

Abstract
Mesenchymal stem cells (MSCs) are promising candidates for the development of cell-based drug delivery systems for autoimmune inflammatory diseases, such as multiple sclerosis (MS). Here, we investigated the effect of Ro-31-8425, an ATP-competitive kinase inhibitor, on the therapeutic properties of MSCs. Upon a simple pretreatment procedure, MSCs spontaneously took up and then gradually released significant amounts of Ro-31-8425. Ro-31-8425 (free or released by MSCs) suppressed the proliferation of CD4+ T cells in vitro following polyclonal and antigen-specific stimulation. Systemic administration of Ro-31-8425-loaded MSCs ameliorated the clinical course of experimental autoimmune encephalomyelitis (EAE), a murine model of MS, displaying a stronger suppressive effect on EAE than control MSCs or free Ro-31-8425. Ro-31-8425-MSC administration resulted in sustained levels of Ro-31-8425 in the serum of EAE mice, modulating immune cell trafficking and the autoimmune response during EAE. Collectively, these results identify MSC-based drug delivery as a potential therapeutic strategy for the treatment of autoimmune diseases.
Keywords: Mesenchymal stem cells, Ro-31-8425, Drug delivery, Multiple sclerosis
Introduction
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS), characterized by inflammatory lesion formation, demyelination, axonal loss, and progressive neurodegeneration [1]. In the majority of patients, MS initially shows a relapsing-remitting course (RRMS), in which acute attacks are followed by a varying degree of recovery [2]. The majority of RRMS patients eventually develop secondary progressive MS (SPMS), characterized by the irreversible accumulation of neurological disability [2, 3].
Immunomodulatory strategies for the relapsing remitting phase of MS have been developed, as exemplified by glatiramer acetate, interferon beta, dimethylfumarate, or natalizumab [4–7]. However, these strategies often come at the price of severe side effects or partial clinical activity. Therefore, there is an unmet need for therapeutic approaches for MS, which not only are highly efficacious but also exhibit minimal side effects. Given these requirements, mesenchymal stem cell (MSC)-based therapies may represent a novel approach for the treatment of MS and other autoimmune inflammatory diseases. MSCs are promising candidates for cell therapy due to their safety, scalability, immune-evasive phenotype, and potent immunomodulatory activities [8, 9]. MSCs modulate both adaptive and innate immune responses, suppressing the activation of T cells, B cells, natural killer cells, and antigen-presenting cells [10, 11], making them of particular interest for the treatment of MS [12, 13]. Indeed, MSCs have shown efficacy in the treatment of the MS animal model, experimental autoimmune encephalomyelitis (EAE), by modulating the activity of dendritic cells and T cells [12, 14, 15]. Moreover, recent reports suggest that MSCs can be used to reduce autoimmune inflammation in MS patients [16, 17]. However, their post-infusion potency is unpredictable [14, 15], raising the need for safe and scalable bioengineering approaches to further maximize the therapeutic potential of MSCs in the context of MS.
One emerging strategy to develop novel and efficacious cellular therapies for inflammation is to harness the scalable, clinically safe immunomodulatory activities of MSCs for cell-based drug delivery. We and others have previously loaded MSCs with anti-cancer drugs and demonstrated their anti-tumor efficacy in different in vivo cancer models [18, 19]. Recently, we developed a multi-step screening platform to identify small molecules that boost MSC anti-inflammatory function [20]. Ro-31-8425, a PKC inhibitor with anti-inflammatory properties [21], also known as bisindoleamine X, upregulates integrin αL (CD11a), a membrane receptor relevant for cell adhesion, tissue homing, and costimulatory signaling at sites of inflammation [22]. Ro-31-8425-pretreated MSCs displayed improved homing to sites of inflammation upon systemic administration and a superior anti-inflammatory impact in a murine ear inflammation model [20].
PKC-governed pathways play a central role in T cell activation and differentiation to regulate the inflammatory response. Inhibiting different PKC isoforms and dependent pathways has thus been proposed as a strategy to combat autoimmune disorders [23–27]. Considering the role of PKC in autoimmune responses and our previous observations of the boosted immunomodulatory effects of Ro-31-8425-pretreated MSCs in inflammatory settings, we sought to explore the impact of Ro-31-8425-MSCs in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS. Herein, we report that with a simple pretreatment protocol, Ro-31-8425 was spontaneously internalized by MSCs and then released from cells to suppress antigen-specific proliferation of CD4+ T cells in vitro. Furthermore, systemically infused Ro-31-8425-pretreated MSCs outperformed the vehicle MSCs and the free Ro-31-8425 group in the EAE mouse model, significantly reducing the clinical score. Ro-31-8425-MSCs led to sustained levels of Ro-31-8425 in the serum, consequently modulating T cell-driven autoimmunity. Collectively, this study demonstrates the potential of MSCs as promising drug delivery vehicles for the treatment of autoimmunity.
Materials and methods
Mesenchymal stem cell culture and compound pretreatment
Human MSCs were purchased from Lonza and expanded in StemPro® MSC SFM CTS™ with 10% SFM supplement (Gibco) (herein referred to as full StemPro medium). Cells were kept at 37 °C with 5% CO2 and media was changed every 3 days. Cells were passaged using 1% trypsin-EDTA solution. MSCs at passages 3-6 were used for all experiments. For compound pretreatment, pre-confluent MSCs were incubated in StemPro medium for 24 h with Ro-31-8425 at the indicated concentrations.
Co-culture system of MSCs and splenocytes
A co-culture system of MSCs and splenocytes was used to directly assess the immunosuppressive properties of Ro-31-8425-pretreated MSCs (cpd-pretreated MSCs). For polyclonal T cell stimulation, bulk splenocytes from naive wild-type C57Bl/6 mice were fluorescently labeled with CFSE (5 μM) and activated with antibodies to CD3 and CD28 (anti-CD3 (clone 17A2) 4 μg/ml plate-bound, anti-CD28 (clone PV-1) 2 μg/ml soluble, both BioXCell). After 72 h, cells were stained for CD3 and CD4 and analyzed by FACS for CFSE dilution in the CD3CD4 double positive population. For antigen-specific proliferation, splenocytes were isolated from 2D2 transgenic mice expressing a transgenic T cell receptor specific for MOG35-55. A total of 100.000 bulk 2D2 splenocytes were activated with MOG35-55 (2 μg/ml) or vehicle and incubated directly with Ro-31-8425 or Ro-31-8425-pretreated MSCs. During the final 16 h, cells were pulsed with 1 Ci [3H]thymidine (PerkinElmer), followed by collection on glass fiber filters and analysis of incorporated [3H]thymidine in a beta-counter (1450 MicroBeta TriLux; PerkinElmer).
Compound storage and release from cpd-pretreated MSCs
A total of 15,000 MSCs/well in a 96 well were pretreated with Ro-31-8425 at 3 μM for 24 h. At 24 h, cellular supernatants were collected and cells were rinsed and then lysed by incubating 1 h at 37 °C with lysis buffer (Cisbio). The other wells were washed to mimic dilution in the blood circulation until the next time point when the process was repeated. Each collected sample was vortexed with 3 vol of acetonitrile, centrifuged 15 min at 3000g and then analyzed for Ro-31-8425 via MassSpec.
Animal models
All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee at Harvard Medical School, and mice were kept in a pathogen-free facility at the Harvard Institutes of Medicine. For EAE model, 8-10-week old female C57Bl/6 mice were immunized with a peptide of amino acids 35–55 of myelin oligodendrocyte glycoprotein (MOG35–55) in an emulsion of complete Freund’s adjuvant with mycobacterium tuberculosis and pertussis toxin as previously described [28–30]. C57Bl/6 mice developed clinical signs of EAE after 9-13 days, with peak disease lasting 1-3 days after EAE onset, and mice completely or partially recover within 7-10 days. Animals were scored daily in a blinded manner using the following scoring system: 1, loss of tone in the tail; 2, hind limb paresis; 3, hind limb paralysis; 4, tetraplegia; and 5, moribund. Animals were sacrificed during the recovery phase for cellular analyses.
MSC administration
MSCs (native or cpd-pretreated) were stained using violet CellTrace and 0.25 × 106 cells (in 100 μl PBS) were administered (via tail vein injection) into each mouse. To elucidate the impact of disease stage on MSC homing to the CNS, cells were administered prior to disease onset (days 3 and 8 in the RR EAE b6 model). Cell presence in the CNS was assessed 24 h post the second cell infusion and at the end of the experiment via flow cytometry.
Quantitative measurement of Ro-31-8425 in mouse serum by liquid chromatography-tandem mass spectrometry
Serum (20 μl) from each group was processed by adding 3 volume of acetonitrile to precipitate protein and the supernatant was subjected to liquid chromatography-tandem mass spectrometry (LC-MS). All analyses were performed using an ACQUITY UPLC system, coupled to an API-6500 mass spectrometer. The analytes were separated using a reverse phase C18 column (Waters, BEH 1.7 μm, 100 Å, 2.1 × 50 mm) at 50 °C and the mobile phase consisted of (A) water containing 0.1% formic acid and (B) acetonitrile containing 0.1% formic acid with a flow rate of 0.8 ml/min. The flow was sprayed to API-6500 mass spectrometer at electrospray ionization (ESI) positive mode in unit resolution and source temperature of 550 °C. Multiple reaction monitoring (MRM) was used to monitor the transition of protonated parent ion 425.1 Da to the product ion 305.0 Da for compound Ro-31-8425, transition of 237.1-194.4 Da for internal standard carbamazepine. The limit of quantification (LOQ) of Ro-31-8425 in the mouse serum was 0.1 ng/ml. The method was linear in the range of 0.1–250 ng/ml with a coefficient of correlation greater than 0.994 in the mouse serum.
Detection of MSCs in the CNS of EAE mice and distribution in other organs and FACS staining
Assessment of MSC presence in the CNS was performed as previously described [31, 32]. Briefly, mice were perfused with ice-cold PBS, and the brain and spinal cord were removed and incubated with collagenase type III and DNase in PBS. Tissues were then homogenized and loaded on a Percoll gradient to enrich CNS infiltrates. Flow cytometry analysis for detection of stained MSCs, as well as DCs, encephalitogenic and regulatory T cells, was performed. Similarly, the kidneys, lungs, spleen, gut, and heart were homogenized and analyzed for MSC presence. Antibodies for flow cytometry were purchased from eBioscience or BD Pharmingen and used at a concentration of 1:100 unless recommended otherwise by the manufacturer. Cells were analyzed on an LSRII or MACSQuant flow cytometer (BD Biosciences and Miltenyi Biotec, respectively). As outlined in the individual figures, Th1 cells were defined as CD3+CD4+IFN-γ+IL-17−IL-10−Foxp3−, Th17 cells as CD3+CD4+IFN-γ−IL-17+IL-10−Foxp3−, Treg cells as CD3+CD4+IFN-γ−IL-17−IL-10−/+Foxp3+, and pro-inflammatory monocytes as CD45hiCD11b+Ly6Chi.
Results
Uptake and gradual release of Ro-31-8425 by MSCs
We previously demonstrated that Ro-31-8425 pretreatment improves homing of systemically administered MSCs to sites of inflammation, resulting in increased suppression of local skin inflammation [20]. To explore the potential of MSCs for drug delivery, we evaluated the uptake and release of Ro-31-8425 by MSCs. We treated MSCs with Ro-31-8425 (3 μM) for 24 h, and after this loading period, we washed the cells, replaced the media, and collected supernatant samples every 24 h for 72 h. Next, the cells were lysed and Ro-31-8425 levels in cell lysate and supernatant samples were measured by liquid chromatography-mass spectrometry (LC-MS). Following a 24 h loading, MSCs retained about 25% of Ro-31-8425 available in the pretreatment solution and released it gradually over time (Fig. 1a). This secretion profile suggests that MSCs may be used for the delivery of immunomodulatory drugs such as Ro-31-8425, releasing them gradually into the blood stream or target organs to maximize their therapeutic efficacy.
Fig. 1.
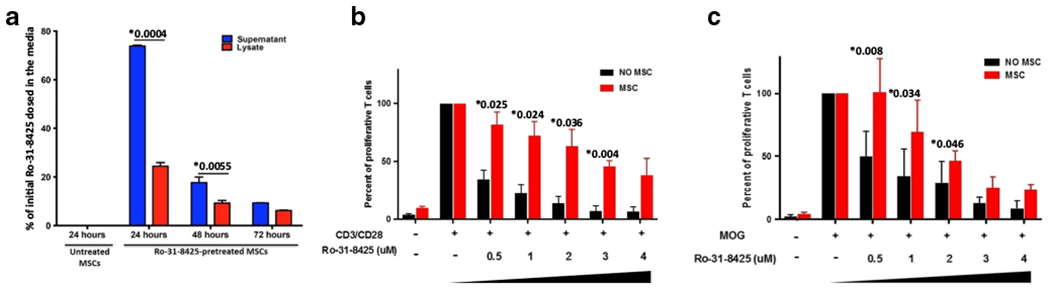
MSCs uptake and then release a significant portion of Ro-31-8425, which displays immune-suppressive properties in vitro. a A total of 15,000 MSCs/well were pretreated with Ro-31-8425 (3 μM for 24 h). At 24 h (right after completion of pretreatment), 48 h, and 72 h, cells were rinsed and levels of Ro-31-8425 in the supernatant and cell lysates were then evaluated via LC-MS. (*p < 0.05, p values are indicated in the graph, one-way ANOVA using Tukey’s HSD, error bars represent SD). b, c Coculture systems of MSCs and splenocytes were established to assess the immunosuppressive properties of Ro-31-8425 w/wo MSCs. b MSCs (vehicle or Ro-31-8425-pretreated at indicated concentrations, 1.5 × 104 cells/well) were co-cultured with CFSE-labeled CD3/CD28-stimulated splenocytes derived from naïve C57Bl/6 mice (1 × 106 cells/well). Other treatment groups included splenocyte incubation for 72 h with Ro-31-8425 alone (without MSCs, at indicated concentrations). Upon 72 h of treatment, CD4+ T cell proliferation was evaluated by measuring CFSE dilution in the CD3CD4 double positive population (n = 3, *p < 0.05 vs. CD3/CD28-activated Ro-31-8425-untreated group, p values are indicated in the graph, one-way ANOVA using Tukey’s HSD, error bars represent SEM). c MSCs (vehicle or Ro-31-8425-pretreated at indicated concentrations, 1 × 104 cells/well) were co-cultured with MOG-stimulated 2D2 splenocytes (1 × 105 cells/well). Another group consisted of splenocytes incubated for 72 h with Ro-31-8425 alone (without MSCs, at indicated concentrations). Upon 72 h of treatment, proliferation was evaluated using 3H-thymidine incorporation (n = 3, *p < 0.05 vs. MOG-activated, Ro-31-8425-untreated group, p values are indicated in the graph, one-way ANOVA using Tukey’s HSD, error bars represent SEM)
Ro-31-8425 suppresses CD4+ T cell proliferation
We next evaluated the immunosuppressive effects of free and MSC-delivered Ro-31-8425 in a polyclonal activation assay and in an antigen-specific system. Therefore, untreated MSCs, Ro-31-8425-loaded MSCs or free Ro-31-8425, were co-cultured with splenocytes from naive wild-type C57Bl/6 mice stimulated with antibodies against CD3/CD28 (Fig. 1b) or instead with splenocytes from 2D2 transgenic mice, which harbor a transgenic T cell receptor specific to myelin oligodendrocyte glycoprotein (MOG35-55) and can be activated by their cognate antigen MOG35-55 presented by antigen-presenting cells [33] (Fig. 1c). In response to CD3/CD28 stimulation, Ro-31-8425 displayed a dose-dependent inhibitory effect on CD4+ T cell proliferation when incubated directly with the splenocytes or when “stored” inside MSCs that were pretreated with different concentrations of Ro-31-8425. Importantly, Ro-31-8425-loaded MSCs resulted in a slower, more sustained suppression of T cell proliferation. Similarly, Ro-31-8425 also inhibited the proliferation of MOG35-55-activated 2D2 transgenic CD4+ T cells (Fig. 1c). Furthermore, we observed a sustained immunosuppressive effect of Ro-31-8425 when loaded into MSCs in this co-culture system. These findings suggest that MSCs store and slowly release Ro-31-8425 to suppress T cell proliferation in a dose-dependent manner in response to both polyclonal and antigen-specific activation. Importantly, Ro-31-8425-loaded MSCs displayed more potent anti-proliferative effects compared to untreated control MSCs, suggesting that Ro-31-8425-MSCs may also show increased anti-inflammatory activity than untreated MSCs in vivo.
Improved therapeutic efficacy of MSC-delivered Ro-31-8425
We evaluated the therapeutic impact of Ro-31-8425-loaded MSCs in EAE induced in C57Bl/6 mice by immunization with MOG35-55 [12]. Vehicle control- or Ro-31-8425-loaded MSCs were administered intravenously on days 3 and 8 following EAE induction; an additional group received free Ro-31-8425, at an amount equivalent to that loaded in the MSCs. Clinical signs of EAE were assessed by daily blinded scoring (Fig. 2a). The administration of Ro-31-8425-loaded MSCs led to a significant amelioration of EAE, and this therapeutic effect was stronger than the one observed following treatment with free Ro-31-8425 or vehicle-loaded MSCs only.
Fig. 2.
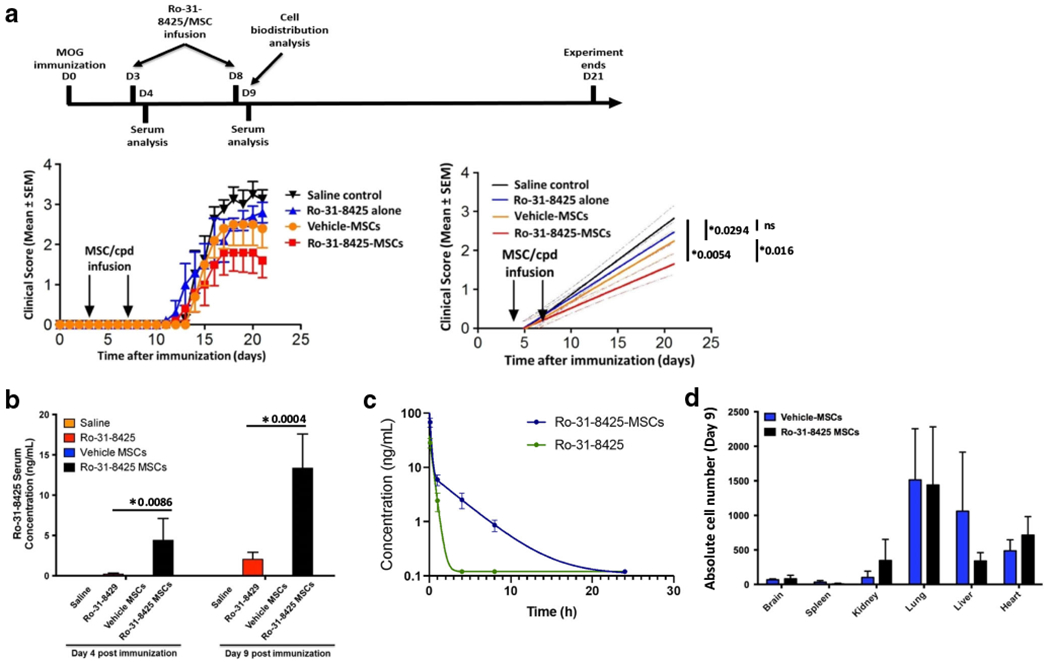
Ro-31-8425 pretreatment improves the therapeutic impact of prophylactic MSC administration in EAE, without altering the MSC global bio-distribution. C57Bl/6 mice were immunized with MOG35–55/CFA and developed remitting-relapsing EAE after 9-13 days. Ro-31-8425 (18.75 μg/kg, 375 ng/mouse) or MSCs (vehicle- or Ro-31-8425-pretreated, 0.25 × 106/mouse) were administered intravenously twice into each mouse on days 3 and 8 post immunization. a Clinical score (right), and linear-regression analysis (left). Clinical scores are mean ± SEM, and representative of two independent experiments, n = 5 mice per group.*p < 0.05, n.s. not significant, p values are indicated in the graph and were determined by two-way analysis of variance (ANOVA). b Twenty-four hours after the second infusion (day 9 post MOG immunization), blood samples were obtained, and Ro-31-8425 serum levels were assessed by LC-MS (*p < 0.05, p values are indicated in the graph, one-way ANOVA using Tukey’s HSD, error bars represent SD). c A single dose of Ro-31-8425 (18.75 μg/kg, 375 ng/mouse) or Ro-31-8425-pretreated MSCs (0.25 × 106/mouse) were administered intravenously into C57Bl/6 mice. The serum concentrations of Ro-31-8425 at indicated time points were quantified by LC-MS. d EAE b6 mice received the above described treatments of Ro-31-8425 alone, vehicle MSCs, or Ro-31-8425-MSC treatments (MSCs were fluorescently stained using violet CellTrace prior to each infusion). Twenty-four hours post the second infusion (day 9), mice were sacrificed, and MSC presence in the CNS (brain and spinal cord), kidneys, lungs, spleen, gut, and heart was assessed by flow cytometry (n = 3 mice per group)
Systemic cell-based delivery of Ro-31-8425 results in prolonged drug circulation
To elucidate the mechanism responsible for the increased therapeutic effect of Ro-31-8425-MSCs, we analyzed serum samples collected 24 h after each MSC infusion or free Ro-31-8425 administration on days 3 and 8 using a validated LC-MS method (Fig. 2b). We found that Ro-31-8425 serum levels in mice infused with Ro-31-8425-MSCs were higher (> 5-fold) than those measured in mice receiving an equivalent amount of free Ro-31-8425. We also analyzed serum samples at multiple time points post intravenous administration of a single dose of Ro-31-8425-MSCs or an equivalent amount of free Ro-31-8425. We detected a 3-fold larger area under the curve (AUC) in samples from Ro-31-8425-MSC treated mice (Fig. 2c), suggesting that MSC-based delivery results in a sustained release of Ro-31-8425 into the circulation.
Ro-31-8425 does not modify MSC bio-distribution
To determine whether Ro-31-8425 affects MSC bio-distribution, we tracked MSCs during EAE in vivo. Fluorescently labeled vehicle- or Ro-31-8425-loaded MSCs were administered intravenously on days 3 and 8 after induction of EAE. Twenty-four hours after the last MSC administration, MSC tissue distribution was determined by FACS. MSCs were detected in the lung, liver, heart, and kidney, and this bio-distribution was not affected by the loading of Ro-31-8425 into MSCs. Indeed, Ro-31-8425 loading did not promote MSC homing to the CNS (Fig. 2d). We also examined the drug concentration in the brain, spleen, and lymph nodes and found the drug concentrations in these organs were not significantly changed by loading Ro-31-8425 in MSCs (Fig. S1). These results indicate that Ro-31-8425 administration in drug-loaded MSCs does not affect MSC homing in vivo, and does not affect drug concentration in major organs.
Ro-31-8425-MSCs modulate the immune response during EAE
We next analyzed the effect of Ro-31-8425-MSCs on the immune response during EAE. Ro-31-8425-MSC treatment led to a decrease in the number of Th17 (CD3+CD4+IL17+) and Th1 (CD3+CD4+IFN-γ+IL-10−) cells in the CNS, as determined by flow cytometry (Fig. 3a). Ro-31-8425-loaded MSCs also decreased the recruitment of pro-inflammatory macrophages (CD11b+CD45+Ly6Chi) to the CNS (Fig. 3b). In addition, Ro-31-8425-MSC administration increased the number of splenic anti-inflammatory IL-10+ type 1 regulatory cells and Foxp3+ T regulatory cells (Fig. 3c), and it also increased the number of pro-inflammatory macrophages and Th1 cells in the spleen (Fig. 3d). Taken together, these findings suggest that Ro-31-8425-MSCs alter immune cell trafficking from the spleen to the CNS, limiting CNS inflammation. In addition, Ro-31-8425 might also alter the priming and maturation of pro-inflammatory T cells, as reflected by an increase of anti-inflammatory T cell phenotypes in the spleen. Overall, our data highlights the potential of Ro-31-8425 pretreatment in guiding the development of MSC-based therapies for MS. Instead, MSCs appear to be acting as a living delivery platform, resulting in sustained serum levels of Ro-31-8425, potentially modulating the immune response by impacting immune cell trafficking between the spleen and CNS.
Fig. 3.
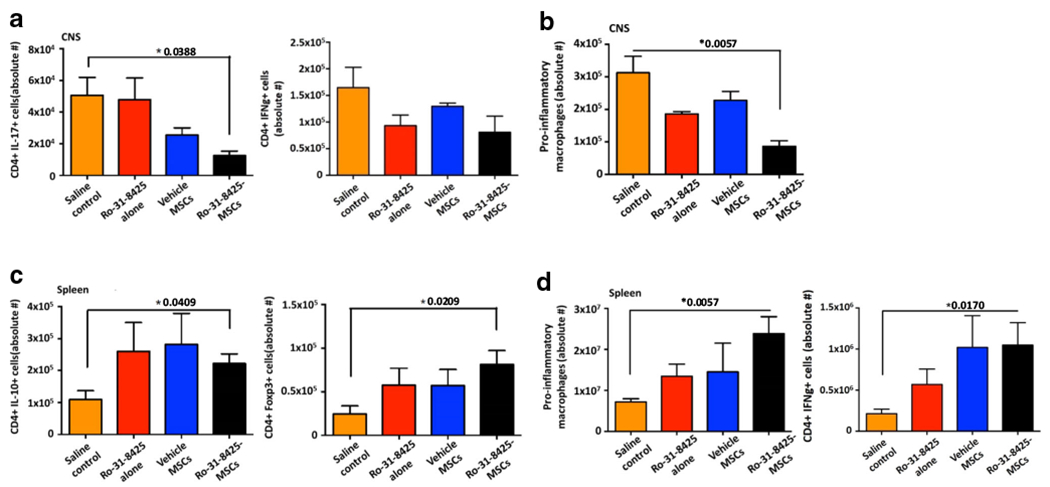
Ro-31-8425-pretreated MSCs modulate the immune response in EAE. C57Bl/6 mice were immunized with MOG35–55 to develop EAE as described above and Ro-31-8425 (18.75 μg/kg, 375 ng/mouse) or MSCs (vehicle- or Ro-31-8425-pretreated, 0.25 × 106/mouse) were administered (via tail vein injection) twice into each mouse on days 3 and 8 post MOG immunization. Mice were sacrificed during the recovery phase of the disease (d22) and the spleen and CNS were harvested and homogenized. Flow cytometry analysis for detection of pro-inflammatory macrophages, CD4+ IL-17+ T cells, CD4+ IFN− + T cells, CD4+ IL-10+ T cells, and CD4+ Foxp3+ T regulatory cells was performed. a CD4+ IL-17+ T cells, CD4+ IFN− + T cells in the CNS. b Pro-inflammatory macrophages in the CNS. c CD4+ IL-10+ T cells, CD4+ Foxp3+ T regulatory in the spleen. d Pro-inflammatory macrophages, CD4+ IFN− + T cells in the spleen. Data presented as ± SEM. *p < 0.05, p values are indicated in the graph, analyzed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons post-test
Discussion
MSCs are considered potential candidates for MS therapy [12, 13, 34, 35]. In this study, we evaluated the use of MSCs for the delivery of the anti-inflammatory compound Ro-31-8425. We have previously shown that Ro-31-8425 increases the recruitment of MSCs to sites of inflammation, boosting the immunosuppressive activity of MSCs [20]. In our current study, we show that free and MSC-loaded Ro-31-8425 suppresses T cell proliferation. MSCs store Ro-31-8425, releasing it in vivo at a constant rate to increase its therapeutic activity. Indeed, Ro-31-8425-MSCs displayed improved therapeutic activity in EAE, outperforming free Ro-31-8425 and vehicle-loaded MSCs. Interestingly, Ro-31-8425 did not affect MSC bio-distribution. In addition, it is also possible that Ro-31-8425 changes the secretome of MSCs, boosting their therapeutic effects. Indeed, in previous studies, we found that Ro-31-8425 pretreatment attenuates the secretion of certain inflammatory cytokines and chemokines (e.g., IL-6 and MCP-1) from MSCs, while the majority of other tested cytokines and chemokines remained unaffected [20], suggesting that Ro-31-8425 has limited effects on the secretome. Thus, future studies are needed to further polarize the secretome towards superior therapeutic potency [36]. Collectively, these data suggest that the improved therapeutic activity of Ro-31-8425-MSCs results from the extended release of Ro-31-8425 in the circulation, resulting in a stronger modulation of immune cell priming and trafficking during EAE.
Ro-31-8425 is a cell-permeable kinase inhibitor that is considered a reversible and selective inhibitor of PKC, a family of kinases that has been shown to participate in the pathogenesis of multiple inflammatory diseases. For instance, PKC-theta is expressed largely in T cells, governing T cell activation and survival [23, 25]. Consequently, PKC-theta signaling in T cells is associated with various inflammatory diseases and murine models of MS, inflammatory bowel disease, arthritis, and asthma [25], identifying the selective inhibition of PKC-theta as a potential therapeutic approach for T cell autoimmunity [25]. Indeed, several PKC-theta inhibitors have shown efficacy in experimental models of MS, inflammatory bowel disease, colitis, and psoriasis [24, 25, 27, 37]. Another PKC isoform, PKC-γ, regulates pain sensitivity and locomotor function and its levels are dependent on the degree of motor function in a murine EAE model[26]. Also, the PKC inhibitor bisindolylmaleimide VII (which belongs to the same chemical family of bisindoleamides as Ro-31-8425) prevented EAE progression in rats [38]. In our study, MSC-delivered Ro-31-8425, a compound which was previously reported to inhibit different PKC isoforms and displayed anti-inflammatory properties in edema and arthritis animal models [21], modulated immune responses by impacting immune cell trafficking and activation. Indeed, when delivered by MSCs, serum levels of Ro-31-8425 (Fig. 2b and c) were above the previously reported IC50 of Ro-31-8425 on different PKC isoforms (for instance, IC50 = 8 nM for PKCα, 8 nM for PKCβI, 14 nM for PKCβII, and 13 nM for PKCγ) [39, 40]. We are currently evaluating the role of different PKC isoforms in our model, but the results presented herein support a potential role of the PKC axis during this autoimmune response.
Our data also highlights the potential of using MSCs to deliver immunomodulatory drugs as a strategy to achieve sustained drug levels in the circulation to maximize their therapeutic efficacy. Pessina et al. previously used MSCs for the delivery of the chemotherapeutic drug paclitaxel, improving its anti-tumor activity in murine models of leukemia and glioblastoma, demonstrating the broad applicability of this approach [19, 41]. While systemic administration of drug-loaded erythrocytes was previously reported to increase the plasma half-life of the loaded drugs [42, 43], our study is the first to demonstrate sustained circulation levels of a drug loaded directly into MSCs. Collectively, our study, along with other reports, highlights the potential use of MSCs to advance effective cell-based delivery of certain drugs. Indeed, this approach, which is cost-effective, simple, and scalable, may be attractive for loading MSCs, or other cell types, with a variety of drugs for treating cancer or autoimmune diseases.
Nevertheless, there are several opportunities to improve the pretreatment approach, namely in controlling the drug release kinetics. Furthermore, this simple pretreatment approach can only be used with drugs that are uptaken spontaneously following cell pretreatment, potentially enabling the use of small, hydrophobic drugs, while excluding the use of entire classes of drugs. In addition to permeabilization strategies including electroporation or low-frequency ultrasound to enhance drug uptake, a potential approach to enable more effective and versatile cell-based drug delivery is to load cells with drug microparticles. Recently, we have reported the encapsulation of a macromolecule anti-prostate cancer pro-drag (G114) in poly(lactic-co-glycolic acid) (PLGA) microparticles, followed by loading of drug microparticles into MSCs [18]. G114-MP-MSCs slowly released G114, and were able to kill prostate cancer cells in vitro as well as suppress prostate cancer tumor growth in a murine in vivo model [18]. This particle-in-a-cell delivery platform can harness the tropic capabilities of cells to sites of disease, while enabling encapsulation of a wide array of drugs (including drugs that cannot be uptaken spontaneously by cells), with tight control over the drug release kinetics from the cells. Furthermore, such a platform can be effective for controlling multiple facets of cell fate post-infusion to advance cell-based therapies, including modulation of cell innate properties (i.e., boost MSC immunomodulatory potential [44, 45]) and cell imaging (i.e., when loading cells with iron oxide NP-loaded MPs [46]). Further studies are needed to evaluate cell fate as well as the PK/PD profiles of MSC-delivered drugs in different disease models and to harness in vivo and in vitro high-throughput screening to identify drugs that activate specific immunoregulatory pathways and regulatory cell populations better matched for the treatment of specific disorders [28, 47–52]. These studies will facilitate the development of MSC-based drug delivery as a potential therapeutic strategy for treating autoimmune inflammation.
Supplementary Material
Key messages.
MSCs can spontaneously take up the ATP-competitive kinase inhibitor Ro-31-8425.
Ro-31-8425-loaded MSCs gradually release Ro-31-8425 and exhibit sustained suppression of T cells.
Ro-31-8425-loaded MSCs have more sustained serum levels of Ro-31-8425 than free Ro-31-8425.
Ro-31-8425-loaded MSCs are more effective than MSCs and free Ro-31-8425 for EAE therapy.
Funding
This work was supported by a research grant from Sanofi-Aventis U.S. to J.M.K. and FJQ, by a grant from King Abdulaziz City for Science and Technology to J.M.K. and FJQ, by National Institutes of Health grants HL095722, and by the Fundação para a Ciência e a Tecnologia through MIT-Portugal-TB/ECE/0013/2013 (to J.M.K.). V.R. received support from an educational grant from Mallinckrodt Pharmaceuticals (A219074) and by a fellowship from the German Research Foundation (DFG RO4866 1/1).
Footnotes
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00109-020-02003-9.
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the studies were conducted and ethical approval was obtained from the Institutional Animal Care and Use Committee at Harvard Medical School.
Conflict of interest Q. W., T. M., C.P., W. S., M.-C. M., and J.R. are employed by Sanofi. JMK has been a paid consultant and or equity holder for companies including Stempeutics, Sanofi, Celltex, LifeVaultBio, Tissium, Takeda, Skintifique, Alivio Therapeutics, Altrix Bio, Ligandal, Vyome, Camden Partners, Stemgent, Gyro Gear, Mirakel, Landsdowne Labs, Biogen, Pancryos, Element Biosciences, Frequency Therapeutics, Molecular Infusions, Quthero, and Mesoblast. JMK is also an inventor on a patent that was licensed to Mesoblast. JMK holds equity in Frequency Therapeutics, a company that has licensed IP generated by JMK that may benefit financially if the IP is further validated. The interests of JMK were reviewed and are subject to a management plan overseen by his institutions in accordance with its conflict of interest policies.
Consent to participate Not applicable.
Consent for publication Not applicable.
Data availability
Data and materials will be shared upon request.
References
- 1.Nylander A, Hafler DA(2012)Multiple sclerosis. J Clin Invest 122:1180–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Compston A, Coles A (2008) Multiple sclerosis. Lancet 372:1502–1517 [DOI] [PubMed] [Google Scholar]
- 3.Rosti-Otajärvi E, Hämäläinen P (2013) Behavioural symptoms and impairments in multiple sclerosis: a systematic review and meta-analysis. Mult Scler J 19:31–45 [DOI] [PubMed] [Google Scholar]
- 4.Ford C, Goodman AD, Johnson K, Kachuck N, Lindsey JW, Lisak R et al. (2010) Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult Scler (Houndmills, Basingstoke, England) 16:342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobs LD, Cookfair DL, Rudick RA, Herndon RM, Richert JR, Salazar AM, Fischer JS, Goodkin DE, Granger CV, Simon JH, Alam JJ, Bartoszak DM, Bourdette DN, Braiman J, Brownscheidle CM, Coats ME, Cohan SL, Dougherty DS, Kinkel RP, Mass MK, Munschauer FE, Priore RL, Pullicino PM, Scherokman BJ, Weinstock-Guttman B, Whitham RH, The Multiple Sclerosis Collaborative Research Group (MSCRG) (1996) Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol 39:285–294 [DOI] [PubMed] [Google Scholar]
- 6.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, Dawson KT, DEFINE Study Investigators (2012) Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 367:1098–1107 [DOI] [PubMed] [Google Scholar]
- 7.Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH et al. (2006) A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354:899–910 [DOI] [PubMed] [Google Scholar]
- 8.Hoogduijn MJ, Popp F, Verbeek R, Masoodi M, Nicolaou A, Baan C, Dahlke MH (2010) The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int Immunopharmacol 10:1496–1500 [DOI] [PubMed] [Google Scholar]
- 9.Singer NG, Caplan AI (2011) Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol Mech 6:457–478 [DOI] [PubMed] [Google Scholar]
- 10.Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F (2003) Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 101:3722–3729 [DOI] [PubMed] [Google Scholar]
- 11.Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC (2003) Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 75:389–397 [DOI] [PubMed] [Google Scholar]
- 12.Uccelli A, Prockop DJ (2010)Why should mesenchymal stem cells (MSCs) cure autoimmune diseases? Curr Opin Immunol 22:768–774 [DOI] [PubMed] [Google Scholar]
- 13.Holloman JP, Ho CC, Hukki A, Huntley JL, Gallicano GI (2013) The development of hematopoietic and mesenchymal stem cell transplantation as an effective treatment for multiple sclerosis. Am J Stem Cells 2:95–107 [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Li Y, Lu M, Cui Y, Chen J, Noffsinger L, Elias SB, Chopp M (2006) Bone marrow stromal cells reduce axonal loss in experimental autoimmune encephalomyelitis mice. J Neurosci Res 84:587–595 [DOI] [PubMed] [Google Scholar]
- 15.Uccelli A, Morando S, Bonanno S, Bonanni I, Leonardi A, Mancardi G (2011) Mesenchymal stem cells for multiple sclerosis: does neural differentiation really matter? Stem Cell Res Ther 6:69–72 [DOI] [PubMed] [Google Scholar]
- 16.Saeed S, Amir Ali S, Oger J (2014) The use of mesenchymal stem cells in the treatment of multiple sclerosis: an overview of open labels and ongoing studies. J Neurol Neurophysiol 5 [Google Scholar]
- 17.Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori J, Kassis I et al. (2010) Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol 67:1187–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levy O, Brennen WN, Han E, Rosen DM, Musabeyezu J, Safaee H, Ranganath S, Ngai J, Heinelt M, Milton Y, Wang H, Bhagchandani SH, Joshi N, Bhowmick N, Denmeade SR, Isaacs JT, Karp JM (2016) A prodrug-doped cellular Trojan Horse for the potential treatment of prostate cancer. Biomaterials. 91:140–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pascucci L, Coccè V, Bonomi A, Ami D, Ceccarelli P, Ciusani E, Viganò L, Locatelli A, Sisto F, Doglia SM, Parati E, Bernardo ME, Muraca M, Alessandri G, Bondiolotti G, Pessina A (2014) Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release 192:262–270 [DOI] [PubMed] [Google Scholar]
- 20.Levy O, Mortensen LJ, Boquet G, Tong Z, Perrault C, Benhamou B, Zhang J, Stratton T, Han E, Safaee H, Musabeyezu J, Yang Z, Multon MC, Rothblatt J, Deleuze JF, Lin CP, Karp JM (2015) A small-molecule screen for enhanced homing of systemically infused cells. Cell Rep 10:1261–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nixon JS, Bishop J, Bradshaw D, Davis PD, Hill CH, Elliott LH, Kumar H, Lawton G, Lewis EJ, Mulqueen M(1991) Novel, potent and selective inhibitors of protein kinase C show oral anti-inflammatory activity. Drugs Exp Clin Res 17:389–393 [PubMed] [Google Scholar]
- 22.Luster A, Alon R, von Andrian U (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 6:1182–1190 [DOI] [PubMed] [Google Scholar]
- 23.Boschelli DH (2009) Small molecule inhibitors of PKCTheta as potential antiinflammatory therapeutics. Curr Top Med Chem 9:640–654 [DOI] [PubMed] [Google Scholar]
- 24.Chand S, Mehta N, Bahia MS, Dixit A, Silakari O (2012) Protein kinase C-theta inhibitors: a novel therapy for inflammatory disorders. Curr Pharm Des 18:4725–4746 [DOI] [PubMed] [Google Scholar]
- 25.Curnock A, Bolton C, Chiu P, Doyle E, Fraysse D, Hesse M, Jones J, Weber P, Jimenez JM (2014) Selective protein kinase Cθ (PKCθ) inhibitors for the treatment of autoimmune diseases. Biochem Soc Trans 42:1524–1528 [DOI] [PubMed] [Google Scholar]
- 26.Lieu A, Tenorio G, Kerr BJ (2013) Protein kinase C gamma (PKCγ) as a novel marker to assess the functional status of the corticospinal tract in experimental autoimmune encephalomyelitis (EAE). J Neuroimmunol 256:43–48 [DOI] [PubMed] [Google Scholar]
- 27.Jimenez J, Boyall D, Brenchley G, Collier P, Davis C, Fraysse D et al. (2013) Design and optimization of selective protein kinase C θ (PKCθ) inhibitors for the treatment of autoimmune diseases. J Med Chem 56:1799–1810 [DOI] [PubMed] [Google Scholar]
- 28.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL (2008) Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 453:65–71 [DOI] [PubMed] [Google Scholar]
- 29.Tsunoda I, Kuang LQ, Theil DJ, Fujinami RS (2000) Antibody association with a novel model for primary progressive multiple sclerosis: induction of relapsing-remitting and progressive forms of EAE in H2s mouse strains. Brain Pathol 10:402–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McRae BL, Kennedy MK, Tan LJ, Dal Canto MC, Picha KS, Miller SD (1992) Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol 38:229–240 [DOI] [PubMed] [Google Scholar]
- 31.Starossom SC, Mascanfroni ID, Imitola J, Cao L, Raddassi K, Hernandez SF, Bassil R, Croci DO, Cerliani JP, Delacour D, Wang Y, Elyaman W, Khoury SJ, Rabinovich GA (2012) Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity. 37:249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai L, Lennon DP, Eaton V, Maier K, Caplan AI, Miller SD, Miller RH (2009) Human bone marrow-derived mesenchymal stem cells induce Th2-polarized immune response and promote endogenous repair in animal models of multiple sclerosis. Glia. 57:1192–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK (2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197:1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cohen JA (2013) Mesenchymal stem cell transplantation in multiple sclerosis. J Neurol Sci 333:43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hauser SL, Chan JR, Oksenberg JR (2013) Multiple sclerosis: prospects and promise. Ann Neurol 74:317–327 [DOI] [PubMed] [Google Scholar]
- 36.Ranganath SH, Levy O, Inamdar MS, Karp JM (2012) Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 10:244–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagahama K, Ogawa A, Shirane K, Shimomura Y, Sugimoto K, Mizoguchi A (2008) Protein kinase C θ plays a fundamental role in different types of chronic colitis. Gastroenterology. 134:459–469 [DOI] [PubMed] [Google Scholar]
- 38.Zhou T, Song L, Yang P, Wang Z, Lui D, Jope RS (1999) Bisindolylmaleimide VIII facilitates Fas-mediated apoptosis and inhibits T cell-mediated autoimmune diseases. Nat Med 5:42–48 [DOI] [PubMed] [Google Scholar]
- 39.Merritt JE, Sullivan JA, Tse J, Wilkinson S, Nixon JS (1997) Different sensitivities of neutrophil responses to a selective protein kinase C inhibitor Ro 31-8425; redundancy in signal transduction. Cell Signal 9:53–57 [DOI] [PubMed] [Google Scholar]
- 40.Wilkinson SE, Parker PJ, Nixon JS (1993) Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J 294(Pt 2):335–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pessina A, Bonomi A, Coccè V, Invernici G, Navone S, Cavicchini L, Sisto F, Ferrari M, Viganò L, Locatelli A, Ciusani E, Cappelletti G, Cartelli D, Arnaldo C, Parati E, Marfia G, Pallini R, Falchetti ML, Alessandri G (2011) Mesenchymal stromal cells primed with paclitaxel provide a new approach for cancer therapy. PLoS One 6:e28321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He H, Ye J, Wang Y, Liu Q, Chung HS, Kwon YM, Shin MC, Lee K, Yang VC (2014) Cell-penetrating peptides meditated encapsulation of protein therapeutics into intact red blood cells and its application. J Control Release 176:123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong X, Niu Y, Ding Y, Wang Y, Zhao J, Leng W, Qin L (2017) Formulation and drug loading features of nano-erythrocytes. Nanoscale Res Lett 12:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ankrum JA, Dastidar RG, Ong JF, Levy O, Karp JM (2014) Performance-enhanced mesenchymal stem cells via intracellular delivery of steroids. Sci Rep 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ranganath SH, Tong Z, Levy O, Martyn K, Karp JM, Inamdar MS (2016) Controlled inhibition of the mesenchymal stromal cell proinflammatory secretome via microparticle engineering. Stem Cell Rep 6:926–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu C, Miranda-Nieves D, Ankrum JA, Matthiesen ME, Phillips JA, Roes I, Wojtkiewicz GR, Juneja V, Kultima JR, Zhao W, Vemula PK, Lin CP, Nahrendorf M, Karp JM (2012) Tracking mesenchymal stem cells with iron oxide nanoparticle loaded poly(lactide-coglycolide) microparticles. Nano Lett 12:4131–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quintana F (2013) The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology. 138:183–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kébir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Prat A, Antel J, Quintana FJ (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 22:586–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintana FJ, Iglesias AH, Farez MF, Caccamo M, Burns EJ, Kassam N, Oukka M, Weiner HL (2010) Adaptive autoimmunity and Foxp3-based immunoregulation in zebrafish. PLoS One 5:e9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mimran A, Mor F, Carmi P, Quintana FJ, Rotter V, Cohen IR (2004) DNA vaccination with CD25 protects rats from adjuvant arthritis and induces an antiergotypic response. J Clin Invest 113:924–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farez Mauricio F, Mascanfroni Ivan D, Méndez-Huergo Santiago P, Yeste A, Murugaiyan G, Garo Lucien P et al. (2015) Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell. 162:1338–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, Pan F, Priel A, Clish CB, Robson SC, Quintana FJ (2015) Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-[alpha]. Nat Med 21:638–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and materials will be shared upon request.