

Abstract
Chromosomal translocation results in development of an Ewing sarcoma breakpoint region 1‐Friend leukemia integration 1 (EWS–FLI1) fusion oncogene in the majority of Ewing sarcoma. The persistent dependence of the tumor for this oncoprotein points to EWS–FLI1 as an ideal drug target. Although EWS–FLI1 transcriptional targets and binding partners are evaluated, the mechanisms regulating EWS–FLI1 protein stability remain elusive. Speckle‐type POZ protein (SPOP) and OTU domain‐containing protein 7A (OTUD7A) are identified as the bona fide E3 ligase and deubiquitinase, respectively, that control EWS–FLI1 protein turnover in Ewing sarcoma. Casein kinase 1‐mediated phosphorylation of the VTSSS degron in the FLI1 domain enhances SPOP activity to degrade EWS–FLI1. Opposing this process, OTUD7A deubiquitinates and stabilizes EWS–FLI1. Depletion of OTUD7A in Ewing sarcoma cell lines reduces EWS–FLI1 protein abundance and impedes Ewing sarcoma growth in vitro and in mice. Performing an artificial‐intelligence‐based virtual drug screen of a 4‐million small molecule library, 7Ai is identified as a potential OTUD7A catalytic inhibitor. 7Ai reduces EWS–FLI1 protein levels and decreases Ewing sarcoma growth in vitro and in a xenograft mouse model. This study supports the therapeutic targeting of OTUD7A as a novel strategy for Ewing sarcoma bearing EWS–FLI1 and related fusions, and may also be applicable to other cancers dependent on aberrant FLI1 expression.
Keywords: 7Ai, CK1, Ewing sarcoma, EWS–FLI1, OTUD7A, SPOP
The fusion oncogene Ewing sarcoma breakpoint region 1‐Friend leukemia integration 1 (EWS–FLI1) drives Ewing sarcoma tumorigenesis. This work identifies Speckle‐type POZ protein (SPOP) and OTU domain‐containing protein 7A (OTUD7A) as the E3 ligase and deubiquitinase, respectively, that regulate EWS–FLI1 protein homeostasis. 7Ai, a putative OTUD7A catalytic inhibitor identified through virtual compound screening, reduces Ewing sarcoma cell growth and migration.
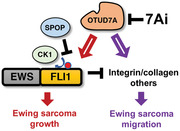
1. Introduction
Ewing sarcoma is an aggressive malignancy that develops in bones or soft tissues of children and young adults. A recurrent chromosomal translocation found in the majority of Ewing sarcoma fuses the Ewing sarcoma breakpoint region 1 or RNA‐binding protein EWS (EWSR1) and Friend leukemia integration 1 transcription factor (FLI1) genes generating an EWS–FLI1 fusion protein. EWS–FLI1 is the critical driver of Ewing sarcoma.[ 1 ] Mechanistically, EWS–FLI1 binds specific GGAA‐containing microsatellite regions to maintain nucleosome depletion.[ 2 , 3 , 4 ] EWS–FLI1 recruits a set of chromatin and transcriptional regulators, including BRG1,[ 5 ] RNA polymerase II,[ 6 ] CREB‐binding protein (CBP)/p300,[ 7 ] RNA helicase A,[ 8 ] and others, to modulate transcription of target genes, including NR0B1, GLI1, FOXOs, LOX, IGF1, and others that maintain properties of malignant transformation.[ 9 ] However, recent studies indicate that EWS–FLI1 does not act in a binary fashion; rather EWS–FLI1 expression levels influence cellular states. High levels of EWS–FLI1 are associated with an immature, proliferative phenotype, whereas reduced levels correlate with decreased proliferation and a more motile cellular phenotype.[ 10 , 11 ]
As the EWS–FLI1 fusion occurs exclusively in the tumor cells, it is considered as an ideal target to treat Ewing sarcoma. Prior efforts to identify and target major EWS–FLI1 downstream genes have not been effective.[ 12 ] Further, direct targeting EWS–FLI1 has been hampered by the lack of enzymatic activity and suitable small molecule interaction domains. Notably, a small molecule enantiomer‐specific EWS–FLI1 inhibitor TK‐216 was identified and being tested in early clinical development.[ 13 ] Recent efforts aim to block EWS–FLI1 interaction with DNA[ 14 ] or modulate its ability to affect chromatin states.[ 15 ] Targeting EWS–FLI1 protein stability constitutes a potential therapeutic strategy. Although proteasome‐mediated[ 16 ] and lysosome‐controlled[ 17 ] EWS–FLI1 degradation have been reported, the identities of E3 ligase(s) and deubiquitinase(s) responsible for EWS–FLI1 protein stability control remain elusive. Ubiquitin carboxyl‐terminal hydrolase 7 (USP7) was identified from a CRISPR screen as a dependency for p53‐wild‐type (WT) Ewing sarcoma[ 18 ] and the deubiquitinase Ubiquitin carboxyl‐terminal hydrolase 19 (USP19) was found to stabilize both EWS–FLI1 and EWSR1 proteins.[ 19 ] However, the multiple roles of USP7 on targeting both tumor suppressors and oncogenes,[ 20 ] as well as the pleiotropy of USP19 [ 21 , 22 , 23 , 24 ] complicate their applications to treat Ewing sarcoma. Inhibitors of USP7 [ 25 , 26 ] and USP19 [ 27 ] have been developed, and their effects on Ewing sarcoma remain to be determined.
2. Results
2.1. The E3 Ligase SPOP Targets EWS–FLI1 for Ubiquitination and Degradation
We found that blocking the 26S proteasome by MG132 significantly increased the protein abundance of EWS–FLI1, but not the wild‐type EWSR1, in two Ewing sarcoma cell lines (A673 and SK‐N‐MC) (Figure 1A,B and Figure S1A,B (Supporting Information)), demonstrating that EWS–FLI1 levels are regulated through protein stability. Inhibition of cullin (CUL) neddylation by MLN4924 also largely stabilized EWS–FLI1 but not EWSR1 in A673 cells (Figure 1C and Figure S1C (Supporting Information)). Antibodies used to detect endogenous EWSR1, FLI1, and EWS–FLI1 fusion in Ewing sarcoma cells were validated by a short hairpin RNA (shRNA) against FLI1‐C‐terminus in A673 cells (Figure S1D, Supporting Information). These data suggest that in Ewing sarcoma EWS–FLI1 protein stability is governed by CUL‐Ring E3 ligases and that the major degron resides in the FLI1‐domain retained in the fusion. By examining EWS–FLI1 binding to a family of CULs, we found EWS–FLI1 associated with CUL3, CUL4A, and CUL5 (Figure S1E, Supporting Information). Examining the sequence of the retained FLI1 segment, we identified a putative degron sequence (VTSSS) for SPOP, a CUL3 family of E3 ligase. The sequence was located between the E26 transformation‐specific, E‐twenty‐six or Erythroblast transformation specific (ETS) DNA‐binding domain and the carboxyl terminus (Figure 1D). Ectopic expression of SPOP promoted EWS–FLI1 protein degradation in cells in a SPOP‐dose‐dependent manner (Figure 1E and Figure S1F (Supporting Information)). Consistent with the presence of the SPOP degron in FLI1, SPOP also destabilized wild‐type FLI1 but not wild‐type EWSR1 (Figure 1F and Figure S1G (Supporting Information)). In support, SPOP bound the fusion protein but not EWSR1 (Figure S1H, Supporting Information). Decreasing the possibility that indirect transcriptional control mediated differences in EWS–FLI1 levels, ectopically expressed EWS–FLI1 was also decreased by SPOP, and this effect was blocked by either MG132 or MLN4924 (Figure 1G).
Figure 1.
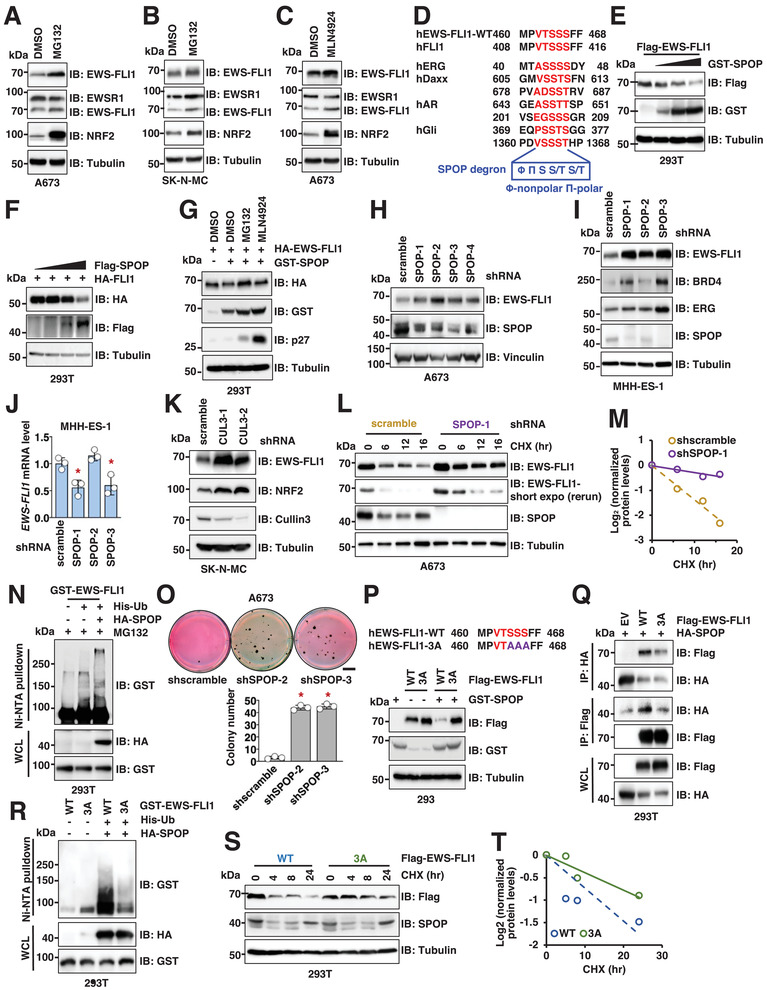
SPOP targets EWS–FLI1 for ubiquitination and degradation depending on a “VTSSS” degron in EWS–FLI1. A,B) Immunoblot (IB) analysis of whole cell lysates (WCL) derived from A673 (A) or SK‐N‐MC (B) cells treated with 10 × 10−6 m MG132 for 4 h. Cells were lysed in EBC buffer unless specifically noted. Notably, the EWS–FLI1 signal was detected by either an EWSR1‐N antibody (A300‐417) that can detect both EWSR1 and EWS–FLI1, or a FLI1‐C antibody (ab180902) that can detect both FLI1 and EWS–FLI1. C) IB analysis of WCL derived from A673 cells treated with 1 × 10−6 m MLN4924 overnight. D) Sequence alignment of indicated EWS–FLI1 species with canonical SPOP substrates. E,F) IB analysis of WCL derived from HEK293T cells transfected with indicated DNA constructs. 100 ng Flag‐EWS–FLI1 or Flag‐FLI1 construct, together with increasing amounts of GST‐SPOP (0, 0.5, 1, 2 µg) or Flag‐SPOP (0, 1, 2, 4 µg) were transfected into cells. Notably, same amounts of DNA were transfected in each reaction and the differences in DNA amounts were supplemented with pCDNA3.0. Cells were collected 48 h post‐transfection unless specified. G) IB analysis of WCL derived from HEK293T cells transfected with indicated DNA constructs. Where indicated, cells were treated with 10 × 10−6 m MG132 or 1 × 10−6 m MLN4924 overnight before cell collection. H,I) IB analysis of WCL derived from A673 (H) or MHH‐ES‐1 (I) cells depleted of endogenous SPOP. Cells were infected with lentiviruses targeting indicated targets and selected with 1 µg mL−1 puromycin for 3 days to eliminate noninfected cells. J) RT‐PCR analyses of EWS–FLI1 mRNA levels in indicated MHH‐ES‐1 cells. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). K) IB analysis of WCL derived from SK‐N‐MC cells depleted of endogenous CUL3. Cells were infected with lentiviruses targeting cullin 3 and selected with 1 µg mL−1 puromycin for 3 days to eliminate non‐infected cells. L,M) IB analysis of WCL derived from control or SPOP‐depleted MHH‐ES‐1 cells. Where indicated, 200 µg mL−1 cycloheximide (CHX) was added to cell culture and cells were harvested at indicated time periods post CHX addition. (M) is a quantification of (L). N) IB analysis of nickel‐nitrilotriacetic acid (Ni–NTA) pulldowns and WCL derived from HEK293T cells transfected with indicated DNA constructs. Cells were treated with 10 × 10−6 m MG132 overnight before cell collection. O) Representative images and quantifications for 3D soft agar assays using indicated cells. Colonies were stained 40 days postinoculation. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). The scale bar represents 5 mm. P) IB analysis of WCL derived from HEK293 cells transfected with 100 ng Flag‐EWS–FLI1‐WT or ‐3A together with 2 µg GST‐SPOP constructs. Q) IB analysis of HA or Flag‐IPs and WCL derived from HEK293T cells transfected with indicated DNA constructs. R) IB analysis of Ni–NTA pulldowns and WCL derived from HEK293T cells transfected with indicated DNA constructs. Cells were treated with 10 × 10−6 m MG132 overnight before cell collection. S,T) IB analysis of WCL derived from HEK293T cells transfected with indicated Flag‐EWS–FLI1 constructs. Where indicated, 200 µg mL−1 CHX was added to cell culture and cells were harvested at indicated time periods post CHX addition. (T) is a quantification of (S).
To further confirm SPOP as a physiological E3 ligase for EWS–FLI1, we depleted endogenous SPOP in 4 Ewing sarcoma cell lines (A673: Figure 1H and Figure S1I (Supporting Information); MHH‐ES‐1: Figure 1I; SK‐N‐MC: Figure S1J (Supporting Information); and EWS894: Figure S1K (Supporting Information)). In each, we observed that SPOP depletion led to increased EWS–FLI1 protein abundance. Notably, depletion of SPOP did not increase EWS–FLI1 messenger RNA (mRNA) levels in MHH‐ES‐1 (Figure 1J) nor A673 cells (Figure S1L, Supporting Information), supporting that SPOP regulates EWS–FLI1 largely through a post‐translational mechanism. Depletion of CUL3, the cullin partner of SPOP, also increased EWS–FLI1 protein levels in SK‐N‐MC cells (Figure 1K). In further support of SPOPCUL3 as a physiological E3 ligase for EWS–FLI1, we observed that SPOP depletion extended the half‐life of EWS–FLI1 proteins (Figure 1L,M), and SPOP expression enhanced EWS–FLI1 ubiquitination in cells (Figure 1N and Figure S1M (Supporting Information)). Demonstrating a functional effect in Ewing cells, we observed that, consistent with previous reports,[ 28 ] depletion of endogenous EWS–FLI1 retarded A673 cell growth in vitro (Figure S2A–E, Supporting Information), while SPOP depletion enhanced clonal proliferation of A673 cells in soft agar, an effect possibly related to increased EWS–FLI1 expression (Figure 1O).
To test whether the VTSSS sequence in the FLI1 segment could function as a degron, we mutated each serine to alanine (S464A/S465A/S466A, 3A‐EWS–FLI1). Compared with WT‐EWS–FLI1, the 3A mutant was resistant to SPOP‐mediated degradation (Figure 1P), largely due to deficiency of 3A‐EWS–FLI1 binding to both exogenous (Figure 1Q) and endogenous SPOP (Figure S3A, Supporting Information). 3A‐EWS–FLI1 also displayed a resistance to MG132 treatment (Figure S3B, Supporting Information), reduced ubiquitination levels (Figure 1R), and a longer protein half‐life (Figure 1S,T).
As SPOP has been characterized as a bona fide E3 ligase that governs BRD4 protein stability in prostate cancer,[ 29 , 30 , 31 ] and BRD4 cooperates with EWS–FLI1 to regulate the EWS–FLI1‐mediated transcriptional programs in Ewing sarcoma,[ 32 ] we examined if BRD4 is involved in SPOP‐depletion‐induced Ewing cell growth control. To this end, we found that SPOP depletion only moderately increased BRD4 proteins in MHH‐ES‐1 cells (Figure 1I) but not in other Ewing sarcoma cells (A673: Figure S1I (Supporting Information) and SN‐N‐MC: Figure S1J (Supporting Information)). Moreover, given that SPOP depletion did not affect EWS–FLI1 mRNA abundance (Figure S1L, Supporting Information), and treatment of MHH‐ES‐1 (Figure S3C, Supporting Information) or A673 (Figure S3D, Supporting Information) cells by a BRD4 inhibitor JQ1 did not affect EWS–FLI1 protein levels, it seems that the SPOP/BRD4 signaling axis identified in prostate cancer may not regulate EWS–FLI1 protein stability in Ewing sarcoma.
2.2. Casein Kinase 1 (CK1) Phosphorylates and Primes EWS–FLI1 for SPOP Recognition and Degradation
Since multiple serines in the “VTSSS” degron could be phosphorylated, and phosphorylation of SPOP degrons can enhance SPOP–substrate binding,[ 29 , 33 ] we next examined whether phosphorylation of the EWS–FLI1‐“VTSSS” degron primes EWS–FLI1 for SPOP recognition and degradation. Pursuing the prediction (by GPS3.0) that the serine residues could be phosphorylated by CKs, we expressed several distinct CK1 isoforms and CK2 kinases and found that most CK1 isoforms, but not CK2, promoted EWS–FLI1 degradation (Figure 2A). In addition, CK1 kinase inhibition with D4476 resulted in the accumulation of EWS–FLI1 in multiple Ewing sarcoma cells (Figure 2B–D and Figure S4A,B (Supporting Information)) without significantly affecting EWS–FLI1 mRNA levels (Figure S4C–E, Supporting Information). Similar to D4476, genetic depletion of CK1α by shRNAs also led to accumulation of endogenous EWS–FLI1 (Figure 2E), as well as extended EWS–FLI1 half‐life (Figure 2F,G). Notably, SPOP levels were unaffected by D4476, and the effect of D4476 on EWS–FLI1 levels was attenuated in the context of SPOP depletion (Figure 2H). This attenuation was explained by reduced EWS–FLI1 binding to SPOP following D4476 treatment (Figure S4F, Supporting Information). In addition, lenalidomide treatment, a CK1α PROTAC that induces CK1 degradation,[ 34 ] increased EWS–FLI1 protein abundance without affecting EWS–FLI1 mRNA levels (Figure 2I and Figure S4G,H (Supporting Information)). We also found that the 3A mutant was resistant to CK1‐mediated degradation (Figure 2J), supporting Ser464/Ser465/Ser466 as functional CK1 phosphorylation sites. Cumulatively, these data suggest that CK1 promotes SPOP‐mediated EWS–FLI1 degradation in a kinase‐activity‐dependent manner (Figure 2K).
Figure 2.
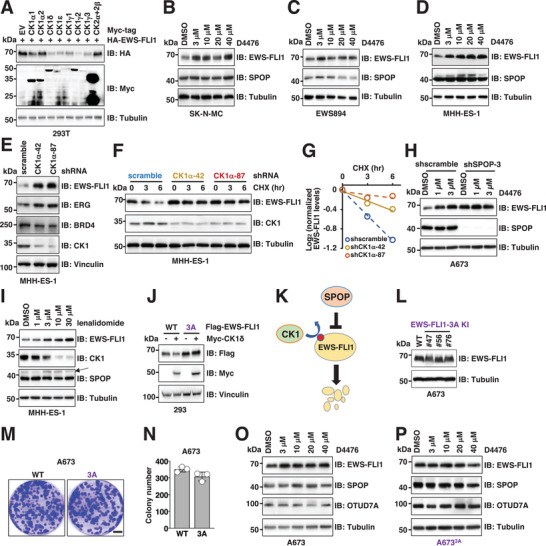
CK1 phosphorylates and primes EWS–FLI1 for SPOP‐mediated degradation. A) IB analysis of WCL derived from HEK293T cells transfected with indicated DNA constructs. Cells were collected 48 h post‐transfection. B–D) IB analysis of WCL derived from SK‐N‐MC (B), EWS894 (C), and MHH‐ES‐1 (D) cells treated with indicated concentrations of CK1 inhibitor D4476 for 16 h. E) IB analysis of WCL derived from MHH‐ES‐1 cells depleted of endogenous CK1α. Cells were infected with lentiviruses targeting CK1α and selected with 1 µg mL−1 puromycin for 3 days to eliminate non‐infected cells. F,G) IB analysis of WCL derived from indicated MHH‐ES‐1 cells treated with 200 µg mL−1 CHX and harvested at indicated time periods. (G) is a quantification of (F). H) IB analysis of WCL derived from control or endogenous SPOP‐depleted A673 cells treated with indicated concentrations of CK1 inhibitor D4476 for 16 h. I) IB analysis of WCL derived from MHH‐ES‐1 cells treated with indicated concentrations of lenalidomide for 16 h. J) IB analysis of WCL derived from HEK293 cells transfected with 100 ng Flag‐EWS–FLI1‐WT or ‐3A together with 2 µg Myc–CK1δ constructs. K) A cartoon illustration of the proposed model: CK1‐mediated EWS–FLI1 phosphorylation primes EWS–FLI1 for SPOP recognition and degradation. L) IB analysis of WCL derived from parental and three isogenic EWS–FLI1‐S464A/S465A/S466A knock‐in A673 cells. M,N) Representative images for 2D colony formation using cells from (L, #76) and quantified in (N). Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). Scale bar represents 10 mm. O,P) IB analysis of WCL derived from parental or an isogenic EWS–FLI1‐S464A/S465A/S466A knock‐in A673 (L, #76) cells treated with indicated concentrations of CK1 inhibitor D4476 for 16 h.
Because 3A‐EWS–FLI1 was resistant to both SPOP (Figure 1P)‐ and CK1 (Figure 2J)‐mediated degradation, we replaced the degron with the 3A mutant by CRISPR mediated knock‐in (KI) in A673 cells (A673 3A ) (Figure S5A–F, Supporting Information). A673 3A cells expressed comparable levels of EWS–FLI1 to the parental cells (Figure 2L) and displayed a similar growth in vitro (Figure 2M,N). Increased EWS–FLI1 protein levels observed in A673 parental cells upon D4476 treatment (Figure 2O) was not observed in the A673 3A KI cells (Figure 2P). In contrast to A673 cells, we observed that neither D4476 treatment (Figure S6A,B, Supporting Information), nor SPOP depletion (Figure S6C, Supporting Information) increased EWS–FLI1 protein abundance in EWS502 cells. Exploring genetic alterations in Ewing sarcoma cells (DEPMAP portal), we noted a point mutation in CUL3 (E358Q) that was present only in EWS502 cells (Figure S6D, Supporting Information). We hypothesized that E358Q might result in a loss‐of‐function mutant such that CUL3–E358Q‐containing SPOP E3 ligases in EWS502 cells cannot degrade physiological SPOP substrates. Consistent with this notion, EWS–FLI1 protein levels did not increase following CUL3 depletion in EWS502 cells (Figure S6E, Supporting Information). Unlike WT‐CUL3, ectopic expression of E358Q–CUL3 failed to promote EWS–FLI1 degradation (Figure S6F, Supporting Information). Interestingly, as a scaffolding subunit in CUL3 E3 ligase complexes (Figure S6G, Supporting Information), the E358Q–CUL3 mutant retained a comparable binding to both SPOP (Figure S6H, Supporting Information) and Rbx1 (Figure S6I, Supporting Information) as WT‐CUL3. In addition, E358Q–CUL3 was also efficiently neddylated to a comparable level as WT‐CUL3 in cells (Figure S6J, Supporting Information), a modification critical for CUL3 E3 ligase activation and function.[ 35 ] These results suggest that the E358Q–CUL3 mutant forms an intact SPOPCUL3 E3 ligase complex. Notably, compared with WT‐CUL3, E358Q–CUL3 was deficient in facilitating SPOP binding to EWS–FLI1 (Figure S6K, Supporting Information), suggesting that the inability of SPOPCUL3–E358Q E3 ligase complexes in degrading EWS–FLI1 might partly be due to that the E358Q–CUL3 mutation weakens SPOP binding to its substrates, including EWS–FLI1. This result offers an additional layer of regulation for SPOP binding to its substrates through CUL3 mutations. Together, these data further support a physiological role of SPOPCUL3 in targeting EWS–FLI1 for degradation and suggest that Ewing sarcoma tumors may inactivate SPOP‐mediated EWS–FLI1 degradation through CUL3 mutations to promote Ewing sarcoma growth.
2.3. The Deubiquitinase OTU domain‐containing protein 7A (OTUD7A) is Identified as a Deubiquitinating Enzyme (DUB) to Control EWS–FLI1 Protein Stability through a Genetic Screen
Since SPOP/CK1 destabilizes EWS–FLI1, activation of SPOP/CK1 could offer a therapeutic strategy to treat Ewing sarcoma. However, potential tumor suppressor functions of SPOP [ 33 , 36 ] and CK1,[ 37 ] as well as the predicted challenge of targeting SPOPCUL3 , led us to evaluate for possible DUBs that would antagonize SPOPCUL3 function to stabilize EWS–FLI1. Among the five families of DUBs,[ 38 ] we focused on ovarian tumor proteases (OTUs) since they recognize specific ubiquitin chain linkages to regulate distinct signaling cascades associated with human tumors.[ 39 ] Thus far, 16 mammalian OTUs have been identified. We and others have reported roles of OTUD7B in maintaining mechanistic target of rapamycin (mTOR) complex homeostasis,[ 40 ] activating NF‐κB signaling[ 41 ] and regulating the cell cycle.[ 42 ] However, the physiological roles for the majority of OTUs are just beginning to be appreciated. Since EWS–FLI1 is necessary for Ewing sarcoma growth,[ 43 ] we reasoned that inhibiting DUBs that stabilize EWS–FLI1 would reduce Ewing sarcoma cell proliferation by downregulating EWS–FLI1. We screened OTU‐directed shRNAs for those that decreased A673 cell proliferation. Three independent shRNAs were used to silence each of 9 OTU genes. Cell viability was monitored at 3 days post‐shRNA infection by 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assays, or at 3‐week after shRNA infection by colony formation assays. We found that depletion of Ubiquitin thioesterase OTUB1 (OTUB1) and OTUD7A reduced A673 cell growth (Figure 3A) and diminished colony formation (Figure S7A,B, Supporting Information). Because alterations in cell growth could also result from non‐EWS–FLI1 OTU targets, we next examined interactions between each individual OTU with EWS–FLI1. We found that OTUD3, OTUD4, OTUD6B, and OTUD7A bound to EWS–FLI1 in cells (Figure 3B). Among these 4 OTUs, ectopic expression of OTUD3 or OTUD7A (Figure S8A,B, Supporting Information), but not OTUD6B nor OTUD4 (Figure S8B,C, Supporting Information), stabilized endogenous EWS–FLI1 proteins in A673 cells. These data support OTUD3 and OTUD7A as candidates to regulate EWS–FLI1 protein stability. Consistently, both OTUD3 and OTUD7A could deubiquitinate EWS–FLI1 in cells (Figure 3C and Figure S8D,E (Supporting Information)). However, in MHH‐ES‐1 cells, depletion of endogenous OTUD3 minimally influenced EWS–FLI1 protein abundance (Figure S8F, Supporting Information) but significantly affected cell growth (Figure S8G,H, Supporting Information). This suggests that shOTUD3‐induced growth reduction may be EWS–FLI1‐independent. Depletion of endogenous OTUD7A by shRNAs (Figure S8I, Supporting Information) led to reduced EWS–FLI1 protein abundance in SK‐N‐MC cells. Moreover, an interaction of OTUD7A with EWS–FLI1 at endogenous levels was observed (Figure S8J, Supporting Information). These data support OTUD7A as a possible EWS–FLI1 deubiquitinating enzyme to control EWS–FLI1 protein stability.
Figure 3.
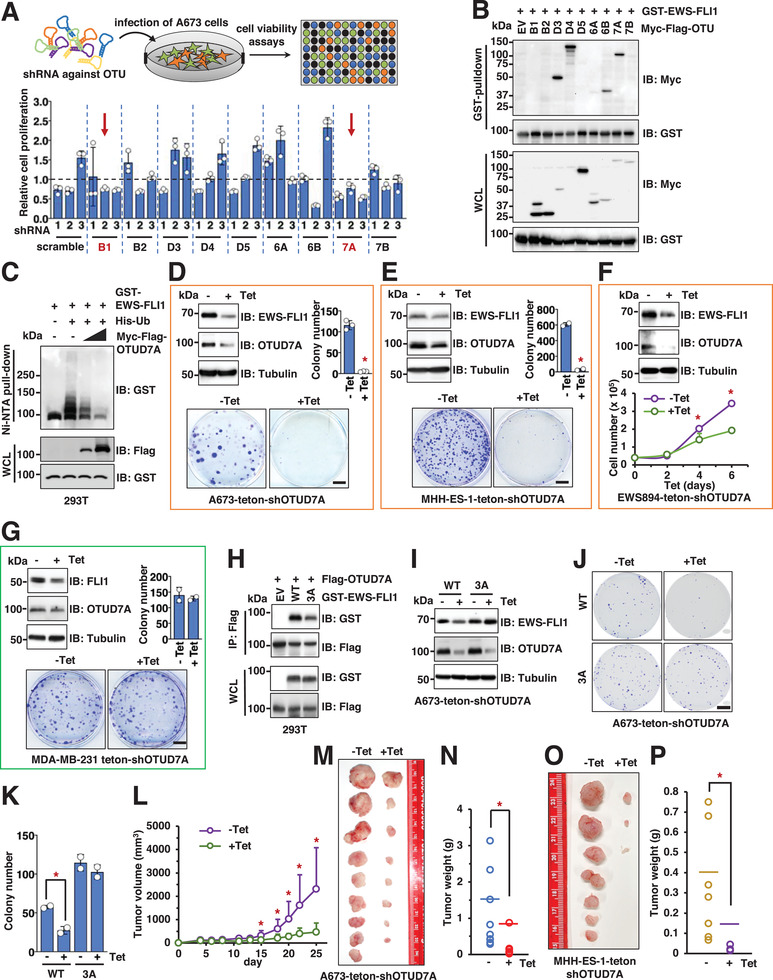
Genetic inactivation of OTUD7A leads to decreased EWS–FLI1 protein abundance and subsequently impeded Ewing sarcoma cell growth in vitro and in mice. A) shRNA‐mediated OTUB1 and OTUD7A depletion attenuates A673 cell viability. Top panel, illustration of the pipeline for shRNA‐guided screen: 3 independent shRNAs targeting each OTU were used to deplete endogenous OTU targets. 3 days postinfection, 1000 cells were plated into 96‐well plates in triplicates and cell viability was monitored 3 days postseeding by MTT assays. Error bars were calculated as mean +/− SD, n = 3. B) IB analysis of GST‐pulldown and WCL derived from HEK293T cells transfected with indicated DNA constructs. C) IB analysis of Ni–NTA pulldown and WCL derived from HEK293T cells transfected with indicated DNA constructs. D–G) Top panels, IB analysis of WCL derived from A673 (D), MHH‐ES‐1 (E), EWS894 (F), or MDA‐MB‐231 (G) cells depleted of endogenous OTUD7A by a tet‐on shRNA against endogenous OTUD7A. 1 µg mL−1 tetracycline was added into cell culture for 72 h before cell collection. Bottom panels, representative colony formation assays (D, E, G) or cell growth assays (F) using cells obtained in the corresponding top panels. Error bars were calculated as mean +/− SD, n = 3 for (D), (F) and n = 2 for (E), (G). *p < 0.05 (one‐way ANOVA test). For (D) and (G), the scale bar represents 5 mm. For (E), the scale bar represents 10 mm. H) IB analysis of Flag‐IPs and WCL derived from HEK293T cells transfected with indicated DNA constructs. I) IB analysis of WCL derived from parental or EWS–FLI1‐3A knock‐in A673 cells expressing teton‐shOTUD7A. Where indicated, 1 µg mL−1 tetracycline was added into cell culture for 72 h before cell collection. J,K) Representative images for 2D colony formation using cells from (I) and quantified in (K). Error bars were calculated as mean +/− SD, n = 2. *p < 0.05 (one‐way ANOVA test). The scale bar represents 10 mm. L–N) Mouse xenograft experiments were performed with indicated A673 cells. 5 days postinjection when tumors were established in mice, either tetracycline dissolved in water with 1% sucrose, or 1% sucrose dissolved in water only, was fed to mice. Tumor volumes were monitored by caliper measurements at indicated days (L). 25 days postinjection, mice were sacrificed, and tumors were dissected (M) and weighed (N). Error bars were calculated as mean +/− SD, n = 9. *p < 0.05 (one‐way ANOVA test). O,P) Mouse xenograft experiments were performed with indicated MHH‐ES‐1 cells. 7 days postinjection when tumors were established in mice, either tetracycline dissolved in water with 1% sucrose, or 1% sucrose dissolved in water only, was fed to mice. 32 days postinjection, mice were sacrificed, and tumors were dissected (O) and weighed (P). Error bars were calculated as mean +/− SD, n = 7. *p < 0.05 (one‐way ANOVA test).
2.4. Genetic Depletion of OTUD7A Impedes Ewing Sarcoma Growth
Stable OTUD7A depletion led to cell death within a week of shRNA or sgRNA infection, preventing us from further analyzing the signaling changes and biological effects of OTUD7A loss. To overcome this, we developed an inducible OTUD7A depletion system. 48 h post‐tetracycline (Tet) addition, we observed a reduction in endogenous OTUD7A and EWS–FLI1 proteins in A673 cells (Figure S9A, Supporting Information), with minimal effects on EWS–FLI1 mRNA (Figure S9B, Supporting Information). Remarkably, induced depletion of OTUD7A led to reduced EWS–FLI1 protein levels in multiple Ewing sarcoma cells, including A673 (Figure 3D), MHH‐ES‐1 (Figure 3E), and EWS894 (Figure 3F). MG132 treatment largely preserved EWS–FLI1 protein levels following OTUD7A depletion (Figure S9C–E, Supporting Information), further supporting a role of OTUD7A in regulating EWS–FLI1 protein stability. Importantly, for all Ewing sarcoma cell lines tested, OTUD7A depletion reduced cell proliferation in vitro (Figure 3D–F). By contrast, depletion of endogenous OTUD7A in non‐Ewing sarcoma cells, such as MDA‐MB‐231 cells, by either tet‐inducible shOTUD7A (Figure 3G) or stable OTUD7A depletion (Figure S9F,G, Supporting Information) did not significantly affect cell growth in vitro, although it reduced endogenous FLI1 protein abundance (Figure 3G and Figure S9H (Supporting Information)).
In further support of a role for OTUD7A in EWS–FLI1 regulation, we observed an interaction of OTUD7A with EWS–FLI1 at endogenous levels (Figure S8J, Supporting Information). The ubiquitination‐deficient 3A‐EWS–FLI1 demonstrated reduced binding ability with OTUD7A (Figure 3H), and depletion of OTUD7A failed to reduce 3A‐EWS–FLI1 protein levels (Figure 3I). These data suggest that OTUD7A stabilizes EWS–FLI1 proteins through the EWS–FLI1‐“VTSSS” motif or EWS–FLI1 ubiquitination. Thus, A673 3A cells offered a model to examine specific effects of inactivating the OTUD7A/EWS–FLI1 signaling. In further support of the inactivation of OTUD7A impeding Ewing sarcoma proliferation, depletion of OTUD7A resulted in significantly reduced colony formation in vitro in A673WT but not A673 3A cells (Figure 3J,K). Moreover, depletion of OTUD7A dramatically reduced tumor growth (Figure 3L) and tumor formation of A673 (Figure 3M,N), but not A673 3A (Figure S10A–C, Supporting Information) cells grown as xenografts. Depletion of OTUD7A also retarded xenografted MHH‐ES‐1 tumor development (Figure 3O,P and Figure S10D,E (Supporting Information)). Further histological analyses of xenografted MHH‐ES‐1 tumors revealed that induced depletion of OTUD7A efficiently reduced EWS–FLI1 protein levels and subsequent cell proliferation (evidenced by Ki67 staining), accompanied by increased cell death (cleaved‐caspase 3) (Figure S10F, Supporting Information). Together, these data demonstrate the dependence of Ewing sarcoma growth in vitro and in a xenografted mouse model on OTUD7A.
Although it is known that FLI1 cannot rescue the loss of EWS–FLI1 in Ewing sarcoma, we detected 3A mutation in both EWS–FLI1 and FLI1 alleles in A673 cells (Figure S5, Supporting Information). To formally demonstrate that the effect of OTUD7A is through the fusion oncoprotein, we expressed EWS–FLI1‐3A by lentiviral infection in A673‐teton‐shOTUD7A cells (Figure S11A, Supporting Information) and observed that EWS–FLI1‐3A was resistant to OTUD7A depletion (Figure S11B, Supporting Information). As a result, unlike WT‐EWS–FLI1, OTUD7A depletion failed to significantly impede 3A‐EWS–FLI1 expressing A673 cell growth in vitro (Figure S11C,D, Supporting Information) and as a xenograft (Figure S11E,F, Supporting Information). As predicted, reconstitution of FLI1‐3A expression in A673‐teton‐shOTUD7A cells (Figure S11G,H, Supporting Information) could not rescue OTUD7A‐depletion‐induced A673 cell growth retardation in vitro (Figure S11I–K, Supporting Information). These data suggest that OTUD7A controls A673 cell growth largely through regulating EWS–FLI1 but not FLI1 protein stability. To further reinforce this notion, we also expressed EWS–FLI1‐3A in both EWS894‐teton‐shOTUD7A (Figure S11L,M, Supporting Information) and MHH‐ES‐1‐teton‐shOTUD7A cells (Figure S11O,P, Supporting Information) and found that EWS–FLI1‐3A largely rescued OTUD7A‐depletion‐induced growth retardation in both cell lines (Figure S11N,Q, Supporting Information). Together, these data support that OTUD7A largely governs Ewing sarcoma growth by maintaining EWS–FLI1 protein stability and support prior studies demonstrating that EWS–FLI1 acts distinctly from FLI1.
2.5. Quantitative Proteomics Supports EWS–FLI1 as an Endogenous OTUD7A Target and Defines a Subset of Characterized EWS–FLI1 Downstream Targets Mediating OTUD7A‐/EWS–FLI1‐Governed Cell Growth
To further understand the pathophysiological function of OTUD7A in Ewing sarcoma, we performed a quantitative proteomics study following genetic OTUD7A inactivation in A673 cells. 72 h following OTUD7A depletion by shRNA induction, we observed significantly reduced EWS–FLI1 protein levels (Figure 4A and Figure S12A (Supporting Information)). At this time, proteins extracted from OTUD7A‐depleted (or control) cells were subjected to nonbiased quantitative mass spectrometry analyses to determine differences in protein abundance (Figure 4B). After excluding common contaminants and proteins nonspecifically enriched from reported microproteins, a total of 7641 nonredundant proteins were identified with protein abundance changes (Table S1, Supporting Information). These data constitute one of the largest Ewing sarcoma‐related proteomic datasets to date. Applying a p‐value < 0.05 and log2 fold change > 0.5 or <‐0.5 threshold for differential protein abundance, we observed that OTUD7A depletion resulted in statistically significant changes of abundance for 890 endogenous proteins, with 283 being upregulated and 607 downregulated (Figure 4C). Notably, our proteomic data were highly reproducible among replicates within the same group (Figure S12B,C, Supporting Information). We found that FLI1 C‐terminus peptides (derived from EWS–FLI1) were significantly decreased (Figure 4C and Table 1 ), a result consistent with our western blot analyses (Figure 4A). To explore whether OTUD7A depletion may modulate the abundance of proteins encoded by EWS–FLI1 transcriptional targets, we compared our proteomic results with a well‐developed transcriptomic study that identified 503 EWS–FLI1 transcriptional targets.[ 44 ] We found that 201 proteins were identified in both our proteomics study and the transcriptomic study. Among them, 33 proteins were significantly downregulated (including EWS–FLI1, Figure 4D and Table 1) and 6 proteins were significantly upregulated (Table 2 ) upon genetic OTUD7A depletion. Another 99 EWS–FLI1 transcriptional targets[ 44 ] demonstrated decreased levels but did not reach statistical significance (p < 0.05) (Table S2, Supporting Information). These data suggest that OTUD7A/EWS–FLI1 signaling modulates a subset of EWS–FLI1 targets.
Figure 4.
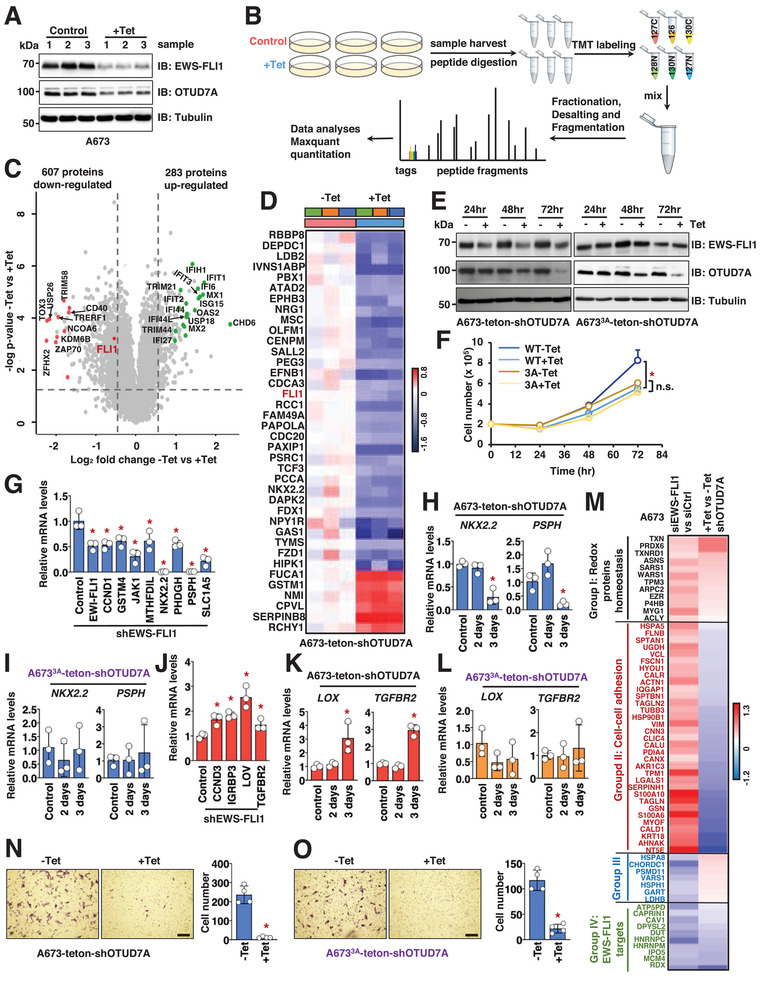
Genetic inactivation of OTUD7A suppresses key EWS–FLI1 downstream signaling. A) IB analysis of WCL derived from A673 cells treated with Tet (tetracycline, 1 µg mL−1) for 72 h. B) A cartoon illustration of the working pipeline for TMT labeling and quantitative mass spectrometry analyses. C) A volcano plot showing down‐ and upregulated genes. EWS–FLI1 is indicated in red color. D) A heatmap summarizing the statistically significantly changed characterized EWS–FLI1 targets in control and OTUD7A‐depleted A673 cells. E) IB analyses of WCL derived from WT or 3A‐A673 cells infected with Tet‐inducible shOTUD7A constructs. Where indicated, cells were collected upon treatment with 1 µg mL−1 tetracycline (Tet) for indicated periods before cell collection. F) A growth curve for cells indicated in (G) for indicated time periods measured by cell number. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). G,J) RT‐PCR analyses of indicated gene changes in control and EWS–FLI1‐depleted A673 cells. Lentiviruses coding shEWS–FLI1 were used to infect A673 cells and selected with 1 µg mL−1 puromycin to eliminate noninfected cells for 72 h before mRNA extraction. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). H,I,K,L) RT‐PCR analyses of mRNAs derived from indicated cells treated with 1 µg mL−1 Tet for indicated periods before cell collection. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). M) A heatmap summarizing the statistically significantly changed proteins upon OTUD7A depletion in A673 cells in (A) that are overlapped with a previous proteomic study[ 45 ] identifying protein changes upon EWS–FLI1 depletion in A673 cells. Group I: common hits from our study and the previous study[ 45 ] that show protein abundance increases upon either EWS–FLI1 or OTUD7A depletion; Group II: cell–cell adhesion proteins showed decreased expression upon OTUD7A depletion but increased expression upon EWS–FLI1 depletion; Group III: proteins showed decreased expression upon EWS–FLI1 depletion but increased expression upon OTUD7A depletion; Group IV: common hits from our study and the previous study[ 45 ] that show protein abundance decreases upon either EWS–FLI1 or OTUD7A depletion. N,O) Representative images for in vitro transwell assays using indicated WT (N) or EWS–FLI1‐3A knock‐in (O) A673‐teton‐shOTUD7A cells treated with 1 µg mL−1 tetracycline for 72 h before cell fixation and staining. Error bars were calculated as mean +/− SD, n = 4. *p < 0.05 (one‐way ANOVA test). The scale bar represents 50 µm.
Table 1.
A list of 33 defined EWS–FLI1 downstream target protein abundance reduced by OTUD7A depletion in A673 cells
| Gene | log2 ‐Tet vs +Tet fold change |
|---|---|
| SRSF protein kinase 1 (SRPK1) | −0.5 |
| cell division cycle‐associated protein 3 (CDCA3) | −0.52 |
| cell division cycle protein 20 homolog (CDC20) | −0.52 |
| thymidylate synthase (TYMS) | −0.52 |
| paternally‐expressed gene 3 protein (PEG3) | −0.53 |
| transcription factor E2‐alpha (TCF3) | −0.55 |
| adrenodoxin, mitochondrial (FDX1) | −0.56 |
| CYFIP‐related Rac1 interactor A (FAM49A) | −0.56 |
| Friend leukemia integration 1 transcription factor (FLI1) | −0.57 |
| ATPase family AAA domain‐containing protein 2 (ATAD2) | −0.57 |
| proline/serine‐rich coiled‐coil protein 1 (PSRC1) | −0.59 |
| pre‐B‐cell leukemia transcription factor 1 (PBX1) | −0.6 |
| poly(A) polymerase alpha (PAPOLA) | −0.61 |
| pro‐neuregulin‐1, membrane‐bound isoform (NRG1) | −0.62 |
| homeobox protein Nkx‐2.2 (NKX2.2) | −0.67 |
| ephrin type‐B receptor 3 (EPHB3) | −0.68 |
| propionyl‐CoA carboxylase alpha chain, mitochondrial (PCCA) | −0.73 |
| sal‐like protein 2 (SALL2) | −0.7 |
| noelin (OLFM1) | −0.76 |
| death‐associated protein kinase 2 (DAPK2) | −0.7 |
| DNA endonuclease RBBP8 (RBBP8) | −0.79 |
| frizzled‐1 (FZD1) | −0.8 |
| centromere protein M (CENPM) | −0.81 |
| regulator of chromosome condensation (RCC1) | −0.84 |
| DEP domain‐containing protein 1A (DEPDC1) | −0.86 |
| LIM domain‐binding protein 2 (LDB2) | −0.88 |
| PAX‐interacting protein 1 (PAXIP1) | −1.08 |
| musculin (MSC) | −1.09 |
| neuropeptide Y receptor type 1 (NPY1R) | −1.1 |
| ephrin‐B1 (EFNB1) | −1.1 |
| homeodomain‐interacting protein kinase 1 (HIPK1) | −1.11 |
| influenza virus NS1A‐binding protein (IVNS1ABP) | −1.24 |
| growth arrest‐specific protein 1 (GAS1) | −1.56 |
Table 2.
A list of 6 defined EWS–FLI1 downstream targets with protein abundance increased by OTUD7A depletion in A673 cells
| Gene | log2 ‐Tet vs +Tet fold change |
|---|---|
| serpin B8 (SERPINB8) | 0.72 |
| tissue alpha‐L‐fucosidase (FUCA1) | 0.64 |
| N‐myc‐interactor (NMI) | 0.60 |
| probable serine carboxypeptidase CPVL (CPVL) | 0.56 |
| glutathione S‐transferase Mu 1 (GSTM1) | 0.52 |
| RING finger and CHY zinc finger domain‐containing protein 1 (RCHY1) | 0.51 |
In addition to characterized EWS–FLI1 target proteins whose protein abundance was controlled by OTUD7A (Figure 4D), there were additional 572 proteins downregulated by OTUD7A genetic depletion (Table S3, Supporting Information), suggesting they are potential targets for OTUD7A or uncharacterized EWS–FLI1 targets. Further DAVID analyses led to identification of enriched biological functions for these hits by plotting the −log p‐value against log2 enrichment (Tables 2 and 3 and Figure S12D (Supporting Information)). Consistent with EWS–FLI1 signaling being a major OTUD7A downstream effector, more than one tenth (62) of the downregulated proteins exert DNA‐binding transcriptional activity (Figure S12D, Supporting Information), many of which have been characterized to associate with EWS–FLI1 on chromatin, including CBP, forkhead box proteins, and zinc finger proteins.
Table 3.
Top enriched functions for downregulated and upregulated proteins in OTUD7A‐depleted A673 cells
| Function | Count | p‐value | Fold enrichment |
|---|---|---|---|
| Disulfide bond | 114 | 3.01 × 10−25 | 2.77 |
| Transcription factor activity, sequence‐specific DNA binding | 62 | 1.93 × 10−10 | 2.36 |
| Homeobox | 21 | 7.90 × 10−10 | 5.09 |
| Immunoglobulin‐like fold | 35 | 1.51 × 10−9 | 3.17 |
| Extracellular matrix organization | 25 | 1.83 × 10−9 | 4.13 |
| Epidermal‐growth‐factor‐like domain | 17 | 4.44 × 10−7 | 4.43 |
| Protein digestion and absorption | 11 | 1.32 × 10−6 | 6.72 |
| Insulin‐like growth factor binding protein, N‐terminal | 13 | 3.04 × 10−6 | 5.08 |
| High mobility group (HMG) box domain | 13 | 8.98 × 10−6 | 4.65 |
| Integrin complex | 6 | 3.10 × 10−4 | 8.31 |
| Antiviral defense | 29 | 8.83 × 10−23 | 11.31 |
| Type I interferon signaling pathway | 21 | 5.96 × 10−20 | 15.66 |
| Innate immunity | 25 | 1.68 × 10−13 | 6.6 |
| Response to virus | 16 | 3.93 × 10−9 | 7.01 |
| 2ʹ‐5ʹ‐Oligoadenylate synthetase activity | 4 | 1.69 × 10−4 | 28.2 |
| retinoic acid‐inducible gene I (RIG‐I)‐like receptor signaling pathway | 8 | 9.57 × 10−4 | 4.88 |
| Thiol protease | 11 | 0.001573 | 3.33 |
| Response to cytokine | 8 | 0.003901 | 3.92 |
| Mitophagy | 6 | 0.004002 | 5.52 |
| Nucleophagy | 16 | 0.007451 | 2.14 |
2.6. Genetic OTUD7A Inactivation Reduces Expression of EWS–FLI1 Transcriptional Targets
To further confirm that the decreased protein levels for a subset of characterized EWS–FLI1 transcription targets following OTUD7A depletion (Figure 4D) were regulated through the OTUD7A/EWS–FLI1 signaling, we examined mRNA abundance. Reduced EWS–FLI1 protein was observed 2 days following OTUD7A shRNA induction (Figure 4E and Figure S12E,F (Supporting Information)). No significant cell growth changes were observed 2 days post‐Tet induction (Figure 4F and Figure S12G (Supporting Information)). Differences in cell proliferation were detected 3 days following OTUD7A depletion, one day following the decrease in EWS–FLI1 (Figure S12H,I, Supporting Information). We hypothesize that this difference results from a lag in the downregulation of EWS–FLI1 targets. To test this possibility, we extracted mRNAs from both A673WT‐teton‐shOTUD7A and A673 3A ‐teton‐shOTUD7A cells 2 and 3 days following tetracycline addition. A673 cells depleted of endogenous EWS–FLI1 by shRNAs served as a control. EWS–FLI1 depletion led to reduced EWS–FLI1 mRNA levels, as well as downregulation of known EWS–FLI1 target genes, with homeobox protein Nkx‐2.2 (NKX2.2) and phosphoserine phosphatase (PSPH) being the most significantly affected targets (Figure 4G). 3 days of treatment resulted in greater suppression of these targets (Figure 4H and Figure S12J–M (Supporting Information)), an effect not detected in A673 3A ‐teton‐shOTUD7A cells (Figure 4I). EWS–FLI1 depletion also increased expression of targets negatively regulated by EWS–FLI1 including lysyl oxidase (LOX) and TGF‐beta receptor type‐2 (TGFBR2) (Figure 4J). OTUD7A depletion for 3 days, but not 2 days, led to significantly increased LOX, TGFBR, and other EWS–FLI1 transcripts in A673WT‐teton‐shOTUD7A (Figure 4K and Figure S12N–P (Supporting Information)) but not A673 3A ‐teton‐shOTUD7A cells (Figure 4L). These data support that changes in EWS–FLI1 following OTUD7A depletion affect a subset of EWS–FLI1 transcriptional targets.
2.7. Quantitative Proteomics Identifies OTUD7A Downstream Targets Mediating Ewing Sarcoma Cell Migration
We compared our proteomic results with a previous study that identified protein abundance changes following EWS–FLI1 silencing.[ 45 ] Our analysis identified 103 out of 105 differentially expressed proteins controlled by EWS–FLI1 in the previous study,[ 45 ] among which 65 were significantly changed upon OTUD7A depletion (Figure 4M). Of the 33 proteins upregulated by siEWS–FLI1 but downregulated by OTUD7A depletion, 12 were associated with cell–cell adhesion (Group II in Figure 4M). Consistent with previous reports showing EWS–FLI1 depletion reduces proliferation but enhances motility,[ 10 ] we found that EWS–FLI1 depletion increased A673 cell migration in vitro (Figure S13A,B, Supporting Information). Intriguingly, OTUD7A depletion significantly reduced cell migration in vitro, in both A673WT and A673 3A cells (Figure 4N,O and Figure S13C,D (Supporting Information)). This result is consistent with the reduced expression of cell–cell adhesion proteins. These data suggest that OTUD7A influences Ewing sarcoma migration independent of EWS–FLI1. Protein candidates related to cell migration that were decreased upon depletion of OTUD7A but increased upon EWS–FLI1 depletion included integrins (Figure S13E,F, Supporting Information) and collagens (Figure S13G,H, Supporting Information) such as ITGAV and COL3A1. Depletion of EWS–FLI1 increased expression of integrin alpha‐V (ITGAV) and collagen alpha‐1(III) chain (COL3A1) (Figure S13I, Supporting Information), whereas OTUD7A depletion significantly reduced levels of both proteins (Figure S13J, Supporting Information). Interestingly, putative SPOP degrons were identified in ITGAV and COL3A1, suggesting that, similar to EWS–FLI1, OTUD7A may cooperate with SPOP to regulate these proteins (Figure S13K, Supporting Information). These data cumulatively suggest that OTUD7A inactivation not only impedes Ewing cell growth largely through reduced EWS–FLI1 protein stability, but also inhibits Ewing sarcoma motility through an EWS–FLI1‐independent manner, possibly by regulating the levels of cell–cell‐adhesion‐related proteins (Figure S13L, Supporting Information). These results motivated us to search for potential small molecules that would inhibit OTUD7A catalytic activity as a possible therapeutic strategy for Ewing sarcoma.
2.8. OTUD7A is Expressed across Tissues, Including Ewing Sarcoma Tumors
We next examined the therapeutic potential of inhibiting OTUD7A in treating Ewing sarcoma. We first demonstrated that, in contrast to WT, the catalytic‐dead C210S–OTUD7A did not stabilize EWS–FLI1 (Figure S14A, Supporting Information). This result confirmed dependence on the OTUD7A deubiquitinase activity in regulating EWS–FLI1 protein stability. We next profiled expression of OTUD7A proteins in a panel of commonly used Ewing sarcoma cell lines commonly used in labs and identified that all Ewing sarcoma cells expressed detectable OTUD7A (Figure S14B, Supporting Information). Evaluation of transcriptomic data for Ewing sarcoma cell lines and Ewing sarcoma tumors[ 46 ] also revealed levels of OTUD7A mRNA expression (Figure S14C, Supporting Information). Following validation of an OTUD7A antibody (Figure S14D, Supporting Information), we assayed a human normal tissue microarray (TMA) and observed varied expression levels of OTUD7A among tissues (Figure S14E, Supporting Information), largely consistent with immunohistochemistry (IHC) staining of tissues provided by Human Protein Atlas. In addition, expression of OTUD7A in mouse tissues including brain and spleen was observed (Figure S14F, Supporting Information). Expression of OTUD7A was also reported in different cancer types by Human Protein Atlas (Figure S15A, Supporting Information). Importantly, we observed OTUD7A expression in metastatic Ewing sarcoma tumors. OTUD7A was detected in tumor cells identified by CD99 and FLI1 antibody staining (Figure 5A and Figure S15B (Supporting Information)). These data suggest that OTUD7A is expressed in Ewing sarcomas and that the enzymatic activity offers a therapeutic target.
Figure 5.
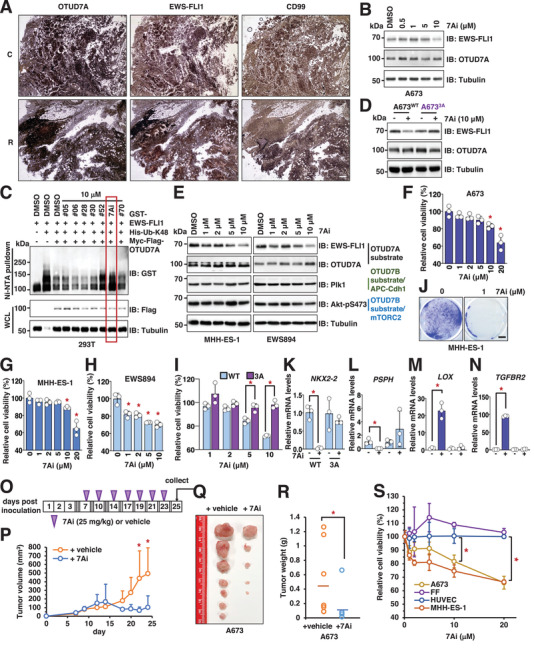
Identification of 7Ai as a lead compound to inhibit OTUD7A activation to suppress Ewing sarcoma growth. A) Representative IHC images for two Ewing sarcoma tumors obtained from patients stained with indicated antibodies. The scale bar represents 25 µm. C: calvarium; R: rib. B) IB analysis of WCL derived from A673 cells treated with indicated doses of compound 7Ai for 12 h before cell collection. C) IB analysis of Ni–NTA pulldowns and WCL derived from HEK293T cells transfected with indicated DNA constructs. Where indicated, indicated compounds were added to cell culture 10 h prior to cell collection. D) IB analysis of WCL derived from indicated A673 cells treated with 10 × 10−6 m compound 7Ai for 12 h before cell collection. E) IB analysis of WCL derived from MHH‐ES‐1 or EWS894 cells treated with indicated doses of compound 7Ai for 12 h before cell collection. F–H) Representative cell viability assays using A673 (F), MHH‐ES‐1 (G), and EWS894 (H) cells treated with indicated doses of compound 7Ai for 72 h before measurements. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). I) Representative cell viability assays using A673WT or A673 3A cells treated with indicated doses of compound 7Ai for 72 h before measurements. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). J) Representative images for 2D colony formation by MHH‐ES‐1 cells treated with indicated doses of compound 7Ai for 14 days. The scale bar represents 10 mm. K–N) RT‐PCR analyses of mRNA level changes of characterized EWS–FLI1 downstream target genes in both WT and EWS–FLI1‐3A knock‐in A673 cells treated with 1 µg mL−1 Tet for 3 days including NKX2‐2 (K), PSPH (L), LOX (M), and TGFBR2 (N). Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test). O) An illustration of the timeline for 7Ai administration into mice. At indicated periods, 25 mg kg−1 7Ai was supplied through IP injection into each mouse. P–R) Mouse xenograft experiments were performed with A673 cells treated with vehicle or 7Ai. 7 days postinjection when tumors were established in mice, 7Ai (25 mg kg−1) was injected through IP route to mice. Tumor volumes were monitored by caliper measurements at indicated days (P). 25 days postinjection, mice were sacrificed and tumors were dissected (Q) and weighed (R). Error bars were calculated as mean +/− SD, n = 7. *p < 0.05 (one‐way ANOVA test). S) Representative cell viability assays using two Ewing sarcoma cells (A673 and MHH‐ES‐1) and two normal control cells (HUVEC and foreskin fibroblast (FF)) treated with indicated doses of compound 7Ai for 72 h before measurements. Error bars were calculated as mean +/− SD, n = 3. *p < 0.05 (one‐way ANOVA test).
2.9. Artificial‐Intelligence‐Aided Virtual Drug Screen Identified 7Ai as an OTUD7A Catalytic Inhibitor
To rapidly assess the binding ability of drug‐like small molecules to OTUD7A, we applied AtomNet, a structure‐based deep convolutional neural network virtual screening technology developed by Atomwise Inc.[ 47 , 48 ] In the absence of a published crystal structure of the OTUD7A–OTU domain, we first generated a homology model of the OTUD7A–OTU domain based on the available crystal structure of the closely related OTUD7B–OTU domain (Protein Data Bank (PDB): 5LRW, 79% sequence identity in this region) (Figure S16A, Supporting Information). Using this generated structure, we performed a virtual screen by sifting through a library of 4 million commercially available, drug‐like compounds that yielded a chemically diverse set of 73 high‐scoring predicted hits. We evaluated these compounds for their ability to reduce EWS–FLI1 protein abundance in both A673 and SK‐N‐MC cells (Figure S16B,C, Supporting Information). One compound that we termed as 7Ai, ranking 44th out of 4 025 533 compounds we screened, reduced EWS–FLI1 protein levels in both Ewing cells without affecting OTUD7A protein levels (Figure S16B,C, Supporting Information). Moreover, 7Ai reduced EWS–FLI1 protein levels in a dose‐dependent manner within 12 h (Figure 5B and Figure S16D (Supporting Information)). Importantly, this activity was not lost following high performance liquid chromatography (HPLC) purification (Figure S16D, Supporting Information), suggesting that 7Ai, rather than contaminants from the chemical synthesis process, mediates OTUD7A suppression. In addition, 7Ai efficiently blocked OTUD7A‐mediated deubiquitination of EWS–FLI1 in cells (Figure 5C and Figure S16E (Supporting Information)). Consistent with the genetic OTUD7A depletion, 7Ai reduced EWS–FLI1 protein abundance in parental A673 but not A673 3A (Figure 5D and Figure S16F (Supporting Information)), highlighting the importance of the OTUD7A/EWS–FLI1 signaling in mediating 7Ai function. 7Ai did not interfere with OTUD7A binding to EWS–FLI1 (Figure S16G, Supporting Information), suggesting this compound might suppress OTUD7A catalytic activity through interaction with the catalytic domain. To explore whether 7Ai directly binds OTUD7A, we purified the bacterially produced His‐tagged OTUD7A OTU domain (aa183‐449) (Figure S16H, Supporting Information). Using isothermal titration calorimetry (ITC), we demonstrated a binding affinity of 7Ai in vitro of ≈1.1 × 10−6 m (stoichiometry is about 1:1) (Figure S16I, Supporting Information). 7Ai efficiently reduced EWS–FLI1 protein expression in multiple Ewing sarcoma cells in addition to A673, including MHH‐ES‐1 and EWS894 (Figure 5E), SK‐N‐MC and EWS502 (Figure S17A, Supporting Information). Notably, 7Ai treatment did not significantly affect OTUD7B activities as indicated by negligible changes in known OTUD7B substrates, including mechanistic target of rapamycin complex 2 (mTORC2)[ 40 ] and anaphase promoting complex (APC)/Cdh1 [ 42 ] (Figure 5E and Figure S17A (Supporting Information)). These data support that compound 7Ai suppresses OTUD7A activity to destabilize EWS–FLI1.
2.10. 7Ai Impedes Ewing Sarcoma Growth In Vitro and In Vivo
We then evaluated the effects of 7Ai treatment on Ewing sarcoma growth. 3 days treatment with 7Ai reduced proliferation of A673, MHH‐ES‐1, and EWS894 cells (Figure 5F–H), which was associated with reduced EWS–FLI1 protein abundance (Figure 5E and Figure S17B,C (Supporting Information)). Notably, this effect was not observed in A673 3A cells (Figure 5I). 7Ai also reduced EWS–FLI1 protein abundance in A673‐teton‐shOTUD7A but not same cells reconstituted with EWS–FLI1‐3A (Figure S17D, Supporting Information). Importantly, 7Ai treatment failed to significantly suppress growth of A673 cells expressing EWS–FLI1‐3A (Figure S17E, Supporting Information). 7Ai treatment reduced transcription of EWS–FLI1 target genes (NKX2.2 and PSPH, Figure 5K,L), and increased transcription of genes negatively regulated by EWS–FLI1 (LOX and TGFBR2, Figure 5M,N). 7Ai did not affect EWS–FLI1 mRNA levels (Figure S17F, Supporting Information). Importantly, 2 week treatment of 7Ai led to reduced colony formation ability of MMH‐ES‐1 (Figure 5J) and A673 cells (Figure S17G, Supporting Information) in vitro. Cumulatively, these data support that 7Ai suppresses Ewing sarcoma growth by reducing EWS–FLI1 protein stability.
We then examined the effect of 7Ai on Ewing sarcoma cells grown as xenografts. Following establishment of ≈0.5 cm A673 tumors in immunocompromised mice, we administrated vehicle or 7Ai (25 mg kg−1, intraperitoneal (IP)) or vehicle every 2–3 days (Figure 5O). Compared with vehicle control group, 7Ai treatment significantly reduced tumor volume (Figure 5P and Figure S17H (Supporting Information)) and tumor growth (Figure 5Q,R). Notably, 7Ai administration over the 3‐week treatment period did not significantly affect body weight (Figure S17I, Supporting Information). 7Ai‐treated tumors demonstrated reduced EWS–FLI1 protein and cell proliferation (Ki67 staining) and increased apoptosis (cleaved‐caspase3 staining) (Figure S17J, Supporting Information). In vitro, 7Ai treatment significantly reduced A673 cell migratory ability (Figure S17K,L, Supporting Information). These data indicate that 7Ai suppresses Ewing sarcoma growth and migration.
To examine if 7Ai exerts selectivity in eradicating Ewing sarcoma cells, we treated two Ewing sarcoma cell lines A673 and MHH‐ES‐1 and two normal control cell lines human umbilical vein endothelial cells (HUVEC) and foreskin fibroblast (FF) with 7Ai for 3 days in vitro. 7Ai treatment efficiently reduced EWS–FLI1 protein abundance in both Ewing sarcoma cells (Figure S18A,B, Supporting Information) but had minimal effects on FLI1 proteins in HUVEC and FF cells (Figure S18C,D, Supporting Information). As observed with A673, 7Ai treatment reduced MHH‐ES‐1 proliferation but exerted neglectable effects in HUVEC and FF cells (Figure 5S). Although preliminary, the limited in vivo side‐effect profile and effect on non‐Ewing sarcoma cells offers the possibility of a therapeutic window for Ewing sarcoma treatment. Subsequent formal in vivo studies will be necessary to support this observation.
2.11. OTUD7A Might Also Control EWS–ERG Fusion Protein Stability in Ewing Sarcoma
In addition to EWS–FLI1 fusion observed in ≈85% Ewing sarcoma tumors, other fusions including EWS–transcriptional regulator ERG (ERG) (≈10% patients) and EWS–protein FEV (FEV) (≈1% patients) have also been observed. Variation in translocation breakpoints result in type I and type II fusions which differ based on included exons (Figure S19A,B, Supporting Information). Our data suggest that SPOP/CK1 and OTUD7A regulate both type I and type II fusions, as the SPOP degron is present in both fusion types (Figure S19A–C, Supporting Information). Moreover, as predicted by the presence of the SPOP degron in the ERG segment in EWS–ERG fusion (Figure S19A, Supporting Information), SPOP also targeted EWS–ERG for degradation (Figure S19D, Supporting Information), and OTUD7A stabilized EWS–ERG (Figure S19E, Supporting Information). EWS–ERG could partially replace EWS–FLI1 in A673 cells to maintain cell growth in vitro (Figure S19F,G, Supporting Information). In this setting, SPOP depletion stabilized EWS–ERG (Figure S19H, Supporting Information). OTUD7A depletion reduced EWS–ERG (Figure S19I, Supporting Information) associated with reduced cell growth (Figure S19J), Supporting Information. 7Ai treatment reduced EWS–ERG protein levels (Figure S19K, Supporting Information). Cumulatively, these data support that the vast majority of Ewing sarcoma would be targets of OTUD7A‐directed treatment. More broadly, this project offers a strategy to therapeutically target a critical oncoprotein initiated by the recognition of a putative protein degron sequence.
3. Discussion
Because it is indispensable for Ewing sarcoma growth, targeting the EWS–FLI1 fusion oncoprotein offers an important and specific therapeutic strategy. Here, we report the identification of a pathophysiologically relevant protein control mechanism. SPOP is the first E3 ubiquitin ligase that targets EWS–FLI1 for ubiquitination and degradation in a CK1‐phosphorylation‐dependent manner. The deubiquitinase OTUD7A antagonizes SPOP function to stabilize EWS–FLI1, revealing OTUD7A as a new Ewing‐sarcoma‐growth‐dependent gene. Applying quantitative proteomic analyses, we confirmed EWS–FLI1 as a bona fide OTUD7A substrate and identified additional OTUD7A substrates that may mediate cellular motility, independent of EWS–FLI1. Since, genetic inactivation of OTUD7A reduced Ewing sarcoma proliferation and motility, sought to target OTUD7A. Using artificial‐intelligence (AI)‐aided virtual drug screening, we identify the first OTUD7A catalytic inhibitor, which limits Ewing sarcoma growth in vitro and in mice by degrading EWS–FLI1.
The FLI1 domain in EWS–FLI1 is targeted by SPOP and OTUD7A. FLI1 has tissue restricted expression and deficiency is associated with thrombocytopenia in humans and mice. FLI1 is not broadly considered an essential gene for cell proliferation (such as MDA‐MD‐231 (Figure 3G and Figure S9F,G (Supporting Information)) although exceptions include certain cancers such as blood and kidney cancer (Figure S20A,B (Supporting Information) from DEPMAP portal). In support of this association, Tet‐induced depletion of OTUD7A in kidney cancer (ACHN) and leukemia (Jurkat and CUTLL1) cells reduced levels of endogenous FLI1 (Figure S20C–E, Supporting Information) accompanied by reduced cell proliferation (Figure S20C–E, Supporting Information). Like genetic OTUD7A depletion, pharmacological inhibition of OTUD7A by the compound 7Ai also decreased proliferation of Jurkat cells (Figure S20F, Supporting Information). Therefore, in addition to Ewing sarcoma, targeted inhibition of OTUD7A may be relevant for other cancers dependent on FLI1 for proliferation, such as leukemia and kidney cancer (Figure S20G, Supporting Information).
Notably, due to the lack of a large cohort of patient data in Ewing sarcoma as a rare cancer, analyzing the Cancer Genome Atlas (TCGA) sarcoma dataset revealed that OTUD7A gene was infrequently altered (Figure S21A, Supporting Information). OTUD7A mRNA levels were not associated with overall patient survival in adult soft tissue sarcomas analyze by TCGA (Figure S21B, Supporting Information). Whether OTUD7A protein abundance predicts Ewing sarcoma patient survival remains to be determined. Interestingly, high OTUD7A mRNA levels were associated with worse patient survival in thymoma (Figure S21C, Supporting Information), uterine corpus endometrial carcinoma (Figure S21D, Supporting Information), and esophageal squamous cell carcinoma (Figure S21E, Supporting Information) patient cohorts, and neared statistical significance for worse breast cancer survival (Figure S21F, Supporting Information). By contrast, high OTUD7A mRNA expression was not associated with survival in ovarian (Figure S21G, Supporting Information) and stomach (Figure S21H, Supporting Information) cancers, and with improved survival of cervical cancer patients (Figure S21I, Supporting Information).
Two deubiquitinases, USP7 [ 18 ] and USP19 [ 49 ] had been reported as vulnerabilities in Ewing sarcoma. Genetic and pharmacologic inactivation of USP7 was shown to reduce Ewing sarcoma growth, although the substrate(s) through which USP7 acted on Ewing sarcoma growth remained unclear.[ 18 ] Genetic depletion of USP19 reduced Ewing sarcoma cell growth in vitro and in mice largely through destabilizing EWS–FLI1 proteins.[ 19 ]
In our study, we find that both genetic and pharmacological inactivation of OTUD7A impede not only Ewing sarcoma growth but also decreased motility. It is possible that the activity of OTUD7A on motility is through EWS–FLI1‐independent substrates such as ITGAV and COL3A. Thus, it seems that inhibiting OTUD7A suppresses both Ewing sarcoma proliferation and may affect its ability to disseminate. Whole‐animal OTUD7A deletion in mice led to decreased dendritic spine density that mimicked neurodevelopmental disorders[ 50 ] associated with 15q13.3 microdeletion syndrome.[ 51 ] Recently, a homozygous OTUD7A–L233F mutation was found in a patient with the 15q13.3 microdeletion syndrome with characterized proteasome dysfunction presumably caused by the loss of function of the OTUD7A deubiquitinase activity.[ 52 ] Although it remains unclear if these neurological disorders caused by OTUD7A dysfunction are limited to changes in dendritic spines, these results offer additional considerations if 7Ai or other OTUD7A inhibitors begin preclinical evaluation for Ewing sarcoma.
Our studies demonstrated efficacy of 7Ai in vivo and in vitro efficacy in the micromolar range. Additional medicinal chemistry studies are needed to further improve its potency and to evaluate pharmacokinetic and side effect properties. Since therapy for initial and relapsed Ewing sarcoma includes cytotoxic chemotherapies, it would be of interest to evaluate the combination of 7Ai with active chemotherapeutic drugs, including the treatment of patients with metastatic disease. Because of the development of other biologically targeted therapies, including those directed at EWS–FLI1 and USP19 and USP7, assaying the activity of 7Ai with these agents would also be of interest.
4. Experimental Section
Cell Culture and Transfection
HEK293, HEK293T, FF, A673, MHH‐ES‐1, MDA‐MB‐231 and ACHN cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Jurkat and CUTLL1 cells were cultured in RPMI‐1640 medium supplemented with 10% FBS. EWS502 and EWS894 cells were maintained in RPMI‐1640 medium supplemented with 15% FBS. SK‐N‐MC were cultured in RPMI‐1640 medium supplemented with 10% FBS, 200 × 10−6 m glutamine (Gibco, 25030081) and nonessential amino acids (Gibco, 11140050). HUVEC cells were cultured in Endothelial Cell Growth Medium 2 (PromoCell, C‐22111) supplemented with 10% FBS. All cell culture media were supplemented with 100 units of penicillin and 100 mg mL−1 streptomycin unless otherwise stated.
Cell transfection was performed using lipofectamine 3000 or polyethylenimine, as described previously.[ 53 , 54 , 55 ] Packaging of lentiviral shRNA or complementary DNA (cDNA) expressing viruses, as well as subsequent infection of various cell lines were performed according to the protocols described previously.[ 56 , 57 ] Following viral infection, cells were maintained in the presence of blasticidin (5 µg mL−1) or puromycin (1 µg mL−1), depending on the viral vector used to infect cells.
MG132 (S2619), MLN4924 (S7109), cycloheximide (S6611), D4476 (S7642), and lenalidomide (S1029) were purchased from Selleck. Tetracycline (87128) and doxycycline (D9891) were purchased from Sigma‐Aldrich. JQ1 was purchased from Sigma (SML0974). Larger quantities of compound 7Ai was purchased from Princetonbio or obtained from Atomwise, Inc.
Plasmid Construction
Flag‐SPOP and CMV‐glutathione‐S‐transferase (GST)‐SPOP were as described previously.[ 29 ] pCDNA3‐HA‐SPOP plasmid was constructed by cloning SPOP into pCDNA3‐HA vector using primers listed below. Myc‐tagged CK1s and CK2s were as described.[ 29 ] His‐ub plasmids were as described.[ 58 ] Myc‐Flag‐OTU plasmids were as described.[ 40 ] His‐OTUD7A was constructed by cloning OTUD7A into pET28a vector using primers listed below. HA‐EWS–FLI1 and HA‐FLI1 were cloned into pCDNA3‐HA vector using primers listed below. pLenti‐HA‐FLI1‐WT and 3A plasmids were cloned into the pLenti‐HA‐hygro vector using primers listed below. pLL5.5‐HA‐EWSR1 and pLL5.5‐HA‐EWS–ERG were previously described.[ 2 ] HA‐SPOP was cloned into pCDNA3‐HA vector using primers listed below. His‐OTUD7A was cloned into pET28a vector using primers listed below. pLenti‐EWS–FLI1‐3A was constructed by cloning EWS–FLI1‐3A into pLenti‐GFP‐hygro vector using primer below from Flag‐EWS–FLI1‐3A plasmid. Myc–cullin plasmids were a generous gift from Yue Xiong lab at University of North Carolina at Chapel Hill.
EWS–FLI‐BglII‐F: GCATAGATCTGCGTCCACGGATTACAGTACC
FLI1‐BglII‐F: GCATAGATCTGACGGGACTATTAAGGAGGC
FLI1‐XhoI‐R: GCATCTCGAGCTAGTAGTAGCTGCCTAAGTG
hSPOP‐BamHI‐F: GCATGGATCCTCAAGGGTTCCAAGTCCTCCAC
hSPOP‐XhoI‐R: GCATCTCGAGTTAGGATTGCTTCAGGCGTTTGCG
OTUD7A‐BglII‐F: GCATAGATCTGTTTCTAGTGTGCTTCCAAACC
OTUD7A‐SalI‐R: GCATGTCGACTCACAGCTCCTCGCGG
EWS–FLI1‐3A, OTUD7A–C210S, and CUL3–E358Q mutants were generated using the QuikChange XL Site‐Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions. Details of plasmid constructions were available upon request.
EWS–FLI‐3A‐F: CCTCCATGCCTGTCACTGCCGCCGCCTTCTTTGGAGCCGCATCAC
EWS–FLI‐3A‐R: GTGATGCGGCTCCAAAGAAGGCGGCGGCAGTGACAGGCATGGAGG
OTUD7A–C210S‐F: CAGGGGATGGGAACTCCCTTTTACATGCTGCTTCACTG
OTUD7A–C210S‐R: CAGTGAAGCAGCATGTAAAAGGGAGTTCCCATCCCCTG
CUL3–E358Q‐F: GTTCGATCGCTTCCTCCTGCAATCATTCAACAATGACCGTCTC
CUL3–E358Q‐R: GAGACGGTCATTGTTGAATGATTGCAGGAGGAAGCGATCGAAC
Reverse transcription PCR (RT‐PCR) primers to examine EWS–FLI1 mRNA changes upon SPOP or OTUD7A depletion were listed below:
EWS‐F: TCCTACAGCCAAGCTCCAAGTC
FLI1‐R: ACTCCCCGTTGGTCCCCTCC
RT‐PCR primers to examine EWS–FLI1 transcriptional targets used in this study were listed below:
EWS–FLI1‐F: CAGTCACTGCACCTCCATCC
EWS–FLI1‐R: TTCATGTTATTGCCCCAAGC
NKX2.2‐F: CTACGACAGCAGCGACAACC
NKX2.2‐R: GCCTTGGAGAAAAGCACTCG
TGFBR2‐F: CATCTGTGAGAAGCCACAGG
TGFBR2‐R: TGCACTCATCAGAGCTACAGG
insulin‐like growth factor‐binding protein 3 (IGFBP3)‐F: CTGCTCAGATTTCCCCAAAG
IGFBP3‐R: TGGCATCAAGCAGGTCATAG
LOX‐F: CATCAAGAAAGGGCATGCTAA
LOX‐R: CTACGGCAGGGACCATATTCT
janus kinase 1 (JAK1)‐F: CAGGTCTCCCACAAACACATCG
JAK1‐R: ACCAGGTCTTTATCCTCCAAGTAGC
G1/S‐specific cyclin‐D1 (CCND1)‐F: CGCACGATTTCATTGAACACTT
CCND1‐R: CGGATTGGAAATACTTCACAT
CCND3‐F: CCTCTGTGCTACAGATTATACCTTTGC
CCND3‐R: TTGCACTGCAGCCCCAAT
glutathione S‐transferase Mu 4 (GSTM4)‐F: TGGAGAACCAGGCTATGGACGT
GSTM4‐R: CCAGGAACTGTGAGAAGTGCTG
D‐3‐phosphoglycerate dehydrogenase (PHGDH)‐F: CTGCGGAAAGTGCTCATCAGT
PHGDH‐R: TGGCAGAGCGAACAATAAGGC
PSPH‐F: GATGCTGTGTGTTTTGATGTTGAC
PSPH‐R: CTTGACTTGTTGCCTGATCACATT
neutral amino acid transporter B(0) (SLC1A5)‐F: CTTGGTAGTGTTTGCCATCGT
SLC1A5‐R: TGCGGGTGAAGAGGAAGTAG
monofunctional C1‐tetrahydrofolate synthase, mitochondrial (MTHFD1L)‐F: GAGCTCTGAAGARGCATGGAG
MTHFD1L‐R: TGCTTCTGGAGGTTACAGCA
shRNAs and sgRNAs
shRNA vectors to deplete endogenous SPOP, CUL3, and various OTUs were purchased from Sigma. Their sequence was listed below:
shSPOP‐1: CCGGCACAGATCAAGGTAGTGAAATCTCGAGATTTCACTACCTTGATCTGTGTTTTTTG
shSPOP‐2: CCGGCAAGGTAGTGAAATTCTCCTACTCGAGTAGGAGAATTTCACTACCTTGTTTTTTG
shSPOP‐3: CCGGCAAACGCCTGAAGCAATCCTACTCGAGTAGGATTGCTTCAGGCGTTTGTTTTTTG
shSPOP‐4: CCGGCTCCTACATGTGGACCATCAACTCGAGTTGATGGTCCACATGTAGGAGTTTTTTG
shCUL3‐1: CCGGCGTGTGCCAAATGGTTTGAAACTCGAGTTTCAAACCATTTGGCACACGTTTTTG
shCUL3‐2: CCGGTTCAGGCTTTACAACGTTTATCTCGAGATAAACGTTGTAAAGCCTGAATTTTTG
shCUL3‐3: CCGGCGTGTGCCAAATGGTTTGAAACTCGAGTTTCAAACCATTTGGCACACGTTTTTG
shOTUB1‐1: CCGGAGGAGTATGCTGAAGATGACACTCGAGTGTCATCTTCAGCATACTCCTTTTTT
shOTUB1‐2: CCGGTGTTTCTATCGGGCTTTCGGACTCGAGTCCGAAAGCCCGATAGAAACATTTTT
shOTUB1‐3: CCGGTGTGGTTGTAAATGGTCCTATCTCGAGATAGGACCATTTACAACCACATTTTT
shOTUB2‐1: CCGGCCTATGTGTCACTGGATTATTCTCGAGAATAATCCAGTGACACATAGGTTTTTG
shOTUB2‐2: CCGGTGGGCTGCTATGTCTCTGTATCTCGAGATACAGAGACATAGCAGCCCATTTTT
shOTUB2‐3: CCGGCCTTCCGTTTACCTGCTCTATCTCGAGATAGAGCAGGTAAACGGAAGGTTTTT
shOTUD3‐1: CCGGGACGTCTGCCATCGCATATTACTCGAGTAATATGCGATGGCAGACGTCTTTTTG
shOTUD3‐2: CCGGTTTGGAAATCAGGGCTTAAATCTCGAGATTTAAGCCCTGATTTCCAAATTTTTG
shOTUD3‐3: CCGGGGGAGTTACACATCGCATATCCTCGAGGATATGCGATGTGTAACTCCCTTTTTG
shOTUD4‐1: CCGGCAAGTCGAGAATCTAACTATTCTCGAGAATAGTTAGATTCTCGACTTGTTTTTG
shOTUD4‐2: CCGGTATGCAATGCCTTAGTCATAACTCGAGTTATGACTAAGGCATTGCATATTTTTG
shOTUD4‐3: CCGGCACTATAGATTCCAAACATAACTCGAGTTATGTTTGGAATCTATAGTGTTTTTG
shOTUD5‐1: CCGGCCATCATTCAAACCAGGGTTTCTCGAGAAACCCTGGTTTGAATGATGGTTTTTTG
shOTUD5‐2: CCGGCCGACTACTTCTCCAACTATGCTCGAGCATAGTTGGAGAAGTAGTCGGTTTTTG
shOTUD5‐3: CCGGAGAACGTCTGAGCCTTCAATGCTCGAGCATTGAAGGCTCAGACGTTCTTTTTTG
shOTUD6A‐1: CCGGCATGATCTACTGCGACAACATCTCGAGATGTTGTCGCAGTAGATCATGTTTTTTG
shOTUD6A‐2: CCGGCACCAACTAAGATTTGGTCATCTCGAGATGACCAAATCTTAGTTGGTGTTTTTTG
shOTUD6A‐3: CCGGGATTTGGTCATGTTGCGTATACTCGAGTATACGCAACATGACCAAATCTTTTTTG
shOTUD6B‐1: CCGGGCAAAGCTACTAACAGGTGTTCTCGAGAACACCTGTTAGTAGCTTTGCTTTTTTG
shOTUD6B‐2: CCGGGCTGACTACTAAGGAGAATAACTCGAGTTATTCTCCTTAGTAGTCAGCTTTTTTG
shOTUD6B‐3: CCGGCGATGAGACTAATGCAGTGAACTCGAGTTCACTGCATTAGTCTCATCGTTTTTTG
shOTUD7A‐1: CGGGCAGCAATTCTAACAGCAATACTCGAGTATTGCTGTTAGAATTGCTGCTTTTTG
shOTUD7A‐2: CCGGCGCACACACTTCAGCAAGAATCTCGAGATTCTTGCTGAAGTGTGTGCGTTTTTG
shOTUD7A‐3: CCGGGCGCGAGAACTGTGCGTTCTACTCGAGTAGAACGCACAGTTCTCGCGCTTTTTG
shOTUD7B‐1: GTACCGGTTGAAGAGTTTCACGTCTTTGCTCGAGCAAAGACGTGAAACTCTTCAATTTTTTG
shOTUD7B‐2: CCGGTGGAAATGCTCACGGTTTATACTCGAGTATAAACCGTGAGCATTTCCATTTTTG
shOTUD7B‐3: CCGGGCAAGGAGGCTAAACAAAGTTCTCGAGAACTTTGTTTAGCCTCCTTGCTTTTT
shCK1α‐42: CCGGGCAGAATTTGCGATGTACTTACTCGAGTAAGTACATCGCAAATTCTGCTTTTT
shCK1α‐87: CCGGGCAAGCTCTATAAGATTCTTCCTCGAGGAAGAATCTTATAGAGCTTGCTTTTTTG
shFLI1: TGCCCATCCTGCACACTTACTTCAAGAGAGTAAGTGTGCAGGATGGGCTTTTTTC (targeting the 3ʹuntraslated region (UTR) of FLI1 as reported in ref. [2])
Teton‐shOTUD7A primers were listed below:
Teton‐shOTUD7A‐F: CCGGGCGCGAGAACTGTGCGTTCTACTCGAGTAGAACGCACAGTTCTCGCGCTTTTT
Teton‐shOTUD7A‐R: AATTAAAAAGCGCGAGAACTGTGCGTTCTACTCGAGTAGAACGCACAGTTCTCGCGC
shOTUD7A‐resistant OTUD7A construct was generated using the QuikChange XL Site‐Directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions.
shOTUD7A‐62‐resistant‐F: CTGCCAGCGGGAAAATTGCGCGTTCTACGG
shOTUD7A‐62‐resistant‐R: CCGTAGAACGCGCAATTTTCCCGCTGGCAG
EWS–FLI1‐3A knock‐in experiment was performed using EWS–FLI1 sgRNAs and single‐stranded donor oligonucleotides (ssoDNA) as listed below:
EWS–FLI1‐3A‐sgRNA‐F: CACCG TGCGGCTCCAAAGAAGCTGG
EWS–FLI1‐3A‐sgRNA‐R: AAAC CCAGCTTCTTTGGAGCCGCA C
EWS–FLI1‐3A‐ssoDNA: GCCCACCAGCAGAAGGTGAACTTTGTCCCTCCCCATCCATCCTCCATGCCTGTCACTGCCGCCGCCTTCTTTGGAGCCGCATCACAATACTGGACCTCCCCCACGGGGGGAATCTACCCC
Knock‐in clones were screened by PCR using primers listed below to search for clone loss of BpmI site after knock‐in.
EWS–FLI‐3A‐KI‐PCR‐F: GTGCACGGCAAAAGATATGCTTAC
EWS–FLI‐3A‐KI‐PCR‐R: CTAGTAGTAGCTGCCTAAGTGTG
sgRNAs to stably deplete endogenous OTUD7A were listed below:
sgOTUD7A‐1A‐F: CACCGAGACTTGTTCGGTCCACGG
sgOTUD7A‐1A‐R: AAACCCGTCCACCGAACAAAGTCTC
sgOTUD7A‐1B‐F: CACCGTGCTGCCCAACACTCAGCCG
sgOTUD7A‐1B‐R: AAACCGGCTGAGTGTTGGGAGCAC
sgOTUD7A‐1C‐F: CACGCAGACCAGGTTCTGCCCCCG
sgOTUD7A‐1C‐R: AAACCGGGGGCAGAACCTGGTCTGC
Immunoblot and Immunoprecipitations Analyses
Cells were lysed in EBC buffer (50 × 10−3 m Tris pH 7.5, 120 × 10−3 m NaCl, 0.5% NP‐40) or Triton X‐100 buffer (50 × 10−3 m Tris pH 7.5, 150 × 10−3 m NaCl, 1% Triton X‐100) supplemented with protease inhibitors (Complete Mini, Roche) and phosphatase inhibitors (phosphatase inhibitor cocktail sets I and II, Calbiochem). The protein concentrations of whole cell lysates were measured by NanoDrop OneC using the Bio‐Rad protein assay reagent as described previously.[ 55 ] Equal amounts of whole cell lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and immunoblotted with indicated antibodies. For immunoprecipitation analysis, unless specified, 1000 µg lysates were incubated with the indicated antibody (1–2 µg) for 3–4 h at 4 °C followed by 1 h incubation with 10 µL Protein A magnetic beads (New England Biolabs). Or, 1000 µg lysates containing tagged molecules were incubated with agarose‐bead‐coupled antibodies for the specific tag for 3–4 h at 4 °C. For endogenous IPs, incubation of cell lysates with antibodies was extended to overnight. The recovered immunocomplexes were washed 5 times with NETN buffer (20 × 10−3 m Tris, pH 8.0, 100 × 10−3 m NaCl, 1 × 10−3 m ethylenediaminetetraacetic acid (EDTA), and 0.5% NP‐40) before being resolved by SDS‐PAGE and immunoblotted with indicated antibodies.
Antibodies
All antibodies were used at a 1:1000 dilution in Tris buffered saline with Tween 20 (TBST) buffer with 5% nonfat milk for western blotting. Anti‐GST antibody (2625), anti‐Cullin3 antibody (2759), anti‐CD99 antibody (20992), anti‐CK1 antibody (2655), anti‐BRD4 antibody (13440), anti‐Plk1 antibody (4513), anti‐Akt–pS473 antibody (4060), anticleaved‐caspase 3 antibody (9661), anti c‐Myc antibody (5605), and anti‐myc‐tag antibody (2278) were obtained from Cell Signaling Technology. Anti‐FLI1 antibody (ab180902), anti‐nuclear factor erythroid 2‐related factor 2 (NRF2) (ab62352), anti‐ERG (ab92513), and anti‐Ki67 antibody (ab254123) were obtained from Abcam. Anti‐SPOP antibody (16750‐1‐AP) was purchased from Proteintech. Anti‐EWSR1 antibody (A300‐417) was purchased from Bethyl Laboratories. Polyclonal anti‐HA antibody (sc‐805), anti‐p27‐antibody (sc1641), anti‐NRF2 antibody (sc81342), anti‐ERG antibody (271048), anti‐ITGAV antibody (376156), anti‐COL3A1 antibody (271249), and anti‐Vinculin antibody (sc‐25336) were obtained from Santa Cruz Biotechnology. Polyclonal anti‐Flag antibody (F‐2425), monoclonal anti‐Flag antibody (F‐3165, clone M2), anti‐Tubulin antibody (T‐5168), anti‐OTUD3 antibody (PA5‐98487), anti‐OTUD7A antibody (SABB04135), anti‐Flag agarose beads (A‐2220), anti‐HA agarose beads (A‐2095), glutathione agarose beads (G4510), peroxidase‐conjugated anti‐mouse secondary antibody (A‐4416), and peroxidase‐conjugated anti‐rabbit secondary antibody (A‐4914) were obtained from MilliporeSigma. Monoclonal anti‐HA antibody (MMS‐101P) was obtained from BioLegend.
Generation of EWS–FLI1‐3A Knock‐In A673 Cells
Parental A673 cells were split into 24‐well plates and transfected with sgRNA against EWS–FLI1 together with EWS–FLI1‐3A‐ssoDNA following protocols as described.[ 59 ] 1 day post‐transfection, cells were selected with 1 µg mL−1 puromycin for 3 days. Surviving cells were counted and each single cell was seeded into 96‐well plates. Each single clone grown up in 96‐well plates was amplified and one copy was used for genomic DNA extraction, followed by PCR and BpmI digestion to screen for potential knock‐in clones. BpmI negative clones were selected and sequenced to verify the knock‐in at the DNA level. 3 isogenic knock‐in clones were selected and saved.
shRNA‐Mediated OTU Screen to Identify OTUs Critical to Maintain Ewing Sarcoma Growth
Three independent shRNAs against each OTU member were selected and lentiviruses expressing each shRNA was individually packaged following protocols as described.[ 55 ] A673 cells were infected with each individual shRNA expressing lentiviruses for 24 h, recovered for 72 h before 1000 surviving cells from each group were seeded in 96‐well plates in triplicates. 72 h later, MTT assays were performed to determine cell viability.
Sample Preparation for Proteomic Analysis
A673 cells were treated with H2O or 1 µg mL−1 tetracycline (to induce a Tet‐inducible OTUD7A depletion) for 72 h (n = 3 biological replicates per time point). Cells were washed 3 times with ice‐cold phosphate‐buffered saline (PBS), then lysed in 8 m urea, Tris‐HCl (pH 7.6) with protease and phosphatase inhibitors (Bimake). Lysates were reduced with 5 × 10−3 m dithiothreitol (DTT), alkylated with 15 × 10−3 m iodoacetamide, then subjected to digestion with LysC (Wako) for 2 h, then trypsin (Promega) overnight at 37 °C at a 1:50 enzyme:protein ratio. The resulting peptide samples were acidified, desalted using Thermo desalting spin columns, then the eluates were dried via vacuum centrifugation. Peptide concentration was determined using Pierce Quantitative Colorimetric Peptide Assay. 40 µg of each sample was reconstituted with 50 × 10−3 m 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) pH 8.5, then individually labeled with 60 µg of TMT (tandem mass tag) 10plex reagent (Thermo Fisher) for 1 h at room temperature. Labeling efficiency was evaluated by liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis of a pooled test mix. Samples were quenched with 50% hydroxylamine to a final concentration of 0.4%. Labeled peptide samples were pooled, desalted using Thermo desalting spin column, and dried via vacuum centrifugation. The dried TMT‐labeled sample was fractionated using high pH reversed phase HPLC.[ 60 ] Briefly, the sample was offline fractionated over a 90 min run, into 96 fractions by high pH reverse‐phase HPLC (Agilent 1260) using an Agilent Zorbax 300 Extend‐C18 column (3.5 µm, 4.6 × 250 mm) with mobile phase A containing 4.5 × 10−3 m ammonium formate (pH 10) in 2% v/v LC–MS grade acetonitrile, and mobile phase B containing 4.5 × 10−3 m ammonium formate (pH 10) in 90% v/v LC–MS grade acetonitrile. The 96 resulting fractions were then pooled in a noncontinuous manner into 24 fractions. The 24 fractions were dried via vacuum centrifugation.
LC/MS/MS Analyses
The 24 fractions were analyzed by LC/MS/MS using an Easy nLC 1200 coupled to a QExactive HF mass spectrometer (Thermo Scientific). Samples were injected onto an EASY‐Spray PepMap C18 column (75 µm id × 25 cm, 2 µm particle size) (Thermo Scientific) and separated over a 150 min method. The gradient for separation consisted of 5–50% mobile phase B at a 250 nL min−1 flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in 80% acetonitrile (ACN). The QExactive HF was operated in data‐dependent mode where the 15 most intense precursors were selected for subsequent higher‐energy collisional dissociation (HCD) fragmentation. Resolution for the precursor scan (m/z 350–1600) was set to 60 000 with a target value of 3 × 106 ions and a maximum injection time of 100 ms. MS/MS scan resolution was set to 60 000 with a target value of 1 × 105 ions and a maximum injection time of 100 ms. Fixed first mass was set to 110 m/z and the normalized collision energy was set to 32% for HCD. Dynamic exclusion was set to 30 s, peptide match was set to preferred, and precursors with unknown charge or a charge state of 1 and ≥8 were excluded.
Proteomics Data Analyses
Raw data files were processed using MaxQuant v1.6.12.0, set to “reporter ion MS2” with “10plex TMT.” Peak lists were searched against a reviewed Uniprot human database (downloaded Feb 2020 containing 20350 sequences), appended with EWS–FLI1 sequences and a common contaminants database, using Andromeda within MaxQuant. All fractions were searched with up to three missed trypsin cleavage sites, fixed carbamidomethylation (C) modification, dynamic oxidation (M), deamidation (NQ), and acetylation (N‐terminal) modifications. Peptide false discovery rate was set to 1%. Data were further analyzed in Perseus and Microsoft Excel. Each reporter ion channel was summed across all quantified proteins and mean‐normalized assuming equal protein loading of all samples.
RNA Extraction and Real‐Time Quantitative Reverse Transcription PCR (qRT‐PCR)
Cells were isolated by dissociation with 0.05% Trypsin, followed by media quenching. The cells were spun down at 300× rcf for 5 min. The media was aspirated, the pellet was suspended with 1× PBS, and then the cells were spun down again. The PBS was aspirated. RNA extraction was performed with a RNA extraction kit (BioBasic BS584). The final elution step was done with 50 µL of RNAse‐free water. The relative enrichment of mRNA was quantified with the NanoDrop OneC (Thermo Fisher Scientific). Three biological replicates were performed for RNA extraction. Quartile analysis was done to exclude outliers and significance was determined by one‐way analysis of variance (ANOVA) tests.
Cell Viability (MTT) Assays
2000 indicated cells were seeded in each well of 96‐well plates for MTT assays to monitor cell viability at indicated time periods using a method adapted from https://www.thermofisher.com/us/en/home/references/protocols/cell‐culture/mtt‐assay‐protocol/vybrant‐mtt‐cell‐proliferation‐assay‐kit.html. Briefly, at indicated time points post‐cell seeding, 10 µL MTT solution was added into each well and incubated in the culture incubator (37 °C with 5% CO2) for 4 h. Then, medium was removed and 100 µL dimethyl sulfoxide (DMSO) was added into each well to dissolve the formazan crystal and incubated for 10 min at 37 °C. After thorough mixing, absorbance at 540 nm was measured using the BioTek Cytation 5 Cell Imaging reader.
Colony Formation Assays
Indicated cells were seeded into 6‐well plates (300 or 600 cells per well) or 6 cm dishes (500 or 1000 cells per dish) and cultured in 37 °C incubator with 5% CO2 for ≈14 days (as indicated in figure legends) until formation of visible colonies. 7Ai was refresed every other day. Colonies were washed with 1× PBS and fixed with 10% acetic acid/10% methanol for 30 min, stained with 20% acidic acid/10% methanol with 0.1% crystal violet until colonies were visibly stained. Colonies were then washed by tap water and air‐dried. Colony numbers were manually counted. At least two independent experiments were performed to generate the error bars.
Soft Agar Assays
The anchorage‐independent cell growth assays were performed as described previously.[ 53 ] Briefly, the assays were preformed using 6‐well plates where the solid medium consisted of two layers. The bottom layer contained 0.8% noble agar and the top layer contained 0.4% agar suspended with 3 × 104 or indicated number of cells. 500 µL complete DMEM medium with 10% FBS was added every 4 days. About 4 weeks later, the cells were stained with iodonitrotetrazolium chloride (1 mg mL−1) (Sigma I10406) overnight for colony visualization and counting. At least two independent experiments were performed to generate the error bar.
Mouse Xenograft Assays
All mouse work was reviewed and approved by UNC Institutional Animal Care and Use Committee under IACUC#19‐031. Mouse xenograft assays were performed as described previously.[ 53 , 54 ] Briefly, for mouse xenograft experiments, 2.5 × 106 parental or EWS–FLI1‐3A knock‐in A673 cells, or A673/MHH‐ES‐1 teton‐shOTUD7A cellsas indicated were mixed 1:1 with matrigel (Corning 354230) and injected into the flank of indicated female nude mice (NCRNU‐M‐M from UNC Animal Facility, 4 weeks old). Six days postinjection, when the tumors were established, 2% sucrose with or without 1 µg mL−1 tetracycline was supplied in water for mice and refreshed every 5 days. Tumor size was measured every two days with a digital caliper, and the tumor volume was determined with the formula: L × W 2 × 0.52, where L is the longest diameter and W is the shortest diameter. After 22 days, mice were sacrificed, and tumors were dissected and weighed.
For 7Ai treatment in nude mice, 2.5 × 106 A673 cells were mixed 1:1 with matrigel (Corning 354230) and injected into the flank of indicated female nude mice (NCRNU‐M‐M from UNC Animal Facility, 4 weeks old). Seven days postinjection, when the tumors were established, 100 µL 7Ai (25 mg kg−1, dissolved in ethanol followed by the addition of sunflower oil and then sonicated at 4 °C in a water bath sonicator until 7Ai was completely dissolved) was given to mice through IP injections and this treatment was repeated every 3 days.
Transwell Assays
1 × 105 cells were plated in an 8.0 mm, 24‐well plate chamber insert (Corning Life Sciences, catalog no. 3422) with serum free DMEM medium at the top of the insert and the same medium containing 20% FBS at the bottom of the insert. Cells were incubated for 24 h and fixed with 4% paraformaldehyde for 15 min. After washing with PBS, cells at the top of the insert were scraped with a cotton swab. Cells adherent to the bottom were stained with 0.5% crystal violet blue for 60 min and then washed with double‐distilled H2O. The positively stained cells were examined under the microscope.
IHC
Freshly dissected xenografted tumors were immediately fixed in 10% formalin for 2 days before transferring to 80% ethanol for one day to prepare tumor blocks. 4 µm sections were cut from each of the tumor blocks by UNC TPL facility and used for IHC study. IHC was performed as described previously.[ 53 ] Normal human tissue TMA was purchased from UNC TPL facility. Human Ewing sarcoma tissues were obtained postmortem and fixed in 10% formalin and embedded in paraffin. This protocol was approved by UNC Office of Human Research Ethics under institutional review board (IRB): 20‐3178.
ITC
ITC measurements were performed using a MicroCal auto‐iTC200 calorimeter (MicroCal, LLC) as described previously.[ 61 ] Briefly, His‐tagged OTU domain of OTUD7A proteins were purified using BL21 strain upon induced by 0.3 × 10−3 m isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) when OD600 around 0.6 overnight at 16 °C. Then, purified human His‐tagged OTU domain of OTUD7A was dialyzed against 50 × 10−3 m HEPES buffer (pH 7.2) with 100 × 10−3 m NaCl for overnight at 4 °C. The concentration of the protein was determined by band intensity in a gel‐cod staining SDS‐PAGE gel. The ITC assay was carried out at 37 °C. The dialyzed His‐OTUD7A‐OTU proteins were diluted to 40 × 10−6 m in the dialysis buffer containing 4% DMSO. Then, 400 × 10−6 m 7Ai, dissolved in the same dialysis buffer with 4% DMSO (50 × 10−3 m HEPES buffer (pH 7.2) with 100 × 10−3 m NaCl) was injected into 0.4 mL of His‐OTUD7A‐OTU protein in the chamber for every 180 s. The dissociation constants and thermodynamic parameters were determined by using the embedded software package Origin7 (Microcal).
OTUD7A OTU Domain Homology Model
A homology model for OTUD7A was built on the homologous protein OTUD7B, for which the catalytic OTU domain was crystallized both in complex with a diubiquitin, monoubiquitin, and as an apo structure (PDB codes 5LRV, 5LRW, 5LRU, respectively.[ 62 ] The paper reported three sites of interest, the distal (S1) and proximal (S1ʹ) ubiquitin‐binding sites, as well as the catalytic center. Substrate recruitment was shown to be primarily driven by the S1 site. In the interest of selectivity, it was desirable to identify a small molecule binding site that was different in the two proteins. In OTUD7B, both S1ʹ and the catalytic site underwent significant conformational changes upon substrate binding. Furthermore, the region around the catalytic site was highly conserved between the two homologs. S1, on the other hand, had a binding pocket formed in part by two alpha helices connected by a loop that appeared suitable for small molecule inhibitors and showed relatively small changes between the three different OTUD7B crystal structures. In OTUD7A, this loop (Q257‐W263) was shorter, suggesting a different shape and smaller size of S1 in the OTUD7A, providing for potential ligand selectivity.
Based on these observations, the S1 region of the OTUD7A model was selected for the virtual screening campaign. The monoubiquitin‐bound OTUD7B crystal structure 5LRW (after removal of the monoubiquitin) seemed the best suited as a modeling template, as it was both in a ligand‐bound conformation and had density for all the residues surrounding the area of interest. The homology model of OTUD7A was built using ICM Pro v3.8.7 (Molsoft L.L.C.). Like in the experimental OTUD7B structures, the long, unstructured V‐loop (residues 276–300) was replaced by QPG.
AI‐Based Small Molecule Virtual Screen
The virtual screen was carried out using the AtomNet neural network, the first deep convolutional neural network for structure‐based drug design.[ 47 , 48 ] A single global AtomNet model was deployed to predict binding affinity of small molecules to a target protein. The model was trained with experimental K i, K d, and the half maximal inhibitory concentration (IC50) values of several million small molecules and protein structures spanning several thousand different proteins, curated from both public databases and proprietary sources. Because AtomNet was a global model, it could be applied to novel binding sites with no known ligands, a prerequisite to most target‐specific machine‐learning models. Another advantage of using a single global model in prospective predictions was that it helped prevent the so‐called model overfitting. The following three‐step procedure was applied to train AtomNet models. The first step was to define the binding site on a given protein structure using a flooding algorithm[ 63 ] based on an initial seed. The initial starting point of the flooding algorithm might be determined using either a bound ligand annotated in the PDB database or crucial residues as revealed by mutagenesis studies, or identification of catalytic motifs previously reported. The second step was to shift the coordinates of the protein–ligand cocomplex to a 3D Cartesian system with an origin at the center‐of‐mass of the binding site. In order to prevent the neural network from memorizing a preferred orientation of the protein structure, data augmentation was then performed by randomly rotating and translating the protein structure around the center‐of‐mass of the binding site. The third step was to sample the conformations or poses of a small molecule ligand within the binding site pocket. For a given ligand, an ensemble of poses were generated, and each of these poses represented a putative cocomplex with the protein. Each generated cocomplex was then rasterized into a fixed‐size regular 3D grid, where the values at each grid point represented the structural features that were present at each point. Similar to a photo pixel containing three separate channels representing the presence of red, green, and blue colors, the grid points represented the presence of different atom types. These grids served as the input to a convolutional neural network, and defined the receptive field of the network. A network architecture of a 30 × 30 × 30 grid with 1 Å spacing was used for the input layer, followed by five convolutional layers of 32 × 33, 64 × 33, 64 × 33, 64 × 33, 64 × 23 (number of filters × filter dimension), and a fully connected layer with 256 ReLU hidden units. The scores for each pose in the ensemble were combined through a weighted Boltzmann averaging to produce a final score. These scores were compared against the experimentally measured pK i or pIC50 (converted from K i or IC50) of the protein and ligand pair, and the weights of the neural network were adjusted to reduce the error between the predicted and experimentally measured affinity using a mean‐square‐error loss function. Training was done using the ADAM[ 64 ] adaptive learning method, the backpropagation algorithm, and minibatches with 64 examples per gradient step.
AtomNet could take any form of 3D protein structures determined by experimental methods including crystallography, NMR, and cryogenic electron microscopy (cryo‐EM) published in PDB format. In case of no available experimental protein structure of the target, the amino acid sequence of the target could be used to build a homology model using the most homologous protein structure as a template as described above. The binding site was identified surrounded by residues R249, W250, R251, W252, Q253, Q254, T255, Q256, Q257, K259, E261, R265, E266, W267, E269, L270, L273, E304, E305, F306, H307, P339, F340, F400 on the OTUD7A homology model.
The Mcule small‐molecule library version v20171018, containing 5 648 837 small organic molecules for drug discovery purchasable from the chemical vendor Mcule, was screened. The library in simplified molecular‐input line‐entry system (SMILES) format was downloaded from Mcule's website (https://mcule.com/). Every compound in the library was pushed through a standardization process including the removal of salts, isotopes, and ions, and conversion to neutral form; conversion of functional groups and aromatic rings to consistent representations. Filters were then applied on some molecular properties including molecular weight between 100 and 700 Da, total number of chiral centers in a molecule ≤ 6, total number of atoms in a molecule ≤ 60, total number of rotatable bonds ≤ 15, and only molecules containing C, N, S, H, O, P, B, halogens were allowed. Other filters such as toxicophores, Eli Lilly's MedChem Rules,[ 65 ] and pan‐assay interference compounds (PAINS) were also applied to remove compounds with undesirable substructures, resulting in a filtered library of 4 025 533 compounds. For each small molecule, a set of 64 poses within the binding site was generated. Each of these poses was scored by the trained model, and the molecules were ranked by their scores. The top 5000 ranking compounds were examined from which a set of 89 compounds containing diverse chemical scaffolds was selected. The selected compounds were sourced from Mcule. Of 88 available compounds, 73 passed quality control with 62 compounds having at least 90% purity measured by LC–MS. Eleven compounds had purity between 78% and 90%. The compound 7Ai had a clean mass spectrum and was at 86.9% purity. After being identified as a hit, compound 7Ai was purified by HPLC and assayed again to confirm its activity.
Activity Assay for 73 Predicted Compounds from AI‐Based Virtual Screen
Each compound was dissolved in DMSO with a concentration of 10 × 10−3 m. The compound samples were assayed in a blinded way (chemical identities unknown to the lab researcher, with two negative control samples containing pure DMSO mixed in). A673 and SK‐N‐MC cells were splitted into 6‐well plates and treated with each compound with a final compound concentration of 10 × 10−6 m for 12 h. Cells were harvested and subjected to western blot analyses.
Statistical Analysis
Statistical analyses were performed using the SPSS 11.5 Statistical Software. p ≤ 0.05 was considered statistically significant. The results were shown as means ± standard deviation (SD) from at least two or three independent experiments as indicated in figure legends. Differences between control and experimental conditions were evaluated by one‐way ANOVA.
Conflict of Interest
Christian Laggner and Kong T. Nguyen are current employees of Atomwise Inc.
Author Contributions
J.C. and Y.J. are co‐first authors. S.S., J.C., and Y.J. contributed equally to this work. I.J.D. and P.L. conceived the project. S.S., J.C., C.L., K.T.N., I.J.D., and P.L. designed experiments. S.S., J.C., Y.J., Y.W., T.V., J.Z., C.L., K.T.N., Z.Z., A.W.P., N.K.B., and L.E.H. performed experiments. S.S., C.L., K.T.N., L.E.H., I.J.D., and P.L. analyzed the data. I.J.D. and P.L. supervised this study. S.S., I.J.D., and P.L. wrote the paper.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
The authors thank Liu and Davis lab members for critical reading of the paper and helpful discussions. The authors also thank Dr. Xiaodong Wang (UNC Chapel Hill) for purifying 7Ai compounds by HPLC. The authors also thank Dr. Xianming Tan (Lineberger Comprehensive Cancer Center at UNC Chapel Hill) for help with statistical analyses. The authors thank Kaixin Liang (UNC Chapel Hill) for her help on this project during her rotation. This work was supported by the NIH Grants (No. R21CA234979, P.L. and I.J.D., and No. R01CA244825, P.L.), Atomwise AIMS Award (No. A17‐034, P.L.), V Scholar Grant (No. V2018‐009, P.L.), Breast Cancer Alliance Young Investigator Grant (P.L.), Gabrielle's Angel Foundation Medical Research Award (P.L.), UNC Lineberger Comprehensive Cancer Center Development Award (P.L.), and UNC University Cancer Research Fund (P.L.). This research was based in part upon work conducted using the UNC Proteomics Core Facility, which was supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The authors also acknowledge the help of Dr. Ashutosh Tripathy, Director of the Macromolecular Interactions Facility in carrying out the ITC experiments and analyzing data. The facility was partly supported by the National Cancer Institute Award No. P30CA016086. The content was solely the responsibility of the authors and did not necessarily represent the official views of the National Institutes of Health.
Su S., Chen J., Jiang Y., Wang Y., Vital T., Zhang J., Laggner C., Nguyen K. T., Zhu Z., Prevatte A. W., Barker N. K., Herring L. E., Davis I. J., Liu P., SPOP and OTUD7A Control EWS–FLI1 Protein Stability to Govern Ewing Sarcoma Growth. Adv. Sci. 2021, 8, 2004846. 10.1002/advs.202004846
Contributor Information
Ian J. Davis, Email: ian_davis@med.unc.edu.
Pengda Liu, Email: pengda_liu@med.unc.edu.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Delattre O., Zucman J., Plougastel B., Desmaze C., Melot T., Peter M., Kovar H., Joubert I., de Jong P., Rouleau G., Aurias A., Thomas G., Nature 1992, 359, 162. [DOI] [PubMed] [Google Scholar]
- 2. Patel M., Simon J. M., Iglesia M. D., Wu S. B., McFadden A. W., Lieb J. D., Davis I. J., Genome Res. 2012, 22, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gangwal K., Sankar S., Hollenhorst P. C., Kinsey M., Haroldsen S. C., Shah A. A., Boucher K. M., Watkins W. S., Jorde L. B., Graves B. J., Graves S. L., Proc. Natl. Acad. Sci. USA 2008, 105, 10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson K. M., Mahler N. R., Saund R. S., Theisen E. R., Taslim C., Callender N. W., Crow J. C., Miller K. R., Lessnick S. L., Proc. Natl. Acad. Sci. USA 2017, 114, 9870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boulay G., Sandoval G. J., Riggi N., Iyer S., Buisson R., Naigles B., Awad M. E., Rengarajan S., Volorio A., McBride M. J., Broye L. C., Zou L., Stamenkovic I., Kadoch C., Rivera M. N., Cell 2017, 171, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L., Chansky H. A., Hickstein D. D., J. Biol. Chem. 2000, 275, 37612. [DOI] [PubMed] [Google Scholar]
- 7. Ramakrishnan R., Fujimura Y., Zou J. P., Liu F., Lee L., Rao V. N., Reddy E. S., Oncogene 2004, 23, 7087. [DOI] [PubMed] [Google Scholar]
- 8. Toretsky J. A., Erkizan V., Levenson A., Abaan O. D., Parvin J. D., Cripe T. P., Rice A. M., Lee S. B., Uren A., Cancer Res. 2006, 66, 5574. [DOI] [PubMed] [Google Scholar]
- 9. Cidre‐Aranaz F., Alonso J., Front. Oncol. 2015, 5, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Franzetti G. A., Laud‐Duval K., van der Ent W., Brisac A., Irondelle M., Aubert S., Dirksen U., Bouvier C., de Pinieux G., Snaar‐Jagalska E., Chavrier P., Delattre O., Oncogene 2017, 36, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Katschnig A. M., Kauer M. O., Schwentner R., Tomazou E. M., Mutz C. N., Linder M., Sibilia M., Alonso J., Aryee D. N. T., Kovar H., Oncogene 2017, 36, 5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uren A., Toretsky J. A., Future Oncol. 2005, 1, 521. [DOI] [PubMed] [Google Scholar]
- 13. Selvanathan S. P., Moseley E., Graham G. T., Jessen K., Lannutti B., Üren A., Toretsky J. A., Cancer Res. 2017, 77, 694. [Google Scholar]
- 14. Caropreso V., Darvishi E., Turbyville T. J., Ratnayake R., Grohar P. J., McMahon J. B., Woldemichael G. M., J. Biol. Chem. 2016, 291, 10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pattenden S. G., Simon J. M., Wali A., Jayakody C. N., Troutman J., McFadden A. W., Wooten J., Wood C. C., Frye S. V., Janzen W. P., Davis I. J., Proc. Natl. Acad. Sci. USA 2016, 113, 3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gierisch M. E., Pfistner F., Lopez‐Garcia L. A., Harder L., Schafer B. W., Niggli F. K., J. Biol. Chem. 2016, 291, 26922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elzi D. J., Song M., Hakala K., Weintraub S. T., Shiio Y., J. Proteome Res. 2014, 13, 3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stolte B., Iniguez A. B., Dharia N. V., Robichaud A. L., Conway A. S., Morgan A. M., Alexe G., Schauer N. J., Liu X., Bird G. H., Tsherniak A., Vazquez F., Buhrlage S. J., Walensky L. D., Stegmaier K., J. Exp. Med. 2018, 215, 2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gorthi A., Romero J. C., Loranc E., Cao L., Lawrence L. A., Goodale E., Iniguez A. B., Bernard X., Masamsetti V. P., Roston S., Lawlor E. R., Toretsky J. A., Stegmaier K., Lessnick S. L., Chen Y., Bishop A. J. R., Nature 2018, 555, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Z., Kang W., You Y., Pang J., Ren H., Suo Z., Liu H., Zheng Y., Front. Pharmacol. 2019, 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coyne E. S., Bedard N., Wykes L., Stretch C., Jammoul S., Li S., Zhang K., Sladek R. S., Bathe O. F., Jagoe R. T., Posner B. I., Wing S. S., Endocrinology 2018, 159, 2966. [DOI] [PubMed] [Google Scholar]
- 22. Jin S., Tian S., Chen Y., Zhang C., Xie W., Xia X., Cui J., Wang R.‐F., EMBO J. 2016, 35, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Altun M., Zhao B., Velasco K., Liu H., Hassink G., Paschke J., Pereira T., Lindsten K., J. Biol. Chem. 2012, 287, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lei C. Q., Wu X., Zhong X., Jiang L., Zhong B., Shu H.‐B., J. Immunol. 2019, 203, 259. [DOI] [PubMed] [Google Scholar]
- 25. Turnbull A. P., Ioannidis S., Krajewski W. W., Pinto‐Fernandez A., Heride C., Martin A. C. L., Tonkin L. M., Townsend E. C., Buker S. M., Lancia D. R., Caravella J. A., Toms A. V., Charlton T. M., Lahdenranta J., Wilker E., Follows B. C., Evans N. J., Stead L., Alli C., Zarayskiy V. V., Talbot A. C., Buckmelter A. J., Wang M., McKinnon C. L., Saab F., McGouran J. F., Century H., Gersch M., Pittman M. S., Marshall C. G., Raynham T. M., Simcox M., Stewart L. M. D., McLoughlin S. B., Escobedo J. A., Bair K. W., Dinsmore C. J., Hammonds T. R., Kim S., Urbe S., Clague M. J., Kessler B. M., Komander D., Nature 2017, 550, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kategaya L., Di Lello P., Rouge L., Pastor R., Clark K. R., Drummond J., Kleinheinz T., Lin E., Upton J. P., Prakash S., Heideker J., McCleland M., Ritorto M. S., Alessi D. R., Trost M., Bainbridge T. W., Kwok M. C. M., Ma T. P., Stiffler Z., Brasher B., Tang Y., Jaishankar P., Hearn B. R., Renslo A. R., Arkin M. R., Cohen F., Yu K., Peale F., Gnad F., Chang M. T., Klijn C., Blackwood E., Martin S. E., Forrest W. F., Ernst J. A., Ndubaku C., Wang X., Beresini M. H., Tsui V., Schwerdtfeger C., Blake R. A., Murray J., Maurer T., Wertz I. E., Nature 2017, 550, 534. [DOI] [PubMed] [Google Scholar]
- 27. Gerald Gavory C. O. D., Rozycka E., Dossang A., Henderson A., Hughes C., Miel H., Barker O., Costa J., Hewitt P., McFarland M., Proctor L., Harrison T., Cancer Res. 2017, AM2017‐1181. [Google Scholar]
- 28. May W. A., Lessnick S. L., Braun B. S., Klemsz M., Lewis B. C., Lunsford L. B., Hromas R., Denny C. T., Mol. Cell. Biol. 1993, 13, 7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dai X., Gan W., Li X., Wang S., Zhang W., Huang L., Liu S., Zhong Q., Guo J., Zhang J., Chen T., Shimizu K., Beca F., Blattner M., Vasudevan D., Buckley D. L., Qi J., Buser L., Liu P., Inuzuka H., Beck A. H., Wang L., Wild P. J., Garraway L. A., Rubin M. A., Barbieri C. E., Wong K. K., Muthuswamy S. K., Huang J., Chen Y., Bradner J. E., Wei W., Nat. Med. 2017, 23, 1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang P., Wang D., Zhao Y., Ren S., Gao K., Ye Z., Wang S., Pan C. W., Zhu Y., Yan Y., Yang Y., Wu D., He Y., Zhang J., Lu D., Liu X., Yu L., Zhao S., Li Y., Lin D., Wang Y., Wang L., Chen Y., Sun Y., Wang C., Huang H., Nat. Med. 2017, 23, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janouskova H., El Tekle G., Bellini E., Udeshi N. D., Rinaldi A., Ulbricht A., Bernasocchi T., Civenni G., Losa M., Svinkina T., Bielski C. M., Kryukov G. V., Cascione L., Napoli S., Enchev R. I., Mutch D. G., Carney M. E., Berchuck A., Winterhoff B. J. N., Broaddus R. R., Schraml P., Moch H., Bertoni F., Catapano C. V., Peter M., Carr S. A., Garraway L. A., Wild P. J., Theurillat J. P., Nat. Med. 2017, 23, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gollavilli P. N., Pawar A., Wilder‐Romans K., Natesan R., Engelke C. G., Dommeti V. L., Krishnamurthy P. M., Nallasivam A., Apel I. J., Xu T., Qin Z. S., Feng F. Y., Asangani I. A., Cancer Res. 2018, 78, 4760. [DOI] [PubMed] [Google Scholar]
- 33. Gan W., Dai X., Lunardi A., Li Z., Inuzuka H., Liu P., Varmeh S., Zhang J., Cheng L., Sun Y., Asara J. M., Beck A. H., Huang J., Pandolfi P. P., Wei W., Mol. Cell 2015, 59, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kronke J., Fink E. C., Hollenbach P. W., MacBeth K. J., Hurst S. N., Udeshi N. D., Chamberlain P. P., Mani D. R., Man H. W., Gandhi A. K., Svinkina T., Schneider R. K., McConkey M., Jaras M., Griffiths E., Wetzler M., Bullinger L., Cathers B. E., Carr S. A., Chopra R., Ebert B. L., Nature 2015, 523, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wimuttisuk W., Singer J. D., Mol. Biol. Cell 2007, 18, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang L., Peng S., Dai X., Gan W., Nie X., Wei W., Hu G., Guo J., Cancer Lett. 2017, 390, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schittek B., Sinnberg T., Mol. Cancer 2014, 13, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Komander D., Clague M. J., Urbe S., Nat. Rev. Mol. Cell Biol. 2009, 10, 550. [DOI] [PubMed] [Google Scholar]
- 39. Mevissen T. E., Hospenthal M. K., Geurink P. P., Elliott P. R., Akutsu M., Arnaudo N., Ekkebus R., Kulathu Y., Wauer T., El Oualid F., Freund S. M., Ovaa H., Komander D., Cell 2013, 154, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang B., Jie Z., Joo D., Ordureau A., Liu P., Gan W., Guo J., Zhang J., North B. J., Dai X., Cheng X., Bian X., Zhang L., Harper J. W., Sun S. C., Wei W., Nature 2017, 545, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu H., Brittain G. C., Chang J. H., Puebla‐Osorio N., Jin J., Zal A., Xiao Y., Cheng X., Chang M., Fu Y. X., Zal T., Zhu C., Sun S. C., Nature 2013, 494, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bonacci T., Suzuki A., Grant G. D., Stanley N., Cook J. G., Brown N. G., MJ ., EMBO J. 2018, 37, e98701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Riggi N., Suva M. L., Suva D., Cironi L., Provero P., Tercier S., Joseph J. M., Stehle J. C., Baumer K., Kindler V., Stamenkovic I., Cancer Res. 2008, 68, 2176. [DOI] [PubMed] [Google Scholar]
- 44. Hancock J. D., Lessnick S. L., Cell Cycle 2008, 7, 250. [DOI] [PubMed] [Google Scholar]
- 45. Tanabe Y., Suehara Y., Kohsaka S., Hayashi T., Akaike K., Mukaihara K., Kurihara T., Kim Y., Okubo T., Ishii M., Kazuno S., Kaneko K., Saito T., Oncotarget 2018, 9, 14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brohl A. S., Solomon D. A., Chang W., Wang J., Song Y., Sindiri S., Patidar R., Hurd L., Chen L., Shern J. F., Liao H., Wen X., Gerard J., Kim J. S., Lopez Guerrero J. A., Machado I., Wai D. H., Picci P., Triche T., Horvai A. E., Miettinen M., Wei J. S., Catchpool D., Llombart‐Bosch A., Waldman T., Khan J., PLoS Genet. 2014, 10, e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Izhar Wallach M. D., Heifets A., arXiv: 2015, p. 1510.02855.
- 48. Hsieh C. H., Li L., Vanhauwaert R., Nguyen K. T., Davis M. D., Bu G., Wszolek Z. K., Wang X., Cell Metab. 2019, 30, 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gierisch M. E., Pedot G., Walser F., Lopez‐Garcia L. A., Jaaks P., Niggli F. K., Schafer B. W., Sci. Rep. 2019, 9, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yin J., Chen W., Chao E. S., Soriano S., Wang L., Wang W., Cummock S. E., Tao H., Pang K., Liu Z., Pereira F. A., Samaco R. C., Zoghbi H. Y., Xue M., Schaaf C. P., Am. J. Hum. Genet. 2018, 102, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Uddin M., Unda B. K., Kwan V., Holzapfel N. T., White S. H., Chalil L., Woodbury‐Smith M., Ho K. S., Harward E., Murtaza N., Dave B., Pellecchia G., D'Abate L., Nalpathamkalam T., Lamoureux S., Wei J., Speevak M., Stavropoulos J., Hope K. J., Doble B. W., Nielsen J., Wassman E. R., Scherer S. W., Singh K. K., Am. J. Hum. Genet. 2018, 102, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garret P., Ebstein F., Delplancq G., Dozieres‐Puyravel B., Boughalem A., Auvin S., Duffourd Y., Klafack S., Zieba B. A., Mahmoudi S., Singh K. K., Duplomb L., Thauvin‐Robinet C., Costa J. M., Kruger E., Trost D., Verloes A., Faivre L., Vitobello A., Clin. Genet. 2020, 97, 567. [DOI] [PubMed] [Google Scholar]
- 53. Liu P., Begley M., Michowski W., Inuzuka H., Ginzberg M., Gao D., Tsou P., Gan W., Papa A., Kim B. M., Wan L., Singh A., Zhai B., Yuan M., Wang Z., Gygi S. P., Lee T. H., Lu K. P., Toker A., Pandolfi P. P., Asara J. M., Kirschner M. W., Sicinski P., Cantley L., Wei W., Nature 2014, 508, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu P., Gan W., Chin Y. R., Ogura K., Guo J., Zhang J., Wang B., Blenis J., Cantley L. C., Toker A., Su B., Wei W., Cancer Discovery 2015, 5, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang Y., Zhang Y., Leung J. Y., Fan C., Popov K. I., Su S., Qian J., Wang X., Holtzhausen A., Ubil E., Xiang Y., Davis I., Dokholyan N. V., Wu G., Perou C. M., Kim W. Y., Earp H. S., Liu P., Nat. Commun. 2019, 10, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu P., Gan W., Guo C., Xie A., Gao D., Guo J., Zhang J., Willis N., Su A., Asara J. M., Scully R., Wei W., Mol. Cell 2015, 57, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guo J., Chakraborty A. A., Liu P., Gan W., Zheng X., Inuzuka H., Wang B., Zhang J., Zhang L., Yuan M., Novak J., Cheng J. Q., Toker A., Signoretti S., Zhang Q., Asara J. M., W. G. Kaelin, Jr. , Wei W., Science 2016, 353, 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu P., Gan W., Su S., Hauenstein A. V., Fu T. M., Brasher B., Schwerdtfeger C., Liang A. C., Xu M., Wei W., Sci. Signaling 2018, 11, eaar8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Science 2013, 339, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mertins P., Tang L. C., Krug K., Clark D. J., Gritsenko M. A., Chen L., Clauser K. R., Clauss T. R., Shah P., Gillette M. A., Petyuk V. A., Thomas S. N., Mani D. R., Mundt F., Moore R. J., Hu Y., Zhao R., Schnaubelt M., Keshishian H., Monroe M. E., Zhang Z., Udeshi N. D., Mani D., Davies S. R., Townsend R. R., Chan D. W., Smith R. D., Zhang H., Liu T., Carr S. A., Nat. Protoc. 2018, 13, 1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang Y., Ma Z., Wang Y., Boyer J., Ni G., Cheng L., Su S., Zhang Z., Zhu Z., Qian J., Su L., Zhang Q., Damania B., Liu P., iScience 2020, 23, 101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mevissen T. E. T., Kulathu Y., Mulder M. P. C., Geurink P. P., Maslen S. L., Gersch M., Elliott P. R., Burke J. E., van Tol B. D. M., Akutsu M., Oualid F. E., Kawasaki M., Freund S. M. V., Ovaa H., Komander D., Nature 2016, 538, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hendlich M., Rippmann F., Barnickel G., J. Mol. Graphics Modell. 1997, 15, 359. [DOI] [PubMed] [Google Scholar]
- 64. Diederik P., Kingma J. B., arXiv: 2014, p. 1412.6980.
- 65. Bruns R. F., Watson I. A., J. Med. Chem. 2012, 55, 9763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.