Scholar]
- 201.Heneka MT, et al. , Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. Journal of Neuroscience, 2002. 22(7): p. 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Jardanhazi-Kurutz D, et al. , Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience, 2011. 176: p. 396–407. [DOI] [PubMed] [Google Scholar]
- 203.Jardanhazi-Kurutz D, et al. , Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochemistry International, 2010. 57(4): p. 375–382. [DOI] [PubMed] [Google Scholar]
- 204.Kalinin S, et al. , Noradrenaline deficiency in brain increases β-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiology of aging, 2007. 28(8): p. 1206–1214. [DOI] [PubMed] [Google Scholar]
- 205.Heneka MT, et al. , Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proceedings of the National Academy of Sciences, 2010. 107(13): p. 6058–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xu H, et al. , Enriched environment enhances β-adrenergic signaling to prevent microglia inflammation by amyloid-β. EMBO molecular medicine, 2018. 10(9): p. e8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Xu H, et al. , Environmental Enrichment Potently Prevents Microglia-Mediated Neuroinflammation by Human Amyloid β-Protein Oligomers. The Journal of Neuroscience, 2016. 36(35): p. 9041–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chai G.-s., et al. , Beta 2-adrenergic receptor activation enhances neurogenesis in Alzheimer’s disease mice. Neural regeneration research, 2016. 11(10): p. 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ni Y, et al. , Activation of β2-adrenergic receptor stimulates y-secretase activity and accelerates amyloid plaque formation. Nature medicine, 2006. 12(12): p. 1390. [DOI] [PubMed] [Google Scholar]
- 210.Dobarro M, Gerenu G, and Ramírez MJ, Propranolol reduces cognitive deficits, amyloid and tau pathology in Alzheimer’s transgenic mice. International Journal of Neuropsychopharmacology, 2013. 16(10): p. 2245–2257. [DOI] [PubMed] [Google Scholar]
- 211.Yu J-T, et al. , Polymorphisms at the β2-adrenergic receptor gene influence Alzheimer’s disease susceptibility. Brain research, 2008. 1210: p. 216–222. [DOI] [PubMed] [Google Scholar]
- 212.Dishy V, et al. , The effect of common polymorphisms of the β2-adrenergic receptor on agonist-mediated vascular desensitization. New England Journal of Medicine, 2001. 345(14): p. 1030–1035. [DOI] [PubMed] [Google Scholar]
- 213.Ukkola O, et al. , Interactions among the α2-, β2-, and β3-adrenergic receptor genes and obesity-related phenotypes in the Quebec Family Study. Metabolism-Clinical and Experimental, 2000. 49(8): p. 1063–1070. [DOI] [PubMed] [Google Scholar]
- 214.Rosenberg PB, et al. , Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. The American Journal of Geriatric Psychiatry, 2008. 16(11): p. 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wagner G, et al. , Antihypertensive treatment and risk of dementia: a retrospective database study. International journal of clinical pharmacology and therapeutics, 2012. 50(3): p. 195–201. [DOI] [PubMed] [Google Scholar]
- 216.Koivunen J, et al. , Amyloid PET imaging in patients with mild cognitive impairment: a 2-year follow-up study. Neurology, 2011. 76(12): p. 1085–1090. [DOI] [PubMed] [Google Scholar]
- 217.Chen Y, et al. , α2A adrenergic receptor promotes amyloidogenesis through disrupting APP-SorLA interaction. Proceedings of the National Academy of Sciences, 2014. 111(48): p. 17296–17301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Zhang F, et al. , The amyloid precursor protein modulates α2A-adrenergic receptor endocytosis and signaling through disrupting arrestin 3 recruitment. The FASEB Journal, 2017. 31(10): p. 4434–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhang F, et al. , β-amyloid redirects norepinephrine signaling to activate the pathogenic GSK3β/tau cascade. Science Translational Medicine, 2020. 12(526). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ehrenberg A, et al. , Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathology and applied neurobiology, 2017. 43(5): p. 393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kaur M and Singh P, Current role of dexmedetomidine in clinical anesthesia and intensive care. Anesthesia, essays and researches, 2011. 5(2): p. 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Jorm C and Stamford J, Actions of the hypnotic anaesthetic, dexmedetomidine, on noradrenaline release and cell firing in rat locus coeruleus slices. BJA: British Journal of Anaesthesia, 1993. 71(3): p. 447–449. [DOI] [PubMed] [Google Scholar]
- 223.Whittington RA, et al. , Dexmedetomidine increases tau phosphorylation under normothermic conditions in vivo and in vitro. Neurobiology of aging, 2015. 36(8): p. 2414–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wisely EV, Xiang YK, and Oddo S, Genetic suppression of β2-adrenergic receptors ameliorates tau pathology in a mouse model of tauopathies. Human Molecular Genetics, 2014. 23(15): p. 4024–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Chalermpalanupap T, et al. , Locus coeruleus ablation exacerbates cognitive deficits, neuropathology and lethality in P301S tau transgenic mice. The Journal of Neuroscience, 2018. 38(1): p. 74–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Busche MA and Hyman BT, Synergy between amyloid-β and tau in Alzheimer’s disease. Nature Neuroscience, 2020: p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Pascoal TA, et al. , Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease. Molecular psychiatry, 2017. 22(2): p. 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Beurel E, Grieco SF, and Jope RS, Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacology & therapeutics, 2015. 148: p. 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Da Mesquita S, et al. , Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature, 2018. 560(7717): p. 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tarasoff-Conway JM, et al. , Clearance systems in the brain—implications for Alzheimer disease. Nature reviews neurology, 2015. 11(8): p. 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wardlaw JM, et al. , Perivascular spaces in the brain: anatomy, physiology and pathology. Nature Reviews Neurology, 2020: p. 1–17. [DOI] [PubMed] [Google Scholar]
- 232.Jessen NA, et al. , The glymphatic system: a beginner’s guide. Neurochemical research, 2015. 40(12): p. 2583–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Plog BA and Nedergaard M, The glymphatic system in central nervous system health and disease: past, present, and future. Annual Review of Pathology: Mechanisms of Disease, 2018. 13: p. 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Iliff JJ, et al. , A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Science translational medicine, 2012. 4(147): p. 147ra 111–147ra 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Iliff JJ, et al. , Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. Journal of Neuroscience, 2014. 34(49): p. 16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hablitz LM, et al. , Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Science advances, 2019. 5(2): p. eaav5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Xie L, et al. , Sleep drives metabolite clearance from the adult brain, science, 2013. 342(6156): p. 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Benveniste H, et al. , Anesthesia with dexmedetomidine and low-dose isoflurane increases solute transport via the glymphatic pathway in rat brain when compared with high-dose isoflurane. Anesthesiology: The Journal of the American Society of Anesthesiologists, 2017. 127(6): p. 976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lilius TO, et al. , Dexmedetomidine enhances glymphatic brain delivery of intrathecally administered drugs. Journal of Controlled Release, 2019. 304: p. 29–38. [DOI] [PubMed] [Google Scholar]
- 240.Haywood J and Vogh B, Some measurements of autonomic nervous system influence on production of cerebrospinal fluid in the cat. Journal of Pharmacology and Experimental Therapeutics, 1979. 208(2): p. 341–346. [PubMed] [Google Scholar]
- 241.Lindvall M, Edvinsson L, and Owman C, Sympathetic nervous control of cerebrospinal fluid production from the choroid plexus. Science, 1978. 201(4351): p. 176–178. [DOI] [PubMed] [Google Scholar]
- 242.Lindvall M, Edvinsson L, and Owman C, Effect of sympathomimetic drugs and corresponding receptor antagonists on the rate of cerebrospinal fluid production. Experimental neurology, 1979. 64(1): p. 132–145. [DOI] [PubMed] [Google Scholar]
- 243.Bekar LK, Wei HS, and Nedergaard M, The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. Journal of Cerebral Blood Flow and Metabolism, 2012. 32: p. 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Gezalian MM, et al. , Cerebrovascular and neurological perspectives on adrenoceptor and calcium channel modulating pharmacotherapies. Journal of Cerebral Blood Flow & Metabolism, 2021.41(4): p. 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Peppiatt CM, et al. , Bidirectional control of CNS capillary diameter by pericytes. Nature, 2006. 443(7112): p. 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.van Veluw SJ, et al. , Vasomotion as a driving force for paravascular clearance in the awake mouse brain. Neuron, 2020. 105(3): p. 549–561. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Szot P, Elevated cerebrospinal fluid norepinephrine in the elderly can link depression and a reduced glymphatic system as risk factors for Alzheimer’s disease. Journal of Aging Science, 2016. [Google Scholar]
- 248.Kang SS, et al. , Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. The Journal of clinical investigation, 2020. 130(1): p. 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhang Z, et al. , Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nature medicine, 2014. 20(11): p. 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mravec B, Lejavova K, and Cubinkova V, Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Current Alzheimer Research, 2014. 11(10): p. 992–1001. [DOI] [PubMed] [Google Scholar]
- 251.Braak H and Del Tredici K, The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain, 2015. 138(10): p. 2814–2833. [DOI] [PubMed] [Google Scholar]
- 252.Trumbore CN, Shear-Induced Amyloid Aggregation in the Brain: V. Are Alzheimer’s and Other Amyloid Diseases initiated in the Lower Brain and Brainstem by Cerebrospinal Fluid Flow Stresses? Journal of Alzheimer’s Disease, 2020(Preprint): p. 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Eser RA, et al. , Selective vulnerability of brainstem nuclei in distinct tauopathies: a postmortem study. Journal of Neuropathology & Experimental Neurology, 2018. 77(2): p. 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kaalund SS, et al. , Locus coeruieus pathology in progressive supranuclear palsy, and its relation to disease severity. Acta Neuropathologica Communications, 2020. 8(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ohm DT, et al. , Degeneration of the locus coeruleus is a common feature of tauopathies and distinct from TDP-43 proteinopathies in the frontotemporal lobar degeneration spectrum. Acta Neuropathologica, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Vermeiren Y and De Deyn PP, Targeting the norepinephrinergic system in Parkinson’s disease and related disorders: The locus coeruleus story. Neurochemistry international, 2017. 102: p. 22–32. [DOI] [PubMed] [Google Scholar]
- 257.Moussaud S, et al. , Alpha-synuclein and tau: teammates in neurodegeneration? Molecular neurodegeneration, 2014. 9(1): p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Ishizawa T, et al. , Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. Journal of Neuropathology & Experimental Neurology, 2003. 62(4): p. 389–397. [DOI] [PubMed] [Google Scholar]
- 259.Seidel K, et al. , The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain pathology, 2015. 25(2): p. 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ghosh A, et al. , An experimental model of Braak’s pretangle proposal for the origin of Alzheimer’s disease: the role of locus coeruleus in early symptom development. Alzheimer’s research & therapy, 2019. 11(1): p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Hatch RJ, et al. , Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta neuropathologica, 2017. 133(5): p. 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Gannon M and Wang Q, Complex noradrenergic dysfunction in Alzheimer’s disease: Low norepinephrine input is not always to blame. Brain research, 2019. 1702: p. 12–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mather M, How arousal-related neurotransmitter systems compensate for age-related decline in Handbook of Cognitive Aging: A Life Course Perspective, Thomas A and Gutchess A, Editors. 2020, Cambridge University Press. [Google Scholar]
- 264.Weinshenker D, Long road to ruin: noradrenergic dysfunction in neurodegenerative disease. Trends in neurosciences, 2018. 41(4): p. 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Adolfsson R, et al. , Changes in the brain catecholamines in patients with dementia of Alzheimer type. The British Journal of Psychiatry, 1979. 135(3): p. 216–223. [DOI] [PubMed] [Google Scholar]
- 266.Herrmann N, Lanctôt KL, and Khan LR, The role of norepinephrine in the behavioral and psychological symptoms of dementia. The Journal of neuropsychiatry and clinical neurosciences, 2004. 16(3): p. 261–276. [DOI] [PubMed] [Google Scholar]
- 267.Mann D, et al. , Changes in the monoamine containing neurones of the human CNS in senile dementia. The British Journal of Psychiatry, 1980. 136(6): p. 533–541. [DOI] [PubMed] [Google Scholar]
- 268.Matthews KL, et al. , Noradrenergic changes, aggressive behavior, and cognition in patients with dementia. Biological psychiatry, 2002. 51(5): p. 407–416. [DOI] [PubMed] [Google Scholar]
- 269.Seals DR and Esler MD, Human ageing and the sympathoadrenal system. The Journal of physiology, 2000. 528(3): p. 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Goldstein DS, Eisenhofer G, and Kopin IJ, Sources and significance of plasma levels of catechols and their metabolites in humans. Journal of Pharmacology and Experimental Therapeutics, 2003. 305(3): p. 800–811. [DOI] [PubMed] [Google Scholar]
- 271.Elrod R, et al. , Effects of Alzheimer’s disease severity on cerebrospinal fluid norepinephrine concentration. American Journal of Psychiatry, 1997. 154(1): p. 25–30. [DOI] [PubMed] [Google Scholar]
- 272.Raskind MA, et al. , Increased plasma and cerebrospinal fluid norepinephrine in older men: differential suppression by clonidine. The Journal of Clinical Endocrinology & Metabolism, 1988. 66(2): p. 438–443. [DOI] [PubMed] [Google Scholar]
- 273.Raskind MA, et al. , Norepinephrine and MHPG levels in CSF and plasma in Alzheimer’s disease. Archives of general psychiatry, 1984. 41(4): p. 343–346. [DOI] [PubMed] [Google Scholar]
- 274.Chiodo LA, et al. , Subtotal destruction of central noradrenergic projections increases the firing rate of locus coeruleus cells. Brain Research, 1983. 264(1): p. 123–126. [DOI] [PubMed] [Google Scholar]
- 275.Szot P, et al. , Depressive-like behavior observed with a minimal loss of locus coeruleus (LC) neurons following administration of 6-hydroxydopamine is associated with electrophysiological changes and reversed with precursors of norepinephrine. Neuropharmacology, 2016. 101: p. 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Szot P, et al. , Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. The Journal of Neuroscience, 2006. 26(2): p. 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Szot P, et al. , Changes in adrenoreceptors in the prefrontal cortex of subjects with dementia: evidence of compensatory changes. Neuroscience, 2007. 146(1): p. 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ding Y-S, Progress in PET imaging of the norepinephrine transporter system. PET and SPECT of Neurobiological Systems, 2021: p. 713–747. [Google Scholar]
- 279.Hoogendijk WJG, et al. , Increased activity of surviving locus ceruleus neurons in Alzheimer’s disease. Annals of Neurology, 1999. 45(1): p. 82–91. [DOI] [PubMed] [Google Scholar]
- 280.Janssens J, et al. , Cerebrospinal fluid and serum MHPG improve Alzheimer’s disease versus dementia with Lewy bodies differential diagnosis. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring, 2018. 10: p. 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.van Hooren RW, et al. , Elevated norepinephrine metabolism is linked to cortical thickness in the context of Alzheimer’s disease pathology. Neurobiology of Aging, 2021. 102: p. 17–22. [DOI] [PubMed] [Google Scholar]
- 282.Riphagen JM, Van Egroo M, and Jacobs HI, Elevated Norepinephrine Metabolism Gauges Alzheimer’s Disease-Related Pathology and Memory Decline. Journal of Alzheimer’s Disease, 2021. (Preprint): p. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ding YS, et al. , PET imaging of the effects of age and cocaine on the norepinephrine transporter in the human brain using (S, S)- [11C] O-methylreboxetine and HRRT. Synapse, 2010. 64(1): p. 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Gulyás B, et al. , The norepinephrine transporter (NET) radioligand (S,S)-[18F]FMeNER-D2 shows significant decreases in NET density in the human brain in Alzheimer’s disease: A post-mortem autoradiographic study. Neurochemistry International, 2010. 56(6): p. 789–798. [DOI] [PubMed] [Google Scholar]
- 285.Tejani-Butt SM, Yang J, and Zaffar H, Norepinephrine transporter sites are decreased in the locus coeruleus in Alzheimer’s disease. Brain research, 1993. 631(1): p. 147–150. [DOI] [PubMed] [Google Scholar]
- 286.Raskind MA, et al. , Patterns of cerebrospinal fluid catechols support increased central noradrenergic responsiveness in aging and Alzheimer’s disease. Biological psychiatry, 1999. 46(6): p. 756–765. [DOI] [PubMed] [Google Scholar]
- 287.Kelly L, et al. , Identification of intraneuronal amyloid beta oligomers in locus coeruleus neurons of Alzheimer’s patients and their potential impact on inhibitory neurotransmitter receptors and neuronal excitability. Neuropathology and Applied Neurobiology, 2020. [DOI] [PubMed] [Google Scholar]
- 288.Somogyi J and Llewellyn-Smith I, Patterns of colocalization of GABA, glutamate and glycine immunoreactivities in terminals that synapse on dendrites of noradrenergic neurons in rat locus coeruleus. European Journal of Neuroscience, 2001. 14(2): p. 219–228. [DOI] [PubMed] [Google Scholar]
- 289.Lanfumey L and Adrien J, Regulation of sleep after neonatal locus coeruieus lesion: functional evidence of β-adrenergic supersensitivity. European journal of pharmacology, 1982. 79(3-4): p. 257–264. [DOI] [PubMed] [Google Scholar]
- 290.Minneman KP, et al. , beta1-and beta2-Adrenergic receptors in rat cerebral cortex are independently regulated. Science, 1979. 204(4395): p. 866–868. [DOI] [PubMed] [Google Scholar]
- 291.Arango V, Underwood MD, and Mann JJ, Fewer pigmented locus coeruleus neurons in suicide victims: preliminary results. Biological psychiatry, 1996. 39(2): p. 112–120. [DOI] [PubMed] [Google Scholar]
- 292.Arango V, et al. , Autoradiographic demonstration of increased serotonin 5-HT2 and β-adrenergic receptor binding sites in the brain of suicide victims. Archives of general psychiatry, 1990. 47(11): p. 1038–1047. [DOI] [PubMed] [Google Scholar]
- 293.Biegon A and Israeli M, Regionally selective increases in β-adrenergic receptor density in the brains of suicide victims. Brain research, 1988. 442(1): p. 199–203. [DOI] [PubMed] [Google Scholar]
- 294.Mann JJ, et al. , Increased serotonin2 and β-adrenergic receptor binding in the frontal cortices of suicide victims. Archives of general psychiatry, 1986. 43(10): p. 954–959. [DOI] [PubMed] [Google Scholar]
- 295.Kalaria R, et al. , Adrenergic receptors in aging and Alzheimer’s Disease: Increased β2-receptors in prefrontal cortex and hippocampus. Journal of neurochemistry, 1989. 53(6): p. 1772–1781. [DOI] [PubMed] [Google Scholar]
- 296.Haberman RP, Branch A, and Gallagher M, Targeting neural hyperactivity as a treatment to stem progression of late-onset Alzheimer’s disease. Neurotherapeutics, 2017. 14(3): p. 662–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Huijbers W, et al. , Tau accumulation in clinically normal older adults is associated with hippocampal hyperactivity. Journal of Neuroscience, 2019. 39(3): p. 548–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wenthur CJ, et al. , Drugs for allosteric sites on receptors. Annual review of pharmacology and toxicology, 2014. 54: p. 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Nguyen V, et al. , Alpha-2 agonists. Anesthesiology clinics, 2017. 35(2): p. 233–245. [DOI] [PubMed] [Google Scholar]
- 300.Ereciń ska M and Silver IA, Metabolism and role of glutamate in mammalian brain. Progress in neurobiology, 1990. 35(4): p. 245–296. [DOI] [PubMed] [Google Scholar]
- 301.Jacob C, et al. , Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. Journal of Alzheimer’s Disease, 2007. 11(1): p. 97–116. [DOI] [PubMed] [Google Scholar]
- 302.Hoshi A, et al. , Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathology and Applied Neurobiology, 2018. 44(6): p. 628–638. [DOI] [PubMed] [Google Scholar]
- 303.Masliah E, et al. , Deficient glutamate tranport is associated with neurodegeneration in Alzheimer’s disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society, 1996. 40(5): p. 759–766. [DOI] [PubMed] [Google Scholar]
- 304.Scott HA, et al. , Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiology of Aging, 2011. 32(3): p. 553.e1–553.e11. [DOI] [PubMed] [Google Scholar]
- 305.Csernansky J, et al. , CSF excitatory amino acids and severity of illness in Alzheimer’s disease. Neurology, 1996. 46(6): p. 1715–1720. [DOI] [PubMed] [Google Scholar]
- 306.Jimenez-Jimenez F, et al. , Neurotransmitter amino acids in cerebrospinal fluid of patients with Alzheimer’s disease. Journal of neural transmission, 1998. 105(2-3): p. 269–277. [DOI] [PubMed] [Google Scholar]
- 307.Pomara N, et al. , Glutamate and other CSF amino acids in Alzheimer’s disease. Am J Psychiatry, 1992. 149(2): p. 251–4. [DOI] [PubMed] [Google Scholar]
- 308.Kaiser E, et al. , Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer’s disease. Neurodegenerative Diseases, 2010. 7(4): p. 251–259. [DOI] [PubMed] [Google Scholar]
- 309.Martinez M, et al. , Amino acid concentrations in cerebrospinal fluid and serum in Alzheimer’s disease and vascular dementia. Journal of neural transmission-Parkinson’s disease and dementia section, 1993. 6(1): p. 1–9. [DOI] [PubMed] [Google Scholar]
- 310.Li S, et al. , Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron, 2009. 62(6): p. 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Matos M, et al. , Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience, 2008. 156(4): p. 898–910. [DOI] [PubMed] [Google Scholar]
- 312.Tong H, et al. , Amyloid-beta peptide decreases expression and function of glutamate transporters in nervous system cells. The International Journal of Biochemistry & Cell Biology, 2017. 85: p. 75–84. [DOI] [PubMed] [Google Scholar]
- 313.Kobayashi S, et al. , Enhanced tau protein translation by hyper-excitation. Frontiers in Aging Neuroscience, 2019. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Zempel H, et al. , Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. Journal of Neuroscience, 2010. 30(36): p. 11938–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Fitzgerald P, Noradrenaline transmission reducing drugs may protect against a broad range of diseases. Autonomic and Autacoid Pharmacology, 2015. 34(3-4): p. 15–26. [DOI] [PubMed] [Google Scholar]
- 316.Betts MJ, et al. , Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain, 2019. 142: p. 2558–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Kelberman M, Keilholz S, and Weinshenker D, What’s that (blue) spot on my MRI? Multimodal neuroimaging of the locus coeruleus in neurodegenerative disease. Frontiers in Neuroscience, 2020. 14: p. 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Dutt S, et al. , Brainstem volumetric integrity in preclinical and prodromal Alzheimer’s disease. Journal of Alzheimer’s Disease, 2020. 77(4): p. 1579–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Joshi S, et al. , Relationships between pupil diameter and neuronal activity in the locus coeruleus, colliculi, and cingulate cortex. Neuron, 2015. 89: p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Vazey EM, Moorman DE, and Aston-Jones G, Phasic locus coeruleus activity regulates cortical encoding of salience information. Proceedings of the National Academy of Sciences, 2018. 115(40): p. E9439–E9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Bachman SL, et al. , Locus coeruleus MRI contrast is associated with cortical thickness in older adults. Neurobiology of Aging, 2021. 100: p. 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Mather M, et al. , Isometric exercise facilitates attention to salient events in women via the noradrenergic system. Neuroimage, 2020. 210: p. 116560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Singh-Curry V and Husain M, The functional role of the inferior parietal lobe in the dorsal and ventral stream dichotomy. Neuropsychologia, 2009. 47(6): p. 1434–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Jacobs HI, et al. , Curvilinear locus coeruleus functional connectivity trajectories over the adult lifespan: a 7T MRI study. Neurobiology of aging, 2018. 69: p. 167–176. [DOI] [PubMed] [Google Scholar]
- 325.Zhang S, et al. , Resting-state functional connectivity of the locus coeruleus in humans: in comparison with the ventral tegmental area/substantia nigra pars compacta and the effects of age. Cerebral Cortex, 2016. 26(8): p. 3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Elman JA, et al. , Task-evoked pupil dilation and BOLD variance as indicators of locus coeruleus dysfunction. Cortex, 2017. 97: p. 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Dahl MJ, et al. , Noradrenergic responsiveness preserves selective attention across the adult lifespan. Journal of Neuroscience, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Neves RM, et al. , Locus coeruleus phasic discharge is essential for stimulus-induced gamma oscillations in the prefrontal cortex. Journal of neurophysiology, 2018. 119(3): p. 904–920. [DOI] [PubMed] [Google Scholar]