

Abstract
Background
Neuroblastoma is the most common extracranial solid tumor of childhood. Patients with high-risk disease undergo extremely aggressive therapy and nonetheless have cure rates below 50%. Treatment with the ch14.18 monoclonal antibody (dinutuximab beta), directed against the GD2 disialoganglioside, improved 5-year event-free survival in high-risk patients when administered in postconsolidation therapy and was recently implemented in standard therapy. Relapse still occurred in 57% of these patients, necessitating new therapeutic options. Bispecific trifunctional antibodies (trAbs) are IgG-like molecules directed against T cells and cancer surface antigens, redirecting T cells (via their CD3 specificity) and accessory immune cells (via their functioning Fc-fragment) toward tumor cells. We sought proof-of-concept for GD2/CD3-directed trAb efficacy against neuroblastoma.
Methods
We used two GD2-specific trAbs differing only in their CD3-binding specificity: EKTOMUN (GD2/human CD3) and SUREK (GD2/mouse Cd3). This allowed trAb evaluation in human and murine experimental settings. Tumor-blind trAb and the ch14.18 antibody were used as controls. A coculture model of human peripheral blood mononuclear cells (PBMCs) and neuroblastoma cell lines was established to evaluate trAb antitumor efficacy by assessing expression of T-cell surface markers for activation, proinflammatory cytokine release and cytotoxicity assays. Characteristics of tumor-infiltrating T cells and response of neuroblastoma metastases to SUREK treatment were investigated in a syngeneic immunocompetent neuroblastoma mouse model mimicking minimal residual disease.
Results
We show that EKTOMUN treatment caused effector cell activation and release of proinflammatory cytokines in coculture with neuroblastoma cell lines. Furthermore, EKTOMUN mediated GD2-dependent cytotoxic effects in human neuroblastoma cell lines in coculture with PBMCs, irrespective of the level of target antigen expression. This effect was dependent on the presence of accessory immune cells. Treatment with SUREK reduced the intratumor Cd4/Cd8 ratio and activated tumor infiltrating T cells in vivo. In a minimal residual disease model for neuroblastoma, we demonstrated that single-agent treatment with SUREK strongly reduced or eliminated neuroblastoma metastases in vivo. SUREK as well as EKTOMUN demonstrated superior tumor control compared with the anti-GD2 antibody, ch14.18.
Conclusions
Here we provide proof-of-concept for EKTOMUN preclinical efficacy against neuroblastoma, presenting this bispecific trAb as a promising new agent to fight neuroblastoma.
Keywords: antibodies, neoplasm, neuroblastoma, CD4-CD8 ratio, immunotherapy, pediatrics
Background
Bispecific trifunctional antibodies are an emerging immuno-oncology strategy for tumor therapy. Innate and adaptive immune responses are jointly activated against tumor cells. These engineered heterodimeric IgG-like antibodies consist of two different heavy and light chains, to combine two antigen-binding sites in one molecule, one directed against a tumor-associated antigen and the other against CD3 in the T-cell receptor complex.1 This construction brings T cells in close proximity to tumor cells to elicit a tumor cytotoxic effect. The intact Fc region binds and activates Fcγ receptors (FcγR) on dendritic cells and macrophages,2 which provide costimulatory signals enhancing T-cell effector function. This third recruited immune component presents tumor-specific antigens internalized from lysed tumor cells either directly at the tumor site to initiate a local tumor-specific T-cell response or in lymphoid organs to create memory T cells providing a long-lasting antitumor response.1 3
Approximately half of all children diagnosed with neuroblastoma, the most common extracranial solid tumor of childhood, present with high-risk disease. Disease relapses in >50% of these patients, in whom long-term survival is <10%, emphasizing the limited success of current treatment protocols. Induction treatment for high-risk neuroblastoma currently includes 6–8 multichemotherapy cycles followed by surgery and radiation therapy. Consolidation therapy involves high-dose myeloablative chemotherapy with autologous stem cell reinfusion. Immunotherapy was sought to improve poor cure rates targeting the GD2 cell surface disialoganglioside, expressed in neuroectodermally derived neuroblastoma,4 with limited expression in normal tissues after birth (restricted to brain, peripheral sensory fibers and skin melanocytes).5 Treatment with the human-mouse chimeric ch14.18 monoclonal antibody (dinutuximab) administered as postconsolidation therapy for minimal residual disease in combination with granulocyte-macrophage colony-stimulating factor and interleukin 2 (IL2) was reported to improve 2-year event-free survival in patients with high-risk neuroblastoma by 20%.6 However, after 5 years, this event-free survival advantage shrank to 10%.7 The SIOPEN High-Risk Neuroblastoma 1 Trial showed 15% improvement in 5-year event-free survival for patients receiving ch14.18 (dinutuximab beta) immunotherapy as postconsolidation therapy to treat minimal residual disease.8 Unfortunately, treatment often requires prolonged hospitalization, since common side effects include severe pain (66%) requiring control with morphine as well as hypersensitivity reactions (53%), capillary leak (23%) and fever (78%).9 The current theory for the cause of the pain is that ch14.18 binds to GD2+ myelin in peripheral nerves causing sensorimotor demyelinating neuropathy through complement activation.10 The ch14.18 mode of action is antibody-dependent cellular cytotoxicity (ADCC)11 and complement-dependent cytotoxicity (CDC)12 mediated by its Fc region. Despite side effects, ch14.18 was introduced into standard-of-care guidelines for postconsolidation treatment of high-risk neuroblastoma to maintain remission of disease.13 14 Treatment options for relapsed neuroblastoma are limited, but currently include chemotherapy with irinotecan and temozolomide among other cytotoxic agents,15 small molecule anaplastic lymphoma kinase inhibitors and agents targeting MDM2, BCL2 and histone deacetylases currently being tested in combination therapy in European pediatric trials for relapsed pediatric cancers.16 Haploidentical stem cell transplantation with subsequent ch14.18 treatment achieved 3-year progression-free survival in 38% of patients with relapsed neuroblastoma, however, side effects prevented 41% of patients from completing the protocol.17 Combining temozolomide and irinotecan with ch14.18 treatment has produced objective responses in 40% of patients with relapsed neuroblastoma.18 None of these therapeutic options is sufficiently established to warrant clear recommendation for relapsed neuroblastoma. Even after introducing ch14.18 into high-risk neuroblastoma therapy, survival in patients remains worse than for patients with other pediatric malignancies, driving the urgent need for new therapeutic options.
We built on the partial success of GD2-directed immunotherapy and evaluated whether this effect could be enhanced with bispecific trifunctional antibodies also targeting GD2. In vitro binding and efficacy against human neuroblastoma cells was compared for the current standard-of-care monoclonal antibody, ch14.18, and the human CD3-targeting bispecific trifunctional antibody (trAb), EKTOMUN. We then compared ch14.18 activity in an immunocompetent mouse model for highly aggressive metastasized neuroblastoma with SUREK, the murine-active version of EKTOMUN, which targets murine Cd3 to activate T cells in mice.
Methods
Neuroblastoma cell lines
The human neuroblastoma cells lines, NB69, SK-N-FI and IMR-5/75, were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany) and cultured in Roswell Park Memorial Institute (RPMI) 1640 media (GIBCO) supplemented with 10% fetal calf serum. SK-N-BE(2) cells were purchased from the American Type Culture Collection (Wesel, Germany), and cultured in Dulbecco’s modified Eagle’s medium (DMEM, GIBCO) supplemented with 10% fetal calf serum. The NXS2 murine neuroblastoma cell line19 was kindly provided by Holger N. Lode (University Medicine Greifswald, Germany) and cultured in DMEM supplemented with 10% fetal calf serum, 100 U/mL penicillin and 100 µg/mL streptomycin. All cell lines were cultured at 37°C in 5% CO2. Cell line identity was assured by short tandem repeat DNA genotyping. Cultures were routinely tested for mycoplasma using the PlasmoTest Kit (Invitrogen).
Bispecific trifunctional antibodies
SUREK, EKTOMUN, TRBs011, TRBs012 and Me361 antibodies were kindly provided by Trion Research or Lindis Biotech (Martinsried, Germany), where they were generated by quadroma technology and purified by affinity and ion exchange chromatography as previously described in detail.20
Quantification of GD2 surface expression and binding analysis of EKTOMUN/SUREK
QuantiBRITE PE calibration beads (BD Biosciences) were used to determine GD2 antigen density on neuroblastoma cells according to the manufacturer’s instructions as previously described.21 To assess binding of EKTOMUN and SUREK to GD2+ neuroblastoma cell lines, we incubated 200 000 SK-N-BE(2) cells with EKTOMUN and 200 000 NXS2 cells with SUREK for 1 hour at 4°C. Subsequently, cells were stained with a PE-labeled secondary antibody directed against mouse IgG2a (cat#407108, Biolegend) for 30 min at 4°C and flow cytometric analysis was performed. To visualize trifunctional effector and target cell binding, we coincubated 2×105 NB69 cells expressing GFP_ffluc with 2×106 PBMCs with or without 1 µg/mL EKTOMUN for 5 min at 37°C. After vortexing, the mixture was fixed immediately using BD cytofix/cytoperm according to the manufacturer’s instructions. Subsequently flow cytometric analysis was performed using antibodies against CD11b (cat#101236, Biolegend), CD4 (cat#357407, Biolegend) and CD8 (cat# 300912, Biolegend). Data were analyzed using FlowJo software (BD).
In vitro assessment of tumor cytotoxicity, cytokine release and T-cell activation
In vitro antibody-mediated tumor cytotoxicity was quantified as previously described22 using a biophotonic luciferase assay. In brief, GFP_ffluc-expressing NB69, SK-N-FI, IMR-5/75 or SK-N-BE(2) neuroblastoma cells were cocultured with freshly thawed peripheral blood mononuclear cells (PBMCs) added 3 hour after neuroblastoma cell seeding in the presence of test antibodies. FcγR blocking was performed using the FcR blocking reagent human (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Bioluminescence was measured 72 hours after adding 28.6 µg of Xenolight D-Luciferin (PerkinElmer, Rodgau, Germany) per 150 µl media, with an exposure time of 180 s. Instrumental background was subtracted. Antibody-mediated tumor cell lysis was calculated using following formula: % specific lysis = [(RLUtumor cells +PBMCs)−(RLUtumor cells + PBMCs + antibody)]/[(RLUtumor cells + PBMCs)−(RLUPBMCs)]×100%. To obtain individual cell populations as required to assess bispecific trAb trifunctionality, T cells were isolated from PBMCs by magnetic-activated cell separation using the Pan T-Cell Isolation Kit (Miltenyi Biotec). Non-T cells retained in the column were eluted and used as accessory immune cells (AICs). IL2 and interferon-γ (IFNG) release by T cells was quantified in media conditioned (24 and 48 hours) by coculture of neuroblastoma cell lines (seeded at 2×105 cells/well in 24-well plates 24 hours before adding PBMCs at an effector ratio of 10:1) using OptEIA enzyme‐linked immunosorbent assays (BD, Heidelberg, Germany) according to the manufacturer’s instructions. T-cell activation and IFNG producing effector cells were flow cytometrically quantified (as previously described22 48 hours after neuroblastoma-PBMC coculture (identical set up to cytokine release assays) using LIVE/DEAD cell stain kit (Invitrogen, Carlsbad, CA, USA) to gate out dead cells and antibodies against CD3 (cat#300328, Biolegend, San Diego, CA, USA), CD4 (cat#300546, Biolegend), CD8a (cat#344742, Biolegend), CD25 (cat#302612, Biolegend), CD56 (cat#318306, Biolegend), CD69 (cat#310904, Biolegend) and IFNG (cat#502532, Biolegend). IFNG was stained intracellularly using cytofix/cytoperm (cat#554722, BD) according to the manufacturer’s instructions. Murine cell surface activation markers were detected using antibodies against Cd3 (cat#100218, Biolegend), Cd4 (cat#100559, Biolegend), Cd8a (cat#100744, Biolegend), Cd25 (cat#101916, Biolegend) and Cd69 (cat#104506, Biolegend). Data were analyzed using FlowJo software (BD). All assays were conducted in triplicates.
Analyzing the method of tumor cell death
The ADCC Reporter Bioassay (Promega, Walldorf, Germany) was used in 96-well format to detect cytotoxicity in NB69 cells (25 000 cells/well, effector to target ratio of 3:1) exposed to antibody and engineered Jurkat effector cells (stably expressing the relevant Fc gamma receptor and a luciferase reporter gene under control of the NFAT response element). CDC in NB69 cells (stably expressing luciferase, 30 000 cells/well) exposed to test antibodies was performed as previously described23 in 96-well format. After 24 hours, background bioluminescence was measured in culture media before adding 28.6 µg of XenoLight D-luciferin (PerkinElmer) per 150 µl media for either assay (ADCC and CDC), and bioluminescence was quantified. Relative light units (RLUs) were reported directly as surrogates for ADCC, and CDC was calculated by [1−(RLUtumor cells + serum + antibody)/(RLUtumor cells + serum)]×100%.
Testing in syngeneic mouse model
Male and female A/J mice (The Jackson Laboratory, Bar Harbor, ME, USA) were bred in-house and group-housed under pathogen-free conditions on a 12-hour day/night cycle with free access to food and water, according to institutional guidelines and compliant with national and EU regulations for animal use in research. Tumor-infiltrating lymphocytes (TILs) were analyzed in A/J mice (8–12-week-old males, 7 per treatment group) intravenously injected with 1×106 NXS2 cells on day 0, then intraperitoneally injected with either 10 µg SUREK in 100 µL phosphate-buffered saline (PBS) or only PBS on day 17. Mice were euthanized on day 21 and macroscopic metastases were dissected from the liver. Metastases were mechanically disaggregated and then enzymatically digested with dispase II (1 mg/mL, Sigma, St. Louis, MO, USA), papain (0.37 mg/mL, Sigma) DNAse I (0.1 mg/mL, AppliChem, Darmstadt, Germany) disolved in calcium-free and magnesium-free Hank’s balanced salt solution (GIBCO, Grand Island, NY, USA) for 20 min at room temperature and filtered through 70 µm nylon cell sieves (Falcon, BD) to obtain a single-cell suspension. Erythrocytes were lysed before flow cytometric analysis of TILs. Spleen tissue was minced, passaged through a 70 µm filter and erythrocytes were lysed before further analysis. Liver metastases were quantified in A/J mice (8–12-week-old females) intravenously injected with 5×105 NXS2 cells on day 0, then intraperitoneally injected with 5 µg of either SUREK, TRBs012 or ch14.18 in 100 µl PBS (or PBS alone) on days 1 and 5, followed by 1 µg of the same agent on day 9. Mice were euthanized on day 21, and macroscopic liver metastases were counted by two independent researchers (blinded to testing group), and results were averaged.
Statistical analysis
Differences between testing groups in in vitro assays were analyzed using unpaired t tests. Differences between testing groups in in vivo assays were compared using the Mann-Whitney test. P values<0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism V.8.3 software (GraphPad Software, San Diego, CA, USA).
Results
EKTOMUN induces neuroblastoma cytotoxicity in vitro
To assess EKTOMUN response against neuroblastoma cells presenting a clinically relevant range of target molecule density, we selected four human neuroblastoma cell lines expressing different GD2 levels from intermediate-risk or high-risk genomic backgrounds, either lacking or harboring MYCN amplifications. Quantification of GD2 molecules on the cell surface in these four human neuroblastoma cell lines and the murine NXS2 neuroblastoma cell line, which is used in our syngeneic mouse model, showed that GD2 molecule density was highest in the MYCN-amplified IMR-5/75 cell line (599 163±5095 molecules/cell) and lowest in the SK-N-FI cell line (4357±327 molecules/cell; figure 1A). The murine NXS2 cell line expressed 19 645±1266 molecules/cell, ranking in the lower half of the tested cell lines. We investigated binding of the bispecific trifunctional antibodies, EKTOMUN (binding of human T cells) and SUREK (binding of murine T cells), and control antibodies (binding specificities and structures in figure 1B) to appropriate target-expressing cells in our cell line panel. Both EKTOMUN and SUREK were able to bind the neuroblastoma cell lines SK-N-BE(2) and NXS2 (figure 1C). The bivalent GD2-directed monoclonal Me361 antibody, the parental antibody to the GD2-directed antibody half of EKTOMUN and SUREK, bound at lower concentrations compared with monovalent EKTOMUN and SUREK. The more effective binding of EKTOMUN to SK-N-BE(2) compared with SUREK binding to NXS2, representing the higher GD2 density on SK-N-BE(2) cells. Both TRBs011 and TRBs012 did not bind to neuroblastoma cells and are used as tumor-blind negative controls. To show trAb-mediated effector and target cell interaction, we coincubated PBMCs together with a GFP+ neuroblastoma cell line with or without EKTOMUN. We stained the mixture with fluorochrome-labeled antibodies against CD4 and CD8 to detect T cells, and against CD11b to detect FcγR+ monocytic AICs. We detected 8.4% triple+effector-target cell clusters (CD11b+/CD4+ or CD8+/GFP+) in the presence of EKTOMUN and only 0.82% without EKTOMUN via flow cytometry (figure 1D). In conclusion, we show differential GD2 expression on neuroblastoma cell lines and provide evidence that both SUREK and EKTOMUN bind to neuroblastoma cells in a concentration-dependent manner. We also show simultaneous interaction of EKTOMUN with all effector and target cell types.
Figure 1.

SUREK and EKTOMUN bispecific trifunctional antibodies bind to GD2 positive neuroblastoma cell lines and form effector-target cell clusters. (A) Flow cytometric quantification of GD2 molecules on the cell surface of five different neuroblastoma cell lines. (B) Schematic description of the antibodies in this study. blue: mouse IgG2a recognizing GD2; gray: mouse IgG2a recognizing the alphavirus glycoprotein E1; red: rat IgG2b recognizing human CD3; yellow: rat IgG2b recognizing mouse Cd3; green-light blue: ch14.18 a humanized chimeric antibody consisting of a human IgG1 Fc fragment attached to a murine Fab region (ligand binding domain recognizing GD2). (C) Flow cytometric binding analysis of the bispecific trifunctional antibodies SUREK, EKTOMUN, TRBs011 and TRBs012 as well as the parental GD2-directed monoclonal antibody Me361 on a human (SK-N-BE(2)) and a murine (NXS2) neuroblastoma cell line. (D) Effector-target cell cluster formation is visualized by representative dot plots showing triple+ cell clusters. Triple+ events are positive for CD4 or CD8 (T cells), CD11b (monocytic accessory immune cells) and GFP (neuroblastoma cells).
In order to evaluate EKTOMUN-dependent neuroblastoma cytotoxicity in vitro, we used human PBMCs from healthy donors in coculture with neuroblastoma cell lines. We first assessed the ‘trifunctionality’ of EKTOMUN by testing, whether all three EKTOMUN specificities (GD2-expressing tumor cells, CD3-expressing T cells and FcγR-expressing AICs) are able and necessary to induce neuroblastoma cell death. The PBMCs were analyzed for their proportion of T cells and AICs by flow cytometry to assess T or AIC contributions to tumor cytolysis. The PBMCs used, consisted of 70% T cells and 30% AICs as determined by flow cytometry (online supplemental figure 1). To differentially investigate each of the three antibody functions, NB69 cells were incubated with (i) T cells at an effector to target (E:T) ratio of 7:1, (ii) AICs (E:T ratio of 3:1) or (iii) or both (E:T ratio of 10:1). PBMC or T cells were not activated prior to incubation. To block trAb–effector cell interaction, we treated one condition with SUREK, which binds murine instead of human CD3 but has the same Fc fragment and GD2 binding arm as EKTOMUN. To prevent FcγR binding, we incubated PBMCs with a FcγR-blocking reagent before adding them to the coculture. The use of SUREK or only AIC completely abolished the CD3-mediated cytotoxic effect and the use of FcγR blocking reagent or only T cells massively decreased tumor cell lysis. EKTOMUN concentration-dependent neuroblastoma cell lysis was only fully induced when both T cells and AICs were present and all EKTOMUN functions were operative (figure 2A), demonstrating EKTOMUN trifunctionality to recognize target cells (via GD2-binding), activate T cells (via CD3-binding) and activate AICs (via FcγR-binding).
Figure 2.

EKTOMUN mediates a cytotoxic effect against neuroblastoma cell lines in the presence of human PBMCs. (A) The NB69 neuroblastoma cell line was stably transduced with a GFP_ffluc construct and cocultivated with T cells, effector:target (E:T)=7:1; accessory immune cells (AIC), E:T=3:1; or PBMCs that include both T cells+AICs, E:T=10 (7+3):1, from the same healthy donor. Cocultures were treated with the trAbs EKTOMUN or SUREK in rising concentrations with or without FcγR-block. Tumor cell lysis was determined by bioluminescent flux relative to an untreated coculture after 72 hours. (B) The MYCN-non-amplified neuroblastoma cell lines SK-N-FI (low GD2 expression) and NB69 (medium GD2 expression) and the MYCN-amplified cells lines SK-N-BE(2) and IMR-5/75 (both high GD2 expression) were cocultured as described in (A) and treated with the trAbs EKTOMUN or TRBs011 or with the monoclonal ch14.18 antibody. Tumor cell lysis was determined by bioluminescent flux relative to an untreated coculture after 72 hours. (C) SK-N-BE(2) and IMR-5/75 cells were cocultured as described in (A) and treated with the trAbs EKTOMUN or TRBs011 or with ch14.18 at indicated concentrations at an E:T ratio of 1:10 (left panels) and at indicated E:T ratios at an antibody concentration of 0.1 ng/mL (right panels). Tumor cell lysis was determined by bioluminescent flux relative to an untreated coculture after 72 hours. (D) NB69 cells were cocultured as described in (A) and treated with either the trAbs EKTOMUN, TRBs011, ch14.18 at 0.1 ng/mL, or without antibody. Micrographs were taken after 72 hours. Results are pooled medians of technical triplicates of three independent experiments. Student’s t-test; *p<0.05; **p<0.01.
jitc-2021-002923supp002.pdf (1.7MB, pdf)
Next, we compared EKTOMUN-mediated lysis of the human NB69 neuroblastoma cell line using PBMCs from three different donors to exclude donor-dependent factors. Very low EKTOMUN concentrations (0.1 ng/mL) already induced tumor cell lysis, which reached nearly 100% using 1 ng/mL at an E:T ratio of 10:1 independent of the PBMC donor (online supplemental figure 2). The half maximal effective concentration (EC50) ranged between 0.04 and 0.15 ng/mL. All following experiments were conducted with PBMCs from donor #2, as they displayed a mid-range cytotoxic potential.
We next compared EKTOMUN efficacy against four different GD2+ neuroblastoma cell lines cocultured with PBMCs to the standard-of-care monoclonal ch14.18 antibody and tumor-blind trAb TRBs011 negative control. Treatment with EKTOMUN in the presence of PBMCs lysed 73%±4.0 and 77%±17.4 of SK-N-FI and NB69 tumor cells, respectively, independent of target antigen expression. TRBs011 mediated only limited NB69 cytotoxicity, and ch14.18 had almost no cytotoxic effect in this experimental setting (figure 2B). The two highly aggressive MYCN-amplified cell lines, IMR-5/75 and SK-N-BE(2), proved more resistant to EKTOMUN, despite very high GD2 surface densities. Resistance was overcome by increasing either the EKTOMUN concentration or E:T ratio (figure 2C), which however, also increased non-specific effects, as seen by the increased cytotoxicity mediated by the tumor-blind trAb, TRBs011. NB69 cytotoxicity in the presence of PBMCs and the indicated antibody was also viewed microscopically. Tumor cells detached when EKTOMUN was added, indicating their imminent death, while NB69 cells remained adherent when exposed to ch14.18, TRBs011 or no antibodies (figure 2D). In a similar experiment green-fluorescent SK-N-FI and NB69 tumor cells were treated with EKTOMUN or the control TRBs011 trAb in the presence of PBMCs. EKTOMUN treatment reduced GFP+ tumor cells, indicating GD2-specific tumor cell death (online supplemental figure 3). Here we show that the GD2-directed trAb, EKTOMUN, mediates a GD2-dependent cytotoxic effect on neuroblastoma cells harboring or lacking MYCN amplifications, and that activity requires the interplay between tumor, T and AICs.
EKTOMUN/SUREK induce less Fc-mediated cytotoxicity than ch14.18
The identical Fc regions in EKTOMUN and SUREK each contain one mouse and one rat heavy chain, in contrast to the fully human-derived Fc region of ch14.18. It is unknown, how effectively the murine-derived Fc regions induce ADCC and CDC. Thus, we evaluated the biological activity of the trAb Fc fragment in comparison to ch14.18. NK cell-mediated ADCC is considered to be the main method through which ch14.18 delivers tumor cell death.24 We used a commercially available ADCC reporter assay in which Jurkat cells stably express the human FcγR IIIa (CD16), which can be bound by human and murine Fc regions with similar affinity.25 FcγR IIIa binding on the engineered Jurkat cells mediates a quantifiable luciferase activation as a surrogate for ADCC activity. SUREK was used to evaluate the Fc region instead of EKTOMUN because it does not recognize human CD3, thus avoiding trAb-CD3 interaction on the surface of CD3+ Jurkat cells. As expected, ch14.18 produced a strong concentration-dependent bioluminescent signal. SUREK also induced Jurkat activation, but to a lesser extent (21% relative to ch14.18 at 25 µg/mL, figure 3A), demonstrating the noticeably weaker ability of the murine Fc region to induce ADCC.
Figure 3.
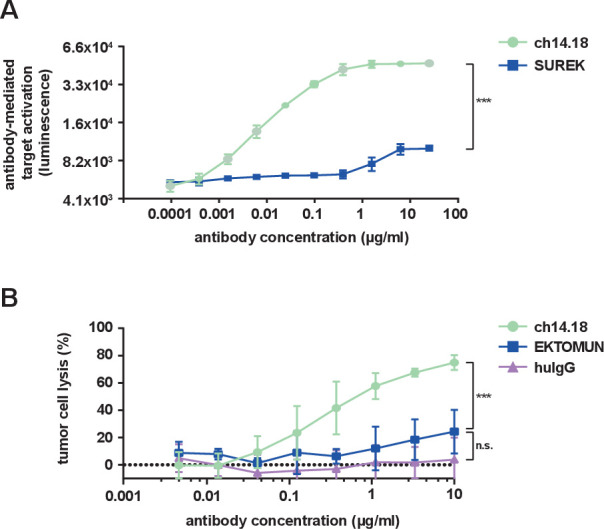
EKTOMUN/SUREK induce less Fc-mediated cytotoxicity than ch14.18. (A) NB69 cells were seeded in the presence of CD16 positive Jurkat cells, which served as effector cells, at an E:T of 3:1. Ch14.18 and the GD2-directed trAb SUREK were added at rising concentrations. The binding of an antibody Fc-region to CD16 induces luciferase expression in Jurkat cells. The bioluminescent signal was measured 6 hours after adding the antibodies to the coculture. (B) The NB69 cell line was stably transduced with a GFP_ffluc construct and treated with rising concentrations of antibodies in the presence of 12.5% human serum from a healthy donor in the culture medium. Tumor cell lysis was determined by bioluminescent flux relative to untreated cells after 24 hours. Student’s t-test; n.s., not significant; ***p<0.001. huIgG, human immunoglobulin G.
CDC is thought to be the main mechanism of action for neuropathic pain, a common side effect of ch14.18. Whether the trAb Fc region induces CDC is also currently unknown, but is interesting in regard to its potential ability to induce neuropathic pain. We performed a CDC assay where the neuroblastoma cell line NB69 was treated with ch14.18 or EKTOMUN in culture medium with 12.5% complement factor-containing human serum. While ch14.18 induced a strong complement-dependent cytotoxic effect after 24 hours (specific lysis: 74.9%±5.4), EKTOMUN induced significantly less CDC (specific lysis: 24.3%±16.0, figure 3B). Our data demonstrate that the cytotoxic effect mediated by the trAb Fc fragment is lower compared with ch14.18, possibly avoiding ch14.18-dependent side effects.
EKTOMUN strongly activates effector cells in vitro
In order to decipher which immune cell subtype is activated on EKTOMUN treatment, we flow cytometrically characterized PBMCs after 48 hours coculture with NB69 neuroblastoma cells and EKTOMUN or control antibodies. CD4+ and CD8+ T cells, NK cells (CD56+CD3−) and NKT (CD56+CD3+) cells were effector cells of this activity. CD3+ cells strongly upregulated activation markers (CD25 and CD69) in the presence of EKTOMUN and, to a lesser extent, in the presence of the unspecific TRBs011 trAb (figure 4A). Similarly, we detected CD25 and CD69 upregulation in CD3−CD56+ cells, indicating that NK cells were engaged and activated by the Fc region (figure 4A). CD4+ T cells expressed the highest CD25 levels (33.6%±6.4 positive cells), while NKT cells expressed the highest CD69 levels (89.0%±2.2 positive cells). After coculturing PBMCs with neuroblastoma cells and EKTOMUN, T-cell effector function was assessed by quantifying IFNG and IL2 cytokine release (figure 4B). EKTOMUN induced PBMCs to release IFNG and IL2 release more strongly than treatment with TRBs011, while ch14.18 induced no cytokine release. Intracellular IFNG staining in effector cells demonstrated that CD4+ and CD8+ T cells, NK (CD56+CD3−) and NKT (CD56+CD3+) cells were IFNG+. While most NKT cells were identified as IFNG-producing (more than 40%), the numbers of CD4+ T cells made them the largest IFN-producing population (figure 4C). However, other immune cell types than those we have analyzed, such as M1 macrophages, could also contribute to IFNG production. Our data demonstrate that treatment with EKTOMUN, strongly activates T, NK and NKT cells as effectors within human PBMCs cocultured with neuroblastoma cells in vitro. Furthermore, T cells exert their effector function by releasing IL2 and IFNG on exposure to EKTOMUN.
Figure 4.

EKTOMUN activates T cells and causes proinflammatory cytokine release. NB69 cells were treated with the trAbs EKTOMUN or TRBs011 or with the monoclonal ch14.18 antibody during coculture with PBMCs from a healthy donor. (A) Representative dot plots of CD69 and CD25 expression on living CD3+/CD56− cells as well as quantification of positive stained cells for CD69 (left panel) and CD25 (right panel) in different effector subpopulations are shown. (B) After 24 and 48 hours, the concentration of Interleukin 2 (IL2) and Interferon-γ (IFNG) in the conditioned media were determined by ELISA. Results are pooled medians of experimental triplicates of three independent experiments. (C) Representative histogram plots of intracellular IFNG production in different effector subpopulations. Definitions of subpopulations: CD4+:CD3+, CD4+, CD8−; CD8+:CD3+, CD4−, CD8+; NK cells: CD3−, CD56+; NKT cells: CD3+, CD56+. Ab: antibody; w/o: without. Student’s t-test; *p<0.05; **p<0.01; ***p<0.001; n.s., not significant.
SUREK treatment shifts Cd4/Cd8 ratio toward cytotoxic Cd8+ T cells in vivo
The presence of activated TIL has been associated with improved outcomes for neuroblastoma patients.26 We investigated whether SUREK has an impact on TILs and their activation status in a highly aggressive murine model mimicking neuroblastoma minimal residual disease. We used the established murine neuroblastoma cell line, NXS2, which is a hybrid of the murine C1300 neuroblastoma cell line (A/J background) and murine dorsal root ganglia cells (C57BL/6 background) in order to achieve GD2 expression.19 We transduced NXS2 cells with GFP to support flow cytometric identification after tumor cell harvest. After intravenous NXS2 cell injection, immunocompetent A/J mice predominantly developed liver metastases.24 Hence, animals were treated as shown in figure 5A, spleens and liver metastases were harvested on day 21 and Cd3+ cells were measured via flow cytometry (gating strategy to identify Cd3+ TILs is shown in online supplemental figure 4). No difference was seen in the frequency of TILs between mice receiving SUREK or the negative control, PBS (online supplemental figure 5A). Interestingly, the composition of T-cell subsets within the metastases changed dramatically on SUREK treatment. The proportion of Cd4+ T cells significantly decreased and there was a trend toward an increase of the Cd8+ T-cell proportion in SUREK-treated animals (online supplemental figure 5B, C). Consequently, the Cd4/Cd8 ratio of TILs was different between mice treated with SUREK (0.59±0.28) and control mice receiving PBS (2.2±0.13), thus, shifting the ratio toward cytotoxic Cd8+ T cells. This effect was also much more pronounced in T cells harvested from tumors, compared with spleens, from SUREK-treated animals (figure 5B). To assess TIL activation, we flow cytometrically determined Cd25 and Cd69 expression on Cd4+ and Cd8+ TILs compared with background activation observed in splenic T cells. Both Cd4+ and CD8+ TILs specifically upregulated the Cd69 activation marker compared with TILs from the PBS control cohort or to splenic T cells from SUREK-treated mice (figure 5C). The Cd25 activation marker was significantly upregulated in Cd4+ TILs from SUREK-treated animals compared with TILs from PBS-treated controls and splenic T cells from SUREK-treated mice (figure 5D). Though Cd25 expression on Cd8+ TILs from SUREK-treated mice was higher than in splenic T cells from the same animals, expression did not significantly differ from CD8+ TILs from PBS-treated mice. Here, we show that SUREK treatment causes a shift in the Cd4-expressing and Cd8-expressing TIL population toward a higher proportion of cytotoxic Cd8+ cells, while also inducing TIL-specific activation of Cd4+ and Cd8+ T cells.
Figure 5.

SUREK reduces the Cd4/Cd8 ratio and leads to activation of Cd4+ and Cd8+ tumor-infiltrating T cells in an immunocompetent murine model for neuroblastoma minimal residual disease. (A) Schematic representation of experimental design and time point of analysis. A/J mice were intravenously (+) injected with 1×106 viable NXS2 cells into a lateral tail vein. Mice received a single intraperitoneal (i.p.) injection of 10 µg SUREK (n=7) or PBS only (n=5) 17 days (d) later. Five days after antibody treatment, mice were sacrificed to tumor infiltrating T cells and splenic T cells. (B) Representative dot plots indicating gating strategy to select tumor infiltrating T cells and determine Cd4/Cd8 ratio in spleen and tumor by flow cytometry. Quantification of (C) Cd69 and (D) Cd25 expression by flow cytometry on Cd4+ and Cd8+ T cells isolated from spleen and tumor. Results are pooled from two independent experiments. Mann-Whitney test; **p<0.01; *p<0.05; n.s., not significant. PBS, phosphate-buffered saline.
SUREK outperforms ch14.18 in a murine model for neuroblastoma minimal residual disease
We used our murine model mimicking neuroblastoma minimal residual disease to directly compare the antitumor potency of SUREK to that of the GD2-directed monoclonal ch14.18 antibody, the current standard-of-care. We intravenously injected NSX2 tumor cells into mice, before treating cohorts every 4 days for a total of three cycles with either SUREK, the unspecific TRBs012 trAb, ch14.18 or PBS starting on day +1. This design mimicks the clinical setting, in which ch14.18 is used as postconsolidation therapy for minimal residual disease (figure 6A). On day +21 post-tumor cell injection, mice were sacrificed to quantify macroscopically visible metastases on the liver surface (examples shown in figure 6B). No superficial liver metastases were detected in four of six mice (67%) belonging to the SUREK treatment group. Single metastasis was detected in each of the other two mice in this group. In contrast, only two of six mice (33%) receiving ch14.18 were free of liver metastases, while the majority developed multiple liver metastases (up to 23) on the liver surface (figure 6C). The tumor-blind trAb, TRBs012, also reduced the number of liver foci compared with the PBS-treated control group, but to a lesser extent compared with SUREK. In conclusion, we provide preclinical evidence that bispecific trifunctional antibodies directed against GD2 show superior efficacy to prevent the outgrowth of liver metastases compared with the standard-of-care monoclonal ch14.18 antibody.
Figure 6.
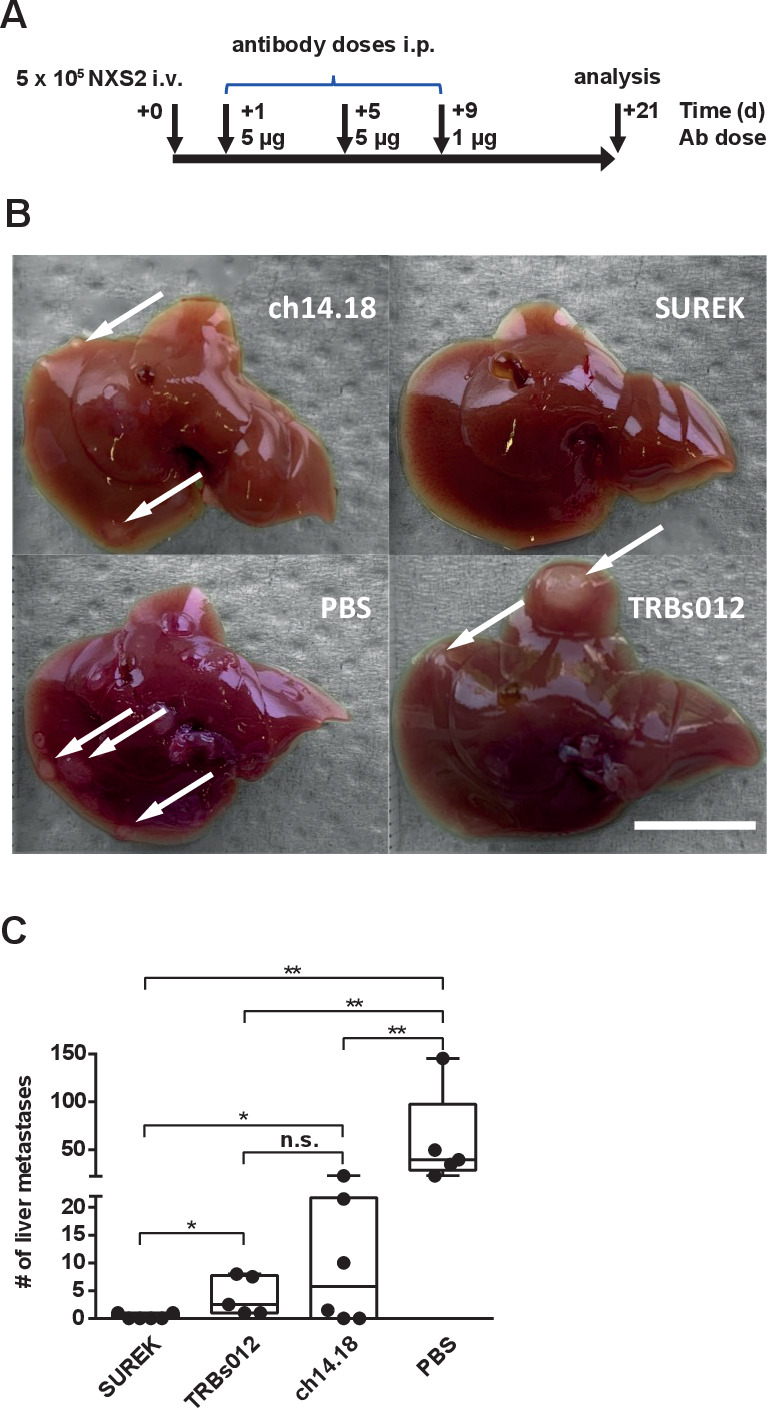
SUREK outperforms ch14.18 in vivo. (A) Treatment and analysis scheme. A/J mice were injected with 5×105 NXS2 cells into a tail vein (intravenous) and received two intraperitoneal (i.p.) injections with 5 µg of SUREK (n=6) or TRBs012 (n=5) or ch14.18 (n=6) on days (d)+1 and +5 and a single injection of 1 µg of the respective antibody on d+9 after tumor cell inoculation. Control mice received three injections with PBS (n=5). (B) Exemplary images of liver specimen in animals treated with SUREK, ch14.18, TRBs012 or PBS. Scale bar equals 10 mm. (C) After 21 d, mice were sacrificed and liver metastases were macroscopically counted. Results are pooled from two independent experiments. Mann-Whitney test; *p<0.05; **p<0.01; n.s., not significant.
Discussion
Although immunotherapy with the chimeric monoclonal ch14.18 antibody targeting the tumor-associated GD2 antigen improved outcomes for patients with high-risk and relapsed neuroblastoma, cure rates for this highly aggressive disease still lag far behind those for other pediatric malignancies.27 28 Since GD2 proved to be a promising target for antibody therapy,6 we set out to investigate whether a GD2-directed bispecific trAb is efficacious against neuroblastoma and how it compares with the current state-of-the-art GD2-directed ch14.18 antibody (dinutuximab beta). In this study, we show that GD2-specific trAb treatment exhibits potent T-cell-mediated killing of neuroblastoma cells independent of target antigen expression level in vitro. Fc fragment-mediated effector mechanisms, such as ADCC and CDC, are significantly lower after trAb treatment compared with ch14.18, potentially preventing the patient from developing severe side effects, such as neuropathic pain. SUREK treatment leads to intratumor activation of TILs, and demonstrates superior direct antitumor efficacy in a murine neuroblastoma minimal residual disease model. Our data suggest that improving conventional antibody therapy by redirecting T cells and AICs toward tumor cells might lead to improved survival in children suffering from relapsed or refractory neuroblastoma.
Recently, bispecific antibodies directed against GD2 on tumor cells and CD3 on T cells have been developed,29–31 and are now being tested in clinical trials.32 33 Cheng and colleagues established a tandem single-chain variable fragment combining GD2 and CD3 specificities and successfully treated subcutaneous and intravenous melanoma and neuroblastoma xenografts.29 31 A tetravalent bispecific antibody, having two binding sites each for GD2 and CD3 in one molecule, also showed promising efficacy in a neuroblastoma xenograft model.30 The main improvement in bispecific trifunctional antibodies over bispecific antibodies is the function directed by the Fc fragment, which activates FcyR-positive immune cells to enhance T-cell activation. This function has the potential to cause cytokine release syndrome. Although bispecific trifunctional antibodies are potent T-cell activators, they were well tolerated by a child in an individual treatment attempt for refractory post-transplant lymphoproliferative disease34 and a cohort of 10 children suffering from recurrent or refractory B‐cell malignancies.35 Adverse events were mainly infusion reactions restricted to fever and chills. Taken together, their administration has so far been well tolerated by children with malignant diseases. Future studies are needed to clarify whether this is also true for heavily pretreated patients with neuroblastoma. The additional activation of FcγR-positive antigen-presenting cells by the trAb Fc fragment forms a tri-cell complex together with T cells and tumor cells and facilitates presentation of tumor antigens. This added functionality creates a polyclonal immune response against tumor antigens not directly targeted by the Fab regions, as demonstrated by tumor rejection and long-lasting immunity through a vaccination effect that was achieved in a melanoma model.3 36 So far, such effects have not been demonstrated for bispecific antibodies. Whether the advantages of the intact Fc fragment in bispecific trifunctional antibodies outweighs risks, such as the potential development of cytokine release syndrome, will need to be clarified in future studies.
Efficacy of a GD2-directed bispecific antibody was shown to correlate with target antigen expression.31 In contrast, we demonstrate potent efficacy of EKTOMUN independently of target antigen expression in vitro and in vivo.37 38 In line with our results, the Bi20 trAb, directed against CD20, and ertumaxomab, a trAb recognizing ERBB2, were shown to be able to kill tumor cells with low target antigen expression. The MYCN-amplified cell lines with high GD2 surface expression seemed to be less susceptible to EKTOMUN-mediated cell death compared with cell lines lacking MYCN amplifications with lower GD2 expression. The cell lines lacking MYCN amplifications proliferate by far more slowly (NB69 doubling time=58 hour, our own data; SK-N-FI doubling time=51 hour, ATCC) than MYCN-amplified cell lines such as IMR5/75 (doubling time=10 hour, our own data) and SK-N-BE(2) (doubling time=30 hour, ATCC). The faster growth of MYCN-amplified cells may have masked the detectable killing effect of effector cells in our experiments because tumor cell growth and death were in balance at a trAb concentration used. However, this could be overcome by increasing either the trAb concentration or the E:T ratio. Brandetti et al showed that MYCN negatively regulates ligands for the NK cell-activing receptors, NKG2D and DNAM-1.39 Whether similar effects negatively influence T-cell activation warrants further studies. Additionally, in silico modeling has predicted that MYCN has an immunosuppressive effect on the tumor microenvironment.40 Furthermore, MYCN amplification is also associated with diminished interferon signaling activity and reduced CXCL10 chemokine expression causing reduced immune cell infiltration in primary metastatic neuroblastomas.41 Our data suggest that the immunosuppressive effects combined with the increased malignancy rather than surface antigen expression are responsible for the lower susceptibility of MYCN-amplified cells to EKTOMUN treatment. Whether this effect is also present in vivo remains a question to be addressed in future studies.
CDC is thought to be the main mechanism causing neuropathic pain in patients undergoing GD2-directed antibody therapy.10 This regularly requires intravenous morphine treatment and prolongs hospitalization. A point mutation (K322A) in the Fc region has been introduced in a new humanized version of ch14.1842 to reduce complement fixation and decrease antibody-induced allodynia. Although mouse IgG2a and rat IgG2b are able to activate human complement fixation as monomers,43 44 their ability to mediate CDC as a heterodimer is unclear. The fact that EKTOMUN induced only limited CDC compared with ch14.18 in our in vitro experiments might be translatable to reduction of this severe side effect and the pain it causes, although this needs to be proven in vivo.
The capability of trAb to induce ADCC has never been compared with monoclonal antibodies. Superior ADCC induction by ch14.18 demonstrated in this study could be explained by the lower target-binding avidity in the bispecific trAb, which has only one target antigen binding site compared with the two Fab regions in monoclonal antibodies. The species origins of the Fc regions in SUREK or EKTOMUN also reduce ADCC induction. Although both mouse IgG2a and rat IgG2b can mediate ADCC in human cells as homodimers,45 46 the constant Fc regions of both mouse and rat isotypes, IgG2a and IgG2b, are only 75% homologous in their amino acid sequences.47 This suggests that antibodies wielding the human IgG1 Fc regions, such as ch14.18, have a species-specific advantage in binding to human FcγR IIIa compared with the heterodimeric mouse IgG2a and rat IgG2b molecules. In line with this, only weak binding of a bispecific trAb to NK cells was reported by Zeidler et al.2 We conclude that bispecific trifunctional antibodies are, in general, less effective at inducing ADCC compared with a humanized monoclonal IgG1 antibody, but that the Fc region in trAbs still mediates the much more important AICs activation via binding to FcγR I, IIa and III.1 2
An advantage of this study is its use of an immunocompetent mouse model mimicking neuroblastoma minimal residual disease. Immunocompetent syngeneic mouse models, as used in this study, provide the best possible crosstalk between tumor, stroma and immune cells to assess immunotherapies. In previous preclinical studies, this model received up to 300 µg ch14.18 per day for up to 5 consecutive days to treat liver metastases after tail vein injection of NXS2 cells.24 In contrast, only 5 µg of SUREK in total was effective in our study, demonstrating its superior efficacy compared with ch14.18. The shift in the Cd4/Cd8 ratio toward cytotoxic Cd8+ T cells as well as the activation of tumor infiltrating T cells on SUREK treatment further supports its strong direct antitumor efficacy. However, whether the shift in the Cd4/Cd8 ratio is due to decreased Cd4+ infiltration, increased Cd8+ infiltration or enhanced Cd8+ proliferation cannot be concluded from our experiments. Since NXS2 cells originate from the GD2-negative C1300 murine neuroblastoma cell line (A/J genetic background) hybridized with dorsal root ganglia cells (C57BL/6J genetic background),19 they are not fully autologous. This cell line is also known to harbor a relatively high mutational burden,48 which is uncommon in neuroblastoma cells,49 a limitation of our study, since it might overestimate the antibody-independent bystander killing by T cells that recognize immunogenic epitopes expressed by the tumor. This effect might even be enhanced by the control trAb TRBs012, since it is able to activate T cells via its CD3 binding arm and its Fc fragment. Similar effects have already been observed by others, although using a different antibody format.50 Eventually, it is not yet entirely understood how bispecific antibodies activate T cells and whether this activation is only due to antibody cross-linking of T-cell and FcγR+ cells (Reviewed in ref.51). A transgenic GD2-expressing neuroblastoma mouse model would provide an alternative model. Voeller et al recently established a neuroblastoma cell line from TH-MYCN transgenic mice (9464D-GD2),48 lentivirally transduced with both, GD2-synthase and GD3-synthase, to express GD2. Future studies will show, whether SUREK is equally efficacious in this less immunogenic tumor model and whether the T unspecific effects produced by TRBs012 are diminished.
Conclusion
This study provides first evidence that bispecific trifunctional antibodies redirecting T cells and AICs against GD2+ neuroblastoma might be a treatment option for children suffering from this appalling disease.
jitc-2021-002923supp001.pdf (931.4KB, pdf)
Acknowledgments
The authors would like to thank Nicole Hübener for critical discussions and Aleixandria McGearey for proofreading the manuscript.
Footnotes
Contributors: FZ conceived the study, designed and performed experiments, interpreted data and wrote the manuscript; SMI, LG, LA, AKl, SS, AKü and KAn designed and performed experiments, HL provided NXS2 cells, PR and HL provided bispecific trifunctional antibodies and interpreted data, JHS and AE coconceived the study and interpreted data, PH and AKü coconceived the study, designed experiments, interpreted data and revised the manuscript. KAs interpreted data and revised the manuscript. All authors revised and approved the last version of the manuscript.
Funding: FZ and AKü are participants in the BIH-Charité Clinician Scientist Program cofunded by the Charité – Universitätsmedizin Berlin and the Berlin Institute of Health. SMI is a recipient of a BIH-MD Scholarship. LG is participant in the “Berlin School of Integrative Oncology”. KAn is funded by the Deutsches Konsortium für Translationale Krebsforschung (DKTK). This work has been funded by the Stiftung Charité.
Competing interests: HL is patent holder for EKTOMUN and HL is the CEO of Trion Research and the inventor or co-inventor of several trifunctional antibody patents. PR is an employee of Trion Research. No other authors disclose potential conflicts of interest.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
Primary patient tissue samples (peripheral blood mononuclear cells) were obtained from healthy donors at Charité Universitätsmedizin Berlin with written informed consent for use in research, as approved by the local ethics committee (Charité’s Ethics Committee; approval number EA2/262/20). All animal experiments were performed in accordance with FELASA guidelines under the supervision and approval of the ethics committee for animal experimentation (Landesamt für Gesundheit und Soziales Berlin; approval number: G0386/17).
References
- 1.Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol 1999;163:1246–52. [PubMed] [Google Scholar]
- 2.Zeidler R, Mysliwietz J, Csánady M, et al. The Fc-region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer 2000;83:261–6. 10.1054/bjoc.2000.1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood 2001;98:2526–34. 10.1182/blood.V98.8.2526 [DOI] [PubMed] [Google Scholar]
- 4.Schulz G, Cheresh DA, Varki NM, et al. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res 1984;44:5914–20. [PubMed] [Google Scholar]
- 5.Lammie G, Cheung N, Gerald W, et al. Ganglioside gd(2) expression in the human nervous-system and in neuroblastomas - an immunohistochemical study. Int J Oncol 1993;3:909–15. 10.3892/ijo.3.5.909 [DOI] [PubMed] [Google Scholar]
- 6.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-Gd2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 2010;363:1324–34. 10.1056/NEJMoa0911123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.AL Y, Gilman AL, Ozkaynak MF. Long-Term follow-up of a phase III study of ch14.18 (Dinutuximab) + cytokine immunotherapy in children with high-risk neuroblastoma: cog study ANBL0032. Clin Cancer Res 2021;27. 10.1158/1078-0432.CCR-20-3909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladenstein R, Pötschger U, Valteau-Couanet D. Investigation of the role of Dinutuximab Beta-Based immunotherapy in the SIOPEN high-risk neuroblastoma 1 trial (HR-NBL1). Cancers 2020;12:309. 10.3390/cancers12020309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ladenstein R, Pötschger U, Valteau-Couanet D, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19:1617–29. 10.1016/S1470-2045(18)30578-3 [DOI] [PubMed] [Google Scholar]
- 10.Yuki N, Yamada M, Tagawa Y, et al. Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J Neurol Sci 1997;149:127–30. 10.1016/S0022-510X(97)05390-2 [DOI] [PubMed] [Google Scholar]
- 11.Mueller BM, Romerdahl CA, Gillies SD, et al. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J Immunol 1990;144:1382–6. [PubMed] [Google Scholar]
- 12.Mujoo K, Kipps TJ, Yang HM, et al. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res 1989;49:2857–61. [PubMed] [Google Scholar]
- 13.Simon T, Hero B, Schulte JH, et al. 2017 GPOH guidelines for diagnosis and treatment of patients with neuroblastic tumors. Klin Padiatr 2017;229:147–67. 10.1055/s-0043-103086 [DOI] [PubMed] [Google Scholar]
- 14.Smith V, Foster J. High-Risk neuroblastoma treatment review. Children 2018;5. 10.3390/children5090114. [Epub ahead of print: 28 08 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kushner BH, Kramer K, Modak S, et al. Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. J Clin Oncol 2006;24:5271–6. 10.1200/JCO.2006.06.7272 [DOI] [PubMed] [Google Scholar]
- 16.Jones DTW, Banito A, Grünewald TGP, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer 2019;19:420–38. 10.1038/s41568-019-0169-x [DOI] [PubMed] [Google Scholar]
- 17.Lang P, Illhardt T, Ebinger M, et al. Haploidentical stem cell transplantation and subsequent immunotherapy with antiGD2 antibody for patients with relapsed metastatic neuroblastoma. JCO 2015;33:10056–56. 10.1200/jco.2015.33.15_suppl.10056 [DOI] [Google Scholar]
- 18.Mody R, Naranjo A, Yu AL, et al. Phase II trial of irinotecan/temozolomide/dinutuximab/granulocyte macrophage colony stimulating factor (I/T/DIN/GMCSF) in children with relapsed/refractory neuroblastoma (NBL): A report from the Children’s Oncology Group (COG). JCO 2018;36:10508–08. 10.1200/JCO.2018.36.15_suppl.10508 [DOI] [Google Scholar]
- 19.Greene LA, Shain W, Chalazonitis A, et al. Neuronal properties of hybrid neuroblastoma X sympathetic ganglion cells. Proc Natl Acad Sci U S A 1975;72:4923–7. 10.1073/pnas.72.12.4923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruf P, Jäger M, Ellwart J, et al. Two new trifunctional antibodies for the therapy of human malignant melanoma. Int J Cancer 2004;108:725–32. 10.1002/ijc.11630 [DOI] [PubMed] [Google Scholar]
- 21.Toews K, Grunewald L, Schwiebert S, et al. Central memory phenotype drives success of checkpoint inhibition in combination with CAR T cells. Mol Carcinog 2020;59:724–35. 10.1002/mc.23202 [DOI] [PubMed] [Google Scholar]
- 22.Ali S, Toews K, Schwiebert S, et al. Tumor-Derived Extracellular Vesicles Impair CD171-Specific CD4+ CAR T Cell Efficacy. Front Immunol 2020;11:531. 10.3389/fimmu.2020.00531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marx S, Wilken F, Wagner I, et al. Gd2 targeting by dinutuximab beta is a promising immunotherapeutic approach against malignant glioma. J Neurooncol 2020;147:577–85. 10.1007/s11060-020-03470-3 [DOI] [PubMed] [Google Scholar]
- 24.Zeng Y, Fest S, Kunert R, et al. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol Immunol 2005;42:1311–9. 10.1016/j.molimm.2004.12.018 [DOI] [PubMed] [Google Scholar]
- 25.Dekkers G, Bentlage AEH, Stegmann TC, et al. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 2017;9:767–73. 10.1080/19420862.2017.1323159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mina M, Boldrini R, Citti A, et al. Tumor-Infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology 2015;4:e1019981. 10.1080/2162402X.2015.1019981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am 2010;24:65–86. 10.1016/j.hoc.2009.11.011 [DOI] [PubMed] [Google Scholar]
- 28.Simon T, Berthold F, Borkhardt A, et al. Treatment and outcomes of patients with relapsed, high-risk neuroblastoma: results of German trials. Pediatr Blood Cancer 2011;56:578–83. 10.1002/pbc.22693 [DOI] [PubMed] [Google Scholar]
- 29.Cheng M, Ahmed M, Xu H, et al. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer 2015;136:476–86. 10.1002/ijc.29007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, Cheng M, Guo H, et al. Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer Immunol Res 2015;3:266–77. 10.1158/2326-6066.CIR-14-0230-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng M, Santich BH, Xu H, et al. Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors. Oncoimmunology 2016;5:e1168557. 10.1080/2162402X.2016.1168557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Modak S. Study of the safety and efficacy of humanized 3F8 bispecific antibody (Hu3F8-BsAb) in patients with relapsed/refractory neuroblastoma, osteosarcoma and other solid tumor cancers, 2019. Available: https://clinicaltrials.gov/ct2/show/NCT03860207 [Accessed Apr 2021].
- 33.Yankelevich M. Activated T cells armed with GD2 bispecific antibody in children and young adults with neuroblastoma and osteosarcoma, 2014. Available: https://clinicaltrials.gov/ct2/show/NCT02173093?term=NCT02173093&draw=2&rank=1 [Accessed Apr 2021].
- 34.Kieslich A, Ruf P, Lindhofer H, et al. Immunotherapy with the trifunctional anti-CD20 × anti-CD3 antibody FBTA05 in a patient with relapsed t(8;14)-positive post-transplant lymphoproliferative disease. Leuk Lymphoma 2017;58:1989–92. 10.1080/10428194.2016.1272687 [DOI] [PubMed] [Google Scholar]
- 35.Schuster FR, Stanglmaier M, Woessmann W, et al. Immunotherapy with the trifunctional anti-CD20 X anti-CD3 antibody FBTA05 (Lymphomun) in paediatric high-risk patients with recurrent CD20-positive B cell malignancies. Br J Haematol 2015;169:90–102. 10.1111/bjh.13242 [DOI] [PubMed] [Google Scholar]
- 36.Eissler N, Ruf P, Mysliwietz J, et al. Trifunctional bispecific antibodies induce tumor-specific T cells and elicit a vaccination effect. Cancer Res 2012;72:3958–66. 10.1158/0008-5472.CAN-12-0146 [DOI] [PubMed] [Google Scholar]
- 37.Stanglmaier M, Faltin M, Ruf P, et al. Bi20 (fBTA05), a novel trifunctional bispecific antibody (anti-CD20 X anti-CD3), mediates efficient killing of B-cell lymphoma cells even with very low CD20 expression levels. Int J Cancer 2008;123:1181–9. 10.1002/ijc.23626 [DOI] [PubMed] [Google Scholar]
- 38.Jäger M, Schoberth A, Ruf P, et al. The trifunctional antibody ertumaxomab destroys tumor cells that express low levels of human epidermal growth factor receptor 2. Cancer Res 2009;69:4270–6. 10.1158/0008-5472.CAN-08-2861 [DOI] [PubMed] [Google Scholar]
- 39.Brandetti E, Veneziani I, Melaiu O, et al. Mycn is an immunosuppressive oncogene dampening the expression of ligands for NK-cell-activating receptors in human high-risk neuroblastoma. Oncoimmunology 2017;6:e1316439. 10.1080/2162402X.2017.1316439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang P, Wu X, Basu M, et al. MYCN Amplification Is Associated with Repressed Cellular Immunity in Neuroblastoma: An In Silico Immunological Analysis of TARGET Database. Front Immunol 2017;8:1473–73. 10.3389/fimmu.2017.01473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Layer JP, Kronmüller MT, Quast T, et al. Amplification of N-myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 2017;6:e1320626. 10.1080/2162402X.2017.1320626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sorkin LS, Otto M, Baldwin WM, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain 2010;149:135–42. 10.1016/j.pain.2010.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Füst G, Medgyesi GA, Bazin H, et al. Differences in the ability of rat IgG subclasses to consume complement in homologous and heterologous serum. Immunol Lett 1980;1:249–53. 10.1016/0165-2478(80)90002-4 [DOI] [Google Scholar]
- 44.Seino J, Eveleigh P, Warnaar S, et al. Activation of human complement by mouse and mouse/human chimeric monoclonal antibodies. Clin Exp Immunol 1993;94:291–6. 10.1111/j.1365-2249.1993.tb03446.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chassoux DM, Linares-Cruz LG, Bazin H, et al. K-cell-mediated cytotoxicity induced with rat monoclonal antibodies. I. antibodies of various isotypes differ in their ability to induce cytotoxicity mediated by rat and human effectors. Immunology 1988;65:623–8. [PMC free article] [PubMed] [Google Scholar]
- 46.Steplewski Z, Sun LK, Shearman CW, et al. Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proc Natl Acad Sci U S A 1988;85:4852–6. 10.1073/pnas.85.13.4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindhofer H, Hess J, Ruf P. Trifunctional Triomab® Antibodies for Cancer Therapy. In: Kontermann R, ed. Bispecific antibodies: Springer. Berlin, Heidelberg, 2011: 289–312. [Google Scholar]
- 48.Voeller J, Erbe AK, Slowinski J, et al. Combined innate and adaptive immunotherapy overcomes resistance of immunologically cold syngeneic murine neuroblastoma to checkpoint inhibition. J Immunother Cancer 2019;7:344. 10.1186/s40425-019-0823-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. 10.1038/nature25480 [DOI] [PubMed] [Google Scholar]
- 50.Belani R, Weiner GJ. T cell activation and cytokine production in anti-CD3 bispecific antibody therapy. J Hematother 1995;4:395–402. 10.1089/scd.1.1995.4.395 [DOI] [PubMed] [Google Scholar]
- 51.Zhukovsky EA, Morse RJ, Maus MV. Bispecific antibodies and cars: generalized immunotherapeutics harnessing T cell redirection. Curr Opin Immunol 2016;40:24–35. 10.1016/j.coi.2016.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-002923supp002.pdf (1.7MB, pdf)
jitc-2021-002923supp001.pdf (931.4KB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information.