

Abstract
Human cells contain hundreds of kinase enzymes that regulate several cellular processes, which likely include transgene delivery and expression. We identified several kinases that influence gene delivery and / or expression by performing a kinome-level screen in which, we identified small-molecule kinase inhibitors that significantly enhanced non-viral (polymer-mediated) transgene (luciferase) expression in cancer cells. The strongest enhancement was observed with several small-molecule inhibitors of Polo-like Kinase 1 (PLK 1) (e.g. HMN-214 and BI 2536), which enhanced luciferase expression up to 30-fold by arresting cells in the G2/M phase of the cell cycle and influencing intracellular trafficking of plasmid DNA. Knockdown of PLK 1 using an shRNA-expressing lentivirus further confirmed enhancement of polymer-mediated transgene expression. In addition, pairwise and three-way combinations of PLK1 inhibitors with the histone deacetylase-1 (HDAC-1) inhibitor Entinostat and the JAK/STAT inhibitor AG-490 enhanced luciferase expression to levels significantly higher than individual drug treatments acting alone. These findings indicate that inhibition of specific intracellular kinases (e.g. PLK1) can significantly enhance non-viral transgene expression for applications in biotechnology and medicine.
Keywords: Kinases, Cell cycle, Polymer Gene Delivery, Transient protein expression, HMN-214, BI 2536
Graphical abstract
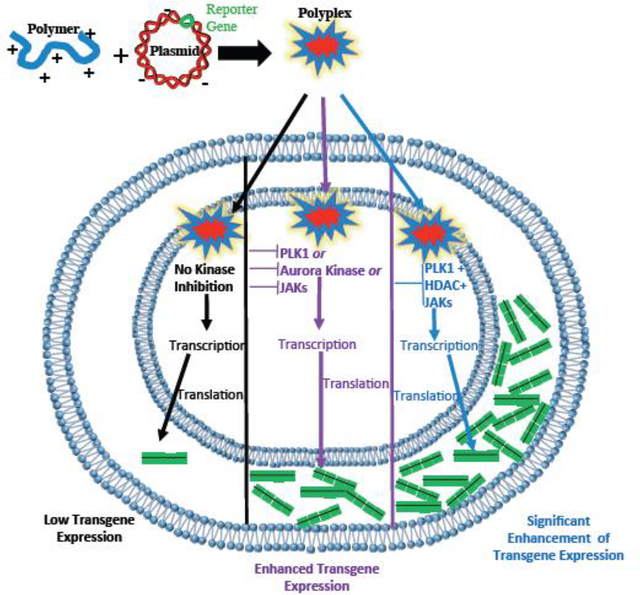
INTRODUCTION
Gene therapy is a promising therapeutic approach for many different genetic disorders. Clinical trials involving the use of viral gene delivery vehicles have already shown promise as potential therapeutic strategies [1, 2]. However, limitations associated with immunogenicity [3], cost [4], and limited cargo load [5] associated with viral vectors motivate the development of non-viral gene delivery methods, including novel polymers as delivery vehicles[6–9]. Unfortunately, current non-viral gene delivery methods demonstrate lower transgene expression levels than those obtained using viral techniques. Most approaches for improving non-viral gene delivery therefore focus on creative approaches towards optimizing the delivery vector to overcome physical barriers within the cell [10, 11]. In particular, cellular uptake and endosomal escape have been significantly improved with the development of novel cationic polymers and lipids [12–14]. At the cellular level, we and others have previously shown that modulating intracellular trafficking by inhibiting cytoplasmic histone deacetylases (HDAC6) leads to enhanced transgene expression [15]. Despite these advances, key intracellular targets (biomarkers) that act as significant barriers to non-viral transgene expression remain poorly understood [16, 17].
In addition to physical barriers (e.g. cell / endosomal / nuclear membranes), transgene delivery may also be limited by specialized biochemical defenses that specifically protect cells against foreign DNA. For example, Toll-Like Receptor 9 (TLR9) binds unmethylated cytosine-guanine (CpG) base pairs within bacterial and viral DNA that are taken up in the endosomes of macrophages and dendritic cells (host CpGs are methylated and do not activate TLR9) [18, 19]. Once TLR9 binds the unmethylated DNA, it initiates a signaling cascade involving several different kinases (IRAK-1, IRAK-4, TAK1, IKK, and MAPK) that activate a set of transcription factors (NF-κB, AP-1, and IRF7). This, in turn, leads to expression of interleukins and interferons leading to the induction of an immune response[20]. TLR9 activation also reduces the magnitude and duration of transgene expression[21], but removing CpGs from plasmid DNA decreases inflammation and enhances transgene expression in lung tissue for up to 56 days[22].
The kinases involved in the TLR9 pathway are just one example of the ≥ 500 kinases in the human kinome [23] that play key roles in intracellular processes, including endocytosis (PI3K [24] and EGFR[25]), cell cycle progression (CDK, PLK, Aurora)[26], and gene transcription (JAK/STAT [27] and JNK[28]). Therefore, we hypothesized that kinases are likely to be involved in cellular uptake and trafficking, nuclear import, and / or transgene expression following plasmid DNA delivery. Some studies in the literature have investigated the role of kinases in delivery. For example, ur Rehman et al. showed that inhibition of Protein Kinase A (PKA) enhances gene delivery 2–3 fold by promoting lipoplex and polyplex (polymer-plasmid DNA complex) uptake by caveolae instead of clathrin-coated pits[29]. Inhibition of Rho kinase by Y-27632 also increases lentiviral transduction by 20% in keratinocytes[30]. In contrast, inhibition of PI3K has been shown to reduce adenoviral transduction, since PI3K plays a key role in α integrin-associated endocytosis of viruses.(11) The tyrosine kinase inhibitor genistein also decreases polyplex uptake by up to 50% by inhibiting cavaeolar uptake[31]. Therefore, there exists some evidence to indicate that kinase inhibition has the potential to either increase or decrease the efficacy of transgene expression.
In this work, we carried out a kinome-level screen of small molecule inhibitors in order to identify kinases that influence the efficacy of transgene expression following non-viral (polymer-mediated) delivery of plasmid DNA. While the screen resulted in the identification of several kinases that enhanced polymer-mediated transgene expression, treatment with small-molecule inhibitors of the cell cycle regulator Polo-Like Kinase 1 (PLK1) resulted in the highest enhancement of transgene expression in cancer cell lines, indicating a pivotal role for this kinase in transgene delivery. Simultaneous inhibition of PLK1, histone deacetylase 1 (HDAC1), and Janus Kinase (JAK, another kinase identified in our screen) resulted in further enhancement of transgene expression relative to inhibition of each individual target. These results demonstrate that inhibition of key intracellular kinase targets using small-molecule inhibitors can enhance transgene expression and potentially improve gene therapy efficacy.
MATERIALS AND METHODS
Cell Culture
PC3-PSMA prostate cancer cells[32], derived from PC3 cells, were kindly provided by Michel Sadelain, MD, PhD, Memorial Sloan -Kettering Cancer Center (New York, NY). MB49 cells were kindly provided by Dr. Christina Voelkel-Johnson (Medical University of South Carolina) as part of an existing collaboration. PC3-PSMA cells were cultured in RPMI 1640 Medium (Hyclone®), containing 10% fetal bovine serum (Hyclone®) and a Penicillin (100 units/mL) – Streptomycin (100 μg/mL) antibiotic combination (MP Biomedicals, LLC). MB49 cells were maintained in Dulbeccos Modifed Eagle’s Medium (Life Technologies) with the same serum and antibiotic content. Cells were grown in an incubator maintained at 37°C and 5% CO2.
Polymers
The 1,4C-1,4bis polymer was synthesized by mixing the monomers 1,4-cyclohexane dimethanoldiglycidyl ether (1,4C, Sigma) and 1,4-bis(3-aminopropyl)-piperazine (1,4Bis, Sigma) at a 1:1 molar ratio as described previously [33]. 25 kDa branched polyethyleneimine (PEI) was purchased from Sigma. Polymers were solubilized in 1X phosphate buffered saline (1X PBS) following 16 hours of polymerization, and the pH was adjusted to 7.4.
Parallel Screening of Small-Molecule Kinase Inhibitors
A library of kinase inhibitors pre-dissolved in DMSO at 10 mM was purchased from Selleck Chem (Cat# L1200, Houston, TX). A complete list of the kinase inhibitors and their known kinase targets can be found in Table S1, supplementary information section. PC3-PSMA cells were seeded in 96 well plates (15,000 cells/well) and incubated overnight (~18 hrs) at 37°C in serum-containing RPMI media (SCM). Polyplexes were prepared by mixing the polymer 1,4C-1,4Bis [33] with pGL3.0 plasmid DNA (Promega; luciferase expression plasmid with a SV40 promoter) at a 10:1 mass ratio and incubating the solution at room temperature for 20 minutes. SCM media was then removed from cells and replaced with SFM media. Polyplexes (60 ng plasmid DNA/well) and kinase inhibitors (final concentration of 10 × IC50 with 0.5% DMSO) were simultaneously added to each well using a Biomek NXP laboratory automated liquid handling station (Beckman-Coulter). Cells were incubated with polyplexes and inhibitors for 6 hours at 37°C. The media was then changed to SCM (with a unique kinase inhibitor in each well) and the cells were incubated at 37°C for an additional 48 hours to allow for transgene (luciferase) expression.
Dose Response Optimization of Kinase Inhibitor Leads
Small molecule PLK1 inhibitors (drugs), BI 2536, BI 6727, HMN-214, and ON01910, used for dose response optimization studies, were all purchased from Selleck Chemicals, and stored at −80°C. Optimization experiments with inhibitor leads were carried out in a similar fashion to screening experiments, but in 24 well plates with 200 ng pGL3.0 or pEGFP-C1 (Clontech; an eGFP expression plasmid with a CMV promoter) plasmid DNA/well, at various concentrations of each inhibitor with various polymers (10:1 w/w 1,4C-1,4Bis or 1:1 w/w PEI) and cell lines (PC3-PSMA or MB49, both seeded at 50,000 cells/well). The Entinostat used in the inhibitor combination transfections was kindly provided by Syndax Pharmaceuticals of Waltham, MA through an agreement with the Cancer Therapeutics Evaluation Program (CTEP) at NIH. The final DMSO concentration in a given well was kept constant at 0.5% in all experiments with inhibitor combinations.
Determination of Cell Viability using the MTT Assay
Cell proliferation in case of different treatment conditions, relative to untreated control cells (treated as 100% viable, or a live control), was quantified using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), a yellow colored reagent which is converted to formazan (a purple dye) by living cells. For screening experiments, transfections were carried out in 96-well cell culture plates, that were seeded with 50,000 cells per well. Following 48 hours of transfection, 10 μL of MTT reagent was added to the cells and incubated at 37°C for 2–4 hours, and the cells were then lysed by adding 20 μL of MTT detergent and incubated for an additional 2 hours at room temperature. Inhibitor dose-optimization transfections were carried out in 24-well plates that were seeded with 50,000 cells per well. After 48 hours, 20 μL MTT reagent was added, followed by 100 μL of MTT detergent for lysis for 2 hours.
In both cases, the concentration of formazan was then determined by measuring the absorbance of each well at 570 nm (A570). Cell proliferation (PRO) was calculated by dividing the A570 of each sample by the A570 of the live cell control (no inhibitor or polyplex added), after subtracting blank absorbance.
Quantification of Luciferase Expression
Luciferase expression was quantified using the Luciferase Assay Kit from Promega (Madison, WI). Media was removed from the plates and cells were washed once with PBS before adding cell culture lysis reagent (Promega) to each well and incubating the plates at 37°C for 20 minutes. Cell lysate (15 μL) was then mixed with luciferin solution (30 μL) and luminescence (LUM) was immediately measured using a Synergy 2 plate reader (Biotek, Winooski, VT). Luminescence units accounting for changes in proliferation (LUV) were calculated by dividing luminescence values (LUM) by relative cell proliferation (PRO). The LUV values of each sample were then divided by the LUV value of the control sample (no drug) to provide the RLUV values shown in each figure. Therefore, the RLUV values presented here account for changes in cell density (e.g. a condition with luminescence similar to the control but with 50% relative cell proliferation will be multiplied by a factor of two) and illustrate the degree of enhancement for each condition relative to the control.
Quantification of EGFP Expression
At 48 hours post-transfection, cells were washed with 1X PBS, trypsinized, and pelleted via centrifugation at 500 X g for 5 minutes. Pellets were resuspended in 1X PBS, and flow cytometry analysis, carried out using a FACSCalibur (Benton Dickinson) machine, was used to quantify intracellular EGFP fluorescence, as detected by the FL1 emission filter. PMT voltage settings were adjusted based on live cell controls. Fluorescence gating was performed such that control samples (at each drug concentration, lacking polyplex treatment) showed 0.1% fluorescence-positive cells. Side and forward scatter plots for the live cell control were also used for live/dead gating to ensure that only live cells were included in the final flow cytometry analyses. Calculations for fluorescence intensity per cell involved subtraction of the drug only conditions (no transfection) arithmetic average cell fluorescence from the transfected counterpart to avoid including drug background fluorescence or autofluorescence in reported values. In PC3-PSMA cells, all experiments involving the BI 2536 inhibitor were carried out three times (listed in Table 1). For all other conditions in PC3-PSMA and MB49 cells, at least two independent experiments (in some cases three) were carried out.
Table 1.
Enhancement of luciferase transgene expression (RLUV) by the 15 most effective small-molecule kinase inhibitors identified from the screen.
| Inhibitor | Kinase Target(s) | RLUV |
|---|---|---|
| BI 6727 | PLK1 | 12.4 ± 13.8 |
| BI 2536 | PLK1 | 12.3 ±5.3 |
| GSK 461364 | PLK1 | 9.9 ± 4.8 |
| HMN-214 | PLK1 | 9.5 ±8.3 |
| SKI-606 | Src/Abl | 6.6 ± 11.0 |
| AG-490 | EGFR/JAK/ErbB2 | 6.4 ± 10.7 |
| PD0332991 | CDK 4,6/Cyclin D1,2 | 6.1 ± 5.6 |
| SNS-314 | Auroras A, B, C | 6.0 ± 6.1 |
| Imatinib | Abl/c-Kit/PDFGR | 5.3 ±4.7 |
| Vinorelbine | p38 MAPK | 4.9 ± 2.5 |
| VX-702 | p38 MAPK | 4.8 ± 4.1 |
| PHA-680632 | Auroras A, B, C | 4.4 ± 5.5 |
| KW 2449 | FLT-3/Abl/Aurora A | 4.4 ± 4.1 |
| NVP-ADW742 | IGF-1R | 4.4 ±5.9 |
| SNS-032 | CDK 2, 7, 9 | 4.1 ± 2.4 |
Production and Evaluation of Lentiviral Particles for PLK1 Knockdown
Plasmids expressing shRNA for knocking down PLK1 (total=20) were selected from the shRNA collection library (The RNA Consortium (TRC)) in Prof. Joshua LaBaer’s laboratory, at The Biodesign Institute, Arizona State University. From these 20 clones, 4 (TRCN0000006246–6249) were selected based on target knockdown efficiency reported in previous publications [34–37]. cDNA was produced (Qiagen Maxiprep kit), and lentiviral particles were produced by transfecting LentiX-293T cells (Clontech) with either PLK1 or scramble control-shRNA and packaging plasmids using our established SOPs[38]. After finalizing viral production, the resulting virus was used to infect PC3-PSMA cells.
To evaluate the silencing of PLK1 using the lentiviral particles, immunoblotting experiments were carried out on PC3-PSMA cell lysates after cells were infected with either a scramble control or each lentiviral clone for PLK1 knockdown (total=4). The GAPDH (loading control) and PLK1 antibodies were purchased from Cell Signaling and used at a working dilution of 1:1000. Following transfer, membranes were blocked with 5% milk in TBS-Tween (TBS-T, 0.2% tween), and incubated in primary antibody overnight at 4°C. Membranes were then washed with TBS-T, and incubated with an HRP-conjugated secondary antibody (1:1000 dilution) for one hour at room temperature. Following additional washes, membranes were treated with SuperSignal West Femto Chemiluminescent substrate (Thermo) for antibody detection. Based on literature and our own results, it was determined that the virus correspondent to clone PLK1-shRNA 6247 would be used for further experiments. This virus successfully knocked down PLK1 in PC3-PSMA cells and affected cell growth.
Combined PLK1 Silencing and Transgene (pGL3 Plasmid) Delivery to PC3-PSMA cells
PC3-PSMA cells were infected with either PLK1 or a scramble control lentivirus. In brief, 12,500 cells were plated in a 24 well plate, and on the following day, polybrene (8 μg/mL) was added, followed by lentivirus (200μl) or no further treatment (i.e. equivalent volume media). Plates were centrifuged for 30 minutes at 2250 rpm and kept in the incubator at 37°C and 5% CO2. Following 48 hours, transfection experiments and luminescence-quantifying assays were carried out as previously described. Experiments were carried out using 1,4C-1,4bis and PEI as polymer carriers at polymer to DNA weight ratios of 10:1 and 1:1, respectively.
Cell Cycle Analysis
Propidium Iodide (PI) staining was carried out in order to determine relative cell cycle proportions of the PC3-PSMA cell population in the presence and absence of PLK1 inhibitors, BI 2536 and HMN 214. Briefly, 250,000 PC3-PSMA cells were plated in a 6 well plate and allowed to attach and grow overnight. Cell culture media was removed and replenished with fresh SCM containing drug or vehicle control. After 48 hours, cells were washed with 1X PBS harvested via trypsinization, and resuspended in a small amount of 1X PBS. Pure ice-cold ethanol was added dropwise to the PBS cell suspension, resulting in a final EtOH concentration of 70%, at which point, cells were stored at 4°C for 1 hour to allow for fixation. Cells were then washed with 1X PBS/2% FBS/0.001% Triton X, followed by a wash with 1X PBS/2% FBS. Cells were then incubated with a PI (Sigma) staining solution prepared in 1X PBS (5% FBS, 50 μg/mL PI, 100 μg/mL RNase A). Cells were then analyzed in a FACS Attune® acoustic focusing flow cytometer, with PI signal detection through the B3 emission filter. Live/Dead gating was carried out using a side/forward scatter dot plot, and only live cells were included in final analysis. Cell doublets were gated out of analysis, as determined by high fluorescent width (B3-W) values, indicative of long detector residence time. Cell cycle percentages given are representative of percentage of cells falling in the subG1 (<2N DNA content; typically apoptotic fraction), G0/G1 (2N DNA content), S (between 2N and 4N DNA content), G2/M cycles (4N DNA content). A small portion of the cell population shows fluorescence greater than that represented by 4N DNA content. Some of these cells may actually have greater than 4N DNA content, while a proportion is likely cell doublets that are not eliminated by gating.
Intracellular Trafficking of Polymer-Plasmid DNA Complexes (Polyplexes)
The Label IT® fluorescein-conjugated plasmid from Mirus was used to monitor plasmid DNA intracellular trafficking. PC3-PSMA prostate cancer cells were plated in 24 well plates at a density of 50,000 cells per well on top of coverslips, and allowed to attach overnight. On the following day, cells were incubated with 10 μg/mL DAPI and allowed to stain overnight at 37°C. The following day, several washes with 1X PBS were carried out in order to remove residual DAPI, thus avoiding false nuclear detection by staining the plasmid DNA. Serum-free media containing the drug or 0.2% DMSO control was added to the cells while polyplexes were formed by mixing PEI with 2 μg of fluorescein-labeled plasmid DNA at a 1:4 polymer to DNA mass ratio. Polyplexes were added to the cells for 6 hours after which, cells were replenished with serum-containing media containing drug or DMSO. Following 48 hours of transfection, cells were washed twice with 1X PBS, and fixed with 2% formaldehyde for 20 minutes. Cells were then washed 5 times with 1X PBS, and mounted using a 90% glycerol solution containing n-propyl gallate. Confocal microscopy was carried out with a Nikon C2 confocal microscope equipped with a 60X water immersion objective, which was utilized to determine polyplex trafficking relative to the nucleus. Laser excitation of 488 nm was used and emission at 525 nm was captured for fluorescein detection. Z-stack images were taken with a step size of 0.330 μm, with the images displayed representing a maximum projection signal through the z-axis, unless otherwise noted.
RESULTS AND DISCUSSION
Kinase Inhibitor Screen
A library of 182 kinase inhibitors was screened in order to identify leads that enhance non-viral transgene expression in cancer cells (please refer Table S1 in the supplementary information for a complete list of the kinase inhibitors). Fifteen different inhibitors enhanced luciferase expression by a factor of 4-fold or higher relative to the polyplex control (1,4C-1,4Bis polymer [33] complexed with pGL3.0 plasmid DNA in 0.5% DMSO) in PC3-PSMA cells [32]; a concentration of 10 times the reported IC50 value for each inhibitor was employed in the screen (please see Table 1 for inhibitor leads, and Table S2 in the supplementary information for a complete list of screening results). Interestingly, some inhibitors, including two PI3K inhibitors (TGX-221 and AS252424), two CDK inhibitors (AZD5438 and Flavopiridol HCl), and a single JAK inhibitor (LY2784544), decreased luciferase expression to less than 50% of the control in cells. The baseline luciferase activity of the 1,4C-1,4Bis polyplex controls was approximately 4×104 luminescence units / mg protein in all screening experiments with 96 well plates, and the relative luciferase expression for transfections with each inhibitor was normalized to this polyplex control to obtain the RLUV values shown in Table 1 and Figure 1.
Figure 1.
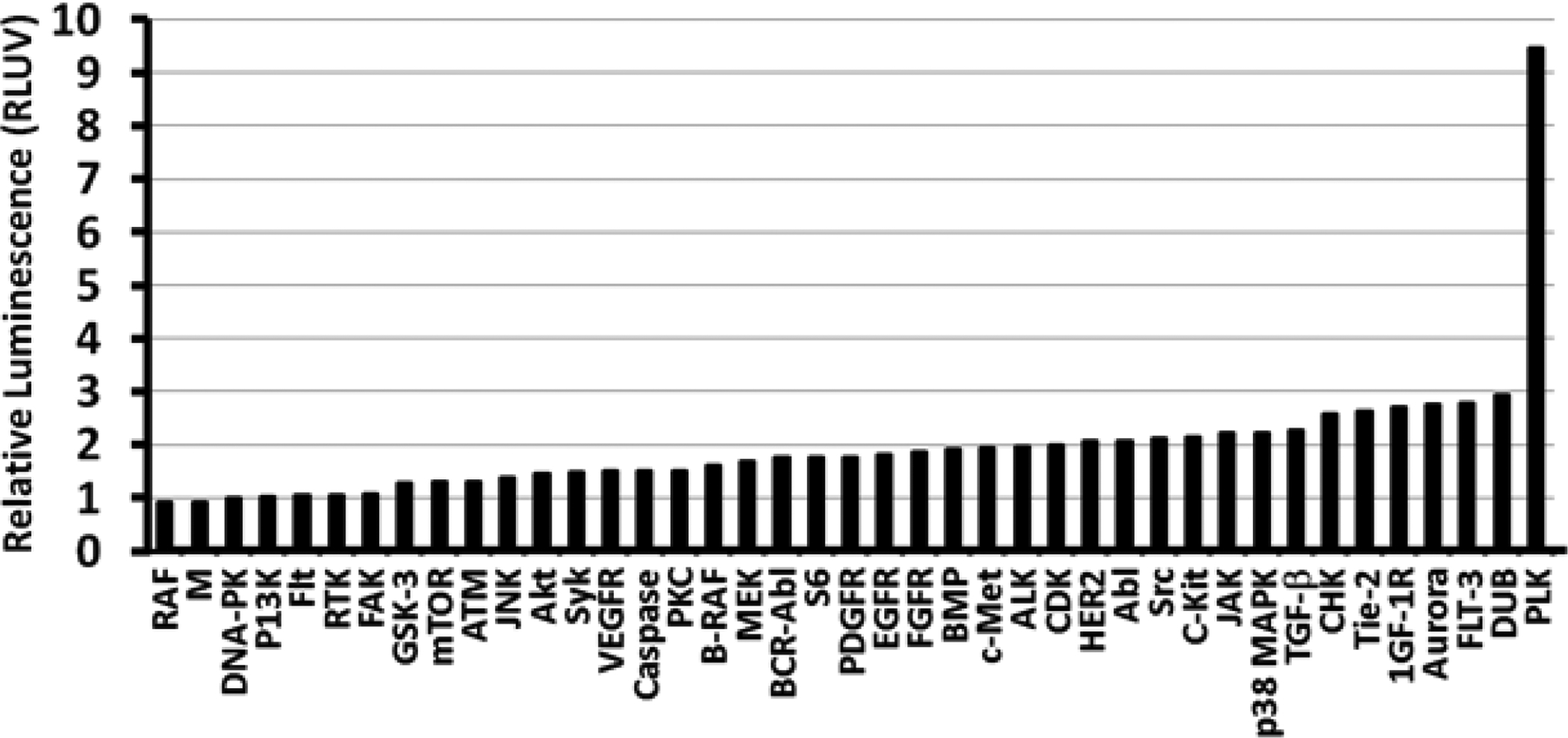
Identification of kinase targets that influence transgene expression. The enhancement values of individual inhibitors with the same kinase target were pooled together to prepare this figure. The y-axis shows enhancement relative to the polyplex control (RLUV = 1), while the x-axis indicates the kinase enzyme targeted by the inhibitor(s).
The relative luminescence values obtained with multiple inhibitors of the same kinase target were averaged in order to determine the effects of inhibiting specific kinases in Figure 1. It is clear that Polo-like Kinase 1 (PLK 1) inhibition enhances luciferase expression strongly at 9.5 ± 3.7 fold relative to the polyplex control, and emerged as the predominant intracellular target. Indeed, the top 4 leads in Table 1 are PLK 1 inhibitors, and the only other PLK 1 inhibitor tested (ON-01910, not shown in Table 1) also displayed 3.2 ± 1.5 fold enhancement. In addition to PLK 1 inhibitors, several other inhibitors for kinases involved in the cell cycle (CDK, Cyclin, p38 MAPK, and Aurora) also showed 2-fold enhancement or higher. These include PD0332991 (CDK/Cyclin inhibitor, RLUV = 6.1 ± 5.6), SNS-314 (Aurora inhibitor, RLUV = 6.0 ± 6.1), and Vinorelbine (p38 MAPK inhibitor, RLUV = 4.9 ± 2.5). Inhibition of the cell cycle kinases also reduced cell proliferation by at least 20% (Aurora kinase) relative to the control, while some PLK1 inhibitors decreased proliferation by up to 36% (see Table S3 in the supplementary information section).
In addition to cell cycle kinases, 4 out of the 9 JAK inhibitors tested showed greater than 2.4-fold enhancement (AZ960, AZD1480, AT9283, and AG-490), with the JAK2 inhibitor AG-490 (IC50 = 10 μM) showing the highest enhancement of 6.4-fold relative to the polyplex control. Interestingly, AG-490 also inhibits the membrane-bound growth factor receptors EGFR (IC50 = 0.1 μM) and HER2 (IC50 = 13.5 μM)[39]. Several other growth factor receptor inhibitors also showed significant enhancement of luciferase expression compared to the polyplex control, including two additional EGFR inhibitors, CI-1033 and Neratinib, which showed 2.7 and 3.8-fold enhancement, respectively. Several different PDGFR inhibitors also enhanced luciferase expression (AP24534, Crenolanib, TSU-68, Masitinib showed 2.1–2.3 fold enhancement), while Imatinib (aka Gleevec) demonstrated the strongest enhancement (5.3-fold). It is important to note that multiple PDGFR inhibitors also inhibit FGFR (AP24534, TSU-68, and Masitinib) and VEGFR (AP24534 and TSU-68). Three VEGFR-specific inhibitors also showed 2.3–2.6 fold enhancement (Axitinib, MGCD-265, and Vatalanib). Finally, the IGF-1R inhibitor NVP-ADW742 and the TGF-β inhibitor SB525334 exhibited 4.4- and 2.3-fold enhancement of luciferase expression, respectively. While these results suggest that growth factor receptor inhibition enhances transgene expression, several other growth factor receptor inhibitors showed no significant enhancement of luciferase expression (RLUV ≤ 1.0).
It is important to note that while our screen revealed several kinases that influence transgene expression (cell cycle kinases, signal transducers, and growth factor receptors), the screen only included inhibitors for 40 of the 518 known human kinases. Therefore, there may be additional kinases which influence transgene expression, but are not identified in our screen. Nonetheless, this screen positively identified one major kinase target, PLK1, which was selected for further investigation.
Optimization of Transgene Expression with the PLK1 Inhibitors HMN-214 and BI 2536
The four most effective PLK1 inhibitors, BI 6727, BI 2536, GSK 461364, and HMN-214, were evaluated at various concentrations (0.1–33 μM) in order to further investigate the effects of PLK1 inhibition on transgene (luciferase) expression. Of these four inhibitors, HMN-214 and BI 2536 consistently showed the highest enhancement of luciferase expression relative to vehicle control with the 1,4C-1,4Bis polymer (11-fold) and PEI (37-fold) at an optimum concentration of 3.3 μM in PC3-PSMA cells (Figure 2). HMN-214 also showed higher enhancement with PEI than 1,4C-1,4Bis in MB49 cells, but to a lesser extent (6 and 12-fold enhancement with 1,4C-1,4Bis and PEI, respectively) at an optimal concentration of 1 μM. As expected for PLK1 inhibition, HMN-214 also significantly reduced cell proliferation, with the lowest viabilities (40–50%) at concentrations above 3.3 μM. Interestingly, a sharp drop in proliferation was observed in MB49 cells at the same HMN-214 concentration which maximally enhances transgene expression (1 μM). It is also important to note that HMN-214 concentrations ≥ 3.3 μM caused considerable changes in cell morphology (please see supplementary information, Figure S1). This anti-proliferative activity of the drug can be useful in cancer gene therapy applications.
Figure 2.
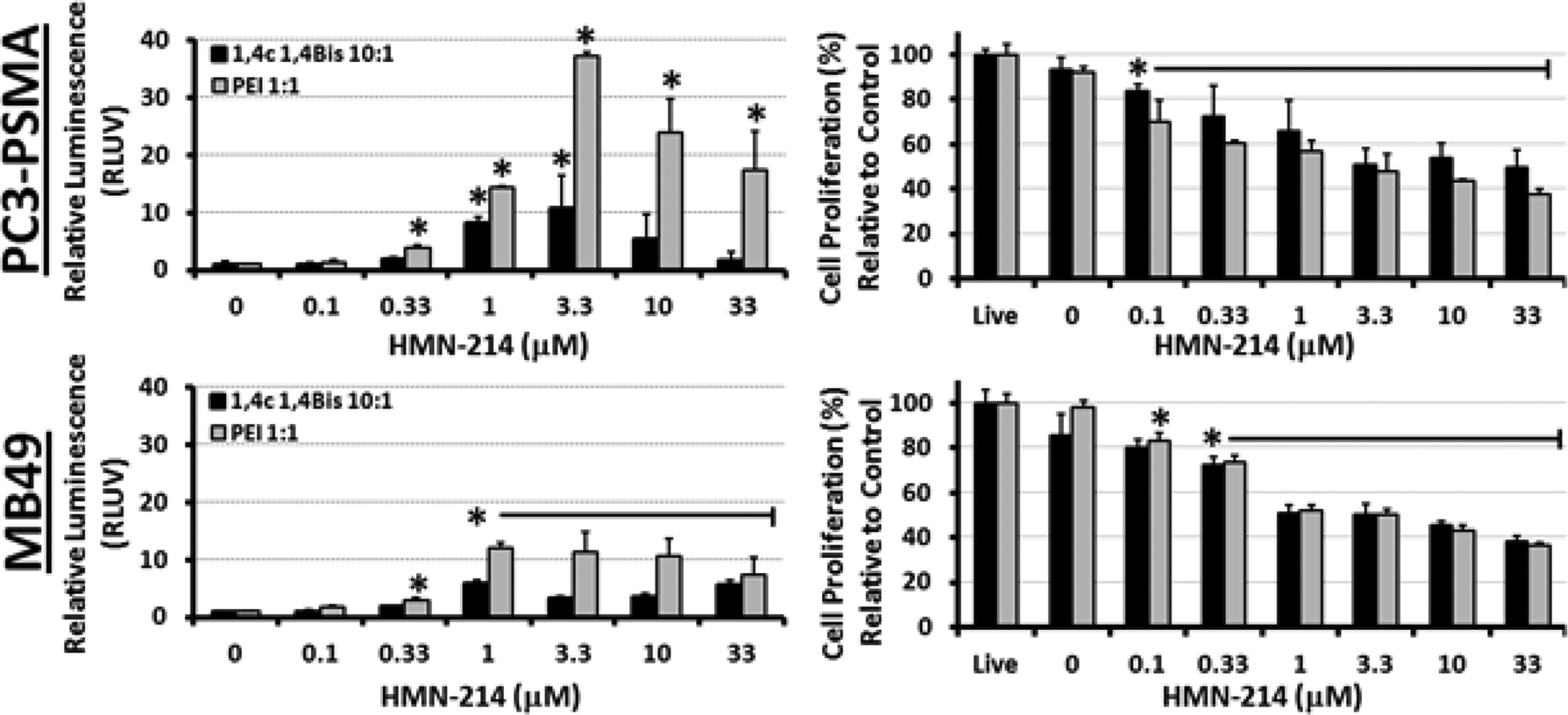
Dose-dependent enhancement of luciferase transgene expression by the PLK1 inhibitor HMN-214 in PC3-PSMA human prostate cancer and MB49 murine bladder cancer cells following delivery of pGL3.0 plasmid DNA using PEI and 1,4C-1,4Bis polymers. Relative enhancement of luciferase expression (RLUV) compared to the polyplex control is shown in the left panels, while effects of the drug on cell proliferation are shown in the panels on the right side. Asterisks (*) indicate statistically significant difference (student’s T-test) from the corresponding polyplex control (n = 3 independent experiments; p < 0.05).
The dose optimization of another effective PLK1 inhibitor, BI 2536, is shown in Figure 3. BI 2536 increased luciferase expression 3–6 fold at concentrations of 10 and 100 nM in PC3-PSMA and MB49 cell lines, respectively. While the optimal concentration for enhancing luciferase expression differs by a factor of 10 between the two cell lines employed, it is interesting to note that the optimal concentration in both cell lines coincides with the point at which cell proliferation is reduced by 40% (Figure 3), suggesting that inhibition of cell division by the drug is necessary for the enhancement of gene delivery. Concentrations of BI 2536 above 1 μM (1000 nM) drastically reduced MB49 cell viability to approximately 20%.
Figure 3.
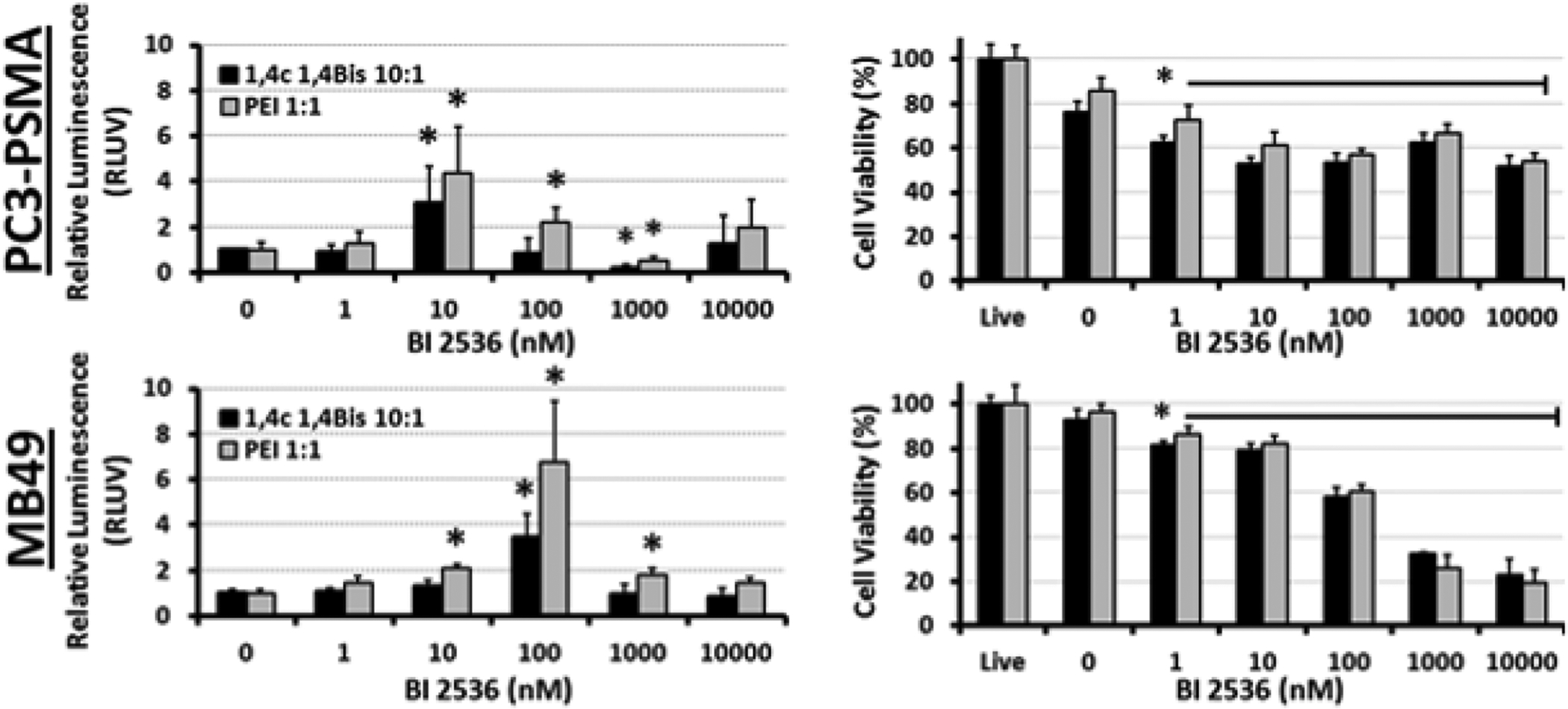
Dose-dependent enhancement of luciferase transgene expression by the PLK1 inhibitor BI 2536 in PC3-PSMA human prostate cancer and MB49 murine bladder cancer cells following delivery of pGL3.0 plasmid DNA using PEI and 1,4C-1,4Bis polymers. Relative enhancement of luciferase expression (RLUV) compared to the polyplex control is shown in the left panels, while effects of the drug on cell proliferation are shown in the panels on the right side. Asterisks (*) indicate p-values <0.05, which show statistically significant enhancement relative to vehicle control (Student’s t-test).
Investigation of PLK1 Inhibitor-mediated Enhancement of EGFP Expression
We expanded on the previous luciferase observations by testing the effects of the PLK1 inhibitors on EGFP transgene expression following plasmid DNA delivery. Table 2 shows the fold-enhancement of EGFP expression per cell (Fluor/cell) and the percentage of cells expressing EGFP (EGFP+%) for the optimum concentration of each drug (please see Figures S2–S8, supporting information section, for full dose range experiments; ON01910 dose responses not shown, as only concentrations of 100 nM and 500 nM were assayed due to high toxicity). Both BI 2536 and HMN-214 showed consistently strong enhancement of transgene expression. BI 2536 strongly enhanced the fluorescence per cell with the 1,4C-1,4Bis polymer (15.8±4.2), but showed a more modest effect with PEI (3.6±1.6). The same phenomenon was also observed with BI 2536 in MB49 cells, where enhancement was strong with 1,4C-1,4Bis (9.7±3.2), but not with PEI (0.4±1.7). In contrast, HMN-214 showed the opposite effect in PC3-PSMA cells, with higher enhancement of transgene expression with PEI than 1,4C-1,4Bis polymers. It is possible that these drugs differentially affect the ability of different polymer carriers to enter cells, perhaps as a function of N to P ratio. It is worth noting that BI 6727 also showed significant enhancement of EGFP and luciferase expression, but this effect was inconsistent (hence the large standard deviation). The effects of ON01910 were also somewhat inconsistent (32.3±17.0) and sharply decreased cell viability. Taken together, it can be seen that inhibition of PLK1 using different small molecules increases GFP expression across the cell population, and also increases fluorescence / cell in cells that express the protein.
Table 2.
Enhancement of polymer-mediated EGFP Expression by PLK1 inhibitors identified as leads from the screen.
| PC3-PSMA Cells | ||||
|---|---|---|---|---|
| 1,4C-1,4bis | PEI | |||
| PLK1 Inhibitor | EGFP+ | Fluor/Cell | EGFP+ | Fluor/Cell |
| BI 2536 (25 nM) | 5.1 ± 1.0* | 15.8 ± 4.2* | 3.6 ± 1.6* | 3.4 ± 1.1* |
| BI 6727 (100 nM) | 4.9 ± 4.0 | 45.8 ± 45.2 | ||
| HMN-214 (3.3 μM) | 4.3 ± 0.5* | 5.6 ± 0.7* | 6.7 ± 4.0* | 15.5 ± 12.8 |
| ON01910 (100 nM) | 6.3 ± 3.3 | 32.3 ± 17.0 | ||
| MB49 Cells | ||||
| 1,4C-1,4bis | PEI | |||
| PLK1 Inhibitor | EGFP+ | Fluor/Cell | EGFP+ | Fluor/Cell |
| BI 2536 (100 nm) | 2.5 ± 0.3* | 9.7 ± 3.2* | 0.5 ± 0.2 | 0.4 ± 1.7 |
| HMN-214 (3.3 μM) | 1.8 ± 0.4* | 9.8 ± 2.3* | ||
p<0.05, Student’s t test (two tailed)
Representative fluorescence microscopy images for PC3-PSMA cells treated with each of the four PLK1 inhibitors and 1,4C:1,4Bis-pEGFP polyplexes are shown in Figure 4. Both BI 2536 and HMN-214 exhibit moderate, yet significant enhancement of EGFP expression in these cells. Interestingly, the baseline EGFP expression using 1:1 PEI was low enough that enhancement in transfection was unobservable using the flow cytometry assay, but results are shown in Table 2 as recorded. Visually, cell fluorescence was almost non-existent with or without drug when PEI was used as the vehicle for pEGFP delivery. It is important to note that an optimal weight ratio of 1:1 PEI to plasmid DNA was employed in these studies.
Figure 4.
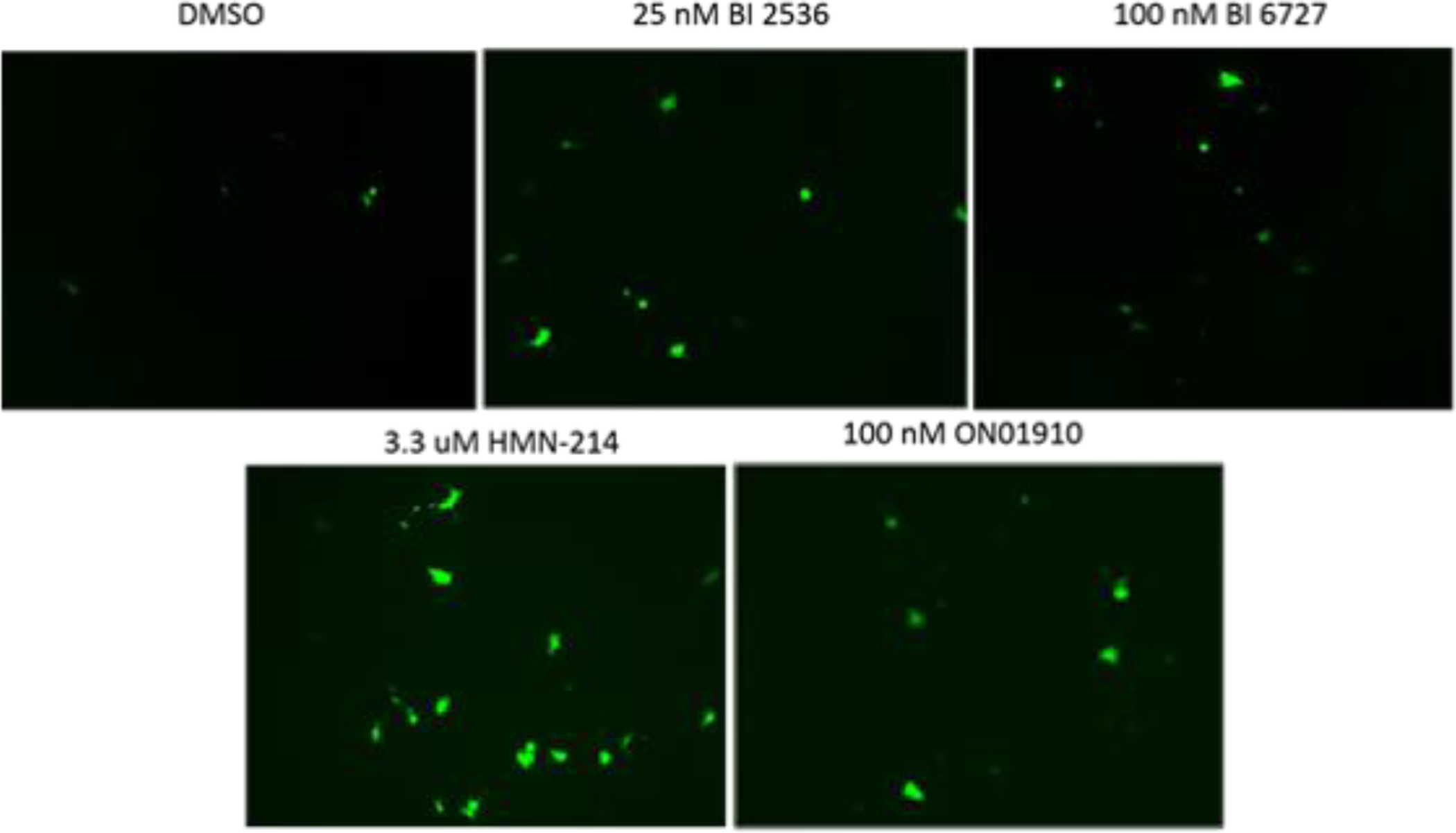
Representative fluorescence microscopy images of PC3-PSMA cells transfected with polyplexes formed with a 10:1 mass ratio of 1,4C-1,4Bis : pEGFP DNA, in the presence of no drug (DMSO) or with PLK1 inhibitors, BI 2536, BI 6727, HMN-214, ON01910. Images were obtained using a Zeiss inverted fluorescence microscope 48 hours post-transfection.
Taken together, the above results with two different reporter genes (firefly luciferase and EGFP) and two different cationic polymers show that several known or putative inhibitors of PLK1 are able to significantly enhance polymer-mediated transgene expression in two different cancer cell lines indicating a wider applicability of this approach.
Effects of PLK1 Silencing on Transgene Expression
Previous results clearly showed that multiple small-molecule inhibitors of PLK1 enhanced the delivery of multiple transgenes with two cationic polymers in different cell lines. We therefore investigated if PLK1 silencing / knockdown using a lentiviral vector could enhance transgene expression in cells. Viruses with four different shRNA constructs were designed and investigated for PLK1 knockdown in PC3-PSMA cells. The efficacy of each construct was tested by immunoblotting cell lysates in order to determine PLK1 expression levels (not shown). Based on PLK1 knockout (PLK1 KO) efficacy, as determined by Western blot against two scramble control viruses (Figure 5 left), PLK1 silencing using the virus expressing PLK1 shRNA 6247 clearly reduced PLK1 expression, as reported previously [34]. We therefore chose this virus for knocking down PLK1 in our subsequent experiments.
Figure 5.
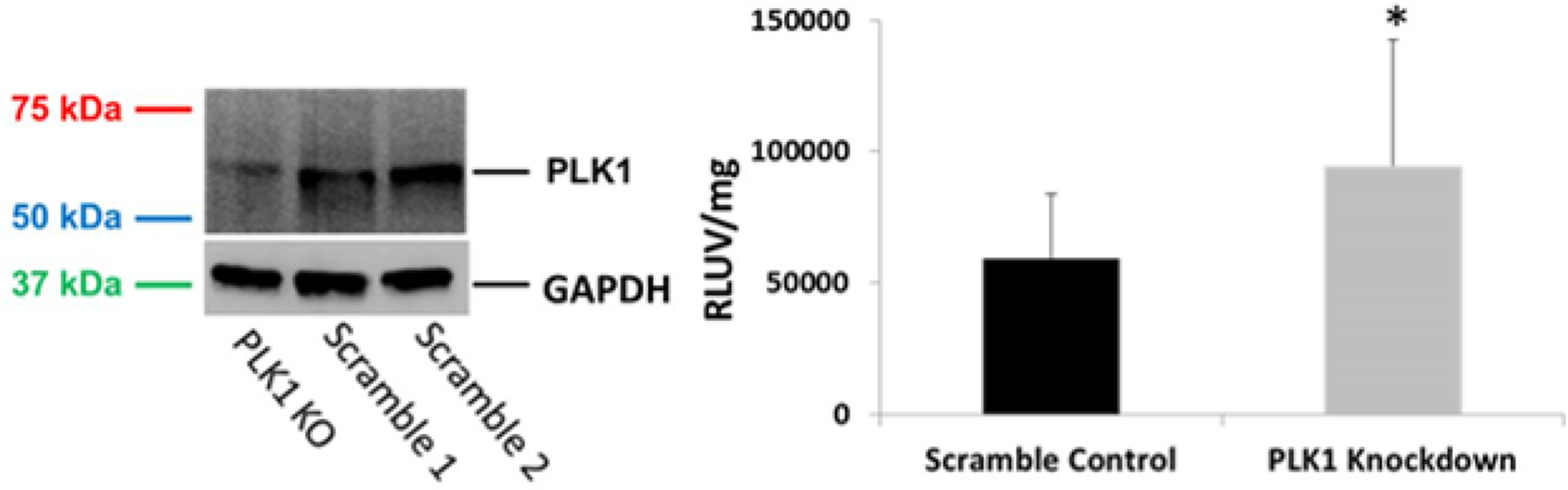
Left – PC3-PSMA cells were infected with a lentivirus expressing scrambled sequence shRNA or lentivirus expressing shRNA construct 6247 against the mRNA of PLK1 (denoted PLK1 KO). Cell lysates, prepared 72 hours after infection, were immunoblotted using a PLK1 antibody probe. Right – Polymer-mediated transfections with polyplexes formed at a 10:1 1,4C-1,4bis : pGL3 plasmid weight ratio, in PC3-PSMA cells, beginning 48 hours after infection with the lentivirus, and proceeding for 48 additional hours to allow for determination of luciferase expression. Transgene expression is reported in relative luminescence units normalized to protein content, and normalized to cell viability (RLUV / mg). * p-value =0.02, Student’s T-test, compared to scrambled control.
Lentivirus-mediated PLK1 silencing resulted in a modest but statistically significant increase in 1,4C-1,4Bis polymer-mediated luciferase expression in PC3-PSMA cells, relative to those treated with a scramble control virus (Figure 5). However, there was no significant increase in transgene expression when using PEI as the polymer (Figure S9, Supplementary Information). The relatively modest enhancement observed in these experiments compared to our inhibitor experiments may be due to partial knockdown of PLK1 by the virus (Figure 5). Surprisingly, the scramble control virus increased transgene expression relative to treatment without any virus when using 1,4C-1,4bis as the polymer carrier (Figure S9). Given that viruses have evolved to be extremely efficient gene delivery vehicles, in part due to their enhanced ability to enter target cell nuclei, it is possible that both scramble control and anti-PLK1 shRNA expressing viruses were able to assist the plasmid in overcoming particular gene delivery barriers faced by polymer-mediated plasmid DNA delivery. This could be due to direct virus-plasmid or virus-polymer binding [40, 41], or indirectly by viral-induced interruption of the target cell’s natural defense(s) against foreign DNA. It is possible that the virus itself was able to assist the plasmid in circumventing particular gene delivery barriers overcome by PLK1 silencing, thus dampening the increase in observed transgene expression enhancement using our method of PLK1 silencing. Interestingly, PEI-mediated gene delivery did not significantly increase in the presence of either virus (Figure S9), suggesting that PEI and the lentiviral vectors used may assist foreign DNA in overcoming similar gene delivery barriers.
Effect of cell cycle on PLK1 inhibitor mediated enhancement of transgene expression
Given the established role of PLK1 in cell cycle regulation, we investigated the effects of HMN-214 and BI 2536 on PC3-PSMA cell cycle progression (in the absence of polymer or plasmid DNA). Nuclear DNA was stained with propidium iodide (PI) in order to determine the amount of DNA using flow cytometry, which allowed us to elucidate the fractions of the cell population in different stages of the cell cycle phase. In all cases, drugs or equivalent volume DMSO (vehicle control) were added to cells, in the absence of polyplexes. Figure 6 and Table 3 indicate that vehicle control (i.e. DMSO)-treated cells were mostly in the G0/G1 phase (63.1 ± 7.6%), with a small fraction of the cells in the S (9.6 ± 2.3%) phase, and approximately a quarter of the cell population (26.6 ±7.0%) in the G2 or M phases of the cell cycle. However, inhibition of PLK1 with 3.3 μM HMN-214 almost completely arrested cells in the G2/M phase (93.6 ± 1.6%), while PLK1 inhibition with 25 nM BI 2536 also resulted in strong accumulation of the cell population (87.1 ± 5.0%) in the G2/M phase of the cell cycle. These results are consistent with the known PLK1 inhibition activity of the drugs [42, 43].
Figure 6.
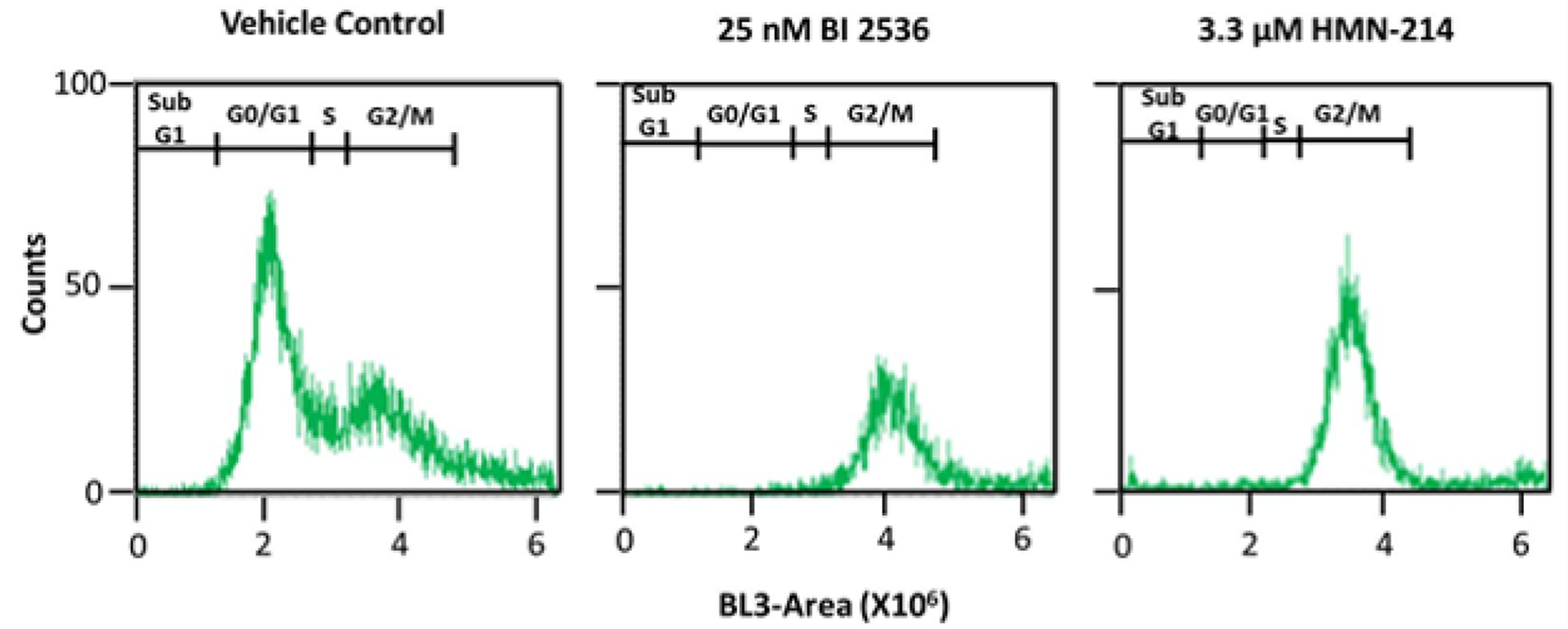
Cell cycle analysis of PC3-PSMA cells treated with vehicle control (DMSO), 25 nM BI 2536, or 3.3 μM HMN-214 using flow cytometry and staining with propidium iodide (PI). Results from one representative experiment out of N = 3 independent experiments are shown. The y-axis (counts) indicates the number of cells with the specific fluorescence intensity shown on the x-axis (BL3-A).
Table 3.
Quantification of PC3-PSMA cells in different stages of the cell cycle in absence and presence of treatment with small molecule PLK1 inhibitors based on N=3 independent experiments.
| Cell Cycle Phase | Vehicle Control | 25 nM BI 2536 | 3.3 μM HMN-214 |
|---|---|---|---|
| Sub-G0/Gl | 0.8 ± 0.3% | 2.0 ± 1.0% | 1.8 ± 0.1% |
| G0/Gl | 63.1 ± 7.6% | 3.6 ± 0.6% | 1.7 ± 0.4% |
| S | 9.6 ± 2.3% | 7.2 ± 4.7% | 2.9 ± 1.2% |
| G2/M | 26.6 ± 7.0% | 87.1 ± 5.0% | 93.6 ± 1.6% |
It is generally accepted that dividing cells, especially cells that are at or near the G2/M transition, are more amenable to transgene expression [44] [45]; indeed, previous studies on cell cycle effects have indicated that transgene expression efficacy is highest for cells in the G2/M phase of the cell cycle. Thus, it is very likely that the observed increase in transgene expression with PLK1 inhibitors is due to the cell cycle arrest in the G2/M phase (Figure 6). It is likely that the compromised nuclear membrane, due to nuclear membrane breakdown (NEBD), in the G2/M phase of the cell cycle, plays a role in the observed enhancement in transgene expression. However, the physicochemical factors responsible for enhancement of transgene expression in the G2/M phase of the cell cycle are not exactly known at this point, although the phenomenon itself has been observed in different studies.
Effects of PLK1 Inhibition on Intracellular Trafficking of Plasmid DNA
Intracellular transport/trafficking of plasmid DNA has been demonstrated to significantly influence transgene expression in cells [15, 46–52]. As shown in Figure 7, we monitored the intracellular distribution of fluorescently labeled plasmid DNA with 25 nM BI 2536 and 3.3 μM HMN-214, and without PLK1 inhibitor treatment using confocal fluorescence microscopy. In the absence of BI 2536 (Figure 7A), plasmid DNA is localized at the Perinuclear Recycling Compartment (PNRC, white arrows) at the microtubule organizing center (MTOC), which is consistent with previous observations of polyplex and nanoparticle transport in these cells [15, 53]. In contrast, treatment with BI 2536 (Figure 7B) disrupts PNRC localization and disperses plasmid DNA throughout the cytoplasm (red arrows).
Figure 7.
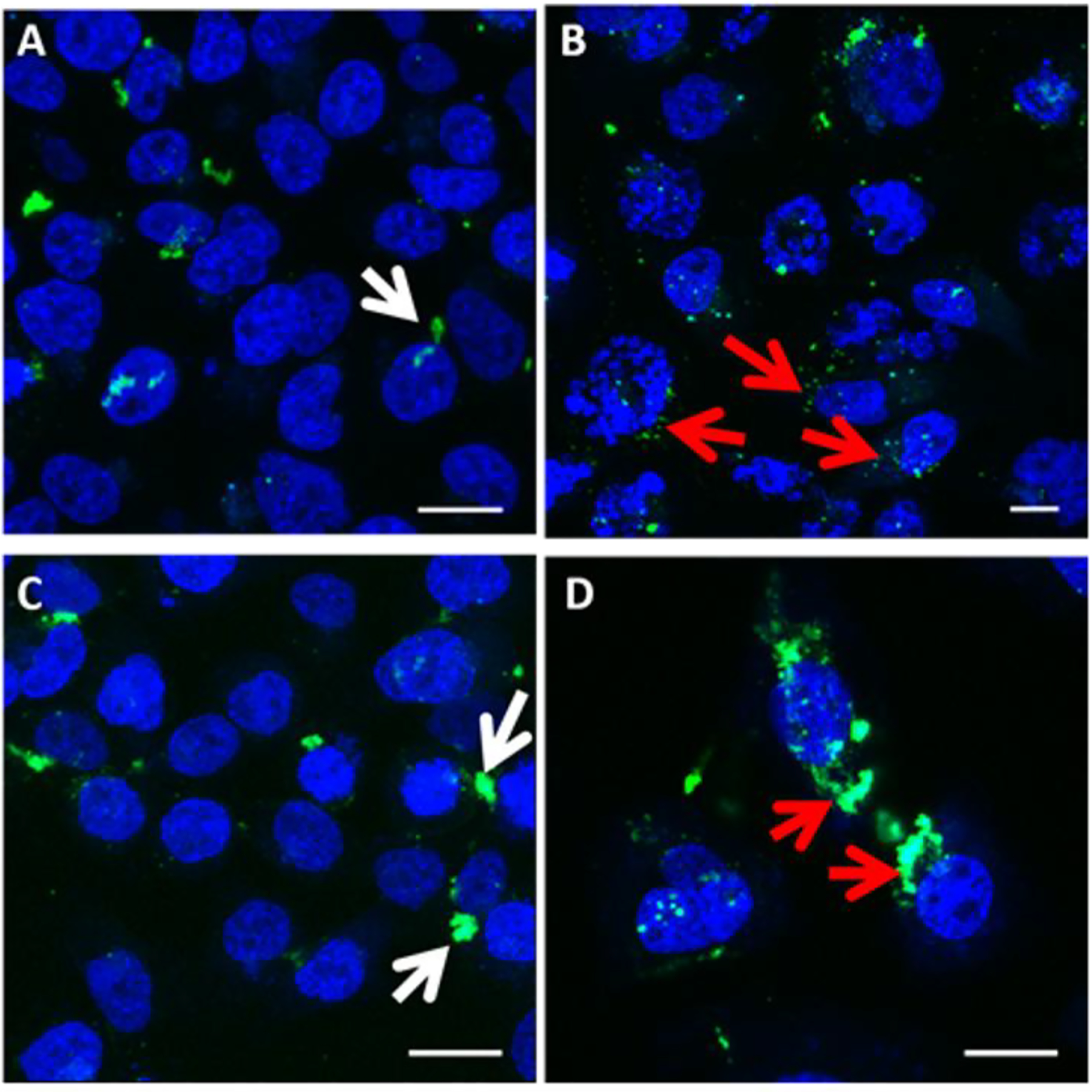
Intracellular trafficking of plasmid DNA in presence and absence of PLK1 inhibitors. PC3-PSMA cells were transfected with polyplexes of PEI:fluorescein (green)-labeled plasmid DNA at a weight ratio of 1:4. Cells treated with A) vehicle control (DMSO) and B) 25 nM BI 2536 are shown 48 hours after co-treatment with polyplexes. Cells treated with C) vehicle control (DMSO) and D) HMN-214 are shown 24 hours after co-treatment with polyplexes. Scale bar = 20 μm. Blue signal indicates DAPI-stained nuclei, and white arrows indicate sequestration of plasmid DNA in the perinuclear recycling compartment (PNRC). Red arrows indicate altered plasmid DNA localization following treatment with PLK1 inhibitors. Images are representative of three independent experiments (N=3).
Images of BI 2536 treated cells (Figure 7B) and cells simultaneously treated with DMSO vehicle control (Figure 7A) were acquired 48 hours following transfection, corresponding temporally to the transgene expression data. Treatment of cells with HMN-214 (3.3 μM), however, resulted in significant detachment and difficulty in imaging beyond 24 hours; thus cells treated with HMN-214 (Figure 7D) and vehicle control (Figure 7C) were imaged 24 hours following transfection. Even though images displayed for HMN-214 treated cells represent a time 24 hours prior to optimal transgene expression, it is still clear that HMN-214 treatment alters intracellular trafficking of the delivered plasmid (Figure 7D), resulting in a similar release from the PNRC as observed with BI 2536 treatment (red arrows). Both drugs appear to disrupt sequestration of the plasmid at the PNRC, and favor the distribution of the plasmid throughout the cytoplasm. This behavior is consistent with our previous observation on increased luciferase expression (20–35 fold enhancement over untreated controls) following treatment with tubacin, a histone deacetylase (HDAC) 6 inhibitor[15], although it is likely that HDAC 6 inhibition increases transgene expression due to increased microtubule stability[50, 54]. Therefore, these results suggest that sequestration within the PNRC has an inhibitory effect, while redistribution of plasmids from this compartment as in the case of treatment with PLK1 inhibitors, may increase polymer-mediated transgene expression.
Enhancement in Transgene Expression using Combinations of Inhibitor Molecules
In addition to PLK1 inhibitors, treatment with inhibitors for other kinases also resulted in enhancement of luciferase transgene expression; for example the JAK inhibitor AG-490 showed a 6-fold enhancement in the kinase inhibitor screen. We therefore hypothesized that combinations of different inhibitors (e.g. HMN-214 and AG-490) of different kinase targets may show even stronger enhancement than the individual drugs by themselves. We also included the histone deacetylase 1 (HDAC 1) inhibitor Entinostat, which has been shown to enhance transgene expression at concentrations of 10–33 μM [55, 56].
When used with 1,4C-1,4Bis polyplexes, individual inhibitor treatments (H = 3.3 μM HMN-214, A = 3.3 μM AG-490, and E = 33 μM Entinostat) increased luciferase expression 10–20 fold higher than the polyplex control, in agreement with Table 1 and Figure 2. However, every pair-wise combination of the inhibitors resulted in significantly higher luciferase expression than any of the individual inhibitor treatments (H+A = 34-fold, E+A = 35-fold, and E+H = 49-fold enhancement; Figure 8). Significantly, the combination of all three inhibitors (H+A+E) resulted in the highest observed enhancement of 86-fold relative to the 1,4C-1,4Bis + pGL3.0 polyplex control.
Figure 8.
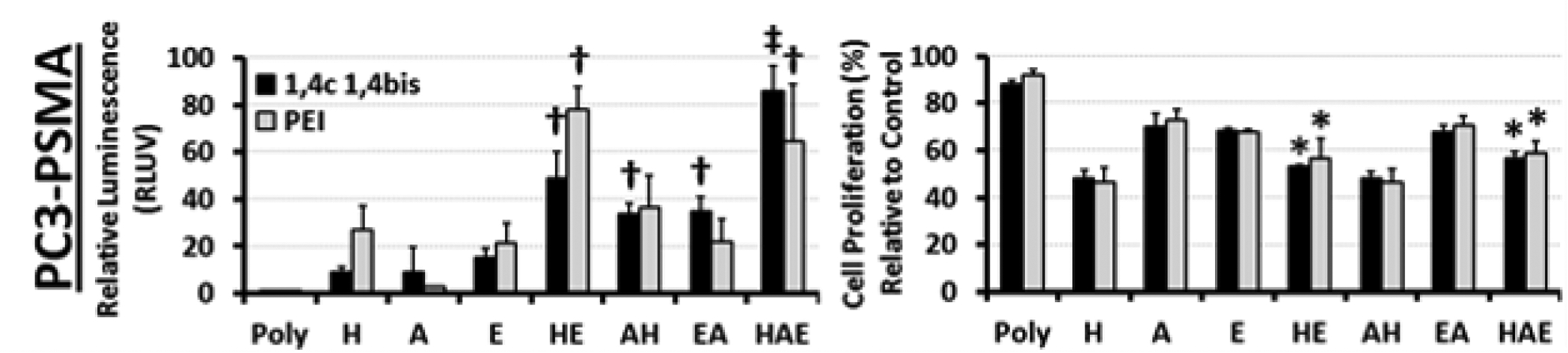
Effects of inhibitor drug combinations on relative luciferase expression (RLUV, left) and cell proliferation (right) in PC3-PSMA cells following transfections with 1,4C-1,4Bis and pGL3.0 plasmid DNA polyplexes. † indicates combinations with RLUV significantly higher than the corresponding individual treatments (e.g. HE compared to H or E), ‡ indicates triple combinations (HAE) with RLUV significantly higher than any pair-wise treatment (e.g. HAE compared to HE, AH, and EA). Asterisks indicate combination treatments containing HMN-214 with significant increases in proliferation compared to HMN-214 alone. In all cases, statistical significance was determined using the Student’s t-test, and a p-value < 0.05 was considered significant. H = 3.3 μM HMN-214 (PLK1 inhibitor), A = 3.3 μM AG-490 (JAK/STAT/ EGFR inhibitor), and E = 33 μM Entinostat (HDAC1 inhibitor, N = 3 independent experiments). Error bars indicate standard deviations.
In contrast, polyplexes formed with PEI did not show a significant increase in transgene expression with inhibitor combinations containing AG-490; 3.3 μM AG-490 (A) alone only shows a slight enhancement of transgene expression (2.4-fold), indicating some dependence on the polymer type for this inhibitor. Enhancement by individual treatment of other inhibitors was similar to previous results; 3.3 μM HMN-214 (H) and 33 μM Entinostat (E) resulted in 29- and 22-fold enhancement, respectively. Pairwise combinations of AG-490 with HMN-214 or Entinostat did not exhibit significant enhancement of transgene expression with PEI polyplexes (no statistically significant differences between HA and H, EA and E, or HAE and HE were observed). Nonetheless, co-treatment of cells with HMN-214 (PLK1 inhibitor) and Entinostat (HDAC1 inhibitor) significantly enhanced transgene expression by up to 78-fold compared to polyplexes alone, and 2–3 times higher than the individual inhibitor treatments (HE ≫ H or E). No significant difference in transgene expression was observed between HAE and HE, further indicating the lack of efficacy of AG-490 when used in combination with PEI.
While inhibitor combinations showed significant enhancement of transgene expression, it is important to mention that the toxicity of the inhibitor combinations was not significantly higher than the individual drug treatments. While all individual drug treatments significantly decreased cell proliferation relative to both polyplex controls (1,4C-1,4Bis and PEI), no significant decrease in cell proliferation was observed between the individual and combination treatments. In contrast, addition of Entinostat to HMN-214 significantly increased cell proliferation by up to 12% more than HMN-214 or HMN-214 with AG-490 (i.e. cell proliferation with HE > H and HAE > H or AH) with both types of polyplexes.
CONCLUSIONS
To our knowledge this is the first study in which parallel screening of small-molecule inhibitors has been employed to identify intracellular kinase targets that play a role in non-viral transgene expression. Our screen, consisting of 182 kinase inhibitors, identified several different inhibitors which significantly enhanced transgene expression. Specifically, we have identified Polo-like Kinase-1 (PLK1) as a key target whose inhibition results in enhancement of non-viral transgene expression. Our results clearly show that the kinase inhibitors HMN-214 and BI 2536 significantly enhance transgene delivery and expression in cancer cell lines. These results are significant since Polo-like kinases are an emerging target for anti-cancer therapies, and BI 2536 is currently in clinical trials [57, 58]. Inhibition of PLK1 by HMN-214 and BI 2536 enhances transgene expression by arresting the cell population in the G2/M phase of the cell cycle (when the nucleus is more permeable) and altering intracellular trafficking of plasmid DNA. These results were reinforced by lentiviral silencing of PLK1, which also significantly enhanced transgene expression. Most importantly, combinations of small-molecule inhibitors resulted in significant enhancement of transgene expression compared to individual inhibitors acting alone. Additionally, the other inhibitors employed in combination drug treatments, AG-490 [59] and Entinostat [60] have been demonstrated to possess anticancer activity. Given the aforementioned anticancer activity of PLK1 inhibition, combination or individual inhibitor treatments may be used to enhance the delivery of a cancer therapeutic gene (i.e. cancer gene therapy) to induce synergistic ablation of tumors.
While treatment with PLK1 inhibitors engendered dramatic reduction in cell proliferation to the cells used in this study (Figures 2 and 3), it is likely that this level of inhibition of cell proliferation would be limited to cancer cells, unlikely to highly affect healthy cells in surrounding tissues. RNA interference to silence PLK1 has demonstrated minimal effects on cell cycle distribution and cell survival of healthy cells in comparison to cancer cells [61, 62]. Additionally, cancer cells have been demonstrated to be more sensitive than healthy cells to the small molecule PLK1 inhibitors BI 2536 [63] and ON01910 [64], both used in this study, although a study conducted by Steegmaier et al suggests that the cell cycle distributions of hTERT-RPE1 immortalized epithelial cells and HeLa cells are equally affected by BI 2536 treatment [65]. Nevertheless, PLK1 is overexpressed in many malignancies and it is expected that, generally, cancerous cells will be more vulnerable to PLK1 inhibition than healthy cells in corresponding tissues.
Taken together, our current results indicate that PLK1 is a promising target for enhancing transgene expression in (cancer) gene therapy, cellular/tissue engineering, and transient protein production. Future work will involve investigation of PLK1 and other lead inhibitors individually, as well as their combinations using appropriate animal models.
Supplementary Material
ACKNOWLEDGEMENTS
Funding from the National Institute of General Medical Sciences (NIGMS), National Institutes of Health (Grant #1R01GM093229-01A1) to KR is gratefully acknowledged. Entinostat used in this study was generously provided by Syndax Pharmaceuticals, Inc. through the Cancer Therapeutics Evaluation Program (CTEP) and the National Cancer Institute, NIH. We would also like to thank Dr. Tong Fu for assistance with training and operation of the FACSCalibur flow cytometer, and Dr. Thrimoorthy Potta and Taraka Sai Pavan Grandhi in the Rege group at ASU for several helpful discussions and generous technical help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR, Effect of gene therapy on visual function in Leber’s congenital amaurosis, N Engl J Med, 358 (2008) 2231–2239. [DOI] [PubMed] [Google Scholar]
- [2].Thomas CE, Ehrhardt A, Kay MA, Progress and problems with the use of viral vectors for gene therapy, Nat Rev Genet, 4 (2003) 346–358. [DOI] [PubMed] [Google Scholar]
- [3].Chan P, Kurisawa M, Chung JE, Yang YY, Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery, Biomaterials, 28 (2007) 540–549. [DOI] [PubMed] [Google Scholar]
- [4].Pack DW, Hoffman AS, Pun S, Stayton PS, Design and development of polymers for gene delivery, Nature reviews, 4 (2005) 581–593. [DOI] [PubMed] [Google Scholar]
- [5].Leong KW, Mao HQ, Truong-Le VL, Roy K, Walsh SM, August JT, DNA-polycation nanospheres as non-viral gene delivery vehicles, J Control Release, 53 (1998) 183–193. [DOI] [PubMed] [Google Scholar]
- [6].Anderson DG, Peng W, Akinc A, Hossain N, Kohn A, Padera R, Langer R, Sawicki JA, A polymer library approach to suicide gene therapy for cancer, Proceedings of the National Academy of Sciences of the United States of America, 101 (2004) 16028–16033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Coll JL, Chollet P, Brambilla E, Desplanques D, Behr JP, Favrot M, In vivo delivery to tumors of DNA complexed with linear polyethylenimine, Human gene therapy, 10 (1999) 1659–1666. [DOI] [PubMed] [Google Scholar]
- [8].Barua S, Ramos J, Potta T, Taylor D, Huang HC, Montanez G, Rege K, Discovery of cationic polymers for non-viral gene delivery using combinatorial approaches, Comb Chem High Throughput Screen, 14 (2011) 908–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Potta T, Zhen Z, Grandhi TSP, Christensen MD, Ramos J, Breneman CM, Rege K, Discovery of antibiotics-derived polymers for gene delivery using combinatorial synthesis and cheminformatics modeling, Biomaterials, 35 (2014) 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wiethoff CM, Middaugh CR, Barriers to nonviral gene delivery, J Pharm Sci, 92 (2003) 203–217. [DOI] [PubMed] [Google Scholar]
- [11].Shim MS, Kwon YJ, Stimuli-responsive polymers and nanomaterials for gene delivery and imaging applications, Advanced Drug Delivery Reviews, 64 (2012) 1046–1059. [DOI] [PubMed] [Google Scholar]
- [12].Yezhelyev MV, Qi L, O’Regan RM, Nie S, Gao X, Proton-sponge coated quantum dots for siRNA delivery and intracellular imaging, J Am Chem Soc, 130 (2008) 9006–9012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tros de Ilarduya C, Sun Y, Düzgüneş N, Gene delivery by lipoplexes and polyplexes, Eur J Pharm Sci, 40 (2010) 159–170. [DOI] [PubMed] [Google Scholar]
- [14].Miyata K, Nishiyama N, Kataoka K, Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses, Chem Soc Rev, 41 (2012) 2562–2574. [DOI] [PubMed] [Google Scholar]
- [15].Barua S, Rege K, The influence of mediators of intracellular trafficking on transgene expression efficacy of polymer-plasmid DNA complexes, Biomaterials, 31 (2010) 5894–5902. [DOI] [PubMed] [Google Scholar]
- [16].Rettig GR, Rice KG, Non-viral gene delivery: from the needle to the nucleus, Expert Opin Biol Ther, 7 (2007) 799–808. [DOI] [PubMed] [Google Scholar]
- [17].Lam AP, Dean DA, Progress and prospects: nuclear import of nonviral vectors, Gene Ther, 17 (2010) 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S, A Toll-like receptor recognizes bacterial DNA, Nature, 408 (2000) 740–745. [DOI] [PubMed] [Google Scholar]
- [19].Ishii KJ, Akira S, Innate immune recognition of, and regulation by, DNA, Trends Immunol, 27 (2006) 525–532. [DOI] [PubMed] [Google Scholar]
- [20].Kumagai Y, Takeuchi O, Akira S, TLR9 as a key receptor for the recognition of DNA, Adv Drug Deliv Rev, 60 (2008) 795–804. [DOI] [PubMed] [Google Scholar]
- [21].Suzuki M, Cerullo V, Bertin TK, Cela R, Clarke C, Guenther M, Brunetti-Pierri N, Lee B, MyD88-dependent silencing of transgene expression during the innate and adaptive immune response to helper-dependent adenovirus, Hum Gene Ther, 21 (2010) 325–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hyde SC, Pringle IA, Abdullah S, Lawton AE, Davies LA, Varathalingam A, Nunez-Alonso G, Green AM, Bazzani RP, Sumner-Jones SG, Chan M, Li H, Yew NS, Cheng SH, Boyd AC, Davies JC, Griesenbach U, Porteous DJ, Sheppard DN, Munkonge FM, Alton EW, Gill DR, CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression, Nat Biotechnol, 26 (2008) 549–551. [DOI] [PubMed] [Google Scholar]
- [23].Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S, The protein kinase complement of the human genome, Science, 298 (2002) 1912–1934. [DOI] [PubMed] [Google Scholar]
- [24].Li E, Stupack D, Klemke R, Cheresh DA, Nemerow GR, Adenovirus endocytosis via alpha(v) integrins requires phosphoinositide-3-OH kinase, J Virol, 72 (1998) 2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Parton RG, Joggerst B, Simons K, Regulated internalization of caveolae, J Cell Biol, 127 (1994) 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Malumbres M, Physiological relevance of cell cycle kinases, Physiol Rev, 91 (2011) 973–1007. [DOI] [PubMed] [Google Scholar]
- [27].Rawlings JS, Rosler KM, Harrison DA, The JAK/STAT signaling pathway, J Cell Sci, 117 (2004) 1281–1283. [DOI] [PubMed] [Google Scholar]
- [28].Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, Davis RJ, Selective interaction of JNK protein kinase isoforms with transcription factors, EMBO J, 15 (1996) 2760–2770. [PMC free article] [PubMed] [Google Scholar]
- [29].ur Rehman Z, Hoekstra D, Zuhorn IS, Protein kinase A inhibition modulates the intracellular routing of gene delivery vehicles in HeLa cells, leading to productive transfection, J Control Release, 156 (2011) 76–84. [DOI] [PubMed] [Google Scholar]
- [30].van den Bogaard EH, Rodijk-Olthuis D, Jansen PA, van Vlijmen-Willems IM, van Erp PE, Joosten I, Zeeuwen PL, Schalkwijk J, Rho kinase inhibitor Y-27632 prolongs the life span of adult human keratinocytes, enhances skin equivalent development, and facilitates lentiviral transduction, Tissue Eng Part A, 18 (2012) 1827–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].van der Aa MA, Huth US, Häfele SY, Schubert R, Oosting RS, Mastrobattista E, Hennink WE, Peschka-Süss R, Koning GA, Crommelin DJ, Cellular uptake of cationic polymer-DNA complexes via caveolae plays a pivotal role in gene transfection in COS-7 cells, Pharm Res, 24 (2007) 1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M, Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen, Neoplasia (New York, N.Y, 1 (1999) 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Barua S, Joshi A, Banerjee A, Matthews D, Sharfstein ST, Cramer SM, Kane RS, Rege K, Parallel Synthesis and Screening of Polymers for Nonviral Gene Delivery, Molecular Pharmaceutics, 6 (2009) 86–97. [DOI] [PubMed] [Google Scholar]
- [34].Grueneberg DA, Degot S, Pearlberg J, Li W, Davies JE, Baldwin A, Endege W, Doench J, Sawyer J, Hu Y, Boyce F, Xian J, Munger K, Harlow E, Kinase requirements in human cells: I. Comparing kinase requirements across various cell types, Proc Natl Acad Sci U S A, 105 (2008) 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Baldwin A, Li W, Grace M, Pearlberg J, Harlow E, Münger K, Grueneberg DA, Kinase requirements in human cells: II. Genetic interaction screens identify kinase requirements following HPV16 E7 expression in cancer cells, Proc Natl Acad Sci U S A, 105 (2008) 16478–16483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bommi-Reddy A, Almeciga I, Sawyer J, Geisen C, Li W, Harlow E, Kaelin WG, Grueneberg DA, Kinase requirements in human cells: III. Altered kinase requirements in VHL−/− cancer cells detected in a pilot synthetic lethal screen, Proc Natl Acad Sci U S A, 105 (2008) 16484–16489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Grueneberg DA, Li W, Davies JE, Sawyer J, Pearlberg J, Harlow E, Kinase requirements in human cells: IV. Differential kinase requirements in cervical and renal human tumor cell lines, Proc Natl Acad Sci U S A, 105 (2008) 16490–16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pearlberg J, Degot S, Endege W, Park J, Davies J, Gelfand E, Sawyer J, Conery A, Doench J, Li W, Gonzalez L, Boyce FM, Brizuela L, Labaer J, Grueneberg D, Harlow E, Screens using RNAi and cDNA expression as surrogates for genetics in mammalian tissue culture cells, Cold Spring Harb Symp Quant Biol, 70 (2005) 449–459. [DOI] [PubMed] [Google Scholar]
- [39].Gazit A, Osherov N, Posner I, Yaish P, Poradosu E, Gilon C, Levitzki A, Tyrphostins. 2. Heterocyclic and alpha-substituted benzylidenemalononitrile tyrphostins as potent inhibitors of EGF receptor and ErbB2/neu tyrosine kinases, J Med Chem, 34 (1991) 1896–1907. [DOI] [PubMed] [Google Scholar]
- [40].Wagner E, Zatloukal K, Cotten M, Kirlappos H, Mechtler K, Curiel DT, Birnstiel ML, Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes, Proc Natl Acad Sci U S A, 89 (1992) 6099–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Curiel DT, Wagner E, Cotten M, Birnstiel ML, Agarwal S, Li CM, Loechel S, Hu PC, High-efficiency gene transfer mediated by adenovirus coupled to DNA-polylysine complexes, Hum Gene Ther, 3 (1992) 147–154. [DOI] [PubMed] [Google Scholar]
- [42].Takagi M, Honmura T, Watanabe S, Yamaguchi R, Nogawa M, Nishimura I, Katoh F, Matsuda M, Hidaka H, In vivo antitumor activity of a novel sulfonamide, HMN-214, against human tumor xenografts in mice and the spectrum of cytotoxicity of its active metabolite, HMN-176, Invest New Drugs, 21 (2003) 387–399. [DOI] [PubMed] [Google Scholar]
- [43].Stewart HJ, Kishikova L, Powell FL, Wheatley SP, Chevassut TJ, The polo-like kinase inhibitor BI 2536 exhibits potent activity against malignant plasma cells and represents a novel therapy in multiple myeloma, Exp Hematol, 39 (2011) 330–338. [DOI] [PubMed] [Google Scholar]
- [44].Brunner S, Sauer T, Carotta S, Cotten M, Saltik M, Wagner E, Cell cycle dependence of gene transfer by lipoplex polyplex and recombinant adenovirus, Gene Therapy, 7 (2000) 401–407. [DOI] [PubMed] [Google Scholar]
- [45].Tseng WC, Haselton FR, Giorgio TD, Mitosis enhances transgene expression of plasmid delivered by cationic liposomes, Biochim Biophys Acta, 1445 (1999) 53–64. [DOI] [PubMed] [Google Scholar]
- [46].Suh J, Wirtz D, Hanes J, Efficient active transport of gene nanocarriers to the cell nucleus, Proceedings of the National Academy of Sciences of the United States of America, 100 (2003) 3878–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dinh AT, Pangarkar C, Theofanous T, Mitragotri S, Understanding intracellular transport processes pertinent to synthetic gene delivery via stochastic simulations and sensitivity analyses, Biophysical journal, 92 (2007) 831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shi J, Chou B, Choi JL, Ta AL, Pun SH, Investigation of Polyethylenimine/DNA Polyplex Transfection to Cultured Cells Using Radiolabeling and Subcellular Fractionation Methods, Molecular pharmaceutics, 10 (2013) 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ingle NP, Xue L, Reineke TM, Spatiotemporal cellular imaging of polymer-pDNA nanocomplexes affords in situ morphology and trafficking trends, Molecular pharmaceutics, 10 (2013) 4120–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Badding MA, Dean DA, Highly acetylated tubulin permits enhanced interactions with and trafficking of plasmids along microtubules, Gene Ther, 20 (2013) 616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reilly MJ, Larsen JD, Sullivan MO, Polyplexes traffic through caveolae to the Golgi and endoplasmic reticulum en route to the nucleus, Molecular pharmaceutics, 9 (2012) 1280–1290. [DOI] [PubMed] [Google Scholar]
- [52].Kulkarni RP, Wu DD, Davis ME, Fraser SE, Quantitating intracellular transport of polyplexes by spatio-temporal image correlation spectroscopy, Proceedings of the National Academy of Sciences of the United States of America, 102 (2005) 7523–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Barua S, Rege K, Cancer-Cell-Phenotype-Dependent Differential Intracellular Trafficking of Unconjugated Quantum Dots, Small, 5 (2009) 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vaughan EE, Geiger RC, Miller AM, Loh-Marley PL, Suzuki T, Miyata N, Dean DA, Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency, Mol Ther, 16 (2008) 1841–1847. [DOI] [PubMed] [Google Scholar]
- [55].Kasman L, Onicescu G, Voelkel-Johnson C, Histone deacetylase inhibitors restore cell surface expression of the coxsackie adenovirus receptor and enhance CMV promoter activity in castration-resistant prostate cancer cells, Prostate Cancer, 2012 (2012) 137163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kasman L, Lu P, Voelkel-Johnson C, The histone deacetylase inhibitors depsipeptide and MS-275, enhance TRAIL gene therapy of LNCaP prostate cancer cells without adverse effects in normal prostate epithelial cells, Cancer Gene Ther, 14 (2007) 327–334. [DOI] [PubMed] [Google Scholar]
- [57].Ellis PM, Chu QS, Leighl N, Laurie SA, Fritsch H, Gaschler-Markefski B, Gyorffy S, Munzert G, A phase I open-label dose-escalation study of intravenous BI 2536 together with pemetrexed in previously treated patients with non-small-cell lung cancer, Clin Lung Cancer, 14 (2013) 19–27. [DOI] [PubMed] [Google Scholar]
- [58].Vose JM, Friedberg JW, Waller EK, Cheson BD, Juvvigunta V, Fritsch H, Petit C, Munzert G, Younes A, The Plk1 inhibitor BI 2536 in patients with refractory or relapsed non-Hodgkin lymphoma: a phase I, open-label, single dose-escalation study, Leuk Lymphoma, 54 (2013) 708–713. [DOI] [PubMed] [Google Scholar]
- [59].Huang C, Yang G, Jiang T, Huang K, Cao J, Qiu Z, Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro, J Exp Clin Cancer Res, 29 (2010) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Marks PA, The clinical development of histone deacetylase inhibitors as targeted anticancer drugs, Expert Opin Investig Drugs, 19 (2010) 1049–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu X, Lei M, Erikson RL, Normal cells, but not cancer cells, survive severe Plk1 depletion, Mol Cell Biol, 26 (2006) 2093–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y, Small interfering RNA-mediated Polo-like kinase 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals, Cancer Res, 65 (2005) 2698–2704. [DOI] [PubMed] [Google Scholar]
- [63].Nappi TC, Salerno P, Zitzelsberger H, Carlomagno F, Salvatore G, Santoro M, Identification of Polo-like kinase 1 as a potential therapeutic target in anaplastic thyroid carcinoma, Cancer Res, 69 (2009) 1916–1923. [DOI] [PubMed] [Google Scholar]
- [64].Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Boomi Nathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP, ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent, Cancer Cell, 7 (2005) 275–286. [DOI] [PubMed] [Google Scholar]
- [65].Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf G, Kraut N, Peters J, Rettig W, BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo, Current Biology, 17 (2007) 316–322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.