

Abstract
Since the outbreak of COVID-19, the pandemic caused by SARS-CoV-2 infection is still spreading at an alarming rate and has caused huge loss of life and economic damage worldwide. Although more than one year has passed, effective treatments for COVID-19 and other pathogenic coronaviruses have not yet been developed. Therefore, the development of SARS-CoV-2 inhibitors is an urgent priority. Given that the Mpro sequences of SARS-CoV-2 and SARS-CoV-1 are 100% identical in the catalytic domain for protein cleavage, the viral main protease (Mpro) is one of the most extensive drug targets in all the drug targets being investigated for SARS-CoV-2. To provide scientific researchers with timely anti-SARS-CoV drug development information for Mpro, we focus on the past and current drug design and development strategies for MPro in this review. We believe that this review will provide meaningful guidance for the design and development of innovative drugs against COVID-19 and other pathogenic coronaviruses in the future.
Since the outbreak of COVID-19, the pandemic caused by SARS-CoV-2 infection is still spreading at an alarming rate and has caused huge loss of life and economic damage worldwide.
1. Introduction
Since the first case was detected in December 2019, coronavirus disease 2019 (COVID-19) has rapidly evolved into a global pandemic.1,2 This rapidly spreading pathogenic virus was soon identified as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a positive-sense single-stranded RNA virus consisting of four structural proteins and an RNA genome.3 Up to now, the origin of SARS-CoV-2 is still under investigation. According to the past reports, the closest strain is the bat coronavirus RaTG13, which has 96% sequence similarity with SARS-CoV-2.3 Humans have encountered many similar fatal virus outbreaks, such as Ebola virus,4 Zika virus5 and Nipah virus,6 as well as various coronaviruses (CoV) including SARS-CoV7 and MERS-CoV.8 However, the recent outbreak of SARS-CoV-2 is the most serious public health crisis since the Spanish influenza pandemic.9,10 As of January 12, 2021, there have been more than 91 million positive cases with nearly 2 million deaths in the world. After in-depth research by scientists, the SARS-CoV-2 genome was shown to encode two large polyproteins, pp1a and pp1ab, which contain overlapping sequences and the main viral protease (Mpro).11 The function of this internally encoded Mpro is essential to the processing of these proteins and critical for viral replication.12 SARS-CoV-2 Mpro is very similar to SARS-CoV-1 Mpro. Specifically, SARS-CoV-1 and SARS-CoV-2 share 96% identity between their respective Mpro sequences and 100% identity in the active site.13
2. Drug design targets
The target of drug action is very important to the design of drug molecules during the development of small molecule drugs. SARS-CoV-2 is an enveloped positive-sense single-stranded RNA virus belonging to the genus β-coronavirus, which also includes SARS-CoV, MERS-CoV, HCoV-OC43 and HCoV-HKU1. Given that SARS-CoV-2 shares about 80% sequence similarity with SARS-CoV-1, most of the antiviral drugs for SARS-CoV-2 were originally developed against SARS-CoV-1 or other related coronaviruses.14 To know the target of SARS-CoV-2, it is necessary to clearly understand its specific life cycle. Specifically, SARS-CoV-2 infects ACE2-expressing cells and enters the cell through either direct cell surface fusion or the endosomal pathway. For direct cell surface fusion, the host membrane protease TMPRSS2 cleaves the viral spike protein, triggering viral membrane fusion with the host cell membrane.15 For endosomal entry, cathepsin L mediates the cleavage of the viral spike protein.16 Once the viral RNA is released in the cytoplasm, it undergoes translation into viral polyproteins pp1a and pp1ab, which are subsequently cleaved by two viral proteases, the main protease (Mpro) and the papain-like protease (PLpro), the main protease also being called 3-chymotrypsin-like protease (3CLpro). Then, the released viral proteins can be assembled to form a viral polymerase RdRp complex to catalyze the replication of viral RNA. Finally, the progeny virions are released from the infected cells through exocytosis and are ready for the next round of infection.
Based on the above introduction to the SARS-COV-2 infection route, we can clearly know that Mpro, PLpro, ACE2 and RdRp play a very critical role in the replication of coronavirus. Among these key targets, Mpro has become the most important target for anti-COVID-19 drug design due to its highly conservative nature and specificity.
3. Main protease (Mpro)
The SARS-COV main protease is excised from the polyprotein by its own proteolytic activity, and then cleaves the polyprotein together with the papain-like protease, resulting in a total of 16 functional non-structural proteins (nsps). According to reports, the Mpro of SARS-COV specifically acts at 11 cleavage sites of large polyprotein 1ab (790 kDa),17 and no human protease has been found to have similar cleavage specificity. The cleaved nsps play essential roles in assembling the viral replication transcription complex (RTC) to initiate the viral replication. Although some companies have developed vaccines that can prevent COVID-19, their preventive effect is not good enough. Therefore, the development of small molecule antiviral drugs that can effectively inhibit SARS-COV-2 is still a top priority.
In view of the high specificity of Mpro and the extremely low toxicity of Mpro inhibitors to host cells, Mpro has become one of the most intriguing drug targets for antiviral drug development.13,18
4. SARS-CoV Mpro inhibitors
Since the SARS outbreak in 2003,19,20 and later on with the COVID-19 outbreak in late 2019,1,2 most of the scientific research teams studying SARS-CoV have devoted themselves to the development of peptidomimetic and non-peptidomimetic Mpro and PLpro inhibitors.21–24 Due to its highly conservative nature and specificity, Mpro has become one of the most popular research targets. Specifically, Mpro is a cysteine protease that cleaves the viral polyprotein at more than 11 sites. It has a unique substrate preference for glutamine at the P1 position, while no host protease is known to have such a preference.25–27 Since the structure of Mpro was published, it has greatly promoted the research on Mpro inhibitors as new antiviral drugs.18,28 From the structural point of view, Mpro has three domains: I, II and III. The active site region spans domains I and II, which are β barrel domains, while domain III presents an α-helical structure. In the active site region, Cys145 acts as a nucleophile, while His41 acts as a general acid/base. Based on the above information on Mpro, scientists have developed many Mpro inhibitors. Although only a small number of inhibitors that have been developed show good affinity for the Mpro target, they provide a good starting point for further Mpro-targeted antiviral drug development.29–32
4.1. SARS-CoV-1 Mpro inhibitors
In 2005, Ghosh and collaborators reported the design, synthesis and biological evaluation of some peptidomimetic SARS-COV-1 Mpro inhibitors. Biological experimental evaluation results show that this type of inhibitor can be used to treat severe acute respiratory syndrome33 These inhibitors exhibited antiviral activity against SARS-CoV-1 in infected cells in the micromolar range; compounds 1 and 2 exhibited excellent inhibitory activity with an IC50 value of 45 μM and 70 μM, respectively. The X-ray crystal structure shows that lead inhibitor 2 has a good binding effect with SARS-CoV-1 Mpro. The results of this study provide an important drug design template for the design of SARS-CoV-1 Mpro small molecule inhibitors (Fig. 1). Meanwhile, it provides reference for the follow-up study of SARS-COV Mpro related inhibitors.
Fig. 1. A, Chemical structures of compounds 1 and 2. B, X-ray crystal structure of compound 2 (thick stick with magenta carbon) with SARS-CoV-1 Mpro. Hydrogen bonds between the inhibitor and SARS-CoV-1 Mpro are shown as green dotted lines.

One year later, Hsu and his collaborators discovered an effective SARS-CoV-1 Mpro inhibitor (TG-0205221, Ki = 53 nM), which showed good anti-SARS coronavirus activity (Fig. 2).34 Analysis of the crystal structure showed that the inhibitor exerts its antiviral activity through the interaction of its 16 carbon atoms with 10 residues on the enzyme, and the extensive hydrophobic contact with the enzyme. The inhibitor binds to SARS-CoV-1 Mpro through 10 hydrogen bonds and 1 covalent bond, forming a very strong binding conformation. The crystal structure of TG-0205221 and SARS-CoV-1 Mpro (resolution = 1.93 Å) shows a unique binding mode, including covalent bonds, hydrogen bonds and many hydrophobic interactions. The results of this study provide a basis for the further development of structure-based peptidomimetic SARS-CoV-1 Mpro inhibitors.
Fig. 2. A, Chemical structure of TG-0205221. B, Stereo-view of SARS-CoV-1 Mpro bound with the TG-0205221 inhibitor shown by electrostatic potential.

In 2009, Hayashi and his colleagues developed a series of peptidomimetic SARS-CoV-1 Mpro inhibitors containing trifluoromethyl, benzothiazolyl and thiazolyl ketones. Some of the compounds show good biological activity.35 Among all the small molecule inhibitors, compound 3 containing P1-pyrrolidone and a warhead thiazolyl unit showed the best inhibitory efficacy against SARS-CoV-1 Mpro with an IC50 value of 2.2 μM. It provides a new direction for the subsequent rapid development of more effective inhibitors (Fig. 3).
Fig. 3. A, Chemical structure of compound 3. B, Molecular dynamics simulated pose of compound 3 (green stick) bound to SARS-CoV-1 Mpro (PDB 1WOF, with red and cyan ribbons with molecular surfaces).

On the basis of previous research work on SARS-COV-1 Mpro inhibitors, four years later, the same authors reported the design and synthesis of a series of dipeptide inhibitors with novel P3 scaffolds. These compounds containing P3 scaffolds are effective against SARS-CoV-1 Mpro and also show good inhibitory activity.36–38 In particular, compound 5 showed the best inhibitory activity with a Ki value of 0.006 mM. Happily, its inhibitory potency against SARS-COV-1 Mpro is 65 times higher than that of the lead compound 4 (Ki = 0.39 μM). In addition, they used isothermal titration calorimetry (ITC) to verify the experimental results (Fig. 4).
Fig. 4. A, Chemical structures of compounds 4 and 5. B, Molecular docking pose and binding interactions of compounds 4 and 5 (orange sticks) bound to SARS-CoV-1 Mpro (PDB ID: 1WOF). Only the residues (yellow color), which are engaged in binding to the ligands (orange sticks), are highlighted. Dotted black lines represent the hydrogen bonding interaction.

While developing peptidomimetic SARS-COV-1 Mpro inhibitors, many medicinal chemists have also made a lot of effort in the design of non-peptidomimetic drug molecules. Vederas and collaborators designed and synthesized many non-peptidyl inhibitors containing pyridyl ester structures.39–41 Some of these compounds have shown high inhibitory activity against SARS-CoV-1 Mpro, and the IC50 value of compound 8 can be up to 50 nM (Fig. 5). They used electrospray mass spectrometry to reveal the mechanism of action of such small molecule inhibitors on SARS-CoV-1 Mpro. These results provide a template for the further development of non-peptidomimetic SARS-COV-1 inhibitors.
Fig. 5. Chemical structures of compounds 6–9.

Subsequently, Ghosh and his co-workers reported a series of 5-chloropyridinyl indolecarboxylates as potent non-peptidyl SARS-CoV-1 Mpro inhibitors (Fig. 6).42 It is worth noting that the position of the ester functional group is critical to the effectiveness of the inhibitor. After investigating the positions of a large number of functional groups, they found that a compound containing 5-chloropyridyl ester 15 showed the most effective inhibitory effect. Its SARS-CoV-1 Mpro IC50 value was 30 nM and its antiviral EC50 value was 6.9 μM. Subsequently, they proved the possible combination of these inhibitors with SARS-CoV-1 Mpro through molecular docking experiments.
Fig. 6. A, Chemical structures of compounds 10–15. B, GOLD docked conformation of 15 (green), covalently linked to Cys-145 of Mpro based on the 2V6N Mpro structure.

Collectively, since the SARS outbreak in 2003, many scientists have made many contributions to the development of SARS-CoV-1 Mpro inhibitors. They have developed many peptidomimetic and non-peptidomimetic SARS-COV-1 Mpro inhibitors. Here, we only list some representative works for your reference. These efforts in the development of SARS-CoV-1 Mpro inhibitors have laid a solid foundation for the subsequent SARS-COV-2 Mpro inhibitors.
4.2. SARS-CoV-2 Mpro inhibitors
In view of the high similarity between SARS-COV-1 Mpro and SARS-COV-2 Mpro, the former provides a lot of reference for the development of SARS-COV-2 Mpro inhibitors. Like SARS-COV-1, SARS-COV-2 Mpro inhibitors can be classified as peptoids and non-peptidomimetics, and the mechanism of action of peptide inhibitors includes two steps. Peptidomimetics that mimic natural peptide substrates initially bind to Mpro and form a noncovalent complex, and the warhead group, which is spatially very close to the catalytic residue of the target protein, undergoes a nucleophilic attack to catalyse the formation of cysteine-participating covalent bonds.43,44 These warheads mainly contain Michael receptors, aldehydes and different types of ketones,45–47 which covalently bind to the Cys145 residue in the Mpro S1′ pocket to exert an inhibitory effect.48,49 At the present moment, many research papers and reviews related to SARS-CoV-2 Mpro inhibitors have been reported.50–52 Next, we will select some representative research work to introduce to you.
Mitsuya and his collaborators developed a series of non-peptidyl SARS-CoV-2 Mpro inhibitors.53 In particular, two indole chloropyridinyl-ester derivatives, GRL-0820 and GRL-0920, exerted potent activity against SARS-CoV-2 in cell-based assays performed using VeroE6 cells and TMPRSS2-overexpressing VeroE6 cells (Fig. 7). They pointed out in the article that although GRL-0820 prevented SARS-CoV-2 infection, viral breakthrough occurred. GRL-0920 not only exerts effective activity on SARS-CoV-2 (EC50 = 2.8 μM), but also significantly reduces the infectivity, replication ability and cytopathic effects of SARS-CoV-2, and no obvious cytotoxicity is seen. Subsequently, structural modeling showed that the indole and chloropyridinyl of the derivatives interact with two catalytic dyad residues of Mpro, Cys145 and His41, resulting in covalent bonding, which was verified by high-performance liquid chromatography–mass spectrometry (HPLC/MS). The research results show that the indole moiety is essential for the anti-SARS-CoV-2 activity of the derivative. This also proves that this type of indole chloropyridine containing active ester has a good anti-SARS-COV-2 effect.
Fig. 7. A, Chemical structures of compounds GRL-0820 and GRL-0920. B, Molecular models of interactions of GRL-0920 with SARS-CoV-2 Mpro.

While developing non-peptidomimetic SARS-COV-2 Mpro inhibitors, many researchers have also made great progress in developing peptidomimetic SARS-COV-2 Mpro inhibitors. Recently, Wang and his co-workers reported some peptidomimetic inhibitors targeting SARS-CoV-2 Mpro.54 The enzymatic assay results show that four inhibitors (boceprevir, GC-376, and calpain inhibitors II and XII) have single-digit to submicromolar IC50 values (Fig. 8). More importantly, they can inhibit SARS-CoV-2 virus replication in cell models with EC50 values of 0.49 to 3.37 μM. Subsequently, they used enzyme kinetic studies, thermal shift binding assays and natural mass spectrometry to further characterize the mechanism of action of these inhibitors. The four compounds provide promising starting points for the further development of SARS-CoV-2 therapeutics.
Fig. 8. Chemical structures of boceprevir, GC-376, calpain inhibitors II and XII.

Based on previous work, Hilgenfeld and colleagues designed an α-ketoamide SARS-CoV-2 Mpro inhibitor (Fig. 9).13 To extend the half-life of the compound in plasma, they incorporated a pyridone ring into the P3–P2 amide bond. Subsequently, a small molecule compound 17 with better activity was screened by a large number of molecular structure optimizations and showed significant inhibition of SARS-CoV-2 Mpro with an IC50 value of 0.67 μM. Meanwhile, they conducted pharmacokinetic experiments on this compound, and the experimental results showed that its pulmonary tropism is obvious and it is suitable for administration through inhalation. In addition, they also obtained the X-ray structure of unliganded SARS-CoV-2 Mpro and its complex with the α-ketoamide inhibitor. The results of this study provide useful guidance for the subsequent development of structure-based α-ketoamide SARS-COV-2 Mpro inhibitors. This type of α-ketoamide inhibitor is one of the most representative SARS-COV-2 Mpro inhibitors found so far.
Fig. 9. A, Chemical structures of compounds 16 and 17. B, Compound 17 in the substrate-binding cleft located between domains I and II of SARS-CoV-2 Mpro in the monoclinic crystal form (space group C2).

Subsequently, Liu and his co-workers proposed peptidomimetic aldehydes as candidates for antiviral drugs.55 Based on this hypothesis, they designed and synthesized compound 18 and compound 19 (Fig. 10), and used a fluorescence resonance energy transfer (FRET)-based cleavage assay to determine their median inhibitory concentration (IC50) values. The test results showed that both compounds showed excellent inhibitory activity, with IC50 values of 0.05 μM and 0.04 μM, respectively. To further confirm the results of enzyme inhibition, they evaluated the ability of these compounds to inhibit SARS-CoV-2 in vitro. Meanwhile, they also demonstrated the effective efficacy of these two compounds against SARS-CoV-2 infection in a cell model, with EC50 values of 0.53 μM and 0.72 μM, respectively. In addition, they also showed good pharmacokinetic properties in animal models: the bioavailability of compound 18 and compound 19via the intraperitoneal route was 87.8% and 80%, respectively. In particular, they proved that the aldehyde group of these two compounds covalently binds to the catalytic cysteine by the crystal structure of the complex of SARS-CoV-2 Mpro with them, thereby inhibiting Mpro activity.
Fig. 10. A, Chemical structures of compounds 18 and 19. B, Schematic diagrams of SARS-CoV-2 Mpro–18 and SARS-CoV-2 Mpro–19 interactions.

Santos-Filho reported two SARS-COV-2 Mpro inhibitors, danoprevir and lopinavir, both of which have a strong interaction with the binding sites of the SARS-CoV-2 main protease (Fig. 11).56 Almost simultaneously, in the current clinical study (NCT04291729) conducted at the Ninth Hospital of Nanchang, Wu and his co-workers evaluated the therapeutic effects of danoprevir, boosted by ritonavir,57 on untreated COVID-19 patients. The data from this small-sample clinical study showed that danoprevir boosted by ritonavir is safe and well tolerated in all patients. After 4 to 12 days of treatment of danoprevir boosted by ritonavir, all eleven patients enrolled were discharged from the hospital as they met all four conditions as follows: 1) normal body temperature for at least 3 days; 2) significantly improved respiratory symptoms; (3) lung imaging shows obvious absorption and recovery of acute exudative lesion; 4) two consecutive RT-PCR negative tests of SARS-CoV-2 nucleotide acid (respiratory track sampling with an interval of at least one day). These clinical experimental data provide strong evidence for the potential of the two molecules in SARS-COV-2 therapy.
Fig. 11. A, Chemical structures of danoprevir and lopinavir. B, Danoprevir and lopinavir docked in the binding site of SARS-CoV-2 Mpro.

Yang and his co-workers also did a lot of beautiful work in the development of anti-SARS-COV-2 Mpro inhibitors since the outbreak of the pandemic. In early 2020, based on the discovery of the N3 inhibitor against SARS-COV-1 Mpro by Rao et al.,58 they identified an N3 inhibitor also suitable for SARS-COV-2, and then determined the crystal structure of Mpro of SARS-CoV-2 in complex with this compound (Fig. 12).11 In addition, Yang and his colleagues used structure-based virtual and high-throughput screening to analyze more than 10 000 compounds including candidate drugs in clinical trials, approved drugs and other pharmacologically active compounds. The test results show that six drug molecules (ebselen, disulfiram, tideglusib, carmofur, shikonin and PX-12) have strong inhibitory effects on SARS-CoV-2 Mpro, showing half-maximal inhibitory concentration values that ranged from 0.67 to 21.4 μM (Fig. 13). Among them, ebselen exhibited promising antiviral activity in cell-based assays. Subsequently, they also characterized the X-ray crystal structure of the composite of Mpro and carmofur, and the crystal structure reveals that the carbonyl reactive group of carmofur showed an antiviral effect by covalently binding to the catalytic Cys145.59
Fig. 12. Chemical structures of the N3 inhibitor, ebselen, disulfiram, tideglusib, carmofur, shikonin, and PX-12.

Fig. 13. Chemical structures of Walrycin B, LLL-12 and Z-FA-FMK.

To explore the mechanism of action of the above six SARS-COV-2 Mpro inhibitors, subsequently, Wang and his co-workers investigated the mechanism of action of the six Mpro inhibitors using a consortium of techniques including the FRET-based enzymatic assay, thermal shift assay, native mass spectrometry, cellular antiviral assays, and molecular dynamics simulations.60 Collectively, the results of this study show that the inhibitory effects of these six compounds on SARS-CoV-2 Mpro are non-specific, and their inhibitory effects can be eliminated or greatly reduced by adding the reducing agent dithiothreitol (DTT). Without DTT, these six compounds inhibit not only Mpro but also a panel of viral cysteine proteases including SARS-CoV-2 PLpro and 2Apro and 3Cpro from enterovirus A71 (EV-A71) and EV-D68.
High-throughput drug screening technology is an effective way to obtain active molecules relatively quickly in drug development. Recently, Zeng and his colleagues used the SARS-CoV-2 Mpro analysis method to perform quantitative high-throughput screening (qHTS) on 10 755 compounds including approved drugs, drugs under investigation, and other biologically active molecules.61 Finally, they obtained 23 SARS-CoV-2 Mpro small molecule inhibitors with IC50 ranging from 0.26 to 28.85 μM. Among them, Walrycin B (IC50 = 0.26 μM), LLL-12 (IC50 = 9.84 μM) and Z-FA-FMK (IC50 = 11.39 μM) are the most effective Mpro inhibitors (Fig. 13). Notably, they used the SARS-CoV-2 cytopathic effect assay to confirm their activity against SARS-CoV-2 virus infection.
To speed up drug discovery and development, Xu and his co-workers investigated the inhibition of SARS-COV-2 Mpro by natural products derived from traditional Chinese medicines.62 In this study, baicalin and baicalein were identified as the first non-covalent, non-peptidomimetic inhibitors of SARS-COV-2 Mpro and exhibited potent antiviral activities in a cell-based system (Fig. 14). Remarkably, X-ray protein crystallography results show that the binding mode of baicalein with SARS-COV-2 Mpro is distinctly different from those of known inhibitors. Baicalein is perfectly ensconced in the core of the substrate-binding pocket by interacting with two catalytic residues, the crucial S1/S2 subsites and the oxyanion loop, acting as a “shield” in front of the catalytic dyad to prevent the peptide substrate from approaching the active site. This binding mode is significantly different from those of known inhibitors. Baicalein's unique mode of action, good in vitro antiviral activity and good safety data from clinical trials all proved its great potential as an anti-coronavirus drug. This also proves why traditional Chinese medicines played an important role in the outbreak of this pandemic.
Fig. 14. A, Chemical structures of baicalin and baicalein. B, Interactions formed between baicalein (green) and surrounding residues (cyan). Residues as well as the ligand are shown as sticks and hydrogen bonds are represented by black-dashed lines.

Recently, Yang and his collaborators designed and synthesized 32 new Mpro inhibitors containing bicyclic proline.63 All the compounds inhibit SARS-CoV-2 Mpro activity in vitro, with IC50 values between 7.6 and 748.5 nM. After evaluating the anti-SARS-CoV-2 virus activity, rat pharmacokinetic properties and safety of these molecules at the cellular level, they finally selected two small molecule compounds (MI-09 and MI-30) with high activity and safety to carry out in vivo antiviral activity tests (Fig. 15). In a transgenic mouse model of SARS-CoV-2 infection, oral or intraperitoneal injection of MI-09 or MI-30 can significantly reduce lung viral load and lung pathological damage. This is the first publicly reported experimental data of Mpro inhibitors in a mouse model of SARS-CoV-2 infection.
Fig. 15. Chemical structures of MI-09 and MI-30.

Mitsuya and his collaborators have long been engaged in the development of antiviral drugs. After continuous exploration, they discovered two small molecule compounds GRL-1720 and 20 that target SARS-CoV-2 Mpro (Fig. 16).64 Based on the detection of VeroE6 cells and RNA-qPCR, the results of cytopathic detection and immunocytochemistry experiments show that these two compounds can prevent SARS-CoV-2 infection. The EC50 values of GRL-1720 and 20 are 15 ± 4 and 4.2 ± 0.7 μM, respectively. Compound 20 completely blocked SARS-CoV-2 infection in vitro, and there was no viral breakthrough or detectable cytotoxicity. In addition, the combination of 20 and remdesivir showed a synergistic effect against SARS-CoV-2. Therefore, they believe that compound 20 can be used as the main inhibitor of Mpro for the development of therapeutic drugs for SARS-CoV-2 infection.
Fig. 16. Chemical structures of GRL-1720, 20 and PF-00835231.

Interestingly, Hoffman and his collaborators reported a hydroxymethyl ketone derivative PF-00835231, which is structurally similar to compound 20 and shows effective inhibition of SARS-CoV-2 in Mpro and antiviral tests.65 Preclinical experiments show that PF-00835231 can be used as an effective inhibitor of SARS-CoV-2 Mpro and has the potential to become an intravenous treatment drug for COVID-19.
4.3. SARS-CoV-2 Mpro inhibitors from computer-aided design
Computer-aided virtual screening technology has played an indispensable role in this COVID-19 pandemic. Many scientists have used this technology to screen out a large number of Mpro inhibitors with potential anti-SARS-COV-2 effects. For example, Lee and his co-workers identified potential inhibitors against COVID-19, using an amalgam of virtual screening, molecular dynamics (MD) simulations, and binding free energy approaches from the Korea Chemical Bank drug repurposing database.66 The screening of the Korea Chemical Bank drug repurposing database resulted in 149 binders. They studied the kinetics of the protein–drug complex formation of the seven highest-scoring drugs through MD simulations. The test results showed that six drugs exhibited good interactions with the active site of SARS-CoV-2 Mpro. Furthermore, binding free energy calculations suggested that the community-acquired bacterial pneumonia drugs ceftaroline fosamil and telaprevir are potent inhibitors against SARS-CoV-2 Mpro (Fig. 17). Molecular dynamics and interaction analysis revealed that ceftaroline fosamil and telaprevir form hydrogen bonds with important active site residues such as Thr24, Thr25, His41, Thr45, Gly143, Ser144, Cys145, and Glu166, which is supported by crystallographic information of known inhibitors.
Fig. 17. A, Chemical structures of atazanavir, ceftaroline fosamil and telaprevir. B, The binding of drugs at subsites S1, S1′, S2, and S3 of SARS-CoV-2 Mpro. Ceftaroline fosamil and telaprevir are shown in stick forms in green and orange, respectively.

The study of Mesecar and his co-workers also proved that telaprevir is a potent inhibitor against Mpro.67 In their study, telaprevir showed significant inhibition of Mpro from SARS-CoV-2 with an IC50 value of 10.7 ± 0.4 μM.
Almost simultaneously, Kang and his co-workers used their pre-trained deep learning-based drug-target interaction model called Molecule Transformer-Drug Target Interaction (MT-DTI) to screen for inhibitors that have a better inhibitory effect on the SARS-CoV-2 main protein.68 The test results showed that atazanavir (Fig. 17), an antiretroviral medication used to treat and prevent the human immunodeficiency virus (HIV), is the best compound, showing an inhibitory potency with a Kd of 94.94 nM against the SARS-CoV-2 main proteinase.
For finding more potential drugs as inhibitors of SARS-COV-2 Mpro, Zhu and his colleagues conducted molecular docking experiments on 1903 small molecule drugs by establishing a homology model based on the structure of SARS-COV-2 Mpro.69 Based on the docking score and the 3D similarity of the binding mode to the known SARS-COV-2 Mpro ligands, four drugs were selected for binding free energy calculations. The calculation results show that both the MM/GBSA and SIE methods vote for nelfinavir, with a binding free energy of −24.69 ± 0.52 kcal mol−1 and −9.42 ± 0.04 kcal mol−1, respectively. Therefore, they proposed that nelfinavir might be a potential inhibitor against SARS-COV-2 Mpro (Fig. 18).
Fig. 18. A, Chemical structure of nelfinavir. B, Interactions between nelfinavir and associated residues in the homology model of SARS-CoV-2 Mpro. The data in red are the interaction distances (Å).

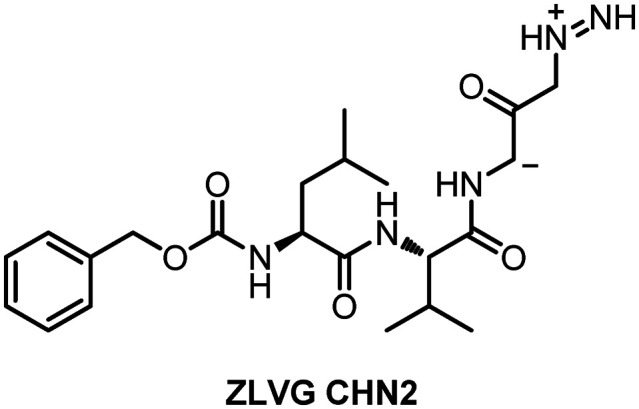
Riva et al. conducted a virtual screening of 12 000 clinical-stage and FDA-approved small molecules in the drug database. The test results showed that six molecules have the characteristics of a cell dose–activity relationship, and showed an effective concentration that may be equivalent to the patient's therapeutic dose.70,71 The six molecules include the PIKfyve kinase inhibitor apilimod, cysteine protease inhibitors MDL-28170, Z LVG CHN2, VBY-825, and ONO 5334, and the CCR1 antagonist MLN-3897. Since many of these molecules have advanced into the clinic, the known pharmacological and human safety profiles of these compounds will accelerate their preclinical and clinical evaluation for COVID-19 therapy. Among them, Z LVG CHN2 exerted its anti-COVID-19 effects by inhibiting the SARS-COV-2 main protease (Fig. 19).
Fig. 19. Chemical structure of Z LVG CHN2.
5. Summary and perspective
The COVID-19 pandemic caused by the highly transmissible SARS-CoV-2 has become the world's largest public health crisis. The pandemic is still spreading at an alarming rate and has caused fatal deaths, especially for the elderly and immunocompromised patients. According to BBC reports, countries such as the United Kingdom and China have found mutant strains that spread faster. Although some people have begun to be vaccinated with vaccines developed by China and the United States, the immunization efficiency is still not high enough. Therefore, the development of antiviral drugs for the treatment of COVID-19 is still urgent. In our current pandemic situation, effective antiviral treatments may have a major impact on reducing morbidity and mortality. At the same time, antiviral drugs can also be used as cheap prevention methods. Computer-aided drug design, especially artificial intelligence, is rapidly emerging. As the accuracy of artificial intelligence in assisted drug design improves, it will provide more help to the development of SARS-COV inhibitors. As a highly conservative SARS-COV target, Mpro should be continuously of concern and studied. The research on Mpro inhibitors is of great significance both for the present and the future development of preventive drugs. In addition, we believe that strengthening the interdisciplinary integration of computer, chemistry and biomedicine will accelerate the development of Mpro inhibitors, and our research team is also working towards this goal.
This review summarizes many representative works in the development of SARS-COV main protease drugs, and focuses on the structure of many small lead compounds and the active sites of action. We hope that this review will provide ideas and help for the future development of anti-COVID-19 and other coronavirus drugs, and help develop a broad-spectrum antiviral drug for the treatment of coronavirus diseases as soon as possible.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
We thank the Taishan Scholars Project of Shandong Province (tsqn202103108 to J. Y.) and Six Talent Peaks Project in Jiangsu Province (131219631004 to J. Y.) for financial support.
Notes and references
- Wu F. Zhao S. Yu B. Chen Y. M. Wang W. Song Z. G. Hu Y. Tao Z. W. Tian J. H. Pei Y. Y. Yuan M. L. Zhang Y. L. Dai F. H. Liu Y. Wang Q. M. Zheng J. J. Xu L. Holmes E. C. Zhang Y. Z. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. Yang X. L. Wang X. G. Hu B. Zhang L. Zhang W. Si H. R. Zhu Y. Li B. Huang C. L. Chen H. D. Chen J. Luo Y. Guo H. Jiang R. D. Liu M. Q. Chen Y. Shen X. R. Wang X. Zheng X. S. Zhao K. Chen Q. J. Deng F. Liu L. L. Yan B. Zhan F. X. Wang Y. Y. Xiao G. F. Shi Z. L. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of V The species severe acute respiratory syndrome- related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewnard J. A. Lancet. 2018;392:189–190. doi: 10.1016/S0140-6736(18)31443-0. [DOI] [PubMed] [Google Scholar]
- Weaver S. C. Costa F. Garcia-Blanco M. A. Ko A. I. Ribeiro G. S. Saade G. Shi P.-Y. Vasilakis N. Antiviral Res. 2016;130:69–80. doi: 10.1016/j.antiviral.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni D. D. Tosh C. Venkatesh G. Senthil K. D. Indian J. Virol. 2013;24:398–408. doi: 10.1007/s13337-013-0171-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. M. Fraser C. Ghani A. C. Donnelly C. A. Riley S. Ferguson N. M. Leung G. M. Lam T. H. Hedley A. J. Philos. Trans. R. Soc., B. 2004;359:1091–1105. doi: 10.1098/rstb.2004.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oboho I. K. Tomczyk S. M. Al-Asmari A. M. Banjar A. A. Al-Mugti H. Aloraini M. S. Alkhaldi K. Z. Almohammadi E. L. Alraddadi B. M. Gerber S. I. Swerdlow D. L. Watson J. T. Madani T. A. N. Engl. J. Med. 2015;372:846–854. doi: 10.1056/NEJMoa1408636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson N. P. Mueller J. Bull. Hist. Med. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- Taubenberger J. K. Morens D. M. Emerging Infect. Dis. 2006;12:15–22. doi: 10.3201/eid1201.050979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z. Du X. Xu Y. Deng Y. Liu M. Zhao Y. Zhang B. Li X. Zhang L. Peng C. Duan Y. Yu J. Wang L. Yang K. Liu F. Jiang R. Yang X. You T. Liu X. Yang X. Bai F. Liu H. Liu X. Guddat L. W. Xu W. Xiao G. Qin C. Shi Z. Jiang H. Rao Z. Yang H. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Liu X. Wang X. J. J. Genet. Genomics. 2020;47:119. doi: 10.1016/j.jgg.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Lin D. Sun X. Curth U. Drosten C. Sauerhering L. Becker S. Rox K. Hilgenfeld R. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. Zhou Q. Li Y. Garner L. V. Watkins S. P. Carter L. J. Smoot J. Gregg A. C. Daniels A. D. Jervey S. Albaiu D. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M. Kleine-Weber H. Schroeder S. Kruger N. Herrler T. Erichsen S. Schiergens T. S. Herrler G. Wu N. H. Nitsche A. Muller M. A. Drosten C. Pohlmann S. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. Luo S. Libby P. Shi G. P. Pharmacol. Ther. 2020;213:107587. doi: 10.1016/j.pharmthera.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel V. Ivanov K. A. Putics Á. Hertzig T. Schelle B. Bayer S. Weißbrich B. Snijder E. J. Rabenau H. Doerr H. W. Gorbalenya A. E. Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Anand K. Ziebuhr J Fau - Wadhwani P. Wadhwani P Fau - Mesters J. R. Mesters Jr Fau - Hilgenfeld R. Hilgenfeld R. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Marra M. A. Jones J. M. Astell C. R. Holt R. A. Brooks- Wilson A. Butterfield Y. S. N. Khattra J. Asano J. K. Barber S. A. Chan S. Y. Cloutier A. Coughlin S. M. Freeman D. Girn N. Griffith O. L. Leach S. R. Mayo M. McDonald H. Montgomery S. B. Pandoh P. K. Petrescu A. S. Robertson A. G. Schein J. E. Siddiqui A. Smailus D. E. Stott J. M. Yang G. S. Plummer F. Andonov A. Artsob H. Bastien N. Bernard K. Booth T. F. Bowness D. Czub M. Drebot M. Fernando L. Flick R. Garbutt M. Gray M. Grolla A. Jones S. Feldmann H. Meyers A. Kabani A. Li Y. Normand S. Stroher U. Tipples G. A. Tyler S. Vogrig R. Ward D. Watson B. Brunham R. C. Krajden M. Petric M. Skowronski D. M. Upton C. Roper R. L. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- Rota P. A. Oberste M. S. Monroe S. S. Nix W. A. Campagnoli R. Icenogle J. P. Penaranda S. Bankamp B. Maher K. Chen M.-H. Tong S. Tamin A. Lowe L. Frace M. DeRisi J. L. Chen Q. Wang D. Erdman D. D. Peret T. C. T. Burns C. Ksiazek T. G. Rollin P. E. Sanchez A. Liffick S. Holloway B. Limor J. McCaustland K. Olsen-Rasmussen M. Fouchier R. Gunther S. Osterhaus A. D. M. E. Drosten C. Pallansch M. A. Anderson L. J. Bellini W. J. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- Harcourt B. H. Jukneliene D. Kanjanahaluethai A. Bechill J. Severson K. M. Smith C. M. Rota P. A. Baker S. C. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim K. P. Ng L. F. P. Liu D. X. J. Virol. 2000;74:1674–1685. doi: 10.1128/jvi.74.4.1674-1685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S. G. Wang N. Chen Z. Tseng M. Barretto N. Lin R. Peters C. J. Tseng C. T. Baker S. C. Li K. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos Y. M. St John S. E. Mesecar A. D. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Yang M. Ding Y. Liu Y. Lou Z. Zhou Z. Sun L. Mo L. Ye S. Pang H. Gao G. F. Anand K. Bartlam M. Hilgenfeld R. Rao Z. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A. Wang Y. Zeng C. Huang X. Xu S. Su C. Wang M. Chen Y. Guo D. Virus Res. 2015;208:56–65. doi: 10.1016/j.virusres.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L. Lin D. Kusov Y. Nian Y. Ma Q. Wang J. von Brunn A. Leyssen P. Lanko K. Neyts J. de Wilde A. Snijder E. Liu H. Hilgenfeld R. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Thiel V. Ivanov K. A. Putics A. Hertzig T. Schelle B. Bayer S. Weissbrich B. Snijder E. J. Rabenau H. Doerr H. W. Gorbalenya A. E. Ziebuhr J. J. Gen. Virol. 2003;84:2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Chen X. Chou C. Y. Chang G. G. Antiviral Chem. Chemother. 2009;19:151–156. doi: 10.1177/095632020901900402. [DOI] [PubMed] [Google Scholar]
- Park J. Y. Ko J. A. Kim D. W. Kim Y. M. Kwon H. J. Jeong H. J. Kim C. Y. Park K. H. Lee W. S. Ryu Y. B. J. Enzyme Inhib. Med. Chem. 2016;31:23–30. doi: 10.3109/14756366.2014.1003215. [DOI] [PubMed] [Google Scholar]
- Zhou Y. Vedantham P. Lu K. Agudelo J. Carrion, Jr. R. Nunneley J. W. Barnard D. Pohlmann S. McKerrow J. H. Renslo A. R. Simmons G. Antiviral Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V. Jung Y. S. Liang P. H. Expert Opin. Ther. Pat. 2013;23:1337–1348. doi: 10.1517/13543776.2013.823159. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K. Xi K. Ratia K. Santarsiero B. D. Fu W. Harcourt B. H. Rota P. A. Baker S. C. Johnson M. E. Mesecar A. D. J. Med. Chem. 2005;48:6767–6771. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- Yang S. Chen S. J. Hsu M. F. Wu J. D. Tseng C. T. Liu Y. F. Chen H. C. Kuo C. W. Wu C. S. Chang L. W. Chen W. C. Liao S. Y. Chang T. Y. Hung H. H. Shr H. L. Liu C. Y. Huang Y. A. Chang L. Y. Hsu J. C. Peters C. J. Wang A. H. Hsu M. C. J. Med. Chem. 2006;49:4971–4980. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- Regnier T. Sarma D. Hidaka K. Bacha U. Freire E. Hayashi Y. Kiso Y. Bioorg. Med. Chem. Lett. 2009;19:2722–2727. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno S. Thanigaimalai P. Yamamoto T. Nakada K. Kakiuchi R. Takayama K. Yamazaki Y. Yakushiji F. Akaji K. Kiso Y. Kawasaki Y. Chen S. E. Freire E. Hayashi Y. Bioorg. Med. Chem. 2013;21:412–424. doi: 10.1016/j.bmc.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanigaimalai P. Konno S. Yamamoto T. Koiwai Y. Taguchi A. Takayama K. Yakushiji F. Akaji K. Kiso Y. Kawasaki Y. Chen S. E. Naser-Tavakolian A. Schon A. Freire E. Hayashi Y. Eur. J. Med. Chem. 2013;65:436–447. doi: 10.1016/j.ejmech.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanigaimalai P. Konno S. Yamamoto T. Koiwai Y. Taguchi A. Takayama K. Yakushiji F. Akaji K. Chen S. E. Naser-Tavakolian A. Schon A. Freire E. Hayashi Y. Eur. J. Med. Chem. 2013;68:372–384. doi: 10.1016/j.ejmech.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Pettersson H. I. Huitema C. Niu C. Yin J. James M. N. Eltis L. D. Vederas J. C. J. Med. Chem. 2007;50:1850–1864. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- Niu C. Yin J. Zhang J. Vederas J. C. James M. N. Bioorg. Med. Chem. 2008;16:293–302. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. P. Pettersson H. I. Zhang J. Aull K. D. Fortin P. D. Huitema C. Eltis L. D. Parrish J. C. James M. N. Wishart D. S. Vederas J. C. J. Med. Chem. 2004;47:6113–6116. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K. Gong G. Grum-Tokars V. Mulhearn D. C. Baker S. C. Coughlin M. Prabhakar B. S. Sleeman K. Johnson M. E. Mesecar A. D. Bioorg. Med. Chem. Lett. 2008;18:5684–5688. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P. Fan K. Chen H. Ma L. Huang C. Tan L. Xi D. Li C. Liu Y. Cao A. Lai L. Biochem. Biophys. Res. Commun. 2006;339:865–872. doi: 10.1016/j.bbrc.2005.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J. Grum-Tokars V. Zhou Y. Turlington M. Saldanha S. A. Chase P. Eggler A. Dawson E. S. Baez-Santos Y. M. Tomar S. Mielech A. M. Baker S. C. Lindsley C. W. Hodder P. Mesecar A. Stauffer S. R. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y. Mandadapu S. R. Groutas W. C. Chang K. O. Antiviral Res. 2013;97:161–168. doi: 10.1016/j.antiviral.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L. George S. Schmidt M. F. Al-Gharabli S. I. Rademann J. Hilgenfeld R. Antiviral Res. 2011;92:204–212. doi: 10.1016/j.antiviral.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz D. H. Choe Y. Hansell E. Chen Y. T. McDowell M. Jonsson C. B. Roush W. R. McKerrow J. Craik C. S. Biochemistry. 2007;46:8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- Zhang H. Z. Zhang H. Kemnitzer W. Tseng B. Cinatl, Jr. J. Michaelis M. Doerr H. W. Cai S. X. J. Med. Chem. 2006;49:1198–1201. doi: 10.1021/jm0507678. [DOI] [PubMed] [Google Scholar]
- Sydnes M. O. Hayashi Y. Sharma V. K. Hamada T. Bacha U. Barrila J. Freire E. Kiso Y. Tetrahedron. 2006;62:8601–8609. doi: 10.1016/j.tet.2006.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A. Chan J. F. Azhar E. I. Hui D. S. Yuen K. Y. Nat. Rev. Drug Discovery. 2016;15:327–347. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T. Meenakshisundaram S. Manickam M. Drug Discovery Today. 2020;25:668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong M. Su H. Zhao W. Xie H. Shao Q. Xu Y. Med. Res. Rev. 2021 doi: 10.1002/med.21783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori S.-I. Higshi-Kuwata N. Raghavaiah J. Das D. Bulut H. Davis D. A. Takamatsu Y. Matsuda K. Takamune N. Kishimoto N. Okamura T. Misumi S. Yarchoan R. Maeda K. Ghosh A. K. Mitsuya H. MBio. 2020;11:e01833-20. doi: 10.1128/mBio.01833-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C. Sacco M. D. Hurst B. Townsend J. A. Hu Y. Szeto T. Zhang X. Tarbet B. Marty M. T. Chen Y. Wang J. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W. Zhang B. Jiang X.-M. Su H. Li J. Zhao Y. Xie X. Jin Z. Peng J. Liu F. Li C. Li Y. Bai F. Wang H. Cheng X. Cen X. Hu S. Yang X. Wang J. Liu X. Xiao G. Jiang H. Rao Z. Zhang L.-K. Xu Y. Yang H. Liu H. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Filho O. A. J. Braz. Chem. Soc. 2020;31:2638–2643. [Google Scholar]
- Chen H. Zhang Z. Wang L. Huang Z. Gong F. Li X. Chen Y. Wu J. J. medRxiv. 2020 doi: 10.1101/2020.03.22.20034041. [DOI] [Google Scholar]
- Yang H. Xie W. Xue X. Yang K. Ma J. Liang W. Zhao Q. Zhou Z. Pei D. Ziebuhr J. Hilgenfeld R. Yuen K. Y. Wong L. Gao G. Chen S. Chen Z. Ma D. Bartlam M. Rao Z. PLoS Biol. 2005;3:e324. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z. Zhao Y. Sun Y. Zhang B. Wang H. Wu Y. Zhu Y. Zhu C. Hu T. Du X. Duan Y. Yu J. Yang X. Yang X. Yang K. Liu X. Guddat L. W. Xiao G. Zhang L. Yang H. Rao Z. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Ma C. Hu Y. Townsend J. A. Lagarias P. I. Marty M. T. Kolocouris A. Wang J. ACS Pharmacol. Transl. Sci. 2020;3:1265–1277. doi: 10.1021/acsptsci.0c00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W. Xu M. Chen C. Z. Guo H. Shen M. Hu X. Shinn P. Klumpp-Thomas C. Michael S. G. Zheng W. ACS Pharmacol. Transl. Sci. 2020;3:1008–1016. doi: 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H. Yao S. Zhao W. Li M. Liu J. Shang W. Xie H. Ke C. Gao M. Yu K. Liu H. Shen J. Tang W. Zhang L. Zuo J. Jiang H. Bai F. Wu Y. Ye Y. Xu Y. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J. Li Y. S. Zeng R. Liu F. L. Luo R. H. Huang C. Wang Y. F. Zhang J. Quan B. X. Shen C. J. Mao X. Liu X. L. Sun W. N. Yang W. Ni X. C. Wang K. Xu L. Duan Z. L. Zou Q. C. Zhang H. L. Qu W. Long Y. H. P. Li M. H. Yang R. C. Liu X. L. You J. Zhou Y. L. Yao R. Li W. P. Liu J. M. Chen P. Liu Y. Lin G. F. Yang X. Zou J. Li L. L. Hu Y. G. Lu G. W. Li W. M. Wei Y. Q. Zheng Y. T. Lei J. Yang S. Y. Science. 2021;37:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori S. Higashi-Kuwata N. Hayashi H. Allu S. R. Raghavaiah J. Bulut H. Das D. Anson B. J. Lendy E. K. Takamatsu Y. Takamune N. Kishimoto N. Murayama K. Hasegawa K. Li M. Davis D. A. Kodama E. N. Yarchoan R. Wlodawer A. Misumi S. Mesecar A. D. Ghosh A. K. Mitsuya H. Nat. Commun. 2021;12:668. doi: 10.1038/s41467-021-20900-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R. L. Kania R. S. Brothers M. A. Davies J. F. Ferre R. A. Gajiwala K. S. He M. Hogan R. J. Kozminski K. Li L. Y. Lockner J. W. Lou J. Marra M. T. Mitchell L. J. Murray B. W. Nieman J. A. Noell S. Planken S. P. Rowe T. Ryan K. Smith G. J. Solowiej J. E. Steppan C. M. Taggart B. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R. Kumar V. Lee K. W. A. Comput. Biol. Med. 2020:104186. doi: 10.1016/j.compbiomed.2020.104186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson B. J., Chapman M. E., Lendy E. K., Pshenychnyi S., Richard T., Satchell K. J. and Mesecar A. D., Broad-spectrum Inhibition of Coronavirus Main and Papainlike Proteases by HCV Drugs, 2020
- Beck B. R. Shin B. Choi Y. Park S. Kang K. Comput. Struct. Biotechnol. J. 2020;18:784–790. doi: 10.1016/j.csbj.2020.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z. Peng C. Shi Y. Zhu Z. Mu K. Wang X. Zhu W. bioRxiv. 2020 doi: 10.1101/2020.01.27.921627. [DOI] [Google Scholar]
- Riva L. Yuan S. Yin X. Martin-Sancho L. Matsunaga N. Pache L. Burgstaller-Muehlbacher S. De Jesus P. D. Teriete P. Hull M. V. Chang M. W. Chan J. F.-W. Cao J. Poon V. K.-M. Herbert K. M. Cheng K. Nguyen T.-T. H. Rubanov A. Pu Y. Nguyen C. Choi A. Rathnasinghe R. Schotsaert M. Miorin L. Dejosez M. Zwaka T. P. Sit K.-Y. Martinez-Sobrido L. Liu W.-C. White K. M. Chapman M. E. Lendy E. K. Glynne R. J. Albrecht R. Ruppin E. Mesecar A. D. Johnson J. R. Benner C. Sun R. Schultz P. G. Su A. I. García-Sastre A. Chatterjee A. K. Yuen K.-Y. Chanda S. K. Nature. 2020;586:113–119. doi: 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L. Yuan S. Yin X. Martin-Sancho L. Matsunaga N. Burgstaller S. Pache L. De Jesus P. Hull M. V. Chang M. bioRxiv. 2020 doi: 10.1101/2020.04.16. [DOI] [Google Scholar]