

Abstract
BACKGROUND-
Epigenetic mechanisms are critical in the pathogenesis of pulmonary arterial hypertension (PAH). Previous studies have suggested that hypermethylation of the Bone Morphogenetic Protein Receptor Type 2 (BMPR2) promoter is associated with BMPR2 downregulation and progression of PAH. Here, we investigated for the first time the role of Switch-Independent 3a (SIN3a), a transcriptional regulator, in the epigenetic mechanisms underlying hypermethylation of BMPR2 in the pathogenesis of PAH.
METHODS-
We used lung samples from PAH patients and non-PAH controls, preclinical mouse and rat PAH models, and human pulmonary arterial smooth muscle cells (hPASMC). Expression of SIN3a was modulated using a lentiviral vector or a siRNA in vitro and a specific Adeno-Associated Virus serotype 1 (AAV1) or a lentivirus encoding for human SIN3a in vivo.
RESULTS-
SIN3a is a known transcriptional regulator; however, its role in cardiovascular diseases, especially PAH, is unknown. Interestingly, we detected a dysregulation of SIN3 expression in patients and in rodent models, which is strongly associated with decreased BMPR2 expression. SIN3a is known to regulate epigenetic changes. Therefore, we tested its role in the regulation of BMPR2 and found that BMPR2 is regulated by SIN3a. Interestingly, SIN3a overexpression inhibited hPASMC proliferation and upregulated BMPR2 expression by preventing the methylation of the BMPR2 promoter region. RNA sequencing analysis suggested that SIN3a downregulated the expression of DNA and histone methyltransferases such as DNMT1 and EZH2 while promoting the expression of the DNA demethylase TET1. Mechanistically, SIN3a promoted BMPR2 expression by decreasing CTCF binding to the BMPR2 promoter. Finally, we identified intratracheal delivery of AAV1.hSIN3a to be a beneficial therapeutic approach in PAH- by attenuating pulmonary vascular and RV remodeling, decreasing RVSP and mPAP pressure, and restoring BMPR2 expression in rodent models of PAH.
CONCLUSIONS-
Altogether, our study unveiled the protective/beneficial role of SIN3a in pulmonary hypertension. We also identified a novel and distinct molecular mechanism by which SIN3a regulates BMPR2 in hPASMC. Our study also identified lung-targeted SIN3a gene therapy using AAV1 as a new promising therapeutic strategy for treating patients with PAH.
Keywords: BMPR2, DNA methylation, epigenetic mechanism, SIN3a, gene therapy, Pulmonary artery hypertension
Introduction
Pulmonary arterial hypertension (PAH) is a rare and fatal lung disease caused by pulmonary vascular remodeling, an increase in pulmonary vascular resistance (PVR), and ultimately respiratory and right ventricle (RV) failure1–3. The vascular remodeling is characterized by a switch from “quiescent ” toward “pro-proliferative ”, “apoptosis-resistant, ” and “pro-inflammatory ” phenotypes of hPASMC and hPAECs. These changes lead to the formation of plexiform lesions, intimal thickening, and vascular muscularization4. Pulmonary vascular pressures and resistance drastically increase and enhance afterload in the RV, promoting RV hypertrophy and ultimately failure5–7. However, our understanding of the PAH pathogenesis remains incomplete. Despite the rapid accumulation of data in the last decades, a full understanding of the molecular mechanisms in PAH is critical to identify novel therapeutic targets and new strategies for treating patients with PAH.
BMPR2 belongs to a family of genes involved in cell growth and differentiation 8, 9. The loss of BMPR2 function is sufficient to potentiate PAH10–12. Germline mutations in the BMPR2 gene occur in ~85% of familial PAH (FPAH) and up to 26% of idiopathic PAH (iPAH) patients. Only 10–20% of people with mutations develop PAH, suggesting that a ‘second hit’ may play a critical role in triggering the disease13–15. Concomitantly, most iPAH patients show a significant reduction in BMPR2 expression, while no mutation has been identified within the BMPR2 gene16–18. A recent study has reported aberrant hypermethylation of the promoter region of BMPR2 in FPAH patients19, suggesting that DNA methylation may repress BMPR2 gene expression. However, the underlying mechanisms driving these changes remain to be unveiled.
Over the last decade, a large body of evidence demonstrated that epigenetic mechanisms, such as DNA or histone modifications, play a critical role in the regulation of gene expression in several diseases20–24. Indeed, DNA methylation may induce structural changes in chromatin structure by recruiting methyl CpG binding proteins (MeCPs) and HDACs. These structural changes prevent the binding of transcription factors (TFs) and subsequent transcription initiations24–26. MeCPs have also been shown to interact with DNA methyltransferases (DNMTs). Abnormal DNMT and MeCP2 expression can affect the DNA methylation level and therefore contribute to disease phenotype27–30.
The SIN3 complex has been previously described as containing the SIN3a and SIN3b corepressors, HDAC1/2, MeCP2, and other associated proteins that regulate the transcriptional machinery31–35. Although SIN3a does not bind DNA on its own, it acts as a scaffold protein for several transcription factors and promotes the recruitment of HDACs, which may repress gene expression. In addition to the transcriptional repression activity, growing evidence suggests that SIN3a may activate transcription of specific target-genes36. Interestingly, a competition between DNMT1 and SIN3a, for the interaction with the repression domain of MeCP2, disrupted the SIN3a/HDAC1/MeCP2 complex and favored DNA methylation37. Moreover, SIN3a has been shown to form complexes with other regulatory proteins, including chromatin regulatory enzymes, to suppress the transcription of genes involved in cell cycle regulation and tumor progression38–40. Histone methylation is known to play an essential role in the regulation of gene expression41, 42. The histone methyltransferase EZH2 (Enhancer of Zeste 2) is the catalytic subunit of the polycomb repressive complex 2 (PRC2) and mediates the trimethylation of histone H3 at lysine 27 (H3K27me3). By facilitating chromatin compaction, EZH2 mediates transcriptional silencing of tumor suppressor genes involved in cell cycle regulation, differentiation, and proliferation43, 44. Numerous studies reported that mutation, amplification, and/or overexpression of the EZH2 gene are implicated in tumorigenesis and correlate with poor prognosis in several cancer types, including prostate cancer, breast cancer, bladder cancer, endometrial cancer, and melanoma45–50. EZH2 overexpression promotes the proliferation of several cancer cell lines, while EZH2 inhibition inhibits tumor growth. In PAH, EZH2 has been previously shown to inhibit migration and proliferation of hPASMC and the development of the disease51.
Interestingly, dysregulation of EZH2 activity is associated with growth and invasion in cancer. Also, emerging evidence suggests an interaction of EZH2 with MeCP2, HDAC1, and SIN3a52, 53. However, the role of SIN3a in PAH has not been investigated yet, and the underlying mechanism of the BMPR2 hypermethylation remains unknown. Therefore, we hypothesized that SIN3a might regulate epigenetic modulators and thus BMPR2 methylation and expression in PAH.
METHODS
“The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results or replicating the procedure.” Detailed Materials and Methods are described in the Data Supplement
Human Lung Tissue Samples
Lung tissue specimens were obtained from patients with PAH at the time of lung transplantation and patients without PAH (used as controls) at the time of thoracic surgery (lobectomy or pneumonectomy for localized lung cancer). Preoperative echocardiography was performed in the control patients to rule out pulmonary hypertension (Table I and II in the Supplement). The study was approved by the local ethics committee (Comité de Protection des Personnes, CPP Ile de France VII, Le Kremlin Bicêtre, France). Informed written consents were obtained from each participant. The samples were de-identified and archived specimens.
Cell Culture
Human pulmonary artery smooth muscle cells (hPASMC) and pulmonary artery endothelial cells (hPAECs) were purchased from Lonza, Inc. (Allendale, NJ). hPASMC were cultured in SmBM medium supplemented with 5% fetal bovine serum (FBS) and SmGM-2 SingleQuots (Lonza). hPAECs were grown in EBM-2 medium supplemented with 5% FBS supplemented with EGM-2 SingleQuots (Lonza). Cells were grown in 5% CO2 at 37°C and passaged at the confluence.
Animal-PAH model and SIN3a Gene transfer
All animal experiments were approved by the Icahn School of Medicine at Mount Sinai institutional animal use and care committee and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. For the PH model induced in rats by monocrotaline (MCT) exposure, male Sprague-Dawley rats (250–300 g body weight) were purchased from Charles River and injected subcutaneously with 60 mg/kg of MCT (Sigma Aldrich). Once PAH was established (after 21 days for MCT), MCT-treated rats were randomly assigned to either receive a single dose of AAV1.LUC (3.5×1011 vg/mL) or AAV1.hSIN3a (3.5×1011 vg/mL) for four weeks. Treatments were intratracheally (IT) aerosolized in 250 µL using an IA-1C Microsprayer (PennCentury, Wyndmoor, PA). For the PH model induced in mice by the combination of SU5416 and chronic hypoxia exposure (SuHx), male mice (aged 8 to 10 weeks) received a weekly subcutaneous injection of SU5416 (20 mg/kg; Sigma Aldrich) for three weeks and were maintained in hypoxia (10% O2). Three weeks later, the mice were randomly assigned to either receive a single dose of AAV1.LUC (1×1011 vg/mL) or AAV1.hSIN3a (1×1011 vg/mL) for four weeks. Treatments were aerosolized in a total volume of 50 µL by IT delivery using an IA-1C Microsprayer (PennCentury, Wyndmoor, PA). For the lentivirus-mediated strategy, 6-week-old male mice received a single dose of lentivirus-shRNA BMPR2 (shBMPR2) alone or in combination with a lenti-SIN3a (total 1.5×108 TU) in 50μL of phosphate-buffered saline (PBS) using an IA-1C Microsprayer for IT delivery (PennCentury, Wyndmoor, PA), as described previously 54. One week later, the mice were randomly assigned to two groups: normoxia or hypoxia. The mice subjected to the hypoxia protocol received a weekly subcutaneous injection of SU5416 (20 mg/kg; Sigma Aldrich) for three weeks and maintained in hypoxia (10% O2) for 3 weeks. Then, mice were maintained in normoxia for one week before sacrifice. Control mice were administered normal saline solution.
Statistical analysis
Results are presented as mean ± standard error of the mean. Data were analyzed using an unpaired t-test for comparisons between means, 1-way analysis of variance with the Bonferroni correction for comparisons between >2 groups, or a two-way ANOVA followed by Bonferroni post-test if necessary. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA).
For detailed methods, see Supplemental Materials and Table III in the Supplement.
RESULTS
Hypermethylation of the BMPR2 promoter region is associated with decreased BMPR2 expression in human patients with PAH.
DNA methylation is a stable and reversible mechanism involved in gene silencing and regulation of the chromatin architecture55. Since the BMPR2 promoter region contains CpG islands19, we first sought to characterize the methylation status of the promoter region of the BMPR2 in iPAH human lung biopsies and non-PAH controls using Targeted-Bisulfite sequencing, one of the most widely used techniques in methylation mapping. Our results showed that PAH patient’s exhibit site-specific methylation in the BMPR2 promoter region compared to non-PAH controls (Figure 1A). Interestingly, we also found that pulmonary BMPR2 mRNA and protein expression levels were both significantly downregulated in PAH patients (Figure 1B–C). These data are in line with other studies19, 56–58, and exhibit positive association between BMPR2 promoter methylation and BMPR2 expression, and suggest that methylation of the BMPR2 promoter region may repress BMPR2 expression in PAH.
Figure 1. Hypermethylation of BMPR2 promoter correlates with decreased SIN3a expression in human PAH and PAH-animal model.
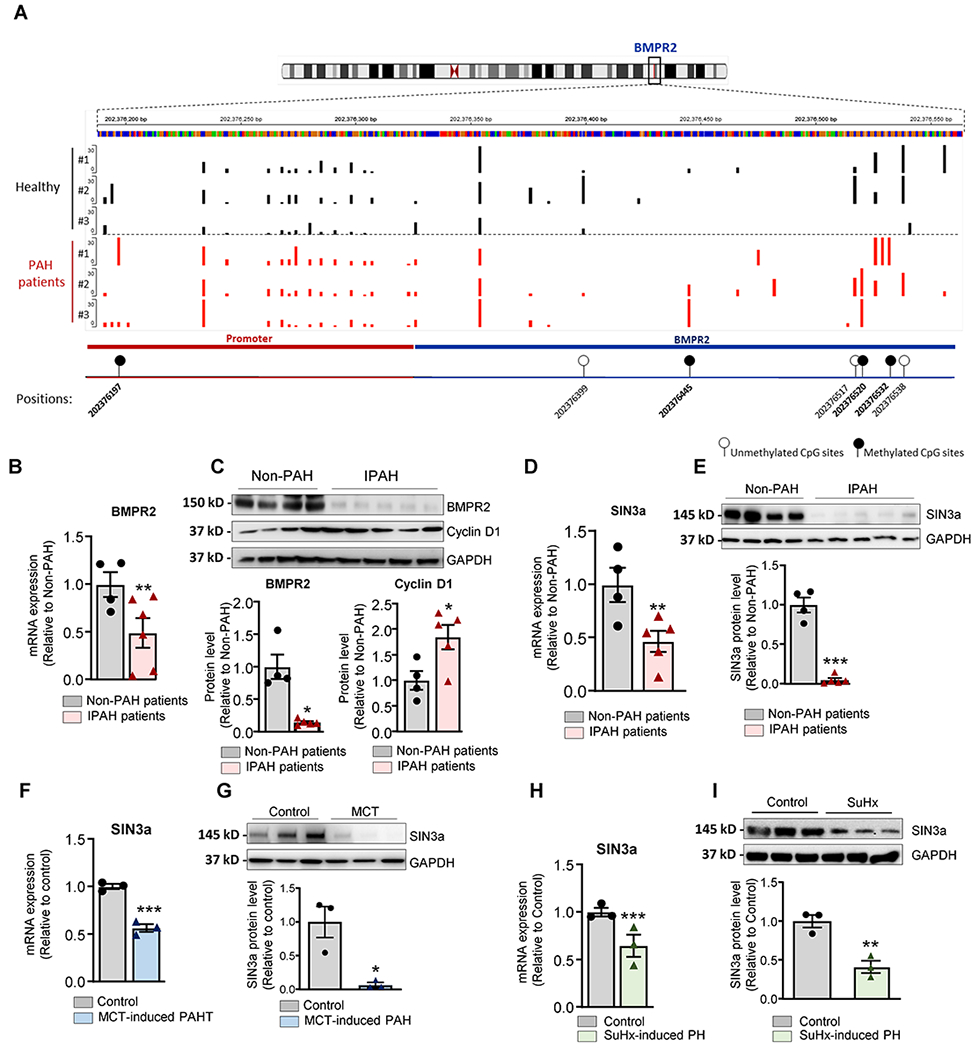
A. Methylation sites within the BMPR2 promoter region were identified by targeted-bisulfite sequencing. B. BMPR2 mRNA expression was analyzed by RT-qPCR in lung tissue from patients with idiopathic PAH (IPAH, n=5) and human non-PAH controls (n= 4). C. Upper panel, Representative BMPR2 and Cyclin D1 Western blot. Lower panel, the bar graph represents the quantification of BMPR2 and Cycin D1after correction for GAPDH by scanning densitometry. Data show fold stimulation with respect to control non-PAH. D. SIN3a mRNA expression was analyzed by RT-qPCR in lung homogenate tissue from patients with idiopathic PAH (IPAH, n=5) and non-PAH control lungs (n= 4). E. A representative blot of SIN3a protein expression (Upper Panel). Lower panel, bar graph represents the quantification of SIN3a correction for GAPDH. Data show fold stimulation with respect to control non-PAH. F. SIN3a mRNA expression was analyzed by RT-qPCR in lung tissue from control and MCT-induced PAH (n=3). G. SIN3a protein expression was assessed by Western blot in lung tissue from control and MCT-induced PAH (n=3). H. SIN3a mRNA expression was analyzed by RT-qPCR in lung tissue from control and Sugen Hypoxia-induced PAH mouse model (n=3). I. SIN3a protein expression was analyzed by western blot in lung tissue from control and Sugen Hypoxia-induced PAH mouse model (n=3). Data are presented as mean ±SEM; * = p<0.05, ** = p<0.01, *** P < 0.001.
Decreased expression of SIN3a in human PAH and experimental models of PH.
Among several chromatin remodeling proteins, we focused on the SIN3 complex in PAH. We examined whether high BMPR2 DNA methylation was associated with changes in SIN3 expression in PAH. Remarkably, real-time qPCR and western blot analysis showed that the expression levels of SIN3a mRNA and protein were both significantly decreased in the same human lung samples from PAH patients compared to non-PAH controls (Figure 1D–E). Next, we analyzed the expression of BMPR2 and SIN3a in the monocrotaline-induced PAH (MCT) model in rats and the Sugen-chronic Hypoxia-induced PH (SuHx) model in mice. Similarly, the results show that reduced BMPR2 expression (Figure I in the Supplement) is associated with low SIN3a expression at the mRNA and protein level in MCT (Figure 1F–G) as well as SuHx models compared to controls (Figure 1H–I). These results demonstrate that the downregulation of SIN3a in PAH occurs across several species in the setting of PAH. Of note, SIN3b transcripts did not change in human PAH and animal models of PH (Figure II in the Supplement). Likewise, mRNA expression of the subunits of the SIN3 complex was not changed in the animal models of PH (Figure III in the Supplement).
Furthermore, we noticed that the BMPR2 expression at the mRNA and protein level was lower in hPASMC compared to hPAECs (Figure 2A), and it was associated with lower protein expression of SIN3a. The analysis of SIN3a expression by immunofluorescence showed that SIN3a is mainly expressed in pulmonary vessel cells and co-localizes with ɑ-smooth muscle actin (α-SMA) in the medial layer of the pulmonary arterial walls of non-PAH controls in comparison with the remodeled vessels of iPAH lungs (Figure 2B). Interestingly, we also noted higher SIN3a expression in pulmonary artery endothelial cells in non-PAH patients, which is consistent with our previous results. Additionally, we also examined the methylation level of the BMPR2 promoter region in both cell types and found a higher basal methylation level of the BMPR2 promoter in hPASMC compared to hPAECs cells (Figure 2C). Therefore, we decided to investigate further the epigenetic role of SIN3a in the methylation of the BMPR2 promoter in PASMCs.
Figure 2. Higher levels of BMPR2 promoter methylation is associated with lower BMPR2 and SIN3a expression in hPASMC.
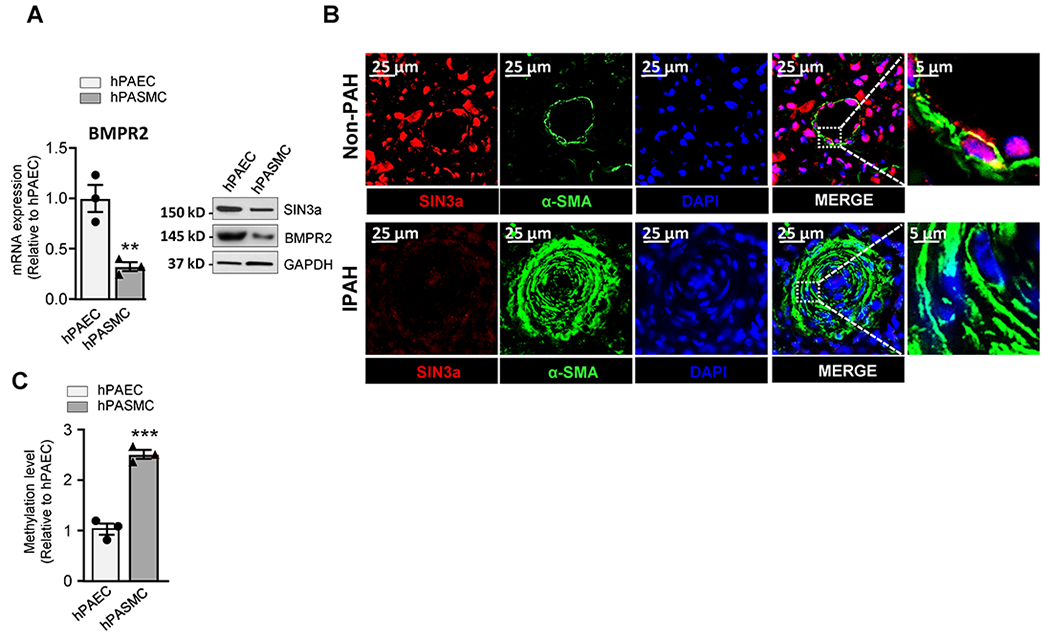
A. BMPR2 (left) mRNA and protein (right) expression of indicated proteins were analyzed by RT-qPCR and immunoblotting in hPASMC and hPAECs. A representative blot is shown. GAPDH was used as a loading control. B. Representative images of Sin3a (Red) and α-SMA (Green) co-immunostaining in lung sections of non-PAH controls and iPAH patients. Nuclei were stained with DAPI (blue). C. Methylation level of the BMPR2 promoter region was analyzed by MS-PCR in hPASMC and hPAECs. Data are presented as mean ±SEM; ns=not significant, ** = p<0.01, *** P < 0.001.
SIN3a decreased the proliferation and migration of hPASMC.
In PAH, vascular remodeling is characterized by enhanced proliferation, migration, and reduced apoptosis of pulmonary vascular cells24. To determine whether SIN3a directly regulates vascular cell growth, we employed a loss-of-function approach (siRNA.SIN3a) to knockdown SIN3a expression (Figure 3A) and a gain-of-SIN3a function using lentivirus vector-mediated overexpression to increase its expression (Figure 3D) without any significant changes in SIN3b expression (data not shown). Importantly, immunoblot analysis revealed that SIN3a silencing represses BMPR2 expression and potentiates Cyclin D1 expression (Figure 3A). Our results show that the depletion of SIN3a significantly increased hPASMC proliferation (Figure 3B) and migration (Figure 3C), whereas SIN3a overexpression reverses these effects (Figure 3E–F). In contrast, SIN3a overexpression increased BMPR2 protein expression in hPASMC while the marker of proliferation Cyclin D1 was decreased (Figure 3D). These results suggest that the loss of SIN3a expression may potentiate hPASMC proliferation in a BMPR2-dependent manner. To elucidate the role of SIN3a in the epigenetic regulation of BMPR2, we overexpressed SIN3a in hPASMC and hPAECs and examined the methylation of BMPR2 by Methyl Specific-PCR. Interestingly, we found that SIN3a overexpression decreased the methylation of the BMPR2 promoter region exclusively in hPASMC (Figure 2D; Figure IV in the Supplement).
Figure 3. SIN3a overexpression inhibits proliferation and potentiates BMPR2 expression in hPASMC.
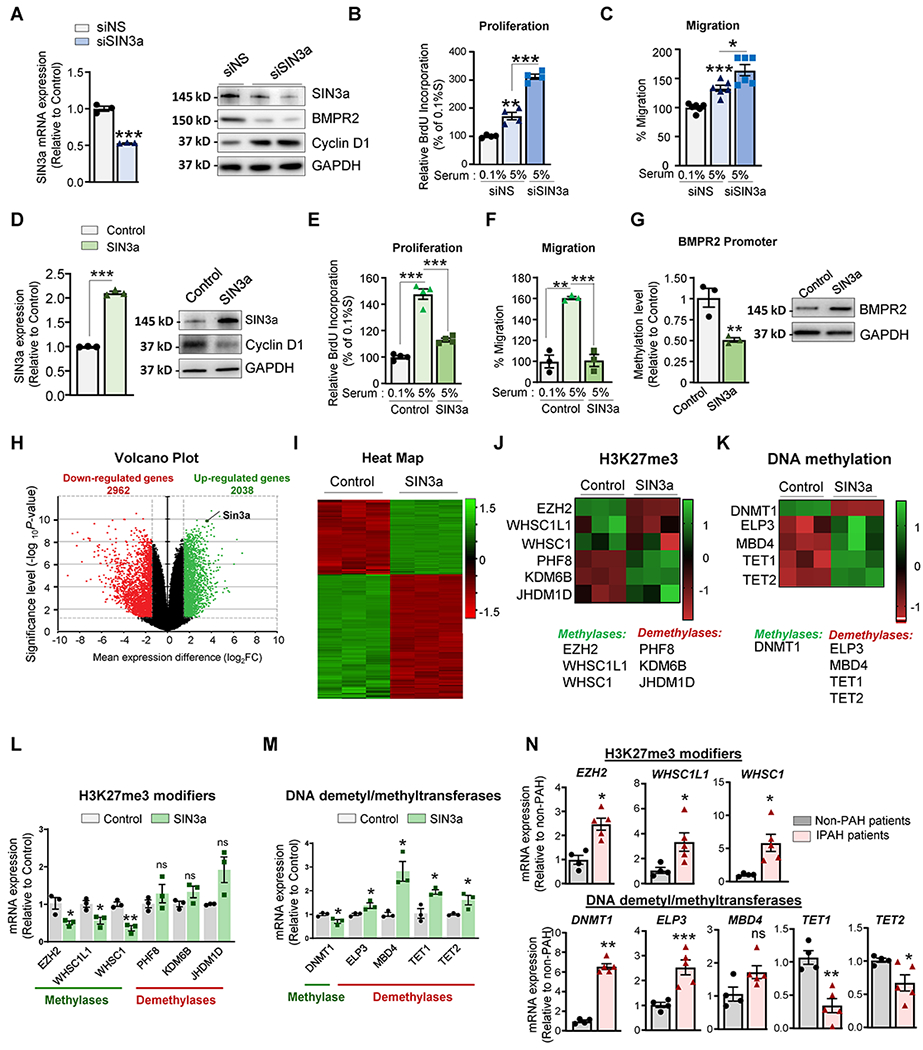
A. SIN3a mRNA (left) or protein (right) expression of the indicated proteins in hPASMC expressing a non-silencing (siNS) or the designated siRNAs against SIN3a (siSIN3a) were analyzed by RT-qPCR and western blot. B-C. Proliferation and migration level were respectively measured by BrdU assay and Boyden chamber-based cell migration assay in hPASMC expressing a non-silencing (siNS) or SIN3a siRNA and cultured either in 0.1% or 5% FBS for 72 hrs. D. SIN3a mRNA (left) or protein (right) expression of the indicated proteins was analyzed by RT-qPCR and immunoblotting in hPASMC overexpressing either an empty vector (Vector) or SIN3a (SIN3a) lentivirus. E-F. Proliferation and migration level were respectively measured by BrdU assay and Boyden chamber-based cell migration assay in hPASMC overexpressing SIN3a or an empty vector as control. Cells were treated either with 0.1% or 5% FBS for 72 hrs. G. Methylation level of the BMPR2 promoter region (left) and protein expression (right) was analyzed by MS-PCR and immunoblotting in hPASMC overexpressing either a control vector or SIN3a. H. Volcano plots showed the log2-fold changes and statistical significance of each gene calculated after differential gene expression analysis. Every point represents a gene. Red points indicate significantly down-regulated genes; green points indicate upregulated genes. I. Heatmap, displaying gene expression for each sample in the RNA-seq dataset, is shown. Each row of the heatmap represents a gene, and each column represents a sample. J-K. Heatmaps illustrating the gene expression of H3K27 modifiers and DNA dimethyl/methyltransferases are shown. L. Transcript levels of EZH2, WHSC1L1, WHSC1, PHF8, KDM6B, and JHDM1 were measured by RT-qPCR in hPASMC overexpressing either an empty vector as a control or SIN3a. M. DNMT1, ELP3, MBD4, TET1, and TET2 mRNA expression level was measured by RT-qPCR in hPASMC overexpressing SIN3a vs. control. N. Transcript levels of the indicated genes were assessed by RT-qPCR lung samples from iPAH patients and non-PAH controls. Data are presented as mean ±SEM; ns=not significant, * = p<0.05, ** = p<0.01, *** P < 0.001.
RNA-Sequencing of SIN3a overexpressing hPASMC revealed significant changes in the epigenetic profile.
To identify the underlying mechanisms and identify new molecular targets regulated by SIN3a, we undertook RNA-seq analysis in hPASMC overexpressing SIN3a. Among the top 5000 genes regulated by SIN3a, 2962 genes were downregulated, and 2038 genes were upregulated upon SIN3a overexpression in hPASMC compared to control cells expressing an empty vector as control (Figure 3H–I). Further analysis revealed that SIN3a overexpression regulated several genes involved in DNA and histone methylation (Figure 3J–K). Indeed, we identified several downregulated DNA and histone methyltransferase genes and upregulated demethyltransferase genes after SIN3a overexpression (Figure 3L–M). Further real-time qPCR analysis individually validated the downregulation of EZH2, WHSC1L1, WHSC1, and upregulation of the histone demethyltransferase PHF8, KDM6B, and JHDM1D (Figure 3L). Interestingly, the DNA methyltransferase gene DNMT1 was significantly repressed, whereas several genes implicated in DNA demethylation, including ELP3, MBD, ten-eleven translocation (TET)1, and TET2, were considerably increased by SIN3a (Figure 3M). Consistent with our previous results, the expression of EZH2, WHSC1L1, WHSC1, ELP3, and DNMT1 was increased in PAH human lung samples compared to non-PAH controls, whereas TET1 and TET2 were both decreased (Figure 3N).
Expression profile of the SIN3a-epigenetic partners in the lungs from PAH patients.
Previous studies have shown that EZH2 may induce hyperactivity of HDAC, which can foster proliferation and impair cell death59. As impairment of the epigenetic profile in PAH may trigger PAH progression, we quantified the expression levels of HDAC1 and EZH2. We found that HDAC1 mRNA and protein expression was markedly increased in human PAH and animal models of PAH (Figure V in the Supplement; Figure 4A, respectively), while the expression of HDAC2 was not changed (Figure V in the Supplement). Consistent with the increase of the EZH2 transcript level previously observed, we also detected an increased abundance of EZH2 levels in lung samples from PAH patients (Figure 4A) and animal models of PAH compared to controls (Figure VI in the Supplement). We also found that SUZ12, another subunit of the PRC2 complex, was significantly increased in iPAH lung samples compared to non-PAH controls (Figure VII in the Supplement). Simultaneously, SUZ12 expression was reduced upon SIN3a overexpression in hPASMC (Figure VII in the Supplement). Altogether, our results suggest that low BMPR2 expression is associated with high DNA methylation level, low SIN3a expression, and a seesaw elevation of HDAC1 and EZH2.
Figure 4. SIN3a regulates BMPR2 expression by modulating the epigenetic landscape in hPASMC.
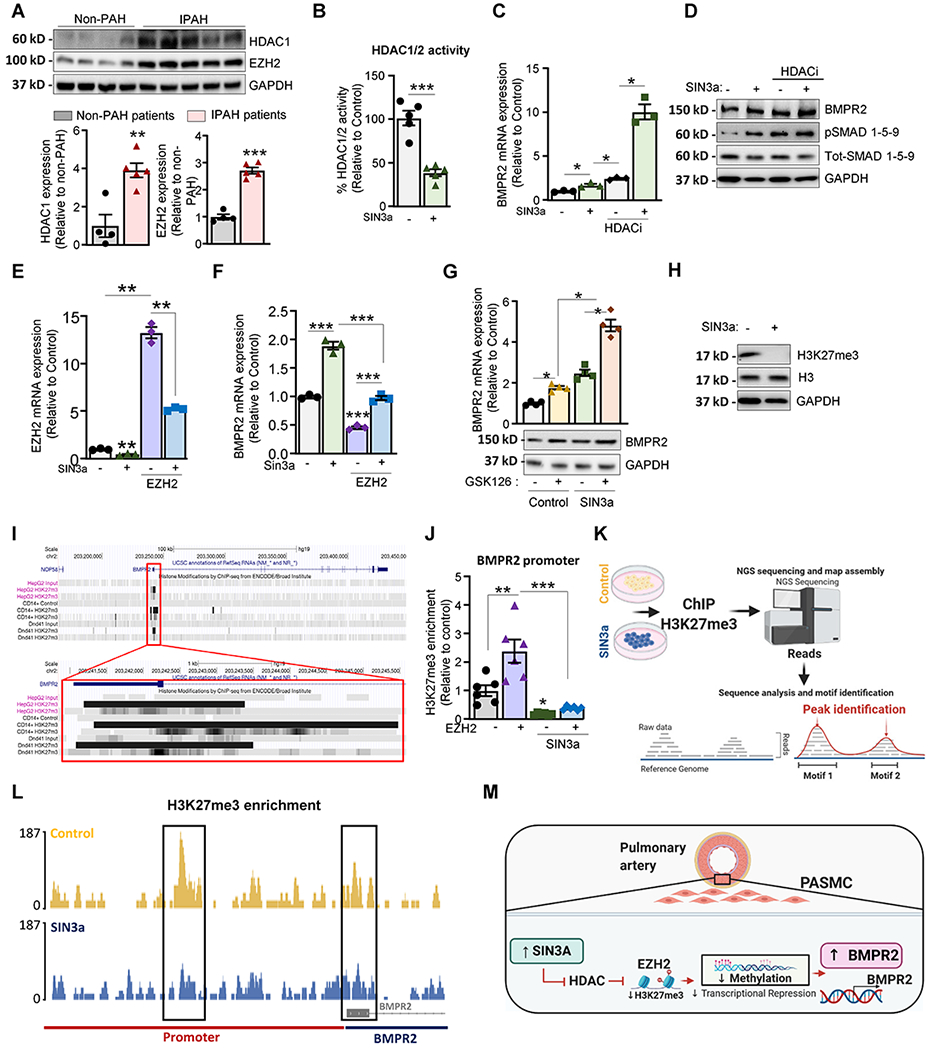
A. Lung homogenates from IPAH patients (n=5) and non-PAH controls (n=4) were analyzed by western blot to assess the protein expression of HDAC1 and EZH2. Upper panel, Representative HDAC1, and EZH2 Western blot. Lower panel, bar graph represents the quantification of HDAC and EZH2 normalized to GAPDH. B. HDAC1/2 activity was determined in hPASMC after the indicated treatments. C. hPASMC overexpressing either an empty vector as control or SIN3a were treated with Romidepsin (FK228, depsipeptide), a potent HDAC1 and HDAC2 inhibitor (HDACi), and BMPR2 mRNA level was determined by RT-qPCR. D. hPASMC overexpressing SIN3a alone or in combination with an HDAC1/2 inhibitor (HDACi) for 48 hr were analyzed by western blot for BMPR2, p-SMAD1/5/9, total-SMAD1/5/9, and GAPDH. E. EZH2 mRNA expression level was measured by RT-qPCR in hPASMC overexpressing SIN3a alone or in combination with lenti-EZH2. F. BMPR2 mRNA level was measured by RT-qPCR in the indicated conditions. G. BMPR2 mRNA (upper panel) and protein expression (lower panel) were analyzed by RT-qPCR and western blot in hPASMC treated with the potent EZH2 inhibitor (GSK126) for 48 hrs. alone or in combination with a lenti-SIN3a. H. hPASMC overexpressing either SIN3a or an empty vector were analyzed by immunoblotting for H3K27me3, Histone H3, and GAPDH. I. ENCODE H3K27me3-ChIP datasets were analyzed using UCSC genome browser to visualize H3K27me3-binding sites (highlighted in grey) at proximity of the promoter region of the human BMPR2 gene. J. ChIP-qPCR analysis of H3K27me3 at the BMPR2 promoter in hPASMC overexpressing either SIN3a or EZH2, alone or in combination. Promoter occupancy levels are expressed as the fold change relative to the control cells infected with an empty vector. K. ChIP-sequencing experimental procedure. L. H3K27me3 profile across the promoter region of BMPR2 on chromosome 2 in hPASMC overexpressing either a control vector or SIN3a. M. Schematic representation of the EZH2/H3K27me3 axis by which SIN3a regulates BMPR2 in hPASMC. Created with BioRender.com. Data are presented as mean ±SEM; * = p<0.05, ** = p<0.01, *** P < 0.001.
SIN3a decreases HDAC activity in hPASMC.
Selective HDAC inhibitors have shown promising results in several preclinical studies, but their use is associated with debilitating side effects60–62. As HDAC may interact with the SIN3 complex, we also tested the effect of SIN3a overexpression on HDAC1/2 activity. We found that SIN3a overexpression decreased HDAC1/2 activity in hPASMC (Figure 4B). Likewise, the loss of SIN3a using a specific siRNA increased HDAC1/2 activity (Figure VIII in the Supplement). Next, we assessed the mRNA and protein expression of BMPR2 in hPASMC overexpressing SIN3a alone or in combination with a potent HDAC1/2 inhibitor (HDACi; Romidepsin-FK228). We found that the level of BMPR2 expression and downstream signaling phospho-SMAD1/5/9 levels were significantly higher in SIN3a-overexpressing cells treated with HDACi (Figure 4C–D). Collectively, these findings indicate that HDAC1 inhibition potentiates SIN3a-induced BMPR2 expression in hPASMC.
SIN3a dynamically regulates BMPR2 DNA methylation and expression by modulating the EZH2 level and decreasing the H3K27me3 content.
Recent studies showed that histone modifications cooperate with DNA methylation to repress gene expression of tumor suppressor genes63, 64. Moreover, studies have suggested that dysregulation of EZH2 and HDAC activity may play a critical role in PAH 50, 65. Therefore, we sought to determine whether SIN3a regulates BMPR2 through EZH2-mediated histone H3K27 trimethylation (H3K27me3). To this end, we first measured the EZH2 transcript levels in hPASMC overexpressing SIN3a alone or in combination with EZH2 overexpression. The results show that SIN3a reduced the EZH2 transcript level (Figure 4E). Furthermore, EZH2 overexpression resulted in a decrease of BMPR2 transcript expression in hPASMC. Our data showed that SIN3a overexpression significantly restored BMPR2 expression and counteracted the repression of BMPR2 expression mediated by EZH2 (Figure 4F). To further validate our findings, we treated hPASMC with a potent and selective EZH2 inhibitor (GSK126) in cells overexpressing either SIN3a or an empty vector as control. Remarkably, EZH2 inhibition increased BMPR2 mRNA and protein levels, yet, in combination with SIN3a overexpression, this effect was potentiated (Figure 4G). Our results suggest that SIN3a restores BMPR2 expression by modulating the repressive mark H3K27me3 in hPASMC. Next, we analyzed the H3K27me3 level in hPASMC overexpressing SIN3a. Interestingly, SIN3a overexpression completely blocked the global H3K27me3 level (Figure 4H). Altogether, our results confirmed that SIN3a potentiates and restores BMPR2 expression by repressing both EZH2 and HDAC1. Previous studies reported that EZH2 works cooperatively with HDACs in the same molecular complex to repress gene transcription66. Furthermore, in line with these results, the EZH2-induced proliferation was abolished by HDACi or GSK126 in hPASMC. Additionally, we found that SIN3a overexpression inhibits hPASMC proliferation induced by EZH2 (Figure VII in the Supplement).
Since EZH2 maintains a transcriptional repression state of target genes by catalyzing the trimethylation of H3K27, we examined the role of SIN3a on the EZH2-induced H3K27me3 within the BMPR2 promoter. Interestingly, the analysis of previously published H3K27me3-ChIP ENCODE datasets showed enrichment of H3K27me3 at the BMPR2 promoter (Figure 4I). As SIN3a overexpression increased BMPR2 gene expression and inhibited the H3K27me3 level in hPASMC, we undertook additional analysis by ChIP-qPCR to measure the level of H3K27me3 in the BMPR2 promoter. We found that EZH2 increased H3K27me3 enrichment within the BMPR2 promoter region in hPASMC (Figure 4J). Moreover, this enrichment was decreased by SIN3a, suggesting that EZH2 may repress BMPR2 expression in an H3K27me3-dependent manner (Figure 4J). Altogether, the co-inhibition of the PRC2 and HDAC1 complex by SIN3a may further facilitate the TFs binding and potentiate the transcriptional activation of BMPR2 in hPASMC. Because previous studies have shown that EZH2 may serve as a recruiting platform for DNMT1 and induce aberrant DNA methylation by upregulating DNMT1 expression59, we also sought to determine whether EZH2 upregulation in PAH triggers hypermethylation of the BMPR2 promoter. To this end, the methylation level of the BMPR2 promoter region was also analyzed by MS-PCR in hPASMC overexpressing either SIN3a or EZH2 alone or in combination. We found that EZH2 overexpression increased the methylation level in the BMPR2 promoter in hPASMC (Figure IX in the Supplement). Our data showed that SIN3a overexpression significantly decreased EZH2-induced methylation and was associated with the restoration of BMPR2 expression in our previous experiments. Next, we analyzed the methylation level of the BMPR2 promoter by MS-PCR in hPASMC treated with HDAC inhibitor alone or in combination with EZH2 inhibitor (GSK126). We found that both HDACi and GSK126 significantly inhibited the methylation level of BMPR2 in hPASMC (Figure IX in the Supplement). Notably, the combined pharmacological inhibition of EZH2 and HDACs markedly upregulated BMPR2 expression in hPASMC (Figure IX in the Supplement), which may reveal a synergic mechanism between both pathways involved in the regulation of BMPR2 expression.
Finally, we further sought to evaluate the role of EZH2, DNMT1 and TET1 on the level of H3K27me3 in the BMPR2 promoter region by ChIP-qPCR in hPASMC. We found that SIN3a knockdown significantly increased the H3K27me3 enrichment within the BMPR2 promoter region (Figure IX in the Supplement) while the pharmacological inhibition of EZH2 or DNMT1 significantly reduced the H3K27me3 content (left panel). Similarly, we found that DNMT1 knockdown decreased the H3K27me3 level while TET1 silencing increased the H3K27me3 enrichment in the BMPR2 promoter (right panel). We further characterized and defined the H3K27me3 profile by ChIP-sequencing in PASMC overexpressing either a control vector or SIN3a (Figure 4K; Figure IX in the Supplement). Analysis of the H3K27me3 profile further confirmed and validated our previous ChIP-qPCR data and provided further insights in regards to other non-targeted genes. As expected, we noticed that SIN3a overexpression affected the H3K27me3 enrichment of BMPR2 as well as other genes (Figure 4L). Collectively, our results suggested that SIN3a overexpression increased BMPR2 expression by decreasing the EZH2-induced H3K27me3 enrichment within the BMPR2 promoter region (Figure 4M).
SIN3a overexpression increases TET1 and inhibits DNMT1 expression in hPASMC.
Recently, it has been shown that EZH2 and TET1 operate in trans to modulate the epigenetic landscape of targeted-genes 67. Additional studies have identified MECP2, DNMT1, HDAC, EZH2, and SIN3a as TET1-interacting proteins68. Therefore, we first assessed the expression level of TET1, DNMT1, and MECP2 in human PAH samples and then investigated the role of TET1 on BMPR2 expression using a shRNA approach to knockdown TET1 expression. The results showed that the TET1 protein level was significantly downregulated, while both DNMT1 and MECP2 were increased in human PAH samples compared to non-PAH controls (Figure 5A). The qRT-PCR analysis revealed that DNMT1 and MECP2 mRNA expression were both upregulated in animal models of PH compared to controls (Figure X in the Supplement). Immunoblotting and qRT-PCR also confirmed a reduction in the TET1 expression level after shTET1-mediated knockdown (Figure 5B). We found that TET1-depleted hPASMC showed a much higher methylation level of the BMPR2 promoter region (Figure 5C), whereas SIN3a overexpression reversed this effect (Figure 5C). Consequently, shTET1 prevented BMPR2 expression at the mRNA and protein levels, while SIN3a restored its expression in TET1-depleted hPASMC (Figure 5E). Inversely, DNMT1 knockdown (Figure 5D) potentiated BMPR2 expression (Figure 5E). The combination of SIN3a overexpression, along with shDNMT1, resulted in a much greater BMPR2 expression (Figure 5E). Thus, these data pinpoint that SIN3a regulates BMPR2 gene expression through methylation-dependent mechanisms tightly regulated by TET1 and the EZH2/DNMT1 axis.
Figure 5. SIN3a overexpression decreased the methylation level of the BMPR2 promoter in hPASMC.
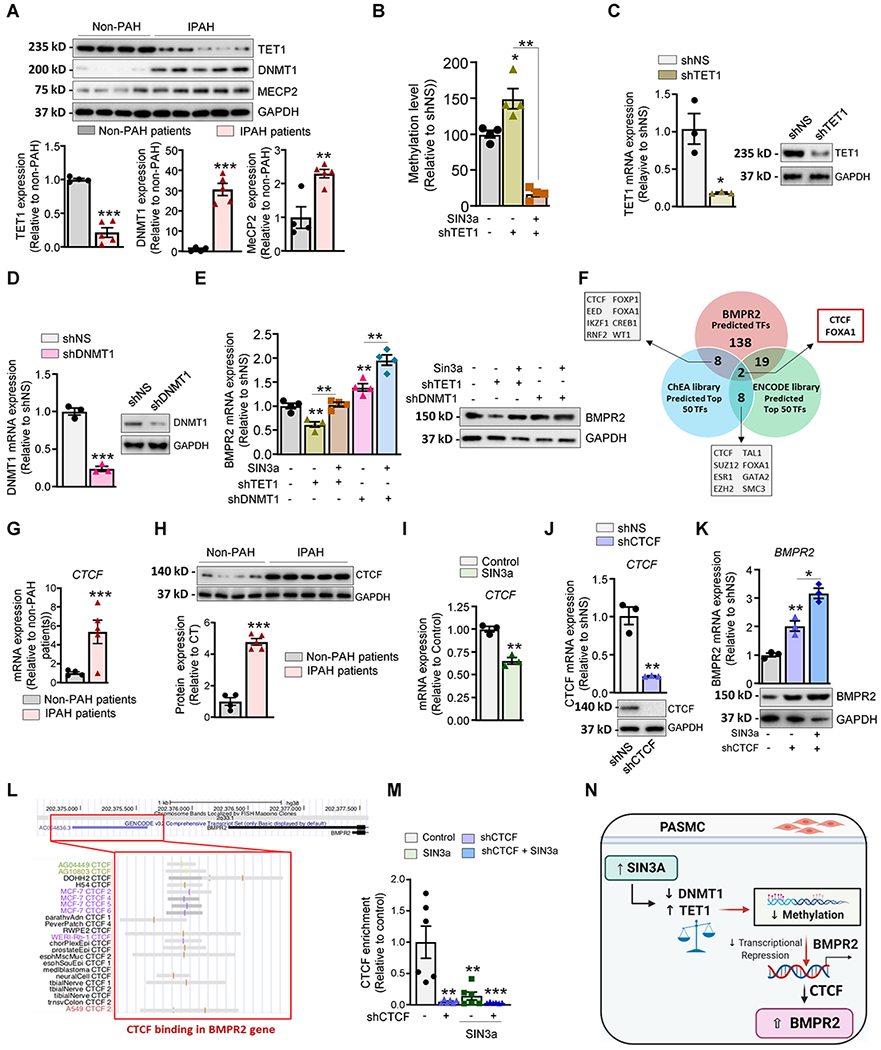
A. Representative immunoblot of TET1, DNMT1, and MECP2 expression in lung homogenates from non-PAH controls (n=4) and IPAH patients (n=5). The graph represents the quantification of the indicated proteins, normalized to GAPDH. B. TET1 mRNA (left) or protein (right) expression was analyzed by RT-qPCR and immunoblotting in hPASMC overexpressing either a non-silencing shRNA (shNS) or a specific shRNA against TET1 (shTET1). C. The methylation level of the BMPR2 promoter region was analyzed by MS-PCR in hPASMC overexpressing shTET1 alone or in combination with lenti-SIN3a. D. DNMT1 mRNA (left) or protein (right) expression was analyzed by RT-qPCR and immunoblotting in hPASMC overexpressing either a non-silencing shRNA (shNS) or a specific shRNA against DNMT1 (shDNMT1). E. BMPR2 mRNA and protein expression levels were measured by RT-qPCR and immunoblotting in the indicated conditions. F. RNA-seq datasets were analyzed using ChEA and encode library to identify the top 50 transcription factors that are regulated by SIN3a (TF). Comparative analysis was performed using the predicted TFs that regulate BMPR2 expression. G-H. CTCF mRNA and protein expression were assessed by RT-qPCR (G) and immunoblot (H) in lung homogenates from non-PAH controls (n=4) and iPAH patients (n=5). I. CTCF mRNA expression was assessed by RT-qPCR in hPASMC overexpressing either an empty vector as control or lenti-SIN3a. J. CTCF mRNA (left) or protein (right) expression was analyzed by RT-qPCR (left) and immunoblotting (right) in hPASMC overexpressing either a non-silencing shRNA (shNS) or a specific shRNA against CTCF (shCTCF). K. BMPR2 mRNA (left) and protein expression (right) were measured by RT-qPCR and immunoblot in hPASMC overexpressing shCTCF alone or in combination with lenti-SIN3a. L. ENCODE CTCF-ChIP datasets were analyzed using UCSC genome browser to visualize CTCF-binding sites (highlighted in grey) at proximity of the promoter region of the human BMPR2 gene. M. ChIP-qPCR analysis monitoring binding of CTCF at the BMPR2 promoter in CTCF-depleted hPASMC alone or in combination with SIN3a overexpression. Promoter occupancy levels are expressed as the fold change relative to the control cells infected with an empty vector. N. Schematic representation of the SIN3a/TET1 and SIN3a/DNMT1 axis promoting the demethylation of the BMPR2 promoter and restoring its expression level in hPASMC. Created with BioRender.com. Data are presented as mean ±SEM; * = p<0.05, ** = p<0.01, *** P < 0.001.
To delineate the transcriptional factors (TFs) regulatory mechanisms of BMPR2 expression, we further analyzed the transcriptome dataset obtained using SIN3a overexpressing hPASMC to identify the top 50 TFs using the ENCODE and ChEA libraries. We then compared these TFs to the predicted TFs based on the BMPR2 promoter sequence (Figure. 5F). We identified the CCCTC-binding factor (CTCF), a ubiquitously, and a highly conserved zinc finger protein as a potential TF candidate. Previous studies have reported that CTCF can act as a transcriptional repressor69, 70. To elucidate the underlying mechanism, we examined CTCF expression in human PAH lung samples and hPASMC overexpressing SIN3a. Remarkably, we found that CTCF mRNA and protein expression levels are increased in human PAH lung samples (Figure 5G–H). As expected, SIN3a overexpression repressed CTCF expression in hPASMC (Figure 5I). We, therefore, hypothesized that the loss of CTCF increases BMPR2 expression. To further test our hypothesis, we silenced CTFC expression using a specific shRNA (Figure 5J) alone or in combination with SIN3a overexpression in hPASMC. We first noted that BMPR2 expression was significantly increased in CTCF-depleted cells (Figure 5K). Interestingly, this effect was potentiated by SIN3a overexpression (Figure 5K). Altogether, the above results suggested that SIN3a may increase BMPR2 expression by relieving CTCF-mediated repression of BMPR2. Interestingly, further bioinformatic analysis of the ENCODE ChIP-seq data datasets allowed us to identify enrichment of CTCF (highlighted in grey) at the proximity of the BMPR2 promoter (Figure 5L). Finally, we provided evidence of CTCF abundance in the BMPR2 promoter in hPASMC by ChIP-qPCR using an anti-CTCF ChIP-grade antibody, which was completely abrogated in CTCF-depleted cells or SIN3a-overexpressing cells (Figure 5M). Collectively, our results suggested that SIN3a decreased the methylation level of the BMPR2 promoter region by upregulating TET1 and repressing DNMT1 expression, which ultimately affects the CTCF binding to the BMPR2 promoter region and potentiates BMPR2 expression in hPASMC (Figure 5N).
Furthermore, the markers of SMC phenotype α smooth muscle actin (α-SMA), Myosin Heavy Chain 11 (Myh11), and Smooth Muscle 22-Alpha (SM-22) were assessed in SIN3a overexpressing or depleted hPASMC (Figure XI in the Supplement). The results show no significant changes in the mRNA expression levels of the SMC markers (Figure XI in the Supplement). Similarly, modulation of DNMT1, TET1, or EZH2 did not alter the expression of these markers. However, further investigation would be necessary to determine the consequences of epigenetic modulations on cell-phenotype in long-term studies.
Therapeutic intratracheal delivery of SIN3a reversed PAH by decreasing pulmonary artery and RV pressures in a monocrotaline-induced PAH rat model.
To evaluate the therapeutic efficacy of lung-targeted gene transfer of SIN3a in vivo, we first used the MCT-induced PAH model. Using a therapeutic strategy, rats were randomly allocated to a control or PAH group that received a single MCT injection. Three weeks later, rats from the MCT group were randomly assigned to intratracheally (IT) receive either aerosolized AAV1 encoding human SIN3a (AAV1.hSIN3a) or AAV1 encoding luciferase as control (AAV1.CT) using an IA-1C microsprayer (Figure 6A). We first assessed the distribution efficiency and specificity of IT delivery of aerosolized AAV1.hSIN3a by analyzing the viral genome copies in lung and RV samples. Our results showed that the number of exogenous SIN3a genome copies in the lungs was significantly higher in the AAV1.hSIN3a-treated group in comparison with the AAV1.CT group. No viral genome copy was detected in the right ventricle (RV) of the AAV1.hSIN3a-treated group (Figure XII in the Supplement). These results demonstrated the specificity of the local IT delivery method toward the lungs and confirmed the on-target specificity of the AAV1.hSIN3a after in vivo gene transfer by IT delivery. To further demonstrate the efficacy of gene transfer, we measured the mRNA expression of SIN3a in the lungs by RT-qPCR using specific primers designed to amplify human (exogenous) and rat (endogenous) SIN3a isoforms. The exogenous SIN3a mRNA levels were highly increased in the lungs of AAV1.hSIN3a-treated animals compared to control animals (Figure 6B). Surprisingly, local IT delivery of AAV1.hSIN3a in the rat lungs also restored the endogenous rat SIN3a transcript in MCT-treated animals compared to AAV1.CT treated animals (Figure XII in the Supplement). Subsequently, the SIN3a protein expression was restored in the MCT animals-treated with AAV1.hSIN3a compared to AAV1.CT (Figure 6C). These findings were further confirmed by immunofluorescent staining of the remodeled pulmonary arteries (Figure 6D). Our data showed that SIN3a expression was significantly downregulated in the MCT-AAV1.CT-treated group. IT delivery of AAV1.hSIN3a successfully restored SIN3a expression in MCT-PAH rats treated with AAV1.hSIN3a (Figure 6D). Next, the pulmonary artery and RV pressures, as well as pulmonary vascular remodeling and RV hypertrophy, were measured four weeks post-AAV instillation. Remarkably, AAV1.hSIN3a therapy significantly decreased the mean pulmonary arterial pressure (mPAP) and distal pulmonary vascular remodeling compared to AAV1.CT-treated MCT group (Figure 6E–F). Moreover, compared to the AAV1 control group, SIN3a overexpression limited adverse hemodynamic profiles by attenuating RV systolic pressure (RVSP) (Figure 6G, left panel) and increasing cardiac output without affecting the heart rate (Figure XII in the Supplement). Subsequently, RV hypertrophy, as determined by the Fulton index, was significantly reduced in AAV1.SIN3a treated-group (Figure 6G, right panel). Similarly, wheat-germ agglutinin staining of RV heart sections showed increased cardiomyocyte cross-sectional area in the MCT treated-group (Figure 6H). AAV1.hSIN3a treatment significantly decreased the RV cardiomyocytes size (Figure 6H) and the transcript levels of ANP, BNP, and βMHC, common markers of the “fetal gene program" (Figure XII in the Supplement). Altogether, these results suggest that lung-targeted instillation of AAV1.hSIN3a reduces RV remodeling.
Figure 6. Therapeutic intratracheal delivery of AAV1.hSIN3a ameliorates MCT-induced PAH in rats.
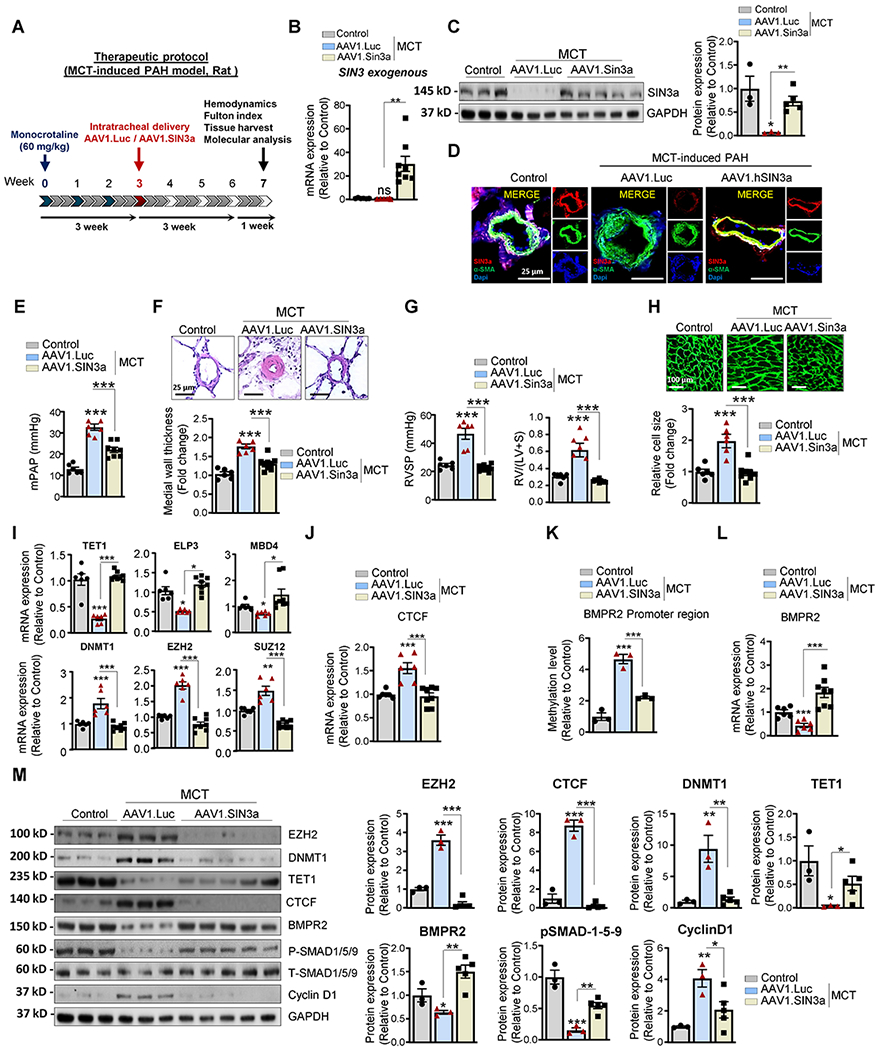
A. Schematic of the experimental design to assess the therapeutic efficacy of AAV1.hSIN3a gene therapy in the rat MCT-induced PAH model. Tissues were collected at week 7 for molecular and histology analysis. B. Exogenous SIN3a mRNA level was assessed in lung tissues by RT-qPCR to determine the efficiency of the IT delivery of AAV1.hSIN3a gene transfer. C. SIN3a protein expression was assessed by immunoblotting in the sham control group, AAV1.CT+MCT and AAV1.hSIN3a+MCT groups. D. SIN3a expression (red) and α SMA (green) in pulmonary arterioles were detected by immunofluorescence using lung sections from the sham control group, AAV1.CT+MCT and AAV1.hSIN3a+MCT groups. Nuclei were counterstained with DAPI (blue). E. Pulmonary artery pressure (PAP) was assessed in control, and MCT-induced PAH rats treated either with AAV1.CT or AAV1.hSIN3a. F. Representative hematoxylin and eosin-stained lung sections of the indicated rats. The graph represents the quantification of the medial thickness. G. The right ventricular systolic pressure (RVSP) (left) and Fulton index RV/(LV + S) (right) were assessed in control and MCT-induced PAH rat treated either with AAV1.CT or AAV1.hSIN3a. H. RV sections were stained with fluorescence-tagged wheat germ agglutinin to examine RV cardiomyocyte cross-sectional area. I. Expression of the transcripts TET1, ELP3, MBD4, DNMT1, EZH2, SUZ12 were measured by RT-qPCR in lungs from control and MCT-induced PAH rats treated either with AAV1.CT or AAV1.hSIN3a. J. Expression of the transcripts CTCF was measured by RT-qPCR in lungs from control and MCT-induced PAH rats treated either with AAV1.CT or AAV1.hSIN3a. K. The methylation level of the BMPR2 promoter region was analyzed by methyl-specific PCR (MS-PCR) in control and MCT-induced PAH rats treated either with AAV1.CT or AAV1.hSIN3a. L. BMPR2 mRNA expression was assessed in the indicated groups by RT-qPCR. M. Lung homogenates were analyzed by western blot for the indicated proteins. Representative western blots (left panel) and respective densitometric quantitation (right panel) for EZH2, BMPR2, DNMT1, TET1, CTCF, pSMAD1/5/9, and Cyclin D1. Protein expression was normalized to Total-SMAD and GAPDH. Data are presented as mean ±SEM; ns=not significant, * = p<0.05, ** = p<0.01, *** P < 0.001.
Furthermore, aerosolized AAV1.hSIN3a markedly restored the transcript levels of TET1, ELP3, and MBD4 and reduced the expression of DNMT1, EZH2, and SUZ12 (Figure 6I). Likewise, AAV1.hSIN3a led to a significant decrease of CTCF expression transcript level compared to AAV1.CT-treated MCT rats (Figure 6J), validating our in vitro results. Concomitantly, AAV1.hSIN3a therapy significantly reduced the methylation level of the BMPR2 promoter region (Figure 6K) and increased BMPR2 mRNA expression level compared to the AAV1.CT treated group (Figure 6L). In addition, the immunoblot analysis validated our previous qPCR results and confirmed that SIN3a gene transfer increased TET1 and BMPR2 protein expression as well as BMPR signaling pSMAD1/5/9 (Figure 6M). In contrast, the protein expression of EZH2, DNMT1, CTCF, and Cyclin D1 was significantly decreased (Figure 6M), which is also consistent with our previous in vitro results.
AAV1.hSIN3a gene transfer decreased RV pressures, vascular and cardiac remodeling in the SuHx induced-PAH mouse model.
To further test the therapeutic potential of AAV1.hSIN3a gene therapy in vivo, we used the combination of a vascular endothelial growth factor receptor antagonist, Sugen 5416 (SU5416), and chronic hypoxia (SuHx) in mice as a PAH model. For the treatment protocol, AAV1 encoding human SIN3a was delivered three weeks after SuHx, and mice were sacrificed four weeks after gene therapy (Figure 7A). For the prevention protocol, AAV1-mediated human SIN3a was IT delivered one week before SuHx, and the animals were sacrificed five weeks post-gene transfer (Figure XIII and XIV in the Supplement). Consistent with the results observed in the rat MCT-induced PAH model, AAV1.hSIN3a was only expressed in the lungs of the AAV1.hSIN3a-treated group in both protocols (Figure XIII and XIV in the Supplement, respectively). Similarly, IT delivery of AAV1.hSIN3a increased exogenous human SIN3a expression and restored endogenous mouse SIN3a mRNA (Figure 7B, Figure XIII and XIV in the Supplement) and SIN3a protein level (Figure 7C). The long-term expression of SIN3a prevented small artery muscularization and remodeling compared to AAV1.CT in both protocols (Figure 7D, Figure XIV in the Supplement). In addition, AAV1.hSIN3a administration attenuated the maladaptive RV remodeling by significantly decreasing the RV systolic pressure (RVSP) and RV hypertrophy (Figure 7E, Figure XIV in the Supplement). Moreover, the treatment with AAV1.hSIN3a significantly decreased RV cardiomyocyte hypertrophy and the expression of the cardiac hypertrophy-associated markers (Figure 7F, Figure XIII and XIV in the Supplement). Similarly, the restoration of SIN3a significantly upregulated TET1, ELP3, and MBD4 mRNA levels and downregulated DNMT1, EZH2, and SUZ12 expression (Figure 7G, Figure XV in the Supplement). By decreasing CTCF expression (Figure 7H, Figure XV in the Supplement) and the methylation level of the BMPR2 promoter region, SIN3a restored the expression of BMPR2 (Figure 7I–J, Figure XV in the Supplement). Immunoblot analysis showed that restoration of SIN3a level by local gene delivery significantly increased TET1 as well as BMPR2 and pSMAD1/5/9. We also noticed a lower expression level of EZH2, DNMT1, CTCF, and Cyclin D1 in the AAV1.hSIN3-treated-group in lung homogenates from SuHx mice treated with AAV1.hSIN3a (Figure 7K).
Figure 7. Therapeutic intratracheal delivery of AAV1.hSIN3a reversed SuHx-induced PAH.
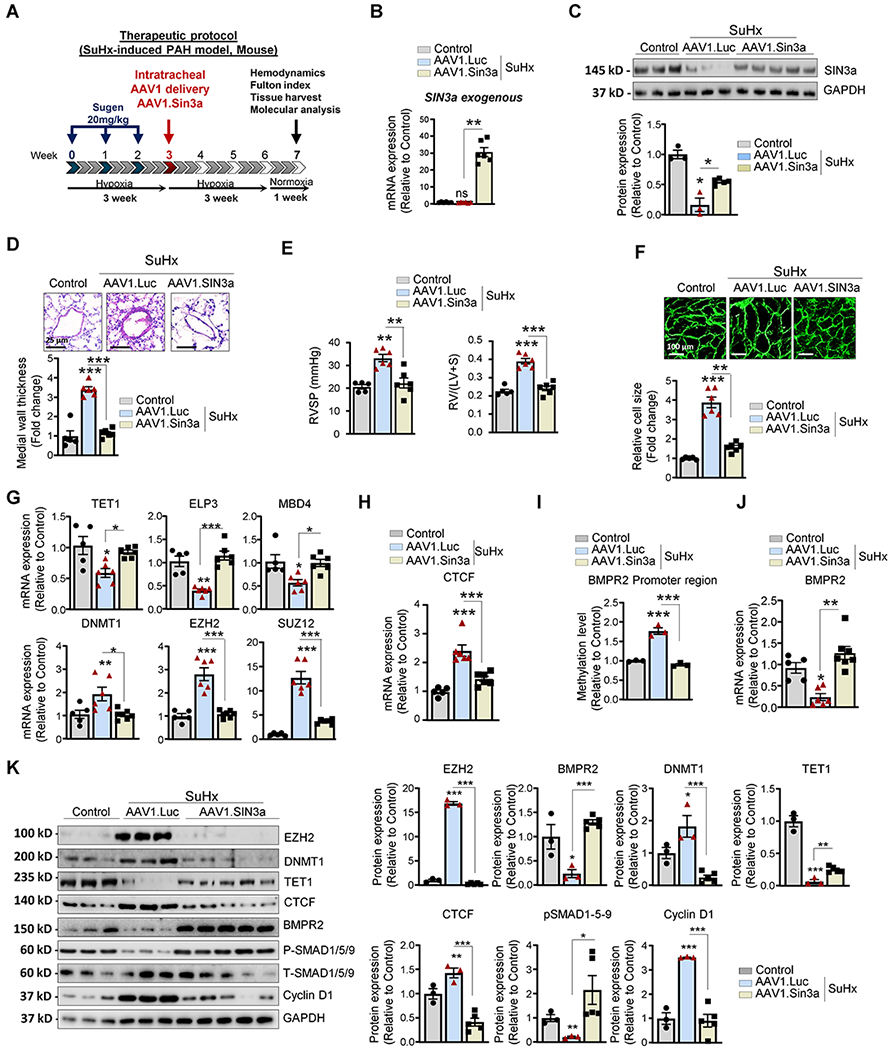
A. Schematic of the experimental design to assess the therapeutic efficacy of AAV1.hSIN3a gene therapy in the SuHx-induced PAH mouse model. Tissues were collected at week 7 for molecular and histology analysis. B. Exogenous SIN3a mRNA level was assessed in lung tissues by RT-qPCR in the mice lungs to determine the efficiency of the IT delivery of AAV1.hSIN3a gene transfer. C. SIN3a protein expression was assessed by immunoblotting. D. Representative hematoxylin and eosin-stained lung sections of the indicated mice. The graph represents the quantification of the medial thickness. E. RVSP (left) and Fulton’s index (right) were determined in control, and SuHx-induced PAH mice treated either with AAV1.CT or AAV1.hSIN3a. F. RV sections were stained with fluorescence-tagged wheat germ agglutinin to measure RV cardiomyocyte cross-sectional area. G. Expression of the transcripts TET1, ELP3, MBD4, DNMT1, EZH2, SUZ12 was measured by RT-qPCR in lungs from control, and SuHx-induced PAH mice treated either with AAV1.CT or AAV1.hSIN3a. H. mRNA expression of CTCF was measured by RT-qPCR. I. The methylation level of the BMPR2 promoter region was analyzed by MS-PCR in control, and SuHx-induced PAH mice treated either with AAV1.CT or AAV1.hSIN3a. J. BMPR2 mRNA expression was assessed in the indicated groups by RT-qPCR. K. Lung homogenates were analyzed by immunoblotting for the indicated proteins. Representative western blots and respective densitometric quantitation for EZH2, BMPR2, DNMT1, TET1, CTCF, pSMAD1/5/9, and Cyclin D1 are shown. Protein expression was normalized to Total-SMAD and GAPDH. Data are presented as mean ±SEM; ns=not significant, * = p<0.05, ** = p<0.01, *** P < 0.001.
We further confirmed our in vivo results using a lentivirus-based approach in the SuHx-induced PAH mice model. To this end, IT delivery of lentiviral vector encoding for human SIN3a was administered alone or in combination with lenti-EZH2 (Figure XVI in the Supplement). We first assessed the expression of SIN3a in mice four weeks post-AAV1.hSIN3a delivery. The results showed that the SIN3a transcript was increased in lenti.SIN3a-treated mice compared to lenti-CT (Figure XVI in the Supplement). Furthermore, instillation of lenti-EZH2 significantly increased the EZH2 level (Figure XVI in the Supplement). Interestingly, the restoration of SIN3a expression decreased EZH2 mRNA expression in mice co-treated with lenti-SIN3a and EZH2 (Figure XVI in the Supplement), which is consistent with our in vitro findings. Remarkably, SIN3a lung treatment decreased RVSP and prevented RV hypertrophy (Figure XVI in the Supplement), as demonstrated by lower Fulton index and decreased cardiomyocyte size (Figure XVI in the Supplement). Moreover, vascular remodeling was also prevented by SIN3a overexpression compared to control or EZH2-treated animals (Figure XVI in the Supplement). Consistently, EZH2 overexpression significantly increased CTCF while decreasing BMPR2 mRNA expression (Figure XVI in the Supplement). The combination gene transfer with lenti.SIN3a markedly decreased CTCF and restored BMPR2 expression (Figure XVI in the Supplement). Moreover, the treatment with lenti.SIN3a alone or in combination with EZH2 increased or restored the protein levels of TET1, BMPR2, and pSMAD1/5/9 signaling. In contrast, the expression levels of EZH2, DNMT1, CTCF, and Cyclin D1 were decreased (Figure XVI in the Supplement), validating our in vitro and in vivo data in different PAH models.
Lentivirus-mediated SIN3a gene transfer attenuated SuHx-induced PAH in shRNA-mediated BMPR2 knockdown mice.
To further validate that the protective effects of SIN3a gene therapy are mediated through BMPR2, we used a specific shRNA lentivirus-based approach via IT delivery to knockdown BMPR2 with a simultaneous overexpression of SIN3a in mice. After one week, the mice were subjected to SuHx for three weeks to induce PAH (Figure 8A). First, we noticed that BMPR2 silencing significantly increased RVSP under normoxia conditions and potentiated the SuHx-increased RVSP and RV hypertrophy (Figure 8B). Markedly, SIN3a overexpression significantly attenuated RVSP and reversed RV hypertrophy despite the loss of BMPR2 in normoxia and SuHx conditions (Figure 8B). In addition, histological analysis showed a significant reduction in distal pulmonary vascular remodeling and cardiomyocyte size in shBMPR2-mice treated with Lenti.SIN3a (Figure 8C–D). At the molecular level, the lentivirus-mediated SIN3a overexpression restored BMPR2 mRNA and protein expression levels in the lungs of animals-treated with sh.BMPR2 in both normoxia and SuHx-induced PAH conditions (Figure 8E–F). Surprisingly, we noticed that the loss of BMPR2 drastically decreased SIN3a expression, suggesting the existence of a possible feedback mechanism (Figure 8E, right panel). Finally, we modulated BMPR2 expression after SIN3a knockdown or overexpression in hPASMC (Figure XVII in the Supplement). Then, we measured Cyclin D1 and BMPR2 expression by qPCR and measured the proliferation level by BrdU assay. Enhanced BMPR2 expression significantly counteracted hPASMC proliferation induced by SIN3a silencing, suggesting that SIN3a regulates proliferation through BMPR2 (Figure XVII in the Supplement). Similar results in BMPR2-depleted cells were observed after SIN3a overexpression (Figure XVII in the Supplement). Consistently, our results confirmed that SIN3a overexpression inhibited hPASMC proliferation (Figure XVII in the Supplement) and found that SIN3a knockdown significantly potentiated hPASMC proliferation induced by a high concentration of serum (Figure XVII in the Supplement). Our results were also consistent with Cyclin D1 levels (Figure XVII in the Supplement).
Figure 8. Lentivirus-mediated SIN3a gene transfer attenuated SuHx-induced PAH in shRNA-mediated BMPR2 knockdown mice.
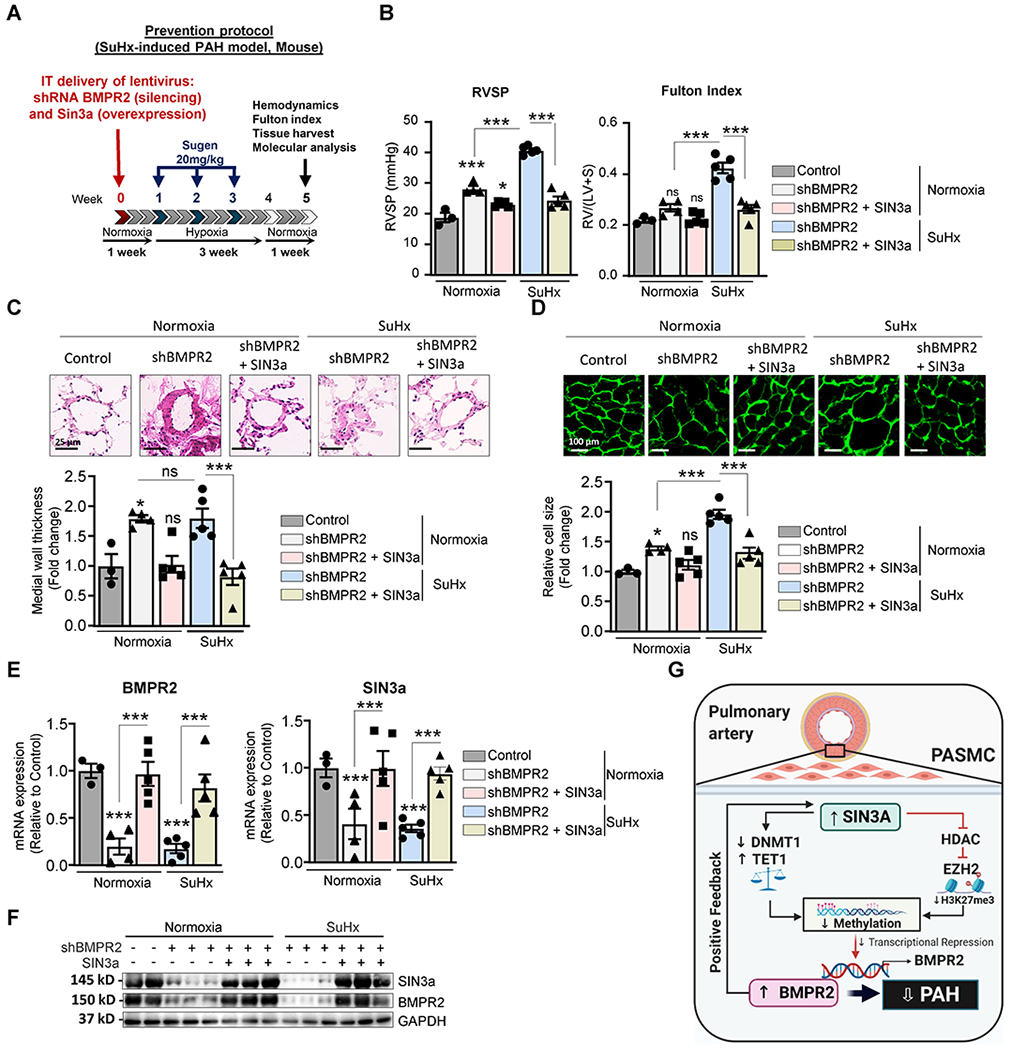
A. Schematic of the experimental design to assess the therapeutic efficacy of SIN3a lentivirus-mediated gene transfer using the SuHx-induced PAH mouse model. Tissues were collected at week 7 for molecular and histology analysis. B. RVSP (left) and Fulton’s index (right) were determined in the indicated conditions. C. Representative hematoxylin and eosin-stained lung sections of the indicated mice. The graph represents the quantification of the medial thickness. D. RV sections were stained with fluorescence-tagged wheat germ agglutinin to measure RV cardiomyocyte cross-sectional area. The graph represents the quantification of the cardiomyocyte size. E. mRNA expression of BMPR2 (left) and SIN3a (right) was assessed by RT-qPCR in the indicated conditions. F. Lung homogenates were analyzed by western blot to assess the protein expression of SIN3a and BMPR2 in control and SuHx-mice treated with shBMPR2 alone or in combination with SIN3a. G. Schematic representation of the molecular mechanisms by which SIN3a inhibits PAH. SIN3a restores BMPR2 expression by a dual mechanism in hPASMC. Our results showed that SIN3a inhibited EZH2 expression and the H3K27me3 contents within the BMPR2 promoter region while decreasing the methylation level of the BMPR2 promoter region by upregulating TET1 and repressing DNMT1 activity, which affects the CTCF binding to the BMPR2 promoter region. Created with BioRender.com. Data are presented as mean ±SEM; ns=not significant, * = p<0.05, *** P < 0.001.
To better define the contribution of each epigenetic modifiers to the regulation of BMPR2 expression, we also used different in vitro approaches to reverse and prevent H3K27me3 and DNA methylation in hPASMC. To this end, we used a potent pharmacological inhibitor of EZH2 (GSK126), DNMT1 (Decitabine), and an RNA interference approach mediated by short hairpin RNA against DNMT1 (shDNMT1) and TET1 (shTET1) in hPASMC. We then assessed the expression of the BMPR2 mRNA by qPCR (Figure XVII in the Supplement), PASMC proliferation by BrdU assay (Figure XVII in the Supplement), and Cyclin D1 mRNA level by qPCR (Figure XVII in the Supplement). Our results showed that TET1 silencing inhibits BMPR2 expression despite SIN3a overexpression. Similarly, EZH2 and DNMT1 significantly impaired the SIN3a shRNA-mediated repression on BMPR2 expression, which resulted in increased BMPR2 levels (Figure XVII in the Supplement). These data suggested that TET1, DNMT1, and EZH2 are all essential downstream mediators in the regulation of BMPR2 expression. Disruption of one of these regulators significantly impairs SIN3a’s capabilities in increasing BMPR2 levels. Consistent with the BMPR2 levels, we found that TET1 knockdown also impaired the anti-proliferative properties of SIN3a. Inversely, pharmacological inhibition of EZH2 and DNMT1 blocked PASMC proliferation in SIN3a-depleted cells.
In summary, our results provided for the first time strong evidence that SIN3a plays a critical role in the regulation of BMPR2 expression and pulmonary vascular remodeling by inhibiting vascular cell proliferation and modulating the lung epigenetic landscape. We found that the restoration of SIN3a regulates several epigenetic pathways in PAH by decreasing HDAC1, EZH2, DNMT1, and CTCF expression. Furthermore, our study demonstrated that SIN3a reduced the methylation of the BMPR2 promoter by promoting TET1 expression and inhibiting the expression of DNMT1 in hPASMC (Figure 8G). These results strongly support that SIN3a may be a clinically relevant target for the treatment of PAH disease.
Discussion
Aberrant promoter hypermethylation or histone modifications are critical in disease initiation and progression71. DNA methylation is described as a potent epigenetic repressor of transcription. Because DNA methylation is reversible, there is much interest in understanding the mechanisms by which it may affect the BMPR2 expression and DNA-binding of transcription factors in PAH disease. In the present study, we discovered that SIN3a might act as an endogenous repressor of HDAC1 activity in pulmonary vascular cells. Concomitantly, we found that SIN3a regulates key regulators of methylation and potentiates BMPR2 expression in hPASMC. Our study also identifies CTCF as a critical transcriptional repressor of BMPR2 expression.
Although SIN3a does not bind DNA independently, it provides a scaffold for several transcription epigenetic partners/modifiers and transcription factors with specific DNA binding activities and consequent activation/repression of specific target genes72. SIN3a knockout mice die during embryogenesis73, suggesting that the SIN3a complex is essential for early embryonic development. Although this molecular complex was initially thought to repress gene expression via histone deacetylation, SIN3a can also facilitate transcriptional activation in a cellular context-dependent manner36, 74. Indeed, we found that the expression of many genes was either upregulated or downregulated in hPASMC overexpressing SIN3a. The loss of SIN3a in PAH impaired the epigenetic machinery, as illustrated by the aberrant EZH2-mediated H3K27me3 and DNA methylation level within the BMPR2 promoter region. Dysregulation of these epigenetic marks represses BMPR2 expression. Herein, we have also shown that SIN3a decreased HDAC1 activity and the enrichment of the repressive mark H3K27me3 at the BMPR2 promoter region by antagonizing the expression of EZH2. Additionally, we have also demonstrated that SIN3a increased TET1-mediated DNA demethylation at the BMPR2 promoter region. Thus, our study identified the novel SIN3a/EZH2-H3K27me3/TET1 pathway as a dynamic epigenetic mechanism underlying the BMPR2 expression and vascular remodeling in PAH disease.
Interestingly, previous studies have demonstrated that the HDAC complex interacts with methylation-regulating proteins such as EZH2, MECP2, DNMT1, and CTCF74, 75. These results are consistent with the previous finding that reducing both mammalian SIN3a and SIN3b leads to increased histone methylation and acetylation in differentiated myotubes cells76. Given that H3K27me3 is associated with gene silencing77, 78, the regulatory role of SIN3a on histone methylation may explain how SIN3a affects transcriptional regulation and modulates biological processes. Our data indicate that SIN3a affects DNA methylation through EZH2 and TET1.
In our study, we found that SIN3a potentiates TET1-mediated DNA demethylation and concomitantly inhibits EZH2/DNMT1-mediated Histone/DNA methylation in hPASMC. We discovered that BMPR2 expression in hPASMC was inversely correlated to DNMT1 and EZH2 levels and depended on its promoter methylation status. CTCF and H3K27me3 were both significantly enriched at the methylated BMPR2 gene promoter in hPASMC. These findings may suggest that the CTCF-dependent recruitment of EZH2 to the BMPR2 gene promoter is likely to participate in the epigenetic silencing of BMPR2 and, therefore, its gene expression. As a catalytic subunit of PRC2, EZH2 can trimethylate histone H3 at lysine 27 on many silenced gene promoters63 and exert critical roles in repressing the initial phase of gene transcription77. Multiple reports have shown that EZH2 is overexpressed in various tumors and is related to the occurrence and poor prognosis of cancer79–82. Inhibition of EZH2 reduced the proliferation of cell lines derived from several types of malignancies, underscoring the potential benefits EZH2 inhibitors possess as cancer therapeutics83. What is clear is that most of our human PAH samples show reduced expression of SIN3a and an increase in EZH2. Heat map analysis showed that the expression of DNA methyltransferase genes is also lower in SIN3a overexpressing hPASMC. These results were also confirmed in animal models of PAH. In our study, ChIP results showed that H3K27me3, a repressive mark, was enriched in BMPR2 promoter in hPASMC, in which the BMPR2 promoter was methylated. In contrast, H3K27me3 was not found at any sites in hPASMC overexpressing SIN3a, in which the BMPR2 promoter was unmethylated. When H3K27me3 and EZH2 were absent, the DNA methylation status was changed from methylation to demethylation. These results demonstrated that the PRC2 complex is involved in regulating the methylation of the BMPR2 gene promoter by SIN3a. Additional regulation of the CTCF-binding to the BMPR2 promoter supports the critical role of SIN3a. It has been reported that CTCF is involved in recruiting PcG complex84, 85. In our study, ChIP results showed that the enrichment of CTCF at the BMPR2 promoter was decreased by SIN3a in hPASMC. These results demonstrated that CTCF may repress BMPR2 in hPASMC in absence of SIN3a. However, the interplay between EZH2 and CTCF involved in the BMPR2 downregulation in PAH is still not clear and would require further investigation.
Previous studies showed that SIN3a acted in cooperation with TET1 to facilitate the transcription of a specific set of genes52. TET1 has been shown to remove DNA methylation by reiterating 5-methylcytosine to 5-hydroxymethylcytosine (5hmC)86. The distribution of 5hmC is affected by histone modifications such as H3K27me3 and H3K4me3, binding proteins of epigenetic marks, and chromatin configuration86. The N-terminal CXXC structure typical for DNA binding of TET1 alone is not sufficient to recognize specific target gene sequences for TET186, implying that other proteins may be involved in the TET1-mediated demethylation at specific DNA sites. Moreover, SIN3a and other epigenetic modifiers such as MeCP2, HDAC1, EZH2, and CTCF have been identified as TET1-interacting proteins 87. Herein, we showed that SIN3a increased TET1 expression and decreased MeCP2, HDAC1, EZH2, and CTCF expression and/or activity.
Furthermore, of therapeutic significance, the present study opens a new avenue for PAH treatment by SIN3a gene therapy by targeting the EZH2-H3K27me3-TET1 pathway in PASMCs that can modulate the epigenetic landscape of pulmonary vascular genes, and therefore pulmonary vascular and RV remodeling. Considering AAV1-mediated SIN3a delivery is highly vascular cells-selective88–90, improvement in pulmonary vascular remodeling (in both prevention and curative experiments) and restoration of BMPR2 expression in lung demonstrated the critical role of SIN3a in PAH pathophysiology and further support our hypothesis that IT delivery of AAV1.hSIN3a gene therapy may be a clinically relevant and promising strategy for treating PAH patients.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
Switch-Independent 3a (SIN3a), an epigenetic modifier, is drastically downregulated in PAH patients and rodent models of PAH, which is strongly associated with decreased BMPR2 expression.
SIN3a overexpression upregulates BMPR2 expression by modulating critical epigenetic pathways and decreasing CTCF transcription factor binding to the BMPR2 promoter in pulmonary vascular smooth muscle cells.
Aerosolized lung targeted-gene transfer of AAV1.hSIN3a reverses and prevents PAH phenotype in preclinical animal models.
What are the clinical implications?
Aerosolized targeted-gene transfer of SIN3a may overcome methylation-mediated BMPR2 silencing by modulating critical epigenetic pathways.
Intratracheal delivery of AAV1 therapies may be used to restore the BMPR2 expression in PAH while improving pulmonary vascular remodeling, lowering pulmonary artery and right ventricular pressures.
This study suggests that SIN3a can be a clinically relevant molecule for the treatment of PAH disease.
Acknowledgments
We thank Dr. Akiko Hata for providing the adenovirus encoding human BMPR2.
Sources of Funding
This work was supported by the National Institutes of Health grant R01 HL133554 and American Heart Association AHA-18IPA34170321 (to LH), NIH-R01HL140469, R01HL124187 and R01HL148786 grant (to SS), NIH 5T32HL007824-22, and Cardiovascular Medical Research and Education Fund (CMREF) (to MB).
Non-standard Abbreviations and Acronyms
- AAV1
Adeno associated Virus 1
- ChIP
Chromatin immunoprecipitation
- DNMT
DNA methyltransferases
- IT
Intratracheal
- LUC
Luciferase
- MCT
Monocrotaline
- PAH
Pulmonary arterial hypertension
- PAEC
Pulmonary artery endothelial cells
- PASMC
Pulmonary artery smooth muscle cells
- PVR
Pulmonary vascular resistance
- RV
Right ventricle
- SuHx
Sugen Hypoxia
- TF
Transcription factor
- WGA
Wheat-germ agglutinin
Footnotes
Disclosures
The authors report no conflicts.
References
- 1.Montani D, Girerd B, Gunther S, Riant F, Tournier-Lasserve E, Magy L, Maazi N, Guignabert C, Savale L, Sitbon O, Simonneau G, Soubrier F and Humbert M. Pulmonary arterial hypertension in familial hemiplegic migraine with ATP1A2 channelopathy. Eur Respir J. 2014;43:641–3. [DOI] [PubMed] [Google Scholar]
- 2.Montani D, Chaumais MC, Guignabert C, Gunther S, Girerd B, Jais X, Algalarrondo V, Price LC, Savale L, Sitbon O, Simonneau G and Humbert M. Targeted therapies in pulmonary arterial hypertension. Pharmacol Ther. 2014;141:172–91. [DOI] [PubMed] [Google Scholar]
- 3.Lai YC, Potoka KC, Champion HC, Mora AL and Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014;115:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thompson AAR and Lawrie A. Targeting Vascular Remodeling to Treat Pulmonary Arterial Hypertension. Trends Mol Med. 2017;23:31–45. [DOI] [PubMed] [Google Scholar]
- 5.Guimaron S, Guihaire J, Amsallem M, Haddad F, Fadel E and Mercier O. Current Knowledge and Recent Advances of Right Ventricular Molecular Biology and Metabolism from Congenital Heart Disease to Chronic Pulmonary Hypertension. Biomed Res Int. 2018;2018:1981568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogaard HJ, Abe K, Vonk Noordegraaf A and Voelkel NF. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest. 2009;135:794–804. [DOI] [PubMed] [Google Scholar]
- 7.Savale L, Weatherald J, Jais X, Vuillard C, Boucly A, Jevnikar M, Montani D, Mercier O, Simonneau G, Fadel E, Sitbon O and Humbert M. Acute decompensated pulmonary hypertension. Eur Respir Rev. 2017;26:170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soon E, Crosby A, Southwood M, Yang P, Tajsic T, Toshner M, Appleby S, Shanahan CM, Bloch KD, Pepke-Zaba J, Upton P and Morrell NW. Bone morphogenetic protein receptor type II deficiency and increased inflammatory cytokine production. A gateway to pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;192:859–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orriols M, Gomez-Puerto MC and Ten Dijke P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol Life Sci. 2017;74:2979–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West J, Fagan K, Steudel W, Fouty B, Lane K, Harral J, Hoedt-Miller M, Tada Y, Ozimek J, Tuder R and Rodman DM. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ Res. 2004;94:1109–14. [DOI] [PubMed] [Google Scholar]
- 11.Yu PB, Deng DY, Beppu H, Hong CC, Lai C, Hoyng SA, Kawai N and Bloch KD. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J Biol Chem. 2008;283:3877–88. [DOI] [PubMed] [Google Scholar]
- 12.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD and Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sztrymf B, Coulet F, Girerd B, Yaici A, Jais X, Sitbon O, Montani D, Souza R, Simonneau G, Soubrier F and Humbert M. Clinical outcomes of pulmonary arterial hypertension in carriers of BMPR2 mutation. Am J Respir Crit Care Med. 2008;177:1377–83. [DOI] [PubMed] [Google Scholar]
- 14.Rosenzweig EB, Morse JH, Knowles JA, Chada KK, Khan AM, Roberts KE, McElroy JJ, Juskiw NK, Mallory NC, Rich S, Diamond B and Barst RJ. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27:668–74. [DOI] [PubMed] [Google Scholar]
- 15.Girerd B, Montani D, Coulet F, Sztrymf B, Yaici A, Jais X, Tregouet D, Reis A, Drouin-Garraud V, Fraisse A, Sitbon O, O’Callaghan DS, Simonneau G, Soubrier F and Humbert M. Clinical outcomes of pulmonary arterial hypertension in patients carrying an ACVRL1 (ALK1) mutation. Am J Respir Crit Care Med. 2010;181:851–61. [DOI] [PubMed] [Google Scholar]
- 16.Rabinovitch M Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morrell NW. Role of bone morphogenetic protein receptors in the development of pulmonary arterial hypertension. Advances in experimental medicine and biology. 2010;661:251–64. [DOI] [PubMed] [Google Scholar]
- 18.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC and Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–8. [DOI] [PubMed] [Google Scholar]
- 19.Liu D, Yan Y, Chen JW, Yuan P, Wang XJ, Jiang R, Wang L, Zhao QH, Wu WH, Simonneau G, Qu JM and Jing ZC. Hypermethylation of BMPR2 Promoter Occurs in Patients with Heritable Pulmonary Arterial Hypertension and Inhibits BMPR2 Expression. Am J Respir Crit Care Med. 2017;196:925–928. [DOI] [PubMed] [Google Scholar]
- 20.Barros SP and Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88:400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballestar E and Esteller M. The impact of chromatin in human cancer: linking DNA methylation to gene silencing. Carcinogenesis. 2002;23:1103–9. [DOI] [PubMed] [Google Scholar]
- 22.Moosavi A and Motevalizadeh Ardekani A. Role of Epigenetics in Biology and Human Diseases. Iran Biomed J. 2016;20:246–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Handy DE, Castro R and Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bisserier M, Janostiak R, Lezoualc’h F and Hadri L. Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary hypertension disease. Vasc Biol 2020;2:R17–R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK and Patra SK. Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem. 2014;62:11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hervouet E The Promising Role of New Generation HDACis in Anti-Cancer Therapies. EBioMedicine. 2018;32:6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Attwood JT, Yung RL and Richardson BC. DNA methylation and the regulation of gene transcription. Cell Mol Life Sci. 2002;59:241–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moore LD, Le T and Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin B, Li Y and Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2:607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ficz G New insights into mechanisms that regulate DNA methylation patterning. J Exp Biol. 2015;218:14–20. [DOI] [PubMed] [Google Scholar]
- 31.Jelinic P, Pellegrino J and David G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol Cell Biol. 2011;31:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverstein RA and Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2005;47:1–17. [DOI] [PubMed] [Google Scholar]
- 33.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH and Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E and Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89:349–56. [DOI] [PubMed] [Google Scholar]
- 35.Kadamb R, Mittal S, Bansal N, Batra H and Saluja D. Sin3: insight into its transcription regulatory functions. Eur J Cell Biol. 2013;92:237–46. [DOI] [PubMed] [Google Scholar]
- 36.Icardi L, Mori R, Gesellchen V, Eyckerman S, De Cauwer L, Verhelst J, Vercauteren K, Saelens X, Meuleman P, Leroux-Roels G, De Bosscher K, Boutros M and Tavernier J. The Sin3a repressor complex is a master regulator of STAT transcriptional activity. Proc Natl Acad Sci U S A. 2012;109:12058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P and Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuzmichev A, Zhang Y, Erdjument-Bromage H, Tempst P and Reinberg D. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1). Mol Cell Biol. 2002;22:835–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sif S, Saurin AJ, Imbalzano AN and Kingston RE. Purification and characterization of mSin3A-containing Brg1 and hBrm chromatin remodeling complexes. Genes Dev. 2001;15:603–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagl NG Jr., Wang X, Patsialou A, Van Scoy M and Moran E Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 2007;26:752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jenuwein T Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–73. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y and Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. [DOI] [PubMed] [Google Scholar]
- 43.Margueron R and Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pande V Understanding the Complexity of Epigenetic Target Space. J Med Chem. 2016;59:1299–307. [DOI] [PubMed] [Google Scholar]
- 45.Chase A and Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–8. [DOI] [PubMed] [Google Scholar]
- 46.Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, Grand FH and Cross NC. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6. [DOI] [PubMed] [Google Scholar]
- 47.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA and Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD and Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA and Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–9. [DOI] [PubMed] [Google Scholar]
- 50.Bisserier M and Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood. 2018;131:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aljubran SA, Cox R Jr., Tamarapu Parthasarathy P, Kollongod Ramanathan G, Rajanbabu V, Bao H, Mohapatra SS, Lockey R and Kolliputi N. Enhancer of zeste homolog 2 induces pulmonary artery smooth muscle cell proliferation. PLoS One. 2012;7:e37712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu F, Zhu Q, Ye D, Zhang Q, Yang Y, Guo X, Liu Z, Jiapaer Z, Wan X, Wang G, Chen W, Zhu S, Jiang C, Shi W and Kang J. Sin3a-Tet1 interaction activates gene transcription and is required for embryonic stem cell pluripotency. Nucleic Acids Res. 2018;46:6026–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li G, Yu M, Weyand CM and Goronzy JJ. Epigenetic regulation of killer immunoglobulin-like receptor expression in T cells. Blood. 2009;114:3422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu T, Shao L, Wang A, Liang R, Lin Y, Wang G, Zhao Y, Hu J and Liu S. CD248 as a novel therapeutic target in pulmonary arterial hypertension. Clin Transl Med 2020;10:e175. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Sharma S, Kelly TK and Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hopper RK, Moonen J-RAJ, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L and Rabinovitch M. In Pulmonary Arterial Hypertension, Reduced BMPR2 Promotes Endothelial-to-Mesenchymal Transition via HMGA1 and Its Target Slug. Circulation. 2016;133:1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M, Vattulainen S, Aho J, Orcholski M, Rojas V, Yuan K, Helenius M, Taimen P, Myllykangas S, De Jesus Perez V, Koskenvuo JW and Alastalo TP. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2014;50:1118–28. [DOI] [PubMed] [Google Scholar]
- 58.Morty RE, Nejman B, Kwapiszewska G, Hecker M, Zakrzewicz A, Kouri FM, Peters DM, Dumitrascu R, Seeger W, Knaus P, Schermuly RT and Eickelberg O. Dysregulated bone morphogenetic protein signaling in monocrotaline-induced pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol. 2007;27:1072–8. [DOI] [PubMed] [Google Scholar]
- 59.Li YX and Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harbor Perspectives in Medicine. 2016;6:a026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eckschlager T, Plch J, Stiborova M and Hrabeta J. Histone Deacetylase Inhibitors as Anticancer Drugs. International Journal of Molecular Sciences. 2017;18(7):1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ganai SA, Ramadoss M and Mahadevan V. Histone Deacetylase (HDAC) Inhibitors - emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol. 2016;14:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang SS, Zhang R, Wang G and Zhang YF. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl Neurodegener. 2017;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, Bergman Y, Simon I and Cedar H. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39:232–6. [DOI] [PubMed] [Google Scholar]
- 64.Gal-Yam EN, Saito Y, Egger G and Jones PA. Cancer epigenetics: modifications, screening, and therapy. Annu Rev Med. 2008;59:267–80. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Q, Dong P, Liu X, Sakuragi N and Guo SW. Enhancer of Zeste homolog 2 (EZH2) induces epithelial-mesenchymal transition in endometriosis. Sci Rep. 2017;7:6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang JP and Ling K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys(27) acetylation in the BIM gene promoter and enhancers. Oncol Lett. 2017;14:5735–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Y, Qi J, Xiong J, Jiang L, Cui D, He J, Chen P, Li L, Wu C, Ma T, Shao S, Wang J, Yu D, Zhou B, Huang D, Schmitt CA and Tao R. Epigenetic Co-Deregulation of EZH2/TET1 is a Senescence-Countering, Actionable Vulnerability in Triple-Negative Breast Cancer. Theranostics. 2019;9:761–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cartron PF, Nadaradjane A, Lepape F, Lalier L, Gardie B and Vallette FM. Identification of TET1 Partners That Control Its DNA-Demethylating Function. Genes Cancer. 2013;4:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yusufzai TM, Tagami H, Nakatani Y and Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13:291–8. [DOI] [PubMed] [Google Scholar]
- 70.Mukhopadhyay R, Yu W, Whitehead J, Xu J, Lezcano M, Pack S, Kanduri C, Kanduri M, Ginjala V, Vostrov A, Quitschke W, Chernukhin I, Klenova E, Lobanenkov V and Ohlsson R. The binding sites for the chromatin insulator protein CTCF map to DNA methylation-free domains genome-wide. Genome Res. 2004;14:1594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim M and Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49:e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bansal N, David G, Farias E and Waxman S. Emerging Roles of Epigenetic Regulator Sin3 in Cancer. Adv Cancer Res. 2016;130:113–35. [DOI] [PubMed] [Google Scholar]
- 73.McDonel P, Demmers J, Tan DW, Watt F and Hendrich BD. Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev Biol. 2012;363:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suganuma T and Workman JL. Chromatin and signaling. Curr Opin Cell Biol. 2013;25:322–6. [DOI] [PubMed] [Google Scholar]
- 75.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J and Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu M and Pile LA. The Transcriptional Corepressor SIN3 Directly Regulates Genes Involved in Methionine Catabolism and Affects Histone Methylation, Linking Epigenetics and Metabolism. J Biol Chem. 2017;292:1970–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Di Croce L and Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013;20:1147–55. [DOI] [PubMed] [Google Scholar]
- 78.Simon JA and Kingston RE. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell. 2013;49:808–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jiang T, Wang Y, Zhou F, Gao G, Ren S and Zhou C. Prognostic value of high EZH2 expression in patients with different types of cancer: a systematic review with meta-analysis. Oncotarget. 2016;7:4584–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pietersen AM, Horlings HM, Hauptmann M, Langerod A, Ajouaou A, Cornelissen-Steijger P, Wessels LF, Jonkers J, van de Vijver MJ and van Lohuizen M. EZH2 and BMI1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008;10:R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sudo T, Utsunomiya T, Mimori K, Nagahara H, Ogawa K, Inoue H, Wakiyama S, Fujita H, Shirouzu K and Mori M. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br J Cancer. 2005;92:1754–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang X, Dai H, Wang Q, Wang Q, Xu Y, Wang Y, Sun A, Ruan J, Chen S and Wu D. EZH2 mutations are related to low blast percentage in bone marrow and -7/del(7q) in de novo acute myeloid leukemia. PLoS One. 2013;8:e61341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Comet I, Riising EM, Leblanc B and Helin K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat Rev Cancer. 2016;16:803–810. [DOI] [PubMed] [Google Scholar]
- 84.Li T, Hu JF, Qiu X, Ling J, Chen H, Wang S, Hou A, Vu TH and Hoffman AR. CTCF regulates allelic expression of Igf2 by orchestrating a promoter-polycomb repressive complex 2 intrachromosomal loop. Mol Cell Biol. 2008;28:6473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yan H, Tang G, Wang H, Hao L, He T, Sun X, Ting AH, Deng A and Sun S. DNA methylation reactivates GAD1 expression in cancer by preventing CTCF-mediated polycomb repressive complex 2 recruitment. Oncogene. 2016;35:3995–4008. [DOI] [PubMed] [Google Scholar]
- 86.Shi DQ, Ali I, Tang J and Yang WC. New Insights into 5hmC DNA Modification: Generation, Distribution and Function. Front Genet. 2017;8:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feinberg AP, Koldobskiy MA and Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hadri L, Bobe R, Kawase Y, Ladage D, Ishikawa K, Atassi F, Lebeche D, Kranias EG, Leopold JA, Lompre AM, Lipskaia L and Hajjar RJ. SERCA2a gene transfer enhances eNOS expression and activity in endothelial cells. Mol Ther. 2010;18:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hadri L, Kratlian RG, Benard L, Maron BA, Dorfmuller P, Ladage D, Guignabert C, Ishikawa K, Aguero J, Ibanez B, Turnbull IC, Kohlbrenner E, Liang L, Zsebo K, Humbert M, Hulot JS, Kawase Y, Hajjar RJ and Leopold JA. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation. 2013;128:512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aguero J, Hadri L, Hammoudi N, Leonardson L, Hajjar RJ and Ishikawa K. Inhaled Gene Transfer for Pulmonary Circulation. Methods Mol Biol. 2017;1521:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.