

Abstract
Several synthetic heterocyclic small molecules like imiquimod, resiquimod, CL097, CL075, bromopirone, tilorone, loxoribine and isatoribine demonstrated TLR7/8 agonistic activity and relatively modest structural changes in such molecules result in major variation in the TLR7 and/or TLR8 activity. A strict dependency of the electronic configuration of the heterocyclic system was also observed to influence the agonistic activity. In the present review, an evolution of imidazole based TLR7/8 agonist from imidazoquinoline based scaffold is delineated along with the elaboration of detailed structure activity relationship (SAR) in each chemotype. The structural and activity details of not only the active compounds but also the related inactive compounds are included to better understand the SAR. TLR7/8 agonists are emerging as promising vaccine adjuvant candidates and the present SAR and structural information will provide a road map towards the identification of more potent and appropriate candidates for further drug discovery.
TLR7/8 agonists are emerging as promising vaccine adjuvant candidates. An evolution of imidazole based TLR7/8 agonist from imidazoquinoline based scaffold is delineated along with the elaboration of detailed structure activity relationship (SAR) in each chemotype.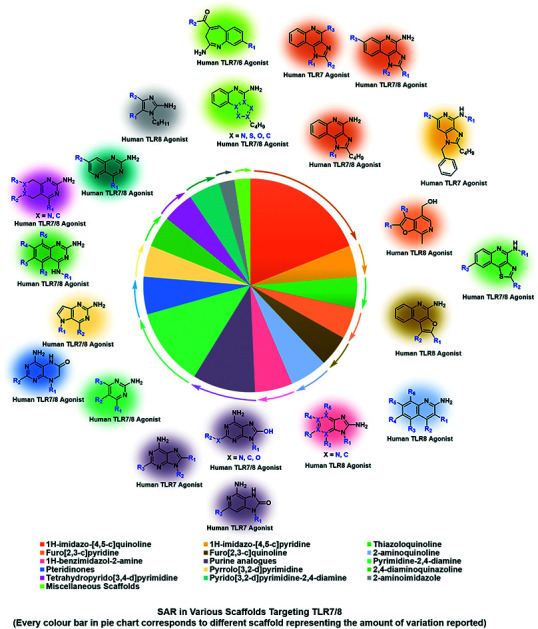
Introduction
Toll-like receptors (TLRs) are pattern recognition receptors present on diverse cell types that recognize specific molecular patterns expressed by pathogens or damaged tissue, referred to as pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), and xenobiotic-associated molecular patterns (XAMPs), that are distinguishable from normal host molecules.1 TLRs act as the “sentinel” of the immune system in humans and other animals to protect the host from invading bacteria, viruses, and other pathogens. There are 10 functional TLRs in the human genome expressed as dimeric trans-membrane proteins with an extracellular domain having leucine-rich repeats (LRR) and a cytosolic domain called the Toll/IL-1 receptor (TIR) domain. TLRs-1, -2, -4, -5, -6 and -10 respond to extracellular stimuli, while TLRs-3, -7, -8 and -9 respond to endosomal PAMPs.2 The ligands for TLRs are highly conserved microbial molecules such as lipopeptides (heterodimer of TLR2 in combination with TLR1 or TLR6), double stranded RNA (TLR3), LPS (recognized by TLR4), flagellin (TLR5), single stranded RNA (TLR7 and TLR8), CpG motif-containing DNA (TLR9), and profilin present on uropathogenic bacteria (TLR 11).3 The activation of TLRs by their cognate ligands leads to the production of inflammatory cytokines and up-regulation of MHC molecules and co-stimulatory signals in antigen-presenting cells as well as activating natural killer (NK) cells (innate immune response), and priming and amplifying T-, and B-cell effect or functions.4 TLR signals serve to link innate and adaptive immunity and can therefore TLR agonists can be exploited as vaccine adjuvants. TLRs are crucial in the recognition of viral and bacterial pathogens and activate innate immune system whose response is characterized by IFNs, inflammatory cytokines and chemokines production. TLR7 and 8 are able to detect GU-rich and AU-rich ssRNA sequences of RNA viruses.5 They are known to serve as endosomal PRRs for a number of ssRNA viruses as influenza, HIV-1, vesicular stomatitis virus, Sendai virus, flaviviruses, and coronaviruses.6 TLR7 agonistic imiquimod is known for its immune-stimulating activity against HSV-2, Sendai virus and HPV.6 TLR7/8 agonists also expressed antiviral activities against respiratory and HBV infections and also enhanced T-cell response towards HIV.7 Many TLR7 agonists have been actively studied in phase 1 and 2 clinical trials aiming to curb the persistent viral load in HIV and HBV-infected individuals.8 TLR7 agonists are emerging as useful candidates in treating hepatitis B infection in phase 1 clinical trial. The results of this trial were promising and paved the way for phase 2 trials.9
Moreover, very recently we observed that a TLR7 agonistic imidazoquinoline analogue, BBIQ showed improved anti-influenza IgG1 and IgG2c responses in mice when administered with recombinant influenza hemagglutinin protein.10 We also explored the application of BBIQ in combination with a suboptimal dose of chloroquine as immunochemotherapeutic agent against Plasmodium berghei ANKA and a strong Th1 immune response against the infection was observed.11 The potent antiviral TLR7/8 agonistic imidazoquinoline derivative resiquimod (R-848) is currently being evaluated for treating hepatitis C and other viral infections.12,13 Resiquimod also activates natural killer cells, indirectly inducing IFNγ, and therefore may promote the development of antigen-specific cell-mediated responses.14 Other low molecular weight synthetic TLR7/8 agonists are in different phase of clinical trial for the potential treatment of hepatitis C infection and cancer.6
Among the human TLRs, TLR2, 4, 7 and 9 can detect plasmodial antigens, thus inducing an antimalarial immune response. Accordingly, many efforts have been made in order to elucidate the involvement of TLRs in severe malaria pathogenesis and of the signaling pathway involving TLRs during infection.15 The recent advances in malaria vaccine formulations involved the incorporation of TLR2 agonists (Pam2Cys and Pam3Cys based lipopeptides), TLR3 agonists (poly(IC:LC)), TLR4 agonists (3-O-deacylated-MPLA (3D-MPLA) and GLA), TLR5 agonists (flagellin), TLR7/8 agonists (imiquimod) and TLR9 agonists (CpG-ODNs).
Chemotherapy represents the mainstay for cancer treatment; however, in the last few decades, immunotherapeutic approaches have shown invaluable potential as antitumor strategies. TLR agonists have established therapeutic benefits as anticancer agents that activate immune cells in the tumor microenvironment and facilitate the expression of cytokines that allow for infiltration of anti-tumor lymphocytes and the suppression of oncogenic signaling pathways.16 TLR7 agonistic imiquimod is one of the most successful drug candidate approved by FDA for the treatment of superficial basal cell carcinoma in 2014. The anti-tumor effect of imiquimod is multifactorial and is hypothesized to act by recruitment of tumor-infiltrating plasmacytoid DCs and macrophages activated by inflammatory cytokines and chemokines (e.g. TNFα, IL-12, IFNα) that signal through TLR7/MYD88-dependent pathways, leading to infiltration of helper T cells.17 Many other TLR7/8 modulators are currently under evaluation in several clinical trials in patients with various tumor conditions.8
In case of allergic diseases (allergic rhinitis, asthma, drug-induced anaphylaxis), the primary reason is strong pro-inflammatory Th2 and IgE-mediated responses against antigens and it can be balanced by activation of TLRs, particularly TLR7, TLR8 and TLR9, which leads to activation of APCs and Th1 response. Among allergic diseases, asthma is an inflammatory disease characterized by airway hyperresponsiveness, eosinophilic infiltration, reversible airflow obstruction, airway remodeling, mucus hypersecretion, and goblet cell hyperplasia. TLR7 ligands play an important role in reduction of these airways problems and acts as novel focused therapeutics to treat asthma and allergies.18,19 The agonists of TLR8 act as potent stimulators of Th1 polarizing cytokines such as IFN-γ and IL-12, and are being developed for the potential treatment of allergic rhinitis. TLR7 agonistic 8-oxoadenine derivative (AZD8848) has anti-inflammatory potential and entered in different phase of clinical trials to demonstrate its ability for long-term remission in allergic disorders such as bronchial asthma and allergic rhinitis.20 The biology and clinical potential of TLR based therapeutics have been extensively reviewed.21–24
Very recently, a TLR7/8 agonistic imidazoquinoline adsorbed to alum (Algel-IMDG) has been used in a vaccine formulation with BBV152 which is a whole virion inactivated SARS-CoV-2. The vaccine was found to be well tolerated in all dose groups with no adverse effects and both humoral and cell mediated immune responses were observed in Algel-IMDG-based vaccine,25 thus indicating the importance of TLR7/8 in generating a protective immune response. The goal of this literature review is to describe the various TLR7/8 agonistic small molecules and their SAR, thereby providing insights into fertile areas of future discovery in respect to TLR7/8 ligand discovery.
Phylogenetic relationship of TLR7 and TLR8 and their ligands
TLR7 and TLR8 are phylogenetically and structurally related to each other and both recognize ssRNA and small heterocyclic molecules in the endosomal compartment. Between TLR7 and TLR8, the crystal structure of TLR8 was reported first in 2013 by Tanji and co-workers.26 It exists as a homodimer and contains 26 leucine rich repeat (LRR) units. The C-terminus of each monomer is separated by 53 Å. In the crystal structure of TLR8 in complex with small heterocyclic ligands, the authors observed that when a ligand is bound to TLR8, its C-terminus is brought together to 30 Å and the loop between LRR14 and LRR15 was cleaved. Further in 2016, Zhang and Co-workers27 successfully co-crystalized TLR7 with three different ligands and observed the formation of an activated m-shaped dimer with two ligand-binding sites. The first site conserved in TLR7 and TLR8 was used for small ligand-binding and there is 72% similarity in the amino acids of first site between TLR7 and TLR8. The second site spatially distinct from that of TLR8 was used for a ssRNA-binding and enhanced the affinity of the first-site ligands.
TLRs 7 and 8 respond to ligands within the endosomal compartment of B cells and dendritic cells by activating NF-κβ in a MyD88-dependent manner and inducing production of various cytokines and chemokines (Fig. 1).28 A comparative study performed by 3M Pharmaceuticals, demonstrated that the TLR7 and TLR8 differ in their target cell selectivity and cytokine induction profile.29,30 TLR7 agonists stimulate virtually all subsets of lymphocytes, and are more effective than TLR8-selective agonists at inducing IFN-α and IFN-γ associated chemokines, whereas TLR8 agonists induce a pro-inflammatory cytokine profile including TNF-α, IL-12 and MIP-1α.29
Fig. 1. TLR7 and 8 signaling pathway.

TLR7 and 8 expressed by human PBMCs respond to guanosine- and uridine-rich oligonucleotides resulting in TNF-α production.31 Diebold et al. showed that influenza virus uridine-rich ssRNA can act as a TLR7 agonist.32 A range of chemical modifications have been made to natural RNA ligands to enhance their selectivity for TLR7 and 8.33–36 TLR7 also responds to guanosine analogues in ssRNA.37 Guanosine or ssRNA individually do not activate TLR7, however their combination leads to receptor activation. Zhang et al. showed that TLR7 recognizes degraded ssRNA with guanosine binding to one site on TLR7 while polyU binds to the other site resulting in synergistic TLR7 activation.38 TLR7 and 8 can also be activated by small interfering RNA (siRNA), tumor-secreted miRNA-21 and miRNA-29.39,40
Several synthetic small heterocyclic molecules also demonstrate TLR7/8 agonistic activity with imiquimod and resiquimod being shown to be activators of the TLR7 pathway.4 Relatively modest structural changes in such molecules result in major variation in TLR7/8 activity. A strict dependency of the electronic configuration of the heterocyclic system was shown for TLR7 or TLR8 activation. Knowledge of the structure–activity relationship (SAR) of various TLR7/8 scaffolds is important for the design and synthesis of additional potent agonists.
Small heterocyclic molecules as TLR7/8 agonists
Initially, a variety of small molecules like imiquimod (1), resiquimod (2), CL097 (4), CL075 (5), bromopirone (7), tilorone (8), loxoribine (9) and isatoribine (10) were shown to have immune stimulatory and antiviral activity via induction of type 1 interferon (IFN-α and β) although their exact mechanism of action was not known. After the discovery of the TLRs in 2002, it was found that these molecules (1–10) induce interferons through TLR activation.4 Imiquimod was approved by FDA in 1997 for the treatment for basal cell carcinoma and actinic keratosis.41 Resiquimod, a second-generation drug is more potent at activating TLR7 with TLR8 agonistic activity, which imiquimod does not. Gardiquimod, like imiquimod, only activates TLR7. CL097 (2-(ethoxymethyl)-1H-imdiazo[4,5-c]quinoline-4-amine) is a water-soluble imidazoquinoline derivative. Similar to resiquimod, CL097 is a dual TLR7 and 8 agonist inducing NF-kβ activation at 0.1 μM in TLR7 transfected HEK293 cells and at 4 μM in TLR8-transfected HEK293 cells (Fig. 2).42
Fig. 2. Structure of various hetero cyclic compounds found to have interferon-inducing activity preceding the discovery of the TLRs.

In 2000, thiazolo[4,5-c]quinoline-4-amine based heterocycles were reported as immune modulators by 3M Pharmaceuticals.43 Among the many related heterocycles, 2-propylthiazolo[4,5-c]quinolin-4-amine (CL075) was reported as the most potent pure TLR8 agonist, which demonstrated activation of hTLR8 transfected HEK293 cells at 3 μM.29 3M-003 is another potent hTLR7/8 agonist inducing TLR7 and 8 activation at 0.3 μM and 3 μM, respectively.29 Interestingly, it was reported to induce cytokines at different sensitivities with IFN-α induced at 0.03 μM and TNF-α and IL-12 only at 0.1 μM.29 Bromopirone (2-amino-5-bromo-6-phenylpyrimidin-4(3H)-one) belongs to a pyrimidine class of heterocycle, with its 6-methyl derivative first reported in 1976 as an interferon inducer.44–46 Bromopirone showed better antiviral activity than its analogues.47,48
In 1970, Krueger and Mayer49,50 reported that tilorone hydrochloride induced antiviral activity in mice after oral administration making it the first synthetic compound which induced an interferon response when administered orally.51 A series of 7,8-disubstituted guanosine derivatives were designed and synthesized by Reitz et al. and were evaluated for their ability to act as B-cell and natural killer cell activators.52 Among them, loxoribine was found to be a potent immune modulator via TLR7/MyD88-dependent pathway.53 Synthesis of isatoribine was reported by Nagahara et al. during their exploration of thiazolo[4,5-d]pyrimidine derivatives as immunotherapeutic agents.54
Structure–activity relationships (SAR)
Based on the preliminary understanding of structural components responsible for TLR7/8 recognition, diverse scaffolds were reported with specific structural features responsible for the TLR7/8 activity (Fig. 3). A detailed SAR in 1H-imidazo[4,5-c]quinoline, imidazo[4,5-c]pyridine, thiazolo[4,5-c]quinoline, furo[2,3-c]pyridine, furo[2,3-c]quinoline, quinoline, 1H-benzimidazol, pyrimidine and imidazole based structures is discussed in the present review. A systematic exploration of the chemical space around each reported TLR7 and -8 agonistic scaffolds is summarized as a case study for medicinal chemists to design and develop novel TLR7 and/or 8 agonists.
Fig. 3. TLR7/8 heterocyclic scaffolds.

SAR in 1H-Imidazo[4,5-c]quinolines
1H-Imidazo[4,5-c]quinolines are the first small heterocyclic molecules to be reported as TLR7/8 agonists.4,55 These imidazoquinolines were found to be potent immune stimulators56 able to induce type-I IFNs in human cells.57 A detailed SAR study on imidazoquinoline was performed in 2010, in which a substituted 1H-imidazo[4,5-c]quinoline library was synthesized with gardiquimod (3) as the lead structure (Fig. 4)58 and the synthesized compounds were evaluated in a human TLR7 reporter gene assay. This SAR study analyzed the derivatization on N-1, C-2 and C-4 position of imidazoquinoline ring (Fig. 4). Initially, the functionalization on secondary amine of the C-2 ethylaminomethylene side chain was carried out to find sites that altered TLR7 activity. Among all the derivatives at the secondary amine, only N-ethyl (32a) and N-ethylenenitrile (32b) analogues retained partial activity while N-propylene amino (32c), N-triethyleneglycol (32d), guanidine (32e) and acyl (32f, 32g) derivatives were completely inactive. A thiourea adduct (32i) was also synthesized from N-propylene amino (32c) derivative, which was also inactive. It was concluded that the secondary amine on the C-2 substituent is not a suitable site for modification. However, the length of the C-2 substituent was found to be an important parameter for TLR7 activity. A complete loss of activity was observed on the replacement of the terminal ethyl group with an n-undecyl group (32h).
Fig. 4. SAR in 1H-imidazo[4,5-c]quinolines.

More lipophilic N-1 benzyl analogues (33a–33l) were synthesized to give better permeability through the dermal barrier. Following a systematic SAR of the C-2 alkyl substituents, a distinct relationship between alkyl chain length and TLR7 activity was observed. Maximum activity was shown by C2-n-butyl derivative (33d) followed by C2-n-pentyl derivative (33e). Substituents with curtailed chain like C2-methyl (33a), C2-ethyl (33b), C2-n-propyl (33c) as well as those with longer chain length like C2-n-hexyl (33f), C2-n-heptyl (33g) and C2-n-nonyl (33h) analogs showed poor activity. The derivatives with an unsaturated hydrocarbon chain in the side chain, for example the C2-n-butyl-3-ene (33l), showed reduced activity and C2-n-butyl-3-yne (33k) analogues were also less active. The lipophilic analogues, C2-n-undecane (33i), C2-n-pentadecane (33j) and compound C2-(n-undecylamino)methyl (32h) showed complete loss of activity, which confirmed that the length of C-2 substituent as an important parameter for TLR7 activity. Overall, this SAR confirmed that the C2-n-butyl substituent (33d) was the substituent with the highest TLR7 activity.
Furthermore, the authors tested the effect of the C-4 position by installing chloro, phenyl, hydroxy, –NHNH2 and –NHOH groups. The 4-Cl (34a), 4-OH (34b) and 4-Ph (34c) as well as the des-amino analogue 34d, were completely inactive, while the 4-NHOH (34e) and 4-NHNH2 (34f) compounds were substantially weaker than their 4-NH2 counterpart (33d). These results indicate the importance of C4–NH2 functionality for maximal TLR7 activity.
Hence, a focused SAR around gardiquimod resulted in a N-1 benzyl-C2-n-butyl analogue 33d which was substantially active and chemically distinct. The exchange of N-1 and C-2 substituents present on the lead molecules 3 and 33d, resulting in compounds 36 and 37, were highly active, with EC50 of 8.6 nM and 0.358 μM respectively. When the imidazole ring was completely replaced with triazole ring and cyclic urea the triazole compounds 35a and 35b, and cyclic urea 35c, were completely inactive, indicating the critical role of the imidazole ring system in the ligand recognition by TLR7.
Overall, a distinct relationship between C2-alkyl chain length and TLR7 activity was observed demonstrating the requirement of the C4-amino group for activity. Another study59 investigate the further modification of N-1 position by the introduction of bulky aryl groups and substituents on the N-1 benzyl group. Specifically, the substituents at N-1 position were varied while keeping the C2-n-butyl and C4–NH2 groups constant. The analogues with bulkier phenyl rings (naphthalene 38 and biphenyl 39) did not show any appreciable improvement in activity. Interestingly, the N-1 amino-methyl substituted benzyl analogues (40, 41) were the most active TLR7 agonists (EC50 = 110 nM and 20 nM, respectively) whereas the para-substituted analogue 41 was found to be five times more active than its regioisomer 40.
Due to the similarity between TLR7 and 8, the potent imidazoquinolines obtained from the above SAR (Fig. 4 and 5) were screened for hTLR8 activity (Table 1).
Fig. 5. SAR in N-1 substituted imidazoquinolines.

hTLR7 and hTLR8 agonistic values of resiquimod, compound 36, 40 and 41.
| Compound | EC50 in hTLR7 cells (ng mL−1) | EC50 hTLR8 cells (ng mL−1) |
|---|---|---|
| Resiquimod 2 | 66.6 | 362.9 |
| Compound 36 | 2.5 | 19.2 |
| Compound 40 | 29.4 | 33.4 |
| Compound 41 | 4.02 | 67.5 |
Resiquimod (R848), compound 36 (Fig. 4), compound 40 (Fig. 5) and compound 41 (Fig. 5) were selected and their activity was measured in hTLR8-transfected HEK-293 cells.60 Compound 36 was the most potent imidazoquinoline in both hTLR7 andhTLR8 transfected cells.
To find out the nature of interaction between TLR8 and compound 36, Tanji et al. crystallized a ligand–receptor complex and showed interactions similar to a resiquimod–hTLR8 complex.61 This crystallographic study demonstrated that the absence of ether oxygen of the C2-ethoxymethyl substituent in compound 36 was favorable for its efficacy and activity as the ether oxygen could engage in unfavorable electrostatic and/or dipolar interactions with the carbonyl oxygen of Gly572 in hTLR8.
Recently, an extension SAR on imidazoquinoline based structures centered on functionalization of N1-benzyl ring by David and coworkers62 showed that the small electronic modification on benzyl ring also affect the relative TLR7/8 activities. It was observed that the incorporation of phenolic hydroxyl groups on the N1-benzyl functionality resulted in improved TLR7 selective compounds with substantial loss of TLR8 activity incorporation of second hydroxyl or amine functionality on benzyl ring improved the TLR8 agonistic activity.
In another study59 fluorescent analogues (Fig. 6) were synthesized to demonstrate internalization and distribution of imidazoquinolines (41) in the endosomal compartments. The free primary amine of N-1 substituent was covalently coupled with fluorescein isothiocyanate and rhodamine B isothiocyanate to afford corresponding compounds 42 and 43.
Fig. 6. Fluorescent analogues of compound 41.

Another fluorescent analogue 45 was also synthesized by converting primary amine first to the isothiocyanate 44, followed by the coupling with bora-diazaindacene dye, BODIPY-TR-cadaverine. All three fluorescent conjugates retained TLR7-activity, although their potencies were slightly less than their parent compound 41.59
Recently, compound 41 was derivatized wherein the free amine group was linked to different n-alkyl chains and cycloalkyl groups through amide linkage and direct amination (Fig. 7).63
Fig. 7. SAR in alkyl chain modified imidazoquinolines.

The first modification in this series involved variation in the linear alkyl chain (4 to 19 carbon) (46a–46p) which were linked via amide bond. All these modifications resulted in a 2–3 fold decrease in TLR7 activity versus the parent compound 41. The compounds with five and seventeen carbons in the alkyl chains (46b and 46n) were the least TLR7 active. The derivatives with carbon chain variants of 5 (46b), 8 (46e), 9 (46f), 10 (46g), 15 (46l) and 17 (46n) had 10–25 fold loss of TLR8 activity as compared to parent compound 41, while derivatives of carbon chain variants 11 (46h), 12 (46i), 13 (46j), 14 (46k) retained activity. The methyl cyclopropane derivative (46r) demonstrated an eight-fold loss of TLR7 activity and seven-fold loss in the TLR8 activity.
Further, similar modifications were performed on amine group of the benzyl methyl moiety (Fig. 7) starting with linear alkyl chains (4 to 19 carbon) (47a–47p) and other different modifications involving alkyl cycloalkanes (47q–47e′). It was observed that with increasing number of carbon atoms in the alkyl chain, the TLR7/8 activity decreased, except for the 5 (47b) and 6 (47c) carbon variants which showed slightly improved TLR8 activity as compared to parent compound 41. The alkyl cycloalkane derivatives (47q–47e′) showed markedly reduced TLR7 activity but unexpectedly showed comparable or greater TLR8 activity than parent compound 41. Among these compounds (47q–47e′), (cyclobutyl)methyl (47z) and (1-methylcyclobutyl)methyl (47a′) showed the most increase in TLR8 activity (5.9 and 7.7 fold, respectively), while (1-methylcyclopropyl)ethyl (47x) and (2-cyclobutyl)-(2,2-dimethyl)ethyl (47c′) demonstrated a small increase in TLR8 activity. Overall, it was concluded that the substitution on amide group of benzyl methyl moiety results in a decrease in both TLR7 and TLR8 activity while the substitution on amine group of benzyl methyl moiety resulted in loss of TLR7 but increase in TLR8, activity.
Bonnert et al.64 generated a library of four different series of imidazoquinoline based compounds by varying substitution at N-1 and C-2 position of imidazoquinoline scaffold (Fig. 8) and evaluated these using an hTLR7 reporter cell assay. Firstly, the 2-methoxyethyl group was kept at C-2 position and variation was performed at the nitrogen of 3-aminopropyl chain at N-1 position of imidazoquinoline. The two series of compounds were synthesized by fixing methyl-3-methylbenzoate group (in blue color) at R2 position in the first series of compounds (48a–48c) and methyl-4-methylbenzoate group in the other series (49a–49k). N-Amide and N-alkyl derivatives were synthesized and screened for TLR7 activity. In both the cases, N-amide derivative with piperidine analogue was found to be potent TLR7 agonists with pEC50 value of 6.8 (48c) and 7.0 (49e), respectively.
Fig. 8. SAR in imidazoquinolines by Bonnert et al.

In a similar SAR of two series (50a–50w and 51a–51h′), where 2-methoxyethyl group was replaced by n-butyl group at C-2 position, an increase in TLR7 activity was observed. In case of methyl-4-methylbenzoate analogues (50a–50w), all the compounds showed similar TLR7 activity, except for piperidine analogues 50n and 50t which showed higher TLR7 activity with pEC50 value 7.1 and 7.4, respectively.
In the fourth series of methyl-3-methylbenzoate analogues, terminal tert-amine (51b–51g), terminal hydroxy (51h), azetidine (51i–51j), pyrrolidine (51k–51m), piperidine (51r–51u), piperazine (51x–51b′), morpholine (51v–51w), azepane (51c′), 1,4-oxazepane (51d′) and 1,4-diazepane (51e′–51h′) derivatives were also synthesized by the authors. It was observed that an electron donating hydroxyl group present on four and five membered ring decreased TLR7 activity. The pyrrolidine (51k), piperidine (51r) and azepane (51c′) analogues without any substitution showed maximum TLR7 activity with pEC50 of 7.2, 7.1 and 7.1, respectively.
Yoshiaki et al. studied the variation at N-1 and C-2 position of imidazoquinoline scaffold (Fig. 9–12).65 Initially, n-butyl group was fixed at C-2 position and the substitution was done at the nitrogen of 3-aminopropyl chain (compound 52a–52g, 53a–53d, 54a–54d, 55a–55d, 56a–56b and 57) as shown in Fig. 9. Among all the compounds, compound 54a was found to have potent TLR7 activity with EC50 value 67.7 nM. However, a similar compound with n-propyl group (compound 58) was not a potent as compound 54a.
Fig. 9. SAR in imidazoquinolines by Yoshiaki et al. (part 1).

Fig. 10. SAR in imidazoquinolines by Yoshiaki et al. (part 2).

Fig. 11. SAR in imidazoquinolines by Yoshiaki et al. (part 3).

Fig. 12. SAR in imidazoquinolines by Yoshiaki et al. (part 4).

In the second phase of SAR, the n-butyl group was replaced with ethoxymethyl group at C-2 position and substitution was done at both the position of nitrogen of 3-aminopropyl chain for the identification of potent TLR7 agonist (Fig. 10 and 11). Initially, one component on nitrogen was kept as methyl-2-(m-tolyloxy)acetate (59a–59e) or ethyl-2-(p-tolyloxy)acetate (60a–60c) at R2 position and different amino carbonyl groups were used as another component at R3 position. Among these compounds, compound 59c with diethyl aminocarbonyl group was identified as a potent TLR7 agonist with EC50 value of 31.7 nM.
In identification of potent TLR7 agonists, more derivatization was done by taking diethyl aminocarbonyl (61a–61k′), dimethylaminocarbonyl (62a–62f), ethylmethylaminocarbonyl (63a–63e), and methoxyethyl-methylaminocarbonyl (64) as one component on nitrogen at R3 position and various substituted tolyloxy acetate derivatives as another component at R2 position (Fig. 10 and 11). It was observed that derivatives (61a–61k′) with diethylaminocarbonyl group were more potent than other substituents.
The alkylamino chain length (Fig. 12) at N-1 position was changed to n = 2 or 4 with 2-aminoethyl group (67) and 4-aminobutyl group (68–72) followed by similar type of substitutions at the nitrogen. The derivatives with shorter chain length (67a–67e) were least active while other derivatives with longer chain length (68a–68c, 69a–69e, 70a–70c, 71a–71c, 72a–72c) were potent as 3-aminopropyl derivatives.
Ferguson and co-workers explored a discrete C-7 position of imidazoquinoline scaffold and synthesized TLR7/8 agonistic C7-methoxycarbonyl derivatives (75, 76) of imidazoquinoline (Fig. 13).66 Imiquimod selectively activates TLR7 and resiquimod triggers both TLR7 and TLR8, with increased activity for TLR7 as compared to imiquimod,58 as per this observation, when C-2 ethoxymethylmoiety of resiquimod was removed (compound 73) TLR7 activity was reduced with complete loss of TLR8 activity. Both TLR7 and 8 activities were restored in compound 74 by the addition of a C-2 butyl group. The same trend appeared in case of C7-methoxycarbonyl derivatives (75, 76) depicting a clear role of the C2-butyl group in TLR7/8 activity.
Fig. 13. Comparative analysis of C-7 substituted imidazoquinolines.

Further, SAR studies on C7-methoxycarbonyl-imidazoquinolines were performed by Ferguson et al.67 The analogues 77–87 (Fig. 14) were screened for TLR-7 and -8 activities using human TLR-7 or -8 transfected HEK-293 cells.
Fig. 14. SAR in C7-methoxycarbonyl imidazoquinolines.

While exploring the chemical space around C7-methoxycarbonylimidazoquinoline, it was observed that TLR7 was more tolerant to substitution than TLR8. Starting with C2-alkyl substitution, C2-n-butyl analogue 76 showed maximum TLR7 activity as compared to analogs with shorter (77–79) or longer (80–81) chain length. TLR8 activity was shown by only C2-n-butyl (76) and C2-n-pentyl (80) analogues. A limited series of N-1 modified analogues containing the C2-butyl group and C7-ester functionality were also synthesized by the authors. The 2-hydroxypropyl stereoisomers (82–84) showed the greatest potencies as dual TLR-7/8 agonists. The observations suggested that the binding to TLR7 and TLR8 is not enantioselective. The des-methyl primary alcohol analogue 85 showed a significant reduction in TLR7 activity which was restored by addition of methyl (compound 86) and benzyl ether substituent (compound 87). It was concluded that the loss of the hydroxyl group of the N-1 substituent has a significant impact on TLR8 activity as shown in compound 86.
A series of TLR7/8 agonistic N-1 substituted C2-n-butyl-C7-methoxycarbonyl-imidazoquinolines (Fig. 15) was reported by the same research group.68 A diverse set of N-1 substitution including N-1 thiols, amines, alcohols, esters and carboxylic acid functionalities were synthesized and screened for TLR7 and -8 activities using HEK blue cells. As reported earlier, an alkyl hydroxyl group at the N-1 position enhanced the TLR8 activity. The esterification of the 2-hydroxypropyl group of compound 82 yielded TLR8 inactive analogues 88 and 89. However, the less bulky ester derivative (88) retained TLR7 activity thereby demonstrating that the presence and position of hydroxyl group are both important for TLR8 activity. In case of aliphatic alkyl hydroxyl group at N-1 position, higher analogues 90 and 91 showed potent TLR7 activity as compared to ethyl hydroxy analogue 85. However, compounds 90 and 91 had no TLR8 activity.
Fig. 15. SAR in N-1 substituted C7-methoxycarbonyl imidazoquinolines.

The amino-alkyl derivatives (92–96) demonstrated a positive correlation in alkyl chain length and TLR7/8 activity. With an increase in chain length from aminoethyl to aminopentyl substitution, TLR8 activity was enhanced and only the aminopentyl derivative showed TLR7 activity. The X-ray crystallographic structure shows that the terminal amino group of an N-1 p-(aminomethyl)benzyl substituted imidazoquinoline (similar structure to 96) extends into a pocket within the TLR8 ectodomain and forms favorable hydrogen bonding interactions with the main-chain carbonyl group of glycine 351, thereby explaining the observed results.69
The activity of sulfides (97–99) and the bulky methyl ester (100–104) derivatives suggested that the binding site in TLR7 is more tolerant to aliphatic substitutions. All sulfides (97–99) and methyl ester (100–104) derivatives showed moderate TLR7, but no TLR8, activity. N-1 substituted acid derivatives (105–107) also had minimal TLR7/8 activity. It was concluded that TLR8 activity correlates with hydrogen bonding donor atom at the N-1 position as demonstrated by the high TLR8 activity and selectivity of compounds containing a terminal amino group.
Recently, Hunt et al.70 reported C7-substituted imidazoquinoline derivatives (Fig. 16).
Fig. 16. SAR in C7-substituted imidazoquinolines.

They explored the effect of electron withdrawing and electron donating groups at C-7 position of the imidazoquinoline scaffold. C7-Methoxy (108a–108b), chloro (109a–109d), nitrile (110a–110d), and hydroxyl (111) analogs were synthesized with varying N-1 substituents from an isobutyl group to the 2-hydroxy-2-methylpropyl and C-2 substituent from n-butyl to ethoxymethyl group. The electron donating groups (EDGs) were stronger activators of TLR7/8 than electron withdrawing groups (EWGs). The C7-hydroxy substituted compound (111) was found to be the most potent TLR7 agonist with EC50value of 73 nM but no TLR8 activity under 100 μM.
Overall, the four positions explored in imidazoquinoline scaffold are C-4 and C-2 position (mostly explored by David and co-workers), N-1 position (by Bonnert et al. and Yoshiaki et al.) and C-7 position (by Ferguson and co-workers). The free amine group at C-4 position and n-butyl group at C-2 position were observed to be the most suitable substituents for TLR7/8 agonistic. The substituents with basic centers at N-1 position and with electron releasing groups at C-7 position were found to be the most effective in improving the TLR7/8 activity.
The majority of TLRs signal via homo- or hetero-dimerization71 as evident in the crystal structures of TLR2 (ref. 72 and 73) and TLR3.74 David et al. examined the dimeric constructs of imidazoquinoline as modulators of TLR7/8.75 Specifically, the dimeric constructs of imidazoquinoline linked at the C-2, C-4, C-8, and N-1 aryl positions were synthesized and evaluated for their hTLR7 and hTLR8 modulatory activities (Fig. 17).
Fig. 17. SAR in dimeric imidazoquinolines.

The imidazoquinoline based dimers linked at C-2 position (112a–112d) showed only antagonistic activities for TLR7 and 8. Compound 112a with a 3-carbon spacer showed maximum antagonistic activity in both TLR7 and TLR8 assays with IC50 values of 3.1 μM and 3.2 μM respectively. Imidazoquinoline dimers linked at the C-4 (113a–113d), C-8 (114a–114c), and N-1 aryl positions (115a–115c) showed only TLR7 activity with no TLR8 agonistic or TLR7/8 antagonistic activities, with an exception of N-1 aryl dimer (115c), with a 12-carbon linker found to be the most potent dual TLR7/8 agonist. Compound 115c with EC50 values of 0.17 μM and 4.78 μM for TLR7 and TLR8 agonism, respectively, showed a long linker is necessary for dual TLR7/8 activity. Compound 113b in which both imidazoquinoline rings are dimerized by 8-carbon linker showed maximum TLR7 activity of 0.56 μM. The linking of quinoline rings at C-8 position was done by introducing amine functionality at C-8 position.
Nitration of compound 41 was carried out to obtain a nitro compound 116a, which on reduction resulted in the amine functionality at C-8 position (116b) in the quinoline ring. Both nitro (116a) as well as amine (116b) derivatives show less TLR7 activity than their parent compound 41 with EC50 value of 0.56 and 0.45 μM, respectively. The C-4, C-8, and N-1 aryl linked dimers were agonists, with the last being most potent and C2-linked dimers were found to be potently antagonistic to both TLR7 and TLR8. It was concluded that there is an important role of C2-n-butyl group for the activity of imidazoquinolines.
David et al. also reported the multimerization effect of potent dual TLR7/8 agonistic compound 41, via the synthesis of dendrimeric compounds 118 and 121 using different linkers (Fig. 18).76 The trimeric dendrimer 118, with three units of imidazoquinoline 41, retained both the TLR7 and 8 activities while the hexameric dendrimer 121 with six units retained substantial TLR7 activity but lost TLR8 activity.
Fig. 18. Synthesis of trimeric and hexameric imidazoquinolines.

SAR in 1H-imidazo[4,5-c]pyridines
1H-Imidazo[4,5-c]pyridine scaffold was evolved from the imidazoquinoline scaffold wherein David et al.77 curtailed the imidazoquinoline scaffold to imidazopyridines and prepared a focused library around the pyridine ring (Fig. 19). The structural library was designed based on the potential activity of 3M-003 (6), loxoribine (9) and isatoribine (10) which all are deprived of the quinoline ring system.
Fig. 19. SAR in imidazo[4,5-c]pyridines.

N-1 benzyl and C2-n-butyl were observed to be the minimal part structure for the potential TLR7/8 activity of imidazoquinolines. So, the SAR in the imidazopyridines (Fig. 19) was initiated with the synthesis of 1-benzyl-2-butyl-1H-imidazo[4,5-c]pyridin-4-amine (122a).77 Compound 122a was found to be a hTLR7-specific agonist (EC50: 1.57 μM), with negligible hTLR8 activity. Acylation and alkylation of C4–NH2 group (122b–122c) and (122d–122e) respectively resulted in the complete loss of TLR7 activity and highlighted the importance of C4–NH2 group for activity.
Later, a total twenty-eight, C6-substituted analogues were synthesized (123a–123z′′), among which compound 123a, with a C6-chloro derivative was totally inactive whereas C6–NH2 derivative 123b displayed potent TLR7 activity comparable to the parent compound 122a. N6-Butyl (123c) and N-6 cyclohexylmethyl (123e) substituents showed nearly equal activity, but activity was diminished in the N-6 heptyl analogue (123d). The N-6 phenyl-substituted compound 123f was marginally weaker than the parent compound 122a, but the activity of the N-6 benzyl analogue 123g was nearly 7.6 times that of compound 122a which prompted authors to go for a detailed SAR investigation on various aryl substituents at the N-6 position. The electron-rich N-6 substituted methoxybenzyl derivatives (123h and 123i) and the pyridinylmethyl compounds (123j and 123k) were marginally more active than the trifluoromethyl (123l) and chloro-(123m) substituted analogues. Compounds 123n and 123o with methyl amine groups at the ortho and para positions were also active in primary screens, with EC50 values of 0.26 and 0.37 μM, respectively. The biphenylmethyl-substituted compound 123p was active, whereas the naphthylmethyl analogue 123q was inactive, confirming the specific role of both steric and electronic effects in governing the TLR7 activity. Methylfuran analogue (123r) was found to be the most active with EC50 value of 0.075 μM.
In the C6-alkyl or -aryl analogues (123s–123z′′), the C6-butyl compound 123s showed 5 times more activity than compound 122a. However, the biaryl containing analogs with direct connection of aryl groups at C-6 position (123u–123w and 123z–123z′) were totally inactive. But the TLR7 activity of this scaffold was restored in the C6-benzyl (123x and 123y) and 6-phenethyl analogues (123z′′). Further, the benzologue 124 was synthesized with naphthalene ring which showed improvement in the activity over the parent imidazopyridine 122a with EC50 of 0.22 μM. Among the dimeric imidazopyridines 125a and 125b, compound 125a synthesized using p-xylylenediamine, demonstrated TLR7 activity while the ortho derivative (125b) was totally inactive. This SAR clearly revealed that the imidazopyridines with appropriate substituents can effectively activate TLR7. N-1 benzyl, C2-butyl and C6-N-benzyl functionalities were observed to be the most suitable substituents for TLR7 activity.
SAR in thiazolo[4,5-c]quinolines
3M Pharmaceuticals first reported the thiazolo[4,5-c]quinoline-4-amine analogues with potential TLR7/8 activity.43 Among the various analogs synthesized, 2-propylthiazolo[4,5-c]quinolin-4-amine (5) was found to be a potent TLR8 agonist at 3 μM.29 Alkoxy, aryloxy and arylalkyleneoxy derivatives of thiazoloquinolines were shown to be active.78,79 To further explore this chemotype, in 2013, Kokatla et al.80 reported a SAR on thiazoloquinolines by considering compound 5 as a lead (Fig. 20). Initially, the alkyl chain length at C-2 position of 5 (Fig. 20) was varied to investigate the relationship between C2-alkyl chain length and TLR7/8 activity. In TLR7-active imidazoquinolines, a distinct relationship was observed between C2-alkyl chain length and TLR7 activity.
Fig. 20. SAR in thiazolo[4,5-c]quinolines.

The C2-methyl (126a), -ethyl (126b), -propyl (5), and -pentyl (126d) analogues displayed comparable potencies in the hTLR7 and hTLR8 reporter assays. TLR7 and TLR8 activity of 0.86 μM and 0.41 μM, respectively, was observed for the n-butyl analogue (126c). The higher homologues (126e–126g) did not exhibit any activity for both TLR7 and TLR8. The isopropyl group at C-2 position (126h) resulted in approximately ten-fold reduction in TLR8- and two-fold reduction in TLR7 activity as compared to compound 5. Several additional branched analogues of both 5 (C2-n-propyl) and 126c (C2-nbutyl) were also synthesized (126i–126n). The methyl substitution at ω-1 (126i) as well as ω-2 (126j) position in C2-n-propylderivative was well tolerated with moderate activity. On the other hand, the ω-2 methyl substituted, 126l was active, whereas the 126k with ω-1 methyl substitution was entirely inactive. Both 126m and 126n with tert-butyl groups at terminal position were also inactive, suggesting that there is poor tolerance for terminal steric bulk in this series of compounds. Bio-isosteric analogues 126o and 126p with trifluoromethyl group were also synthesized and a decrease in activity in TLR8 agonism was observed in the long chain homologues. Substitution at the C-8 position of CL075 (5) was also examined by the authors. Selective electrophilic substitution at C-8 position of the thiozoloquinoline ring afforded 8-nitro analogue 127a and bromo analogue 127b, both of which were inactive. The 8-amino (127c) and 8-azido (127d) analogues were obtained from 127a and both showed attenuated activity relative to the compound 5. Further selective N-alkylation and N-acylation of 8-amino analogue 127c provided analogues 127e and 127f, both of which were inactive. The triazole derivatives 127g–127m synthesized were also inactive except 127m with trimethyl silyl group showed selective TLR8 activity.
C4-Amine of the compound 5 was further alkylated and acylated, yielding analogues 128a–128l (Fig. 20). The C4-N-alkylated compounds 128a–128c, were totally inactive, while some, but not all of the C4-N-acylated derivatives with short acyl groups were found to be active. Specifically, the formyl (128d), and butyryl (128f) analogues, but not the acetyl analogue (128e) had TLR8 activity. The higher homologues (128g and 128h) were completely inactive for TLR7 and TLR8. However, the C4-amine substituted azidoacetamide 128i, azidopropionamide 128j and pentynamide 128k analogues displayed weak TLR8 activity. Aromatic amides at C-4 position were not tolerated, as the benzamide analogue 128l was completely inactive. A series of carbamates (128m–128p) and sulfonamides (128q–128u) were also synthesized. The carbamate derivatives showed very weak activity, while the sulfonamide analogues were completely inactive in the primary reporter gene assays. A few other analogs like urea (128v), phosphoramidate (128w) and guanidine (128x) functional groups at C4 were also examined, but none of them were found to be TLR7 or TLR8 active. Moreover, the formamide 129a, acetamide 129b, and butyramide 129c derivatives were also synthesized and the same pattern of activity was observed as was observed in analogues of compound 5. To conclude, the C2-n-butyl thiazoloquinoline (126c) was observed to be a more potent TLR7 and TLR8 agonist than C2-n-propyl thiazoloquinoline 5.
SAR in furo[2,3-c]pyridines
David et al.81 designed and synthesized an imidazo[1,2-a]pyrazine (131) scaffold (Fig. 21) having structural similarity with other known TLR7/8 ligands. The synthesis of imidazo[1,2-a]pyrazine was achieved by the Groebke–Blackburn–Bienaymé (GBB)82 multicomponent reaction using aldehyde, isocyanide and amidine as building blocks. A small test-library of imidazo[1,2-a]pyridine (130), and 8-amino-imidazo[1,2-a]pyrazine (132) based compounds with appropriate substituents was also synthesized (Fig. 21), but none of these compounds were active for human TLR7 or TLR8 nor TLR3 or TLR9 up to a concentration of 250 μM.
Fig. 21. Scaffolds synthesized by using GBB multicomponent reaction.

Interestingly, when the pyridoxal was used as aldehyde component in the GBB reaction, a novel furo[2,3-c]pyridine (133, Fig. 21) scaffold was identified. Six furo[2,3-c]pyridines (134a–134c and 135a–135c) were synthesized by using two amidines (2-aminopyrazine and 2-aminopyridine), three isonitriles [2-isocyano-2-methylpropane, isocyanocyclohexane and (isocyanomethyl)benzene] and pyridoxal as aldehyde component. Only four compounds 134b, 134c, 136b and 136c with isocyanocyclohexane and (isocyanomethyl)benzene were found to specifically activate TLR8 but not TLR-7 (Fig. 22). Furo[2.3-c]pyridines derived from 2-aminopyridine (135b, 135c) were more potent and to further extend the SAR, a small set of compounds was designed using 2-aminopyridine, pyridoxal and 13 different isonitriles, including linear aliphatic (135e–135f), branched aliphatic (135g–135i), linear aliphatic with silyl group (135j), morpholine ring (135k), ester (135l and 135m), and phosphate ester (135n) group, as well as aromatic substituents (135o–135q). In linear aliphatic isonitriles, C2-N-pentyl (135f) and C2-N-(trimethylsilyl)methyl analogues (135j) displayed maximum TLR8 activity and the branching in aliphatic chain diminished the activity. Compound 135d with a free NH2 group at C-2 position as well as 135k, derived from a morpholine ring containing isonitrile and 135l, derived from a phosphate ester bearing isonitrile were totally inactive except the respective tert-butyl ester derivative (135m). Additionally, the compounds derived from aromatic isonitriles (135o–135q) were also bereft of any TLR8 activity.
Fig. 22. SAR in furo[2,3-c]pyridines.

In the whole series, the compound 135f with N-pentyl group was observed to be the most potent. So, N-pentyl group was fixed and three additional furo[2,3-c]pyridines were synthesized using aniline 136a, 3-fluoroaniline 136b, and 3-nitroaniline 136c. The fluoro derivative 136b was observed to be more active than 135f, with EC50 of 0.37 μM. Further replacement of 2-aminopyridne by aniline in compound 135c resulted in loss of TLR8 activity (137; EC50 1.68 μM). Overall, the GBB reaction protocol with pyridoxal as an aldehyde component resulted in the identification of moderately active TLR8 specific novel furo[2,3-c]pyridine scaffold. Specifically, the C2-N-pentyl (135f) and C2-N-(trimethylsilyl)methyl (135j) furo[2,3-c]pyridines were found to be the most potent TLR8 agonists in this series.
SAR in furo[2,3-c]quinolines
After the discovery of furo[2,3-c]pyridines, David et al.83 explored its benzologues (Fig. 23) which closely resemble the known TLR7/8-active compounds. A library of 4-amino-furo[2,3-c]quinolines were constructed with variation at C-2 position (140b–140y). Compounds 140b–140f showed pure TLR8 activity, with maximal activity exhibited by compound 140d with a C2-butyl group (EC50 = 1.6 μM). The short chain (140b, 140c) as well as long chain (140e, 140f) homologues at C-2 position, displayed lower activity, while the short chain analogue 140a was observed to be totally inactive.
Fig. 23. SAR in furo[2,3-c]quinolines.

The C2-substitued furo[2,3-c]quinolines with branched alkyl groups (140g–140i) showed abrogated TLR8 activity and the analogues with cycloalkane (140j–140m) or aromatic (140n–140o) substituents were totally inactive, indicating the intolerance of steric bulky group at C-2 position for TLR8 activity.
Further to improve TLR8 activity, hydroxyl groups and amine functionality were introduced in the substituents attached at C-2 position. These polar H-bond donor groups were introduced to enhance the interaction of ligand with TLR8 binding pocket. However, all the analogues either with free hydroxyl functionality (140p–140v), terminal primary amine bearing analogs (140x–140y) or the compounds with an ether (H-bond acceptor) containing alkyl group (140w) at C-2 position were inactive.
Shukla et al.84,85 observed a regioisomerism dependent switch in TLR7 activity in imidazoquinolines. So, the key regioisomeric furo[3,2-c]quinolines (141a–141c) were screened but none of the synthesized analogs displayed any TLR7/8 activity. The structurally simpler bicyclic analogues (142a and 142b), were also synthesized and the analogues 142a, 142b retained TLR8-selective activity but were substantially weaker (EC50 = 24.4 and 46.2 μM, respectively). Likewise, the C1-bromo (143a) and C1-benzyl (143b) analogues were synthesized along with the furo[2,3-c]isoquinoline (144) analogue with a C2-butyl substituent. However, all these analogues (143a, 143b and 144) were inactive. Overall, a pure TLR8 agonist, C2-butyl-furo[2,3-c]quinoline (140d) emerged out to be the most potent analogue in this series.
SAR in 2-aminoquinolines
Co-crystallization of previously discussed C2-butyl-furo[2,3-c]quinoline with TLR8 revealed that the hydrogen bond between the oxygen atom of the furanyl ring of the C2-butyl-furo[2,3-c]quinoline and Thr574 of TLR8 was missing. This analysis suggested that the furan ring is optional for TLR8 activity. Based on these observations, 3- and 4-substituted aminoquinoline derivatives were synthesized and screened for TLR8 activity (Fig. 24).86 Initially, the 3-alkoxy-2-aminoquinoline derivatives (145a–145j) at C-3 position were synthesized and screened in a TLR8-specific assay. In this homologous series, 3-butoxy analogue (145d) showed maximum activity with EC50 of 2.18 μM and the clear dependence of substituent chain length with TLR8 activity was observed. Similar dependence of alkyl chain length was observed even in case of branched alkoxy analogues (145g–145j). Further to understand the effect of electronegativity of hetero atom at C-3 position, N-3 butylquinoline and 3-(butylthio)quinoline analogues (145k and 145l, respectively) were synthesized while these compounds were less active than 3-butoxy analogue 145d. So, the C3-alkylquinolin-2-amines (145m–145r) without heteroatom in alkyl chain length were screened and among the various analogues synthesized, 3-pentyl quinoline (145n) possessed maximum TLR8 activity which was nearly ten-fold greater than that of 3-butoxy analogue 145d. Additional substitution at C-4 position (146a–146c) of compound 145d was done to examine further alteration in activity. These compounds were totally inactive for TLR8, suggesting that there is a poor tolerance of substitution at C-4 position of quinoline ring. The result was also confirmed by inactivity of C-4 substituted quinoline-2-amines (147a–147d) at up to 500 μM.
Fig. 24. SAR in 2-aminoquinolines.

Beesu et al.69 observed a key interaction of 3-pentylquinoline-2-amine (145n) in the binding site of hTLR8 and installed a alkylamino group at all the possible positions on quinoline ring of 3-pentylquinoline-2-amine (145n). Authors continued the SAR from C-4 position and synthesized C4-substituted 3-pentylquinolin-2-amines (148a–148d, R1 position), but only slight improvement in TLR8 activity was observed (Fig. 25).
Fig. 25. SAR in 3-pentylquinoline-2-amines.

Next the 2-amino-3-pentylquinoline was substituted at C-5 position (R2 position) with 2-aminomethylbenzyl (149a), 3-aminomethylbenzyl (149b), and 4-aminomethylbenzyl (149c) groups. An improvement in activity was observed for both compounds 149b and 149c with EC50 value of 49 and 38 nM respectively, whereas the 2-aminomethylbenzyl-substituted compound 149a showed weaker TLR8 activity than the parent compound, 3-pentylquinolin-2-amine (145n). The inactivity of benzyl (149d), carboxamide (149e) and cyanide analogues (149f–149g) for TLR8 activity proved the important role of terminal amine group in binding hTLR8. The aryl coupled 5-(aminomethyl)phenyl analogues 149h and 149i were also synthesized via Suzuki coupling reaction but none of the analogs showed prominent TLR8 activity. Moreover, alkylamine substituted homologues (149j–149m) also demonstrated the dependence of TLR8 activity on the length of the alkylamine substituent, wherein a progressive increase in activity from the aminopropyl (149j, 91 nM), aminobutyl (149k, 27 nM) and aminopentyl (149l, 9 nM) analogues was observed. However, the activity decreased with further increase in alkyl amine length (149m, aminohexyl). No improvement in the TLR8 activity was observed in carboxamide (149n) and guanidine (149o) derivatives.
An amino alkyl substitution was introduced at the C-6 (150a–150c, R3), C-7 (151a–151c, R4) and C-8 (152, R5) position of quinoline ring. Compounds 150a–150c showed slight decrease in the activity while compounds 151a–151c showed slight gain in the activity as compared to parent compound, 3-pentylquinolin-2-amine (145n). The C8-substitued compound 152 was totally inactive, whereas a dual C5 and C7-substituted compound 153 was found to be weaker agonist (621 nM) than the parent compound (145n). It was concluded that the substitution on C-5 position of 3-pentyl-quinoline-2-amine is well tolerated.
SAR in 1H-benzimidazol-2-amines
After the discovery of 3-pentyl-quinolin-2-amine as a structurally simple and highly potent pure TLR8-specific agonist, Beesu et al.87 reported a detailed SAR in the ring contracted 1-alkyl-1H-benzimidazol-2-amines as TLR8 agonists (Fig. 26). Initially, the ring-contracted 2-amino-3-alkylindole analogue 154a was synthesized but was observed to be unstable and resulted in the rapid formation of oxidized 3-ol derivative 154b. Both the hydrochloride salt of compound 154a and oxidized derivative 154b were found to be TLR8 inactive. Due to the stability issue of 2-aminoindole scaffold, structurally similar 1-alkyl-2-aminobenzimidazole analogues were synthesized (155a–155f). Initially effect of chain length at N-1 position of 2-aminobenzimidazole scaffold was investigated with the synthesis of N1-butyl (155a), -pentyl (155b) and -hexyl analogues (155c). N1-Pentyl substituted compound 155b was observed to be a potent TLR8 agonist with EC50 value 3.23 μM.
Fig. 26. SAR in 1H-benzimidazol-2-amines.

Benzyl and 3-aminomethylbenzyl substituted imidazoquinoline scaffolds have been reported as pure TLR7 or mixed TLR7/8 agonists. N1-Substituted benzyl (155e) and 3-aminomethylbenzyl (155f) analogues of 2-aminobenzimidazole were synthesized but none of these analogs showed any TLR8 activity. The substitution of N1-pentyl group with butoxycarbonyl group (155d) and acylation of the N-2 amine (154c) on lead compound 155b resulted in the complete loss of activity. These results demonstrated that both free amines at C-2 and n-pentyl group at N-1 position of benzimidazole scaffold are important for the TLR8 activity.
Yoo et al.77 observed that the benzo analogues of imidazo[4,5-c]pyridines have TLR7 specific activity. So, all the possible regioisomeric benzo analogues (154d–154f) of compound 155b were synthesized. Among these compounds, only compound 154f showed moderate TLR8 activity and other two were totally inactive. The observation suggested that the substitution on C-4 and C-5 position of 2-aminobenzimidazole scaffold can be tolerated. Along with this, all the possible regioisomers of imidazopyridines (154g–154j) were also screened for TLR7/8 activity, but none of these analogs were active.
To understand the steric effect on a benzimidazole ring, systematically methyl substituted analogs at C-4 to C-7 positions (154k–154n) were synthesized and only the C4-methyl derivative 154k (EC50 1.13 μM) was observed to be more potent than the parent compound 155b. Furthermore, the variation in the TLR8 activity was observed with alteration of the electronic environment at C-4 position of 2-aminobenzimidazole scaffold using different electron-donating and electron-withdrawing substituents (156a–156m).
The methoxy analogue 156a with EC50 value of 3.74 μM showed comparable activity to that of parent compound 155b but was less active than its 4-methyl analogue (154k). The substitutions by electron-withdrawing groups such as –F, –Cl, –CF3, –Br and –NO2 (156b–156f) at C-4 resulted in the complete loss of TLR8 activity. Ethyl group (156g) substitution led to a slight diminished TLR8 agonism as compared to its lower homologue (154k) showing that the long alkyl substituents or bulkier alkyl groups are not favorable at the C-4 position. The electron-deficient analogues (156b–156f) were observed to be totally inactive, while the analogues with electron donating groups at C-4 position (156h–156j) show moderate activity. Additional analogs with phenyl (156k), benzyl (156l), and benzyloxy (156m) groups at C-4 position of 2-aminobenzimidazole scaffold also resulted in the complete loss of TLR8 activity. All the analogues were screened for activity using reporter cell lines specific for human TLR2, TLR3, TLR4, TLR5, TLR7, TLR9, NOD1, and NOD2 but no activity was detected, confirming the specificity of the active analogues for human TLR8.
SAR in purine analogues
After the discovery of loxoribine (9) and isatoribine (10) as type 1 interferon (IFN-α and β) inducers, various SAR studies were performed on the adenine scaffold. In 2002, Hirota and co-workers88 synthesized a set of adenine derivatives by varying substitution at C-2, C-8 and N-1 positions. The investigation resulted in the understanding that the hydroxyl group at C-8 position and benzyl group at N-9 position are the most efficient substituents for IFN-α induction (Fig. 27).
Fig. 27. Minimum effective concentration (MEC) of 8-hydroxy-9-benzyladenines for the induction of IFN-α.

A large increase in activity was observed due to the introduction of alkyl, alkylthio, alkylamino and alkoxy groups at C-2 position wherein the butoxy derivative (157a) was observed to be the most potent in the series with a minimum effective concentration (MEC) value of 1 nM, followed by butylthio (157b), butyl (157c) and butylamino (157d) derivatives.89 Further, various alkoxy moieties were explored at C-2 position for interferon induction90 and observed that compound 158 with 2-methoxyethoxy group at C-2 position was about 100-fold more potent than the imiquimod. Cottam and co-workers91 also synthesized a series of 8-substituted amino adenines (159) and observed that the C-8 substitution with amines decreased the interferon-inducing activity of the adenine scaffold (Fig. 28).
Fig. 28. SAR in 9-benzyladenines.

Later, Filippov et al.92 reported 7-hydro-8-oxoadenine derivatives (160 and 161) with an azido functionalized alkoxy chain at the C-2 position. The azido group failed to improve the TLR7 activity of the scaffold and all the derivatives were less active than the corresponding butoxy derivative 157a.
To improve TLR7 activity and water solubility, Isobe and co-workers93 reported a series of 8-oxoadenines with substitution on the benzyl ring at the N-9 position of the adenine scaffold (Fig. 29). Among the synthesized library, the dimethylamine analogue (162b) was observed to be approximately 20-fold more active than the parent compound 6-amino-9-benzyl-2-(butylamino)-7,9-dihydro-8H-purin-8-one (162a). The piperidine analogue (162c) showed comparable activity with compound 162b, while the substitution with morpholine derivative (162d) resulted in the loss of TLR7 activity, suggesting the role of basic nitrogen at the benzylic position for the higher activity. This observation was further confirmed with the high hTLR7 activity of the N-methyl-piperazine (162e) derivative. The meta-substituted piperidine (163a) and N-methyl-piperazine (163b) derivatives were observed to be weaker agonists than their corresponding para-substituted (162c and 162e) analogs.
Fig. 29. SAR in N-9 substituted 8-oxoadenines.

SAR of N-9 pyridylmethyl adenines was also performed by the same research group.94 This showed 3-pyridyl derivatives as the most potent interferon inducers followed by 4-pyridyl and 2-pyridyl derivatives. 6-substitued-3-pyridyl derivatives (164a–164l) plus a dimethylaminoethoxy analogue (164c) showed single figure nano-molar activity and improved solubility compared to its methyl (164a) and methoxy (164b) analogues. When the methyl chain length was increased (164d–164e), approximately two-fold decrease in activity was observed. The mono-methyl analogue 164f also showed a significant decrease in the activity. In compound 164c, the replacement of dimethylamino group with morpholine (164g) resulted in the loss of activity as observed in compound 162d. Among the nitrogen atom linked 3-pyridyl derivatives (164h–164l), N-methyl analogue (164h) showed a four-fold loss of activity compared to the oxygen linked compound 164c while the cyclic amino compounds (164i–164k) showed high activity with single figure nano molar EC50 values. The des-methyl analogue (164l) showed a nine-fold loss of activity as compared to N-methyl-piperazine derivative 164k and indicated the importance of tertiary amine at this position for the activity.
After analyzing improved solubility and activity of N-9 benzyl and pyridylmethyl adenine derivatives, Bazin and co-workers95 reported a series of N-9 heterocycloalkyl substituted 8-oxoadenine derivatives (165a–165f). During the SAR investigation, authors analyzed the effects of variation in the chain length of N-9 alkyl linker on TLR7 and TLR8 activity (Fig. 30).
Fig. 30. SAR in 9-(4-piperidinylalkyl)-8-oxoadenines.

The SAR studies clearly highlighted the importance of linker length between the adenine and the 4-piperidinyl ring as evident by the observation that compound 165a wherein the 4-piperidinyl ring was directly attached with N-9 position of adenine was inactive whereas all the other analogs (165b–165g) with linker were active. The increase in chain length positively enhanced the TLR7 activity but no linear correlation between linker length and TLR7 activity was observed. The 5-carbon linker 8-oxoadenine derivative (165f) was observed to be the most potent TLR7 agonist and IFN-α inducer in the series while 1-carbon linker derivative (165b) was the most potent TLR8 agonist. The 8-oxoadenine derivatives with longer chain linker demonstrated many folds decrease in TLR8 activity.
Recently, Biggadike et al.96 installed a five and six membered saturated oxygen and nitrogen containing heterocyclic substituents at the N-9 position of 8-oxoadenine scaffold. The heterocycles were linked via methylene chain of varying length and all the derivatives were screened for IFN-α and TNF-α induction. IFN-α was used as a measure of TLR7 activity, and TNF-α was used as a measure of TLR8 activity. Initially, the oxygen containing saturated heterocyclic tetrahydropyran ring was attached and the impact of N-9 chain length was explored (166a–166f). In the second series (167a–167f), the size of heterocyclic ring and position of attachment to the oxygen bearing heterocycle was investigated. Among 4′-linked tetrahydropyran (THP) derivatives (166a–166f), the n-propyl (166c), n-butyl (166d) and n-pentyl (166e) chain linked analogues were observed to be potent IFN-α inducers than the benchmark 6-amino-9-benzyl-2-butoxy-7,9-dihydro-8H-purin-8-one (157a, Fig. 27) (pEC50 6.9). The examination of alternatively linked THP (166d, 167c and 167d) and tetrahydrofuran (THF) derivatives (167a–167b and 167e–167f) showed that the ring size and position of attachment has very little impact on activity. In case of N-9 linked nitrogen heterocyclic derivatives (168a–168h), the C- and N-linked piperidine (168a–168d) and N-linked piperazine (168e) analogues were observed to be the most potent in the series. The N-linked pyrrolidine derivative (168f) demonstrated selective TLR7 activity but less activity as compared to its 6-membered counterpart 168a. The expansion to the 7-membered azepane analogue (168g) resulted in a slight enhancement in activity. Moreover, the low activity of the morpholine derivative (168h) highlighted the importance of basic amine to enhance IFN-α activity. The N-linked piperidine analogue was further explored to study the effect of the length of the linking chain. A significant reduction in IFN-α activity was observed in analogues (169a–169b) which has linking chain of less than four carbon atoms while those with 4–6 carbon atom chain (169c–169e) showed a high activity and selectivity for TLR7.
For the optimization of C-2 substituents, the N-linked piperidine analogues attached through n-butyl (170a–170c) or n-pentyl chain (171a–171c) were explored. The 2-(S)-pentyloxy derivatives (170a and 171a) were observed to be nearly 10-fold more active than their n-butyloxy counterparts (169c and 169d). The longer 2-(S)-hexyloxy derivative (170b) did not show a significant difference in activity while a slight drop in the activity was observed in the 2-(S)-butyloxy derivative (171b). The substitution with cyclopentyloxy (170c) and isopropyloxy (171c) groups resulted in a major loss in activity (Fig. 31).
Fig. 31. SAR in 8-oxoadenines containing 5 and 6-membered saturated rings.

Encouraged by these results, the SAR of THF and THP analogues was further explored by taking five different C-2 groups (n-butyloxy, n-butylamino, (R)-2-pentyloxy, (S)-2-pentyloxy and cyclo-propylethoxy) and alkyl chains of different length (n-ethyl, n-propyl and n-butyl). Among these, a 2-(S)-pentyloxy 4′-linked THP analogue 172 was potent for IFN-α and TNF-α induction with pEC50 in human PBMCs of 7.9 and 6.9, respectively.
The researchers at the Bristol-Myers Squibb, New Jersey, USA also performed a similar SAR around 8-oxoadenine scaffold by varying aromatic ring substitutes at N-9 position (173a–173e) and observed the importance of heteroatom around core scaffold for hydrogen bonding with the receptors.97–99
After various SAR studies around 8-oxoadenine scaffold, Pryde et al.100 studied how the changes in the electronic environment of adenine scaffold alter TLR7 activity. The nitrogen atom in the adenine scaffold was substituted with carbon atom or shifted to other positions, which resulted in a library of purines as well as non-purine derivatives (Fig. 32). 6-Amino-9-benzyl-2-methyl-7,9-dihydro-8H-purin-8-one (174) with hTLR7 agonistic EC50 value of 829 nM was chosen as the lead compound and electronic variations in the scaffold were studied. Among 3-deazapurines (175–176), 1-deazapurine (177) and N-7 methylene substituted derivative (178), only compounds 175 and176 showed EC50 values below 2 μM, whereas all the other derivatives were weak TLR7 agonists (Fig. 33).
Fig. 32. SAR in 8-oxoadenine with aromatic ring substituents.

Fig. 33. Diverse set of purine and non-purine derivatives by Pryde, D. C. et al.

When the purine ring system was replaced with pyrazolo-triazine (179), thiolate purine (180), pyrazolo-pyrimidine (181 and 182) and ring expanded derivative (183), a many fold decrease in TLR7 activity was observed as compared to the parent compound 174. Recently, an extension SAR study was also performed on pyrazolo[4,3-d]pyrimidine scaffold101 wherein the addition of basic amine at the benzylic group increased the TLR7 activity. Further, C-2 substituted 3-deazapurine derivatives (184–189) were explored, wherein the branched alkyl substituent (184) and n-propyl substituent (185) at C-2 position showed 3–5 fold improved TLR7 activity, while complete removal of the C-2 alkyl group (187) or N-6 amino group (189), or extending the length of C-2 alkyl ether group (186) resulted in loss of TLR7 activity. Interestingly, C2-trifluoromethyl-3-deazapurine derivative (188) was observed as the most potent TLR7 agonist in the series with EC50 value 102 nM, but with low solubility.
Further SAR studies (Fig. 34a and b) were performed to improve the solubility as well as activity of compound 188.102,103 Starting with N-1 derivatives (190a–190k), the substitution with alkoxy side chain (190a) or cyclic ethers (190b) resulted in many folds decrease in activity compared to the parent compound 188. Further, various aromatic substituents (190c–190k) were also explored to study the SAR and alter the solubility of the compounds. Among the pyridine derivatives (190c–190e), compound 190c was identified as a potent TLR7 agonist with EC50 value of 50 nM with improved aqueous solubility over the parent compound 188. The introduction of a second heteroatom (190f–190h) in the pyridine ring resulted in a complete loss of activity. Both the isoxazole (190i) and thiazolo (190j) analogues showed good activity whereas the oxazole (190k) derivative showed high activity with 35-fold improvement in aqueous solubility.
Fig. 34. a SAR in 2H-imidazo[4,5-c]pyridin-2-ones. b SAR in N1-pyridinylmethyl substituted 2H-imidazo[4,5-c]pyridin-2-ones.

Substitutions were also performed at C-2 position (190l–190s) while keeping N-9 pyridine group of the most potent compound 190c, wherein the methyloxazole (190m) and triazole (190n) analogues were observed to be more potent than oxazole (190l) and pyrazole derivatives (190o). The introduction of ether side chain (190p–190q) resulted in loss of TLR7 activity while tetrahydrofuran (190r) and tetrahydro-2H-pyran (190s) derivatives showed promising results. The above SAR led to the identification of N-9 pyridylmethyl-8-oxo-3-deazapurine analogues as potent TLR7 agonists. As an extension to this study, substituted isomeric N-9 pyridinylmethyl derivatives (Fig. 34b) were further investigated.
Among the synthesized analogs, the 2,5-isomers (193a–193f) demonstrated high activity with EC50 value <10 nM, compared to 2,3-substituted isomers (191a–191f) and 2,4-substituted isomers (192a–192f). A large number of 2,5-isomers were further synthesized (194a–194o) to find the most potent derivative with good aqueous solubility. The N-linked (194b and 194d) derivatives were observed to be more active than the corresponding O-linked (194a and 194c) analogues. In the N-linked series, all the three secondary (194d), tertiary (194e), and cyclic tertiary amine (194f) derivatives retained good activity. The addition of polar or basic groups on the cyclic tertiary amine (194g–194h) was also well tolerated. The 2,5-substituted isomers with a substituent containing basic center (194i–194o) were the most active analogues and compound 194l with EC50 value of 0.2 nM was the most active analogue (Fig. 34b).
SAR in pyrimidine-2,4-diamines
In 2009, Bennett and co-workers104 reported the synthesis of the TLR7-active pyrimidine-2,4-diamine scaffold. A set of derivatives with TLR7 activity was synthesized by varying substitution at different positions of pyrimidine scaffold (Fig. 35–38) and the synthesized compounds evaluated in a human TLR7 reporter assay.
Fig. 35. SAR in pyrimidine-2,4-diamines by Bennett et al. (part 1).

Fig. 36. SAR in pyrimidine-2,4-diamines by Bennett et al. (part 2).

Fig. 37. SAR in pyrimidine-2,4-diamines by Bennett et al. (part 3).

Fig. 38. SAR in pyrimidine-2,4-diamines by Bennett et al. (part 4).

In the first set, substitution was done at the nitrogen of 3-aminopropyl group of 5-(3-aminopropyl)-6-methyl-N4-pentylpyrimidine-2,4-diamine (Fig. 35). By keeping methyl-2-(m-tolyl)acetate (in 195a–195m) and methyl-2-(p-tolyl)acetate (in 196a–196i) as one component and carbonyl derivatives as another component, two series of compounds were synthesized. In both the series 2-(dimethylamino)acetaldehyde derivatives (195b and 196b) were observed to be potent TLR7 agonists with pEC50 values of 6.2 and 6.4, respectively. Among the methyl-2-(m-tolyloxy)acetate (197a), methyl-2-(3-(2-oxoethyl)phenyl)acetate (197b) and methyl-2-(4-(2-oxoethyl)phenyl)acetate (197c) derivatives, no significant difference in TLR7 activity was observed. The substitution of C-4 pentylamine with (S)-3-aminoheptanol (198a–198b) resulted in the loss of TLR7 activity as compared to their parent compounds (196a and 196b).
Additional analogues were further synthesized with substituted benzyl derivatives at C-5 and amino alkanols at C-2 position of pyrimidine scaffold (Fig. 35–37). In the case of para-alkyl acetate derived benzyl derivatives, when substitution was done at alkyl group (R2 group shown in blue color) a marginal variation in TLR7 activity was observed in both N-4 butyl (199a–199b) and N-4pentyl (200a–200r) derivatives.
Interestingly, in case of methyl-2-phenylacetate derivatives (201a–201c and 202a–202c), compound 201b, 201c and 202b with electron donating groups showed more activity for TLR7 as compared to the other analogues. The methoxy (203a–203h) and hydroxy (204a–204c) groups at R1 position (Fig. 35) was fixed while varying the substituents at R2 position. Among these derivatives, 1-butylpyrrolidine derivative (203f) was the most potent TLR7 agonist with pEC50 value 8.3 (Fig. 36).
Similar SAR extension study was performed with (S)-2-aminopentanol (205a–205d and 206a–206b), (S)-2-aminohexanol (205e–205f), (S)-3-aminohexanol (207a–207d, 208a–208d and 210) and 3-aminoheptanol (207e–207g) derivatives (Fig. 37). Compounds 207c, 207d and 207g with electron donating groups were observed as most active in the series with pEC50 values of 8.2, 8.7, and 8.2, respectively. The insertion of sulphur atom between pyrimidine and phenyl ring (211a–211b) resulted in slight improvement in TLR7 activity as compared to the parent compounds 199a and 199b but no significant improvement was observed in (S)-1-aminobutan-2-ol analogs (212a and 212b).
In case of meta-alkyl acetate derived benzyl derivatives (213–217), similar TLR7 activity trend was observed. The compounds (213b, 213e, 215a, 215b and 216) with methoxy group at R2 position were observed to be the most active in the series (Fig. 38).
Mcinally et al.105 reported C-5 ortho-methoxybenzyl substituted pyrimidin-2,4-diamines as dual TLR7 and TLR8 agonists (Fig. 39). Two series of compounds were synthesized, one with (S)-2-aminopentanol (218a–218h) at C-4 position of pyrimidine ring and different methylamines at para-position of benzyl ring, while in the other series installation of (S)-3-aminohexanol (219a–219m) at C-4 position resulted in enhanced TLR7/8 activity.
Fig. 39. SAR in C5-ortho-methoxybenzyl substituted pyrimidin-2,4-diamines.

Apros therapeutics reported a class of 2,4-diaminopyrimidine106 derivatives with branched chiral amine at C-4, ortho-methoxybenzyl substituent at C-5 and propanoic acid at C-6 position of pyrimidine ring. The absolute configuration of C-4 amino functionality directly impact the TLR7 agonistic potency, where S-configured amine analogues were 10 time more potent than R-configured derivatives.
In 2016, David et al.107 identified N-4 butyl-5-iodo-6-methylpyrimidine-2,4-diamine (220b, Fig. 39) as a pure TLR8 agonist from high-throughput screening of 123 943 compounds108 and C-4 alkyl chain length (220a–220d) was further varied to test its effect on activity. Compound 220b with N-4 butyl chain length showed high TLR8 activity (EC50: 1.64 μM). The substitution on C4-amine (220e and 220f) resulted into a complete loss of TLR8 activity, which indicated that C4-amine act as hydrogen bond donor and is essential for the TLR8 activity. Bio-isosteric 4-butoxy (220g) and 4-thiobutyl (220h) analogues of the compound 220b, showed no TLR8 activity. To examine the effect of alkyl chain at C-4 position, n-pentyl (220i) as well as other N-alkyl derivatives (221a and 221b) were also screened for TLR8 activity and all the compounds were totally inactive, indicating the importance of hydrogen bond donor atom at this position for TLR8 activity.
A substantial loss of activity was observed among 5-desiodo (222a), 5-fluoro (222b), 5-chloro (222c) and 5-bromo (222d) analogues of the compound 220b at R2 position. The EC50value of the C5-des-iodo analogue 222a was observed to be less than C5-iodo analogue 220b while des-iodo and des-methyl analogue 223a was slightly active and the des-methyl analogue 224a was totally devoid of TLR8-agonism. Moreover, various analogues (223b–223d and 224b–224d) with electron-donating group or electron-withdrawing groups at C-6 position were screened but none of them showed any promising results. Interestingly, the C6-phenyl analogue (225a) showed TLR8 activity whereas its iodo derivative 225b was totally inactive. It has been observed in previous SAR that the insertion of an aminoalkyl group on an aminoquinoline scaffold resulted in a 20-fold enhancement of TLR8 activity.69 Therefore, 5-propylamino (222e), -butylamino (222f) and -pentylamino (222g) analogues of parent compound 220b were also synthesized with the activity of compound 222f and 222g nearly 73-fold higher than 220b. A similar result was observed among 6-phenyl-5-alkylamino derivatives (225c–225e), where 5-butylamino (225d) and 5-pentylamino (225e) analogues were observed to have higher activity in the series (Fig. 40).
Fig. 40. SAR in pyrimidin-2,4-diamines by David and co-workers.

The 5-butylamine substituted pyrimidine-2,4-diamine (222f) (Fig. 40) was found to be a pure human TLR8 agonist with an activity of 300 nM. Hence, the C-5 derivatives were further explored by Beesu, M. et al.109 where in the 5-alkyl substituted analogues (226–229) were found to be weak dual TLR7/8 agonists (Fig. 41). The bioisosteric replacement of ω-amines at C-5 position in the parent compound 222f (Fig. 40) resulted in a primary alcohol (230) and tertiary alcohol bearing analogues (231, 232), which did not show any substantial change in TLR7 and TLR8 activity. The 5-pent-1-yne-substituted analogue (233) as well as 5-ethynyl (234) and its trimethylsilyl substituted precursor (235) were weak TLR8 agonists with no TLR7 activity. The 5-benzyl analogue (236) was observed as TLR7 inactive and weak TLR8 agonist (EC50 value 1.2 μM), while when it's homologous 5-phenylethyl (237), 5-phenylpropynyl (238), 5-phenylpropyl (239), 5-phenylbutyl (240) and 5-phenylpentyl (241) were screened, the compound 239 was observed to be a dual TLR7/8 agonist with EC50 value of 0.19 and 0.75 μM, respectively.
Fig. 41. SAR in C5-substituted pyrimidin-2,4-diamines.

A 6-fold decrease in TLR7 activity and complete loss of TLR8 activity was observed for the 5-cyclohexylpropyl analogue 242, indicating the importance of π electron system and trimethylene spacer for optimal activity. Therefore, a series of hetero-aromatic substituted pyrazolylpropyl (243), 1,2,4-triazolylpropyl (244), indolylpropyl (245), 2-methyl-benzo[d]imidazolylpropyl (246), propyl-isoindoline (247), and propyl-isoindoline-1,3-dione (248) analogues were synthesized and analyzed for TLR7/8 activity. An increase in TLR8 activity was observed for compound 247 as compared to its di-keto (248) and 2-methyl (246) derivatives. Compound 249 which is a part structure of 247 showed dual TLR7/8 activity demonstrating that in the presence of heteroatom in the ring, aromatic substitution at the C-5 position is not necessary for TLR7/8 activity. To further explore this hypothesis, piperidinyl propyl (250), piperidinyl propynyl (251) and azepanylpropyl (252) analogues as well as acyclic aliphatic amine bearing analogues 253 and 254 were screened. TLR8 activity was retained in all these analogues but a decrease in TLR7 activity was observed for compounds 250, 252 and 253, and a complete loss of TLR7 activity for compounds 251 and 254. On screening of di-heteroatom containing saturated heterocyclic substituents, including piperazinyl (255–258), thiomorpholinyl (259), and morpholinyl (260 and 261) analogues, the Boc-protected piperazinyl compound (255) was observed to be a potent pure TLR8 agonist with EC50 value of 30 nM while morpholinyl (261) analogue was a potent dual TLR7/8 agonist with EC50 value of 46 and 280 nM, respectively. An analogue of compound 261 with an open chain morpholine mimic showed reduced TLR7 activity but retained TLR8 activity. The decrease in TLR7 activity in morpholinylbutyl analogue 263 confirmed the importance of propyl spacer as a requirement for maximum activity. This systematic SAR at C-5 position in pyrimidine-2,4-diamine resulted in the identification of compound 261 as a potent TLR7/8 agonist.
McGowan et al. also performed a high throughput screen of approximately 400 000 compounds, which resulted in a single hit, N-4 butyl-5-ethoxypyrimidine-2,4-diamine (264a) as a potent hTLR7 scaffold.110 Compound 264a (Fig. 42) was synthesized and observed to be a dual TLR7/8 agonist. In the further SAR study, a series of C5-ether substituted N-4 butyl analogues (264b–264t) were synthesized and C5-n-propylether (264b) and isopropylether (264c) derivatives observed to be weak TLR7/8 agonists while the benzyl ether derivative (264d) showed dual TLR7/8 activity suggesting that the analogues with one carbon spacer were more potent than three carbon spacer derivative (264e) and simple alkyl ether derivatives (264b–264c).
Fig. 42. SAR in substituted pyrimidin-2,4-diamines by McGowan and co-workers.

The heterocyclic ring as C5-ether substituent showed alteration in the activity and selectivity for the TLR7 and TLR8 activity. The activity variation was dependent upon the size of ring and the position of heteroatom in five membered rings (264f–264k), six-member ring (264l–264n) and bicyclic ring (264o–264t) systems. In general, it was observed that the heterocycles lacking a nitrogen at the 2-position, relative to the carbon that connects the heterocycle to the ether, showed activity in the micromolar range (264f–264g), while heterocycles with these attributes (264i–264t) showed sub-micromolar lowest effective concentrations (LECs) for TLR7 and 8 activity except 2-oxazole analog (264h). The iso-quinoline derivative (264p) showed maximum TLR7 and TLR8 activity with LEC value of 0.008 μM and 0.14 μM, respectively.
Another set of compounds (265a–265n) was synthesized by holding ether moiety and varied amines at the C-4 position from straight and branched chain alkyl amines to those containing heteroatom. Structurally similar butylamine derivatives showed reduced TLR7 activity due to the increased π character (265a), cycloalkyl ring (265b) at the end of the chain, increased lipophilic character (265c) and branching at the alpha and beta position (265d and 265e). The analogue containing the (S)-2-aminopentanol moiety (265g) showed more agonistic potential as compared to its R-isomer (265f), with 30-fold increased selectivity for TLR8. The same selectivity was observed for the corresponding homologue 265j. The alkyl substituted analogs with methyl branching at the terminus (265h) decreased the activity while the branching in the middle of alkyl chain (265i) with a methyl group improved the activity for both TLR7 and TLR8. The branched γ-amino alcohol derivatives 265k, 265l and 265m showed less TLR7 activity and the compound 265n containing an (S)-3-aminoheptanol moiety, a homolog of 265m, showed equal TLR7 and TLR8 activity.
SAR in pteridinones
In 2013, Roethle et al.111 during the investigation of TLR7-active 8-oxopurine analogs, tested expanding the imidazole ring of the oxopurine scaffold to create a new series of TLR7 ligands. An imidazolone ring of the oxo-purine scaffold (6,5-ring system) was expanded to 6,6-ring and 6,7-ring system resulting in a new TLR7 scaffold (Fig. 43).
Fig. 43. Basic skeleton of 6,5-ring, 6,6-ring and 6,7-ring system.

The 6,7-ring system in the pyrimido(diazepinone) core had no TLR7 activity while the pteridinone scaffold (6,6-ring system) was found to be TLR7 active and selective. IFN-α was used as a measure of TLR7 activity, and TNF-α was used as a surrogate measure of TLR8 activity. The ratio of the MEC values for IFN-α and TNF-α was used as a general measure of selectivity for TLR7 versus TLR8 for each analogue (Fig. 44). Initially, based on the previous SAR, C-2 position was fixed with butoxy group and variation was done at N-8 position of 4-amino-2-butoxy-7,8-dihydropteridin-6(5H)-one scaffold (266a–266k). Among N-8 benzyl (266a) and its pendent basic amine derivatives (266b–266d), the meta (266b) and para (266c) linked derivatives showed better activity and selectivity than ortho (266d) linked and its neutral analogue (266a). Both the meta-linked piperidine (266e) and morpholine (266f) derivatives were observed as less active and TLR7 selective than pyrrolidine derivative (266b). The bicyclic tertiary amine derivatives (266g and 266h) also demonstrated good activity with 3 nM MEC for IFN-α induction. The extension of pyrrolidino group, as propylene analogue (266i) and the biaryl analogues (266j and 266k), showed good activity but less selectivity than their parent compound 266b.
Fig. 44. SAR in pteridinones.

Further, the N-8 position was fixed with potent meta-linked pyrrolidinobenzyl derivative and further variation was done at the C-2 position (267a–267n).
The C-2 methoxyethoxy derivative (267a) and the amine linked n-butyl analogue (267b) were observed to be TLR7 active with an MEC of 10 nM for IFN-α induction and selectivity ratio of 1000-fold (Fig. 44). The amino methoxyethyl (267c), its mercapto derivative (267d) as well as carbon linked analogue (267e) were less active than their respective alkoxy derivative (266b). The increase in the alkoxy chain length (267f) at the C-2 position resulted in the loss of TLR7 activity. The cyclopropyl group at the C-2 alkoxy terminal (267g) demonstrated better TLR7 activity with MEC value of 30 nM. Among the branched alkoxy derivatives (267h–267j), compound 2j with α-methyl substitution was equipotent to compound 266b with slightly decreased TNF/IFN ratio. Among benzyl (267k), cyclohexyl (267l) and 4-tetrahydropyran (267m) analogues, only the compound 267m was a potent inducer of IFN-α with a MEC of 10 nM and selectivity ratio of 100-fold, while the amide linked analogue (267n) showed poor TLR7 activity.
The C-7 methyl substitution of compounds 266b and 266c resulted in 100-fold (268a) and 10-fold (268b) loss in the TLR7 activity, respectively. Compound 269a with a spiro cyclopropyl group showed very less activity while the tetracyclic compound 269b showed nearly equal activity to its related analogue 268a. The migration of benzylic group from N-8 position of pteridinone scaffold to C-7 position (compound 270a) also resulted in the loss of activity, which was restored to some extent by N-8 methylation as observed in compound 270b and 270c.
Overall, the methylation on pteridinone at C-7 position was observed to be poorly tolerated, while the oxidation at this position resulted in dioxopteridine derivatives (271a–271d) which were potent TLR7 agonists. A direct comparison of pteridinones to their respective dioxopteridine analogues showed a general trend, where dioxopteridines exhibited better activity and slightly lowers selectivity.
The importance of nitrogen atom at N-1 position of the pteridinone scaffold on TLR7 activity and selectivity was determined by screening a series of deaza-analogues (272a–272c and 273a–273d) in which nitrogen atom was replaced with a carbon atom (Fig. 44). The deaza modified compounds 272a and 272b showed less activity as compared to their corresponding compound 266b and 266c, respectively. The compound 272c with C2-methoxyethyl group showed excellent activity (MEC = 0.32 nM for IFN-α induction) with 10-fold selectivity for IFN-α. The dioxo analogues (273a and 273b) showed the increased activity relative to their respective mono-oxo derivatives (272a and 272b). The biaryl analogues 273c and 273d were effective at stimulating IFN-α production with MEC values of 30 and 10 nM, respectively. However, these analogues had minimal selectivity relative to induction of TNF-α. Overall, compound 266b with a novel scaffold was observed as the most potent and selective TLR7 agonist with EC50 value 290 nM for TLR7 and EC50 value 9.0 μM for TLR8.
SAR in pyrrolo[3,2-d]pyrimidines
McGowan and co-workers112 based upon the structural analysis of imidazoquinolines, 8-hydroxyadenines, pyrimidines and pteridinones proposed pyrrolo[3,2-d]pyrimidin-2-amine derivatives as novel TLR7 agonists. To confirm the hypothesis, N5-benzyl and C4-n-butylamine pyrrolo[3,2-d]pyrimidine (274a) was synthesized and observed to be a TLR7 selective agonist. To better understand the SAR in the pyrrolo[3,2-d]pyrimidine scaffold, a library of three different series of compounds by varying substitutions at C-4 and N-5 positions of pyrrolo[3,2-d]pyrimidine scaffold was synthesized (Fig. 45).
Fig. 45. SAR in pyrrolo[3,2-d]pyrimidines.

In the first series (274b–274o), n-butylamine was conserved at C-4 and variation was done at N-5 position. Increase in the distance of phenyl ring from the scaffold (274b–274c) resulted in the significant improvement of TLR8 activity but this trend was not observed for the TLR7 activity. The replacement of phenyl ring with an amide group (274d) led to the loss of TLR7 activity but improvement in TLR7 activity was observed in isobutene (274e), thiophene (274f) and 2-thiazole (274g) derivatives. Imidazo[1,2-a]pyridine (274h) and pyridine analogues (274i–274k) demonstrated increased TLR7 activity due to the presence of nitrogen atom of C-4 heterocycle adjacent to the methylene group. 2-Pyridyl ring was further modified (274l–274o) and all the analogues were observed to be TLR7 selective agonists with the exception of the 2-pyridyl-6-methoxy derivative (274o) which exhibited dual TLR7/8 activity.
In the second series (275a–275r), the alternative of C4-n-butylamine was explored by keeping 2-pyridyl at N-6 position. A significant loss in TLR7 activity was observed in the C4-ether substituted analogues (275a–275c) while moderate decline in the activity was observed for amino alcohol derivative 275d. Heterocyclic substituents at C-4 position (275e–275r) showed a variety of TLR7 activity, which were found to depend upon the ring size and position of the hetero-atoms. 2-Pyridyl (275e–275f), thiazolo (275g) and 1,3,4-oxadizole (275h) derivatives showed lower TLR7 activity compared to parent butyl derivative (274i). Among oxazole (275i–275k), thiazole (275l–275m) and isoxazole (275n–275q) derivatives, when alkyl group was placed next to the oxygen or sulfur atom an improved TLR7 activity was observed.
Regioisomer of 5-methylisoxazole analogue (275o) with reversed N–O bond (275r) showed nearly 10-fold decrease in the TLR7 activity. As 5-methylisoxazole is the most tolerated group at this position, so by keeping this group constant, authors again explored 2-pyridyl derivatives (276a–276h) at N-5 position, which results in the identification of several potent TLR7 agonists, especially when substituted by electron donating group on pyridine ring (276a and 276b).
In the third series (277a–277d and 278a–278b), the effect of introduction of an oxygen atom at C-4 position was explored. The 5-methyl substituted isoxazole (277a and 278a), oxazole (277b and 278b) and thiazole (277c) analogues were observed as potent TLR7 agonist while 1,2,4-oxadiazole (277d) analogue was observed to be a weak TLR7 agonist. So, pyrrolo[3,2-d]pyrimidine was found to be a novel TLR7 scaffold in which 2-pyridyl derivatives at N-5 and 5-methylisoxazole methylamine derivatives at C-4 positions showed maximum activity.
In continuation of structural modification on pyrrolopyrimidine core, the same research group identified thienopyrimidines as novel TLR7/8 agonists.113 The α-branched aminoalcohol at C-4 position and lacking of benzyl substituent at 5-position (279) showed maximum activity, particularly for TLR8 (Fig. 46).
Fig. 46. Structure of thienopyrimidine and pyrrolopyrimidine analogues.

Chia Tai Tinging Pharmaceuticals reported a series of TLR7 agonistic pyrrolopyrimidine analogues (280–282).114,115 Among the synthesized analogs, n-butoxy group at C-2 and a pendent benzyl group bearing a basic amine at C-7 position were kept constant and a variety of cyclic and acyclic tertiary amines were observed to be tolerable as amine substituents (280). An increase in TLR7 activity was observed due to the incorporation of nitrile group at C-6 position (281). Replacement of phenyl ring with thiazole (282) also demonstrated good TLR7 activity.
SAR in 2,4-diaminoquinazolines
After the discovery of pyrrolo[3,2-d]pyrimidines as TLR7/8 agonists, the pyrrole ring of the lead scaffold and 2,4-diaminoquinazoline derivatives were explored.116 Initially, the effect of alkylamine substitution (283a–283g) was studied at C-4 position of 2-aminoquinazoline scaffold, in which n-butylamine (283d) and n-pentylamine (283e) derivatives were observed as the most potent TLR7/8 agonists, while smaller alkyl chain (283a–283c) and branched alkyl chain (283f–283g) derivatives were less potent (Fig. 47). Increase in TLR8 selectivity was observed due to insertion of oxygen atom in the alkyl chain (283h). Further, by keeping n-butylamine at the C-4 position, substitution was explored at C-5 (R2 substituent), C-6 (R3 substituent), C-7 (R4 substituent) and C-8 (R5 substituent) positions of the 2-aminoquinazoline scaffold. First, the C-8 substituted N-4 butyl-8-fluoroquinazoline-2,4-diamine (284) was synthesized and observed as dual TLR7 and TLR8 agonist without any enhancement in their potencies compared to that of 283d.
Fig. 47. SAR in 2,4-diaminoquinazolines.

In the case of C-7 substituted analogues (285a–285e) no significant improvement in TLR7/8 activity was observed compared to un-substituted 283d. Methoxy (285a) and tert-methylamine (285b) derivatives, being dual TLR7/8 agonists, were slightly more TLR8 selective while the amide derivatives (285d–285e) were more selective towards TLR7 and the C7-hydroxylmethyl (285c) derivative was equipotent for both TLR7 and TLR8 with a lowest effective concentration (LEC) value of 0.1 μM. As an extension to these studies, the C-6 position of N4-butyl-quinazoline-2,4-diamine was further functionalized with fluoro (286a), methoxy (286b), hydroxymethyl (286c), cyano (286d), keto (286e) and corresponding alcohol (286f) groups. The fluoro-substituted compound 286a was observed as the most potent dual TLR7 and 8 agonists with comparable potencies to the un-substituted 283d, while the other analogues were found to be many folds weaker, suggesting the tolerance of fluorine atom at the C-6 position of the quinazoline ring for the TLR7/8 activity. The difluoro analogue (287a) was many fold more potent than dimethoxy analogue (287b), confirming this observation. Compounds (286g–286h) with aromatic ring substitution at C-6 position also showed good activity for TLR7/8.
Investigation at the C-5 position of N-4 butyl-quinazoline-2,4-diamine showed that the substitution with smaller groups such as chlorine (288a), methyl (288b) and methoxy (288c) made the scaffold TLR8 selective while larger cyclopentylmethyl group (such as in 288d) totally reversed the selectivity. No improvement in TLR7/8 activity was observed in case of C-5 phenylethyl substituted analogue (288e) and 2-methoxyethoxy (288f) derivatives compared to un-substituted 283d. But gives the idea that a sterically hindered moiety at a distance of one or two atoms from quinazoline scaffold and adjacent to the n-butylamine can enhance TLR7 selectivity and activity. Based on these observations, a set of benzyl ether (288g–288i) and pyridyl ether (288j–288l) derivatives were prepared and it was observed that the position of nitrogen in pyridyl ring also played an important role in determining the activity and selectivity. Both compounds 288i and 288l showed maximum TLR7 selectivity with LEC value of 0.03 μM and >25 μM for TLR7 and TLR8, respectively.
The effect of the carboxamide functionality with different alkyl amines was studied at C-5 position and among the N-alkyl derivatives (289a–289c), methyl amide analogue (289a) was observed to be a potent TLR7/8 agonist. In N-benzylamide analogues (289d–289i), enantiospecific activity was observed, as S-isomer (289g) is more potent TLR7 agonist as compared to its R-isomer (289f). Installation of methoxy group (289h–289i) in S-isomer (289g) decreases its TLR7 activity.
To further explore the enantio-specific TLR7/8 activity in 2,4-diaminoquinazoline scaffold, authors have performed another SAR study around 2,4-diaminoquinazoline scaffold117 (Fig. 48). Several (S) and (R)-configured β- and γ-amino alcohols (290a–290j) were explored at the C-4 position of quinazoline moiety and compared with N4-butylquinazoline-2,4-diamine (283d). Comparison of (S)-2-aminobutanol derivative (290a) and its higher homologues 290b and 290c showed the direct dependency of TLR7 activity on chain length as (S)-2-aminohexanol derivative (290c) has highest TLR7 agonism and all three have the same activity range against TLR8. The enantio specific TLR7/8 activity was confirmed, as R-isomers (290d and 290e) were observed less potent compared to their corresponding (S)-isomers (290b and 290c). 4-Methyl substitution (290f) on compound 290b resulted in the loss of TLR7/8 agonistic activities and the isoleucinol derivative (290g) showed the importance of 3-methyl substitution for TLR7/8 agonism over 4-methyl substituted analogue 290f. With increased chain length (S)-3-aminohexanol (290h) and (S)-3-aminoheptanol (290i) were observed as potent dual TLR7/8 agonists and the methyl substitution (290j) at the terminal hampered the activity. So, by keeping (S)-3-aminohexanol and (S)-3-aminoheptanol groups at C-4 position, variation was carried out at the C-5 (291a–291d), C-6 (292a–292f), C-7 (293a–293f) and C-8 (294a–294b) positions of 2-aminoquinazoline.
Fig. 48. SAR in isomeric 2,4-diaminoquinazolines.

No significant improvement in TLR7 activity was observed in C-5 substituted methyl (291a–291b) and fluoro (291c–291d) derivatives and an improved TLR8 activity was observed in compounds 291b and 291c. The TLR7/8 agonistic potencies were decreased in C-6 substituted analogues (292a–292f) while the C-7 substituted analogs (293a–293f) retained the activities. The C7-methyl (293a–293b) and fluoro derivatives (293c–293d) were observed to be the most potent in the series. The TLR7/8 agonistic activities of 8-fluoro derivatives (294a–294b) were observed in the same range of un-substituted compounds 290h and 290i. Overall, these SAR studies showed the importance of (S)-isomeric substituent on quinazoline scaffold for TLR7/8 activity and C-7 substituted derivatives identified as the most potent TLR7/8 agonists.
SAR in tetrahydropyrido[3,4-d]pyrimidines
Based on the comparative structural analysis of TLR7/8 agonistic imidazoquinolines, imidazopyridines and pyrimidine derivatives, McGowan and co-workers118 in 2018 proposed that the benzene ring in 2,4-diaminoquinazoline scaffold can be replaced with piperidine without compromising TLR7/8 activity. A set of tetrahydropyrido[3,4-d]pyrimidine derivatives were synthesized and explored as a new class of TLR7/8 agonists (Fig. 49). Initially, based on the position of nitrogen atom in piperidine ring and substitution on the nitrogen atom, two regioisomeric series A (295a–295g) and B (296a–296g) were screened for TLR7/8 activity. Among the synthesized compounds, the un-substituted regioisomer A (295a) was observed as less potent as compared to regioisomer B (296b) while the methyl (295b), benzyl (295c), phenethyl (295d), acetamide (295e), and benzamide (295f) derivatives of A series were more potent than regioisomer B derivatives (296b–296f). Among the sulfonamide derivatives (295g and 296g), compound 296g was observed to be a potent TLR7/8 agonist with many folds selectivity for TLR8.
Fig. 49. SAR in tetrahydropyrido[3,4-d]pyrimidines.

Due to overall better activity of compound 295c with benzyl substitution of A series, further differently substituted benzyl (297a–297i), biphenyl (297j), naphthyl (297k), pyridyl (297l–297n) and other heterocyclic (297o–297q) derivatives of regioisomer A were explored and observed that the benzyl derivatives having substitution at C-2 position of the benzene ring (297a–297d) showed better activity while derivatives with substitution at C-4 position (297f–297j) were less potent than the un-substituted compound 295c. 1-Napthyl analog (297k) was observed to be an equipotent TLR7 agonist with 7-fold less TLR8 activity while remaining analogues (297l–297q) showed weaker TLR7/8 agonistic activities compared to that of compound 295c.
Based on the previous observations of a carboxamide and (S)-configured aminoalcohol derivatives of 2,4-diaminoquinazolines, which showed promising activities, a library of amide analogues of regioisomer A (298a–298p) were also explored by the authors. Among the synthesized analogs, alkylamides (298a–298b) and cycloalkylamide (298c) derivatives were observed to be potent agonists with TLR8 selectivity, while pyridyl (298d–298e) and pyrazine (298f–298g) derivatives showed weak TLR7/8 activity. Among thiophene (298h), thiazole (298i–298m), 1,2,3-thiadiazole (298n) and oxazole (298o–298p) derivatives, compound 298h was the most potent agonist with the LEC value of 0.2 and 0.4 μM for TLR7 and 8, respectively. Addition of 2-amino group in thiazole ring (298j) hampered the TLR7/8 activity which was regained in 2-methyl analogue (298k). By keeping thiophene and 2-methylthiazole amide in the pyridine ring, N-4 n-butane group was substituted with (S)-3-aminohexanol (299a–299b) and (S)-3-aminoheptanol (300a–300b) moieties.
The analogues containing (S)-3-aminoheptanol (300a and 300b) were observed to be more potent TLR7 and 8 agonists than their lower homologues 299a and 299b. Overall, it was concluded that like benzene ring, piperidine ring attached to pyrimidine also showed similar activity for TLR7/8 agonism and resulted in tetrahydropyrido[3,4-d]pyrimidines as a novel class of TLR7/8 agonists.
SAR in pyrido[3,2-d]pyrimidin-2-amines
Very recently after analyzing TLR7/8 agonistic pyrrolo[3,2-d]pyrimidine (pyrrole ring attached to pyrimidine), 2,4-diaminoquinazoline (benzene ring attached to pyrimidine) and tetrahydropyrido[3,4-d]pyrimidine (piperidine ring attached to pyrimidine) scaffolds, Mackman et al.119 tested the possibility of pyrido[3,2-d]pyrimidine (pyridine ring attached to pyrimidine) scaffold as possible new scaffold with TLR7/8 activity. A library of pyrido[3,2-d]pyrimidine derivatives were synthesized with substituents at the C-4 and C-7 positions (Fig. 50) and tested for induction of IFN-α (TLR7) and IL-12p40 (TLR8).
Fig. 50. SAR in pyrido[3,2-d]pyrimidines.

A lack of TLR7/8 activity was observed for butoxy analogue (301a) while its corresponding amino analogue (301b) was observed to be active and similar TLR7/8 activity was observed in its α-methyl substituted amino analogue (301c) suggesting a tolerability of α-substitution on the butyl chain attached at C-4 position. Therefore, a set of α-substituted derivatives (301d–301s) were further analyzed by varying the length of N-4 alkyl chain and substituents at the α-position with hydroxyl, amine, acid and amide groups (Fig. 48). Among the (R)-2-aminopenatnol (301d) and (S)-2-aminopenatnol (301e) derivatives, the S-isomer was observed to have more potent TLR7/8 activity and a similar trend emerged in their higher homologues (301f and 301g). C4-Alkyl chain length also played an important role in TLR7/8 activity as reduction in activity was observed with extension of α-alkyl chain length (301h) or hydroxyl group distance (301i). The reduction in activity was also observed when hydroxyl group in 301f, was substituted with an amine (301j) or other hydrogen bond donor groups (301k–301m).
Among α,α-disubstituted derivatives (301n–301s), the α-methyl-(R) analogue (301n) showed nearly 5-fold improvement in TLR8 activity compared to that of compound 301f and also retained >80-fold selectivity over TLR7, while its (S)-isomer (301o) showed lesser TLR8 activity but improved TLR7 activity. The C4-alkyl chain length and the chirality of this substituent played an important role in determining the activity as shortening (301p) and extension of alkyl chain length (301q–301r) reduced the activity. The presence of two alkyl chains 301s without a chiral carbon resulted in complete loss of the TLR7/8 activity.
Further, the authors also compared the effect of change in the electronic environment over activity by screening quinazoline (290c and 290e) and quinoline (302a–302b) derivatives. The quinazoline derivatives showed similar TLR7/8 agonistic trends like pyrido[3,2-d]pyrimidine derivatives (301f–301g) while the removal of additional nitrogen atom from core scaffold resulted in complete loss of activity (302a–302b). A further modification at C-7 position of compound 301n with chloro (303a), methyl (303b) and fluoro group (304a) did not affect the TLR8 activity but improved its selectivity over TLR7. A complete loss in activity was observed in compound 304c and 304d, having hydroxyl group and carboxylic acid functionality, respectively at the terminal of the alkyl chain.
Overall the authors reported a pyrido[3,2-d]pyrimidine scaffold with TLR7/8 activity and the 7-fluoro substituted analogue 304a was observed to be the most potent TLR8 agonist in the series with nearly 100 fold selectivity over TLR7.
SAR in 2-aminoimidazoles
David and co-workers120 in search of a new TLR7/8 scaffold disclosed a 1-pentyl-2-aminoimidazole as the smallest known TLR7/8 agonist. Its free base was unstable in DMSO but as an HCl salt (305a) was observed to have TLR8 activity with EC50 value of 28.40 μM. To identify an appropriate substitution site on the imidazole ring its 4-phenyl (305b), 5-phenyl (305c) and 4,5-biphenyl (305d) derivatives were screened. Approximately 10-fold improvement in for TLR8 activity was observed in 4-phenyl analogue (305b) while 5-phenyl analogue (305c) was totally inactive for TLR8. A di-substituted analogue (305d) also exhibited very low TLR8 activity. A detailed SAR was further observed at C-4 position of imidazole ring with different phenyl derivatives (306a–306n′) (Fig. 51). The majority of analogues were observed with similar EC50 values but variation was observed in their maximal relative responses in the human TLR8-specific reporter gene assays.
Fig. 51. SAR in 2-aminoimidazoles.

Mono and di-methyl substituted phenyl derivatives (306a–306h) showed similar TLR8 activities as compared to un-substituted compound 305b while the insertion of nitrogen atom in the phenyl (306i) and furan (306j) ring resulted in many folds decrease in activity. The role of H-bond donating or -accepting functional groups was also examined on the C4-phenyl ring by screening 2-phenolic (306k), 2-hydroxymethyl (306l), 4-aminophenyl (306m), 4-benazamide (306n), 4-methylbenzoate (306o), and 2-methylbenzoate (306p) derivatives but none of them showed improved activity as compared to the un-substituted phenyl derivative 305b. Interestingly, the 2-methoxyphenyl analogue (306q) showed little improved TLR8 activity. Therefore, to extend the SAR, its regioisomer (306r and 306s), methyl (306t and 306u), dimethoxy (306v) and trimethoxy (306w) derivatives were also synthesized screened for the TLR8 activity. None of these derivatives showed any significant improvement in the TLR8 activity.
Similarly, the substitution of phenyl ring with electron withdrawing groups i.e. chloro (306x–306z), fluoro (306a′–306d′) and trifluoromethyl (306e′–306g′) substituents showed the inability for significant enhancement of TLR8 activity. The phenylethyl analogue with additional methylene residue (306h′) resulted in slight loss of the activity while the incorporation of two carbon atoms (306i′) resulted in improved TLR8 activity. No improvement in TLR8 activity was observed in case of naphthyl (306j′–306k′), biphenyl (306l′) and benzyloxyphenyl (306m′) derivatives.
All the active analogues were pure TLR8 agonists but TLR8 inhibition was observed for compounds 305c and 306m′ with further investigation confirming inhibition of TLR2, TLR3, TLR4, TLR5, TLR7, and TLR9 responses by 306m′, suggesting an off-target effect. Overall, the investigation confirmed the TLR8 specific activity of 2-aminoimidazoles.
SAR in miscellaneous scaffolds
Among all the reported TLR7/8 agonists, VTX-analogues are a part of a structurally very distinct series containing 3H-benzo[b]azepin-2-amine as the core structure (307). The synthetic route of benzazepine derivatives121 was first reported by Ventrix Pharmaceuticals in 2009. Motolimod or VTX-2337 (308) is a selective and potent TLR8 agonist with EC50 of 100 nM and nearly 50-fold selectivity over TLR7.122 VTX-2337 induces TNF-α and IL-12 secretion from monocytes and myeloid DCs. TL8-506 (309) is also a TLR8 agonistic ligand that is more potent than resiquimod. It is nearly 500-fold more selective over TLR7 with EC50 value of 30 nM and 15 μM for TLR7 and TLR8, respectively (Fig. 52).123,124
Fig. 52. Structure of core VTX and derivatives.

The effect of macrocyclization on the TLR7/8 activity in the imidazoquinolines,125 purine126,127 and related des-aza-purine128 scaffold (Fig. 53) was investigated. In imidazoquinoline analogue (310) macrocylization was achieved by alkyl linkage between C-5 and N-1 position and no significant improvement in the activity was observed. In case of purine based structures (311–313), macrocyclic ring was constructed from the alkoxy chain on the C-2 position with N-9 benzyl group. Linker length of six atoms between the phenyl ring and pyrimidine ring were the most permissive. The improved TLR7 activity over TLR8 was observed with the incorporation of a double bond in to the macrocycle and a similar SAR trend was also observed in the des-aza-purine analogues.
Fig. 53. Structure of macrocyclic TLR7/8 agonists.

In 2014, a series of dual TLR7/8 agonistic 1,2,4-trizine analogues were reported for the treatment of viral infection.129 During the SAR investigation, a variety of 5-substituted amines and 6-substituted ethers was analyzed over TLR7/8 activity (Fig. 54). In the first series (314a–314j) n-butylamine was kept constant at C-5 and ether functionality with aryl (314e–314g) or heteroaryl (314h–314i) rings were well tolerated and observed to be equipotent for TLR7 and 8, whereas analogues with linear ether substituents showed higher selectivity for TLR8. Similar activity trend was also observed in compound 315 and 316, whereas heteroaryl substituted ether analogue was observed more potent than alkyl ether.
Fig. 54. SAR in 1,2,4-trizine analogues.

After a very close observation of various SAR on TLR7/8 agonistic scaffolds, David and co-workers130 noted a strict dependency on the electronic environment of heterocyclic system for TLR7 as well as TLR8 activation. To examine this hypothesis, the authors synthesized and compared 13 electronically distinct chemotypes (Fig. 55). Among them 2-butyloxazolo[4,5-c]quinolin-4-amine 317 was observed as a potent dual TLR7/8-agonistwith EC50 values of 0.55 and 0.18 μM in TLR7 and TLR8 assays, respectively. The exchange of oxazole ring with thiazole (126c), resulted in loss of activity towards both TLR7 as well as TLR8 whereas the nitrogen insertion atom in the C2-butyl chain led to a totally inactive analogue N-propyloxazolo[4,5-c]quinoline-2,4-diamine (318). However, N-propylthiazolo[4,5-c]quinoline-2,4-diamine (319) exhibited nearly same TLR7 and reduced TLR8 activity as compared to compound 126c. The C2-N-acyl derivative (320) of thiazolo[4,5-c]quinoline series was also observed to be totally inactive.
Fig. 55. SAR in tricyclic ring system.

Among the regioisomeric 2-butylthiazolo[5,4-c]quinolin-4-amine (321) and 2-butyloxazolo[5,4-c]quinolin-4-amine (322), compound 321 was observed to be totally inactive for TLR7/8 while compound 322 showed very low TLR8 activity. These observations showed that type and position of heteroatom in the scaffold play an important role for activity. So, further by the repositioning of nitrogen atom in imidazole ring, two novel scaffolds imidazo[1,2-c]quinazoline (323) and imidazo[1,2-a]quinoxaline (324) were synthesized, among them compound 324 was found to be selective TLR8 agonists while compound 323 was totally inactive. Authors with the idea that additional nitrogen in ring system may restore activity also screened three trizole derivatives (325–327), but none of them was found active in TLR7 and TLR8 reporter gene assay. However, 2-butyl-2H-pyrazolo[3,4-c]quinolin-4-amine (328) was found to be a highly potent TLR7 and TLR8 agonist with EC50 of 0.19 and 0.056 μM, respectively. Overall, it was concluded that the TLR7/8 activities of the structurally similar scaffolds have strict dependency on the electronic environment of the molecules, in which a very small change in the electronic environment can alter the activity with a very high impact.
Conclusion
In our systematic literature survey, eighteen TLR7 and/or TLR8 agonistic scaffolds were identified, namely imidazo[4,5-c]quinoline, thiazolo[4,5-c]quinoline, oxazolo[4,5-c]quinoline, pyrazolo[3,4-c]quinoline, furo[2,3-c]quinoline, imidazo[4,5-c]pyridine, 8-hydroxyadenine, pteridinone, furo[2,3-c]pyridine, 2-aminoquinoline, pyrido[3,2-d]pyrimidine, 2,4-diaminoquinazoline, tetrahydropyrido[3,4-d]pyrimidine, pyrrolo[3,2-d]pyrimidine, benzo[b]azepine, pyrimidine, benzimidazole and imidazole. The most active representative analogues of each scaffold is shown in the Fig. 56.
Fig. 56. Representative potent TLR7 and/or TLR8 agonists.

Among all the scaffolds, imidazoquinoline derivatives are mostly dual TLR7/8 agonists, in which free amine group at the C-4 position and n-butyl group at C-2 position (41) were observed to be the most suitable substituents to obtain the best TLR7/8 activity. Substitutions at N-1 and C-7 positions were found to be the most effective in improving TLR7/8 activity. TLR7/8 activities have strict dependency on the electronic environment of the imidazoquinoline ring, as a change in the electronic environment either by shifting or substituting nitrogen atoms altered the TLR7 and/or TLR8 activity. Thiazolo[4,5-c]quinoline, oxazolo[4,5-c]quinoline, pyrazolo[3,4-c]quinoline and furo[2,3-c]quinoline resulting from the SAR on imidazoquinolines were also potent scaffolds and followed a similar SAR pattern (126c, 317, 328, 140d). The structural simplification of the imidazoquinoline ring into imidazopyridine and 8-oxoimidazopyridine ring resulted in pure TLR7 scaffolds. N-1 benzyl, C2-butyl and C6-N-benzyl functionalities on the imidazopyridine (123g), and N1-methylpyridyl and C6-trifluoromethyl functionalities in 8-oxoimidazopyridines (189c) were observed to be the most suitable substituents for TLR7 activity. Similar trend in TLR7 activity was observed in 8-oxoadenine derivatives with C2-butylamino and C2-butoxy groups. On the other hand, the expansion of imidazole ring in 8-oxoadenine scaffold introduced a TLR7 selective activity in pteridinone scaffold (266b). A weak TLR8 activity was observed in furo[2,3-c]pyridine analogues, C2-N-pentyl and C3-pyridinylamino groups (135f) were the minimal part required for maximum activity.
The complete removal of imidazole ring from imidazoquinoline resulted in a TLR8 specific 2-aminoquinoline system. C3-n-Pentyl and C5-alkylamine as well as -benzylamine are optimal substituents for maximum activity in this scaffold (149l). Further modification in quinoline ring system by the insertion of heteroatom and/or expansion or contraction of ring size added novel TLR7 and/or 8 scaffolds. An important effect of isomeric substituent on activity was observed in both pyrido[3,2-d]pyrimidine (304a) and 2,4-diaminoquinazoline analogues (293d).The potent TLR7/8 activity of tetrahydropyrido[3,4-d]pyrimidine analogue (298h) gave the idea that the benzene ring in 2,4-diaminoquinazolines can be altered without compromising activity. Pyrrolo[3,2-d]pyrimidines demonstrated TLR7 activity in which N5-2-pyridyl and C4-5-methylisoxazole methylamine substituents were observed to be most suitable for maximum activity (275o). The expansion of pyridine ring in quinoline scaffold was also performed, which resulted in a pure TLR8 agonistic benzo[b]azepine scaffold (309). Further reduction in ring size resulted in a pyrimidine scaffold with dual TLR7/8 agonistic activities. In the identification of smallest possible TLR7 and/or TLR8 agonistic scaffold, substituted benzo[d]imidazole and imidazole-based structures were observed to be TLR8 scaffolds. In both of these scaffolds a free amine group at the C-2 position and n-butyl group at N-1 position were observed to be the most suitable substituents for activity (154k, 305b).
Overall, it was observed that due to the immune modulatory effect of TLR7 and TLR8 agonists, number of research articles in this area are coming rapidly with a diverse array of applications, including treatment for cancer and viral infection, and use as vaccine adjuvants. The development of TLR7/8 agonists are now focused on design of novel small molecular entities with enhanced potency and selectivity, and use of potent agonists in novel pharmaceutical formulation, combinations and delivery systems. The development of efficacious molecules with adequate safety margin is major challenge in this field. Several clinical trials on small TLR7 and 8 agonistic molecules are going on to better understand safety and efficacy.6
In the last few years, better understandings of chemical environment around TLR7/8 agonistic small molecules have given researchers the ability to incorporate the diverse chemical modification strategies which resulted in the discovery of new active scaffolds. A comprehensive collection of all small TLR 7 and 8 agonistic molecules with their activity is very important for rapid development of potent agonists as done in this review. So together, current chemical environment knowledge and ongoing clinical studies, will help in rapid exploration of TLR 7 and 8 agonists for therapeutic use.
The future directions in this field involve the improvement of efficacy and safety of vaccines containing TLR 7/8 agonists. Usually, the strategies utilized are the use of delivery systems or chemical modification to slow dissemination from site of injection. The chemical modification with lipidation such as in 3M-052 has demonstrated promising effects.131 In the delivery systems, encapsulating nanoparticles (e.g. polymersomes, niosomes) are the modern approaches developed for prolonged release.132 However, the adsorption over alum adjuvants are traditional approaches where TLR7/8 agonists are linked or physically adsorbed on the surface through ligand exchange mechanism.133 Besides these complex strategies, the synergistic combination of TLR7/8 agonists with other TLR2, TLR4 and TLR9 have shown promising results. Combinatorial delivery of multiple TLR adjuvants with antigen doses are also employed with hypothesis of mimicking pathogenic infections.134
TLR7/8 agonist–antigen conjugates have shown prominent T and B cell responses than unconjugated mixture of adjuvant and antigen. For an instance, 3M-012, an amine functionalized resiquimod coupled to HIV Gag protein or influenza virus via PEG linker generated efficacious immunity against infections.135,136 Conjugation of TLR7/8 agonist to thermo-responsive polymer elicited high titers of anti-RSV F antibodies and retained protection against RSV intranasal challenge.137 Imidazoquinoline based TLR7/8 agonist CL3707 coupled to a TLR2 agonistic lipopeptide fragment Pam2Cys have also demonstrated enhanced humoral responses in mice against HIV infection.138 Imidazoquinoline derivative has also been utilized for linked tri-agonist combinations with Pam2CSK4 (TLR2 agonist), pyrimido-indole derivative (TLR4 agonist) and ODN 1826 CpG DNA (TLR9 agonist). Each tri-agonistic combination containing TLR7/8 agonist elicited distinct immune responses in presence of TLR2 agonist and TLR9 agonist highlighting the importance of targeting TLR7/8 in vaccine development.139
An evolution of imidazole from imidazoquinoline scaffold along with the complete TLR7 and TLR8 SAR studies of all the intermediate scaffolds presented in this review clearly highlight the possibility to develop new chemical entities with improved activities. TLR7/8 agonists are emerging as promising vaccine adjuvant candidates and the present SAR and structural information provides a road map towards the identification of even more potent vaccine adjuvant candidates.
Conflicts of interest
There is no conflict of interest to declare.
Acknowledgments
DBS and NP are thankful to Ministry of Human Resource Development (MHRD), Govt. of India for the award of Scheme for Promotion of Academic and Research Collaboration (SPARC/2018-2019/P386/SL). This project was also supported by funding from National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Contracts HHS-N272201400053C and HHS-N272200800039C and also by a Global Connections Bridging Grant number 511803900 to NP and DBS from the Australian Global Connections Fund administered by the Australian Academy of Technology & Engineering. D. K. and A. K. are thankful to UGC, New Delhi, for the award of Junior and Senior Research Fellowships. This publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
References
- Takeda K. Kaisho T. Akira S. Toll-Like Receptors. Annu. Rev. Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Takeuchi O. Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Warshakoon H. J. Hood J. D. Kimbrell M. R. Malladi S. Wu W. Y. Shukla N. M. Agnihotri G. Sil D. David S. A. Potential adjuvantic properties of innate immune stimuli. Hum. Vaccines. 2009;5:381–394. doi: 10.4161/hv.5.6.8175. [DOI] [PubMed] [Google Scholar]
- Hemmi H. Kaisho T. Takeuchi O. Sato S. Sanjo H. Hoshino K. Horiuchi T. Tomizawa H. Takeda K. Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88–dependent signaling pathway. Nat. Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- Diebold S. S. Kaisho T. Hemmi H. Akira S. Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Patinote C. Karroum N. B. Moarbess G. Cirnat N. Kassab I. Bonnet P. A. Deleuze-Masquéfa C. Agonist and antagonist ligands of toll-like receptors 7 and 8: Ingenious tools for therapeutic purposes. Eur. J. Med. Chem. 2020;193:112238. doi: 10.1016/j.ejmech.2020.112238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico S. Pozzetti L. Papa A. Carullo G. Gemma S. Butini S. Campiani G. Relitti N. Modulation of the Innate Immune Response by Targeting Toll-like Receptors: A Perspective on Their Agonists and Antagonists. J. Med. Chem. 2020;63:13466–13513. doi: 10.1021/acs.jmedchem.0c01049. [DOI] [PubMed] [Google Scholar]
- Anwar M. A. Shah M. Kim J. Choi S. Recent Clinical Trends in Toll-like Receptor Targeting Therapeutics. Med. Res. Rev. 2019;39:1053–1090. doi: 10.1002/med.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- https://clinicaltrials.gov/ct2/show/NCT02015715
- Kaushik D. Dhingra S. Patil M. T. Piplani S. Khanna V. Honda-Okubo Y. Li L. Fung J. Sakala I. G. Salunke D. B. Petrovsky N. BBIQ, a pure TLR7 agonist, is an effective influenza vaccine adjuvant. Hum. Vaccines Immunother. 2020;16:1989–1996. doi: 10.1080/21645515.2019.1710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroa R. Kaushik D. Bagai U. Kaur S. Salunke D. B. Efficacy of TLR7 agonistic imidazoquinoline as immunochemotherapeutic agent against P. Berghei ANKA infected rodent host. Bioorg. Med. Chem. Lett. 2019;29:1099–1105. doi: 10.1016/j.bmcl.2019.02.029. [DOI] [PubMed] [Google Scholar]
- Mark K. E. Corey L. Meng T. C. Magaret A. S. Huang M. L. Selke S. Slade H. B. Tyring S. K. Warren T. Sacks S. L. Leone P. Bergland V. A. Wald A. Topical resiquimod 0.01% gel decreases herpes simplex virus type 2 genital shedding: a randomized, controlled trial. J. Infect. Dis. 2007;195:1324–1331. doi: 10.1086/513276. [DOI] [PubMed] [Google Scholar]
- Pockros P. J. Guyader D. Patton H. Tong M. J. Wright T. McHutchison J. G. Meng T. C. Oral resiquimod in chronic HCV infection: safety and efficacy in 2 placebo-controlled, double-blind phase IIa studies. J. Hepatol. 2007;47:174–182. doi: 10.1016/j.jhep.2007.02.025. [DOI] [PubMed] [Google Scholar]
- Caron G. Duluc D. Frémaux I. Jeannin P. David C. Gascan H. Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-γ production by memory CD4+ T cells. J. Immunol. 2005;175:1551–1557. doi: 10.4049/jimmunol.175.3.1551. [DOI] [PubMed] [Google Scholar]
- Kaur A. Kannan D. Mehta S. K. Singh S. Salunke D. B. Synthetic Toll-like Receptor Agonists for the Development of Powerful Malaria Vaccines: A Patent Review. Expert Opin. Ther. Pat. 2018;28:837–847. doi: 10.1080/13543776.2018.1530217. [DOI] [PubMed] [Google Scholar]
- Braunstein M. J. Kucharczyk J. Adams S. Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018;13:583–598. doi: 10.1007/s11523-018-0589-7. [DOI] [PubMed] [Google Scholar]
- Drobits B. Holcmann M. Amberg N. Swiecki M. Grundtner R. Hammer M. Colonna M. Sibilia M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Invest. 2012;122:575–585. doi: 10.1172/JCI61034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S. C. Guenounou M. Toll-like receptors and immune response in allergic disease. Clin. Rev. Allergy Immunol. 2004;26:115e125. doi: 10.1007/s12016-004-0006-0. [DOI] [PubMed] [Google Scholar]
- Matesic D. Lenert A. Lenert P. Modulating toll-like receptor 7 and 9 responses as therapy for allergy and autoimmunity. Curr. Allergy Asthma Rep. 2012;12:8e17. doi: 10.1007/s11882-011-0233-4. [DOI] [PubMed] [Google Scholar]
- Hennessy E. J. Parker A. E. O'Neill L. A. J. Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discovery. 2010;9:293e307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- Parkinson T. The future of toll-like receptor therapeutics. Curr. Opin. Mol. Ther. 2008;10:21–31. [PubMed] [Google Scholar]
- Wang Y. Zhang S. Li H. Wang H. Zhang T. Hutchinson M. R. Yin H. Wang X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020;53:1046–1055. doi: 10.1021/acs.accounts.9b00631. [DOI] [PubMed] [Google Scholar]
- Hussein W. M. Liu T. Y. Skwarczynski M. Toth I. Toll-like receptor agonists: a patent review (2011 - 2013) Expert Opin. Ther. Pat. 2014;24:453–470. doi: 10.1517/13543776.2014.880691. [DOI] [PubMed] [Google Scholar]
- Chi H. Li C. Zhao F. S. Zhang L. Ng T. B. Jin G. Sha O. Anti-tumor Activity of Toll-Like Receptor 7 Agonists. Front. Pharmacol. 2017;8:304. doi: 10.3389/fphar.2017.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ella R. Reddy S. Jogdand H. Sarangi V. Ganneru B. Prasad S. Das D. Raju D. Praturi U. Sapkal G. Yadav P. Reddy P. Verma S. Singh C. Redkar S. V. Gillurkar C. S. Kushwaha J. S. Mohapatra S. Bhate A. Rai S. Panda S. Abraham P. Gupta N. Ella K. Bhargava B. Vadrevu K. M. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBV152: interim results from a double-blind, randomised, multicentre, phase 2 trial, and 3-month follow-up of a double-blind, randomised phase 1 trial. Lancet Infect. Dis. 2021 doi: 10.1016/S1473-3099(21)00070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; , https://www.thelancet.com/journals/laninf/article/PIIS1473-3099(21)00070-0/fulltext
- Tanji H. Ohto U. Shibata T. Miyake K. Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science. 2013;339:1426–1429. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Ohto U. Shibata T. Krayukhina E. Taoka M. Yamauchi Y. Tanji H. Isobe T. Uchiyama S. Miyake K. Shimizu T. Structural Analysis Reveals that Toll-like Receptor 7 is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity. 2016;45:737–748. doi: 10.1016/j.immuni.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Akira S. Takeda K. Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Gorden K. B. Gorski K. S. Gibson S. J. Kedl R. M. Kieper W. C. Qiu X. Tomai M. A. Alkan S. S. Vasilakos J. P. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J. Immunol. 2005;174:1259–1268. doi: 10.4049/jimmunol.174.3.1259. [DOI] [PubMed] [Google Scholar]
- Gorski K. S. Waller E. L. Bjornton-Severson J. Hanten J. A. Riter C. L. Kieper W. C. Gorden K. B. Miller J. S. Vasilakos J. P. Tomai M. A. Alkan S. S. Distinct indirect pathways govern human NK-cell activation by TLR-7 and TLR-8 agonists. Int. Immunol. 2006;18:1115–1126. doi: 10.1093/intimm/dxl046. [DOI] [PubMed] [Google Scholar]
- Heil F. Hemmi H. Hochrein H. Ampenberger F. Kirschning C. Akira S. Lipford G. Wagner H. Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Diebold S. S. Kaisho T. Hemmi H. Akira S. Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Diebold S. S. Massacrier C. Akira S. Paturel C. Morel Y. Reis e Sousa C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006;36:3256–3267. doi: 10.1002/eji.200636617. [DOI] [PubMed] [Google Scholar]
- Forsbach A. Nemorin J. G. Montino C. Müller C. Samulowitz U. Vicari A. P. Jurk M. Mutwiri G. K. Krieg A. M. Lipford G. B. Vollmer J. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008;180:3729–3738. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- Lan T. Kandimalla E. R. Yu D. Bhagat L. Li Y. Wang D. Zhu F. Tang J. X. Putta M. R. Cong Y. Trombino A. F. Sullivan T. Agrawal S. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl. Acad. Sci. U. S. A. 2007;104:13750–13755. doi: 10.1073/pnas.0706059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T. Dai M. Wang D. Zhu F.-G. Kandimalla E. R. Agrawal S. Toll-like Receptor 7 Selective Synthetic Oligoribonucleotide Agonists: Synthesis and Structure−Activity Relationship Studies. J. Med. Chem. 2009;52:6871–6879. doi: 10.1021/jm901145s. [DOI] [PubMed] [Google Scholar]
- Shibata T. Ohto U. Nomura S. Kibata K. Motoi Y. Zhang Y. Murakami Y. Fukui R. Ishimoto T. Sano S. Ito T. Shimizu T. Miyake K. Guanosine and its modified derivatives are endogenous ligands for TLR7. Int. Immunol. 2016;28:211–222. doi: 10.1093/intimm/dxv062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Ohto U. Shibata T. Krayukhina E. Taoka M. Yamauchi Y. Tanji H. Isobe T. Uchiyama S. Miyake K. Shimizu T. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity. 2016;45:737–748. doi: 10.1016/j.immuni.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Hornung V. Guenthner-Biller M. Bourquin C. Ablasser A. Schlee M. Uematsu S. Noronha A. Manoharan M. Akira S. de Fougerolles A. Endres S. Hartmann G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- Judge A. D. Sood V. Shaw J. R. Fang D. McClintock K. MacLachlan I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005;23:457–462. doi: 10.1038/nbt1081. [DOI] [PubMed] [Google Scholar]
- Miller R. L. Gerster J. F. Owens M. L. Slade H. B. Tomai M. A. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int. J. Immunopharmacol. 1999;21:1–14. doi: 10.1016/s0192-0561(98)00068-x. [DOI] [PubMed] [Google Scholar]
- Butchi N. B. Pourciau S. Du M. Morgan T. W. Peterson K. E. Analysis of the neuroinflammatory response to TLR7 stimulation in the brain: comparison of multiple TLR7 and/or TLR8 agonists. J. Immunol. 2008;180:7604–7612. doi: 10.4049/jimmunol.180.11.7604. [DOI] [PubMed] [Google Scholar]
- Gerster J., Lindstrom K., Marszalek G., Merrill B., Mickelson J. and Rice M., Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof, WO2000006577, 2000
- Hunter J. H. and Skulnick H. I., Substituted pyrimidines, US Pat., US3956302A, 1976
- Nichol F. R. and Underwood G. E., Interferon Induction, US Pat., US3932617, 1976
- Nichol F. R. Weed S. D. Underwood G. E. Stimulation of Murine Interferon by a Substituted Pyrimidine. Antimicrob. Agents Chemother. 1976;9:433–439. doi: 10.1128/aac.9.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga W. Skulnick H. I. Stringfellow D. A. Weed S. D. Renis H. E. Eidson E. E. 5-substituted 2-amino-6-phenyl-4(3H)-pyrimidinones. Antiviral- and interferon-inducing agents. J. Med. Chem. 1980;23:237–239. doi: 10.1021/jm00177a004. [DOI] [PubMed] [Google Scholar]
- Li L. H. Wallace T. L. Wierenga W. Skulnick H. I. DeKoning T. F. Antitumor activity of pyrimidinones, a class of small-molecule biological response modifiers. J. Biol. Response Modif. 1987;6:44–55. [PubMed] [Google Scholar]
- Krueger R. E. Mayer G. D. Tilorone hydrochloride: an orally active antiviral agent. Science. 1970;169:1213–1224. doi: 10.1126/science.169.3951.1213. [DOI] [PubMed] [Google Scholar]
- Mayer G. D. Krueger R. F. Tilorone Hydrochloride: Mode of Action. Science. 1970;169:1214–1215. doi: 10.1126/science.169.3951.1214. [DOI] [PubMed] [Google Scholar]
- Stringfellow D. A. Glasgow L. A. Tilorone Hydrochloride: an Oral Interferon-Inducing Agent. Antimicrob. Agents Chemother. 1972;2:73–78. doi: 10.1128/aac.2.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz A. B. Goodman M. G. Pope B. L. Argentieri D. C. Bell S. C. Burr L. E. Chourmouzis E. Come J. Goodman J. H. Small-Molecule Immunostimulants. Synthesis and Activity of 7,8-Disubstituted Guanosines and Structurally Related Compounds. J. Med. Chem. 1994;37:3561–3578. doi: 10.1021/jm00047a014. [DOI] [PubMed] [Google Scholar]
- Lee J. Chuang T. H. Redecke V. She L. Pitha P. M. Carson D. A. Raz E. Cottam H. B. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc. Natl. Acad. Sci. U. S. A. 2003;100:6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara K. Anderson J. D. Kini G. D. Dalley N. K. Larson S. B. Smee D. F. Jin A. Sharma B. S. Jolley W. B. Thiazolo[4,5-d]pyrimidine nucleosides. The synthesis of certain 3-.beta.-D-ribofuranosylthiazolo[4,5-d]pyrimidines as potential immunotherapeutic agents. J. Med. Chem. 1990;33:407–415. doi: 10.1021/jm00163a064. [DOI] [PubMed] [Google Scholar]
- Jurk M. Heil F. Vollmer J. Schetter C. Krieg A. M. Wagner H. Lipford G. Bauer S. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 2002;3:499. doi: 10.1038/ni0602-499. [DOI] [PubMed] [Google Scholar]
- Hood J. D. Warshakoon H. J. Kimbrell M. R. Shukla N. M. Malladi S. S. Wang X. David S. A. Immunoprofiling toll-like receptor ligands: Comparison of immunostimulatory and proinflammatory profiles in ex vivo human blood models. Hum. Vaccines. 2010;6:322–335. doi: 10.4161/hv.6.4.10866. [DOI] [PubMed] [Google Scholar]
- Gerster J. F. Lindstrom K. J. Miller R. L. Tomai M. A. Birmachu W. Bomersine S. N. Gibson S. J. Imbertson L. M. Jacobson J. R. Knafla R. T. Maye P. V. Nikolaides N. Oneyemi F. Y. Parkhurst G. J. Pecore S. E. Reiter M. J. Scribner L. S. Testerman T. L. Thompson N. J. Wagner T. L. Weeks C. E. Andre J.-D. Lagain D. Bastard Y. Lupu M. Synthesis and Structure−Activity-Relationships of 1H-Imidazo[4,5-c]quinolines That Induce Interferon Production. J. Med. Chem. 2005;48:3481–3491. doi: 10.1021/jm049211v. [DOI] [PubMed] [Google Scholar]
- Shukla N. M. Malladi S. S. Mutz C. A. Balakrishna R. David S. A. Structure-activity relationships in human toll-like receptor 7-active imidazoquinoline analogues. J. Med. Chem. 2010;53:4450–4465. doi: 10.1021/jm100358c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N. M. Mutz C. A. Ukani R. Warshakoon H. J. Moore D. S. David S. A. Syntheses of fluorescent imidazoquinoline conjugates as probes of Toll-like receptor 7. Bioorg. Med. Chem. Lett. 2010;20:6384. doi: 10.1016/j.bmcl.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi L. Van Haren S. Dowling D. J. Bergelson I. Shukla N. M. Malladi S. S. Balakrishna R. Tanji H. Ohto U. Shimizu T. David S. A. Levy O. The Imidazoquinoline Toll-Like Receptor-7/8 Agonist Hybrid-2 Potently Induces Cytokine Production by Human Newborn and Adult Leukocytes. PLoS One. 2015;10:e0134640. doi: 10.1371/journal.pone.0134640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H. Ohto U. Shibata T. Miyake K. Shimizu T. Structural Reorganization of the Toll-Like Receptor 8 Dimer Induced by Agonistic Ligands. Science. 2013;339:1426–1429. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- David S. A., Yupeng L., Brush M., Trautman K., Gustafson C., Maurer D. and Balaji P., Therapeutic compounds and methods of use thereof, WO2020009946A1, 2020
- Chipman S. D., Demattei J., Kiwan R. and Kachura M. A., Alkyl chain modified imidazoquinoline TLR7/8 agonist compounds and uses thereof, WO2019040491, 2019
- Bonnert R. V., Mcinally T. and Thom S., Imidazoquinolines with immuno-modulating properties, WO2008135791, 2008
- Yoshiaki I., Mai K., Tomoaki N., Shingo T. and Hirotaka K., Imidazoquinolines which act via Toll like receptors, WO2011068233, 2011
- Shi C. Xiong Z. Chittepu P. Aldrich C. C. Ohlfest J. R. Ferguson D. M. Discovery of Imidazoquinolines with Toll-Like Receptor 7/8 Independent Cytokine Induction. ACS Med. Chem. Lett. 2012;3:501–504. doi: 10.1021/ml300079e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffo C. E. Shi C. Xiong Z. Olin M. Ohlfest J. R. Aldrich C. C. Ferguson D. M. Structure–Activity Relationship Analysis of Imidazoquinolines with Toll-like Receptors 7 and 8 Selectivity and Enhanced Cytokine Induction. J. Med. Chem. 2014;57:339–347. doi: 10.1021/jm4004957. [DOI] [PubMed] [Google Scholar]
- Larson P. Kucaba T. A. Xiong Z. Olin M. Griffith T. S. Ferguson D. M. Design and Synthesis of N1-Modified Imidazoquinoline Agonists for Selective Activation of Toll-like Receptors 7 and 8. ACS Med. Chem. Lett. 2017;8:1148–1152. doi: 10.1021/acsmedchemlett.7b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesu M. Caruso G. Salyer A. C. D. Khetani K. K. Sil D. Weerasinghe M. Tanji H. Ohto U. Shimizu T. David S. A. Structure-Based Design of Human TLR8-Specific Agonists with Augmented Potency and Adjuvanticity. J. Med. Chem. 2015;58:7833–7849. doi: 10.1021/acs.jmedchem.5b01087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt J. R. Kleindl P. A. Moulder K. R. Prisinzano T. E. Forrest M. L. Further exploration of the structure-activity relationship of imidazoquinolines; identification of potent C7-substituted imidazoquinolines. Bioorg. Med. Chem. Lett. 2020;30:126788. doi: 10.1016/j.bmcl.2019.126788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I. Segal D. M. Davies D. R. The structural biology of Toll-like receptors. Structure. 2011;19:447–459. doi: 10.1016/j.str.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M. S. Kim S. E. Heo J. Y. Lee M. E. Kim H. M. Paik S. G. Lee H. Lee J. O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Jin M. S. Lee J. O. Structures of TLR-ligand complexes. Curr. Opin. Immunol. 2008;20:414–419. doi: 10.1016/j.coi.2008.06.002. [DOI] [PubMed] [Google Scholar]
- Liu L. Botos I. Wang Y. Leonard J. N. Shiloach J. Segal D. M. Davies D. R. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–381. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N. M. Mutz C. A. Malladi S. S. Warshakoon H. J. Balakrishna R. David S. A. Toll-Like Receptor (TLR)-7 and −8 Modulatory Activities of Dimeric Imidazoquinolines. J. Med. Chem. 2012;55:1106–1116. doi: 10.1021/jm2010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N. M. Salunke D. B. Balakrishna R. Mutz C. A. Malladi S. S. David S. A. Potent adjuvanticity of a pure TLR7-agonistic imidazoquinoline dendrimer. PLoS One. 2012;7:e43612. doi: 10.1371/journal.pone.0043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo E. Crall B. M. Balakrishna R. Malladi S. S. Fox L. M. Hermanson A. R. David S. A. Structure-activity relationships in Toll-like receptor 7 agonistic 1H-imidazo[4,5-c]pyridines. Org. Biomol. Chem. 2013;11:6526–6545. doi: 10.1039/c3ob40816g. [DOI] [PubMed] [Google Scholar]
- Prince R. B., Merrill B. A., Heppner P. D., Kshirsagar T. A., Wurst J. R., Manske K. J. and Rice M. J., Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines, WO2006086449, 2006
- Prince R. B., Rice M. J., Wurst J. R., Merrill B. A., Kshirsagar T. A. and Heppner P. D., Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridies, WO2006009826, 2006
- Kokatla H. P. Yoo E. Salunke D. B. Sil D. Ng C. F. Balakrishna R. Malladi S. S. Fox L. M. David S. A. Toll-like receptor-8 agonistic activities in C2, C4, and C8 modified thiazolo[4,5-c]quinolines. Org. Biomol. Chem. 2013;11:1179–1198. doi: 10.1039/c2ob26705e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke D. B. Yoo E. Shukla N. M. Balakrishna R. Malladi S. S. Serafin K. J. Day V. W. Wang X. David S. A. Structure–Activity Relationships in Human Toll-like Receptor 8-Active 2,3-Diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012;55:8137–8151. doi: 10.1021/jm301066h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaaban S. Abdel-Wahab B. F. Groebke-Blackburn-Bienaymé multicomponent reaction: emerging chemistry for drug discovery. Mol. Diversity. 2016;20:233–254. doi: 10.1007/s11030-015-9602-6. [DOI] [PubMed] [Google Scholar]
- Kokatla H. P. Sil D. Malladi S. S. Balakrishna R. Hermanson A. R. Fox L. M. Wang X. Dixit A. David S. A. Exquisite Selectivity for Human Toll-Like Receptor 8 in Substituted Furo[2,3-c]quinolines. J. Med. Chem. 2013;56:6871–6885. doi: 10.1021/jm400694d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N. M. Kimbrell M. R. Malladi S. S. David S. A. Regioisomerism-dependent TLR7 agonism and antagonism in an imidazoquinoline. Bioorg. Med. Chem. Lett. 2009;19:2211–2214. doi: 10.1016/j.bmcl.2009.02.100. [DOI] [PubMed] [Google Scholar]
- Shukla N. M. Malladi S. S. Day V. David S. A. Preliminary evaluation of a 3H imidazoquinoline library as dual TLR7/TLR8 antagonists. Bioorg. Med. Chem. 2011;19:3801–3811. doi: 10.1016/j.bmc.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokatla H. P. Sil D. Tanji H. Ohto U. Malladi S. S. Fox L. M. Shimizu T. David S. A. Structure-based design of novel human Toll-like receptor 8 agonists. ChemMedChem. 2014;9:719–723. doi: 10.1002/cmdc.201300573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesu M. Malladi S. S. Fox L. M. Jones C. D. Dixit A. David S. A. Human Toll-Like Receptor 8-Selective Agonistic Activities in 1-Alkyl-1H-benzimidazol-2-amines. J. Med. Chem. 2014;57:7325–7341. doi: 10.1021/jm500701q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K. Kazaoka K. Niimoto I. Kumihara H. Sajiki H. Isobe Y. Takaku H. Tobe M. Ogita H. Ogino T. Ichii S. Kurimoto A. Kawakami H. Discovery of 8-Hydroxyadenines as a Novel Type of Interferon Inducer. J. Med. Chem. 2002;45:5419–5422. doi: 10.1021/jm0203581. [DOI] [PubMed] [Google Scholar]
- Isobe Y. Tobe M. Ogita H. Kurimoto A. Ogino T. Kawakami H. Takaku H. Sajiki H. Hirota K. Hayashi H. Synthesis and structure-activity relationships of 2-substituted-8-hydroxyadenine derivatives as orally available interferon inducers without emetic side effects. Bioorg. Med. Chem. 2003;11:3641–3647. doi: 10.1016/s0968-0896(03)00369-9. [DOI] [PubMed] [Google Scholar]
- Kurimoto A. Ogino T. Ichii S. Isobe Y. Tobe M. Ogita H. Takaku H. Sajiki H. Hirota K. Kawakami H. Synthesis and evaluation of 2-substituted 8-hydroxyadenines as potent interferon inducers with improved oral bioavailabilities. Bioorg. Med. Chem. 2004;12:1091–1099. doi: 10.1016/j.bmc.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Jin G. Wu C. C. Tawatao R. I. Chan M. Carson D. A. Cottam H. B. Synthesis and immunostimulatory activity of 8-substituted amino 9-benzyladenines as potent Toll-like receptor 7 agonists. Bioorg. Med. Chem. Lett. 2006;16:4559–4563. doi: 10.1016/j.bmcl.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Weterings J. J. Khan S. van der Heden van Noort G. J. Melief C. J. M. Overkleeft H. S. van der Burg S. H. Ossendorp F. van der Marel G. A. Filippov D. V. 2-Azidoalkoxy-7-hydro-8-oxoadenine derivatives as TLR7 agonists inducing dendritic cell maturation. Bioorg. Med. Chem. Lett. 2009;19:2249–2251. doi: 10.1016/j.bmcl.2009.02.095. [DOI] [PubMed] [Google Scholar]
- Nakamura T. Wada H. Kurebayashi H. McInally T. Bonnert R. Isobe Y. Synthesis and evaluation of 8-oxoadenine derivatives as potent Toll-like receptor 7 agonists with high water solubility. Bioorg. Med. Chem. Lett. 2013;23:669–672. doi: 10.1016/j.bmcl.2012.11.114. [DOI] [PubMed] [Google Scholar]
- Isobe Y. Kurimoto A. Tobe M. Hashimoto K. Nakamura T. Norimura K. Ogita H. Takaku H. Synthesis and Biological Evaluation of Novel 9-Substituted-8-Hydroxyadenine Derivatives as Potent Interferon Inducers. J. Med. Chem. 2006;49:2088–2095. doi: 10.1021/jm051089s. [DOI] [PubMed] [Google Scholar]
- Bazin H. G. Li Y. Khalaf J. K. Mwakwari S. Livesay M. T. Evans J. T. Johnson D. A. Structural requirements for TLR7-selective signaling by 9-(4-piperidinylalkyl)-8-oxoadenine derivatives. Bioorg. Med. Chem. Lett. 2015;25:1318–1323. doi: 10.1016/j.bmcl.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggadike K. Ahmed M. Ball D. I. Coe D. M. Dalmas Wilk D. A. Edwards C. D. Gibbon B. H. Hardy C. J. Hermitage S. A. Hessey J. O. Hillegas A. E. Hughes S. C. Lazarides L. Lewell X. Q. Lucas A. Mallett D. N. Price M. A. Priest F. M. Quint D. J. Shah P. Sitaram A. Smith S. A. Stocker R. Trivedi N. A. Tsitoura D. C. Weller V. Discovery of 6-Amino-2-{[(1S)-1-methylbutyl]oxy}-9-[5-(1-piperidinyl)pentyl]-7,9-dihydro-8H-purin-8-one (GSK2245035), a Highly Potent and Selective Intranasal Toll-Like Receptor 7 Agonist for the Treatment of Asthma. J. Med. Chem. 2016;59:1711–1726. doi: 10.1021/acs.jmedchem.5b01647. [DOI] [PubMed] [Google Scholar]
- Sanjeev G., Liqi H., Shoshana P., Yam P. and Prasanna S., 6-Amino-7,9-dihydro-8H-purin-8-one derivatives as toll-like receptor 7 (TLR7) agonists as immunostimulants, WO2019035968, 2019
- Sanjeev G., Shoshana P., Yam P., Prasanna S. and Ian Y., Toll-like receptor 7 (TLR7) agonists having a tricyclic moiety, conjugates thereof, and methods and uses therefor, WO2019035969, 2019
- Sanjeev G., Liqi H., Shoshana P., Yam P. and Prasanna S., 6-Amino-7,9-dihydro-8H-purin-8-one derivatives as immunostimulant toll-like receptor 7 (TLR7) agonists, WO2019035970, 2019
- Pryde D. C. Tran T.-D. Jones P. Parsons G. C. Bish G. Adam F. M. Smith M. C. Middleton D. S. Smith N. N. Calo F. Hay D. Paradowski M. Proctor K. J. W. Parkinson T. Laxton C. Fox D. N. A. Horscroft N. J. Ciaramella G. Jones H. M. Duckworth J. Benson N. Harrison A. Webster R. The discovery of a novel prototype small molecule TLR7 agonist for the treatment of hepatitis C virus infection. MedChemComm. 2011;2:185–189. [Google Scholar]
- Matthias B., Naidu C. S., Matthew C., Sanjeev G., Liqi H., Yam P. B., Prasanna S., Christine T. M. and Qian Z., 1H-pyrazolo[4,3-d]pyrimidine compounds as toll-like receptor 7 (TLR7) agonists and methods and uses therefor, WO2020028608, 2020
- Tran T. D. Pryde D. C. Jones P. Adam F. M. Benson N. Bish G. Calo F. Ciaramella G. Dixon R. Duckworth J. Fox D. N. Hay D. A. Hitchin J. Horscroft N. Howard M. Gardner I. Jones H. M. Laxton C. Parkinson T. Parsons G. Proctor K. Smith M. C. Smith N. Thomas A. Design and optimisation of orally active TLR7 agonists for the treatment of hepatitis C virus infection. Bioorg. Med. Chem. Lett. 2011;21:2389–2393. doi: 10.1016/j.bmcl.2011.02.092. [DOI] [PubMed] [Google Scholar]
- Jones P. Pryde D. C. Tran T. D. Adam F. M. Bish G. Calo F. Ciaramella G. Dixon R. Duckworth J. Fox D. N. Hay D. A. Hitchin J. Horscroft N. Howard M. Laxton C. Parkinson T. Parsons G. Proctor K. Smith M. C. Smith N. Thomas A. Discovery of a highly potent series of TLR7 agonists. Bioorg. Med. Chem. Lett. 2011;21:5939–5943. doi: 10.1016/j.bmcl.2011.07.076. [DOI] [PubMed] [Google Scholar]
- Bennett N. J., Mcinally T., Mochel T., Thom S. and Tiden A., Pyrimidine derivatives for the treatment of asthma, COPD, allergic rhinitis, allergic conjunctivitis, atopic dermatitis, cancer, hepatitis B, hepatitis C, HIV, HPV, bacterial infections and dermatosis, WO2009067081, 2009
- Mcinally T., Mochel T., Hasegava F. and Hori S., Benzylamine compounds as Toll-like receptor 7 agonists, WO2012066336, 2012
- Wu T. Y., Pyrimidine compounds containing acidic groups, WO2018106606, 2018; Wu T. Y. and Miller A. T., Pyrimidine compounds containing acidic groups useful to treat diseases connected to the modulation of tlr7, WO2019236496, 2019
- Beesu M. Salyer A. C. Trautman K. L. Hill J. K. David S. A. Human Toll-like Receptor (TLR) 8-Specific Agonistic Activity in Substituted Pyrimidine-2,4-diamines. J. Med. Chem. 2016;59:8082–8093. doi: 10.1021/acs.jmedchem.6b00872. [DOI] [PubMed] [Google Scholar]
- Salyer A. C. D. Caruso G. Khetani K. K. Fox L. M. Malladi S. S. David S. A. Identification of Adjuvantic Activity of Amphotericin B in a Novel, Multiplexed, Poly-TLR/NLR High-Throughput Screen. PLoS One. 2016;11:e0149848. doi: 10.1371/journal.pone.0149848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesu M. Salyer A. C. D. Brush M. J. H. Trautman K. L. Hill J. K. David S. A. Identification of High-Potency Human TLR8 and Dual TLR7/TLR8 Agonists in Pyrimidine-2,4-diamines. J. Med. Chem. 2017;60:2084–2098. doi: 10.1021/acs.jmedchem.6b01860. [DOI] [PubMed] [Google Scholar]
- McGowan D. Herschke F. Pauwels F. Stoops B. Last S. Pieters S. Scholliers A. Thoné T. Van Schoubroeck B. De Pooter D. Mostmans W. Khamlichi M. D. Embrechts W. Dhuyvetter D. Smyej I. Arnoult E. Demin S. Borghys H. Fanning G. Vlach J. Raboisson P. Novel Pyrimidine Toll-like Receptor 7 and 8 Dual Agonists to Treat Hepatitis B Virus. J. Med. Chem. 2016;59:7936–7949. doi: 10.1021/acs.jmedchem.6b00747. [DOI] [PubMed] [Google Scholar]
- Roethle P. A. McFadden R. M. Yang H. Hrvatin P. Hui H. Graupe M. Gallagher B. Chao J. Hesselgesser J. Duatschek P. Zheng J. Lu B. Tumas D. B. Perry J. Halcomb R. L. Identification and optimization of pteridinone Toll-like receptor 7 (TLR7) agonists for the oral treatment of viral hepatitis. J. Med. Chem. 2013;56:7324–7333. doi: 10.1021/jm400815m. [DOI] [PubMed] [Google Scholar]
- McGowan D. C. Herschke F. Pauwels F. Stoops B. Smyej I. Last S. Pieters S. Embrechts W. Khamlichi M. D. Thoné T. Van Schoubroeck B. Mostmans W. Wuyts D. Verstappen D. Scholliers A. De Pooter D. Dhuyvetter D. Borghys H. Tuefferd M. Arnoult E. Hong J. Fanning G. Bollekens J. Urmaliya V. Teisman A. Horton H. Jonckers T. H. M. Raboisson P. Identification and Optimization of Pyrrolo[3,2-d]pyrimidine Toll-like Receptor 7 (TLR7) Selective Agonists for the Treatment of Hepatitis B. J. Med. Chem. 2017;60:6137–6151. doi: 10.1021/acs.jmedchem.7b00365. [DOI] [PubMed] [Google Scholar]
- McGowan D. C. and Raboissonr P. J. B., Thieno[3,2-d]pyrimidines derivatives for the treatment of viral infections, WO2015014815A1, 2015
- Zhaozhong D., Fei S., Hao W., Lifang W. and Ling Y., Pyrrolopyrimidine compounds used as TLR7 agonist, WO2016023511A1, 2016
- Shuhui C., Zhaozhong D., Fei S., Hao W., Lifang W. and Ling Y., 7-(thiazol-5-yl) pyrrolopyrimidine compound as tlr7 agonist, WO2017076346, 2017
- Pieters S. McGowan D. Herschke F. Pauwels F. Stoops B. Last S. Embrechts W. Scholliers A. Mostmans W. Van Dijck K. Van Schoubroeck B. Thoné T. De Pooter D. Fanning G. Rosauro M. L. Khamlichi M. D. Houpis I. Arnoult E. Jonckers T. H. M. Raboisson P. Discovery of selective 2,4-diaminoquinazoline toll-like receptor 7 (TLR 7) agonists. Bioorg. Med. Chem. Lett. 2018;28:711–719. doi: 10.1016/j.bmcl.2018.01.014. [DOI] [PubMed] [Google Scholar]
- Embrechts W. Herschke F. Pauwels F. Stoops B. Last S. Pieters S. Pande V. Pille G. Amssoms K. Smyej I. Dhuyvetter D. Scholliers A. Mostmans W. Van Dijck K. Van Schoubroeck B. Thone T. De Pooter D. Fanning G. Jonckers T. H. M. Horton H. Raboisson P. McGowan D. 2,4-Diaminoquinazolines as Dual Toll-like Receptor (TLR) 7/8 Modulators for the Treatment of Hepatitis B Virus. J. Med. Chem. 2018;61:6236–6246. doi: 10.1021/acs.jmedchem.8b00643. [DOI] [PubMed] [Google Scholar]
- McGowan D. C. Herschke F. Khamlichi M. D. Rosauro M. L. Benedicto S. M. P. Pauwels F. Stoops B. Pande V. Scholliers A. Van Schoubroeck B. Mostmans W. Van Dijck K. Thoné T. Horton H. Fanning G. Jonckers T. H. M. Raboisson P. Design and synthesis of tetrahydropyridopyrimidine based Toll-Like Receptor (TLR) 7/8 dual agonists. Bioorg. Med. Chem. Lett. 2018;28:3216–3221. doi: 10.1016/j.bmcl.2018.08.015. [DOI] [PubMed] [Google Scholar]
- Mackman R. L. Mish M. Chin G. Perry J. K. Appleby T. Aktoudianakis V. Metobo S. Pyun P. Niu C. Daffis S. Yu H. Zheng J. Villasenor A. G. Zablocki J. Chamberlain J. Jin H. Lee G. Suekawa-Pirrone K. Santos R. Delaney W. E. Fletcher S. P. Discovery of GS-9688 (Selgantolimod) as a Potent and Selective Oral Toll-Like Receptor 8 Agonist for the Treatment of Chronic Hepatitis B. J. Med. Chem. 2020;63:10188–10203. doi: 10.1021/acs.jmedchem.0c00100. [DOI] [PubMed] [Google Scholar]
- Beesu M. Caruso G. Salyer A. C. Shukla N. M. Khetani K. K. Smith L. J. Fox L. M. Tanji H. Ohto U. Shimizu T. David S. A. Identification of a Human Toll-Like Receptor (TLR) 8-Specific Agonist and a Functional Pan-TLR Inhibitor in 2-Aminoimidazoles. J. Med. Chem. 2016;59:3311–3330. doi: 10.1021/acs.jmedchem.6b00023. [DOI] [PubMed] [Google Scholar]
- Howbert J. J., Kusukuntla V. R., Tretyakov A., Nielson N., Krasik P., Jiang J. and Yang H. W., Methods of synthesis of Banzazepine derivatives, WO2010054215, 2010
- Lu H. Dietsch G. N. Matthews M. A. Yang Y. Ghanekar S. Inokuma M. Suni M. Maino V. C. Henderson K. E. Howbert J. J. Disis M. L. Hershberg R. M. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin. Cancer Res. 2012;18:499–509. doi: 10.1158/1078-0432.CCR-11-1625. [DOI] [PubMed] [Google Scholar]
- Yamamoto T. Kanuma T. Takahama S. Okamura T. Moriishi E. Ishii K. J. Terahara K. Yasutomi Y. STING agonists activate latently infected cells and enhance SIV-specific responses ex vivo in naturally SIV controlled cynomolgus macaques. Sci. Rep. 2019;9:5917. doi: 10.1038/s41598-019-42253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J. Sun M. Shi G. Xu Y. Han Y. Li X. Dong W. Zhan L. Qin C. Toll-Like Receptor 8 Agonist Strengthens the Protective Efficacy of ESAT-6 Immunization to Mycobacterium tuberculosis Infection. Front. Immunol. 2017;8:1972. doi: 10.3389/fimmu.2017.01972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lixin L. and Huiping G., 2-Amino-quinoline derivatives, WO2018196823, 2018
- Chowdari N. S., Gangwar S., He L., Posy S. L., Poudel Y. B., Purandare A. V., Sivaprakasam P. and Wan H., Macrocyclic toll-like receptor 7 (TLR7) agonists, WO2019209811, 2019
- Alexander A. P., Jean-Francois B., Maurice D. F. M., Claude F. J. M., Philippe M. and Bernard R. P. J., Macrocyclic purines for the treatment of viral infections, WO2014009509, 2014
- Alexander A. P., Jean-Francois B., Maurice D. F. M., Claude F. J. M., Philippe M. and Bernard R. P. J., Macrocyclic deaza-purinones for the treatment of viral infections, WO 2014154859, 2014
- Jean-François B., Vincent G., Georges G. J. E., Christophe H., Philippe J., Vincent L. and McGowan D. G., 1,2,4-triazine derivatives for the treatment of viral infections, WO2014053516, 2014
- Yoo E. Salunke D. B. Sil D. Guo X. Salyer A. C. D. Hermanson A. R. Kumar M. Malladi S. S. Balakrishna R. Thompson W. H. Tanji H. Ohto U. Shimizu T. David S. A. Determinants of Activity at Human Toll-like Receptors 7 and 8: Quantitative Structure–Activity Relationship (QSAR) of Diverse Heterocyclic Scaffolds. J. Med. Chem. 2014;57:7955–7970. doi: 10.1021/jm500744f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnov D. Schmidt J. J. Capecchi J. T. Wightman P. D. Vaccine adjuvant activity of 3M-052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine. 2011;29:5434–5442. doi: 10.1016/j.vaccine.2011.05.061. [DOI] [PubMed] [Google Scholar]
- Rathee J. Kanwar R. Kaushik D. Salunke D. B. Mehta S. K. Niosomes as efficient drug delivery modules for encapsulation of Toll-like receptor 7 agonists and IDO-inhibitor. Appl. Surf. Sci. 2020;505:144078. [Google Scholar]
- Fox C. B. Orr M. T. Hoeven N. V. Parker S. C. Mikasa T. J. Phan T. Beebe E. A. Nana G. I. Joshi S. W. Tomai M. A. Elvecrog J. Fouts T. R. Reed S. G. Adsorption of a synthetic TLR7/8 ligand to aluminum oxyhydroxide for enhanced vaccine adjuvant activity: a formulation approach. J. Controlled Release. 2016;244:98–107. doi: 10.1016/j.jconrel.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A. Kaushik D. Piplani S. Mehta S. K. Petrovsky N. Salunke D. B. TLR2 Agonistic Small Molecules: Detailed Structure–Activity Relationship, Applications, and Future Prospects. J. Med. Chem. 2021;64:233–278. doi: 10.1021/acs.jmedchem.0c01627. [DOI] [PubMed] [Google Scholar]
- Wille-Reece U. Flynn B. J. Loré K. Koup R. A. Kedl R. M. Mattapallil J. J. Weiss W. R. Roederer M. Seder R. A. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15190–15194. doi: 10.1073/pnas.0507484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbrook B. C. D’Agostino R. B. Aycock S. T. Jorgensen M. J. Hadimani M. B. Bruce King S. Alexander-Miller M. A. Adjuvanting an inactivated influenza vaccine with conjugated R848 improves the level of antibody present at 6months in a nonhuman primate neonate model. Vaccine. 2017;35:6137–6142. doi: 10.1016/j.vaccine.2017.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francica J. R. Lynn G. M. Laga R. Joyce M. G. Ruckwardt T. J. Morabito K. M. Chen M. Chaudhuri R. Zhang B. Sastry M. Druz A. Ko K. Choe M. Pechar M. Georgiev I. S. Kueltzo L. A. Seymour L. W. Mascola J. R. Kwong P. D. Graham B. S. Seder R. A. Thermoresponsive Polymer Nanoparticles Co-deliver RSV F Trimers with a TLR-7/8 Adjuvant. Bioconjug. Chem. 2016;27:2372–2385. doi: 10.1021/acs.bioconjchem.6b00370. [DOI] [PubMed] [Google Scholar]
- Gutjahr A. L. Papagno F. Nicoli A. Lamoureux F. Vernejoul T. Lioux E. Gostick D. A. Price G. Tiraby E. Perouzel E. Appay V. Verrier B. Paul S. Cutting edge: a dual TLR2 and TLR7 ligand induces highly potent humoral and cell-mediated immune responses. J. Immunol. 2017;198:4205–4209. doi: 10.4049/jimmunol.1602131. [DOI] [PubMed] [Google Scholar]
- Albin T. J. Tom J. K. Manna S. Gilkes A. P. Stetkevich S. A. Katz B. B. Supnet M. Felgner J. Jain A. Nakajima R. Jasinskas A. Zlotnik A. Pearlman E. Davies D. H. Felgner P. L. Burkhardt A. M. Esser-Kahn A. P. Linked Toll-Like Receptor Triagonists Stimulate Distinct, Combination-Dependent Innate Immune Responses. ACS Cent. Sci. 2019;5:1137–1145. doi: 10.1021/acscentsci.8b00823. [DOI] [PMC free article] [PubMed] [Google Scholar]