

Abstract
Background
The whole exome sequencing (WES) with targeted gene analysis is an effective diagnostic tool for cardiomyopathy. The early‐onset sudden cardiac death (SCD) was commonly associated with dilated cardiomyopathy (DCM) induced by pathogenic genetic mutations.
Methods
In a Chinese Han family, the patient of 24 years old occurred with early‐onset and DCM and died of SCD associated with ICD storms induced by repetitive ventricular tachycardia/fibrillation (VT/F). Genomic DNA samples of peripheral blood were conducted for WES and Sanger sequence. Then, we performed bioinformatics analysis for 200 genes susceptible to cardiomyopathies and arrhythmias. Further, we analyzed how the potential pathogenic mutations affecting the secondary structure, hydrophobicity, and phosphorylation of amino acids, protein properties, and their joint pathogenicity by ProtParam, SOPMA, and ORVAL algorisms. The protein–protein interaction was analyzed by STRING algorism.
Results
The mutations of LDB3 p.M456R, MYH6 p.S180Y, and SYNE1 p.S4607F were identified as “Damaging/Deleterious.” The SYNE1 (p.S4607F) increased one of alpha helix and decreased one of beta sheet. The LDB3 (p.M456R) reduced one of beta sheet and increased one of beta turn. The MYH6 (p.S180Y) decreased two of beta sheets and four of beta turns, but significantly increased twelve coils. The hydrophobicity of amino acid residues and their adjacent sequences were decreased by LDB3 (p.M456R) and MYH6 (p.S180Y), and significantly increased by SYNE1 (p.S4607F). The mutations of LDB3 (p.M456R), SYNE1 (p.S4607F), and MYH6 (p.S180Y) resulted in the phosphorylation changes of the corresponding amino acid sites or the nearby amino acid sites. The pairwise combinations of LDB3, MYH6, and SYNE1 mutations have the high probability of causing disease, especially the highest probability for SYNE1 and LDB3 mutations. There was obviously indirect interaction of the proteins encoded by SYNE1, LDB3, and MYH6.
Conclusions
The multiple heterozygous mutations of SYNE1, LDB3, and MYH6 may be associated with young and early‐onset of DCM and SCD.
Keywords: arrhythmia, cardiomyopathy, gene, sudden cardiac death
What is new?
We firstly reported that the multiple heterozygous mutations of SYNE1 (p.S4607F), LDB3 (p.M456R), and MYH6 (p.S180Y) may be associated with young and early‐onset dilated cardiomyopathy, malignant ventricular arrhythmia, and sudden cardiac death.
The mutations of LDB3, MYH6, and SYNE1 were identified as “Damaging/Deleterious,” and abnormally changed the secondary structure, hydrophobicity, and phosphorylation of amino acids, which therefore increased the joint pathogenicity.
There was obviously indirect interaction of the proteins encoded by SYNE1, LDB3, and MYH6.
1. BACKGROUND
Cardiomyopathies are classified as dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy (RCM), and arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D). DCM is a rare cardiac disease characterized by left ventricular enlargement, reduced left ventricular contractility, and impaired systolic function, which can lead to sudden cardiac death (SCD), with a reported frequency of 46% (Cecchi et al., 2012; Fu & Eisen, 2018). Hereditary DCM is present in almost 50% of reported cases, mostly inherited in an autosomal dominant pattern, although some cases reported with X‐linked, autosomal recessive, and mitochondrial DNA inheritance (McNally et al., 2013). The hereditary DCM is complicated by incomplete penetrance, high variability in age of onset and disease progression, and high genetic heterogeneity (Haas et al., 2015). Cardiomyopathies are associated with >100 known genes, including genes encoding for sodium and potassium channel in sarcolemma, dystrophin proteins in cytoplasm, myofibril with sarcomere/Z‐disk, thin/thick filament, RNA‐binding protein, cytoskeletal proteins, desmosome mitochondria, sarcoplasmic reticulum, and nucleus envelope (McNally et al., 2013; Reichart et al., 2019). Following with the advantage of targeted sequencing technology, multiple mutations are increasingly reported and are often associated with high disease severity. Notably, 12.8% of DCM has at least three known mutations (Haas et al., 2015).
The whole exome sequencing (WES) with targeted gene analysis is an effective diagnostic tool for patients with DCM in a clinical setting. The pathogenic or likely pathogenic mutations were identified in 12% of DCM by WES (Ramchand et al., 2020). The genetic testing was considered valuable for future SCD risk factor stratification and genetic counseling for DCM. For example, desmosome and LAMA gene mutations increased the risk of arrhythmia and SCD for DCM (Gigli et al., 2019; Li et al., 2020). Here, we aimed to identify novel mutations and genes responsible for young and early‐onset DCM with SCD using WES. In doing so, we identified multiple potential pathogenic mutations of LDB3, MYH6, and SYNE1 genes in this patient.
2. METHODS
2.1. Ethical compliance
This study was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No. GDREC2016001H (R1)]. Detailed clinical information was collected. The clinical information included family history, age of presentation, initial symptoms of ventricular tachycardia, physical examination, electrocardiograms (ECGs), and echocardiograms based on their informed consents.
2.2. Whole exome sequencing
Genomic DNA samples of patients were isolated from peripheral blood using a standard DNA extraction protocol. The isolated genomic DNA was then fragmented into 150–200 bp and subjected to DNA library preparation using established Illumina paired‐end protocols. Adaptor‐ligated libraries were amplified via PCR. A portion of each library was used to create an equimolar pool. Each pool was amplified to enrich targets sequenced by the Agilent SureSelectXT Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). Whole exome capture was performed with the Agilent SureSelectXT Human All Exon 50 Mb Kit (Agilent Technologies Inc.) following the manufacturer's protocol. The exome‐enriched libraries were sequenced with the Illumina Hiseq 2000 platform (Illumina, San Diego, CA, USA) according to the manufacturer's instructions, and 100 bp paired‐end sequencing reads were generated. Each sample was sequenced per lane to obtain an average theoretical depth of 100×.
2.3. Read mapping, variant detection, and functional annotation
Raw reads were collected for quality control, in which low‐quality reads were filtered, and 3′/5′ adapters were trimmed using the Trim Galore program. Clean reads were aligned to the human reference genome (University of California Santa Cruz, UCSC build hg19) using the Burrows‐Wheeler Aligner (BWA) program. The quality scores were recalibrated, and reads were realigned to the reference genome using the Genome Analysis Toolkit (GATK) software package. Following the exclusion of duplicate reads, insertion‐deletions (InDels) and single‐nucleotide polymorphisms (SNPs) were called using the GATK or Sequence Alignment/Map tools (SAM tools).
SNPs and InDels were annotated using a pipeline in which all insertion, and deletion mutations were occurring in coding regions. InDels were considered damaging, and nonsynonymous SNPs predicted by SIFT (http://sift.jcvi.org/www/) (Kumar et al., 2009) and PolyPhen‐2 (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2010). The mutations were screened in approximately 200 genes (shown in our previous study (Lin et al., 2017)), associated with hereditary arrhythmias and cardiomyopathies. The filtering criteria for mutation inclusion were as follows: same mutations in the WES data; missense, nonsense, and InDel mutations; and SNPs with a minor allele frequency not more than 0.01, according to the NCBI SNP database (Smigielski et al., 2000; Via et al., 2010). We subsequently focused the genetic investigation on rare functional mutations, defined as previously unreported or with a very low of minor allele frequency (MAF < 0.1%) in the European population of the ExAC database. Meanwhile, these mutations predicted to alter the protein sequence (Di Resta et al., 2015).
2.4. Sanger sequencing
When the suspected pathogenic mutations were obtained in each step, they were screened again using Sanger sequencing in the other members of the family. The following primers designed with Primer Premier 5.0 were used and showed as follows (forward primer, reverse primer, length, annealing temperature).
MYH6: AGGGTGTAGTTGGGAGGAGAG, GAGGGGAGGGTTAGGGGTA, 406 bp, 57.0°C.
LDB3: CTTCCTGATTTTCTTTATTGT, GAGTTGCTTTCTCTGTTACC, 497 bp, 48.5℃.
SYNE1: AAGAGTGAAGTGCTGGGGAAG, AGGGGTCAAGGTGTGGGG, 961 bp, 58.5°C.
COL3A1: GCATTCCTTCGACTTCTC, TATTTCAACTGGTTTTCATCT, 404 bp, 49.5°C.
PSEN2: AGGTCCTTGTGCTCCTTTTTC, TCATCATTACTTCCCTTCTCCC, 301 bp, 57.5°C.
OBSCN: CATCCTCCAGTCTCCTTTCCC, CCCCACCTTACCTGCCCTT, 608 bp, 61.5°C.
PKP2: CAAGTCTCCAGGTGTCCGC, TTTCCACAAGCAAGTCGGTC, 671 bp, 58.4°C.
TTN, CAACAGGTCTTCGTCGGATT, CCTCTTGCTTGGGTATTTTCAT, 877 bp, 57.5°C.
TTN: TAAAATGGGAGAAAGATGGTCA,TTGGGATGTGATAGGTTGAATA, 1,023 bp, 56.5°C.
COL1A2: AGCCCCTCCCACTAAAGAC,AAAATACACCACACGATACAACT, 909 bp, 55.0°C.
A 50 μl polymerase chain reaction (PCR) mix was made up of 10× PCR buffer (100 mM Tris‐HCl, pH 8.3, 500 mM KCl), 125 μM dNTPs, 10 pmol of each primer, 2 U of Taq DNA polymerase, and 100 ng genomic DNA templates. The following steps were used for the PCR: 94°C for 3 min and 35 cycles (of 94°C for 30 s, 55°C for 1 min, and 72°C for 1 min), followed by 72°C for 10 min. The PCR products were purified using a Wizard Miniprep Kit. The purified PCR products were sequenced with an ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit using standard conditions. The sequencing products were purified using ethanol/EDTA/sodium acetate precipitation and analyzed on an ABI 3730xl Automated DNA Sequencer.
2.5. Myocardial biopsy
The right internal jugular vein was punctured, and a 7‐Fr vascular sheath was inserted. A long, flexible catheter with forceps in the head end was inserted into a vein and threaded up into the heart while monitored by X‐ray. Five small snips of muscle at the apical septum of the right ventricle were grabbed. Some cardiac tissues, which were fixed with formalin solutions, then dehydrated, paraffin‐embedded and sliced, were used for HE and Masson staining. Some cardiac tissues were fixed with glutaraldehyde and osmic acid, dehydrated, resin embedded and sliced, and then prepared for uranyl acetate and lead nitrate staining and analyzed using a transmission electron microscope.
2.6. Physical‐chemical analysis and secondary structure prediction
ProtParam is a tool which allows the computation of various physical and chemical parameters for a given protein for a user entered protein sequence. The computed parameters include the molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half‐life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY) (Wilkins et al., 1999). The secondary structures of wild‐type and potential pathogenic mutations were predicted by SOPMA algorism (Geourjon & Deléage, 1995).
2.7. Joint pathogenicity and protein–protein interaction analysis
ORVAL (https://orval.ibsquare.be) is the first web bioinformatics platform for the exploration of predicted candidate disease‐causing variant combinations, aiming to aid in uncovering the causes of oligogenic diseases (i.e., diseases caused by variants in a small number of genes). This tool integrates innovative machine learning methods for combinatorial variant pathogenicity prediction, further external annotations and interactive and exploratory visualization techniques (Renaux et al., 2019). The STRING online tool (https://string‐db.org/) was used to extract the target protein information from PPI database to explore any direct and indirect protein–protein interactions (PPIs) in the target genes (Szklarczyk et al., 2019).
3. RESULTS
3.1. Familial characteristics
In a Chinese Han family, a 24‐year‐old female patient repeatedly (II: 1, proband) presented with palpitations, dizziness, fatigue, sweating, and then syncope after exertion for 1 year. She woke up on her own for about 1 min (Figure 1a). During this period, ventricular tachycardia occurred and was converted into sinus rhythm by intravenous application of amiodarone. She was hospitalized in January 2016. The dynamic electrocardiogram (Figure 1f‐i) illustrated that: rSr' type of QRS waves in II, III and aVF leads, and obvious notch in S waves; incomplete right bundle branch block, poor increases of R waves in precordial lead, rS and Qr type of QRS waves in V4‐V6 leads; T wave inversion in II, III, aVF, and V3‐V6 leads; short episodes of atrial tachycardia (Figure 1f); multi‐source premature ventricular contraction (Figure 1g); ventricular tachycardia originated from the apical septum of right and left inferior wall (Figure 1h,i). Echocardiography showed that the ventricular apical aneurysm, mild mitral and tricuspid regurgitation, and decreased left ventricular systolic function. Cardiac magnetic resonance imaging (Figure 2) indicated that bilateral ventricular myocardial thickening and scattered fibrosis, especially the myocardium in middle and apical free wall of left ventricle was the most serious, showing extensive fibrosis and local thinning; severe left ventricular enlargement and severe reduction of global systolic function; mild enlargement of right ventricle and mild reduction of global systolic function; normal size of right atrium and slight enlargement of left atrium; mild regurgitations were seen in tricuspid, mitral, and pulmonary valves. In January 2016, she was implanted with implantable cardioverter‐defibrillator (ICD) and suffered from palpitation and ICD discharge twice, followed by vomiting, dizziness, shortness of breath, fatigue, and numbness at the end of limbs in July 2016. Although therapy with beta blocker and amiodarone, she still occurred with SCD in October 2017, due to frequent ICD storms induced by repetitive ventricular tachycardia/fibrillation (VT/F). Her father (I: 1) and mother (I: 2) had normal echocardiography and electrocardiogram with no clinical symptoms, indicating no history and clinical evidence of cardiomyopathy and arrhythmia.
FIGURE 1.
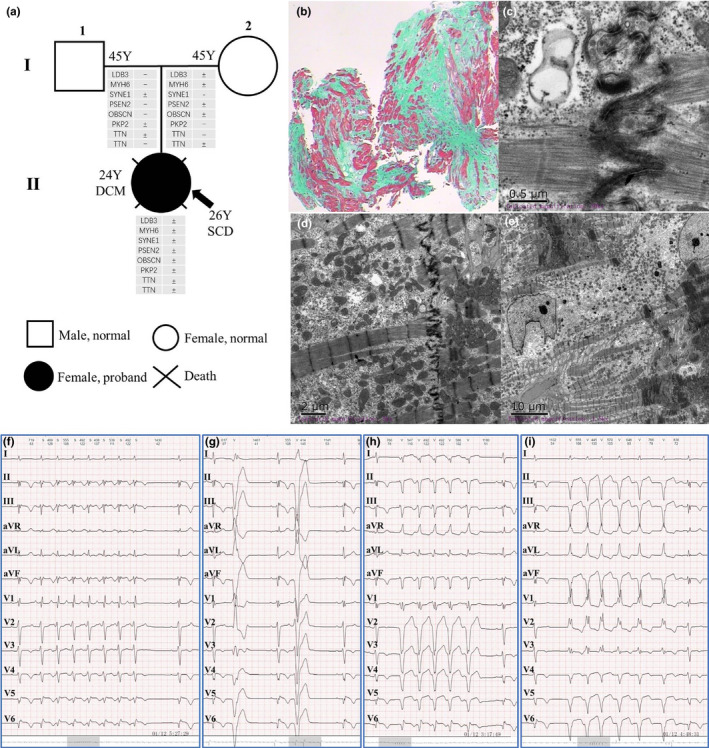
The pedigree, myocardial biopsy, and electrocardiogram. (a) The potential pathogenic mutations susceptible to cardiomyopathy and arrhythmia among the family members. (b) Myocardial HE staining. (c‐e) Myocardial electron‐microscopic examination. (f‐i) Electrocardiograms
FIGURE 2.
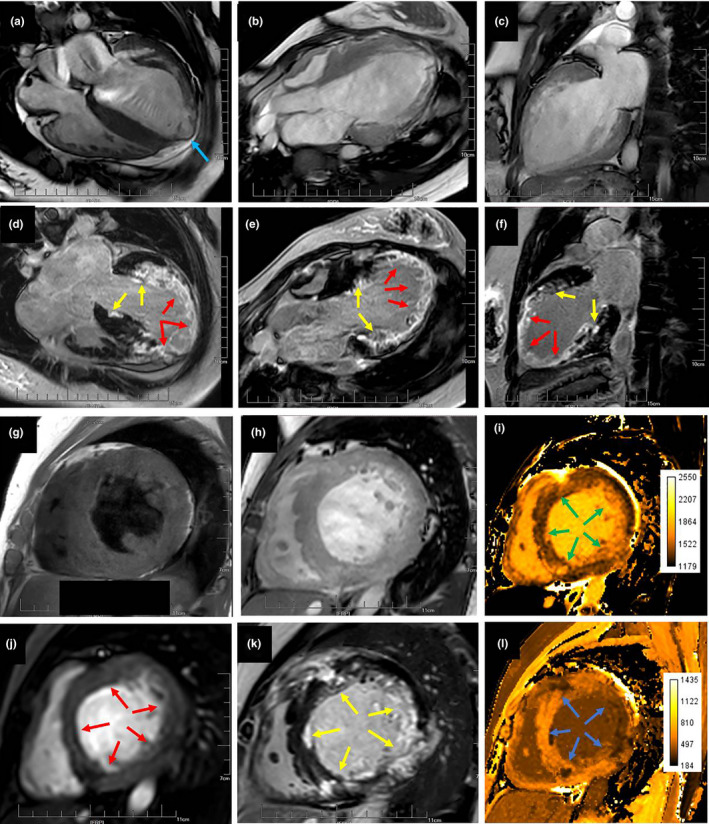
Cardiac magnetic resonance imaging. The first line (a‐c) was the film sequence (balance TFE). The second line (d‐f) was delay enhanced sequence (DCE PSIR). Figure (a‐f) showed that the anterior wall, lateral wall, inferior wall, and septum of left ventricle were extensively involved. Some districts were transmurally involved (red arrow). Part of districts was mainly involved in endocardium (yellow arrow). The middle and apical segment were most obviously involved. The apical myocardium was obviously thinned and ventricular aneurysm was formed (blue arrow). Figure (g), T1W TSE. Figure (h), Balance TFE. Figure (i), MOLLI T1 native. Figure (j), Dynamic sTFE. Figure (k), PSIR TFE. Figure (l), MOLLI T1 DCE. The short‐axis view of middle segment showed thickening of interventricular septum (19 mm), scattered and multiple lesions in the anterior wall, lateral inferior wall and inferior interventricular septum of left ventricle. On plain scan, T1W showed low signal, while T1 value was significantly prolonged (green arrow). The first pass perfusion showed low perfusion (red arrow). The delayed enhancement showed the high enhancement (yellow arrow). The delayed enhancement T1 value was significantly shortened (blue arrow)
3.2. Pathological features of myocardium
The pathological examination showed that some muscle fibers of myocardial tissue were atrophied and reduced in volume, some muscle fibers were hypertrophic; the nucleus of myocardium was large and deeply stained, and obvious interstitial fiber hyperplasia was found (Figure 1b). The intercalated disk of cardiomyocytes was not connected (Figure 1c‐e), and the local space was widened. The sizes of mitochondria were different, and part of mitochondria was slightly focal vacuolar degeneration. The local myocardial fibrils were dissolved and disappeared, and many glycogen particles were accumulated and filled.
3.3. Genetic screening
In this family (Figure 1a), a set of candidate genes associated with cardiomyopathies and arrhythmias were screened using the WES data of II: 1. The results (Table 1) showed that II: 1 carried the heterozygous mutations of PSEN2 p.G34S (rs200636353), OBSCN p.N393D, LDB3 p.M456R (rs566463138), PKP2 p.K52R (rs549598534), MYH6 p.S180Y, TTN p.I25488T, TTN p.Y3760H (rs377740664), SYNE1 p.S4607F, COL3A1 p.Q1381H, COL3A1 p.K1391R (rs373015577), and COL1A2 p.F1341I. The mutations of COL3A1 and COL1A2 were verified to be false positive by Sanger sequencing. The mutations of PSEN2 p.G34S, OBSCN p.N393D, PKP2 p.K52R, TTN p.I25488T, and TTN p.I25613T were predicted as “Tolerated/Benign” by SIFT and Polyphen‐2 algorithms. The mutations of LDB3 p.M456R, MYH6 p.S180Y, and SYNE1 p.S4607F were predicted as “Damaging/Deleterious.” According to the population of 1000 Genomes Project (2015 release version), the minor allele frequency (MAF) of LDB3 p.M456R was less than 0.001, while the mutations of MYH6 p.S180Y and SYNE1 p.S4607F were not found. For II: 1 proband, the SYNE1 p.S4607F mutation was inherited from her father (I: 1), while the mutations of LDB3 p.M456R and MYH6 p.S180Y were inherited from her mother (I: 2). The parents of II: 1 had no clinical evidence of cardiomyopathy based on electrocardiogram and echocardiogram examination. Conservative analysis demonstrated that LDB3 p.M456, MYH6 p.S180, and SYNE1 p.S4607 were very conservative among species (Figure 3). The verifications of Sanger sequencing for LDB3 p.M456R, MYH6 p.S180Y, and SYNE1 p.S4607F were showed in Figures 1a and 3.
TABLE 1.
The potential pathogenic mutations of II: 1
| Chr | Location | Gene | Amino acid change | All | AFR | AMR | EAS | EUR | SAS | SIFT | Polyphen2 | Protein domain |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| chr1 | 227069708 | PSEN2 | NM_000447:exon4:c.G100A:p.G34S | 0.0011 | – | – | 0.005 | – | 0.001 | 0.44(T) | 0.01(B) | – |
| chr1 | 228401330 | OBSCN | NM_001098623:exon3:c.A1177G:p.N393D | – | – | – | – | – | – | 1.00(T) | 0.10(B) | Immunoglobulin subtype |
| chr10 | 88477741 | LDB3 | NM_001080114:exon10:c.T1367G:p.M456R | 0.0002 | – | – | 0.001 | – | – | 0.00(D) | 0.97(D) | Zinc finger, LIM‐type |
| chr12 | 33049511 | PKP2 | NM_001005242:exon1:c.A155G:p.K52R | 0.0002 | – | – | 0.001 | – | – | 0.17(T) | 0.12(B) | – |
| chr14 | 23874023 | MYH6 | MYH6:NM_002471:exon7:c.C539A:p.S180Y | – | – | – | – | – | – | 0.00(D) | 0.97(D) | Myosin head, motor domain |
| chr2 | 179397684 | TTN | NM_003319:exon186:c.T76463C:p.I25488T | – | – | – | – | – | – | 0.06(T) | 0.00(B) | Fibronectin type III |
| chr2 | 179615849 | TTN | NM_133379:exon46:c.T11278C:p.Y3760H | 0.0012 | – | – | 0.006 | – | – | 0.60(T) | 0.00(B) | |
| chr2 | 189875505 | COL3A1 | NM_000090:exon50:c.G4143T:p.Q1381H | – | – | – | – | – | – | 0.07(T) | 0.00(B) | Fibrillar collagen, C‐terminal |
| chr2 | 189875534 | COL3A1 | NM_000090:exon50:c.A4172G:p.K1391R | – | – | – | – | – | – | 0.04(D) | 0.00(B) | |
| chr7 | 94059625 | COL1A2 | NM_000089:exon52:c.T4021A:p.F1341I | – | – | – | – | – | – | 1.00(T) | 0.11(B) | |
| chr6 | 152651787 | SYNE1 | NM_033071:exon77:c.C13820T:p.S4607F | – | – | – | – | – | – | 0.00(D) | 0.99(D) | – |
The minor allele frequency (MAF) in all population (ALL) or the species of African (AFR), American (AMR), East Asian (EAS), European (EUR), and South Asian (SAS) from 1000genomes (2015 version).
Abbreviations: –, no report; B, benign; Chr, chromosome; D, damaging; T, tolerated.
FIGURE 3.
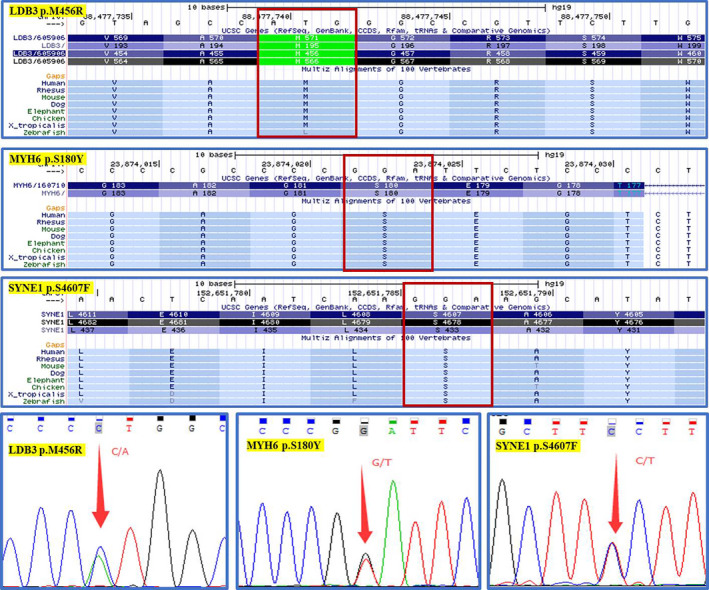
The conservative analysis of the risk genetic mutations
3.4. The prediction of secondary structure and protein properties
Compared with wild type (WT) of MYH6, the total number of positively charged residues (Arg + Lys) was obviously reduced by MYH6 p.S180Y (362 vs. 316). The total number of positively charged residues (Arg + Lys) increased from 54 of LDB3 (WT) to 55 of LDB3 p.M456R (Table 2). The SYNE1 p.S4607F increased one of alpha helix and decreased one of beta sheet. The LDB3 p.M456R reduced one of beta sheet and increased one of beta turn. MYH6 p.S180Y decreased two of beta sheets and four of beta turns, but significantly increased twelve coils (Table 3).
TABLE 2.
The effects of mutations on the protein properties
| Genotype/protein properties | SYNE1 (WT) | SYNE1 (p.S4607F) | LDB3 (WT) | LDB3 (p.M456R) | MYH6 (WT) | MYH6 (p.S180Y) |
|---|---|---|---|---|---|---|
| Number of amino acids | 8,748 | 617 | 1,939 | |||
| Molecular weight | 1,005,126.19 | 1,005,186.29* | 66,671.22 | 66,696.21* | 223,734.71 | 223,810.81* |
| Theoretical pI | 5.36 | 5.36 | 8.29 | 8.39* | 5.58 | 5.58 |
| NO. of negatively charged residues (Asp + Glu) | 1,429 | 1,429 | 49 | 49 | 361 | 361 |
| No. of positively charged residues (Arg + Lys) | 1,114 | 1,114 | 54 | 55* | 362 | 316* |
| Carbon (C) | 43,928 | 43,934* | 2,951 | 2,952* | 9,749 | 9,755* |
| Hydrogen (H) | 70,825 | 70,829* | 4,544 | 4,547* | 15,839 | 15,843* |
| Nitrogen (N) | 12,347 | 12,347 | 808 | 811* | 2,777 | 2,777 |
| Oxygen (O) | 13,931 | 13,930* | 898 | 898 | 3,104 | 3,104 |
| Sulfur (S) | 321 | 321 | 30 | 29* | 66 | 66 |
| Total number of atoms | 141,352 | 141,361* | 9,231 | 9,237* | 31,535 | 31,545* |
| Ext. coefficient‐1 | 933,830 | 933,830 | 71,375 | 71,375 | 116,965 | 118,455* |
| Abs 0.1% (1 g/L) | 0.929 | 0.929 | 1.071 | 1.070* | 0.523 | 0.529* |
| Instability index | 51.65 | 51.65 | 59.09 | 58.95 | 46.98 | 46.84 |
| Aliphatic index | 88.80 | 88.80 | 60.81 | 60.81 | 81.56 | 81.56 |
| Grand average of hydropathicity (GRAVY) | −0.62 | −0.62 | −0.42 | −0.43* | −0.81 | −0.81 |
Extinction coefficients are in units of M−1 cm−1, at 280 nm measured in water. Ext. coefficient‐1 and Abs‐1 [Abs 0.1% (=1 g/L)], assuming all pairs of Cys residues form cystines. Ext. coefficient‐2 and Abs‐2[Abs 0.1% (=1 g/L)], assuming all Cys residues were reduced.
Abbreviation: WT, wild type.
Showed the changes of the parameters.
TABLE 3.
The effects of mutations on the secondary structure of protein
| Genotype/properties | SYNE1 (WT) | SYNE1 (p.S4607F) | LDB3 (WT) | LDB3 (p.M456R) | MYH6 (WT) | MYH6 (p.S180Y) |
|---|---|---|---|---|---|---|
| αhelix | 3,845 (77.16%) | 3,846 (77.18%)* | 132 (21.39%) | 132 (21.39%) | 1,479 (76.28%) | 1,473 (75.97%)* |
| ß sheet | 269 (5.40%) | 268 (5.38%)* | 107 (17.34%) | 106 (17.18%)* | 125 (6.45%) | 123 (6.34%)* |
| ß Turn | 230 (4.62%) | 230 (4.62%) | 51 (8.27%) | 52 (8.43%)* | 73 (3.76%) | 69 (3.56%)* |
| Coil | 639 (12.82%) | 639 (12.82%) | 327 (53.00%) | 327 (53.00%) | 262 (13.51%) | 274 (14.13%)* |
Showed the changes of the parameters.
3.5. Hydrophobicity and phosphorylation
The mutations of LDB3 p.M456R, SYNE1 p.S4607F, and MYH6 p.S180Y located in the non‐transmembrane districts of proteins. The hydrophobicity analysis (Table 4) demonstrated that the hydrophobicity of amino acid residues and their adjacent sequences of LDB3 p.M456R and MYH6 p.S180Y decreased, and the change of hydrophobicity induced by LDB3 p.M456R was much greater than that induced by MYH6 p.S180Y. The hydrophobicity of amino acid residues and their adjacent sequences of SYNE1 p.S4607F significantly increased. The change of hydrophobicity score from high to low was LDB3 > SYNE1 > MYH6.
TABLE 4.
The changes of Hydrophobicity and phosphorylation
| LDB3 (p.M456R) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | 450 | 451 | 452 | 453 | 454 | 455 | 456 | 457 | 458 | 459 | 460 | 461 | 462 | |
| AA | W | G | P | F | L | V | A | M | G | R | S | W | H | P |
| M | G | P | F | L | V | A | R* | G | R | S | W | H | P | |
| HP | W | 1.06 | 1.64 | 1.39 | 0.84 | 0.84 | 0.80 | 0.88 | 0.21 | −0.39 | −1.24 | −1.83 | −1.73 | −1.77 |
| M | 1.06 | 1.64 | 0.68* | 0.13* | 0.13* | 0.09* | 0.17* | −0.50* | −1.10* | −1.96* | −2.54* | −1.73 | −1.77 | |
| PP | W | – | – | – | – | – | – | – | – | – | No | – | – | – |
| M | – | – | – | – | – | – | – | – | – | Yes | – | – | – | |
| MYH6 (p.S180Y) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | 174 | 175 | 176 | 177 | 178 | 179 | 180 | 181 | 182 | 183 | 184 | 185 | 186 | |
| AA | W | I | L | I | T | G | E | S | G | A | G | K | T | V |
| M | I | L | I | T | G | E | Y* | G | A | G | K | T | V | |
| HP | W | 0.04 | 0.04 | 0.34 | 0.69 | 0.98 | 0.43 | −0.42 | −1.00 | −0.46 | −0.80 | −0.49 | −0.83 | −1.29 |
| M | 0.04 | 0.04 | 0.29* | 0.63* | 0.92* | 0.38* | −0.48* | −1.06* | −0.51* | −0.86* | −0.54* | −0.83 | −1.29 | |
| PP | W | – | – | – | – | – | – | Yes | – | – | – | – | – | – |
| M | – | – | – | – | – | – | No | – | – | – | – | – | – | |
| SYNE1 (p.S4607F) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | 4,607 | 4,608 | 4,609 | 4,610 | 4,611 | 4,612 | 4,613 | 4,614 | 4,615 | 4,616 | 4,617 | 4,618 | 4,619 | |
| AA | W | S | L | I | E | L | T | T | Q | S | L | S | E | L |
| M | F* | L | I | E | L | T | T | Q | S | L | S | E | L | |
| HP | W | −0.78 | −0.53 | 0.06 | −0.23 | −0.31 | −0.23 | −0.82 | 0.10 | 0.46 | 0.77 | 0.52 | 0.23 | 0.74 |
| M | −0.78 | −0.53 | 0.06 | 0.17* | 0.09* | 0.17* | −0.42* | 0.50* | 0.86* | 1.17* | 0.92* | 0.63* | 0.74 | |
| PP | W | Yes | – | – | – | – | – | – | – | – | – | – | – | – |
| M | No | – | – | – | – | – | – | – | – | – | – | – | – | |
Abbreviations: AA, amino acids; HP, hydrophobicity; PP, phosphorylation; W, wild type; M, mutation .
Showed the changes of the parameters.
The phosphorylation analysis showed that the mutations of LDB3 p.M456R, SYNE1 p.S4607F and MYH6 p.S180Y resulted in the phosphorylation changes of the corresponding amino acid sites. The LDB3 p.M456R led to the transformation of 456th amino acid from the non‐phosphorylation site to phosphorylation site. The SYNE1 p.S4607F and MYH6 p.S180Y led to the disappearance of phosphorylation of the corresponding amino acid sites. The reliability of phosphorylation from high to low was SYNE1 > LDB3 > MYH6.
3.6. Joint pathogenicity and protein–protein interaction
We analyzed the joint pathogenicity of candidate pathogenic sites (Table 5), including the genes of LDB3, MYH6, PSEN2, and SYNE1. The pairwise combinations of LDB3 p.M456R, MYH6 p.S180Y, and SYNE1 p.S4607F had the high probability of causing disease, among which the combination of SYNE1 and LDB3 mutations had the highest joint pathogenic probability, and the other two combinations were also very high. The supporting scores of joint pathogenicity of SYNE1/LDB3, SYNE1/MYH6 and LDB3/MYH6 mutations were 99.80, 99.60, and 97.60, respectively. The probability of other genes or other combinations was relatively low. The three proteins corresponding to SYNE1, LDB3, and MYH6 had a real protein–protein interaction relationship (Figure 4), which could form an indirect protein–protein interaction network, while the proteins corresponding to OBSCN and PSEN2 genes had no protein–protein interaction with the proteins corresponding to other genes.
TABLE 5.
The risk prediction of pairwise‐gene combination
| Gene A | Gene B | Gene A alleles | Gene B alleles | CS | SS | PC |
|---|---|---|---|---|---|---|
| LDB3 | SYNE1 | 10:88477741:T:G | 6:152651787:G:A | 0.83 | 99.80 | D |
| MYH6 | SYNE1 | 14:23874023:G:T | 6:152651787:G:A | 0.78 | 99.60 | D |
| LDB3 | MYH6 | 10:88477741:T:G | 14:23874023:G:T | 0.68 | 97.60 | D |
| PSEN2 | SYNE1 | 1:227069708:G:A | 6:152651787:G:A | 0.57 | 80.00 | D |
| LDB3 | OBSCN | 10:88477741:T:G | 1:228401330:A:G | 0.55 | 73.20 | D |
| PSEN2 | LDB3 | 1:227069708:G:A | 10:88477741:T:G | 0.51 | 60.00 | D |
| SYNE1 | OBSCN | 6:152651787:G:A | 1:228401330:A:G | 0.45 | 36.20 | N |
| MYH6 | OBSCN | 14:23874023:G:T | 1:228401330:A:G | 0.35 | 8.20 | N |
| PSEN2 | MYH6 | 1:227069708:G:A | 14:23874023:G:T | 0.25 | 1.00 | N |
| PSEN2 | OBSCN | 1:227069708:G:A | 1:228401330:A:G | 0.13 | 0.00 | N |
Classification score represented the pathogenicity probability of this combination in Random Forest algorithms. Support score represented the pathogenicity support of this combination in in Random Forest algorithms.
Abbreviations: CS, classification score; SS, support score; PC, predicted class; D, disease‐causing; N, neutral .
FIGURE 4.
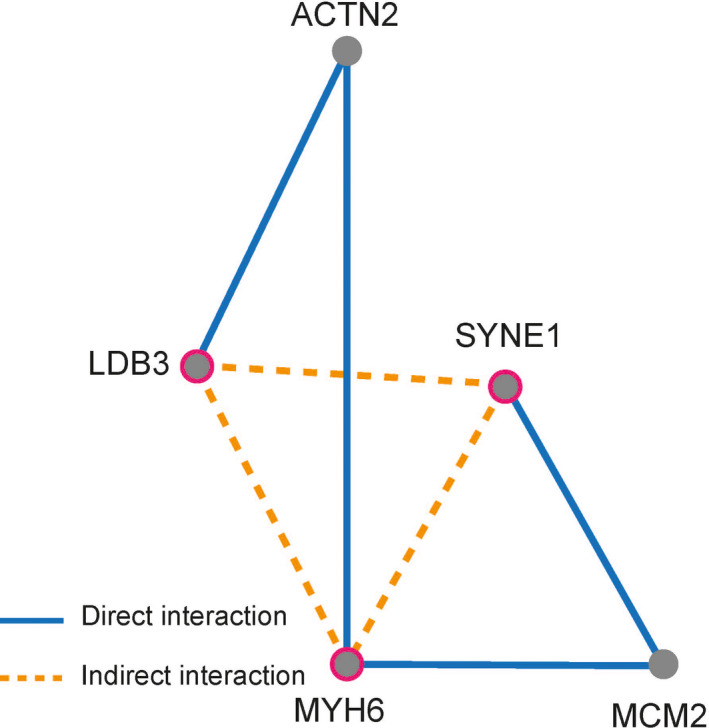
The protein–protein interactions for the risk genes associated with cardiomyopathy. ACTN2, actinin alpha 2; MCM2, minichromosome maintenance complex component 2
4. DISCUSSION
4.1. LDB3 and cardiomyopathy
LDB3 encodes the ZASP protein in humans and cypher protein in mice. It expresses as a cardiac and skeletal protein in the cytoplasm, which not only stabilizes the Z‐line, but also modifies the calcium and sodium currents of cardiomyocytes (Zheng et al., 2009). The suppression of ZASP/cypher protein induced by LDB3 pathogenic mutation was associated with DCM, left ventricular noncompaction (LVNC), arrhythmogenic cardiomyopathy (ACM) (Lopez‐Ayala et al., 2015), HCM (Fratev et al., 2014), heart failure (HF), and SCD (Vatta et al., 2003). The mutation of LDB3 p.M456R with MAF less than 0.001 located in exon 10 and predicted as “Damaging/Deleterious.” LDB3 p.M456R not only reduced its hydrophobicity of amino acid residues and their adjacent sequences, but also led to the transformation of 456th amino acids from the non‐phosphorylation site to phosphorylation site, which therefore might change the function of ZASP protein.
The heart‐specific domain of ZASP/Cypher encoded by exon 10 of LDB3, preferentially expresses in the heart and binds to phosphoglucomutase 1 (PGM1) (Arimura et al., 2004). The pathogenic mutations in exon 10 of LDB3 may be associated with DCM via impaired the ZASP/Cypher binding to PGM1 (Arimura et al., 2009). PGM1 is a key enzyme in cellular glucose utilization and energy homeostasis, which support cardiac contractile activity and pathway of glucose metabolism (including glucose uptake and phosphorylation, glycolysis, and glucose oxidation) to produce an essential fuel under both normal and stress conditions in the heart (Depré et al., 1998; Sambandam et al., 2002). PGM1 translocating from cytoplasm and binding to ZASP/Cypher in the Z‐disks was enhanced in cardiomyocytes under cultured conditions of stress (Arimura et al., 2009). Therefore, we speculated that the mutation of LDB3 p.M456R in exon 10 would impair the cardiac glucose utilization and energy homeostasis, which therefore induced DCM, malignant ventricular tachycardia, and SCD as one of the key risk factors for this patient.
4.2. MYH6 and cardiomyopathy
Cardiac muscle myosin, along with actin, is one of the major components of sarcomere, the building block of cardiac contractile system. Myosin consists of two heavy chain subunits (alpha and beta), two light chain subunits and two regulatory subunits. MYH6 encodes the alpha myosin heavy chain subunit (α‐MHC) that encompasses ~26,000 bp and consists of 39 exons, 37 of which contain coding information. The α‐MHC consisting of head, neck, and tail domains plays a vital role in myofibril assembly and proper heart development. The tail domains of α‐MHC form a coiled coil that stabilizes the molecule so that the head domain is able to generate force through its interaction with actin (England & Loughna, 2013). MYH6 mutations cause HCM, DCM, and congenital heart disease with incomplete penetrance (Carniel et al., 2005; Ching et al., 2005; Granados‐Riveron et al., 2010; Gruner et al., 2011; Hershberger et al., 2010; Posch et al., 2011). The MYH6 p.R721T was associated with high risk of sick sinus syndrome (Holm et al., 2011). In three generations of Australian family, the MYH6 p.R654T resulted in early‐onset sinus node dysfunction, ventricular arrhythmias, and subsequent cardiac arrest (Lam et al., 2015). The compound heterozygosis for recessive MYH6 mutations (I704N/T1379M, D588A/E1207K) were demonstrated in patients with hypoplastic left heart and reduced right ventricular ejection fraction. The MYH6 p.I704N and p.D588A were located in the head domain and predicted to impair power stroke recovery and its interaction with actin, respectively. Additionally, the MYH6 p.T1379M and p.E1207K were located in the tail domain and predicted to affect the local structure of the coiled coil domain (Theis et al., 2015). The MYH6 p.A230P and p.A1366D disrupted myofibril formation, whereas the MYH6 p.H252Q enhanced myofibril assembly (Granados‐Riveron et al., 2010), which therefore induced atrial septal defects. Moreover, MYH6 knockdown quickly and efficiently induced DCM and HF (Carroll et al., 2016). In our study, the MYH6 p.S180Y were predicted as “Damaging/Deleterious” and not found in the population of 1000 Genomes Project. The MYH6 p.S180Y was located in the motor domain of myosin head, which associated with power stroke recovery and interaction with actin. The mutation reduced the hydrophobicity of α‐MHC, and meanwhile led to the disappearance of potential phosphorylation of the corresponding amino acid site, which may induce weaken force and interaction with actin, and subsequently participate into the pathogenicity of DCM.
4.3. SYNE1 and cardiomyopathy
SYNE1 encodes the nesprin, including 8,797 amino acids in human. Its N‐terminus is localized to cardiac sarcomeres, while the C‐terminus is localized to the nuclear envelope participating in a complex that links the nucleoskeleton to the cytoskeleton (LINC) (Zhang et al., 2002). The nesprin is involved in a force transmission between cytoskeleton and the nucleus and transduces the mechanical signal into transcriptional response through protein–protein interactions (Gu et al., 2017). SYNE1 mutations are associated with different cardiac phenotypes as DCM with conduction system defects (Puckelwartz et al., 2010), but also with slight left ventricular basal and septal hypertrophy with mild diastolic dysfunction (Zhang et al., 2007). According to previous reports, the SYNE1 p.P3992R (NM_033071: exon73:c.C11975G) was associated with emery‐dreifuss muscular dystrophy and HCM (Sandra et al., 2019). The SYNE1 mutations (p.R8272Q, p.S8381C, p.N8406K and p.R374H) increased levels of nesprin‐1α and Lamin A/C protein and disrupted LINC, which contributed to the pathogenesis of DCM (Puckelwartz et al., 2010; Zhou et al., 2017). The splice mutation (c.6403‐1) of SYNE1 inducing physical disruption of LINC complex interactions may be the plausible biological mechanism underlying the familial DCM (Haskell et al., 2017). The SYNE1 p.S4607F predicted as “Damaging/Deleterious” in our study was not found in the population of 1000 Genomes Project. It significantly increased the hydrophobicity of amino acid residues and their adjacent sequences, and meanwhile led to the disappearance of phosphorylation of the corresponding amino acid site. These changes of protein property and secondary structure of nesprin would lead to LINC disruption, which might participate into the pathogenesis of DCM and SCD in our study.
In the family of our study, the parents carrying with only SYNE1 p.S4607F or LDB3 p.M456R/MYH6 p.S180Y mutations presented with no phenotype of cardiomyopathy or arrhythmia. When fusing with these three mutations in the patient of II: 1, she occurred with young early‐onset and severe DCM, and subsequently died of VT/F even with ICD and anti‐arrhythmia therapy. The pairwise combinations of these three mutations have obvious and high joint pathogenicity, especially the combination of SYNE1/LDB3 and SYNE1/MYH6 mutations. It was worth noting that, the participation of SYNE1 p.S4607F may be the most important risk factor for DCM, VT/F and SCD, when carrying with LDB3 and MYH6 mutations. These three mutations induced early‐onset, exacerbate, progressive, and deteriorated phenotype of DCM involving in the bilateral ventricles, characterized by obviously atrophied, dissolved and disappeared of myocardial fibers; hyperplasia interstitial fibers, disrupted intercalated disk, vacuolar degeneration of mitochondria, accumulated and filled glycogen particles. The abnormal indirect protein–protein interaction induced by these three mutations through changing their hydrophobicity and phosphorylation of amino acids may contribute into the mechanism of young early‐onset DCM, VT/F and SCD.
4.4. Study limitations
Further animal or cell experiments are needed to confirm the mechanisms how these three mutations abnormally change the cardiac structure and function and result in the DCM and SCD.
5. CONCLUSIONS
We firstly reported that the multiple heterozygous mutations of SYNE1 p.S4607F, LDB3 p.M456R, and MYH6 p.S180Y with high joint pathogenicity may induce abnormal indirect protein–protein interactions through changing their hydrophobicity and phosphorylation of amino acids, which therefore led to young early‐onset and severe DCM, and subsequent SCD associated with VT/F.
CONFLICT OF INTEREST
The authors declare that they have no competing interest.
AUTHOR CONTRIBUTIONS
TZ and YBL: Pedigree analysis and writing; YTM: Bioinformatics analysis; ZQZ, JZX, and FW: Imaging analysis; ZY and XJG: Case collection and follow‐up; JNH: Sanger sequencing; YL and YBL: Quality control of clinical data and clinical design.
ETHICAL APPROVAL
This study was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [No. GDREC2016001H (R1)]. With the consent of the ethics committee, we followed up the patients and their family members under the condition of informed consents and obtained blood samples for genetic analysis.
ACKNOWLEDGMENTS
We thank Professor Shulin Wu, Yumei Xue, and Fang Rao from Guangdong Cardiovascular Institute. We appreciate Jinsheng Tao and Zhipeng Cao for assistance in data analysis and visualization.
Zhao T, Ma Y, Zhang Z, et al. Young and early‐onset dilated cardiomyopathy with malignant ventricular arrhythmia and sudden cardiac death induced by the heterozygous LDB3, MYH6, and SYNE1 missense mutations. Ann Noninvasive Electrocardiol. 2021;26:e12840. 10.1111/anec.12840
Ting Zhao and Yuting Ma contributed equally to this manuscript.
Funding information
These fundings supported this study: The Foundation Project of Medical Science and Technology Research in Guangdong Province (A2018043); Science and Technology Project of Zhuhai (20191210E030072); Natural Science Foundation of Guangdong (2016A030313279); the Construction Foundation (ZHLCZD011) of the Key Clinical Specialties in Zhuhai
Contributor Information
Yi Luo, Email: yiluo63@126.com.
Yubi Lin, Email: linyubi88@qq.com, Email: linyb23@mail.sysu.edu.cn.
DATA AVAILABILITY STATEMENT
The data used in this study are not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura, T. , Hayashi, T. , Terada, H. , Lee, S.‐Y. , Zhou, Q. , Takahashi, M. , Ueda, K. , Nouchi, T. , Hohda, S. , Shibutani, M. , Hirose, M. , Chen, J. , Park, J.‐E. , Yasunami, M. , Hayashi, H. , & Kimura, A. (2004). A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. Journal of Biological Chemistry, 279(8), 6746–6752. 10.1074/jbc.M311849200 [DOI] [PubMed] [Google Scholar]
- Arimura, T. , Inagaki, N. , Hayashi, T. , Shichi, D. , Sato, A. , Hinohara, K. , Vatta, M. , Towbin, J. A. , Chikamori, T. , Yamashina, A. , & Kimura, A. (2009). Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovascular Research, 83(1), 80–88. 10.1093/cvr/cvp119 [DOI] [PubMed] [Google Scholar]
- Carniel, E. , Taylor, M. R. , Sinagra, G. , Lenarda, A. D. , Ku, L. , Fain, P. R. , Boucek, M. M. , Cavanaugh, J. , Miocic, S. , Slavov, D. , Graw, S. L. , Feiger, J. , Zhu, X. Z. , Dao, D. , Ferguson, D. A. , Bristow, M. R. , & Mestroni, L. (2005). Alpha‐myosin heavy chain: A sarcomeric gene associated with dilated and hypertrophic phenotypes of cardiomyopathy. Circulation, 112(1), 54–59. [DOI] [PubMed] [Google Scholar]
- Carroll, K. J. , Makarewich, C. A. , McAnally, J. , Anderson, D. M. , Zentilin, L. , Liu, N. , Giacca, M. , Bassel‐Duby, R. , & Olson, E. N. (2016). A mouse model for adult cardiac‐specific gene deletion with CRISPR/Cas9. Proceedings of the National Academy of Sciences of the United States of America, 113(2), 338–343. 10.1073/pnas.1523918113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi, F. , Tomberli, B. , & Olivotto, I. (2012). Clinical and molecular classification of cardiomyopathies. Global Cardiology Science and Practice, 2012(1), 4. 10.5339/gcsp.2012.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching, Y. H. , Ghosh, T. K. , Cross, S. J. , Packham, E. A. , Honeyman, L. , Loughna, S. , Robinson, T. E. , Dearlove, A. M. , Ribas, G. , Bonser, A. J. , Thomas, N. R. , Scotter, A. J. , Caves, L. S. D. , Tyrrell, G. P. , Newbury‐Ecob, R. A. , Munnich, A. , Bonnet, D. , & Brook, J. D. (2005). Mutation in myosin heavy chain 6 causes atrial septal defect. Nature Genetics, 37(4), 423–428. 10.1038/ng1526 [DOI] [PubMed] [Google Scholar]
- Depré, C. , Rider, M. H. , & Hue, L. (1998). Mechanisms of control of heart glycolysis. European Journal of Biochemistry, 258(2), 277–290. 10.1046/j.1432-1327.1998.2580277.x [DOI] [PubMed] [Google Scholar]
- Di Resta, C. , Pietrelli, A. , Sala, S. , Bella, P. D. , Bellis, G. D. , Ferrari, M. , Bordoni, R. , & Benedetti, S. (2015). High‐throughput genetic characterization of a cohort of Brugada syndrome patients. Human Molecular Genetics, 24(20), 5828–5835. [DOI] [PubMed] [Google Scholar]
- England, J. , & Loughna, S. (2013). Heavy and light roles: Myosin in the morphogenesis of the heart. Cellular and Molecular Life Sciences, 70(7), 1221–1239. 10.1007/s00018-012-1131-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratev, F. , Mihaylova, E. , & Pajeva, I. (2014). Combination of genetic screening and molecular dynamics as a useful tool for identification of disease‐related mutations: ZASP PDZ domain G54S mutation case. Journal of Chemical Information and Modeling, 54(5), 1524–1536. 10.1021/ci5001136 [DOI] [PubMed] [Google Scholar]
- Fu, Y. , & Eisen, H. J. (2018). Genetics of dilated cardiomyopathy. Current Cardiology Reports, 20(11), 121. 10.1007/s11886-018-1061-0 [DOI] [PubMed] [Google Scholar]
- Geourjon, C. , & Deléage, G. (1995). SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Computer Applications in the Biosciences, 11(6), 681–684. [DOI] [PubMed] [Google Scholar]
- Gigli, M. , Merlo, M. , Graw, S. L. , Barbati, G. , Rowland, T. J. , Slavov, D. B. , Stolfo, D. , Haywood, M. E. , Dal Ferro, M. , Altinier, A. , Ramani, F. , Brun, F. , Cocciolo, A. , Puggia, I. , Morea, G. , McKenna, W. J. , La Rosa, F. G. , Taylor, M. R. G. , Sinagra, G. , & Mestroni, L. (2019). Genetic risk of arrhythmic phenotypes in patients with dilated cardiomyopathy. Journal of the American College of Cardiology, 74(11), 1480–1490. 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granados‐Riveron, J. T. , Ghosh, T. K. , Pope, M. , Bu'Lock, F. , Thornborough, C. , Eason, J. , Kirk, E. P. , Fatkin, D. , Feneley, M. P. , Harvey, R. P. , Armour, J. A. L. , & Brook, J. D. (2010). Alpha‐cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Human Molecular Genetics, 19(20), 4007–4016. [DOI] [PubMed] [Google Scholar]
- Gruner, C. , Care, M. , Siminovitch, K. , Moravsky, G. , Wigle, E. D. , Woo, A. , & Rakowski, H. (2011). Sarcomere protein gene mutations in patients with apical hypertrophic cardiomyopathy. Circulation: Cardiovascular Genetics, 4(3), 288–295. 10.1161/CIRCGENETICS.110.958835 [DOI] [PubMed] [Google Scholar]
- Gu, Q. , Mendsaikhan, U. , Khuchua, Z. , Jones, B. C. , Lu, L. , Towbin, J. A. , Xu, B. , & Purevjav, E. (2017). Dissection of Z‐disc myopalladin gene network involved in the development of restrictive cardiomyopathy using system genetics approach. World Journal of Cardiology, 9(4), 320–331. 10.4330/wjc.v9.i4.320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas, J. , Frese, K. S. , Peil, B. , Kloos, W. , Keller, A. , Nietsch, R. , Feng, Z. , Müller, S. , Kayvanpour, E. , Vogel, B. , Sedaghat‐Hamedani, F. , Lim, W.‐K. , Zhao, X. , Fradkin, D. , Köhler, D. , Fischer, S. , Franke, J. , Marquart, S. , Barb, I. , … Meder, B. (2015). Atlas of the clinical genetics of human dilated cardiomyopathy. European Heart Journal, 36(18), 1123–1135. [DOI] [PubMed] [Google Scholar]
- Haskell, G. T. , Jensen, B. C. , Samsa, L. A. , Marchuk, D. , Huang, W. , Skrzynia, C. , Tilley, C. , Seifert, B. A. , Rivera‐Muñoz, E. A. , Koller, B. , Wilhelmsen, K. C. , Liu, J. , Alhosaini, H. , Weck, K. E. , Evans, J. P. , & Berg, J. S. (2017). Whole exome sequencing identifies truncating variants in nuclear envelope genes in patients with cardiovascular disease. Circulation: Cardiovascular Genetics, 10(3). ), e001443 10.1161/CIRCGENETICS.116.001443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger, R. E. , Norton, N. , Morales, A. , Li, D. , Siegfried, J. D. , & Gonzalez‐Quintana, J. (2010). Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circulation: Cardiovascular Genetics, 3(2), 155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, H. , Gudbjartsson, D. F. , Sulem, P. , Masson, G. , Helgadottir, H. T. , Zanon, C. , Magnusson, O. T. , Helgason, A. , Saemundsdottir, J. , Gylfason, A. , Stefansdottir, H. , Gretarsdottir, S. , Matthiasson, S. E. , Thorgeirsson, G. , Jonasdottir, A. , Sigurdsson, A. , Stefansson, H. , Werge, T. , Rafnar, T. , … Stefansson, K. (2011). A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nature Genetics, 43(4), 316–320. 10.1038/ng.781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lam, L. , Ingles, J. , Turner, C. , Kilborn, M. , Bagnall, R. D. , & Semsarian, C. (2015). Exome sequencing identifies a novel mutation in the MYH6 gene in a family with early‐onset sinus node dysfunction, ventricular arrhythmias, and cardiac arrest. HeartRhythm Case Reports, 1(3), 141–145. 10.1016/j.hrcr.2015.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z. , Chen, P. , Li, C. , Tan, L. , Xu, J. , Wang, H. , Sun, Y. , Wang, Y. , Zhao, C. , Link, M. S. , Wilde, A. A. M. , Wang, D. W. , & Wang, D. W. (2020). Genetic arrhythmias complicating patients with dilated cardiomyopathy. Heart Rhythm: the Official Journal of the Heart Rhythm Society, 17(2), 305–312. 10.1016/j.hrthm.2019.09.012 [DOI] [PubMed] [Google Scholar]
- Lin, Y. , Zhang, Q. , Zhong, Z. A. , Xu, Z. , He, S. , Rao, F. , Liu, Y. , Tang, J. , Wang, F. , Liu, H. , Xie, J. , Wu, H. , Wang, S. , Li, X. , Shan, Z. , Deng, C. , Liao, Z. , Deng, H. , Liao, H. , … Wu, S. (2017). Whole genome sequence identified a rare homozygous pathogenic mutation of the DSG2 gene in a familial arrhythmogenic cardiomyopathy involving both ventricles. Cardiology, 138(1), 41–54. [DOI] [PubMed] [Google Scholar]
- Lopez‐Ayala, J. M. , Ortiz‐Genga, M. , Gomez‐Milanes, I. , Lopez‐Cuenca, D. , Ruiz‐Espejo, F. , Sanchez‐Munoz, J. J. , Oliva‐Sandoval, M. J. , Monserrat, L. , & Gimeno, J. R. (2015). A mutation in the Z‐line Cypher/ZASP protein is associated with arrhythmogenic right ventricular cardiomyopathy. Clinical Genetics, 88(2), 172–176. [DOI] [PubMed] [Google Scholar]
- McNally, E. M. , Golbus, J. R. , & Puckelwartz, M. J. (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. Journal of Clinical Investigation, 123(1), 19–26. 10.1172/JCI62862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posch, M. G. , Waldmuller, S. , Müller, M. , Scheffold, T. , Fournier, D. , Andrade‐Navarro, M. A. , De Geeter, B. , Guillaumont, S. , Dauphin, C. , Yousseff, D. , Schmitt, K. R. , Perrot, A. , Berger, F. , Hetzer, R. , Bouvagnet, P. , & Özcelik, C. (2011). Cardiac alpha‐myosin (MYH6) is the predominant sarcomeric disease gene for familial atrial septal defects. PLoS One, 6(12), e28872. 10.1371/journal.pone.0028872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckelwartz, M. J. , Kessler, E. J. , Kim, G. , DeWitt, M. M. , Zhang, Y. , Earley, J. U. , Depreux, F. F. S. , Holaska, J. , Mewborn, S. K. , Pytel, P. , & McNally, E. M. (2010). Nesprin‐1 mutations in human and murine cardiomyopathy. Journal of Molecular and Cellular Cardiology, 48(4), 600–608. 10.1016/j.yjmcc.2009.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchand, J. , Wallis, M. , Macciocca, I. , Lynch, E. , Farouque, O. , Martyn, M. , Phelan, D. , Chong, B. , Lockwood, S. , Weintraub, R. , Thompson, T. , Trainer, A. , Zentner, D. , Vohra, J. , Chetrit, M. , Hare, D. L. , & James, P. (2020). Prospective evaluation of the utility of whole exome sequencing in dilated cardiomyopathy. Journal of the American Heart Association, 9(2), e013346. 10.1161/JAHA.119.013346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichart, D. , Magnussen, C. , Zeller, T. , & Blankenberg, S. (2019). Dilated cardiomyopathy: From epidemiologic to genetic phenotypes: A translational review of current literature. Journal of Internal Medicine, 286(4), 362–372. 10.1111/joim.12944 [DOI] [PubMed] [Google Scholar]
- Renaux, A. , Papadimitriou, S. , Versbraegen, N. , Nachtegael, C. , Boutry, S. , Nowé, A. , Smits, G. , & Lenaerts, T. (2019). ORVAL: A novel platform for the prediction and exploration of disease‐causing oligogenic variant combinations. Nucleic Acids Research, 47(W1), W93–W98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambandam, N. , Lopaschuk, G. D. , Brownsey, R. W. , & Allard, M. F. (2002). Energy metabolism in the hypertrophied heart. Heart Failure Reviews, 7(2), 161–173. [DOI] [PubMed] [Google Scholar]
- Sandra, M. , Maria Pia, L. , Stefano, C. , Pietro, P. , Crociani, P. , Aldo, R. , Giuseppe, D. S. , & Massimo, C. (2019). Emery‐Dreifuss muscular dystrophy type 4: A new SYNE1 mutation associated with hypertrophic cardiomyopathy masked by a perinatal distress‐related spastic diplegia. Clinical Case Reports, 7(5), 1078–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smigielski, E. M. , Sirotkin, K. , Ward, M. , & Sherry, S. T. (2000). dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Research, 28(1), 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, D. , Gable, A. L. , Lyon, D. , Junge, A. , Wyder, S. , Huerta‐Cepas, J. , Simonovic, M. , Doncheva, N. , Morris, J. , Bork, P. , Jensen, L. , & von Mering, C. (2019). STRING v11: Protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Research, 47(D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis, J. L. , Zimmermann, M. T. , Evans, J. M. , Eckloff, B. W. , Wieben, E. D. , Qureshi, M. Y. , O'Leary, P. W. , & Olson, T. M. (2015). Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circulation: Cardiovascular Genetics, 8(4), 564–571. [DOI] [PubMed] [Google Scholar]
- Vatta, M. , Mohapatra, B. , Jimenez, S. , Sanchez, X. , Faulkner, G. , Perles, Z. , Sinagra, G. , Lin, J.‐H. , Vu, T. M. , Zhou, Q. , Bowles, K. R. , Lenarda, A. D. , Schimmenti, L. , Fox, M. , Chrisco, M. A. , Murphy, R. T. , McKenna, W. , Elliott, P. , Bowles, N. E. , … Towbin, J. A. (2003). Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non‐compaction. Journal of the American College of Cardiology, 42(11), 2014–2027. [DOI] [PubMed] [Google Scholar]
- Via, M. , Gignoux, C. , & Burchard, E. G. (2010). The 1000 Genomes Project: New opportunities for research and social challenges. Genome Medicine, 2(1), 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, M. R. , Gasteiger, E. , Bairoch, A. , Sanchez, J. C. , Williams, K. L. , Appel, R. D. , & Hochstrasser, D. F. (1999). Protein identification and analysis tools in the ExPASy server. Methods in Molecular Biology, 112, 531–552. [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Bethmann, C. , Worth, N. F. , Davies, J. D. , Wasner, C. , Feuer, A. , Ragnauth, C. D. , Yi, Q. , Mellad, J. A. , Warren, D. T. , Wheeler, M. A. , Ellis, J. A. , Skepper, J. N. , Vorgerd, M. , Schlotter‐Weigel, B. , Weissberg, P. L. , Roberts, R. G. , Wehnert, M. , & Shanahan, C. M. (2007). Nesprin‐1 and ‐2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Human Molecular Genetics, 16(23), 2816–2833. 10.1093/hmg/ddm238 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. , Ragnauth, C. , Greener, M. J. , Shanahan, C. M. , & Roberts, R. G. (2002). The nesprins are giant actin‐binding proteins, orthologous to Drosophila melanogaster muscle protein MSP‐300. Genomics, 80(5), 473–481. 10.1006/geno.2002.6859 [DOI] [PubMed] [Google Scholar]
- Zheng, M. , Cheng, H. , Li, X. , Zhang, J. , Cui, L. , Ouyang, K. , Han, L. , Zhao, T. , Gu, Y. , Dalton, N. , Bang, M. , Peterson, K. , & Chen, J. (2009). Cardiac‐specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Human Molecular Genetics, 18(4), 701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, C. , Li, C. , Zhou, B. , Sun, H. , Koullourou, V. , Holt, I. , Puckelwartz, M. J. , Warren, D. T. , Hayward, R. , Lin, Z. , Zhang, L. , Morris, G. E. , McNally, E. M. , Shackleton, S. , Rao, L. , Shanahan, C. M. , & Zhang, Q. (2017). Novel nesprin‐1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Human Molecular Genetics, 26(12), 2258–2276. 10.1093/hmg/ddx116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in this study are not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.