

Abstract
Optimal ex vivo expansion protocols of tumor specific T-cells followed by adoptive cell therapy must yield T-cells able to home to tumors and effectively kill them. Our previous study demonstrated ex vivo activation in the presence of IL-12 induced optimal CD8+ T-cell expansion and melanoma regression, however, adverse side effects including autoimmunity can occur. This may be due to transfer of high avidity self-specific T-cells. In this study, we compared mouse low and high avidity T-cells targeting the tumor antigen tyrosinase related protein-2 (TRP-2). Not surprisingly, high avidity T-cells provide superior tumor control, yet low avidity T-cells can promote tumor regression. The addition of IL-12 during in vitro expansion boosts low avidity T-cell responsiveness, tumor regression, and prevents T-cell exhaustion. Herein we demonstrate that IL-12 primed T-cells are resistant to PD-1/PD-L1 mediated suppression and retain effector function. Importantly, IL-12 pre-conditioning prevented exhaustion as LAG-3, PD-1, and TOX were decreased, while simultaneously increasing KLRG1. Using intravital imaging, we also determined that high avidity T-cells have sustained contacts with intratumoral DCs and tumor targets compared to low avidity T-cells. However, with antigen overexpression this defect is overcome and low avidity T-cells control tumor growth. Taken together, these data illustrate that low avidity T-cells can be therapeutically beneficial if co-cultured with IL-12 cytokine during in vitro expansion, and highly effective in vivo if antigen is not limiting. Clinically, low avidity T-cells provide a safer alternative to high avidity TCR engineered T-cells as IL-12 primed low avidity T-cells cause less autoimmune vitiligo.
Introduction
Immune checkpoint inhibition targeting Programmed Death-1 (PD-1) and Programmed Death Ligand-1 (PD-L1) has revolutionized cancer immunotherapy 1. This approach has been highly successful, with an overall five year survival rate of 34.2% in melanoma patients 2. However the majority of patients still do not significantly benefit from targeting this pathway. Additionally, checkpoint inhibition is not able to prevent or reverse exhaustion of all T-cells within the tumor nor are all T-cells in the tumor highly cytotoxic 3, 4. Finally, severe adverse events including autoimmunity can result following checkpoint inhibition 5, identifying a critical need for alternative approaches.
Adoptive cell therapy has been a highly sought after cancer immunotherapy approach for many years 6. The adoptive transfer of tumor specific T-cells has shown significant clinical response rates 7. In fact, for melanoma patients there has been a large effort to isolate melanoma specific T-cells from tumors for expansion with IL-2 and reinfusion into patients 8. This approach has yielded some success (median survival 53.5 versus 3.5 months for melanoma without cellular therapy), but is not without off-target side effects and long term T-cell exhaustion 9, 10. Thus far, researchers have focused on the isolation and optimization of high avidity T-cell receptors (TCRs) for use in adoptive T-cell therapy 11. The possibility of these high avidity T-cells recognizing self-antigens and eliciting pathology has been demonstrated in the MAGE A3 clinical trials, where up to 40% of patients died from cardiac and brain pathology 9, 12. Therefore, the use of low-to-medium avidity TCRs may yield a safer, but no less efficacious clinical tumor therapy with less autoimmunity 13–15. Many groups, including ours, have investigated the impact of IL-12 priming on anti-tumor effects 16–22. In addition, alternative cytokines or combinations may be preferred as we and others have shown that IL-2 priming alone yields poor effector function from CD8+ T-cells resulting in an exhausted or anergic phenotype 17, 23. To overcome this poor effector function, we previously reported IL-2 plus IL-12 co-culture was superior to limit tumor growth compared to IL-2 plus type 1 IFN during initial T-cell priming 22. Using the high avidity ovalbumin specific, OT-1 CD8+ T-cell model and a B16 Ova-expressing melanoma tumor cell line, we reported that stimulation with IL-2 plus IL-12 or type 1 interferon (IFN) enhanced adoptively transferred T-cell survival and development of optimal effector functions 22. We determined that IL-12 differentially programmed CD8+ T-cells to express less PD-1, compared to IL-2 alone or IL-2 plus type 1 interferon, allowing IL-12 primed cells superior effector function including long term tumor control 22.
While previous reports, including our own, illustrated the significant benefit of adding IL-12 to the in vitro co-culture conditions, several questions remained. Thus, the objective of this current study was to address these questions using a physiologically relevant tumor antigen normally expressed in healthy tissue, and compare both a high and low avidity T-cell response and exhaustion profile in the context of IL-2 plus IL-12 co-culture. We also extended our analysis to evaluate if there was an additive benefit of checkpoint blockade, or enhanced tumor clearance with differential target antigen expression following IL-12 pre-conditioning, and determined the extent of T-cell exhaustion. Finally, we evaluated the risk for autoimmunity using both high and low avidity T-cells for adoptive T cell immunotherapy. We hypothesized that low avidity T-cells if optimally activated and expanded, could provide durable clinical responses, escape exhaustion, and limit immune related adverse events (IRAE) including autoimmunity. To accomplish these goals, we used a model system to more closely approximate clinical trials investigating self-antigen specific T-cells using TCR transgenic mice that produce CD8+ T-cells of either low or high functional avidity: TRP2 high avidity (TRP2high) and the TRP2 low avidity (TRP2low) mice 24, 25. These TCR transgenic mice produce CD8+ T-cells that are specific for tyrosinase related protein 2 (TRP2), ubiquitously expressed in both healthy melanocytes and B16F10 melanoma cells. We hypothesized that IL-12 cytokine priming would render the lower avidity T-cells resistant to exhaustion, with minimal off-tissue pathology. In the current study, we determined that IL-2 plus IL-12 priming enhanced low avidity CD8+ T-cell proliferation, inflammatory cytokine production and tumor destruction, whereas IL-2 pretreatment alone did not. Importantly, IL-12 cytokine priming decreased the levels of the exhuastion transcription factor TOX and exhaustion surface markers LAG-3 and PD-1 in both TRP2low and TRP2high CD8+ T-cells. Using intravital microscopy, we showed that high avidity T-cells made stable contacts with intratumoral DCs following co-culture with IL-12 plus IL-2. Stable interactions of low avidity T-cells required high tumor antigen levels. When TRP2 was overexpressed in vivo, and IL-12 plus IL-2 were used in vitro, low avidity T-cells had improved activation, trafficking, stable contacts with tumor targets and exerted tumor control to similar levels as high avidity T-cells. Finally, PD-1 blockade therapy did not have an added benefit following adoptive therapy of CD8+ T-cells expanded with IL-2 plus IL-12, suggesting this approach would be applicable for patients failing checkpoint therapy and could avoid potential autoimmune sequelae. Taken together, low avidity self-specific CD8+ T-cells stimulated in vitro with IL-2 plus IL-12 can target and destroy tumors expressing sufficient antigen and are resistant to tumor induced exhaustion, thus maintaining long term effector function while limiting the induction of autoimmunity.
Materials and Methods:
Mice
C57BL/6 TRP2low and TRP2high TCR transgenic mice were obtained from Dr. Arthur Hurwitz [13, 14]. B6(D2)-Tg(CAG-Brainbow1.0)2Eggn/J (Stock: 021011) were crossed to C57BL/6 TRP2low, C57BL/6-Tg(UBC-GFP)30Scha/J (Stock: 004353) were crossed to TRP2high C57BL/6 mice and CD11cVenus B6.Cg-Tg(Itgax-Venus)1Mnz/J (Stock: 008829) were purchased from The Jackson Laboratory. Experiments were conducted under specific pathogen-free conditions and performed in compliance with relevant laws and guidelines, and with approval of the Institutional Animal Care and Use Committee at the University of Minnesota.
Tumor cell lines
B16F10 melanoma cells (RRID:CVCL_0159, ATCC CRL-6475) were maintained in complete RPMI 1640 medium (10% FBS + 1X Glutamax™, 1X MEM NEAA, 1 mM Sodium Pyruvate, 100 units/mL Penicillin, 100 μg/mL Streptomycin and 55 μM 2-ME). B16F10 cells were obtained directly from ATCC and passaged less than 10 times, and were Mycoplasma negative. The TRP2-teal fluorescent protein (TFP) vector was generated by sequential cloning of DCT-pCMV6-ORF vector (Sino Biological) and pCMV6-TFP fusion vector creating a TRP2-mTFP expression vector. This construct was introduced to B16F10 parental cells by lipofectamine transfection and subsequent dilutional cloning, puromycin selection and FACS sorting. Clone 2C4 was isolated by flow cytometry based on high levels of mTFP expression. A PD-L1-pCMV6-ORF (Sino Biological) was introduced to B16F10 parental cells by lipofectamine and clone 11G2 was isolated after dilutional cloning and hygromycin selection.
In vitro stimulation and adoptive transfer of TRP2high and TRP2low T-cells
Naive TRP2high or TRP2low CD8+ T-cells were purified from spleen and lymph nodes by negative selection following the manufacturer’s protocol (StemCell) (Supplemental Fig. C.) 0.5-2x106 enriched cells were stimulated in flat-bottom 12-24 well plates with anti-CD3 (Clone 17A3, 50 μg/mL, BioXcell) and rB7-1/Fc chimeric protein (0.8μg/mL, R&D Systems) immobilized on the surface (coated 12 hours before adding cells) 26 in the presence of 5 U/ml IL-2 (R&D Systems) with or without 10 ng/mL murine rIL-12 (R&D Systems), as these cytokine concentrations stimulated optimal development of effector function (Supplemental Fig. D). After 72 hours, 1x106 – 5x106 cells were harvested and intravenously transferred to recipient mice.
Tumor growth and flow cytometric analysis of T-cells
Mice were injected s.c. with 2.5 × 105 B16F10 cells in the flank, and tumor growth was monitored by determining the area with calipers. Tumor-bearing mice received 1x106 – 5x106 activated TRP2-specific T-cells by adoptive transfer on day 10 post tumor injection. Mice were sacrificed at the indicated times, spleens and tumors were isolated and homogenized, and lymphocytes from tumor homogenates were enriched on Ficoll gradients as previously described 22. Samples were stained with Ghost viability dye (Tonbo Biosciences), and antibodies against CD11b (clone M1/70, Thermo Fisher Scientific), B220 (clone RA3-6B2, Thermo Fisher Scientific), CD19 (clone 1D3, Thermo Fisher Scientific), CD4 (GK1.5, Tonbo Biosciences), Thy1.2 (clone 53-2.1, BD Biosciences) CD8α (53–6.7), PD-1 (J43) (Thermo Fisher Scientific), LAG-3 (clone C9B7W), KLRG1 (clone 2F1), CD44 (clone IM7), CD127 (clone A7R34) (Biolegend), fixed and permeabilized with subsequent BD Cytofix/Cytoperm™ (BD Biosciences) (2 min at RT, to preserve GFP and tdtomato fluorescence) and FOXP3 Fix/Perm Kit (15 min at 4°C, Tonbo Biosciences) and stained with antibodies against TOX (clone TXRX10, Thermo Fischer) and T-bet (clone 4B10, Biolegend). In separate experiments, tumor and spleen homogenates were incubated for 4 h on anti-CD3 coated plates (50μg/mL) in the presence of Golgi block stained with antibodies against CD8α (53–6.7), PD-1 (J43) (Thermo Fisher Scientific), LAG-3 (clone C9B7W), KLRG1 (clone 2F1), CD44 (clone IM7), CD127 (clone A7R34) (Biolegend), fixed and permeabilized with BD Cytofix/Cytoperm™ (BD Biosciences) (to preserve GFP and tdtomato fluorescence) and subsequent FOXP3 Fix/Perm Kit stained for IFN-γ (XMG1.2, Tonbo), TNFα (clone MP6-XT22), and Ki67 (clone 16A8)(Biolegend) staining was performed. Transferred CD8+ T-cells were identified as live, CD8+, GFP+ (TRP2high) or tdTomato+ (TRP2low). Cell counts were obtained using PKH26 reference microbeads (Sigma-Aldrich). Data were acquired on FACS Fortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software (version 10).
Anti-PD-1 injections
Anti—PD-1 mAb (clone J43) was purchased from BioXcell. Mice received 200 μg (i.p.) anti—PD-1 or PBS control on D1 post TRP2 cells transferred, and then every other day for a total of 3 injections.
Cytotoxicity assay
5x104 B16F10 or 2C4 tumor cells were plated in 12 well tissue culture treated plates overnight. 5x105 activated TRP2low or TRP2high T-cells (5:1 ratio) were added to the wells with tumor targets. 12 hours later, tumor cells and T-cells were removed from the plates and stained with antibodies directed at PD-L1 (MIH5, Thermo Fisher Scientific), CD44 (IM7), CD8α (53–6.7), Thy1.2 (53-2.1) as well as a Live/Dead stain (Tonbo Biosciences). Cells were fixed and permeabilized with the Tonbo FOXP3 staining kit (TNB-0607) and stained intracellularly for active Caspase-3 (BD Biosciences). Percentage of tumor cells that were killed was determined as the percentage of active Caspase-3 positive gated events. Data were collected on a BD Fortessa and analyzed using FlowJo software (version 10).
2-Photon tumor imaging
Large tumors were harvested on day 3 post T-cell transfer into warm RPMI 1640 containing 10% FBS and mounted on a coverslip. Movies were acquired using an MP5 two-photon microscope TCS (Leica) equipped with a Mai Tai HP DeepSee lasers (SpectraPhysics), an 8,000-Hz resonant scanner, a 25× 0.95 NA objective, two non-descanned detectors and two HyD detectors. During imaging, continuous oxygenated DMEM high glucose media lacking phenol red (Hyclone) was exchanged in the 37 degree heated chamber containing the sample. Tissue was excited at 930nm and multiple fluorophores were imaged using the custom dichroic mirrors with the following collection filters: mTFP and second harmonic generation (435-485nm), GFP (500-520nm), Venus (520-555nm), and tdTomato (565-605nm). Data were spectrally unmixed with LASF software (Lecia version 3.1.0) and processed with Imaris software (Bitplane version 9.2.1) [16]. TRP2high and TRP2low cell motility were tracked by Imaris Spots and manually confirmed. mTFP tumors and Venus CD11c dendritic cells were identified by Imaris surfaces and TRP2 contacts were determined by distance transformations of the surfaces. For each individual animal, we collected 30 min movies of at least two different locations within the tumor separated by at least 1 mm. Within each of these macroscopic locations, we imaged 3-4 positions concurrently. These individual positions were separately analyzed and averaged to identify typical cell behavior. Data presented in the figures are representative for the typical cell behavior observed across all mice for each condition and cell transfer situation. Each dot in the motility measurement graphs represents one cell.
Autoimmune Vitiligo
Autoimmune vitiligo was assessed using two distinct approaches. In the first protocol, OT-1xUBC-GFP or WT B6 mice were given 5x106 TRP2highGFP or TRP2lowtdtomato T-cells primed in vitro with IL-2 plus IL-12, and seven days later given 200μg poly I:C subcutaneously in the flank. Fourteen days after transfer, mice were given 50 μg of TRP2 peptide subcutaneously in the flank with 200μg polyI:C. Mice were monitored weekly for vitiligo induction. In the second protocol, OT-1xUBC-GFP or WT B6 mice were given 5x106 TRP2highGFP or TRP2lowtdtomato T-cells primed in vitro with IL-2 plus IL-12 and the mice were monitored weekly for vitiligo.
Statistical significance
Statistical analyses (paired or unpaired Student’s t-test; One-Way ANOVA with Tukey post-hoc test) were performed in Prism 8.0.1 (GraphPad). P-values lower than 0.05 were considered significant. Graphs show mean ± standard error of the mean.
Results
IL-2 plus IL-12 co-culture during initial T-cell priming improves high and low avidity TCR transgenic CD8+ T-cell inflammatory cytokine production and tumor killing
Previous studies have demonstrated poor CD8+ T-cell effector function and tumor clearance after adoptive transfer following in vitro priming in the presence of IL-2 alone 27. Our previous study used T-cell receptors (TCRs) specific for the model antigen ovalbumin (Ova) expressed in tumor cells and determined that the addition of IL-12 together with IL-2 during the in vitro priming phase overcame the defect and provided superior anti-tumor effects 22. However, previous studies did not determine if IL-12 differentially impacted low or high avidity T-cells, and whether IL-12 primed T-cells could eliminate tumors expressing self-antigens. Thus, in the current study we evaluated T-cells of high and low TCR avidity targeting a protein naturally expressed in both healthy melanocytes and in B16F10 melanoma cells called tyrosinase related protein 2 (TRP2). To test this, we harvested T-cells from TCR transgenic mice that produce CD8+ T-cells of either low (TRP2low) or high (TRP2high) avidity 24, 25. TRP2high T-cells displayed approximately an 8-fold higher sensitivity to antigen when cultured with IL-2 (Supplemental Fig. 1A). This increased antigen sensitivity was not due to increased TCR expression from TRP2high T-cells as TCRbeta levels were similar between TRP2low and TRP2high T-cells (Supplemental Fig. 1B, left). We first investigated whether IL-2 or IL-2 plus IL-12 cytokine treatment differentially impacted TRP2high or TRP2low TCR CD8+ T-cells. To test this, enriched naïve CD8+ T-cells (>97% pure, Supplemental Fig. 1B, right) were expanded in vitro with anti-CD3 and recombinant B7-1 in the presence of IL-2, with or without IL-12 co-culture. We determined that IL-12 significantly increased CD44 expression, albeit only a slight change was noted (Fig. 1A). The expression of TH1 transcription factor T-bet increased significantly with IL-2 plus IL-12 co-culture in both high and low avidity T-cells (Fig. 1B). The high avidity cells produced more IFNγ when cultured with IL-2 alone compared to low avidity T-cells (Fig. 1C, Supplmental Fig. 1C). However, IL-2 plus IL-12 cytokine co-culture during priming enhanced IFNγ production in both low avidity T-cells and high avidity T-cells compared to IL-2 alone (Fig. 1C). As we previously reported, co-culture with IL-2 plus IL-12 did not substantially increase the expression of the inhibitory receptor PD-1 on high avidity CD8+ T-cells 22 (Fig. 1D). However there was an increase in PD-1 expression in low avidity T-cells following IL-2 plus IL-12 co-culture compared to IL-2 alone (Fig. 1D). The low avidity T-cells had less PD-1 compared to the high avidity T-cells with IL-2 plus IL-12 directly post in vitro activation (Fig. 1D). Next, we investigated the cytolytic potential of TRP2 specific T-cells against melanoma (B16F10) target cells, as the B16F10 cell line endogenously expresses the TRP2 melanocyte target antigen. We investigated direct cytolytic potential of TRP2low and TRP2high CD8+ T-cells in vitro by developing a killing assay to simultaneously analyze both the T-cells and the target tumor cells undergoing active apoptosis. While B16F10 cells die in culture over time, only a small (~2.3%) percentage of cells express active caspase-3 (Supplemental Fig. 1D). This indicates that B16F10 cells did not die by apoptosis when cultured alone, but rather by another form of cellular death such as necrosis. When T-cells were added to the culture, 1.7±0.6 % of target cells were killed by TRP2low T-cells, while TRP2high T-cells killed tumor cells killed at a significantly higher rate of 4.8±2% (Fig. 1E). IL-2 plus IL-12 co-culture during priming increased TRP2low T-cells ability to kill tumor targets (6±2.6%), making them more comparable to TRP2high T-cells (Fig. 1E). IL-12 co-culture also significantly improved the TRP2high T-cells ability to kill targets in vitro (8.3±3.2%) (Fig. 1E). We next tested the ability of antigen specific T-cells to kill tumor targets in vivo after adoptive transfer of T-cells following in vitro stimulation with IL-2 plus IL-12. B16F10 tumor mass was first measured at 10 days post inoculation in C57BL/6 mice. TRP2low or TRP2high avidity T-cells expanded for three days in vitro with IL-2 with or without IL-12 were adoptively transferred to tumor bearing mice on day 10 post tumor inoculation. At this stage the B16F10 tumors are large (>50mm2) and vascularized 28. Without intervention, these tumors result in uniform death of the recipient mice by approximately D20 (Supplemental Fig. 1E). Transfer of IL-2 primed TRP2low cells did not control tumors and mice had to be euthanized by day 20 post tumor inoculation (Fig. 1F). IL-2 primed TRP2high cell transfer resulted in delayed B16F10 tumor growth, but not enhanced survival (Fig. 1G and 1H). Importantly, adoptive transfer of high avidity TRP2 CD8+ T-cells co-cultured with IL-12 significantly delayed tumor growth while low avidity TRP2 CD8+ T-cells trended towards protection (Fig. 1I–1K). However, high avidity T-cells were superior to low avidity T-cells in controlling tumor growth with increased survival (Fig. 1J–1K). We additionally observed increased infiltration for both TRP2high and TRP2low with IL-12 cytokine priming (Supplemental Fig. 1F). Taken together TRP2high CD8+ T-cells co-cultured with IL-12 plus IL-2 delayed B16F10 melanoma growth and enhanced recipient survival.
Figure 1: IL-12-primed CD8+ T-cells have superior effector function and tumor killing capacity compared to IL-2-primed cells.
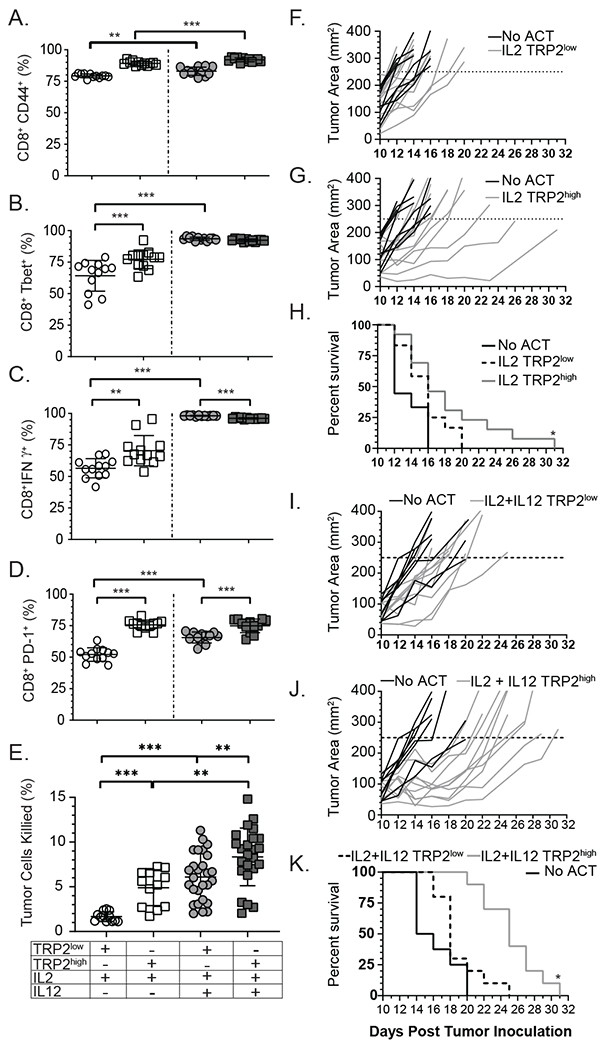
Naïve CD8+ T-cells were enriched from spleen and lymph nodes from TRP2low or TRP2high mice and cultured on plates coated with 50 μg/mL anti-CD3 and 0.8 μg/mL recombinant B7-1 and 5 IU/mL recombinant human IL-2 and/or 10 ng/mL IL-12 for three days. Cells were analyzed for (A) CD44, (B) T-bet, (C) IFNγ or (D) PD-1 by flow cytometry. (A-D) n=12 for each condition from one representative experiment. Similar results were observed in three independent experiments (E) Activated T-cells were co-cultured for 12 hours with B16F10 tumor target cells at a ratio of 5:1 (Effectors:Targets) to determine in vitro killing capacity. B16F10 target cells were analyzed for anti-active caspase-3. Compiled data from 6 independent experiments. (F) B16F10 tumor bearing mice were treated with 5x106 IL-2 activated TRP2low (G) TRP2high cells , or (I) TRP2low , (J) TRP2high activated in the presence of IL-2 plus IL-12. Mice were followed for tumor growth and survival (H and K). Individual mice are shown in panels F, G, I, J, with the combined survival shown in panels H and K. Data are from two independent experiments. ns = not statistically different, **=p<0.001, ***=p<0.0001.
Pre-conditioning low and high avidity T-cells with IL-12 co-culture prevents T cell exhaustion.
We next assayed tumor infiltrating T-cell effector functionality. Following a 4-hour re-stimulation ex vivo with anti-CD3, half of the TRP2low T-cells cultured in IL-2 exhibited an exhausted profile as early as 3 days post transfer, expressing high levels of PD-1 (Supplemental Fig. 1G) and very little IFNγ production directly ex vivo or with anti-CD3 re-stimulation (Fig. 2A and 2B, Supplmental Fig. 1H). The addition of IL-12 co-culture during priming increased IFNγ production ex vivo to some extent, but more significantly following anti-CD3 (Figure 2A and 2B). A similar trend was seen at day 9 (Fig. 2C and 2D). Further investigation into the effector status of TRP2low and TRP2high T-cells revealed altered expression of the transcription factor T-bet. Both TRP2high and TRP2low T-cells showed increased T-bet expression when IL-12 was present during priming (Fig. 2E). We then investigated production of TNFα of both TRP2low and TRP2high T-cells post anti-CD3 re-stimulation. We observed a significant increase in TNFα production with IL-12 addition during priming (Fig. 2F). IL-2 primed TRP2low T-cells produced very little TNFα, but this increased significantly with IL-12 priming (Fig. 2F). A similar trend was noted in TRP2high T-cells. Both TRP2high and TRP2low T-cells increased KLRG-1 expression with IL-12 priming (Fig. 2G). We assessed the percentage of transferred cells producing both IFNγ and TNFα, an indicator of strong effector function with high cytolytic potential, and noted that IL-12 priming significantly increased the percentage of multi-cytokine producing T-cells for both TRP2low and TRP2high cells (Fig. 2H). Taken together, IL-12 primed TRP2low and TRP2high cells had increased cytokine production and activation marker expression compared to IL-2 primed counterparts. We next wanted to investigate the extent of T-cell exhaustion imposed on both high and low avidity T-cells following tumor infiltration.
Figure 2: IL-12 priming prevents exhaustion in the tumor environment.
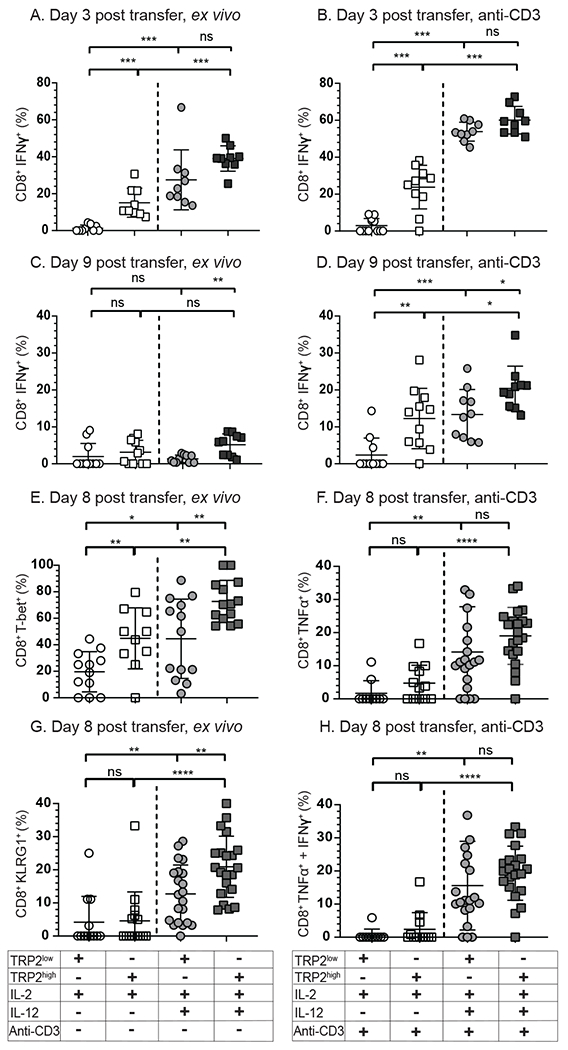
(A-D) IFNγ production by TRP2 reactive T-cells on (A,B) day 3 or (C, D) day 9 post transfer into tumor bearing mice. TRP2highUBQ-GFP T-cells and TRP2lowACT-tdTomato T-cells were isolated from the tumor environment and analyzed directly ex vivo for (A, B) IFNγ or (E) T-bet or (G) KLRG-1 or after 4 hour stimulation with plate bound anti-CD3 for (B, D) IFNγ, (F) TNFα, or dual expression of (H) TNFα and IFNγ. Culture conditions and subsequent ex vivo stimulation are shown in the table below the plots. Compiled data from three experimental replicates, n= 3-6 per condition. Ns = not statistically different, **=p<0.001, ***=p<0.0001.three isolated FACS experiments.
IL-12 enhanced anti-tumor effects are not improved with PD-1 blockade.
PD-1 signaling has been implicated in functional deficits in tumor infiltrating T-cells and anti-PD-1 immunotherapy has been shown to re-invigorate anti-tumor immunity 29. Signaling through PD-1 reduces T-cell contact time with target cells by preventing the TCR induced stop signal 30. To investigate if PD-1 was responsible for the deficits of TRP2low T-cells we first compared PD-1 levels of IL-2 alone or IL-12 plus IL-2 co-culture primed TRP2 specific T-cells. In vivo, we observed that early (day 2-3) post transfer, PD-1 levels were reduced from pre-transfer in vitro levels, and IL-12 cytokine co-culture had no impact on the expression levels (Fig. 3A). However, at day 9 post adoptive transfer, TRP2low and TRP2high T-cells primed with IL-2 plus IL-12 had a significant reduction in PD-1 compared to IL-2-primed T-cells alone (Fig. 3B). Upon further investigation we observed a significant decrease in the inhibitory receptor LAG-3 in both tumor infiltrating TRP2low and TRP2high T-cells (Fig. 3C). We next investigated intracellular levels of TOX in IL-2 versus IL-12 primed cells, as TOX has been shown to be expressed by exhausted cells, promote LAG-3 and PD-1, and inhibit KLRG-1 expression 31, 32. Consistent with reduced exhaustion, we observed a significant decrease in TOX with IL-12 plus IL-2 co-culture (Fig. 3D). Taken together, these results suggested that IL-12 co-culture enhances anti-tumor immunity by limiting and/or preventing T-cell exhaustion.
Figure 3: IL-12 co-culture prevents T-cell exhaustion which is not enhanced by PD-1 blockade.
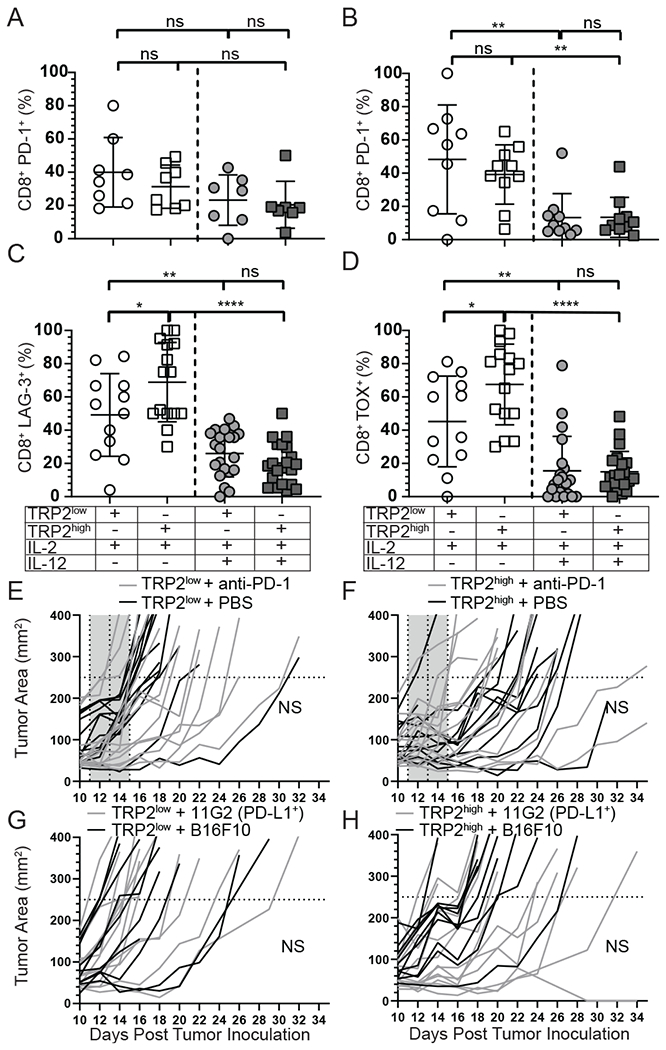
(A-B) PD-1 expression on TRP2 Low and High T-cells isolated from the tumor from three independent experiments on either D2-3 (A) or D9 (B). (C) LAG-3 and (D) TOX expression in TRP2low and TRP2high T-cells isolated from the tumor on D9 post adoptive transfer. (E-F) Mice were treated with 1x106 IL-2 + IL-12 primed TRP2low or TRP2high T-cells and then given 200μg of anti-PD1 or PBS control on D1, 3 and 5 post transfer. Mice were followed for tumor growth. (G-H) Mice were inoculated with B16F10 or PD-L1 overexpression (11G2) tumor cells and adoptively transferred with 1x106 IL-2 + IL-12 primed T-cells and followed for tumor growth over time. Data are from two independent replicates. ns = not statistically different. **=p<0.001, ***=p<0.0001.
To test whether PD-1/PD-L1 pathway regulates effector function of TRP2high or TRP2low cells, we investigated if PD-1 blockade provided any additional benefit for tumor clearance. Transgenic T-cells were activated with IL-2 plus IL-12 as before and transferred into mice bearing D10 tumors. Starting on D1 post transfer, anti-PD-1 was given and mice were monitored for tumor growth and survival. We determined that PD-1 blockade did not provide any additional benefit to TRP2low or TRP2high recipient mice (Fig. 3E and 3F). In fact there were no differences in survival when mice were treated with TRP2 high or low avidity cells with/without PD-1 inhibition (data not shown). Additionally we addressed PD-1/PD-L1 inhibition using a tumor target with constitutively high PD-L1 expression to mitigate off target effects with the anti-PD-1 antibody and directly test the interactions of PD-L1 expression on the tumor cells with PD-1 on the T-cells. The parental B16F10 cell line expresses very little PD-L1 in vitro at baseline (Supplemental Fig. 2A), thus we generated a B16F10 cell line that expressed constitutive PD-L1 driven by the cytomegalovirus (CMV) promoter. We isolated a clone (11G2) which expressed nearly 1,000 fold PD-L1 compared to parental cell line and remained stable over multiple passages (Supplemental Fig. 2A). We then inoculated mice with 11G2 and waited 10 days for the tumors to become established. After 10 days, in vitro activated TRP2low or TRP2high T-cells co-cultured with IL-2 plus IL-12 were adoptively transferred into 11G2- or parental B16F10-tumor bearing mice. We observed no differences in survival between transfer of TRP2low or TRP2high T-cells into 11G2 or B16F10-tumor bearing mice (Fig. 3G and 3H and data not shown). Taken together, these data suggest that IL-2 plus IL-12 pre-conditioning of both low and high avidity T cells reduces PD-1 expression at late time points following adoptive transfer, and that this reduction of PD-1 renders IL-12 primed tumor reactive CD8+ T-cells resistant to PD-1:PD-L1 mediated suppression.
IL-12 co-culture restores the ability of low avidity T-cells to traffic to the tumor site but does not promote strong interactions with intratumoral DCs or tumor cells
Given that IL-12 priming improved CD8+ T-cell functionality both in vitro and in vivo, we next evaluated the effect of IL-12 on T-cell proliferation, trafficking, and dynamic functionality in vivo. We hypothesized that TRP2low T-cells might have a proliferation or trafficking defect, or poor cellular interactions with either intratumoral dendritic or tumor cells compared to TRP2high T-cells. To test this, we first measured BrdU incorporation in TRP2low and TRP2high transferred cells after day 2 in the tumor itself or in the periphery. Supplemental Fig. 2B demonstrates that there are similar levels of BrdU in both sites indicating equal proliferation. To track low and high avidity TRP2 T-cells in vivo directly, we co-transferred TRP2highUBQ-GFP T-cells (green fluorescent protein) and TRP2lowACT-tdTomato T-cells (red) into CD11c-YFP (yellow fluorescent protein) recipient mice that had been inoculated with B16F10-mTFP (monomeric teal fluorescent protein) tumor cells ten days prior. We then harvested the mice on D13 post tumor inoculation (at day 3 post T cell infusion) and performed ex vivo two-photon microscopy. We first quantified TRP2high and TRP2low cells that migrated to the tumor to determine if TCR avidity had an impact on cellular trafficking to the tumor site. We did not find a defect in the TRP2low T-cells ability to traffic and infiltrate the B16F10 tumors with IL-12 plus IL-2 priming (Supplemental Fig. 1F). However, TRP2low T-cells and TRP2high T-cells cultured with IL-2 alone did not effectively traffic to the tumor as tumor infiltriation was equally poor in both groups (Supplemental Fig. 1F). Using two-photon microcopy we analyzed T-cell velocity, track displacement, and quantified the contact time of TRP2high and TRP2low T-cells with both CD11c+ DCs and mTFP+B16F10 tumor cells given the finding that IL-12 co-culture could correct the trafficking defect of TRP2low T cells. We observed no significant differences in average velocity within the tumor microenvironment between TRP2high and TRP2low CD8+ T-cells after IL-12 priming (Fig. 4A). We then analyzed all of the contacts made by TRP2high and TRP2low T cells we measured a significant decrease in the contact time between TRP2low T-cells and tumor infiltrating DCs and with B16F10 tumor cells compared to TRP2high CD8+ T-cells (Fig. 4B). However, TRP2high and TRP2low cells exhibited different types of contacts with both CD11c+ DCs and B16F10 tumor cells (Fig. 4C and 4D). There were instances of stable contacts of TRP2high and TRP2low where the cells were confined and engaged with the tumor/DC throughout the imaging time (Fig. 4C and supplemental movie 1). There were also occurrences where TRP2high and TRP2low cells made transient contacts with targets and were less confined in their migratory path (Figure 4D and supplemental movie 1). This potentially indicates that the decreased tumor control we observed with TRP2low CD8+ T-cells could be due to less productive contact with targets to provide a killing signal.
Figure 4: Antigen overexpression restores the ability of IL-12 primed low avidity T-cells to make sustained contacts with tumor targets.
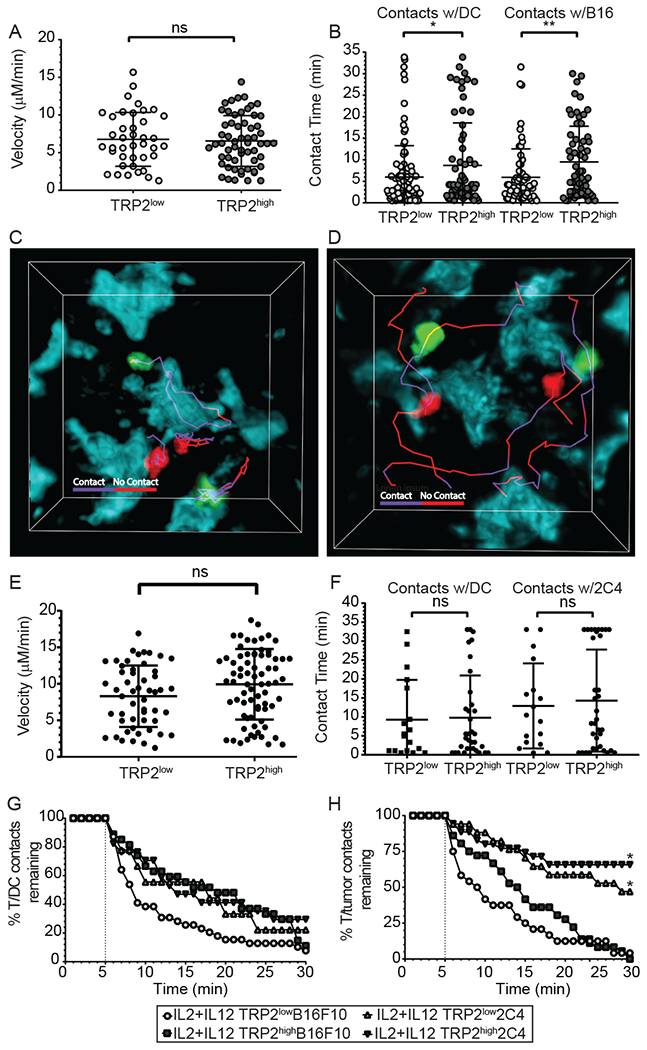
(A-H) CD11c-YFP mice received B16F10-TFP or 2C4 tumors. Ten days after inoculation, mice received 5-10x106 IL-2 plus IL-12 TRP2low and TRP2high T-cells. 72 hours after adoptive T-cell transfer, mice were euthanized and tumors were removed and analyzed by two-photon microscopy. (A, B) T-cells were identified with Brainbow tdTomato (TRP2low) or UBC-GFP (TRP2high) expression and analyzed using Imaris software for average velocity and contact time in B16F10 tumors. (C, D) Representative images showing TRP2low and TRP2high T-cell contacts with tumor cell surfaces using tracks. Purple tracks represent sustained contact with the T-cell and tumor cell, while red tracks represent no-contact between T-cells and tumor targets at the timepoint on the track. Two photon microscopy was performed on ex vivo 2C4 tumors and (E) velocity and (F) contact time with dendritic cells or tumor targets. (G-H) Contact decay was analyzed between TRP2low or TRP2high and (E) YFP+ DCs (F) or TFP+ tumor targets. Contacts lasting less than five minutes were excluded from analysis. Data are from two independent replicates. ns = not statistically different, **=p<0.001, ***=p<0.0001.
Antigen overexpression combined with IL-12 co-culture of low avidity T-cells prevents exhaustion and allows effective T-cell activation and stable contacts with tumor targets.
We next tested the hypothesis that low avidity CD8+ T-cells have a competitive disadvantage when target antigen is limited in the tumor microenvironment. To address this concept, we generated a tumor cell line with increased and constitutive TRP2 expression. We used the CMV promoter and enhancer to drive the expression of a TRP2-mTFP fusion protein. After transfection of the B16F10 parental line and subsequent dilutional cloning, we identified a stable clone, termed 2C4. For this activation assay we used splenocytes from either TRP2low or TRP2high mice and co-cultured the T-cells with plate bound 2C4 or parental B16F10 cells. We observed that 2C4 co-culture significantly increased activation and proliferation of both TRP2low and TRP2high T-cells (Supplemental Fig. 2C). mTFP was included in the vector to allow visualization of the tumor cells by two-photon microscopy, along with simultaneous imaging of GFP (TRP2high), tdTomato (TRP2low) and Venus (CD11c+ DCs). We then investigated in vitro cytotoxicity of TRP2low and TRP2high CD8+ T-cells targeting 2C4 tumor cells compared to parental B16F10 tumor cells. Similar to parental B16F10 with 2C4 we measured a significant increase in the cytotoxic potential of TRP2low T-cells when cells were primed with IL-12 compared to IL-2 alone (Supplemental Fig. 2D, Left). Interestingly, TRP2high and TRP2low T-cells killed 2C4 tumor target cells equally well (Supplemental Fig. 2D, Right). We next investigated whether antigen overexpression would restore TRP2low T-cells ability to establish and maintain tumor cell and DC contacts within the tumor environment. We inoculated CD11c-Venus mice with 2C4 tumors and adoptively transferred in vitro activated TRP2low and TRP2high CD8+ T-cells on D10 post tumor inoculation. We harvested the mice on D3 post T cell transfer and used ex vivo two photon microscopy to analyze dynamic functionality of CD8+ T-cells within 2C4 tumors. Similar to B16F10 we observed significant increases in infiltration with IL-12 addition during priming in 2C4 tumors for both TRP2low and TRP2high T-cells (Supplemental Fig. 2E). Results in Figure 4 demonstrate that there is no difference in the T-cell velocity for TRP2low or TRP2high CD8+ T-cells in the 2C4 tumor environment (Fig. 4E). Increased expression of TRP2 erased the contact deficiency between TRP2low and tumor targets or intratumoral DCs compared with TRP2high T-cells (Fig. 4F), this is in contrast to previous experiments with endogenous levels of antigen (Fig. 4B). We next compared all the contacts between 2C4 and parental B16F10-mTFP that lasted longer than 5 min in duration as those would be more likely to be a productive contact (Figure 4G and 4H). We found, there was no difference with TRP2high cells ability to interact with intratumoral DCs (Fig. 4G). However, TRP2low T-cell interactions with tumor infiltrating DCs there was a significant increase in the T/DC contacts when TRP2 was overexpressed (Fig. 4G). Most striking was the difference observed between TRP2low and TRP2high CD8+ T-cells had a significantly increased ability to form stable contacts with the TRP2 overexpressing 2C4 tumors targets compared to parental B16F10 (Fig. 4H), indicating that IL-12 co-culture along with high and constitutive tumor antigen expression corrected the TRP2low defect. Taken together, these data demonstrate that the overexpression of TRP2 antigen on the tumor cell allows stable contacts for IL-2 plus IL-12 primed TRP2low CD8+ T-cells.
Antigen overexpression combined with IL-12 co-culture of low avidity T-cells prevents exhaustion and allows effective tumor clearance and has decreased risk for autoimmunity.
Since overexpression of TRP2 improved the ability of low avidity CD8+ T-cells to form stable contacts, we investigated if this prolonged interaction correlated with increased survival and enhanced tumor clearance. To test this, mice were implanted with 2C4 tumors and treated with IL-2 plus IL-12 in vitro activated TRP2low and TRP2high CD8+ T-cells. TRP2low cells significantly delayed tumor growth (Fig. 5A) and enhanced survival (Fig. 5C). Transfer of TRP2high CD8+ T-cells into 2C4 tumor-bearing mice also protected recipient mice (Fig. 5B and 5C). Both low and high avidity T cell transfer resulted in enhanced survival of 36% (5/14) and 36% (5/14) of mice at day 30 post tumor inoculation (Fig. 5C). In both groups some mice were cured and eradicated the tumor, but the majority of the mice had to be euthanized by day 42 (Fig. 5C). Given this result, we next investigated the phenotype of tumor infiltrating lymphocytes present in 2C4 tumors. KLRG1 expression was significantly higher in TRP2high T-cells with IL-2 priming alone, and was not further increased with IL-12 priming in TRP2high T-cells contrary to B16F10 tumors, although it was increased in TRP2low T-cells (Figure 5D). With IL-2 alone, T-bet expression was higher in TRP2high T-cells compared to TRP2low T-cells, but interestingly, T-bet expression was not significantly different between TRP2high and TRP2low T-cells with IL-12 priming. This is in contrast to parental B16F10 tumors (Figure 5E). We found that TRP2low and TRP2high T-cells expressed high levels of TOX with IL-2 priming alone in 2C4 tumors. Again, this was significantly reduced with IL-12 present during priming in both populations of T-cells, suggesting IL-12 prevented the exhaustion phenotype (Figure 5F). IL-12 cytokine was therefore necessary for optimal TRP2low T-cell priming in 2C4 tumors, but not necessary for TRP2high T-cell priming. Taken together, IL-12 plus sufficient tumor antigen levels corrected the defects for low avidity cells to become fully activated, control tumors, prolong survival and prevent exhaustion. Finally, we wanted to assess the potential for autoimmunity following IL-12 primed adoptive T-cell therapy, as high avidity T-cells primed with IL-12 have previously been shown to have enhanced self-reactivity 13. Recipient mice received IL-2 plus IL-12 primed TRP2low or TRP2high cells and were examined for development of autoimmune vitiligo by monitoring fur and skin depigmentation as a loss of melanocytes. Shown in Figure 5G are representative mice from TRP2high, TRP2low, and no transfer recipient mice illustrating significantly less autoimmunity in TRP2low (1/12) compared to (10/14) for TRP2high recipients. We determined that there was a lower risk for autoimmune vitiligo following transfer for IL-2 plus IL-12 primed TRP2low cells compared to TRP2high cells (Figure 5G and Supplemental Fig. 2F).
Figure 5: Antigen overexpression ameliorates effector function differences of IL-12 primed low or high avidity T-cells.
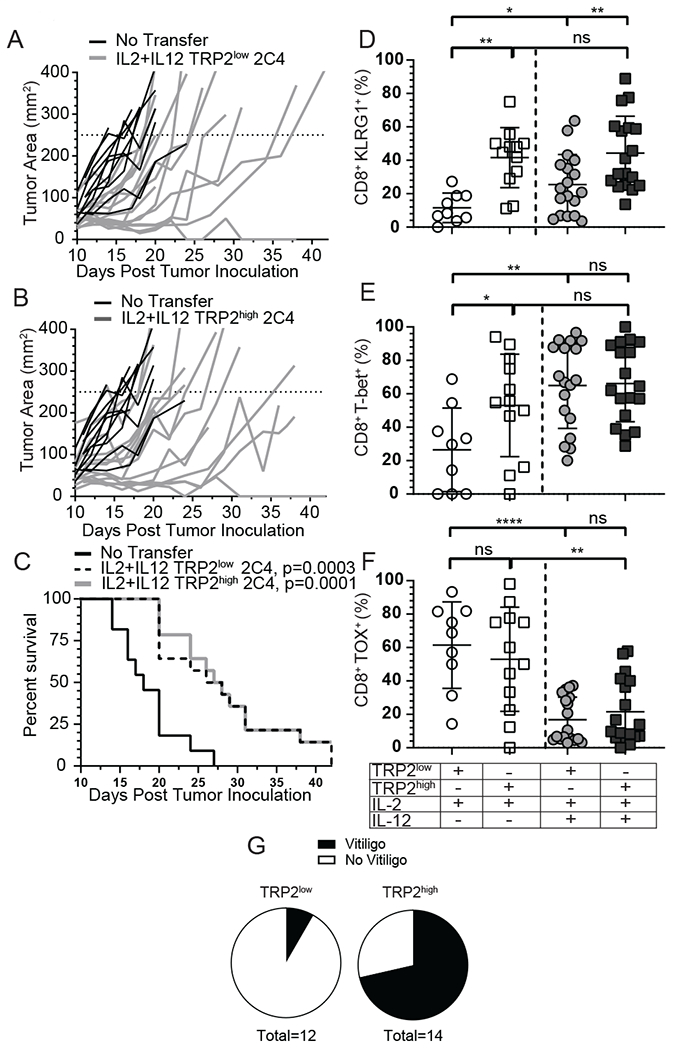
(A-C) A murine TRP2-TFP tumor clone that expressed stable and high levels of TRP2-TFP (2C4) was inoculated into recipient mice and 1x106 IL-2 + IL-12 primed TRP2low or TRP2high T-cells or PBS control (no T cell transfer) were adoptively transferred on D10 post tumor inoculation. Mice were followed for (A-B) tumor growth and (C) survival. Data from three independent experiments. (D-F) 2C4 tumors and spleen were harvested on D8 post adoptive T-cell transfer and lymphocytes were analyzed for (D) KLRG-1 and intracellular expression of (E) T-bet and (F) TOX by flow cytometery. Data are from three independent replicates. ns = not statistically different, **=p<0.001, ***=p<0.0001. (G) OT-1xUBC-GFP or WT B6 mice were given 5x106 IL-2 plus IL-12 in vitro primed TRP2highGFP or TRP2lowtdtomato T-cells mice were followed for vitiligo induction. Incidence is shown 3 months after adoptive transfer. Data from two independent experiments pooled incidence.
Discussion:
This study advances our understanding of the mechanisms of tumor induced T-cell exhaustion and how adoptive CD8+ T-cell therapy can be optimized for maximal cancer survival benefit. Herein we demonstrate that low avidity tumor reactive CD8+ T-cells are defective compared to high avidity T-cells when stimulated with IL-2 alone. Low avidity T-cells with IL-2 provide poor tumor control, have limited effector capability, exhibit poor cytotoxicity, fail to infiltrate tumors effectively and have an exhausted phenotype in the tumor environment. In contrast, high avidity T-cells are able to provide moderate tumor control, and survive within the tumor environment, however, they too exhibit an exhausted phenotype. Co-culturing CD8+ T-cells with IL-12 cytokine plus IL-2 during in vitro priming erases the deficits for low avidity T-cells and generates strong effector CD8+ T-cells that are resistant to exhaustion. IL-12 primed low avidity CD8+ T-cells are able to destroy tumor tissue, provide host survival and sustain themselves within the tumor environment, especially when the antigen is abundantly expressed. Finally, adoptive T-cell transfer of low avidity cells results in lower frequency of irAE including autoimmune vitiligo. High avidity CD8+ T-cells also have enhanced anti-tumor effects and decreased exhaustion when co-cultured with IL-12, however, subsequent autoimmune vitiligo is more prevelant with high avidity T-cells. Taken together, IL-12 plus IL-2 pre-conditions low avidity T-cells for enhanced proliferation, trafficking, activation, cytokine production, stable contacts with tumor targets, tumor clearance, and decreased T-cell exhaustion while still remaining safe.
IL-12 suppression of PD-1 within the tumor environment has been previously reported for high avidity T-cells 22, but the impact on low avidity T-cells had not been explored. Moreover, the mechanism by which IL-12 cytokine signaling suppresses PD-1 expression is not completely understood. It is hypothesized that T-bet suppresses the transcription of PD-1 by preventing transcriptional machinery from binding the PD-1 locus 33. Indeed, IL-12 primed low and high avidity CD8+ T-cells express more T-bet following in vitro activation (Fig. 1B). Normally PD-1 is upregulated following T-cell activation and the translocation of NF-ATc1 to the nucleus where it interacts with the Pdcd1 promoter 34. Upon activation, naïve T-cells transition to effector cells and the Pdcd1 locus is demethylated to facilitate transcription. The site is remethylated as antigen is cleared, but remains demethylated in exhausted CD8+ T-cells during chronic viral infection 35. IL-12 primed low and high avidity CD8+ T-cells also express more T-bet when isolated from the tumor environment at later time points (Fig. 2E). This increased T-bet expression could disrupt PD-1 transcription and translation over time, thus preventing T-cell enhaustion. A potential mechanism for differential regulation of PD-1 re-expression might be due to the differences in the level of demethylation/re-methylation of the pdcd1 locus by IL-12. At early time points after in vivo transfer, this level of PD-1 remains high on both IL-2 and IL-12 plus IL-2 primed T-cells. However, at late time points, T-cells receiving IL-12 cytokine have significantly lower PD-1 expression compared to IL-2 alone primed T-cells (Supplemental Fig. 1G). This decreased PD-1 correlated with increased T-cell effector function and ability to respond to TCR stimuli and tumor antigen. IL-12 priming increases effector function by both increasing effector cytokines as well as preventing exhaustion within the tumor environment. This reduction of PD-1 was associated with reduced LAG-3 and TOX expression. The transcription factor TOX has recently been shown to be integral for inducing and maintaining T-cell exhaustion by upregulating inhibitory receptors LAG-3, PD-1, and TIGIT and inhibiting activation markers such as KLRG-1 31. Our data is consistent with a recent report by Page et al. 36 demonstrating that the DNA-binding factor TOX was induced in CD8+ T-cells during an lymphocytic choriomeningitis virus infection. In this study, Page and colleagues showed that TOX expression was inhibited by the addition of interleukin-12 in a T-bet and Eomes dependent fashion. This result is consistent with our findings and offers a potential mechanism for the effects of IL-12 in vitro priming.
It was surprising that neither PD-1 blockade nor PD-L1 overexpression altered tumor clearance after IL-12 co-culture (Fig. 3). We would have predicted a benefit of targeting this inhibitory pathway given the residual, ableit low level of PD-1 on IL-12 primed tumor specific T-cells (Fig. 3). However, this was not the case and these data suggest that there is no additive benefit of checkpoint therapy following adoptive T-cell therapy with IL-12 pre-conditioned cells in the B16F10 tumor model. This result could obviate the need to give checkpoint blockade in addition to IL-12 primed T-cell adoptive therapy in the clinic, thus limiting the unintended autoimmunity and irAE associated with PD-1/PD-L1 checkpoint blockade. It also suggests that patients that have failed PD-1 immunotherapy would be potential candidates for an IL-12 plus IL-2 co-culture adoptive T-cell therapy.
Antigen availability is a major factor impacting T-cell functionality within the tumor environment. It is interesting that TRP2 antigen overexpression increased low avidity T-cells ability to control tumor growth and ameliorated the differences between high and low avidity T-cell effector function and expression of T-bet, KLRG1 and TOX. We attributed these to altered T-cell trafficking and dynamic cellular interactions (direct contact time) within the tumor microenvironment between the effector T-cells and the targeted tumor cells themselves 37. This result is in contrast to the study by Dougan et al. where low and high avidity T-cells provided comparable tumor control 38. Their model utilized the adoptive transfer of naïve low or high avidity T-cells and a different target antigen, tyrosinase related protein-1 (TRP1). Differences in antigen availability could explain our contrasting findings. TRP1 and TRP2 could be expressed at distinct levels in melanocytes or within the B16F10 tumors 39. There may also be a different level of transcription for TRP2 compared to TRP1. In fact, during melanogenesis the transcript level of TRP2 is very low in unstimulated melanocytes 40. Moreover, cell culture conditions can significantly alter transcript levels 41. Previous studies have demonstrated differential regulation of melanogenesis with different hormones or cytokines 40. In fact, IFNγ can decrease melanocyte protein expression, and we do not yet know if this has any differential effect on TRP2 compared to TRP1 expression or transcript control within tumor cells or healthy melanocytes 42. If this is true, it would suggest that TRP1 antigen is expressed at higher levels in B16F10 melanoma compared to the TRP2 antigen. In this situation, both low and high avidity T-cells specific for TRP1 would respond optimally and clear tumors, as was reported 38. In our situation, TRP2 overexpression enhanced tumor killing and host survival after low and high avidity T cell transfer, mirroring the findings with cells targeting TRP1 (Fig. 4 and 5). Additional tumor microenvironment conditions could be explored to enhance MHCI and tumor antigen presentation. Taken together, these data would suggest a threshold effect, where sufficient antigen could yield maximal T-cell function. An alternative explanation could be peptide binding to MHCI, and TRP1 may be more stable or persist longer in the context of MHCI compared to TRP2. This enhanced stability and MHCI presentation would allow sustained or enhanced availability for responding low and high avidity T-cells. Finally, differential Aire regulation of TRP1 and TRP2 in the thymus may allow more effective immune rejection of melanoma for these two targets 43. Our study highlights the importance of testing for sufficient antigen availability and stability for optimal T-cell function and tumor clearance. This information could be highly informative and predictive of a successful adoptive T-cell transfer.
The clinical benefits of IL-12 have been previously explored to boost anti-tumor immune responses 22, 44–46. While systemic IL-12 therapy was promising in mouse models of cancer, clinical delivery of systemic IL-12 therapy in humans has been complicated by extreme toxicity and even death 46. This toxicity prevented systemic IL-12 use in the clinic and hampered further exploration of this cytokine in adoptive cell therapy. Strauss and colleagues have tested an alternative strategy to administer IL-12. In a Phase I safety study, they administered a fusion protein containing bioactive IL-12 with an antibody that binds to histones on free DNA fragments, targeting regions of tumor necrosis 47. We propose an alternative and only add IL-12 while pre-conditioning T-cells during the in vitro priming of T-cells, not systemically. Currently in the clinic, most therapy protocols use IL-2 alone during activation, generation of effector T-cells and during infusion to increase survival and homeostasis of adoptively transferred T-cells (reviewed in 48–50). Along this line, several groups have investigated the use of differential cytokine priming or in vivo expression of cytokines within the tumor microenvironment with variable success 16, 18–21, 51. In most cases the field has focused on high avidity T-cell studies using IL-12. High avidity T-cells also pose a significant risk for autoimmunity 15, especially if the tumor target contains non-mutated self-antigens 52. Our data suggests that IL-2 alone generates exhausted CD8+ T-cells with poor effector function (Fig. 1). Therefore, the addition of IL-12 with IL-2 is necessary, especially for low avidity T-cells (Fig. 5). Pre-conditioning with IL-12 improves proliferation, trafficking, activation, cytokine production, stable contacts with tumor targets, tumor clearance, and decreased T-cell exhaustion. This method would eliminate potential toxicity of systemic IL-12 while still providing the therapeutic efficacy that IL-12 cytokine promises with decreased risk of developing autoimmunity.
Supplementary Material
Key Points.
Low Avidity T-cells provide poor tumor control, and exhibit an exhausted phenotype.
IL-12 cytokine priming corrects low avidity T-cell defects and prevents exhaustion.
Low and high avidity T- cells function equivalent with increased tumor antigen.
Acknowledgements
The authors thank Dr. Arthur Hurwitz for providing mice. The authors declare no competing financial interests.
This work was supported by NIH R01 AI106791 (BTF), P01 AI35296 (BTF, MFM), T32 AI007313 (CGT), and the Minnesota Partnership for Biotechnology and Medical Genomics MNP#18.01 (BTF).
References
- 1.Wei SC, Duffy CR, and Allison JP, Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discovery, 2018. 8(9): p. 1069–1086. [DOI] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Drilon A, Wolchok JD, Carvajal RD, McHenry MB, Hosein F, Harbison CT, Grosso JF, and Sznol M, Five-Year Survival and Correlates Among Patients With Advanced Melanoma, Renal Cell Carcinoma, or Non-Small Cell Lung Cancer Treated With Nivolumab. JAMA Oncol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, and Ribas A, PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 2014. 515(7528): p. 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, and Wherry EJ, Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science, 2016. 354(6316): p. 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, and Wolchok JD, Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol, 2015. 26(12): p. 2375–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baruch EN, Berg AL, Besser MJ, Schachter J, and Markel G, Adoptive T cell therapy: An overview of obstacles and opportunities. Cancer, 2017. 123(S11): p. 2154–2162. [DOI] [PubMed] [Google Scholar]
- 7.Hinrichs CS and Rosenberg SA, Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev, 2014. 257(1): p. 56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, Levy D, Kubi A, Hovav E, Chermoshniuk N, Shalmon B, Hardan I, Catane R, Markel G, Apter S, Ben-Nun A, Kuchuk I, Shimoni A, Nagler A, and Schachter J, Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res, 2010. 16(9): p. 2646–2655. [DOI] [PubMed] [Google Scholar]
- 9.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, McMahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, and Rosenberg SA, Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother, 2013. 36(2): p. 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernatchez C, Radvanyi LG, and Hwu P, Advances in the treatment of metastatic melanoma: adoptive T-cell therapy. Seminars in oncology, 2012. 39(2): p. 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.June CH and Sadelain M, Chimeric Antigen Receptor Therapy. N Engl J Med, 2018. 379(1): p. 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, Binder-Scholl GK, Smethurst DP, Gerry AB, Pumphrey NJ, Bennett AD, Brewer JE, Dukes J, Harper J, Tayton-Martin HK, Jakobsen BK, Hassan NJ, Kalos M, and June CH, Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood, 2013. 122(6): p. 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markiewicz MA, Wise EL, Buchwald ZS, Cheney EE, Hansen TH, Suri A, Cemerski S, Allen PM, and Shaw AS, IL-12 enhances CTL synapse formation and induces self-reactivity. J Immunol, 2009. 182(3): p. 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan DJ, Kreuwel HT, Fleck S, Levitsky HI, Pardoll DM, and Sherman LA, Activation of low avidity CTL specific for a self epitope results in tumor rejection but not autoimmunity. J Immunol, 1998. 160(2): p. 643–651. [PubMed] [Google Scholar]
- 15.Zhong S, Malecek K, Johnson LA, Yu Z, de Miera E. Vega-Saenz, Darvishian F, McGary K, Huang K, Boyer J, Corse E, Shao Y, Rosenberg SA, Restifo NP, Osman I, and Krogsgaard M, T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A, 2013. 110(17): p. 6973–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubinstein MP, Cloud CA, Garrett TE, Moore CJ, Schwartz KM, Johnson CB, Craig DH, Salem ML, Paulos CM, and Cole DJ, Ex vivo interleukin-12-priming during CD8(+) T cell activation dramatically improves adoptive T cell transfer antitumor efficacy in a lymphodepleted host. J Am Coll Surg, 2012. 214(4): p. 700–707; discussion 707-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valenzuela J, Schmidt C, and Mescher M, The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol, 2002. 169(12): p. 6842–6849. [DOI] [PubMed] [Google Scholar]
- 18.Kunert A, Chmielewski M, Wijers R, Berrevoets C, Abken H, and Debets R, Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology, 2017. 7(1): p. e1378842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan C, Dannull J, Nair SK, Ding E, Tyler DS, Pruitt SK, and Lee WT, Local secretion of IL-12 augments the therapeutic impact of dendritic cell-tumor cell fusion vaccination. J Surg Res, 2013. 185(2): p. 904–911. [DOI] [PubMed] [Google Scholar]
- 20.Miguel A, Herrero MJ, Sendra L, Botella R, Algas R, Sanchez M, and Alino SF, Comparative antitumor effect among GM-CSF, IL-12 and GM-CSF+IL-12 genetically modified tumor cell vaccines. Cancer Gene Ther, 2013. 20(10): p. 576–581. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, and Morgan RA, Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther, 2011. 19(4): p. 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gerner MY, Heltemes-Harris LM, Fife BT, and Mescher MF, Cutting edge: IL-12 and type I IFN differentially program CD8 T cells for programmed death 1 re-expression levels and tumor control. J Immunol, 2013. 191(3): p. 1011–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, Rosenberg SA, and Morgan RA, Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother, 2013. 62(4): p. 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh V, Ji Q, Feigenbaum L, Leighty RM, and Hurwitz AA, Melanoma progression despite infiltration by in vivo-primed TRP-2-specific T cells. J Immunother, 2009. 32(2): p. 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu Z, Cuss SM, Singh V, Gurusamy D, Shoe JL, Leighty R, Bronte V, and Hurwitz AA, CD4+ T Cell Help Selectively Enhances High-Avidity Tumor Antigen-Specific CD8+ T Cells. J Immunol, 2015. 195(7): p. 3482–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Curtsinger JM, Johnson CM, and Mescher MF, CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol, 2003. 171(10): p. 5165–5171. [DOI] [PubMed] [Google Scholar]
- 27.Inoue M, Plautz GE, and Shu S, Treatment of intracranial tumors by systemic transfer of superantigen-activated tumor-draining lymph node T cells. Cancer Res, 1996. 56(20): p. 4702–4708. [PubMed] [Google Scholar]
- 28.Langenkamp E, Vom Hagen FM, Zwiers PJ, Moorlag HE, Schouten JP, Hammes HP, Gouw AS, and Molema G, Tumor Vascular Morphology Undergoes Dramatic Changes during Outgrowth of B16 Melanoma While Proangiogenic Gene Expression Remains Unchanged. ISRN Oncol, 2011. 2011: p. 409308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, and Wherry EJ, Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science, 2012. 338(6111): p. 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, and Bluestone JA, Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol, 2009. 10(11): p. 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, and Wherry EJ, TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature, 2019. 571(7764): p. 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, and Wherry EJ, Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol, 2009. 10(1): p. 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, and Wherry EJ, Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol, 2011. 12(7): p. 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oestreich KJ, Yoon H, Ahmed R, and Boss JM, NFATc1 regulates PD-1 expression upon T cell activation. J Immunol, 2008. 181(7): p. 4832–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM, and Ahmed R, Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8(+) T cells. Immunity, 2011. 35(3): p. 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Page N, Klimek B, De Roo M, Steinbach K, Soldati H, Lemeille S, Wagner I, Kreutzfeldt M, Di Liberto G, Vincenti I, Lingner T, Salinas G, Bruck W, Simons M, Murr R, Kaye J, Zehn D, Pinschewer DD, and Merkler D, Expression of the DNA-Binding Factor TOX Promotes the Encephalitogenic Potential of Microbe-Induced Autoreactive CD8(+) T Cells. Immunity, 2018. 48(5): p. 937–950 e938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boldajipour B, Nelson A, and Krummel MF, Tumor-infiltrating lymphocytes are dynamically desensitized to antigen but are maintained by homeostatic cytokine. JCI Insight, 2016. 1(20): p. e89289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dougan SK, Dougan M, Kim J, Turner JA, Ogata S, Cho HI, Jaenisch R, Celis E, and Ploegh HL, Transnuclear TRP1-specific CD8 T cells with high or low affinity TCRs show equivalent antitumor activity. Cancer Immunol Res, 2013. 1(2): p. 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kameyama K, Sakai C, Kuge S, Nishiyama S, Tomita Y, Ito S, Wakamatsu K, and Hearing VJ, The expression of tyrosinase, tyrosinase-related proteins 1 and 2 (TRP1 and TRP2), the silver protein, and a melanogenic inhibitor in human melanoma cells of differing melanogenic activities. Pigment Cell Res, 1995. 8(2): p. 97–104. [DOI] [PubMed] [Google Scholar]
- 40.Kippenberger S, Loitsch S, Solano F, Bernd A, and Kaufmann R, Quantification of tyrosinase, TRP-1, and Trp-2 transcripts in human melanocytes by reverse transcriptase-competitive multiplex PCR--regulation by steroid hormones. J Invest Dermatol, 1998. 110(4): p. 364–367. [DOI] [PubMed] [Google Scholar]
- 41.Kippenberger S, Bernd A, Bereiter-Hahn J, Ramirez-Bosca A, Kaufmann R, and Holzmann H, Transcription of melanogenesis enzymes in melanocytes: dependence upon culture conditions and co-cultivation with keratinocytes. Pigment Cell Res, 1996. 9(4): p. 179–184. [DOI] [PubMed] [Google Scholar]
- 42.Le Poole IC, Riker AI, Quevedo ME, Stennett LS, Wang E, Marincola FM, Kast WM, Robinson JK, and Nickoloff BJ, Interferon-gamma reduces melanosomal antigen expression and recognition of melanoma cells by cytotoxic T cells. Am J Pathol, 2002. 160(2): p. 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu ML, Nagavalli A, and Su MA, Aire deficiency promotes TRP-1-specific immune rejection of melanoma. Cancer Res, 2013. 73(7): p. 2104–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao W, Li Y, Zeng L, Zhang X, Zhou Z, Zheng M, and Wan H, Intratumoral injection of dendritic cells overexpressing interleukin12 inhibits melanoma growth. Oncol Rep, 2019. 42(1): p. 370–376. [DOI] [PubMed] [Google Scholar]
- 45.Lin L, Rayman P, Pavicic PG Jr., Tannenbaum C, Hamilton T, Montero A, Ko J, Gastman B, Finke J, Ernstoff M, and Diaz-Montero CM, Ex vivo conditioning with IL-12 protects tumor-infiltrating CD8(+) T cells from negative regulation by local IFN-gamma. Cancer Immunol Immunother, 2019. 68(3): p. 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, and Ryan JL, Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood, 1997. 90(7): p. 2541–2548. [PubMed] [Google Scholar]
- 47.Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, McMahon S, Lamping E, Marte JL, Madan RA, Bilusic M, Silver MR, Bertotti E, Schlom J, and Gulley JL, First-in-Human Phase I Trial of a Tumor-Targeted Cytokine (NHS-IL12) in Subjects with Metastatic Solid Tumors. Clin Cancer Res, 2019. 25(1): p. 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choudhry H, Helmi N, Abdulaal WH, Zeyadi M, Zamzami MA, Wu W, Mahmoud MM, Warsi MK, Rasool M, and Jamal MS, Prospects of IL-2 in Cancer Immunotherapy. Biomed Res Int, 2018. 2018: p. 9056173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mortara L, Balza E, Bruno A, Poggi A, Orecchia P, and Carnemolla B, Anti-cancer Therapies Employing IL-2 Cytokine Tumor Targeting: Contribution of Innate, Adaptive and Immunosuppressive Cells in the Anti-tumor Efficacy. Front Immunol, 2018. 9: p. 2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foley KC, Nishimura MI, and Moore TV, Combination immunotherapies implementing adoptive T-cell transfer for advanced-stage melanoma. Melanoma Res, 2018. 28(3): p. 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alsaieedi A, Holler A, Velica P, Bendle G, and Stauss HJ, Safety and efficacy of Tet-regulated IL-12 expression in cancer-specific T cells. Oncoimmunology, 2019. 8(3): p. 1542917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeh S, Karne NK, Kerkar SP, Heller CK, Palmer DC, Johnson LA, Li Z, Bishop RJ, Wong WT, Sherry RM, Yang JC, Dudley ME, Restifo NP, Rosenberg SA, and Nussenblatt RB, Ocular and systemic autoimmunity after successful tumor-infiltrating lymphocyte immunotherapy for recurrent, metastatic melanoma. Ophthalmology, 2009. 116(5): p. 981–989 e981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.