

Abstract
Introduction:
Systemic sclerosis (SSc), also known as scleroderma, is a complex orphan disease characterized by early inflammatory features, vascular hyper-reactivity, and fibrosis of the skin and internal organs. Although substantial progress has been made in the understanding of the pathogenesis of SSc, there is still no disease-modifying drug that could significantly impact the natural history of the disease.
Areas covered:
This review discusses the rationale, preclinical evidence, first clinical evidence and pending issues concerning new promising therapeutic options that are under investigation in SSc. The search strategy was based on PubMed database and clinicaltrial.gov, highlighting recent key pathogenic aspects and phase I or II trials of investigational drugs in SSc.
Expert opinion:
The identification of new molecular entities that potentially impact inflammation and fibrosis may constitute promising options for a disease modifying-agent in SSc. The early combinations of antifibrotic drugs (such as pirfenidone) with immunomodulatory agents (such as mycophenolate mofetil) may also participate to achieve such a goal. A more refined stratification of patients, based on clinical features, molecular signature, and identification of subpopulations with distinct clinical trajectories, may also improve management strategies in the future.
Keywords: Autoimmunity, Fibrosis, Investigational drugs, Macrophages, Myofibroblasts, Scleroderma, Systemic sclerosis, Vasculopathy
1. INTRODUCTION
Systemic sclerosis (SSc) also called scleroderma, is a complex orphan disease characterized by vascular manifestations, early inflammatory features and fibrosis of the skin and internal organs such as lung (1–4). SSc is the rheumatic disease with the highest case-specific mortality and has a major impact on quality of life (4). Although substantial progress has been made in the understanding of the pathogenesis of SSc there is still no disease-modifying drug that could significantly impact the natural history of the disease as a whole (5–7).
1.1. Overview of the pathogenesis of SSc
The pathogenesis of SSc involves a classical triad of key mechanisms that combines endothelial dysfunction and apoptosis of endothelial cells (8), uncontrolled activation of adaptive and innate immunity (notably including M1 inflammatory and M2 pro-fibrotic macrophages) (9), and over-production of extracellular matrix (ECM) components by chronically activated myofibroblasts leading to the build-up of a rigid and fibrotic extracellular matrix in multiple organs that disrupts their function (10). The main cytokines and cellular subsets are presented in Figure 1 and the interactions between pro-fibrotic pathways are illustrated in Figure 2.
Figure 1: Main cellular types, pathogenic mechanisms and hypotheses in systemic sclerosis.
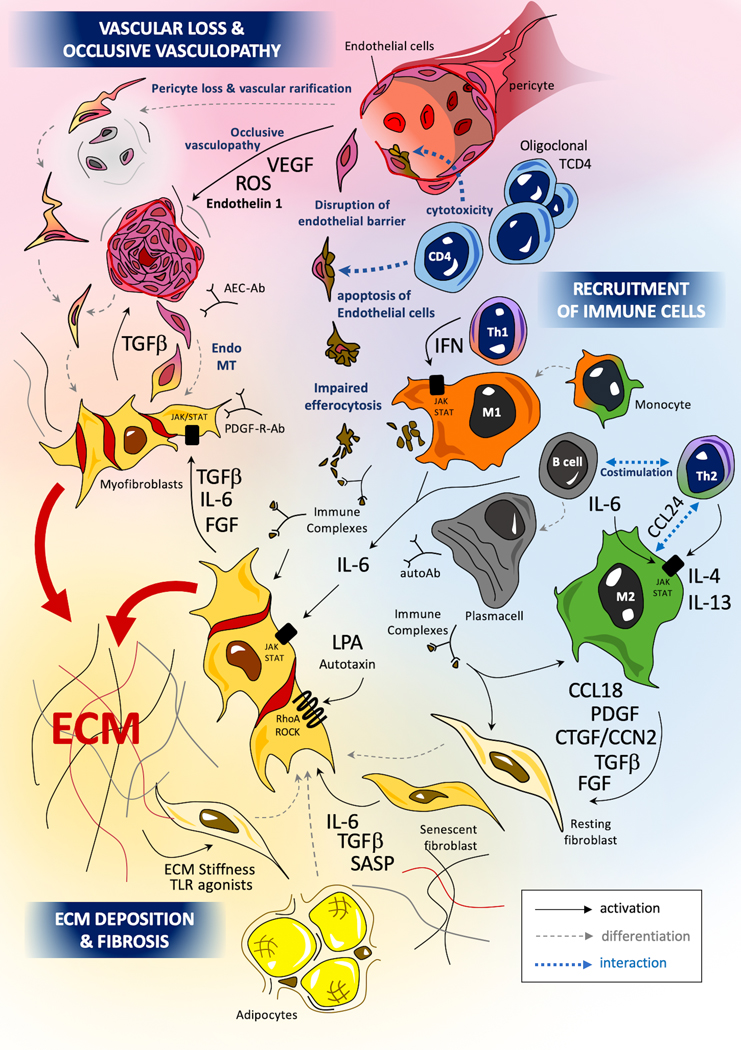
The pathogenesis of SSc involves 3 main mechanisms: occlusive microangiopathy, early inflammatory processes and uncontrolled extra-cellular matrix (ECM) production with resultant fibrosis. Recent studies highlight the role of oligoclonal cytotoxic T-CD4+, driving the apoptosis of endothelial cell (EC) (175,176). Innate immunity, and notably monocytes and macrophages, play a key role in the pathogenesis of SSc (177). Macrophages can adopt various activation profiles depending on their surrounding micro-environment. Interferon (IFN) type II signaling, involving JAK1/TYK2/STAT-1 or TLR-4 agonists induce a classical M1 pro-inflammatory polarization. Th2 cytokines such as IL-4 or IL-13 can induce an alternative profibrotic M2 activation through a STAT3/6 dependent signaling (181): IL-6 also potentiates M2 polarization, notably through the up-regulation of the IL-4 receptor (182). A concomitant excess of CD163highM2 and M1 macrophages has been identified in skin tissues of SSc patients (183) (9). SSc-macrophages show impaired capacities efferocytosis apoptotic cells (phagocytosis of apoptotic cells) with the potential release of internuclear components from these un-eliminated cell debris. The impact of immune complexes composed by autoantibodies and intra-nuclear proteins (topoisomerase, centromere proteins) may also participate to macrophage and fibroblast activation. Myofibroblasts are the major effectors of fibrosis. Myofibroblasts in SSc originate from a variety of tissue-resident mesenchymal progenitor cell types, including fibroblasts, pericytes, microvascular endothelial cells and vascular pre-adipocytes (12). The trans-differentiation of resting fibroblasts and other progenitor cells into pro-fibrotic and inflammatory myofibroblasts is driven by canonical smad-dependent (smad2/3 and 4) and non-canonical smad-independent tumoral growth factor (TGF)-β signaling. Activated myofibroblasts also produce profibrotic mediators such as IL-6 or connective tissue growth factor (CTGF)/CCN2, leading to an autocrine profibrotic pathogenic loop maintaining sustained cellular activation. IL-6 mediates its effects through JAK1/2/TYK2 with subsequent phosphorylation of STAT3 (predominantly) and STAT 1. STAT3 notably promotes the production of key ECM components such as col1a1, col1a2, and profibrotic markers such as CTGF/CCN2 (10). CTGF/CCN2 exerts profibrotic properties notably as a co-factor of TGF-β signaling. CTGF/CCN2 can interact with specific receptors (such as integrins or lipoprotein receptor-related proteins), ECM proteins (such as fibronectin or perlecan) and growth-factors (such as VEGF and TGF-β), with subsequent activation of fibroblast proliferation and myofibroblasts activation (184,185). Uncontrolled production of extra-cellular ECM components such as collagens, tenascin C or fibronectin can in turn activate myofibroblasts either through a direct process involving innate immune sensors such as TLR-4, or through an indirect activation notably depending on mechano-sensing of increased matrix stiffness by integrins (23,24).
IFN= interferon, endoMT=endothelial to mesenchymal transition, Autoab=autoantibodies, PDGF-R-Ab= autoantibodies with agonist effects on PDGF-Receptor, AEC-ab=anti-endothelial cell antibodies, notably including anti endothelin-receptor antibodies with agonists properties, ROS=reactive oxygen species
Figure 2: Non approved pharmacological targets and interaction of selected profibrotic pathways in SSc myofibroblasts that are currently being evaluated in clinical trials.

Latent TGF-β can notably be activated by integrins (notably from the αV class) or thrombospondins. In its active form, TGF-β can interact with a specific heterodimeric receptor (TGF-β-R-I and -II). Two TGF-β-dependent signalization pathways are described: a canonical pathway involving the interaction of TGF-β-R-I with smad 2/3 and 4; and a non-canonical pathway, Smad independent, that notably involves (but is not limited to) c-Abl, TAK1, p38, JNK, SRC, RhoA/ROCK and JAK2-STAT-3. TGF-β1 also increases SSc-related oxidative stress notably through the up-regulation of NADPH oxidase (NOX) 4. This upregulation of NOX4 by TGF-β1 is smad3 dependent and results in increased collagen type I, alpha-SMA and fibronectin 1 gene expression in dermal fibroblasts. These effects are suppressed by pan-NOX inhibitors such as diphenyleneidonium (not represented) or specific NOX1/4 inhibitor such as GKT-137831 (not represented), highlighting that NOX4 may constitute a relevant therapeutic target (152,186). IL-6 mediates its effects through its receptor (composed of IL-6Ra or the soluble form of IL-6R in association with a 130kDA signaling transducer (gp130)) that activates JAK1/2/TYK2 with subsequent phosphorylation of STAT3 (predominantly) and STAT 1. STAT-3 acts as a key integrator of profibrotic signals. STAT3 notably promotes the production of key extra-cellular matrix components such as col1a1, col1a2, and profibrotic markers such as CTGF/CCN2 (10). CTGF/CCN2 exerts profibrotic properties notably as a co-factor of TGF-β signaling and as a target of YAP. CTGF/CCN2 can interact with specific receptors (such as integrins or lipoprotein receptor-related proteins), extra-cellular matrix proteins (such as fibronectin or perlecan) and growth-factors (such as VEGF and TGF-β), with subsequent activation of fibroblast proliferation and myofibroblasts activation, although the specific mechanisms involved are still to be determined (184,185). Rho serves as another integrator of profibrotic pathways and can be activated by TGF-β, LPA agonists or integrin signaling in a FAK dependent manner. Rho subsequently activates ROCK that participates in a cascade sustaining fibrotic response and induces cytoskeleton remodeling. In return, increased extra-cellular matrix stiffness participates in the activation of latent TGF-β perpetuating a pro-fibrotic pathogenic loop. IL-4 and IL-13 participate in fibrosis by inducing the proliferation of fibroblasts and by increasing their production of TGF-β and CTGF/CCN2 in a STAT6 dependent manner (103). On the contrary, α-MSH and MC1-R agonists may exert anti-fibrotic properties by suppressing TGF-β signaling although the precise sub-cellular mechanisms are still to be determined.
FAK=focal adhesion kinase, LPA= lysophosphatidic acid; LPA-R=LPA-Receptor; IL-6R=IL-6 receptor; TGF-βRI & II= TGF-β receptor I & II; PAI-1=plasminogen activator inhibitor 1; ROS=Reactive Oxygen Species; YAP=Yes Associated Protein; α-MSH =α-Melanocyte-stimulating hormone; IL-4Rα=IL-4 receptor α; IL-13Rα1= IL-13 receptor α1; ECM=Extra-cellular matrix
Myofibroblasts are the major effectors of ECM production and fibrosis (11). Myofibroblasts in SSc originate from a variety of tissue-resident mesenchymal progenitor cell types, including fibroblasts, pericytes, microvascular endothelial cells and vascular preadipocytes (12,13). Activated myofibroblasts in fibrotic tissue have mesenchymal properties with an enhanced RhoA/ROCK (Rho associated protein kinase)-dependent contractility and a high expression of α-smooth muscle actin (SMA) (2). The trans-differentiation of resting fibroblasts and other progenitor cells into pro-fibrotic and inflammatory myofibroblasts is driven by canonical smad-dependent and non-canonical smad-independent tumoral growth factor (TGF)-β signaling (14). Activated myofibroblasts also produce profibrotic mediators such as IL-6 or connective tissue growth factor (CTGF)/CCN2 (15), leading to an autocrine profibrotic pathogenic loop maintaining sustained cellular activation (15). CTGF/CCN2 can synergize with TGF-β signaling and is a target of key pro-fibrotic transcription factors such as the Yes-Associated Protein (YAP) (Figure 2), participating to myofibroblast differentiation (16–18). Myofibroblasts in SSc demonstrate extensive epigenetic remodeling, and metabolic alterations including enhanced glycolysis and altered NAD+ homeostasis (19,20). Additionally, myofibroblasts in SSc are characterized by apoptosis resistance and uncontrolled production of extra-cellular matrix (ECM) components such as collagens, tenascin C or fibronectin (21,22). In turn, these secreted extracellular components can activate myofibroblasts either through a direct process involving innate immune sensors such as TLR-4, or through an indirect activation notably depending on mechano-sensing of increased matrix stiffness by integrins in a FAK dependent manner (23–26). Evasion of apoptosis is a hallmark of activated myofibroblasts in skin and lung biopsies from SSc patients, and can be targeted for selective elimination (27).
Beyond the classical cellular subsets described in Figure 1, single-cell RNA-sequencing has also identified new SSc-associated sub populations: endothelial cells from SSc patients showed a specific pattern of gene expression identifying vascular injury and activation, ECM generation and negative regulation of angiogenesis as key processes. HSPG2 and APLNR were among the key markers of endothelial cell SSc signature (28). In the lung, a specific population of SPP1-high macrophages with proliferating properties has also been identified by Single-cell RNA-sequencing. This population did not exactly overlap with CD163+ M2 macrophages and was over-represented in SSc patients as compared to healthy controls (29). The same team has highlighted the high heterogeneity of fibroblast sub-populations notably in the lungs, suggesting that SPINT2hi and MFAP5hi fibroblasts in addition to myofibroblasts could play an important role in SSc-interstitial Lung disease (SSc-ILD) (29). A diverse activation and over-representation of these new cellular subsets may participate to explain the heterogeneity of the phenotype of patients with SSc.
1.2. Heterogeneity of SSc as a therapeutic challenge
SSc is characterized by a diverse range of clinical manifestations with various trajectories regarding skin fibrosis, organ involvement and mortality (30,31). Capturing the heterogeneity of this systemic disease in a practical approach, feasible in daily practice, remains challenging (32,33). Nonetheless, the identification of homogeneous subsets of SSc patients, sharing predominant pathogenic pathways may help to improve the design of randomized controlled trials (RCTs). Early stages of the disease have also been identified as a window of opportunity, for the management of diffuse cutaneous systemic sclerosis (dcSSc) (9,34), but also for the treatment of interstitial lung disease (ILD) (5,35).
Recent RCTs have especially focused on drugs targeting fibrotic manifestations of the disease, such as skin or lung involvement, as these manifestations reflect overall disease progression or impact survival (5,35,36). Recent reviews have discussed the results of phase II or III RCTs and the therapeutic progress allowed by better identification of molecular targets in SSc (37,38). The tyrosine kinase inhibitor nintedanib, notably targeting VEGF, PDGF-bb and FGF-dependent signals, attenuated forced vital capacity (FVC) decline in patients SSc-ILD in a phase III trial and is now FDA-approved in this context (5,39). Inhibiting IL-6 signaling with tocilizumab resulted in a stabilization of FVC in a phase II and a phase III trial including patients with early progressive skin disease and elevated markers of inflammation (35,40). For these reasons, tocilizumab has also been recently FDA-approved for SSc-ILD. Abatacept, a fusion protein inhibiting co-stimulation through CTLA-4 signaling had a numerically positive impact on mRSS change in a phase II trial (41,42). All these recent trials demonstrate that a better understanding of the pathogenesis of the disease has led to substantial progress and emphasize the important role of these targets in the pathogenesis of SSc (43). Nonetheless the potential disease-modifying effects of these drugs has not been demonstrated to date, and the identifications of new therapeutic targets may therefore help reaching such a goal.
Several recent RCTs have thus failed to reach their primary outcome in SSc in the past. This could be explained as the candidate drug may have preferential effects on the wrong target based on wrong pathogenic hypotheses, as this could have been the case for IFNα as a therapeutic option (44), since recent transcriptomic analyses from the skin of dcSSc patients have highlighted the role of IFN type I signature as part of the pathogenic process (45). Impacting the wrong target may also be responsible for safety issues as demonstrated by initial attempts of targeting all isoforms of TGF-β, including those involved in hemostasis, with subsequent hemorrhagic adverse events (46). Off-targets effects or impact on macrophages may explain the failure of PPARγ agonists as antifibrotic agents in SSc, as PPARγ is also involved in enhanced M2 polarization with subsequent detrimental effects due to this sub-population (47). The selection of the wrong outcome may also have participated in the negativity of the Phase II and III trials of tocilizumab using skin fibrosis as their primary objective, since on the contrary the effects on FVC and ILD as secondary objectives have led to the FDA approval of this drug for SSc-ILD (35,40,48). Lastly, the heterogeneity of SSc highlighted by the variable molecular signatures identified in the skin of SSc patients may also have participated in limiting the impact of abatacept on skin fibrosis in the overall dcSSc population, whereas it showed encouraging results in patients with an inflammatory pattern (41).
The present review will discuss the rationale, preclinical evidence, first clinical evidence and pending issues concerning a selection of therapeutic options under investigation in SSc with emphasis on candidate drugs with Phase I or II trials published or planned (Table 1). In this review, a classification of these drugs based on their main impact (i.e. effects on fibrosis, inflammation/immunomodulation, other pathways) is proposed although some of these may have widespread effects that may not be limited to one component of SSc pathogenesis. The expert opinion section will focus on possible strategies to manage the heterogeneity of the SSc and its impact on drugs under investigation in SSc.
Table 1:
Investigational drugs in systemic sclerosis with published or announced Phase I, II or III clinical trials
| Molecules | Characteristics | Potential mechanisms of action in SSc | Development advancement in SSc | Reference on clinicaltrial.gov |
|---|---|---|---|---|
| Abituzumab | Mono-clonal antibody | Inhibitor of integrins from the αV class, with impact on activation of latent TGF-β and inhibition of FAK dependent activation of Rho dependent profibrotic pathways | Phase II in SSc-ILD | NCT02745145 |
| Abatacept | fusion protein | CTLA-4 dependent inhibition of co-stimulation leading to decreased Th1, 2, and B cell activation. | Phase II in early dcSSc | NCT02161406 |
| AVID200 | fusion protein | Selective inhibitor of TGF-β1 and 3 | Phase I in early dcSSc | NCT03831438 |
| Belimumab | Mono-clonal antibody | Selective inhibition of BAFF/BLyS, with impact on B-cell survival and maturation. | Phase II in early dcSSc | NCT01670565 |
| Belumosudil (KD025) | selective oral inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2) | Selective ROCK-2 inhibitor that may inhibit myofibroblast activation and increase pro-resolving properties/efferocytosis of macrophages. | Phase II in early dcSSc |
NCT03919799 NCT04680975 |
| CM-101 | Mono-clonal antibody | Selective targeting of CCL-24, with subsequent inhibition of the homing of immune cells to tissue thus obtaining an anti-fibrotic effect | Phase II awaited in dcSSc and lcSSc | NA |
| Dasatinib | Oral small molecule | Senolytic agents that target senescent fibroblasts, responsible for an excessive production of IL-6 and TGF-β | Phase I/II in SSc-ILD | NCT00764309 |
| Fasudil | cyclic nucleotide-dependent protein kinase inhibitor | RhoA/ROCK inhibitor that may inhibit myofibroblast activation and increase pro-resolving properties/efferocytosis of macrophages. | Phase III | NCT00498615 |
| Fresolimumab | Mono-clonal antibody | Inhibitor of TGF-β1, 2 and 3 | Phase I in early dcSSc | NCT01284322 |
| IVIg | serum IgG fraction pooled from several thousands of donors | Targets and neutralizes auto-antibodies/immune complexes, inhibits monocyte activation, limits M1 and M2 activation | Phase II in early dcSSc |
NCT04137224 NCT04138485 NCT01785056 |
| JAK inhibitors: Tofacitinb (pan-JAK inhibitor) Itacitinib (JAK1 inhibitor) | Oral small molecule | Inhibits the signaling of key cytokines and mediators of fibroblast activation (TGF-β non-canonical pathway, IL-6) and macrophage activation (IFNg, IL-13, IL-4), B-effector cell activation. | Phase I/II in early dcSSc |
NCT03274076 NCT04789850 |
| Metelimumab (CAT-192) | Mono-clonal antibody | Inhibitor of TGF-β1 | Phase I/II in early dcSSc | NCT00043706 |
| MT-7117 | synthetic, oral, non-peptide small molecule | selective agonist of MC1R, that decrease the expression and impact of IL-6 and TGF-β, increase pro-resolving properties/efferocytosis of macrophages. | Phase II in early dcSSc | NCT04440592 |
| Pirfenidone | Oral small molecule | Suspected inhibition of TGF-β signaling | Phase II in SSc-ILD |
NCT03221257 NCT01933334 |
| Rituximab | Mono-clonal antibody | Anti-CD20 mab targeting B-cell, with subsequent impact of IL-6 release and potential impact on autoantibody production | Phase II in PAH-associated SSc | NCT01086540 |
| Romilkimab (SAR156597) | Mono-clonal antibody | Inhibits IL-13 and IL-14 signaling, leading to limited M2 and myofibroblasts activation | Phase II in early dcSSc | NCT02921971 |
| SAR100842 | selective oral antagonist of LPA1 | may inhibit myofibroblast activation through its impact on RhoA-ROCK signaling and increase pro-resolving properties/efferocytosis of macrophages. | Phase II in early dcSSc | NCT01651143 |
| Ziritaxestat (GLPG1690) | Oral autotaxin inhibitor | Inhibit the production of LPA producing enzyme autotaxin with subsequent inhibition of myofibroblast activation through its impact on TGF-β non-canonical signaling and increase pro-resolving properties/efferocytosis of macrophages. | Phase II in early dcSSc | NCT03798366 |
2. PROMISING INVESTIGATIONAL DRUGS IN SYSTEMIC SCLEROSIS
2.1. Anti-fibrotic drugs
2.1.1. Inhibition of TGF-β signaling
TGF-β is considered as one of the key growth factors driving fibrosis in SSc (49). It is the main factor inducing the trans-differentiation of fibroblasts into myofibroblasts (10). Three isoforms of TGF-β are classically described (TGF-β1, 2 and 3). TGF-β is notably secreted by platelets, monocytes, macrophages, T lymphocytes and fibroblasts/myofibroblasts with paracrine and autocrine effects (Figure 1). TGF-β is sequestrated in the ECM in an inactivated form, that can be activated by various signals including increased MEC stiffness, actions of integrins (notably from the αV class) or thrombospondins (50). Key actors involved in TGF-β canonical and non-canonical pathways are provided in Figure 2 (14). TGF-β induces the production of key components of the ECM such as the col1A1 or fibronectin by myofibroblasts (49). TGF-β could also participate in SSc-associated vasculopathy due to its effects on angiogenesis, notably through a TGF-β-R II/ALK1/endoglin-dependent mechanism (14).
TGF-β or downstream associated pathways are up-regulated in the fibrotic tissues of all main mouse models of SSc, including the tsk1, fra-2, bleomycin and HOCl models (51,52). In patients with SSc, immunohistochemical analyses show that the expression of all three isoforms of TGF-β are increased in the skin and serum, but a large proportion remains in the latent form (53–55). Some in vitro studies have nonetheless reported that peripheral blood mononuclear cells (PBMCs) from patients with SSc could release high levels of active TGF-β (56).
Pirfenidone is an oral antifibrotic therapy approved in the treatment of IPF (57). The precise mechanisms of action of pirfenidone are still to be determined but one of its key effects is the inhibition of TGF-β signaling, notably reducing TGF-β1-dependent trans-differentiation of fibroblasts (58). The tolerability and safety of pirfenidone in patients with SSc-ILD were evaluated in an open-label, phase II study including 36 patients (59). This study demonstrated an acceptable tolerability profile of Pirfenidone in SSc-ILD. Tolerability was not affected by concomitant treatment with mycophenolate mofetil (MMF), suggesting that pirfenidone could be used as add-on therapy. The efficacy of pirfenidone in SSc-ILD has been evaluated in a double-blind, randomized, placebo-controlled, pilot study including 34 patients. In this study with limited sample size, pirfenidone failed to improve/stabilize FVC in comparison with placebo over 6 months (60). The scleroderma lung study (SLS) III is a phase II multi-center, double-blind, parallel group, randomized and placebo-controlled clinical trial assessing the efficacy of Pirfenidone versus placebo, as an add-on therapy with MMF in patients with SSc-ILD (NCT03221257). The primary outcome will be the change from baseline FVC (% predicted) over 18 months, measured at 3-month intervals. This study is still ongoing.
Selective inhibitors of TGF-β have also been evaluated or are under investigation in SSc. Metelimumab (CAT-192) is a recombinant human antibody that neutralizes TGF-β1. It showed no evidence of efficacy on the evolution of mRSS in a multicenter, randomized, placebo-controlled phase I/II in patients with early dcSSc. Moreover, there was significant morbidity and mortality, including one death in the group receiving 0.5 mg/kg of metelimumab and three deaths in the group receiving 5 mg/kg of metelimumab, although none were considered related to the treatment (61). Fresolimumab is a human monoclonal antibody neutralizing all mammalian isoforms of TGF-β. In a pilot open-label phase I study including 15 dcSSc patients, mRSS improved after fresolimumab treatment (median decrease of −5 (7.2), p=0.05)). Nonetheless, several patients had epistaxis, gum bleeding, or subconjunctival eye hemorrhage as well as anemia. This raised concerns about the safety of a non-selected targeting of all TGF-β isoforms (46). AVID200 is a fusion protein resulting from the fusion of the TGF-β-R ectodomain to an IgG Fc region, acting as a potent TGF-β trap, with high selectivity against TGF-β1 and TGF-β3 and limited activity against TGF-β2. As TGF-β2 is involved in physiological hematopoiesis, and myocardial functioning, selective inhibition of TGF-β1 and TGF-β3 may show a better tolerance and safety profile. A Phase I trial designed to evaluate the safety, tolerability and preliminary efficacy of AVID200 in dcSSc has shown that AVID200 at doses of 1 and 3 mg/kg was well-tolerated. Recruitment for additional doses and an extension cohort are currently ongoing (62).
The inhibition of integrin-dependent activation of latent TGF-β could also constitute a relevant approach. Abituzumab is a humanized monoclonal antibody that inhibits the activity of integrins from the αV class. A phase II, double-blind, parallel-group, multicenter trial (NCT02745145) was designed to assess the effects of abituzumab as add-on therapy to MMF in SSc-ILD. Patients were randomized (2:2:1) to receive abituzumab 1500mg, abituzumab 500mg or placebo every 4 weeks for 104 weeks. The annual rate of change in absolute FVC was the primary outcome. This study was unfortunately prematurely terminated due to slow enrollment, and no robust conclusion on efficacy could be drawn from this study although no safety signals were detected (63).
A better understanding of both canonical and non-canonical TGF-β signaling may also offer new therapeutic perspectives, such as Takinib, a TAK1 inhibitor that can limit p38 phosphorylation and could impair TGF-β non-canonical pathway (64). ALK5 that participates to the ligand-dependent phosphorylation of smad2/3 in the canonical pathways could constitute another target as it may participate to the fibrotic phenotype of scleroderma fibroblasts, although no clinical data in SSc is available to date (65). Moreover, solely targeting ALK5 may not be sufficient to block key features of the persistent myofibroblast phenotype in SSc. Thus, antagonism of canonical TGF-β signaling without targeting non-canonical pathways and/or concomitant FAK-dependent pro-fibrotic signals (Figure 2) may not be clinically relevant approaches in SSc (66). The selective inhibition of TGF-β may have no impact on inflammatory manifestations of the disease, as TGF-β mainly impacts myofibroblasts. To date the evaluation of selective TGF-β inhibitor is limited to skin involvement and its impact on SSc-associated ILD is still to be explored. This is a major point to demonstrate the potential disease-modifying effect of TGF-β inhibition or to discuss its combination with immunomodulatory therapies like MMF.
2.1.2. Rho/ROCK inhibitors
TGF-β, CTGF/CCN2, lysophosphatidic acid (LPA) receptors and integrins can activate intra-cellular signaling through a common mediator (Rho GTPase), leading to the activation of the Rho-associated protein kinase ROCK (67). The RhoA/ROCK signal is a major pathway involved in many biological processes, including mechano-sensing, cell contractility and fibrosis. RhoA/ROCK activation is involved in the non-canonical TGF-β signaling, and contributes to the contractile phenotype of activated myofibroblasts (Figure 2) (68,69). STAT-3 phosphorylation in myofibroblast is activated by ECM stiffness in a ROCK-dependent manner (70). RhoA/ROCK activation also limits pro-resolving properties of macrophages, notably by impairing their efferocytosis capacities, i.e. their ability to phagocyte apoptotic cells (71). Significant associations between ROCK1/2 and RhoA gene polymorphisms and SSc have also been reported (72). ROCK inhibitors on the contrary exert anti-inflammatory properties and can shift M1 macrophages into M2 (73). They also have vasodilating effects through their impact on vascular smooth muscle cell contractility (74).
RhoA/ROCK inhibitors Y-27632 and Fasudil can improve the efferocytosis capacities of blood monocyte-derived-macrophages from patients with SSc (71). In the HOCl SSc mouse model, the ROCK inhibitor Fasudil, significantly prevented skin fibrosis, the expression of α-SMA, as well as the activity of ROCK in the fibrotic skin of HOCl-treated mice, through the inhibition of the phosphorylation of Smad2/3 and ERK1/2. Fasudil also reduced lung fibrosis and macrophage lung infiltrates in this model (75). Novel orally bioavailable Rho inhibitors have been recently designed. In the bleomycin mouse model, such inhibitors could significantly limit skin thickness and collagen content in comparison with vehicle control (67). The ROCK2 inhibitor KD025 also showed promising results in the mouse model of chronic GVHD (76).
A single-center, double-blind, placebo-controlled, randomized, 3-period study has investigated the efficacy of fasudil to reverse cold-induced vasospasm in patients with SSc-associated Raynaud’s phenomenon. Only 17 patients completed the study, but fasudil showed no significant benefit in terms of skin temperature recovery time or digital blood flow assessed by laser Doppler after cold challenge (77). Two studies are currently registered to evaluate the relevance of ROCK-2 inhibition by KD025 (belumosudil) in patients with dcSSc. NCT04680975 is a phase 2, open-label, single-cohort, multicenter, 24-week trial including 15 participants with early dcSSc. NCT03919799 will include 60 adult subjects with early dcSSc randomized into 3 groups (1:1:1) to either receive orally administered belumosudil (200 mg once daily and 200 mg twice daily) or matched placebo for 28 weeks. This will be a double-blind study for the first 28 weeks with an open-label extension period of 24 weeks. The announced primary outcome of these two studies is the ACR-CRISS after 24 weeks of therapy. These trials are currently ongoing. Belumosudil (KD025) has been recently approved for cGHVD by the FDA, strengthening the relevance of its evaluation in dcSSc.
Rho/ROCK inhibition may also exert vasodilating properties that could positively impact the vascular manifestations of the disease (78). The most relevant target, i.e. select inhibition of Rho, ROCK-1 or ROCK-2 is still to be determined, as differential effects may be observed depending on the targeted kinase (79).
2.1.3. LPA-1 pathway and Autotaxin inhibitors.
Lysophosphatidic acid (LPA) is a lipid mediator that binds to specific G protein-coupled receptors and signals notably through RhoA/ROCK activation (80). LPA receptors are designated from LPA1 to LPA6 and their expression varies according to tissues and cell types (81). LPA is produced in case of inflammation and cell injury and derived from lysophosphatidylcholine or other lysophospholipids under the action of lysophospholipase D, also called autotaxin. LPA1 signaling participates in myofibroblast differentiation through its impact on RhoA/ROCK (82,83). The LPA-producing enzyme autotaxin may also play an important role in the fibrotic manifestations of SSc through a pathogenic loop involving IL-6 (84). Plasma and serum levels of LPA are elevated in SSc patients. LPA1 receptor is overexpressed in isolated dermal fibroblasts and skin biopsies from SSc patients (82,85).
Ziritaxestat (GLPG1690), an oral autotaxin inhibitor and SAR100842, a potent selective oral antagonist of LPA1 are under investigation in SSc. Using a therapeutic protocol, SAR100842 significantly decreased dermal thickening, inhibited myofibroblast differentiation and reduced the amount of skin collagen content in tsk1 mice. SAR100842 also inhibited the secretion of CXCL1, CCL-2 and IL-6 in SSc dermal fibroblasts and of CXCL1 and CCL-2 in mice (82). Both autotaxin and LPA1 receptor inhibition abrogated bleomycin-induced lung fibrosis (86).
An 8-week double-blind, randomized, placebo-controlled study followed by a 16-week open-label extension was performed to assess the safety and efficacy of SAR100842 in patients with early dcSSc. The primary endpoint was safety during the double-blind period of the trial and mRSS served as a surrogate marker for efficacy. Seventeen patients were included in the placebo arm and 15 in the SAR100842 arm. Overall, SAR100842 was well tolerated. At week 8, the reduction in mRSS was greater in the SAR100842 group than in the placebo group although this result did not reach statistical significance (−3.57 (4.18) in the treatment group versus −2.76 (4.85) for the placebo; p=0.46) (87).
The efficacy, safety and tolerability of ziritaxestat in patients with early dcSSc was evaluated in a phase 2a randomized, double-blind, placebo-controlled trial including 21 and 12 patients in the treatment and placebo arm respectively. The primary endpoint was the change from baseline mRSS at 24 weeks (88). A statistically significant difference was observed between groups for the primary endpoint with a mean difference of –2.8 (IC95% –5.6, –0.1) in favor of ziritaxestat (p=0.0411). The American college of Rheumatology-Combined Response Index in diffuse Systemic Sclerosis (ACR-CRISS) showed trends toward a numerically higher probability of improvement with ziritaxestat.
2.2. Immunomodulatory drugs
2.2.1. JAK inhibitors
Janus Kinase (JAK) inhibitors are a class of oral tyrosinase inhibitors (TKIs) that target one or more of the four members of the JAK family i.e. JAK1, JAK2, JAK3 and/or tyrosine kinase 2 (TYK2). These JAK kinases mediate the signaling of numerous cytokines and growth factors, notably through the phosphorylation of transcription factors belonging to the STAT (Signal Transducer and Activator of Transcription) family. The signaling of major cytokines participating in the pathogenesis of SSc involves JAK/STAT pathways (Table 2). JAK/STAT pathways notably participate in fibroblast activation through IL-6 signaling, and could also indirectly participate to TGF-β signaling via a JAK2 dependent non-canonical activation (Figure 2) (89). JAK/STAT pathways are also involved in the polarization of M1(IFNγ) and M2(IL-4/IL-13) macrophages, both overexpressed in the skin of patients with early dcSSc (9). JAK inhibitors could thus have combined anti-inflammatory and anti-fibrotic properties (90) (Table 1).
Table 2:
JAK/STAT signaling in the pathogenesis of systemic sclerosis
| Cytokines notably involved in the pathogenesis of SSc | JAK associated to their receptor | STAT | Main cellular targets in the context of systemic sclerosis | Main effects |
|---|---|---|---|---|
| IL-4 | JAK1/3 | STAT6 | -Induces M2 polarization in Macrophages -Fibroblast proliferation |
Pro-fibrotic Immuno-modulatory |
| IL-10 | JAK2/TYK2 | STAT3 | -Could participate in fibrosis through fibroblast activation -Exerts anti-inflammatory effects on B-cell and macrophages |
Pro-fibrotic anti-inflammatory |
| IL-13 | JAK1/2/3/TYK2 | STAT6 | -Induced M2 polarization in Macrophages -Fibroblast proliferation and TGF-β secretion |
Pro-fibrotic Immuno-modulatory |
| IL-6 | JAK1/2/TYK2 | STAT1/3 | -Favor IL-4 receptor expression an M2 polarization -Induce myofibroblast activation |
Pro-fibrotic Pro-inflammatory |
| Interferon α/β (Type 1 IFN) | JAK1/TYK2 | STAT1/2/4 | -Inhibit the pro-resolving properties of macrophages -participates to endothelial dysfunction |
Pro-inflammatory Anti-angiogenic |
| Interferon γ (Type II IFN) | JAK1/TYK2 | STAT1 | -Induces M1 polarization in macrophages -Indirect effects on endothelial cells through increased secretion of CXCL10 by M1 -Indirect effects on fibroblasts through increased secretion of IL-6 by M1 |
Pro-inflammatory Anti-angiogenic |
JAK1/2 inhibition by ruxolitinib can limit fibrosis in bleomycin-induced lung fibrosis and in a mouse model of skin fibrosis triggered by constitutive activation of type I TGF-β receptor (89). The combined anti-inflammatory and anti-fibrotic properties of tofacitinib, ruxolitinib and itacitinib can decrease both M1 and M2 polarization in human monocyte-derived macrophages in vitro (91). Ruxolitinib could prevent skin and lung fibrosis notably through its impact on M1 and M2 polarization, in the hypochlorous acid (HOCl)-induced SSc mouse model (91). Tofacitinib also prevents skin and lung fibrosis in the bleomycin and tsk1 mouse models when tofacitinib and bleomycin injection are concomitantly initiated (92,93). Nonetheless, tofacitinib fails to limit these manifestations when introduced 9 days after the beginning of bleomycin injections (92).
The safety of tofacitinib in 15 patients with early dcSSc was evaluated in a 6 month, 2 center, double-blind, randomized placebo-controlled Phase I/II trial (94). Tofacitinib was well tolerated with no Grade 3 or higher adverse events before or at 6 months. There were trends for efficacy in favor of tofacitinib notably on skin fibrosis evaluated by mRSS and on the probability of overall improvement evaluated by the ACR-CRISS. Case reports in SSc and morphea (95,96) and the efficacy of ruxolitinib in acute and chronic graft versus host disease (GVHD) (97,98) also support the need for further evaluation of JAK inhibitors in SSc.
Although the selection of the most relevant JAK inhibitor based on the targeted JAK(s) may still deserve further investigations, the selectivity of JAK inhibition in vivo is controverted. Tofacitinib or ruxolitinib are now considered by some authors as pan-JAK inhibitors, i.e. targeting all JAKs (99). This lack of selectivity could be both considered as a strength, regarding the widespread impact they could have in this systemic disease, and a limitation when considering a less efficient and selected impact on more specific SSc-associated pathways. Nonetheless, the phosphorylation of JAK1, 2 and 3 is increased in the skin and lung of SSc patients, suggesting that pan-JAK-inhibition should be prioritized, notably in dcSSc (91,92). Considering their impact on IL-6 (for tofacitinib, baricitinib and ruxolitinib), similarly to tocilizumab, their role in the prevention of progression in SSc-ILD should be discussed in patients with high risk of progression (38,100). The impact of selective inhibition of JAK1 with itacitinib will also be evaluated in early dcSSc with mRSS as primary endpoint (NCT04789850).
2.2.2. Combined targeting of IL-4 and IL-13 with romilkimab
Romilkimab (SAR156597) is an engineered, humanized, bispecific immunoglobulin-G4 antibody that binds and neutralizes the immuno-modulatory and pro-fibrotic Th2 cytokines IL-4 and IL-13 (101). Patients with SSc have elevated serum levels of IL-4 (102). This cytokine participates in fibrosis by inducing the proliferation of fibroblasts and by increasing their production of TGF-β and CTGF/CCN2 (103). IL-4 can also induce a profibrotic polarization of macrophages (M2(IL-4)). IL-13 induces similar effects, notably as IL-4 and IL-13 receptors share common subunits and downstream signaling pathways.
In the Tsk1 mouse model of SSc, knocked-out mice for the IL-4Rα do not develop dermal thickness (104). In the mouse model of pulmonary fibrosis induced by intra-tracheal instillation of bleomycin, the blockade of the IL-13/IL-4 pathway by an inhibiting recombinant fusion protein derived from IL-13, significantly reduces collagen content (105).
The safety and efficacy of romilkimab was evaluated in a randomized, double-blind, placebo-controlled, 24-week, phase II, proof of concept study in patients with early diffuse cutaneous SSc (106). Background concomitant immunomodulatory therapies were allowed and 48 patients were included in the romilkimab arm versus 49 in the placebo arm. The primary outcome was the change from baseline to week 24 in mRSS. The primary outcome was met as least-squares mean change in mRSS was −4.76 (0.86) for romilkimab versus −2.45 (0.85) for placebo yielding a mean (90% CI) difference of −2.31 (1.21) (−4.32 to −0.31; p=0.0291, one-sided). The change in FVC and DLco also numerically favored active therapy. Romilkimab showed disappointing results in IPF, as a phase II trial demonstrated a lack of efficacy of active therapy in reducing FVC decline in comparison with placebo (101).
The results from the phase II proof-of-concept trial in dcSSc highlight that early dcSSc patients may constitute a relevant targeted population for this new drug. Early dcSSc patients is a subpopulation with a more active, inflammatory disease. The positive results of romilkimab on mRSS in early dcSSc may represent new evidence for a shift of paradigm in dcSSc: targeting specific pro-fibrotic pathways in this population of more inflammatory patients may prevent the shift from inflammation to fibrosis that occurs in the natural history of skin disease (9). Therefore, patients with early inflammatory SSc may benefit from early anti-fibrotic therapy and not only from early anti-inflammatory drugs. Nonetheless, the impact of background immunomodulatory therapy in this phase II trial is still to be further explored, as they may also have participated in limiting skin inflammation.
2.2.3. The melanocortin-1 receptor agonist MT-7117
α-Melanocyte-stimulating hormone (α-MSH) is a tri-decapeptide derived from pro-opiomelanocortin (POMC) by proteolytic cleavage. α-MSH is a major pigment-inducting factor regulating skin color through its effect on melanocytes (107). Although α-MSH was originally isolated from the pituitary gland, it can also be produced in the skin itself, exerting local effects (108). Beyond its pigment-inducting properties, α-MSH may also exert anti-fibrotic effects by suppressing TGF-β1-induced collagen synthesis in human dermal fibroblasts in vitro (109). α-MSH has anti-inflammatory properties notably through the downregulation of pro-inflammatory mediators such as IFN-γ and IL-6 (110,111). These effects of α-MSH are mediated by melanocortin receptors (MCR). Five MCR are described (MC-1 to 5-R). MC1R is especially expressed in melanocytes and skin fibroblasts but also in monocytes, macrophages (including alveolar macrophages), lymphocytes and neutrophils (112). MCR agonists also enhance the pro-resolving efferocytosis capacities of macrophages (113,114). MT-7117 is a new synthetic, orally-administered, non-peptide small molecule, which acts as a selective agonist of MC1R with potential immunomodulatory properties.
In a mouse model of dermal fibrosis induced by repetitive TGF-β skin injections, collagen deposition and the number of α-SMA positive dermal cells were significantly reduced by injection of α-MSH (108). α-MSH also reduced skin fibrosis and collagen content after intradermal injection of bleomycin in mice (109). In this model, α-MSH increased the tissue levels of antioxidant superoxide dismutase 2 and heme-oxygenase 1, suggesting that its anti-fibrotic effects could be at least in part due to its antioxidant properties (109). α-MSH limited the apoptosis of vascular endothelial cells in a rat model of acute respiratory distress syndrome (115). After intratracheal instillation of bleomycin, α-MSH analogs notably decreased the expression of IL-6 and TGF-β (116).
NCT04440592 is a phase II, multicenter, randomized, double-blind, placebo-controlled, parallel-group study that aims to evaluate efficacy, safety, and tolerability of MT-7117 in patients with dcSSc. This is a 52 week-study with an estimated enrollment of 72 participants with disease duration ≤3 years from the first non-Raynaud’s phenomenon (RP) manifestation. Inclusion criteria also involve enrichment based on inflammatory markers such as an increase in mRSS ≥3 units within the past 9 months, or presence of tendon friction rubs or C-reactive protein (CRP) levels ≥ 6 mg/L in patients with a disease duration of more than 18 months. The primary outcome will be the ACR-CRISS at Week 52. This study is currently ongoing.
MT-7117 is a selective agonist of MC1R, an important receptor mediating the effects of α-MSH in the skin but this receptor may not be the most expressed in other tissues such as lungs, where MC3R is also identified, notably on macrophages in mice (112). Pro-resolving properties of α-MSH, such as enhanced efferocytosis are mediated by MC3R in mice (114), and thus MT-7117 may not show similar pro-resolving effects. Patients with dcSSc also present skin hyperpigmentation (117), suggesting that some effects of α-MSH may already be up-regulated and a plateau effect of MC1R agonist could be observed.
2.2.4. Targeting B cells and combination therapy targeting both innate and adaptive immunity.
Adaptive immunity plays a key role in the pathogenesis of SSc, as the B-cell lineage is responsible for autoantibody production with subsequent formation of immune complexes (IC) that can activate fibroblasts and monocytes/macrophages (118,119). Higher number of memory B cells expressing CD19 and CD95 has been identified in SSc, and this sub-population is a key source of IL-6 (120). Patients with SSc have increased serum levels of BAFF (B cell-activating factor, also known as BLyS for B Lymphocyte Stimulator)), a mediator facilitating survival and maturation of B cells (121). Histological analyses of lung tissues from patients with SSc-ILD show a prominent B-cell infiltration frequently arranged in lymphoid aggregates (122).
B-cell depletion with an anti-CD20 antibody in the Tsk/+ mouse model reduced skin fibrosis with lower levels of serum immunoglobulin and anti-topo I antibodies (123). In the bleomycin mouse model, skin and lung fibrosis, as well as hypergammaglobulinemia and autoantibody production, are significantly decreased by CD19 deficiency (124). BAFF antagonists also attenuated skin and lung fibrosis in the bleomycin mouse model (121). Treatment with abatacept, an inhibitor of co-stimulation notably targeting B and T cell co-activation, decreased B cell count in bleomycin-induced skin fibrosis (125). Concomitantly, this decrease of B-cells was associated with decreased dermal thickness, collagen content and myofibroblasts count in the skin both in a preventive and curative protocol (125). In vitro, ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor that specifically targets effector B cells, was able to decrease the production of cytokines IL-6 by B cells from patients with SSc (126).
In a phase II randomized, double blind, placebo controlled-trial, abatacept showed promising results with numerical greater improvement of mRSS as compared to placebo in patients with early dcSSc without background immunomodulatory therapies (41,42). A recent meta-analysis of 20 studies, has highlighted that rituximab (anti-CD20, targeting B cells) improved pulmonary outcomes (FVC and DLco) in the first year of treatment (127) and, an analysis of the EUSTAR cohort suggested positive effects of rituximab on mRSS in comparison with matched SSc-controls (128). A randomized, double-blind, placebo-controlled, phase II multicenter trial evaluating the efficacy of rituximab in 57 patients with SSc-PAH has demonstrated that six-minute walk distance (6MWD) at 24 weeks as primary outcome was not different in the rituximab and placebo arms but that this difference was statistically significant in favor of rituximab based on a secondary analysis model including 6MWD data out to week 48 (p=0.03) (129). Belimumab is a fully humanized IgG1γ monoclonal antibody targeting BAFF. A recent single-center, double-blind, placebo-controlled, pilot study has evaluated 20 patients with dcSSc recently started on MMF, showing an improvement of mRSS in both arms, but with higher median difference in comparison with baseline mRSS in the belimumab arm (−10 (IQR −13, −9) and −3.0 (IQR −15, −1) in the belimumab and placebo groups respectively), although the difference was not statistically significant (P = 0.411) (130). The drug was well-tolerated and additional studies are needed to better define the place of BAFF inhibition in the treatment of SSc. Clinical evidence for the relevance of BTK inhibitors in SSc are still needed, but ibrutinib showed promising results in a phase Ib/II trial in chronic GVHD (131), strengthening the relevance of evaluating this therapeutic option in SSc.
A 52-week, single-center, randomized, double-blind, placebo-controlled phase II trial evaluating the combined effect of rituximab and belimumab as add-on therapy with MMF in patients with early dcSSc is currently ongoing (NCT03844061). This will be the first clinical trial evaluating the association of two monoclonal antibodies in SSc, both targeting adaptive immunity. This may constitute a major breakthrough, that could pave the way for other combinations in the future. Such combinations could allow to target both innate and adaptive immunity with simultaneous inhibition of pro-inflammatory and pro-fibrotic pathways.
2.3. Other mechanisms of action
2.3.1. Intravenous Immunoglobulins (IVIg)
IVIg preparations are mainly based on the serum IgG fraction pooled from several thousands of donors. IVIg exert immunomodulatory effects which can be either F(ab)- or Fc-dependent (132). Through their F(ab) portion, IVIg can directly target and neutralize autoantibodies. In an Fc-dependent manner, IVIg can modulate FcγR expression and can prevent monocyte activation through their interaction with the FcγRIIB which exerts immunomodulatory effects. IVIg can also limit and disrupt the interaction of IC with their target cells. Such IC, notably composed of SSc-associated autoantibodies and autoantigens have been recently identified as activators of skin fibroblasts and monocytes in SSc (118,119). Immune complexes could also participate to endothelial damage in SSc, and IVIg could thus limit these effects (133). IVIg can simultaneously impair the differentiation of human monocyte-derived macrophages into anti- and pro-inflammatory macrophages notably by inhibiting GM-CSF-driven STAT-5 activation and TLR signaling (134). IVIg could thus participate in limiting both M1 and M2 activation (135).
The effects of IVIg on skin have been evaluated in vivo in the bleomycin mouse model (136). Human IVIg were injected for 5 consecutive days immediately (preventive) or after 28 days (therapeutic) of subcutaneous bleomycin injections. Thirty-five days after the onset of the experiment, IVIg significantly reduced collagen content both in the preventive and curative protocols. Seven days after the onset of the preventive protocol, IVIg significantly prevented the expression of TGF-β in the skin as well as dermal macrophage infiltrate (136).
The assessment of the impact of IVig in SSc is mainly based on the results of uncontrolled, open-label observational studies. In seven women with SSc (5 limited and 2 diffuse), IVIg significantly reduced joint pain and tenderness measured with the VAS, and increased hand function and quality of life (137). An observational study on 15 patients reported a significant improvement in GI involvement assessed with the GIT 2.0 as well as muscle disease parameters such as median creatine kinase level (138). This significant impact on muscle-related outcomes was confirmed in a multicenter observational cohort, that also reported a trend for an improvement of GI involvement (139). Two cohort studies have highlighted that steroid consumption was decreased at the end of IVIg in patients with SSc (139,140). A Japanese double-blind, placebo-controlled, multicenter trial including 63 patients with dcSSc demonstrated that the change in mRSS was similar 12 weeks after administration or at discontinuation of IVIg. This trial included various administration protocols and subgroups based on mRSS progression within each arm precluding firm conclusions regarding the primary outcome (141).
A randomized, multicenter, double-blind, placebo-controlled, phase II study to evaluate the efficacy and safety of IVIg in patients with early dcSSc was recently withdrawn due to business reasons (NCT04138485; (clinical-trial.gov)). The primary endpoint of the study was the difference in ACR-CRISS over 48 weeks. A new RCT is needed to assess the impact of IVIg in this population of early dcSSc. Another phase II trial is referenced on clinicaltrial.gov with safety as primary outcome (NCT04137224). The promising result on GI involvement may also suggest that some patients with lcSSc could also be responders to IVIg. The lack of availability of IVIg, which relies on serum donor banks, may thus constitute an issue for wide use of this therapeutic option in SSc.
2.3.2. Targeting the chemokine CCL24
Chemokines are small signaling proteins that play a key role in the migration and activation of cells. Chemokines have been hypothesized to be significant contributors to the early inflammatory molecular signature in SSc (142,143). Patients with SSc notably have increased serum levels of Chemokine c-c motif ligand 24, eotaxin-2 (CCL24). CCL24, and its receptor CCR3 (C-C chemokine receptor type 3), may contribute to the type 2 immune reaction involving Th2 lymphoctytes and M2 macrophages (Figure 1) (144). CCL24 is also involved in the migration of inflammatory cells and fibroblasts. In this perspective, it has been thought to block the homing of immune cells to tissue thus obtaining an anti-fibrotic effect. CCL24 also contributes to endothelial dysfunction being thus involved in all major pathogenic mechanisms of SSc. CM-101 is a humanized IgG1 monoclonal antibody against CCL24 with potential therapeutic impact both in lcSSc and dcSSc.
In preclinical models of SSc, including bleomycin-induced scleroderma in mice, CM-101 was significantly effective in mitigating the development of skin fibrosis and lung inflammation, characteristic of human SSc, with a good safety profile (145). For this reason, CM-101 might be useful for blocking the evolution SSc and similar clinical settings that involve lung and skin inflammation. A multicenter Phase II clinical trial, is planned in early dcSSc and lcSSc patients that will be randomized in three arms to receive one of the 2 highest doses found to be safe in the phase 1 study or placebo. The first clinical evidence of efficiency in patients with SSc is still to be further explored.
3. CONCLUSION: LESSONS AND PROSPECTS INHERITED FROM PULMONARY HYPERTENSION AND IDIOPATHIC PULMONARY FIBROSIS
Recent breakthroughs in the management of pulmonary hypertension, with a positive impact on survival, have been achieved by early association of existing drugs rather than the design of new treatments. This should inform the research on investigational drugs in SSc, especially considering that immunomodulatory drugs are now broadly used in patients with early dcSSc. Therefore, the identification of new therapeutic options has to be considered on top of existing immunomodulators already in use as background therapies. The association of monoclonal antibodies could also constitute a change of paradigm in the management if SSc, if such combinations prove to be well tolerated. The simultaneous inhibition of profibrotic and pro-inflammatory pathways by a single drug (JAK inhibitors) or through combination strategies (pirfenidone and MMF, or romilkimab and immunomodulators) may constitute a promising option for disease-modifying management of SSc. The introduction of anti-fibrotic drugs at the early inflammatory phase of the disease, in addition to immunomodulatory treatments, may also allow clinically meaningful results for patients with dcSSc in the future. Nonetheless, the relevance and safety of such associations are still to be determined in RCT.
Some recently FDA-approved drugs in SSc-ILD, such as nintedanib, or under investigation in Phase III trials in the same indication, such as pirfenidone, were initially approved in idiopathic pulmonary fibrosis (IPF). Thus, drugs under investigation in phase III trials for IPF may also represent new promising therapeutic options in SSc. A phase III trial of Pamrevlumab, a monoclonal antibody targeting CTGF/CCN2 is ongoing in IPF (NCT039551346). Interestingly, in a scleroderma mouse model of angiotensin II-induced skin fibrosis, targeting CTGF with a monoclonal antibody reduced skin fibrosis, suggesting that beyond IPF, CTGF/CCN2 targeting with Pamrevlumab may also show beneficial effects in SSc (146). Serum amyloid P (also called pentraxin-2, PTX-2) is an endogen mediator that may exert anti-inflammatory and antifibrotic properties notably through the enhancement of pro-resolving phagocytic properties of macrophages and inhibition of TGF-β secretion by CD204+ M2 (147,148). PTX-2 knockout mice show persistent inflammatory response and increase lung fibrosis after bleomycin challenge (149). Although the role of PTX-2 in SSc remains controversial (150,151), the phase III trial evaluating the efficacy and safety of recombinant human PTX-2 in IPF may reinforce the relevance of PTX-2 for SSc-ILD (NCT04552899). GKT-137831 is a NOX1/4 inhibitor that showed anti-fibrotic properties in preclinical models of SSc, as the production of ROS by NOX4 could participate in profibrotic TGF-β1 signaling (Figure 2) (152). The phase II trial of GKT-137831 in IPF may thus demonstrate the clinical relevance of targeting this pathway in pulmonary fibrosis which may represent another therapeutic opportunity for SSc-ILD (NCT03865927).
4. EXPERT OPINION: PRACTICAL CONSIDERATIONS FOR PERSONALIZED MEDICINE AND PROMISING CANDIDATE DRUGS
Beyond the selection of patients with a high-risk of progression, the next challenge may be the identification and stratification of patients according to the main underlying mechanism or main involved pathway to demonstrate the efficacy of therapeutic targets adapted to specific subpopulations. Three levels of stratification could guide this identification of new targets and new SSc subsets for personalized medicine: a molecular, cellular and phenotypic level.
4.1. The molecular level
Substantial progress has been made for the identification and stratification of global molecular signatures in the skin of patients with early SSc (153). Applying in-depth molecular analysis of skin biopsies, four distinct molecular signatures have been consistently identified by different research teams in observational studies and clinical trials. These are: an inflammatory pattern, a fibro-proliferative pattern, a normal-like pattern and a limited cutaneous pattern (41). The long-term natural history of these patterns is still to be determined but in short-term trials, they may predict treatment response (41,154). In patients with early dcSSc, the phase II trial assessing safety and efficacy of abatacept, has demonstrated that patients with an inflammatory pattern had a better response to therapy based on change of mRSS as compared to patients with a fibroproliferative pattern. This may suggest that inflammatory patients may benefit the most from anti-inflammatory drugs, and that JAK inhibitors or IVIg may be more relevant for this subset of patients. On the contrary, agents with direct anti-fibrotic properties (Selective TGF-β inhibitor or ROCK inhibitor) could be discussed for patients with a fibroproliferative gene signature. Additional gene signatures identified in skin biopsies could help for further enrichment in RCTs.
Cellular senescence is an age-related normal process characterized by irreversible exit of cells from the cell cycle. While cellular senescence is a normal part of biological aging and is likely to have beneficial homeostatic roles, the premature, excessive or sustained accumulation of senescent cells in fibrotic tissues can have detrimental effects. In particular, senescent cells can overexpress pro-fibrotic cytokines such as IL-6 and TGF-ß (a secretome called senescence-associated secretory pattern, SASP) (Figure 1) (155). Senolytic agents such as dasatinib can specifically target senescent fibroblasts. Dasatinib has been shown to limit bleomycin-induced lung fibrosis in mice (156). A recent pilot study sought to evaluate the effects of dasatinib in SSc-ILD. In this open-label clinical trial, improvers had higher skin gene signature of senescence at baseline; decrease in skin expression of SASP and other senescence-related gene sets upon treatment was associated with clinical improvement with decreasing skin score and stable or improving lung fibrosis scores (157). These results suggest that other informative gene signatures exist beyond the well-identified inflammatory, fibro-proliferative and normal-like gene signatures. Dedicated signature-based pilot studies allowing the identification of specific responders for a given treatment may be discussed for enrichment strategies for phase II or III trials.
4.2. The cellular level:
The results of hierarchical cluster analysis of flow cytometric characterization of peripheral blood mononuclear cells from SSc patients have allowed the identification of new subgroups of SSc patients. Three groups were thus defined in this study, a group with few immune abnormalities, another with high proportions of activated T and T reg cells and a last one where T-follicular helpers and plasmablasts were predominant (158). Interestingly this last group was associated with the progression of micro-vasculopathy. Simple cellular markers used in daily practice may also have a predictive value for organ involvement such as SSc-ILD, as increased monocyte count allows the identification of a subgroup of patients with lower survival (159).
At this cellular level, a better understanding of epigenetic regulations of myofibroblasts may also bring new treatment opportunities regarding fibrotic manifestations, as suggested by the recent identification of the transcription factor PU.1 as an essential regulator of the pro-fibrotic gene expression program in fibroblasts (160). The pharmacological and genetic inactivation of PU.1 led to the disruption of the fibrotic network and enabled reprogramming of fibrotic fibroblasts into resting fibroblasts with regression of the fibrotic features in vivo. Similarly, the specific targeting of myofibroblasts through death receptors such as the TRAIL/DRs pathways could also reverse established skin fibrosis to near-normal skin architecture in the bleomycin mouse model (161).
Beyond the identification of new cellular targets, a better understanding of cellular metabolism could also lead to new therapeutic options. In fibrosis, myofibroblasts show metabolic alterations, including enhanced glycolysis and disrupted NAD homeostasis (19,20). Macrophage polarization is associated with major metabolic changes. Inflammatory M1 have an anaerobic metabolism whereas M2 classically use aerobic mitochondrial respiration (162). Therefore, pro-inflammatory polarization of macrophages causes impaired mitochondrial respiration resulting in the accumulation of endogenous metabolites such as itaconate. Such metabolites can, in return, suppress the expression of proinflammatory cytokines. Recent studies identify itaconate as a key antifibrotic mediator (163). Itaconate is decreased in BAL fluids from patients with IPF and inhaled itaconate could ameliorate lung fibrosis in mice (164). These results suggest that itaconate acts as an endogenous pulmonary regulatory pathway that can limit fibrosis and could represent a new therapeutic lever.
4.3. The phenotypic level and the association with clinical trajectories
The precise association of these gene signatures or new prominent cellular subpopulations with skin trajectories or visceral involvement is still needed (30). These molecular and cellular considerations are still poorly included in our understanding of the natural history of the disease and their association with survival or trajectories of skin and organ involvement has not been validated. On the contrary, recent innovative approaches have proposed new stratification strategies based on simple biomarkers, such as antibody status, in association with disease cutaneous subtypes (dcSSc and lcSSc), which successfully predict clinical trajectory and survival (31). More innovative classification technics, based on cluster analyses of patients based on clinical manifestation during the entire course of the disease, have also demonstrated that SSc patients from the European EUSTAR cohort could be subclassified into 6 clusters with distinct prognoses (165). Simple and practical early predictive markers for detection of future severe patients are nonetheless still needed, and autoantibodies in combination with clinical, radiological and functional parameters could be promising candidates. Including molecular signatures in association with such parameters may also constitute a relevant strategy, although to date this approach remains less practical and less accessible (33). A key aspect of this phenotypic approach is the early detection of subgroups that will share common clinical trajectories. The evolution of skin and organ involvement is still partly unpredictable when only referring to the dcSSc and lcSSc subsets (166). Deciphering the early predictors of FVC decline or skin trajectories (167), but also identifying unified determinants of the severity of vascular phenotype are still key issues (168). Although fibrosis is considered as the end-stage of the disease process, all fibrotic features of the disease (e.g. fibrotic ILD or skin disease) may not share the exact same pathogenic process and/or the same natural history. The wrong assumption that SSc-associated fibrosis in all organs would share the exact same mechanisms with a similar natural history and trajectory may lead to inadequate therapeutic choices or selection of patients for RCTs (32). Beyond fibrosis, some patients also have specific inflammatory manifestations of the disease, such as inflammatory synovitis or pericardial effusion, which notably concern patients with anti-RNA polymerase III antibodies (169). In such subgroups of patients solely targeting fibrosis may not show promising results and therapeutic strategies based on immunomodulatory agents with combined anti-inflammatory and anti-fibrotic properties such as tocilizumab or JAK inhibitors may appear more relevant (48).
4.4. The experts’ selection of promising candidate drugs
Myeloablative stem cell-transplantation have demonstrated positive effects on survival and quality of life in SSc, demonstrating that a disease-modifying therapeutic approach may be possible (170,171). In this respect, the simultaneous targeting of multiple cytokines may be especially promising. The significant effect of the anti-IL-4 and IL-13 Romilkimab on mRSS in a phase II trial demonstrates that impacting Th2 signaling in dcSSc is a promising option and this should foster a future phase 3 trial for this drug in this population. Although targeting TGF-β could be considered as an “old” strategy, its central role in the pathogenesis of the disease and the design of new drugs targeting specific isoforms with potential better safety profile in addition with the selection of more adapted targeted subpopulations (early dcSSc) may help to revisit this pathway as a therapeutic option in SSc. The selective TGF-β1 and TGF-β3 antagonist AVID200 may thus constitute an especially promising candidate. Pan-JAK inhibitors such as Tofacitinib or Ruxolitinib may also be of specific interest in the future. Their widespread impact may participate to mimic the combined effects of drugs like tocilizumab, targeting IL-6 signaling, and Romilkimab, targeting IL-4 and 13 signaling, with thus potential impact on ILD and skin involvement in dcSSc (91). Moreover, the potential effects of JAK inhibitors on PAH in preclinical models may suggest that this therapeutic class may impact vasculopathy, providing a rationale for its relevance in lcSSc, a disease subset that represents more than 70% of SSc patients but that is still neglected to date despite its detrimental impact on quality of life (172–174).
Article Highlights.
Recent advances in the understanding of systemic sclerosis (SSc) pathogenesis have allowed the FDA approval of two drugs for the treatment of SSc-ILD (tocilizumab and nintedanib) but no drug has been approved as a disease-modifying agent in SSc so far.
TGF-β plays a key role in fibroblast activation and myofibroblast trans-differentiation through non-canonical and canonical pathways. Selective inhibition of TGF-β1 and TGF-β3 by AVID200, showed good tolerance and safety profile (NCT03831438) fostering the relevance of future trials evaluating the efficacy of this drug in SSc.
Considering the multiplicity of pathways involved in the pathogenesis of SSc, simultaneous targeting of multiple cytokines by the same drug may be especially promising. The concomitant targeting of the immunomodulatory and profibrotic IL-4 and IL-13 with Romilkimab has shown promising results on skin involvement in dcSSc in a phase II trial (NCT02921971)
Considering the recent demonstration that both M1 (pro-inflammatory) and M2 (pro-fibrotic) macrophages were over-activated in the skin of patients with diffuse SSc, JAK inhibition with pan-JAK inhibitors such as Tofacitinib (NCT03274076) or Ruxolitnib may be especially promising with potential impact on IL-6 (STAT-1/3 dependent) pro-inflammatory & profibrotic signaling, and IL-4/IL-13 (STAT6 dependent) pro-fibrotic & immunomodulatory signaling.
Combination therapies may offer the opportunity of targeting multiple pathways at the same time. In this respect, the ongoing Scleroderma-Lung-Study III, a phase III trial assessing the efficacy of Pirfenidone versus placebo, as add-on therapy with mycophenolate mofetil in patients with SSc-ILD (NCT03221257), may demonstrate the proof of concept of the combined efficacy of an immunomodulatory drug with an anti-fibrotic molecule in SSc. This may pave the way for further evaluation of combination therapies for SSc in the future.
These potential new therapeutic options will still need to be adapted to relevant subpopulations of patients, highlighting the importance of pursuing the effort for better stratification and precise phenotyping of patients with SSc. Identification of molecular signatures that could predict treatment response as identified in the abatacept trial (NCT02161406), may constitute a promising strategy.
Acknowledgments
Funding
A Lescoat was funded by the French network of the University Hospitals HUGO (Hôpitaux Universitaires du Grand Ouest-GIRCI) (AAP JCM2020) and by grant support from the Rennes University Hospital (CORECT Visiting Grant 2020). D Khanna was funded by grant support from the NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases (K24-AR-063129)
Declaration of interest
J Varga reports consulting agreements/advisory for Boehringer Ingelheim, TeneoBio, Mitobridge, Emerald Pharma, Horizon Therapeutics, Formation Biologics, Astellas, Forbius, Abingworth and research funding from Pfizer, Bristol-Myers-Squibb, Teneo-Bio, Takeda, and Sun Bio.
M Matucci-Cerinic has received consulting fees or honorarium from Actelion, Janssen, Inventiva, Bayer, Biogen, Boehringer, CSL Behring, Corbus, Galapagos, Mitsubishi, Samsung, Regeneron, Acceleron, MSD, Chemomab, Lilly, Pfizer, and Roche. D Khanna reports the following conflicts of interest: Grant support from NIH, Immune Tolerance Network, Bayer, BMS, Horizon, Pfizer. Consultant: Acceleron, Actelion, Abbvie, Amgen, Bayer, Boehringer Ingelheim, CSL Behring, Corbus, Gilead, Galapagos, Genentech/Roche, GSK, Horizon, Merck, Mitsubishi Tanabe Pharma, Sanofi-Aventis, and United Therapeutics. He also holds stocks in Eicos Sciences, Inc (less than 5%) and has Leadership/Equity positions – Chief Medical Officer, CiviBioPharma/Eicos Sciences, Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Abbreviations
- FAK
focal adhesion kinase
- LPA
lysophosphatidic acid
- LPA-R
LPA-Receptor
- IL-6R
IL-6 receptor
- TGF-βRI & II
TGF-β receptor I & II
- PAI-1
plasminogen activator inhibitor 1
- ROS
Reactive Oxygen Species
- YAP
Yes Associated Protein
- α-MSH
α-Melanocyte-stimulating hormone
- IL-4Rα
IL-4 receptor α
- IL-13Rα1
IL-13 receptor α1
- ECM
Extra-cellular matrix
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers 2015;1:15002. [DOI] [PubMed] [Google Scholar]
- 2.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007;117:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes M, Allanore Y, Chung L, Pauling JD, Denton CP, Matucci-Cerinic M. Raynaud phenomenon and digital ulcers in systemic sclerosis. Nat Rev Rheumatol 2020;16:208–221. [DOI] [PubMed] [Google Scholar]
- 4.Denton CP, Khanna D. Systemic sclerosis. The Lancet 2017;390:1685–1699. [DOI] [PubMed] [Google Scholar]
- 5.Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, Mayes MD, et al. Nintedanib for Systemic Sclerosis–Associated Interstitial Lung Disease. New England Journal of Medicine 2019;380:2518–2528.** This phase III trial demonstrates the efficacy of FDA-approved nintedanib to limit FVC decline in SSc-ILD
- 6.Wollin L, Distler JH, Denton CP, Gahlemann M. Rationale for the evaluation of nintedanib as a treatment for systemic sclerosis–associated interstitial lung disease. Journal of Scleroderma and Related Disorders 2019;4:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuwana M, Distler O. Recent progress and missing gaps to achieve goal in the care of systemic sclerosis–associated interstitial lung disease. Journal of Scleroderma and Related Disorders 2020;5:3–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum 2013;65:1953–1962. [DOI] [PubMed] [Google Scholar]
- 9.Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 2020;79:379–386.* This study demonstrates that M1, M2 and fibroblast associated skin signatures are key features in patients with early dcSSc
- 10.Chakraborty D, Šumová B, Mallano T, Chen C-W, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun 2017;8:1130.* This study demonstrates that STAT3 is a key integrator of profibrotic signaling in SSc.
- 11.Walraven M, Hinz B. Therapeutic approaches to control tissue repair and fibrosis: Extracellular matrix as a game changer. Matrix Biol 2018;71–72:205–224. [DOI] [PubMed] [Google Scholar]
- 12.Shaw TJ, Rognoni E. Dissecting Fibroblast Heterogeneity in Health and Fibrotic Disease. Curr Rheumatol Rep 2020;22:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat M-L, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170:1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lafyatis R Transforming growth factor β--at the centre of systemic sclerosis. Nat Rev Rheumatol 2014;10:706–719. [DOI] [PubMed] [Google Scholar]
- 15.Parapuram SK, Shi-wen X, Elliott C, Welch ID, Jones H, Baron M, et al. Loss of PTEN expression by dermal fibroblasts causes skin fibrosis. J Invest Dermatol 2011;131:1996–2003. [DOI] [PubMed] [Google Scholar]
- 16.Liu S, Shi-wen X, Abraham DJ, Leask A. CCN2 is required for bleomycin-induced skin fibrosis in mice. Arthritis Rheum 2011;63:239–246. [DOI] [PubMed] [Google Scholar]
- 17.Toyama T, Looney AP, Baker BM, Stawski L, Haines P, Simms R, et al. Therapeutic Targeting of TAZ and YAP by Dimethyl Fumarate in Systemic Sclerosis Fibrosis. J Invest Dermatol 2018;138:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi-Wen X, Racanelli M, Ali A, Simon A, Quesnel K, Stratton RJ, et al. Verteporfin inhibits the persistent fibrotic phenotype of lesional scleroderma dermal fibroblasts. J Cell Commun Signal 2021;15:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Psarianos P, Ghoraie LS, Yip K, Goldstein D, Gilbert R, et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat Metab 2019;1:147–157. [DOI] [PubMed] [Google Scholar]
- 20.Shi B, Wang W, Korman B, Kai L, Wang Q, Wei J, et al. Targeting CD38-dependent NAD+ metabolism to mitigate multiple organ fibrosis. iScience 2021;24:101902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lerbs T, Cui L, King ME, Chai T, Muscat C, Chung L, et al. CD47 prevents the elimination of diseased fibroblasts in scleroderma. JCI Insight 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nihtyanova SI, Denton CP. Pathogenesis of systemic sclerosis associated interstitial lung disease. Journal of Scleroderma and Related Disorders 2020;5:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun 2016;7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest 2013;123:1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagares D, Busnadiego O, García-Fernández RA, Kapoor M, Liu S, Carter DE, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum 2012;64:1653–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi-wen X, Thompson K, Khan K, Liu S, Murphy-Marshman H, Baron M, et al. Focal adhesion kinase and reactive oxygen species contribute to the persistent fibrotic phenotype of lesional scleroderma fibroblasts. Rheumatology (Oxford) 2012;51:2146–2154. [DOI] [PubMed] [Google Scholar]
- 27.Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apostolidis SA, Stifano G, Tabib T, Rice LM, Morse CM, Kahaleh B, et al. Single Cell RNA Sequencing Identifies HSPG2 and APLNR as Markers of Endothelial Cell Injury in Systemic Sclerosis Skin. Front Immunol 2018;9:2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Bittar HT, et al. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Annals of the Rheumatic Diseases 2019;78:1379–1387.* This analysis demonstrates the heterogeneity of skin fibroblasts in SSc using single cell RNA seq.
- 30.Ledoult E, Launay D, Béhal H, Mouthon L, Pugnet G, Lega J-C, et al. Early trajectories of skin thickening are associated with severity and mortality in systemic sclerosis. Arthritis Res Ther 2020;22:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nihtyanova SI, Sari A, Harvey JC, Leslie A, Derrett‐Smith EC, Fonseca C, et al. Using Autoantibodies and Cutaneous Subset to Develop Outcome‐Based Disease Classification in Systemic Sclerosis. Arthritis Rheumatol 2020;72:465–476.* This study uses autoantibodies to propose a new disease classification of patients with SSc.
- 32.Sobanski V, Lescoat A, Launay D. Novel classifications for systemic sclerosis: challenging historical subsets to unlock new doors. Curr Opin Rheumatol 2020;32:463–471. [DOI] [PubMed] [Google Scholar]
- 33.Hinchcliff M, Mahoney JM. Towards a new classification of systemic sclerosis. Nature Reviews Rheumatology 2019;34:1–2. [DOI] [PubMed] [Google Scholar]
- 34.Dobrota R, Maurer B, Graf N, Jordan S, Mihai C, Kowal-Bielecka O, et al. Prediction of improvement in skin fibrosis in diffuse cutaneous systemic sclerosis: a EUSTAR analysis. Ann Rheum Dis 2016;75:1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khanna D, Lin CJF, Furst DE, Goldin J, Kim G, Kuwana M, et al. Tocilizumab in systemic sclerosis: a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet Respiratory Medicine 2020:S2213260020303180.** This phase III trial demonstrates the efficacy of FDA-approved Tocilizumab to limit FVC decline in early dcSSc patients with SSc-ILD
- 36.Volkmann ER, Tashkin DP, LeClair H, Roth MD, Kim G, Goldin J, et al. Treatment With Mycophenolate and Cyclophosphamide Leads to Clinically Meaningful Improvements in Patient-Reported Outcomes in Scleroderma Lung Disease: Results of Scleroderma Lung Study II. ACR Open Rheumatology 2020;2:362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roofeh D, Lescoat A, Khanna D. Emerging drugs for the treatment of scleroderma: a review of recent phase 2 and 3 trials. Expert Opin Emerg Drugs 2020:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roofeh D, Khanna D. Management of systemic sclerosis: the first five years. Curr Opin Rheumatol 2020;32:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y, Distler JH. Therapeutic molecular targets of SSc-ILD. Journal of Scleroderma and Related Disorders 2020;5:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khanna D, Denton CP, Jahreis A, Laar JM van, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016;387:2630–2640.* This phase II trial demonstrates the efficacy of FDA-approved Tocilizumab to limit FVC decline in early dcSSc patients with SSc-ILD
- 41.Khanna D, Spino C, Johnson S, Chung L, Whitfield ML, Denton CP, et al. Abatacept in Early Diffuse Cutaneous Systemic Sclerosis: Results of a Phase II Investigator-Initiated, Multicenter, Double-Blind, Randomized, Placebo-Controlled Trial. Arthritis & Rheumatology 2020;72:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung L, Spino C, McLain R, Johnson SR, Denton CP, Molitor JA, et al. Safety and efficacy of abatacept in early diffuse cutaneous systemic sclerosis (ASSET): open-label extension of a phase 2, double-blind randomised trial. The Lancet Rheumatology 2020;2:e743–e753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung MP, Chung L. Drugs in phase I and phase II clinical trials for systemic sclerosis. Expert Opinion on Investigational Drugs 2020;29:349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Black CM, Silman AJ, Herrick AI, Denton CP, Wilson H, Newman J, et al. Interferon-alpha does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 1999;42:299–305. [DOI] [PubMed] [Google Scholar]
- 45.Skaug B, Assassi S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine 2020;132:154635. [DOI] [PubMed] [Google Scholar]
- 46.Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest 2015;125:2795–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang C, Zhang Y, Zhang C, Liu Y, Liu Y, Xu G. Pioglitazone increases VEGFR3 expression and promotes activation of M2 macrophages via the peroxisome proliferator‑activated receptor γ. Mol Med Rep 2019;19:2740–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roofeh D, Lin CJF, Goldin J, Kim GH, Furst DE, Denton CP, et al. Tocilizumab Prevents Progression of Early Systemic Sclerosis Associated Interstitial Lung Disease. Arthritis Rheumatol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J 1987;247:597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, et al. Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J Biol Chem 1995;270:7304–7310. [DOI] [PubMed] [Google Scholar]
- 51.Morin F, Kavian N, Batteux F. Animal models of systemic sclerosis. Curr Pharm Des 2015;21:2365–2379. [DOI] [PubMed] [Google Scholar]
- 52.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A 1986;83:4167–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sfikakis PP, McCune BK, Tsokos M, Aroni K, Vayiopoulos G, Tsokos GC. Immunohistological demonstration of transforming growth factor-beta isoforms in the skin of patients with systemic sclerosis. Clin Immunol Immunopathol 1993;69:199–204. [DOI] [PubMed] [Google Scholar]
- 54.Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C. Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud’s phenomenon. Arthritis Rheum 1994;37:278–288. [DOI] [PubMed] [Google Scholar]
- 55.Ozbilgin MK, Inan S. The roles of transforming growth factor type beta3 (TGF-beta3) and mast cells in the pathogenesis of scleroderma. Clin Rheumatol 2003;22:189–195. [DOI] [PubMed] [Google Scholar]
- 56.Hasegawa M, Sato S, Takehara K. Augmented production of transforming growth factor-beta by cultured peripheral blood mononuclear cells from patients with systemic sclerosis. Arch Dermatol Res 2004;296:89–93. [DOI] [PubMed] [Google Scholar]
- 57.King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–2092. [DOI] [PubMed] [Google Scholar]
- 58.Hall CL, Wells AR, Leung KP. Pirfenidone reduces profibrotic responses in human dermal myofibroblasts, in vitro. Lab Invest 2018;98:640–655. [DOI] [PubMed] [Google Scholar]
- 59.Khanna D, Albera C, Fischer A, Khalidi N, Raghu G, Chung L, et al. An Open-label, Phase II Study of the Safety and Tolerability of Pirfenidone in Patients with Scleroderma-associated Interstitial Lung Disease: the LOTUSS Trial. J Rheumatol 2016;43:1672–1679. [DOI] [PubMed] [Google Scholar]
- 60.Acharya N, Sharma SK, Mishra D, Dhooria S, Dhir V, Jain S. Efficacy and safety of pirfenidone in systemic sclerosis-related interstitial lung disease-a randomised controlled trial. Rheumatol Int 2020;40:703–710. [DOI] [PubMed] [Google Scholar]
- 61.Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum 2007;56:323–333. [DOI] [PubMed] [Google Scholar]
- 62.Lafyatis R, Spiera R, Domsic R, Papazoglou A, Ligon C, Morse CMZ, et al. Thu0329 Safety, Target Engagement, and Initial Efficacy of Avid200, a First-in-Class Potent and Isoform-Selective Inhibitor of Tgf-Beta 1 and 3, in Patients with Diffuse Cutaneous Systemic Sclerosis (dcssc): A Phase 1 Dose Escalation Study. Annals of the Rheumatic Diseases 2020;79:394–395. [Google Scholar]
- 63.Khanna D, Tashkin DP, Wells AU, Seibold JR, Wax S, Vazquez-Mateo C, et al. STRATUS: A Phase II Study of Abituzumab in Patients With Systemic Sclerosis–associated Interstitial Lung Disease. The Journal of Rheumatology 2021;in press. [DOI] [PubMed] [Google Scholar]
- 64.Da Q, Yan Z, Li Z, Han Z, Ren M, Huang L, et al. TAK1 is involved in sodium L-lactate-stimulated p38 signaling and promotes apoptosis. Mol Cell Biochem 2020. [DOI] [PubMed] [Google Scholar]
- 65.Chen Y, Shi-wen X, Eastwood M, Black CM, Denton CP, Leask A, et al. Contribution of activin receptor-like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum 2006;54:1309–1316. [DOI] [PubMed] [Google Scholar]
- 66.Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 2003;278:12384–12389. [DOI] [PubMed] [Google Scholar]
- 67.Kahl DJ, Hutchings KM, Lisabeth EM, Haak AJ, Leipprandt JR, Dexheimer T, et al. 5-Aryl-1,3,4-oxadiazol-2-ylthioalkanoic Acids: A Highly Potent New Class of Inhibitors of Rho/Myocardin-Related Transcription Factor (MRTF)/Serum Response Factor (SRF)-Mediated Gene Transcription as Potential Antifibrotic Agents for Scleroderma. J Med Chem 2019;62:4350–4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilles G, McCulloch AD, Brakebusch CH, Herum KM. Maintaining resting cardiac fibroblasts in vitro by disrupting mechanotransduction. PLoS One 2020;15:e0241390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Akhmetshina A, Dees C, Pileckyte M, Szucs G, Spriewald BM, Zwerina J, et al. Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum 2008;58:2553–2564. [DOI] [PubMed] [Google Scholar]
- 70.Oh RS, Haak AJ, Smith KMJ, Ligresti G, Choi KM, Xie T, et al. RNAi screening identifies a mechanosensitive ROCK-JAK2-STAT3 network central to myofibroblast activation. J Cell Sci 2018;131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lescoat A, Ballerie A, Lelong M, Augagneur Y, Morzadec C, Jouneau S, et al. Crystalline Silica Impairs Efferocytosis Abilities of Human and Mouse Macrophages: Implication for Silica-Associated Systemic Sclerosis. Front Immunol 2020;11:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pehlivan Y, Yolbas S, Cetin GY, Alibaz-Oner F, Cagatay Y, Yilmaz N, et al. Investigation of the association between Rho/Rho-kinase gene polymorphisms and systemic sclerosis. Rheumatol Int 2016;36:421–427. [DOI] [PubMed] [Google Scholar]
- 73.Xie Y, Zhao D, Dong P, Lai L. Macrophage-targeting Fasudil treatment protects liver from the ischemia/reperfusion injury by promoting M2 macrophage polarization. Biosci Rep 2018. [DOI] [PubMed] [Google Scholar]
- 74.Novelli D, Fumagalli F, Staszewsky L, Ristagno G, Olivari D, Masson S, et al. Monocrotaline-induced pulmonary arterial hypertension: Time-course of injury and comparative evaluation of macitentan and Y-27632, a Rho kinase inhibitor. Eur J Pharmacol 2019;865:172777. [DOI] [PubMed] [Google Scholar]
- 75.Bei Y, Hua-Huy T, Nicco C, Duong-Quy S, Le-Dong N-N, Tiev K-P, et al. RhoA/Rho-kinase activation promotes lung fibrosis in an animal model of systemic sclerosis. Exp Lung Res 2016;42:44–55. [DOI] [PubMed] [Google Scholar]
- 76.Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016;127:2144–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fava A, Wung PK, Wigley FM, Hummers LK, Daya NR, Ghazarian SR, et al. Efficacy of Rho kinase inhibitor fasudil in secondary Raynaud’s phenomenon. Arthritis Care Res (Hoboken) 2012;64:925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y, Wu S. Effects of fasudil on pulmonary hypertension in clinical practice. Pulm Pharmacol Ther 2017;46:54–63. [DOI] [PubMed] [Google Scholar]
- 79.De Silva TM, Kinzenbaw DA, Modrick ML, Reinhardt LD, Faraci FM. Heterogeneous Impact of ROCK2 on Carotid and Cerebrovascular Function. Hypertension 2016;68:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Y, Natarajan V. Lysophosphatidic acid (LPA) and its receptors: role in airway inflammation and remodeling. Biochim Biophys Acta 2013;1831:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol 2014;171:3575–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ledein L, Léger B, Dees C, Beyer C, Distler A, Vettori S, et al. Translational engagement of lysophosphatidic acid receptor 1 in skin fibrosis: from dermal fibroblasts of patients with scleroderma to tight skin 1 mouse. Br J Pharmacol 2020;177:4296–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sato M, Shegogue D, Hatamochi A, Yamazaki S, Trojanowska M. Lysophosphatidic acid inhibits TGF-beta-mediated stimulation of type I collagen mRNA stability via an ERK-dependent pathway in dermal fibroblasts. Matrix Biol 2004;23:353–361. [DOI] [PubMed] [Google Scholar]
- 84.Castelino FV, Bain G, Pace VA, Black KE, George L, Probst CK, et al. An Autotaxin/Lysophosphatidic Acid/Interleukin-6 Amplification Loop Drives Scleroderma Fibrosis. Arthritis & Rheumatology (Hoboken, NJ) 2016;68:2964–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tokumura A, Carbone LD, Yoshioka Y, Morishige J, Kikuchi M, Postlethwaite A, et al. Elevated serum levels of arachidonoyl-lysophosphatidic acid and sphingosine 1-phosphate in systemic sclerosis. Int J Med Sci 2009;6:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ninou I, Kaffe E, Müller S, Budd DC, Stevenson CS, Ullmer C, et al. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm Pharmacol Ther 2018;52:32–40. [DOI] [PubMed] [Google Scholar]
- 87.Allanore Y, Distler O, Jagerschmidt A, Illiano S, Ledein L, Boitier E, et al. Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for Patients With Diffuse Cutaneous Systemic Sclerosis: A Double-Blind, Randomized, Eight-Week Placebo-Controlled Study Followed by a Sixteen-Week Open-Label Extension Study. Arthritis Rheumatol 2018;70:1634–1643. [DOI] [PubMed] [Google Scholar]
- 88.Khanna D, Denton C, Furst D, Mayes M, Matucci-Cerinic M, Smith V, et al. A Phase 2a Randomized, Double-blind, Placebo-controlled Study of Ziritaxestat in Early Diffuse Cutaneous Systemic Sclerosis (NOVESA). Arthritis & Rheumatology 2020;72 (suppl 10). [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Liang R, Chen C-W, Mallano T, Dees C, Distler A, et al. JAK1-dependent transphosphorylation of JAK2 limits the antifibrotic effects of selective JAK2 inhibitors on long-term treatment. Ann Rheum Dis 2017;76:1467–1475. [DOI] [PubMed] [Google Scholar]
- 90.Talotta R The rationale for targeting the JAK/STAT pathway in scleroderma-associated interstitial lung disease. Immunotherapy 2020;13:241–256. [DOI] [PubMed] [Google Scholar]
- 91.Lescoat A, Lelong M, Jeljeli M, Piquet-Pellorce C, Morzadec C, Ballerie A, et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem Pharmacol 2020;178:114103. [DOI] [PubMed] [Google Scholar]
- 92.Wang W, Bhattacharyya S, Marangoni RG, Carns M, Dennis-Aren K, Yeldandi A, et al. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. Journal of Scleroderma and Related Disorders 2020;5:40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aung WW, Wang C, Xibei J, Horii M, Mizumaki K, Kano M, et al. Immunomodulating role of the JAKs inhibitor tofacitinib in a mouse model of bleomycin-induced scleroderma. J Dermatol Sci 2020. [DOI] [PubMed] [Google Scholar]
- 94.Khanna D, Nagaraja V, Koenig A, Khanna P, Young A, Moore J, et al. Tofacitinib in Early Diffuse Cutaneous Systemic Sclerosis— Results of Phase I/II Investigator-Initiated, Double-Blind Randomized Placebo-Controlled Trial. ACR Meeting Abstracts. Available at: https://acrabstracts.org/abstract/tofacitinib-in-early-diffuse-cutaneous-systemic-sclerosis-results-of-phase-i-ii-investigator-initiated-double-blind-randomized-placebo-controlled-trial/. Accessed August 17, 2020. [Google Scholar]
- 95.Komai T, Shoda H, Hanata N, Fujio K. Tofacitinib rapidly ameliorated polyarthropathy in a patient with systemic sclerosis. Scand J Rheumatol 2018;47:505–506. [DOI] [PubMed] [Google Scholar]
- 96.Damsky W, Patel D, Garelli CJ, Garg M, Wang A, Dresser K, et al. Jak Inhibition Prevents Bleomycin-Induced Fibrosis in Mice and Is Effective in Patients with Morphea. Journal of Investigative Dermatology 2020;140:1446–1449.e4. [DOI] [PubMed] [Google Scholar]
- 97.Zeiser R, Bubnoff N von, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N Engl J Med 2020;382:1800–1810. [DOI] [PubMed] [Google Scholar]
- 98.Saidu NEB, Bonini C, Dickinson A, Grce M, Inngjerdingen M, Koehl U, et al. New Approaches for the Treatment of Chronic Graft-Versus-Host Disease: Current Status and Future Directions. Front Immunol 2020;11:578314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goll GL, Kvien TK. New-generation JAK inhibitors: how selective can they be? Lancet 2018;391:2477–2478. [DOI] [PubMed] [Google Scholar]
- 100.Roofeh D, Distler O, Allanore Y, Denton CP, Khanna D. Treatment of systemic sclerosis–associated interstitial lung disease: Lessons from clinical trials. Journal of Scleroderma and Related Disorders 2020;5:61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raghu G, Richeldi L, Crestani B, Wung P, Bejuit R, Esperet C, et al. SAR156597 in idiopathic pulmonary fibrosis: a phase 2 placebo-controlled study (DRI11772). Eur Respir J 2018;52. [DOI] [PubMed] [Google Scholar]
- 102.Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol 1997;24:328–332. [PubMed] [Google Scholar]
- 103.Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res 2020;312:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McGaha T, Saito S, Phelps RG, Gordon R, Noben-Trauth N, Paul WE, et al. Lack of skin fibrosis in tight skin (TSK) mice with targeted mutation in the interleukin-4R alpha and transforming growth factor-beta genes. J Invest Dermatol 2001;116:136–143. [DOI] [PubMed] [Google Scholar]
- 105.Jakubzick C, Choi ES, Joshi BH, Keane MP, Kunkel SL, Puri RK, et al. Therapeutic attenuation of pulmonary fibrosis via targeting of IL-4- and IL-13-responsive cells. J Immunol 2003;171:2684–2693. [DOI] [PubMed] [Google Scholar]
- 106.Allanore Y, Wung P, Soubrane C, Esperet C, Marrache F, Bejuit R, et al. A randomised, double-blind, placebo-controlled, 24-week, phase II, proof-of-concept study of romilkimab (SAR156597) in early diffuse cutaneous systemic sclerosis. Annals of the Rheumatic Diseases 2020;79:1600–1607.** This phase II trial demonstrates the potential effects of Romilkimab to limit skin fibrosis in dcSSc.
- 107.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science 1992;257:1248–1251. [DOI] [PubMed] [Google Scholar]
- 108.Böhm M, Raghunath M, Sunderkötter C, Schiller M, Ständer S, Brzoska T, et al. Collagen metabolism is a novel target of the neuropeptide alpha-melanocyte-stimulating hormone. J Biol Chem 2004;279:6959–6966. [DOI] [PubMed] [Google Scholar]
- 109.Kokot A, Sindrilaru A, Schiller M, Sunderkötter C, Kerkhoff C, Eckes B, et al. alpha-melanocyte-stimulating hormone suppresses bleomycin-induced collagen synthesis and reduces tissue fibrosis in a mouse model of scleroderma: melanocortin peptides as a novel treatment strategy for scleroderma? Arthritis Rheum 2009;60:592–603. [DOI] [PubMed] [Google Scholar]
- 110.Taherzadeh S, Sharma S, Chhajlani V, Gantz I, Rajora N, Demitri MT, et al. alpha-MSH and its receptors in regulation of tumor necrosis factor-alpha production by human monocyte/macrophages. Am J Physiol 1999;276:R1289–1294. [DOI] [PubMed] [Google Scholar]
- 111.Delgado R, Carlin A, Airaghi L, Demitri MT, Meda L, Galimberti D, et al. Melanocortin peptides inhibit production of proinflammatory cytokines and nitric oxide by activated microglia. J Leukoc Biol 1998;63:740–745. [DOI] [PubMed] [Google Scholar]
- 112.Dinparastisaleh R, Mirsaeidi M. Antifibrotic and Anti-Inflammatory Actions of α-Melanocytic Hormone: New Roles for an Old Player. Pharmaceuticals (Basel) 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 2004;304:1147–1150. [DOI] [PubMed] [Google Scholar]
- 114.Montero-Melendez T, Patel HB, Seed M, Nielsen S, Jonassen TEN, Perretti M. The melanocortin agonist AP214 exerts anti-inflammatory and proresolving properties. Am J Pathol 2011;179:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miao Y-L, Deng X-M, Li J-B, Zhao X-K, Cao J-M, Tian Y-P. [Effect of alpha-melanocyte stimulating hormone on the apoptosis of the vascular endothelial cell of the lung in two-hit acute respiratory distress syndrome in rat]. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 2004;16:596–598. [PubMed] [Google Scholar]
- 116.Colombo G, Gatti S, Sordi A, Turcatti F, Carlin A, Rossi C, et al. Production and effects of alpha-melanocyte-stimulating hormone during acute lung injury. Shock 2007;27:326–333. [DOI] [PubMed] [Google Scholar]
- 117.Leroy V, Henrot P, Barnetche T, Cario M, Darrigade A-S, Manicki P, et al. Association of skin hyperpigmentation disorders with digital ulcers in systemic sclerosis: Analysis of a cohort of 239 patients. J Am Acad Dermatol 2019;80:478–484. [DOI] [PubMed] [Google Scholar]
- 118.Raschi E, Chighizola CB, Cesana L, Privitera D, Ingegnoli F, Mastaglio C, et al. Immune complexes containing scleroderma-specific autoantibodies induce a profibrotic and proinflammatory phenotype in skin fibroblasts. Arthritis Res Ther 2018;20:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao X, Jia G, Guttman A, DePianto DJ, Morshead KB, Sun K-H, et al. Osteopontin Links Myeloid Activation and Disease Progression in Systemic Sclerosis. Cell Rep Med 2020;1:100140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum 2004;50:1918–1927. [DOI] [PubMed] [Google Scholar]
- 121.Matsushita T, Kobayashi T, Mizumaki K, Kano M, Sawada T, Tennichi M, et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci Adv 2018;4:eaas9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lafyatis R, O’Hara C, Feghali-Bostwick CA, Matteson E. B cell infiltration in systemic sclerosis-associated interstitial lung disease. Arthritis Rheum 2007;56:3167–3168. [DOI] [PubMed] [Google Scholar]
- 123.Hasegawa M, Hamaguchi Y, Yanaba K, Bouaziz J-D, Uchida J, Fujimoto M, et al. B-lymphocyte depletion reduces skin fibrosis and autoimmunity in the tight-skin mouse model for systemic sclerosis. Am J Pathol 2006;169:954–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yoshizaki A, Iwata Y, Komura K, Ogawa F, Hara T, Muroi E, et al. CD19 regulates skin and lung fibrosis via Toll-like receptor signaling in a model of bleomycin-induced scleroderma. Am J Pathol 2008;172:1650–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ponsoye M, Frantz C, Ruzehaji N, Nicco C, Elhai M, Ruiz B, et al. Treatment with abatacept prevents experimental dermal fibrosis and induces regression of established inflammation-driven fibrosis. Ann Rheum Dis 2016;75:2142–2149. [DOI] [PubMed] [Google Scholar]
- 126.Einhaus J, Pecher A-C, Asteriti E, Schmid H, Secker K-A, Duerr-Stoerzer S, et al. Inhibition of effector B cells by ibrutinib in systemic sclerosis. Arthritis Res Ther 2020;22:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Goswami RP, Ray A, Chatterjee M, Mukherjee A, Sircar G, Ghosh P. Rituximab in the treatment of systemic sclerosis-related interstitial lung disease: a systematic review and meta-analysis. Rheumatology (Oxford) 2020. [DOI] [PubMed] [Google Scholar]
- 128.Elhai M, Boubaya M, Distler O, Smith V, Matucci-Cerinic M, Alegre Sancho JJ, et al. Outcomes of patients with systemic sclerosis treated with rituximab in contemporary practice: a prospective cohort study. Ann Rheum Dis 2019;78:979–987. [DOI] [PubMed] [Google Scholar]
- 129.Zamanian RT, Badesch D, Chung L, Domsic RT, Medsger T, Pinckney A, et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis Associated Pulmonary Arterial Hypertension: A Multi-center, Double-blind, Randomized, Placebo-controlled Trial. Am J Respir Crit Care Med 2021. Available at: http://www.atsjournals.org/doi/10.1164/rccm.202009-3481OC. Accessed March 13, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gordon JK, Martyanov V, Franks JM, Bernstein EJ, Szymonifka J, Magro C, et al. Belimumab for the Treatment of Early Diffuse Systemic Sclerosis: Results of a Randomized, Double-Blind, Placebo-Controlled, Pilot Trial. Arthritis Rheumatol 2018;70:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Waller EK, Miklos D, Cutler C, Arora M, Jagasia MH, Pusic I, et al. Ibrutinib for Chronic Graft-versus-Host Disease After Failure of Prior Therapy: 1-Year Update of a Phase 1b/2 Study. Biol Blood Marrow Transplant 2019;25:2002–2007. [DOI] [PubMed] [Google Scholar]
- 132.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol 2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 133.Raschi E, Privitera D, Bodio C, Lonati PA, Borghi MO, Ingegnoli F, et al. Scleroderma-specific autoantibodies embedded in immune complexes mediate endothelial damage: an early event in the pathogenesis of systemic sclerosis. Arthritis Res Ther 2020;22:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Domínguez-Soto Á, Simón-Fuentes M, Las Casas-Engel M de, Cuevas VD, López-Bravo M, Domínguez-Andrés J, et al. IVIg Promote Cross-Tolerance against Inflammatory Stimuli In Vitro and In Vivo. J Immunol 2018;201:41–52. [DOI] [PubMed] [Google Scholar]
- 135.Kozicky LK, Zhao ZY, Menzies SC, Fidanza M, Reid GSD, Wilhelmsen K, et al. Intravenous immunoglobulin skews macrophages to an anti-inflammatory, IL-10-producing activation state. J Leukoc Biol 2015;98:983–994. [DOI] [PubMed] [Google Scholar]
- 136.Kajii M, Suzuki C, Kashihara J, Kobayashi F, Kubo Y, Miyamoto H, et al. Prevention of excessive collagen accumulation by human intravenous immunoglobulin treatment in a murine model of bleomycin-induced scleroderma. Clin Exp Immunol 2011;163:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nacci F, Righi A, Conforti ML, Miniati I, Fiori G, Martinovic D, et al. Intravenous immunoglobulins improve the function and ameliorate joint involvement in systemic sclerosis: a pilot study. Annals of the Rheumatic Diseases 2007;66:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Raja J, Nihtyanova SI, Murray CD, Denton CP, Ong VH. Sustained benefit from intravenous immunoglobulin therapy for gastrointestinal involvement in systemic sclerosis. Rheumatology (Oxford) 2016;55:115–119. [DOI] [PubMed] [Google Scholar]
- 139.Sanges S, Rivière S, Mekinian A, Martin T, Le Quellec A, Chatelus E, et al. Intravenous immunoglobulins in systemic sclerosis: Data from a French nationwide cohort of 46 patients and review of the literature. Autoimmun Rev 2017;16:377–384. [DOI] [PubMed] [Google Scholar]
- 140.Chaigne B, Rodeia S, Benmostefa N, Bérézné A, Authier J, Cohen P, et al. Corticosteroid-sparing benefit of intravenous immunoglobulin in systemic sclerosis-associated myopathy: A comparative study in 52 patients. Autoimmun Rev 2020;19:102431. [DOI] [PubMed] [Google Scholar]
- 141.Takehara K, Ihn H, Sato S. A randomized, double-blind, placebo-controlled trial: intravenous immunoglobulin treatment in patients with diffuse cutaneous systemic sclerosis. Clin Exp Rheumatol 2013;31:151–156. [PubMed] [Google Scholar]
- 142.Bellando Randone S, George J, Mazzotta C, Guiducci S, Furst DE, Mor A, et al. Angiostatic and Angiogenic Chemokines in Systemic Sclerosis: An Overview. Journal of Scleroderma and Related Disorders 2017;2:1–10. [Google Scholar]
- 143.Mathes AL, Christmann RB, Stifano G, Affandi AJ, Radstake TRDJ, Farina GA, et al. Global chemokine expression in systemic sclerosis (SSc): CCL19 expression correlates with vascular inflammation in SSc skin. Ann Rheum Dis 2014;73:1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Forssmann U, Uguccioni M, Loetscher P, Dahinden CA, Langen H, Thelen M, et al. Eotaxin-2, a novel CC chemokine that is selective for the chemokine receptor CCR3, and acts like eotaxin on human eosinophil and basophil leukocytes. J Exp Med 1997;185:2171–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mor A, Salto MS, Katav A, Barashi N, Edelshtein V, Manetti M, et al. Blockade of CCL24 with a monoclonal antibody ameliorates experimental dermal and pulmonary fibrosis. Annals of the Rheumatic Diseases 2019;78:1260–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Makino K, Makino T, Stawski L, Lipson KE, Leask A, Trojanowska M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res Ther 2017;19:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nouno T, Okamoto M, Ohnishi K, Kaieda S, Tominaga M, Zaizen Y, et al. Elevation of pulmonary CD163+ and CD204+ macrophages is associated with the clinical course of idiopathic pulmonary fibrosis patients. J Thorac Dis 2019;11:4005–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cox N, Pilling D, Gomer RH. Serum amyloid P: a systemic regulator of the innate immune response. J Leukoc Biol 2014;96:739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pilling D, Gomer RH. Persistent lung inflammation and fibrosis in serum amyloid P component (APCs−/−) knockout mice. PLoS One 2014;9:e93730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tennent GA, Dziadzio M, Triantafillidou E, Davies P, Gallimore JR, Denton CP, et al. Normal circulating serum amyloid P component concentration in systemic sclerosis. Arthritis Rheum 2007;56:2013–2017. [DOI] [PubMed] [Google Scholar]
- 151.Pilling D, Buckley CD, Salmon M, Gomer RH. Serum amyloid P and fibrosis in systemic sclerosis: comment on the article by Tennent et al. Arthritis Rheum 2007;56:4229; author reply 4229–4230. [DOI] [PubMed] [Google Scholar]
- 152.Dosoki H, Stegemann A, Taha M, Schnittler H, Luger TA, Schröder K, et al. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp Dermatol 2017;26:73–81. [DOI] [PubMed] [Google Scholar]
- 153.Hinchcliff M, Huang C-C, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol 2013;133:1979–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chakravarty EF, Martyanov V, Fiorentino D, Wood TA, Haddon DJ, Jarrell JA, et al. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthritis Res Ther 2015;17:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Volkmann ER, Varga J. Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol 2019;15:208–224. [DOI] [PubMed] [Google Scholar]
- 156.Kanemaru R, Takahashi F, Kato M, Mitsuishi Y, Tajima K, Ihara H, et al. Dasatinib Suppresses TGFβ-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Inhibits Pulmonary Fibrosis. Lung 2018;196:531–541. [DOI] [PubMed] [Google Scholar]
- 157.Martyanov V, Whitfield ML, Varga J. Senescence Signature in Skin Biopsies From Systemic Sclerosis Patients Treated With Senolytic Therapy: Potential Predictor of Clinical Response? Arthritis & Rheumatology 2019;71:1766–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kubo S, Nakayamada S, Miyazaki Y, Yoshikawa M, Yoshinari H, Satoh Y, et al. Distinctive association of peripheral immune cell phenotypes with capillaroscopic microvascular patterns in systemic sclerosis. Rheumatology (Oxford) 2019;58:2273–2283. [DOI] [PubMed] [Google Scholar]
- 159.Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med 2019;7:497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, et al. PU.1 controls fibroblast polarization and tissue fibrosis. Nature 2019;566:344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Park J-S, Oh Y, Park YJ, Park O, Yang H, Slania S, et al. Targeting of dermal myofibroblasts through death receptor 5 arrests fibrosis in mouse models of scleroderma. Nat Commun 2019;10:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Galván-Peña S, O’Neill LAJ. Metabolic Reprograming in Macrophage Polarization. Front Immunol 2014;5. Available at: https://www.frontiersin.org/articles/10.3389/fimmu.2014.00420/full. Accessed September 20, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tian F, Wang Z, He J, Zhang Z, Tan N. 4-Octyl itaconate protects against renal fibrosis via inhibiting TGF-β/Smad pathway, autophagy and reducing generation of reactive oxygen species. Eur J Pharmacol 2020;873:172989. [DOI] [PubMed] [Google Scholar]
- 164.Ogger PP, Albers GJ, Hewitt RJ, O’Sullivan BJ, Powell JE, Calamita E, et al. Itaconate controls the severity of pulmonary fibrosis. Sci Immunol 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sobanski V, Giovannelli J, Allanore Y, Riemekasten G, Airò P, Vettori S, et al. Phenotypes Determined by Cluster Analysis and Their Survival in the Prospective European Scleroderma Trials and Research Cohort of Patients With Systemic Sclerosis. Arthritis Rheumatol 2019;71:1553–1570.* This study proposes a new deciphering of the heterogeneity of SSc using a cluster analysis.
- 166.Wirz EG, Jaeger VK, Allanore Y, Riemekasten G, Hachulla E, Distler O, et al. Incidence and predictors of cutaneous manifestations during the early course of systemic sclerosis: a 10-year longitudinal study from the EUSTAR database. Ann Rheum Dis 2016;75:1285–1292. [DOI] [PubMed] [Google Scholar]
- 167.Herrick AL, Peytrignet S, Lunt M, Pan X, Hesselstrand R, Mouthon L, et al. Patterns and predictors of skin score change in early diffuse systemic sclerosis from the European Scleroderma Observational Study. Ann Rheum Dis 2018;77:563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Allanore Y, Distler O, Matucci-Cerinic M, Denton CP. Review: Defining a Unified Vascular Phenotype in Systemic Sclerosis. Arthritis & Rheumatology (Hoboken, NJ) 2018;70:162–170. [DOI] [PubMed] [Google Scholar]
- 169.Lescoat A, Ballerie A, Belhomme N, Cazalets C, Carlan M de, Droitcourt C, et al. Synovial involvement assessed by power Doppler ultra-sonography in systemic sclerosis: results of a cross-sectional study. Rheumatology (Oxford) 2018;57:2012–2021. [DOI] [PubMed] [Google Scholar]
- 170.Laar JM van, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014;311:2490–2498. [DOI] [PubMed] [Google Scholar]
- 171.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med 2018;378:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Frantz C, Huscher D, Avouac J, Hachulla E, Balbir-Gurman A, Riemekasten G, et al. Outcomes of limited cutaneous systemic sclerosis patients: Results on more than 12,000 patients from the EUSTAR database. Autoimmun Rev 2020;19:102452. [DOI] [PubMed] [Google Scholar]
- 173.Allanore Y Limited cutaneous systemic sclerosis: the unfairly neglected subset. Journal of Scleroderma and Related Disorders 2016;1:241–246. [Google Scholar]
- 174.Lescoat A, Murphy SL, Roofeh D, Pauling JD, Hughes M, Sandler R, et al. Considerations for a combined index for limited cutaneous systemic sclerosis to support drug development and improve outcomes. Journal of Scleroderma and Related Disorders 2020:2397198320961967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Maehara T, Kaneko N, Perugino CA, Mattoo H, Kers J, Allard-Chamard H, et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J Clin Invest 2020;130:2451–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fox DA, Lundy SK, Whitfield ML, Berrocal V, Campbell P, Rasmussen S, et al. Lymphocyte subset abnormalities in early diffuse cutaneous systemic sclerosis. Arthritis Res Ther 2021;23:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stifano G, Christmann RB. Macrophage Involvement in Systemic Sclerosis: Do We Need More Evidence? Curr Rheumatol Rep 2016;18:2. [DOI] [PubMed] [Google Scholar]
- 178.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lescoat A, Ballerie A, Jouneau S, Fardel O, Vernhet L, Jego P, et al. M1/M2 polarisation state of M-CSF blood-derived macrophages in systemic sclerosis. Ann Rheum Dis 2019;78:e127. [DOI] [PubMed] [Google Scholar]
- 180.Lescoat A, Jégo P, Lecureur V. M-CSF and GM-CSF monocyte-derived macrophages in systemic sclerosis: the two sides of the same coin? Ann Rheum Dis 2019;78:e19. [DOI] [PubMed] [Google Scholar]
- 181.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008;8:958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 2014;15:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, Carvalho CR de, Kairalla R, et al. Association of Interferon- and transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis & Rheumatology (Hoboken, NJ) 2014;66:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jun J-I, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nature Reviews Drug Discovery 2011;10:945–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tam AYY, Horwell AL, Trinder SL, Khan K, Xu S, Ong V, et al. Selective deletion of Connective Tissue Growth Factor attenuates experimentally -induced pulmonary fibrosis and pulmonary arterial hypertension. The International Journal of Biochemistry & Cell Biology 2021:105961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Murphy-Marshman H, Quensel K, Shi-Wen X, Barnfield R, Kelly J, Peidl A, et al. Antioxidants and NOX1/NOX4 inhibition blocks TGFβ1-induced CCN2 and α-SMA expression in dermal and gingival fibroblasts. PLoS One 2017;12:e0186740. [DOI] [PMC free article] [PubMed] [Google Scholar]