

Abstract
Herein we report a new high-denticity chelator based on the iron siderophore desferrioxamine (DFO). Our new chelator — DFO2 — was designed and synthesized with the purpose of improving the coordination chemistry and radiolabeling performance with radioactive zirconium-89. The radionuclide zirconium-89 ([89Zr]-Zr4+) has found wide-usage for positron emission tomography (PET) imaging when coupled with proteins, antibodies, and nanoparticles. DFO2 has a potential coordination number of twelve, which provides theranostic potential for binding large oxophilic metal ions. Following synthesis of the DFO2 chelator and the [natZr]Zr-DFO2 complex, we performed density functional theory calculations to study its coordination sphere, and zirconium-89 radiolabeling experiments for comparisons with the “gold standard” chelator DFO. DFO (CN = 6) is thought to coordinate with zirconium in a hexadentate fashion leaving two open coordination sites where water is thought to coordinate (total CN = 8). DFO2 (potential CN = 12, dodecadentate) saturated the coordination sphere of zirconium with four hydroxamate groups (CN = 8) with no room left for water to directly coordinate, and only binds a single atom of zirconium per chelate. Following quantitative radiolabeling with zirconium-89, the preformed [89Zr]Zr-(DFO) and [89Zr]Zr-(DFO2) radiometal-chelate complexes were subjected to a battery of in vitro stability challenges including human blood serum, apo-transferrin, serum albumin, iron, hydroxyapatite, and EDTA. One objective of these stability challenges was to determine if the increased denticity of DFO2 over DFO imparted improved chelate stability, and another was to determine which of these assays is most relevant to perform with future chelators. In all assays DFO2 showed superior stability with zirconium-89, except for the iron challenge where both DFO2 and DFO were identical. Substantial differences in stability were observed for human blood serum using a precipitation method of analysis, apo-transferrin, hydroxyapatite, and EDTA. These results suggest that DFO2 is a promising next-generation scaffold for zirconium-89 chelators and holds promise for radiochemisty with even larger radionuclides, which will expand to utility of DFO2 into theranostic applications.
Keywords: 89Zr, zirconium-89, DFO, desferrioxamine, antibody, chelator, bifunctional chelator, theranostic, thorium, radiometal, radionuclide, PET, immuno-PET, radioimmunotherapy
Graphical Abstract

TOC synopsis: DFO2 is a new acyclic chelator containing six hydroxamic acid moieties and a potential coordination number of twelve (dodecadentate). This chelator presents double the potential coordination number as desferrioxamine (DFO, hexadentate), providing interesting coordination options beyond the standard 6–8 coordination numbers that are typical for zirconium(IV). This additional coordinating ability has imbued DFO2 with superior radiochemical properties to DFO, as demonstrated by zirconium-89 experiments, and will enable theranostic applications with even larger radiometal ions.
INTRODUCTION
The radionuclide zirconium-89 has gained popularity over the last decade due to its favorable decay characteristics ([89Zr]Zr4+, t1/2 = 78.4 hr, β+ ratio = 22.7%, Eβ+(mean)= 396 keV) for positron emission tomography (non-invasive nuclear imaging modality, PET).1–3 The radiometal zirconium-89 has a half-life that is well matched with large and slowly distributing biological vectors such as antibodies and nanoparticles.4–16 Previously, the only chelator capable of effectively coordinating with zirconium-89 (radiolabeling) and forming a complex with sufficient stability and kinetic inertness in vivo was the iron siderophore desferrioxamine (DFO).17–21 DFO is typically non-site-selectively conjugated to lysine residues (primary amine) on antibodies to form thiourea linkages, using the bifunctional chelator (BFC) p-SCN-Bn-DFO (commercially available), although amide linkages formed via DFO-activated esters are also used.22, 23 Zirconium-89 is known to accumulate in bone when released from a chelate-complex in vivo, and experiments in mice have consistently shown a substantial amount of bone uptake (~5–10% ID/g after ~3–7 days).2, 9, 24 Despite the apparent instability of the [89Zr]Zr-(DFO) complex in mice, human immuno-PET imaging studies using zirconium-89 do not appear to suffer from the same degree of high bone uptake.6, 25–27 In recent years, a number of promising new chelators have been published which all aim to improve radiolabeling performance and in vivo stability with zirconium-89 (Figure 1).3, 18, 28–42
Figure 1.
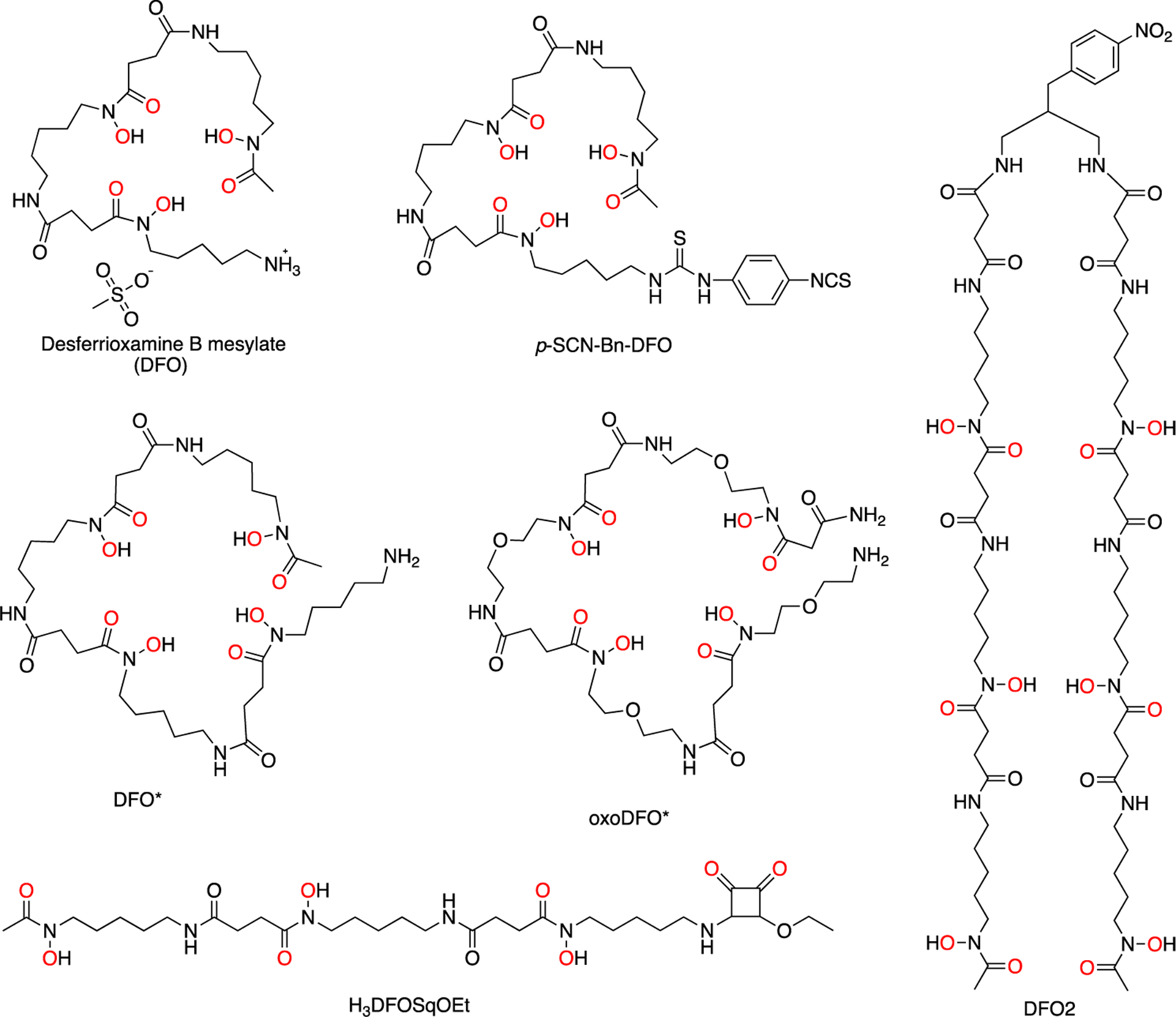
Chemical structures of a selection of recently published bifunctional chelators based on desferrioxamine (DFO) for zirconium-89, including p-SCN-Bn-DFO, DFO*, oxoDFO*, DFO squarate ester (H3DFOSqOEt), and DFO2.
Over the previous few years we have been working on the design, synthesis, and evaluation of a new family of DFO-based chelators. The first of these new chelators is presented herein and named DFO2, as it is comprised of two desferrioxamine molecules tethered together using a bifunctional linker. The resulting chelator is potentially 12-coordinate (six hydroxamic acid groups, CN=12), with an octadentate (CN=8) coordination sphere expected for Zr4+ with DFO2. One drawback of many new chelators is their cumbersome synthesis and difficult purification — typically a result of the intrinsic need of chelators to contain a large number of oxygen- and nitrogen-rich polar functional groups with high hydrogen bonding potential. We have achieved simple synthesis and purification by using commercial DFO as our major building block, although low water solubility of the unconjugated DFO2 was encountered.
The long-term goals of this new “DFO2” chelator family is a rapid and modular synthesis platform to enable fast evaluation of a variety of chelator derivatives, and then rapid synthetic iteration of the lead ligand scaffolds. The priorities of our chelate system are thus simple and fast synthesis, with the requirement to contain a modular design where we can easily change the linker group, add different chelating moieties, change the bifunctional conjugation chemistry, and tune physical properties such as polarity/solubility. The first example of this new chelator family, DFO2, has been synthesized, characterized, radiolabeled with zirconium-89, and evaluated via in vitro radiochemical stability assays. We have additionally evaluated the coordination sphere using density functional theory (DFT) calculations as we did not obtain diffractable crystals and therefore no X-ray crystallographic data. We have performed many different types of in vitro stability assays with the purpose of determining which assays are most relevant for probing 89Zr-chelate stability. We hope that the results of these assays will make this process of screening chelators more efficient in the future by identifying which in vitro assays best predict in vivo stability differences.
RESULTS AND DISCUSSION
To expand the denticity of DFO from a potential coordination number of six (three hydroxamic acid groups), we chemically tethered two molecules of commercially available DFO together. To achieve this, we required a linker that could both tether two DFO molecules with suitable chemistry, but also provide orthogonal reactivity in the bifunctional component of the chelator. The bifunctional linker will act as a handle to facilitate future conjugations with common targeting vectors used in molecular imaging such as peptides, antibodies, and nanoparticles. We utilized a common p-NO2-phenyl group, which in a follow-up study we will hydrogenate to the aniline form, followed by reaction with thiophosgene or a related reagent to form the bifunctional-ready p-SCN-phenyl group. This electrophile is commonly used in bioconjugation chemistry and easily forms a thiourea linkage with an available primary amine (e.g. ε-amine of lysine). To obtain this p-NO2-phenyl group in the backbone of our chelator we must perform three synthetic steps, which has provided a challenge due to low yields and difficult purification. In the future we plan to optimize the design of DFO2 by exploring alternative linker options to the p-NO2-phenyl propylenediamine linker.
Synthesis of DFO2 revolved around a propylenediamine linker with a p-NO2-phenyl group extending from the backbone. We synthesized this fragment in three steps starting from p-NO2-benzylbromide and diethylmalonate (Scheme 1). An optimization was made to a previously published synthetic method for this linker fragment, where during step two of the synthesis to form compound 2 we used ammonium hydroxide as the source of amine instead of ammonia gas. This route does not require access to a compressed cylinder of ammonia gas, or the additional hazards of ammonia gas.43 The functional linker moiety 3 was synthesized in three chemical steps with a cumulative yield of ~8%.
Scheme 1.

Synthesis of the linker molecule 3, which was used to tether two DFO molecules together to form DFO2.
To attach commercially available desferrioxamine B mesylate (DFO, Scheme 2) to our linker fragment (3), we had to modify DFO through a ring-opening reaction with succinic anhydride. This step formed DFO-COOH (4), which extended the length of the DFO chain by four atoms (from 31 to 35 atoms long), and also converted the terminal functional group from a primary amine to a carboxylic acid. The backbone linker 3 was then reacted with DFO-COOH (4) to form DFO2 (5) using standard peptide coupling conditions. As with the bifunctional chelators p-SCN-Ph-DFO or p-NO2-Ph-DFO, the solubility of DFO2 is poor. During the synthetic step to produce DFO2 (5), the solvent DMF was required to achieve solubility and suitable reaction yields. The poor solubility of the resulting DFO2 (5) chelator enabled us to purify DFO2 by only precipitation and washing, where we tested a large number of solvent combinations and found cold ethyl acetate was most effective. Coordination of DFO2 with ZrCl4 salt rapidly formed the coordinated Zr(DFO2) complex under mild ambient conditions. A small amount of strong polar solvent dimethylformamide (DMF) was required to solubilize DFO2 at the macro-scale and facilitate this non-radioactive coordination chemistry. At the low concentrations of chelator required for radiochemistry (μM-nM), sufficient solubility can be achieved by transferring a small volume of a DFO2 stock solution dissolved in DMF or DMSO into aqueous radiolabeling buffer. The non-radioactive Zr(DFO2) complex was characterized by standard techniques including 1H/13C-NMR spectroscopy and low- and high-resolution mass spectrometry. Despite the poor water solubility, we have not been able to grow X-ray crystallography quality crystals.
Scheme 2.

Synthesis of DFO2 from modified desferrioxamine B (4) and linker group (3), followed by non-radioactive Zr4+ coordination showing one possible conformational isomer (6).
In lieu of crystallographic data, we performed density functional theory (DFT) calculations to evaluate the coordination sphere of the Zr(DFO2) complex (Figure 2). Calculations were performed using Materials Studio software with the DMol3 functional and PBE basis set, with solvent effects modeled by COSMO. All the calculations were performed in solution with water as the solvent. Due to the large structure of the Zr(DFO2) complex, it took substantial time to complete calculations. The DFO2 chelator with many polar hydroxamic acid and carbonyl groups was prone to getting stuck in local minimums, potentially due to intramolecular hydrogen bonding between the two DFO chains. The calculated structure of the Zr(DFO2) complex at the global minimum energy suggests an octadentate structure with no bound water molecules (Figure 2).
Figure 2.
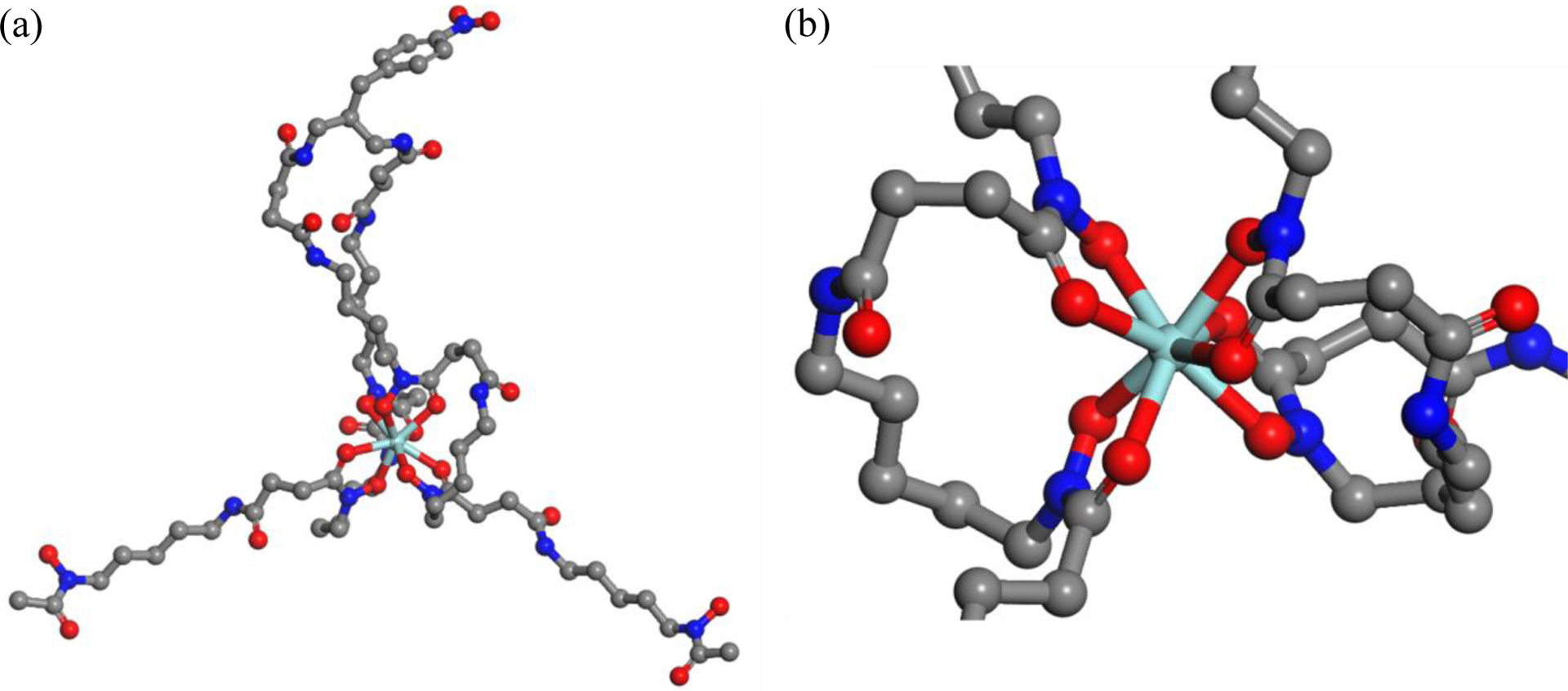
Energy-minimized and geometry-optimization molecular structures calculating using DFT (Materials Studio, DMol3/PBE, solvent as water with COSMO) showing (a) the Zr(DFO2) complex, and (b) the close-up of Zr(IV) ion’s geometry in Zr(DFO2) complex. Hydrogen atoms are removed from figure for visual clarity but are included for calculation.
Hydroxamic acid groups are known as being exceptionally well suited for binding Zr4+, more so than endogenous biological ligands that would be available to compete (e.g. transferrin, albumin, chloride, phosphate). Brechbiel et al. in an investigation of the complexation of Zr4+ with hydroxamates showed that Zr4+ forms an eight coordinated complex with four bidentate hydroxamates.44, 45 As ligands they used two hydroxamate derivatives, acetohydroxamic acid (AHA) and N-methyl acetohydroxamic acid (Me-AHA). Single crystal X-ray diffraction studies of Zr(Me-AHA)4 revealed the average bond length of 2.193 Å for the Zr-O bonds. Also, data obtained via DFT calculations for the Zr(Me-AHA)4 complex suggested an average bond length of 2.248 Å for eight Zr-O bonds. Table 1 shows our calculated bond lengths for the Zr(DFO2) complex. The calculated average bond length of 2.244 Å for Zr-O bonds in Zr(DFO2) complex is in agreement with those of the single crystal X-ray diffraction and DFT calculations of Zr(Me-AHA)4 complex.44, 45 The calculated structure of the Zr(DFO2) complex in Figure 2 shows the bottom hydroxamic acid of each DFO tail in the DFO2 structure remaining un-bound. It is possible that other hydroxamates in DFO2 could bind instead of one or both of the bottom hydroxamate moieties and form alternative geometric isomers/conformers. It is also possible that these hydroxamates could switch during any reorganization of the coordination sphere, thereby forming an equilibrium between geometric isomers. More detailed DFT studies in combination with advanced spectroscopy techniques will likely be required to better elucidate the precise details of the coordination sphere. At this stage of research, the in vitro and in vivo stability data is the most important as we are seeking a practical and functional improvement over existing chelators for use in molecular imaging and targeted radionuclide therapy.
Table 1.
Calculated Zr-O bond lengths of Zr(DFO2) complex.*
| Zr(DFO2)* | ||
|---|---|---|
| Bond Type | Bond Length (Å) DFT* | Avg. Bond Length (Å) |
| N…O-Zr | 2.223 | 2.217 |
| N…O-Zr | 2.201 | |
| N…O-Zr | 2.228 | |
| N…O-Zr | 2.217 | |
| C…O-Zr | 2.263 | 2.270 |
| C…O-Zr | 2.254 | |
| C…O-Zr | 2.290 | |
| C…O-Zr | 2.273 | |
| Avg. of all 8 (O-Zr) bond lengths | 2.244 | |
| O=C…Zr | 3.107 | 3.086 |
| O=C…Zr | 3.079 | |
| O=C…Zr | 3.084 | |
| O=C…Zr | 3.075 | |
| O–N…Zr | 3.094 | 3.086 |
| O–N…Zr | 3.101 | |
| O–N…Zr | 3.08 | |
| O–N…Zr | 3.07 | |
Density Functional Theory calculated using Materials Studio software via DMol3/PBE.
It is possible that the two hydroxamate groups that are not strictly needed in the octadentate Zr(DFO2) complex could still improve the stability of the complex when compared with strictly octadentate chelators such as Zr(DFO*) through a process that could be roughly analogized to avidity. Although the resulting zirconium(IV) complexes for both DFO2 and DFO* are octadentate and should have comparable complex stability (both utilize hydroxamate groups), DFO2 could offer improved overall stability and in vivo kinetic inertness as a result of the two additional hydroxamic acid groups being covalently attached in close physical proximity to the coordination sphere. During any reorganization or transchelation event at the inner-coordination sphere, these extra two hydroxamate groups in DFO2 could potentially “out-compete” any in vivo ligand competition ligands such as water, chloride, phosphate, or proteins, and effectively increase stability. We radiolabeled both our new chelator DFO2 and the “gold standard” chelator DFO with zirconium-89 and have performed many in vitro stability assays. In the future it would be useful to obtain DFO* to serve as a comparison, and/or to synthesize an octadentate derivative of DFO2. DFO* was not commercially available at the time of performing these experiments. If the radiochemical stability of [89Zr]Zr-DFO2 was compared with zirconium-89 complexes of structurally similar chelators (e.g. DFO*) with a denticity of only 8, and if [89Zr]Zr-DFO2 demonstrated improved stability, it could hypothetically be attributed to the presence of the extra two hydroxamate groups.
Although it is possible that DFO2 could simply chelate two separate atoms of zirconium-89 at the same time with an expected coordination environment for each zirconium atom of three hydroxamate groups (CN = 6, with two H2O total CN = 8), the huge molar excess of chelator over the radiometal used for radiolabeling experiments makes this very unlikely to occur. Even when performing coordination chemistry with DFO2 and ~0.9 molar equivalents of non-radioactive Zr4+ salt, there was no Zr2-DFO2 observed via mass spectrometry. Radiolabeling experiments with zirconium-89 suggest the same result, where no appreciable difference in radiochemical yields (RCYs) or specific activity were observed when using 500 nmol, 50 nmol, 5 nmol, or 0.5 nmol of DFO or DFO2 (Table 2). If each molar equivalent of DFO2 was binding two molar equivalents of zirconium-89, one would expect precisely two-times higher specific activity. The difference in polarity between [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) was assessed by logD partition coefficients, determined using standard shake-flask methods with octanol and phosphate buffered saline (Table 2; pH=7.4). These results suggest that the [89Zr]Zr(DFO) complex is more water soluble with a logD value of −2.70 ± 0.06; however, it is important to note that this DFO is not the bifunctional derivative containing a phenyl ring, but rather non-functionalized desferrioxamine B mesylate which is far more water soluble. The logD value obtained for the [89Zr]Zr(DFO2) complex was −0.71 ± 0.01; however, this version contains a p-NO2-Ph moiety which will substantially decrease water solubility. Regardless, improved water solubility is a priority for future iterations of this new DFO2 family of chelators.
Table 2.
Results from radiolabeling DFO and DFO2 with zirconium-89.*
| Complex | Chelator (nmol) | [89Zr]Zr-oxalate (μCi) / (MBq) | RCY 60 min (%) | SA (mCi•μmol−1) / (MBq•μmol−1) | logDPBS pH 7.4 |
|---|---|---|---|---|---|
| [89Zr]Zr- (DFO2) | 500 | 459 / 17.0 | 99 | 0.9 / 33.6 | −0.71 ± 01 |
| 50 | 282 / 10.4 | 99 | 5.6 / 207 | ||
| 5 | 290 / 10.7 | 7.4 | 4.3 / 159 | ||
| 0.5 | 283 / 10.5 | 1 | ~ | ||
| [89Zr]Zr-(DFO) | 500 | 273 / 10.1 | 99 | 0.5 / 20.0 | −2.70 ± 06 |
| 50 | 276 / 10.2 | 99 | 5.5 / 202 | ||
| 5 | 269 / 10.0 | 2.3 | ~ | ||
| 0.5 | 282 / 10.4 | 1.4 | ~ |
At chelator concentrations of 500−0.5 nmol, radiochemical yields (RCY) determined via radio-iTLC (n=3), and logD octanol-buffer partition coefficient values (phosphate buffered saline pH=7.4) (n=5).
One of the most common in vitro stability assays performed for radiolabeled molecules is some form of a blood serum incubation/competition. For zirconium-89, DFO is known to form a stable complex that is not appreciably transchelated by human or murine blood serum, even out beyond seven days incubation time. For zirconium-89 stability assays in blood serum, radio-iTLC is most commonly used to assess the degree of transchelation to serum proteins due to its simple and fast execution. Spotting the iTLC strips and eluting with mobile phase takes ~10–15 minutes, and many can be eluted simultaneously. Then using a standard radio-TLC reader (Bioscan/EZ AR2000) or even an automated gamma counter (cutting strips into pieces), these eluted radio-iTLC strips can be measured at a rate of about 1–3 minutes each. When monitoring the radiolabeling of large proteins such as antibodies, size-exclusion HPLC chromatography provides more reliable information, including the detection of antibody aggregates, but it requires ~20–50 minutes per HPLC run.
We radiolabeled both DFO and DFO2 with zirconium-89 quantitatively (>99% RCY) to form [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes (no free radiometal), and then without purification transferred aliquots of these complexes to human blood serum and assessed their stability for 7 days (Figure 3). We evaluated their serum stability using two versions of the same assay: standard radio-iTLC eluted with EDTA mobile phase (50 mM, pH=5–5.5) (Figure 3A), and by precipitating the serum proteins with cold acetonitrile, centrifugation to pellet the proteins, and decanting the water/acetonitrile which contained the intact [89Zr]Zr(chelator). This was followed by rinsing the protein pellet with more ice-cold acetonitrile/water mix (~70:30) and measuring the radioactivity in each decanted fraction (Figure 3B). The results of both assays suggested superior stability of the [89Zr]Zr(DFO2) radiometal complex by demonstrating less zirconium-89 associated with serum proteins. The precipitation method resulted in the largest difference in stability, with the percent intact radiometal complexes after 7 days incubation being 81% ± 4% for [89Zr]Zr(DFO) complex and 87% ± 1% for [89Zr]Zr(DFO2) complex.
Figure 3.

Results from in vitro stability assays (n=3) comparing the stability of zirconium-89 complexes of the new chelator DFO2 to the gold standard chelator DFO, with A) competition against human blood serum over a 7-day duration and evaluated via radio-iTLC, and B) the same blood serum stability assay evaluated by precipitating proteins (cold acetonitrile) and decanting supernatant via centrifugation.
The iron transport protein transferrin is the most obvious culprit for zirconium-89 transchelation due to the similarities in binding properties between Zr4+ and Fe3+.46–48 As such, we performed an apo-transferrin (apo = metal free, holo = metal bound) competition challenge, which should be more strict than blood serum for two reasons: 1) blood serum contains transferrin that is partially bound by iron (holo) and therefore not all binding sites are available, and 2) we used super-physiological concentrations of ~250 μM. For reference, human blood serum contains ~36 μM transferrin (~80 kDa), which is partially iron-bound (holo).49 Another major blood protein that can bind to a variety of metal ions is serum albumin (~66.5 kDa), which is typically around 70 μM in the blood. For this assay we used human serum albumin (Aldrich, >96%) at a concentration of ~1.125 mM for a super-physiological quantity, but this was not the apo-form (Figure 4B). The results of these radiometal-chelator stability assays again suggest that the [89Zr]Zr(DFO2) complex possesses superior stability and inertness to transchelation by blood serum proteins than the [89Zr]Zr(DFO) complex (Figure 4). The difference in stability (percent intact radiometal-chelator complex) was very small between both chelators (~1–3%), with the exception of the 9-day timepoint for the apo-transferrin challenge (Figure 4A). Only at this long timepoint did a substantial difference in stability manifest, with the percent intact radiometal complexes after 9 days incubation being 65% ± 11% for [89Zr]Zr(DFO) complex and 80% ± 1% for [89Zr]Zr(DFO2) complex.
Figure 4.

Results from in vitro stability assays comparing [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes. Both evaluated via radio-iTLC, with A) competition against the human iron transport protein apo-transferrin (5-fold molar excess), over a 9-day duration, and B) human serum albumin (45-fold molar excess) over a 7-day duration.
The chelator desferrioxamine is an iron siderophore produced by bacteria, and since our new chelator DFO2 is based on this siderophore, it is reasonable to assume that they will both have high selectivity and stable binding for Fe3+.50, 51 Following the methods of Deri et al,32 we mixed the [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes with a 10-fold molar excess of ferric chloride (FeCl3) and monitored stability via radio-iTLC for 11 days (Figure 5A). Within error, there appeared to be no difference in the ability of these two zirconium-89 chelate complexes to resist transchelation with ferric iron. Because iron homeostasis is very tightly regulated in the body, there is essentially no “free” iron in the body and so we don’t believe the lack of stability against transchelation by “free” iron is a practical issue for these hydroxamate-based zirconium-89 chelators.
Figure 5.

Results from in vitro stability assays comparing [89Zr]Zr(DFO) complex and [89Zr]Zr(DFO2) complex, with A) competition against free iron(III) chloride over an 11-day duration, evaluated via radio-iTLC, and B) hydroxyapatite (HTP, BioRad Bio-Gel®) competition over a 24 hour duration, evaluated via centrifugation and decanting supernatant from the HTP pellet.
High uptake of zirconium-89 is routinely observed in the bones of mice, and therefore stability of these complexes in the presence of hydroxyapatite is potentially very relevant. We mixed hydroxyapatite resin (BioRad biogel HTP) in TRIS/HCl buffer (50 mM, pH=7.4) with pre-complexed [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) to determine the degree of transchelation and/or radiometal-chelate adsorption to the hydroxyapatite. After 24 hours incubation, competition mixtures were centrifuged to pellet the hydroxyapatite and the remaining solution was decanted via pipet. The hydroxyapatite pellet was re-suspended and then re-pelleted, and the solution decanted. The amount of radioactivity present in the pellet vs supernatant (plus rinse) was used to determine the percent stability, which was expressed as the percent of radiometal-chelate complex remaining in solution and not associated with the hydroxyapatite pellet. These results showed a substantial difference in stability, where the most stringent competition containing ~40 mg of hydroxyapatite revealed stability of 71% ± 1% for [89Zr]Zr(DFO) and 90% ± 1% for [89Zr]Zr(DFO2) (Figure 5B). A control of [89Zr]Zr-oxalate (most zirconium-89 is delivered in 1 M oxalic acid, then neutralized to pH=7–7.4 for radiolabeling) was also added to ~40 mg of hydroxyapatite where only 1.4% ± 0.4% remained in solution and nearly all zirconium-89 was tightly associated with the hydroxyapatite.
As a final in vitro stability experiment we again followed a method by Deri et al,32 where pre-formed radiometal-chelate complexes were mixed with a 100-fold molar excess of the chelator ethylenediaminetetraacetic acid (EDTA). Competition mixtures ranging from pH=5.0–8.0 (pH increments of 0.5) were incubated with shaking (as with all stability assays discussed) and monitored via radio-iTLC for 7 days (Table 3). This stability assay revealed a large difference in stability between [89Zr]Zr(DFO) and the [89Zr]Zr(DFO2) complex, particularly at physiologically relevant pH values of 6.5–7.5. After 7-days incubation and at pH 7.5, the [89Zr]Zr(DFO) complex remained 23.7% ± 9.6% intact where the [89Zr]Zr(DFO2) complex was 95.0% ± 0.5% intact.
Table 3.
Results from in vitro EDTA transchelation stability assays.*
| Starting complex | Ligand challenge | pH | % Intact starting species by incubation time | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Initial | 1 h | 3 h | 1 d | 3 d | 5 d | 7 d | |||
| 89Zr(DFO2) | 100 fold excess EDTA | 8.0 | 100 | 100 | 100 | 99.9 ± 0.1 | 94.3 ± 2.2 | 88.6 ± 2.8 | 81.4 ± 1.4 |
| 7.5 | 100 | 100 | 100 | 100 | 98.9 ± 1.0 | 97.6 ± 1.2 | 95.0 ± 0.5 | ||
| 7.0 | 100 | 100 | 100 | 99.8 ± 0.1 | 98.7 ± 0.3 | 92.5 ± 6.4 | 85.3 ± 2.7 | ||
| 6.5 | 100 | 100 | 99.5 ± 0.2 | 99.9 ± 0.2 | 98.0 ± 1.5 | 95.5 ± 1.9 | 90.4 ± 4.9 | ||
| 6 | 100 | 98.6 ± 0.4 | 96.6 ± 0.7 | 77.6 ± 2.9 | 42.1 ± 8.0 | 27.1 ± 8.1 | 20.6 ± 5.9 | ||
| 5.5 | 100 | 95.6 ± 1.0 | 90.2 ± 0.5 | 35.4 ± 1.1 | 7.0 ± 2.0 | 3.9 ± 1.8 | 1.5 ± 0.7 | ||
| 5.0 | 100 | 74.3 ± 0.9 | 43.9 ± 3.0 | 1.6 ± 0.1 | 1.0 ± 0.4 | 1.0 ± 0.6 | 0.6 ± 0.2 | ||
| 89Zr(DFO) | 100 fold excess EDTA | 8.0 | 98.1 ± 0.2 | 96.8 ± 1.3 | 97.5 ± 0.4 | 91.8 ± 1.3 | 75.9 ± 1.7 | 53.7 ± 1.4 | 33.6 ± 4.3 |
| 7.5 | 97.3 ± 0.6 | 96.6 ± 0.3 | 95.8 ± 0.5 | 84.9 ± 2.1 | 59.7 ± 5.5 | 36.5 ± 11.2 | 23.7 ± 9.6 | ||
| 7.0 | 96.2 ± 0.7 | 95.9 ± 0.7 | 95.5 ± 0.7 | 79.0 ± 1.6 | 50.2 ± 3.6 | 28.1 ± 8.1 | 16.1 ± 4.4 | ||
| 6.5 | 95.5 + 0.9 | 96.4 + 2.2 | 92.7 + 0.4 | 58.2 ± 3.6 | 21.6 + 1.5 | 11.6 + 0.7 | 8.5 + 1.4 | ||
| 6 | 99.3 + 0.2 | 94.9 + 0.2 | 84.6 + 2.2 | 20.4 ± 1.5 | 4.5 + 0.2 | 3.6 + 0.5 | 3.4 + 1.3 | ||
| 5.5 | 99.1 ± 0.2 | 89.6 ± 0.7 | 69.9 ± 1.5 | 5.3 ± 0.8 | 2.4 ± 0.3 | 2.7 ± 0.3 | 1.9 ± 0.1 | ||
| 5.0 | 97.0 ± 0.2 | 63.5 ± 1.3 | 27.3 ± 3.3 | 1.9 ± 0.7 | 1.1 ± 0.1 | 1.2 ± 0.1 | 1.1 + 0.0 | ||
| 100–99% | 98–90% | 89–80% | ≤ 79% | ||||||
Pre-radiolabeled [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes were mixed with a 100-fold molar excess of EDTA chelator at various pH values, and monitored by radio-iTLC for 7 days.
All together, these results suggest that our new chelator — DFO2 — offers a superior chelation environment for zirconium-89 when compared with DFO by offering a higher coordination number and improved stability and inertness to a wide variety of in vitro challenges. The next stage of this research will explore the synthesis of a bifunctional DFO2 derivative, bioconjugation with a model antibody, radiolabeling, and in vivo stability assessment in mice. Long-term, we hope to link in vivo results back to this study and determine which of these in vitro stability assays were ultimately the most predictive of the chelators in vivo behavior.
Based on the simple synthetic pathway we have developed for DFO2 and its excellent in vitro stability, we believe there is a lot of potential in these dual-DFO based chelators for expansion to other radiometals and theranostic applications. In addition to zirconium-89, there are even larger metal ions of interest to the radiochemistry community that we plan to test in the future, which these high-denticity chelators might be uniquely suitable for. We are working on next-generation DFO2 derivatives with further simplified synthesis, improved solubility, and different bifunctional linker groups with these goals in mind.
CONCLUSIONS
The new chelator DFO2 presents a promising new modular and high-denticity chelator scaffold, which we will build upon using its modular synthesis scheme to produce highly stable and customizable bifunctional chelators for zirconium-89 and other interesting radiometals. With double the potential coordination number of the “gold standard” chelator for zirconium-89 — DFO (CN = 12 vs 6) — DFO2 appears to saturate the octadentate coordination sphere of zirconium and prevents the coordination of water. This conclusion is supported by high-resolution mass spectrometry of the non-radioactive Zr(DFO2) complex, density functional theory calculations, and radiolabeling results (specific activity). In contrast, DFO is thought to coordinate with zirconium in a hexadentate fashion leaving two open coordination sites where two molecules of water are thought to coordinate (total CN = 8). Following quantitative radiolabeling with zirconium-89, the [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes were compared using a large number of in vitro stability challenges including human blood serum, apo-transferrin, serum albumin, iron, hydroxyapatite, and EDTA. The results of these numerous stability assays demonstrated that our new chelator DFO2 formed a more stable zirconim-89 complex than DFO, except for the iron challenge where both chelators were identical. Substantial differences in stability were not observed for human blood serum using radio-iTLC, but when using a protein precipitation method of analysis the stability values after 7 days were 81% ± 4 % for [89Zr]Zr(DFO) complex and 87% ± 1% for [89Zr]Zr(DFO2) complex. The stability challenge against the iron transport protein apo-transferrin revealed values of 65% ± 11% for [89Zr]Zr(DFO) and 80% ± 1% for [89Zr]Zr(DFO2) after 9 days incubation. To simulate transchelation/adsorption with bone, we developed a hydroxyapatite resin challenge which revealed stability values of 71% ± 1% for [89Zr]Zr(DFO) and 90% ± 1% for [89Zr]Zr(DFO2) after 24 hours incubation. Finally, competition against a 100-fold molar excess of EDTA showed a stark difference in stability, with values of 23.7% ± 9.6% for [89Zr]Zr(DFO) and 95.1% ± 0.5% for [89Zr]Zr(DFO2) after 7 days incubation (pH = 7.5). Taken together, these suggest that our new DFO2 chelator is a promising next-generation scaffold for the elaboration of zirconium-89 chelators. We plan to use the modular synthesis of the DFO2 chelator to tune its polarity, conjugation chemistry, and coordination chemistry.
EXPERIMENTAL SECTION
Reagents.
All solvents and reagents were purchased from commercial suppliers (Sigma Aldrich, St. Louis, MO; TCI America, Portland, OR; Fisher Scientific, Waltham, MA) and were used as received unless otherwise indicated. DMSO used for chelator stock solutions was of molecular biology grade (>99.9%: Sigma Aldrich). DFO.mesylate was purchased from Abcam. 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) was purchased from AK Scientific. N,N-diisopropylethylamine (DIEA), diethyl malonate, 4-nitrobenzyl bromide, and borane-tetrahydrofuran solution (BH3.THF) were purchased from Sigma-Aldrich. ZrCl4, potassium carbonate anhydrous, and ammonium hydroxide were purchased from Fisher Scientific. All the chemicals were used directly without purification.
Radiochemistry Instrumentation.
89Zr was produced at Memorial Sloan Kettering Cancer Center using an EBCO TR19/9 variable-beam energy cyclotron (Ebco Industries Inc., British Columbia, Canada) via the 89Y(p,n)89Zr reaction. 89Zr was purified in accordance with previously reported methods to create 89Zr with a specific activity of 5.3 – 13.4 mCi/μg (195 – 497 MBq/μg).2 Radiolabeling reactions were monitored using silica-gel impregnated glass-microfiber instant thin layer chromatography paper (iTLC-SG, iTLC-SA, Varian, Lake Forest, CA) and analyzed on a Bioscan AR-2000 radio-TLC plate reader using Winscan Radio-TLC software (Bioscan Inc., Washington, DC). All radiolabeling chemistry was performed with ultrapure water (>18.2 MΩ cm−1 at 25°C, Milli-Q, Millipore, Billerica, MA), and treatment of water or buffers with Chelex-100 resin was performed as indicated (1.2 g/L, 24 h) (BioRad Laboratories, Hercules, CA). Radioactivity in samples was measured using a Capintec CRC-15R dose calibrator (Capintec, Ramsey, NJ). For [89Zr]Zr4+ logD experiments, a Perkin-Elmer (Waltham, MA) Automated Wizard Gamma Counter was used for counting radioactivity.
Chemical Characterization Methods.
1H and 13C NMR spectra were recorded at the University of Saskatchewan on a 500 MHz Bruker Avance, and a 500 MHz Bruker Avance III HD NMR spectrometer at 25 °C in (CD3)2SO and CD3OD. 1H chemical shifts were referenced to the residual protons of the deuterated solvents (δ = 2.50 ppm for (CD3)2SO; δ = 3.31 ppm for CD3OD); 13C chemical shifts were referenced to the (CD3)2SO signal at δ = 39.52 ppm and the CD3OD signal at δ = 49.00 ppm. Coupling constants are reported to the nearest 0.5 Hz (1H NMR spectroscopy) or rounded to integer values in Hz (13C NMR spectroscopy). Assignments were supported by additional NMR experiments, DEPT, HMQC, and COSY, for compounds 3, 4, 5, and 6. High resolution mass spectra were measured on a JEOL AccuTOF GCv 4G using field desorption ionization (FDI). For the isotopic pattern only the mass peak of the isotope with the highest natural abundance is given.
Density Functional Theory (DFT) Calculations.
DFT geometry optimizations were carried out using DMol3 and Biovia Materials Studio, version 2017 R2,52, 53 using the generalized gradient approximation (GGA) employing the Perdew-Burke-Ernzerhof (PBE)54 functional both for the potential during the self-consistent field (SCF) procedure and for the energy.55 DMol3 double numerical plus d-function (DND) basis set using 4.4 basis file included polarization functions for all atoms with all-electron core treatments. Quantum simulation of solvated molecules were modeled using the conductor-like screening model (COSMO) solvation model56, 57 in DMol3, with a dielectric value representing water (ε = 78.54).
Synthesis of diethyl-2-[(4-nitrophenyl)methyl]–propanedioate (1):
This synthesis was adapted from Pandya et al.58 Diethyl malonate (8.6 mL, 56.378 mmol) and potassium carbonate anhydrous (2.808 g, 20.318 mmol) were dissolved in acetone (8.0 mL). The mixture was stirred for 1 hour at room temperature. Then 4-nitrobenzyl bromide (2.213 g, 10.244 mmol) was added and stirred for 2 hours at 45°C. After cooling to room temperature, the yellow reaction mixture was filtered to remove the remaining potassium carbonate and was washed with acetone (20 mL). The filtrate was collected and the solvent was reduced by rotary evaporation in vacuo, ethanol was added (~10 mL), and then kept at −20 °C in the freezer overnight. Crystals formed and were separated by filtration and the filtrate was placed back in the freezer at −20 °C overnight to produce more crystals. This process was completed a total of three times. White crystals of (1) (1.393 g, 46.13%) was obtained from three rounds of precipitation and filtration. 1H NMR (CDCl3, 500 MHz) δ =1.22 [t, 6H, J = 7.1 Hz, CH2CH3], 3.32 [d, 2H, J = 7.8 Hz, =CCH2CH], 3.66 [t, 1H, J = 7.8 Hz, =CCH2CH], 4.14 – 4.21 [m, 4H, OCH2CH3], 7.39 [d, 2H, J = 8.7 Hz, CH=CCH2 Ar], 8.15 [d, 2H, J = 8.7 Hz, CH=CNO2 Ar]
Synthesis of 2-(4-nitrobenzyl) malonamide (2):
This synthesis was adapted from Price et al.43 Compound 1 (295 mg, 0.999 mmol) was dissolved in methanol (5 mL), followed by addition of ammonium hydroxide (5 mL). The reaction mixture was stirred for 45 hours at room temperature. A yellow precipitate was formed and isolated by filtration, and then washed with methanol and boiling acetonitrile. Solvent was removed by rotary evaporation in vacuo. Yellow crystals of compound (2) (180.78 mg, 61.7%) was obtained. 1H NMR [(CD3)2SO, 500 MHz]: δ = 3.10 [d, 2H, J = 7.6 Hz, =CCH2CH], 3.38 [t, 1H, J = 7.7 Hz, =CCH2CH], 7.07 [s, 2H, CONH2], 7.27 [s, 2H, CONH2], 7.48 [d, 2H, J = 8.8 Hz, CH=CCH2 Ar], 8.14 [d, 2H, J = 8.8 Hz, CH=CNO2 Ar]
Synthesis of (2-(4-nitrobenzyl)-1,3-propylenediamine (3):
This synthesis was adapted from Price et al.43 A mixture of compound 2 (1.005 g, 4.237 mmol) in dry THF (15 mL) was cooled in an ice bath, followed by dropwise addition of a diborane (BH3.THF) solution (8.14 mL, 1.0 M in THF) under nitrogen gas. After stirring for half an hour, the solution was left to warm up to room temperature, then it was heated to reflux under nitrogen gas overnight. Before HCl (12 M; 15 mL) was added, the reaction was cooled to room temperature. The reaction was then warmed to reflux with no inert gas for one hour. Then the solvent was evaporated by rotary evaporator and NaOH (6 M; 30 mL) was added to the residue, which was then extracted with dichloromethane (5 × 15 mL). The organic fractions were collected and dried with anhydrous Na2SO4. The solvent was then removed by rotary evaporation in vacuo, and ethanol (20 mL) was added followed by HCl (12 M; 4 mL). The solution was placed in the freezer at −20 °C overnight. A white solid formed, was filtered, and dried. Compound 3 (250 mg, 28.20%) was obtained. 1H NMR (D2O, 500 MHz): δ = 2.57 [sept, 1H, J = 6.9 Hz, CH2CHCH2], 2.99 [d, 2H, J = 7.6 Hz, CCH2CH], 3.07 [dd, 2H, J = 6.7 Hz, J = 13.7 Hz, CH2NH2], 3.19 [dd, 2H, J = 6.7 Hz, J = 13.7 Hz, CH2NH2], 7.55 [d, 2H, J = 8.8, NO2CCH], 8.28 [d, 2H, J =8.8, CCH=CH]. 13C NMR (D2O, 500 MHz): δ 34.7, 36.5, 39.9, 124.0, 130.1, 145.2, 146.7. HRMS (ESI; m/z): [M+H]+ calcd for C10H15N3O2: 210.1242; found: 210.1241.
Synthesis of DFOCOOH (4).
Compound 4 (DFOCOOH) was prepared according to a published procedure.59 To a solution of succinic anhydride (0.857 g, 8.56 mmol) in pyridine (4.0 mL), desferrioxamine (DFO) mesylate salt (0.250 g, 0.380 mmol) was added. It formed a white suspension. The reaction mixture was stirred at room temperature overnight. To this mixture was added an aqueous solution of sodium hydroxide (0.16 M; 45 mL) which was stirred for 5 hours and formed a clear solution. Hydrochloric acid (12 M; 6.0 mL) was added dropwise to this solution until the pH of the solution reached 2.0 (pH strip). The solution turned cloudy upon addition of hydrochloric acid. The mixture was left at 5 °C in the fridge for further precipitation. After vacuum filtration the precipitate was washed several times with a 0.01 M solution of hydrochloric acid, followed by lyophilization. Compound 4 was obtained as a white solid (0.232 g, 93%). 1H NMR [(CD3)2SO, 500 MHz]: δ = 1.20 (m, 7H, CH), 1.37 (m, 7H, CH), 1.49 (m, 6H, CH), 2.27 (m, 6H, CH), 2.40 (t, 2H, CH), 2.57 (t, 4H, CH), 2.99 (q, 6H, CH), 3.45 (t, 6H, CH), 7.80 (m, 3H, NH), 9.62 (s, 2H, N-OH), 9.66 (s, 1H, N-OH), 12.07 (m, 1H, COOH) ppm; 13C[1H] NMR [(CD3)2SO, 126 MHz]: δ = 20.4, 23.5, 26.1, 27.6, 28.8, 29.2, 29.9, 30.0, 38.4, 46.8, 47.1 (CH), 170.1, 170.7, 171.3, 172.0, 173.9 (CO) ppm.
Synthesis of p-NO2-Bn-DFO2 (5).
Compound 4 (0.348 g, 0.527 mmol) is only sparingly soluble in DMF (10.5 mL) at room temperature and was dissolved at 80 °C. N,N-diisopropylethylamine (DIEA; 0.072 g, 0.557 mmol) was added to the solution of 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU; 0.211 g, 0.555 mmol) in DMF (11.0 mL) in a separate vial. This solution then was added to the solution of compound 4 in DMF followed by addition of a solution of compound 3 (0.052 g, 0.248 mmol) in DMF (5.3 mL). The reaction mixture was stirred at room temperature for 24 hours. Volatiles were evaporated to reduce the volume to ~1/6. The crude product was then precipitated by slow addition of ice-cold ultra-pure water (70.0 mL) into this residue. The product was separated by centrifugation, followed by two rounds of re-suspension in ice-cold ultra-pure water and centrifugation. Finally, product was dissolved in a minimum volume of DMF, followed by addition of ice-cold ethyl acetate to precipitate product, then centrifugation and decanting. The final powder was mixed with ultrapure water and lyophilized. The p-NO2-Bn-DFO2 (5) product was obtained as a bone white powder (0.2216 g, 60%). 1H NMR [(CD3)2SO, 500 MHz]: δ = 1.21 (m, 17H, CH), 1.38 (m, 17H, CH), 1.49 (m, 15H, CH), 2.26 (m, 11H, CH), 2.57 (m, 8H, CH), 2.99 (m, 17H, CH), 3.45 (m, 12H, CH), 7.51 (d, 2H, J = 8.5 Hz, C6H4), 7.76 (m, 6H, NH), 7.82 (m, 2H, NH), 8.14 (d, 2H, J = 8.1 Hz, C6H4), 9.59 (s, 4H, N-OH), 9.63 (s, 2H, N-OH) ppm; 13C[1H] NMR [(CD3)2SO, 126 MHz]: δ = 20.3, 23.5, 26.0, 27.5, 28.0, 28.7, 28.8, 29.8, 29.9, 30.8, 30.9, 38.4, 46.8, 47.1 (CH), 123.2, 130.3, 145.9, 149.0 (C6H4), 170.0, 170.1, 171.1, 171.3, 171.7, 171.9, 177.7 (CO) ppm; HRMS (TOF): m/z calcd for C68H115N15O22+H+: 1494.8413 [M+H]+; found: 1494.8565; calcd for [C68H115N15O22+2H]2+/2: 747.9243 [M+2H]2+/2; found: 747.9241.
Synthesis of Zr(DFO2) complex (6).
Compound 5 (50.00 mg, 33.45 μmol) was dissolved in a solvent mixture of DMF:H2O (1:5; 1.3 mL) at 80 °C. A solution of ZrCl4 (7.64 mg, 32.78 μmol) in H2O (0.7 mL) was slowly added to the resulting mixture. The reaction was stirred at room temperature for 24 hours. After removing all volatiles under high vacuum, the product was obtained as a thin yellow glassy film. 1H NMR [(CD3)2SO, 500 MHz]: δ = 1.23, 1.27, 1.39, 1.49, 1.63, 1.96, 2.12, 2.26, 2.31, 2.58, 2.61, 2.99, 3.09, 3.45, 3.61 (m, CH), 7.53 (m, C6H4), 7.80, 7.88, 7.95 (m, NH), 8.14 (d, 2H, J = 8.3 Hz, C6H4), 9.73 (br, N-OH) ppm; 13C[1H] NMR [(CD3)2SO, 126 MHz]: δ = 16.8, 20.4, 22.3, 22.5, 23.2, 23.5, 24.9, 25.7, 26.0, 26.6, 26.9, 27.7, 28.0, 28.8, 28.9, 29.2, 29.3, 30.0, 34.1, 38.4, 46.8, 47.1, 49.7, 50.0, 50.3, 50.6, 63.8 (CH), 119.1, 123.2, 123.4, 127.5, 130.4, 145.9, 146.1, 148.0, 149.1 (C6H4), 160.9, 161.0, 162.7, 162.8, 170.1, 170.4, 170.7, 170.8, 171.0, 171.17, 171.23, 171.3, 171.4, 171.8, 171.9, 173.6, 173.8, 173.9, 177.8 (CO) ppm; HRMS (TOF): m/z calcd for C68H111N15O22Zr+H+: 1580.7153 [M+H]+; found: 1580.7109; calcd for [C68H111N15O22Zr+2H]2+/2: 790.8610 [M+2H]2+/2; found: 790.8643.
[89Zr]Zr4+ Radiolabeling Studies.
Zirconium-89 was received after target dissolution and purification as the [89Zr]Zr(oxalate) complex in 1.0 M oxalic acid. Phosphate buffered saline (PBS, pH=7.4, pre-treated with Chelex resin) was used for radiolabeling buffer, sodium carbonate (1.0 M) was used to neutralize aliquots of zirconium-89 in oxalic acid to pH=6.8–7.2 (checked by pH strip), and an EDTA solution (50 mM, pH=5–5.5) was used as mobile phase for eluting radio-iTLC (Varian iTLC-SG) to check RCY and purity. For radio-iTLC analysis, intact [89Zr]Zr(chelate) (chelate=DFO or DFO2) remained at the baseline, while the [89Zr]Zr4+ that remained unbound or was transchelated to serum proteins eluted with the solvent front. Radio-iTLC were analyzed using a BioScan AR2000 and the integration of activity at the baseline and solvent front were used to calculate yields and degree of transchelation during stability assays. The commercially available chelator DFO mesylate (Macrocyclics) was used as a control alongside our new chelator DFO2. Stock solutions of both chelators were prepared in DMSO at a concentration of 17.0 mg/mL. Aliquots of these stock solution were transferred to radiolabeling buffer (Chelex treated PBS pH=7.4) to prepare separate solutions of DFO and DFO2 chelators at final volumes of 2.5 mL (100 μM). For each chelate solution, aliquots of 4–5 mCi (148–185 MBq) of [89Zr]Zr(oxalate) was neutralized with sodium carbonate as described above, and diluted to a total volume of 2.5 mL in PBS. This solution was combined with the chelate solution to produce the final radiolabeling solution containing 5 mL totol volume with 50 μM chelate and 4–5 mCi of zirconium-89. The radiolabeling reaction mixture was allowed to mix on an Eppendorf thermomixer (800 rpm, 60 minutes, 37 °C). Radiochemical yields via radio-iTLC for both DFO and DFO2 chelators were >99% after 60 minutes reaction time. These pre-formed [89Zr]Zr(chelate) solutions were used without purification for stability assays. Radiolabeled complexes of DFO and DFO2 chelators with zirconium-89 were also prepared at 100 μM final concentration in the same manner as described above, and used for some of the stability assays described below.
Serum Stability Assay.
[89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes were prepared as described above, and 400 μL aliquots (100 μM) were transferred to microcentrifuge tubes (Eppendorf Low-Bind, 1.5 mL) containing 600 μL of previously frozen human blood serum (Sigma Aldrich). Samples were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. Assay progress was monitored by radio-iTLC at times of 1 day, 5 days, and 7 days. As with other assays performed for this study, zirconium-89 transchelated by blood serum components elutes to the solvent front in an EDTA mobile phase (50 mM, pH=5.0–5.5), where [89Zr]Zr(chelate) remains at the baseline.
An alternative analysis method was performed, where aliquots (300 μL) of the [89Zr]Zr(chelate)/human blood serum competition mixture was removed, mixed with ice-cold acetonitrile (700 μL) to precipitate proteins, and then centrifuged at 10,000 rpm for 10 minutes (4 °C). The supernatant was decanted via pipet and measured by dose calibrator, and the process was repeated with ice-cold water/acetonitrile mixture (30:70). The amount of zirconium-89 remaining stuck to the serum proteins in the original microcentrifuge tube was assumed to be transchelated, and the amount of radioactivity in the supernatant was assumed to be chelate-bound. This ratio was used to calculate the % stability of the [89Zr]Zr(chelate) complexes. As a further control, 300 μL of only [89Zr]Zr(chelate) complex with no blood serum was put through the same process (ice-cold acetonitrile, centrifugation) and it was found that for both [89Zr]Zr(chelate) complexes ~10% of the activity in solution remained stuck in the microcentrifuge tubes after centrifugation and decanting twice. The stability values reported were not corrected for this sticking factor.
Apo-Transferrin Assay.
[89Zr]Zr(chelate) complexes were prepared as described above. For this assay, a 5-fold molar excess of apo-transferrin (Sigma Aldrich) was used relative to [89Zr]Zr(chelate) complex, from a stock solution of apo-transferrin in PBS (Chelex treated, pH=7.4, 500 μM). The final competition solutions (n=3) for each of [89Zr]Zr(DFO) complex and [89Zr]Zr(DFO2) complex contained 200 μL total volume, 50 μM of [89Zr]Zr(chelate) complex (PBS, 100 μL of 100 μM), and 250 μM of apo-transferrin (PBS, 100 μL at 500 μM). Samples were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. Assay progress was monitored by radio-iTLC at times of 0, 1 day, 3 days, 5 days, 7 days, and 9 days.
Serum Albumin Assay.
[89Zr]Zr(chelate) complexes were prepared as described above. Human serum albumin (Sigma Aldrich, >96%) was dissolved in PBS (chelex treated, pH=7.4) at a concentration of 100 mg/mL (~1.5 mM), and 150 μL aliquots were transferred to microcentrifuge tubes (n=3). [89Zr]Zr(chelate) complex was then added (50 μL, 100 μM, ~50 μCi) to each tube for a final concentration of 25 μM [89Zr]Zr(chelate) complex and ~1.125 mM albumin (~45-fold molar excess). Samples were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. Assay progress was monitored by radio-iTLC at times of 0, 1 day, 3 days, 5 days, and 7 days.
EDTA Transchelation Assay.
These transchelation challenges were setup following the methods of Deri et al.,32 with a molar ratio of 100-fold molar excess of EDTA to pre-formed [89Zr]Zr(DFO) complex or [89Zr]Zr(DFO2) complex. The solutions prepared for this assay were: 1) 2.5 mL of [89Zr]Zr(DFO) complex and [89Zr]Zr(DFO2) complex (50 μM each) as described above, 2) EDTA stock solution (5 mM) split and then each sub-stock solution adjusted by pH meter to pH 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0, and 3) ammonium acetate buffer (500 mM) split and then each sub-stock solution adjusted by pH meter to pH 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0. These three stock solutions were combined in microcentrifuge tubes (1.5 mL tubes, n=3) for each chelator and pH range in ratios of [89Zr]Zr(chelate) complex (200 μL, 50 μM), EDTA (200 μL, 5 mM), and ammonium acetate (100 μL, 500 mM), with the EDTA and ammonium acetate solutions matching pH (e.g. pH=7.0 each for the pH=7.0 challenge, n=3). The final EDTA transchelation assays solutions contained a total volume of 500 μL with [89Zr]Zr(chelate) complex (20 μM, ~100 μCi, ~3.7 MBq) and EDTA solution (2 mM) for a 100-fold molar excess of EDTA challenge. Transchelation was monitored by radio-iTLC as described above (EDTA mobile phase, 50 mM, pH=5.0–5.5), where zirconium-89 associated with DFO or DFO2 chelators remained at the baseline and zirconium-89 transchelated to EDTA eluted with the solvent front. Samples were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. Assay progress was monitored at times 0, 1 hour, 3 hours, 1 day, 3 days, 5 days, and 7 days.
Iron(III) Competition Study.
Radiolabeled [89Zr]Zr(DFO) complex and [89Zr]Zr(DFO2) complex were prepared as described above. The metal salt iron(III) chloride was prepared as a stock solution (1.0 mM, chelex-treated millipure water). Aliquots of [89Zr]Zr(chelate) complex (200 μL, 100 μM) were combined with iron(III) solution (200 μL, 1.0 mM) to yield final completion mixtures (n=3) containing 50 μM of [89Zr]Zr(chelate) complex and 500 μM of iron(III) solution (10-fold molar excess). Samples of [89Zr]Zr(chelate) complexes in competing iron solutions were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. Assay progress was monitored by radio-iTLC as described above at times 0, 1 day, 3 days, 5 days, 7 days, and 9 days.
Hydroxyapatite (HTP) Stability Challenge.
Hydroxyapatite resin (HTP, BioRad Bio-Gel®) was weighed into microcentrifuge tubes (20 mg and 40 mg sets, n=3 per weight and per chelator/control). To each tube containing HTP, TRIS/HCl buffer was added (50 mM, 900 μL, pH=7.4). [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes were radiolabeled as described above, with the exception that TRIS/HCl buffer (50 mM, pH=7.4) was used. and aliquots were transferred to each tube (100 μM, 100 μL, ~100 μCi, ~3.7 MBq), as well as a set of unchelated but neutralized [89Zr]Zr(oxalate) complex as controls (~100 μCi, ~3.7 MBq, n=3). The final concentration of [89Zr]Zr(chelate) complex in each competition was 10 μM. HTP competition mixtures were placed on Eppendorf Thermomixers at 37 °C and 800 rpm agitation. After 24 hours, these samples were centrifuged (10,000 rpm, 10 minutes) to pellet the HTP, the supernatant decanted via pipet, and the pellet was washed with TRIS/HCl buffer (1.0 mL) twice, followed by centrifugation and decanting. The amount of zirconium-89 adsorbed/bound by the HTP and the amount remaining chelate-bound in the supernatant and washes was measured via dose calibrator. The percent stability was determined from a ratio of the percent radioactivity in the supernatant/washes and the HTP pellet.
LogD Octanol/Buffer Partition Coefficient.
Radiolabeled [89Zr]Zr(DFO) and [89Zr]Zr(DFO2) complexes were prepared as described above (100 μM). After radiolabeling, logD values were determined by transferring an aliquot of each radiolabeled chelator (~ 5 μCi, ~5–10 μL) to 3 mL of PBS (pH=7.4) in 15 mL falcon centrifuge tubes (n = 5). An equal volume of 1-octanol (3 mL) was then added, tubes were capped, and then vortex mixed for 60 seconds each. The tubes were then centrifuged for 10 minutes (3000 rpm) to aid in phase separation. Samples were removed from the centrifuge, allowed to sit for 10 minutes, and 1.0 mL aliquots of each phase were removed carefully via pipet and transferred to small microcentrifuge tubes (1.5 mL) and radioactivity measured on a gamma counter. The ratio of radioactivity (counts) in each phase was used to calculate the logD values (equation 1). The amount of radioactivity in the octanol phase was used in place of solute concentrations (ionized and un-ionized), and the amount in the PBS (pH=7.4) phase was used in place of solute concentration (ionized and un-ionized).
| (1) |
Supplementary Material
ACKNOWLEDGEMENTS
EWP was supported by the Natural Sciences and Engineering Research Council (NSERC) (Discovery Grant, RGPIN-2017-03952 2017-2022), the Canada Research Chairs program (CRC in Radiochemistry, 2016-2021, #231072), the Canadian Institutes of Health Research (CIHR Project Grant #388243), the Canadian Foundation for Innovation John Evans Leadership Fund (2016 #35162), the Sylvia Fedoruk Canadian Centre for Nuclear Innovation, the Saskatchewan Health Research Foundation (SHRF Establishment grant), and startup funds from the University of Saskatchewan Department of Chemistry and College of Arts and Sciences. EKS was supported by a Saskatchewan Health Research Foundation (SHRF) postdoctoral fellowship (2017-2019). JSL was supported by R01 CA204167 and R35CA232130 as well as the Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics at Memorial Sloan Kettering Cancer Center.
ABBREVIATIONS
- PET
positron emission tomography
- 89Zr
zirconium-89
- RCY
radiochemical yield
Footnotes
The authors declare no competing financial interests.
Disclosures: None.
Supporting Information
The Supporting Information is available online and includes NMR spectra and DFT coordinates.
REFERENCES
- 1.Holland JP; Williamson MJ; Lewis JS Unconventional nuclides for radiopharmaceuticals. Mol. Imaging 2010, 9 (1), 1–20. [PMC free article] [PubMed] [Google Scholar]
- 2.Holland JP; Sheh Y; Lewis JS Standardized methods for the production of high specific-activity zirconium-89. Nucl. Med. Biol 2009, 36 (7), 729–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhatt NB; Pandya DN; Wadas TJ Recent Advances in Zirconium-89 Chelator Development. Molecules 2018, 23 (3), 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escorcia FE; Steckler JM; Abdel-Atti D; Price EW; Carlin SD; Scholz WW; Lewis JS; Houghton JL Tumor-Specific Zr-89 Immuno-PET Imaging in a Human Bladder Cancer Model. Mol. Imaging Biol 2018, (20), 808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price EW; Carnazza KE; Carlin SD; Cho A; Edwards KJ; Sevak KK; Glaser JM; de Stanchina E; Janjigian YY; Lewis JS 89Zr-DFO-AMG102 Immuno-PET to Determine Local HGF Protein Levels in Tumors for Enhanced Patient Selection. J. Nucl. Med 2017, 58 (9), 1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulaner GA; Hyman DM; Lyashchenko SK; Lewis JS; Carrasquillo JA 89Zr-Trastuzumab PET/CT for Detection of Human Epidermal Growth Factor Receptor 2-Positive Metastases in Patients With Human Epidermal Growth Factor Receptor 2-Negative Primary Breast Cancer. Clin. Nucl. Med 2017, 42 (12), 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price EW; Orvig C In The Chemistry of Molecular Imaging; John Wiley & Sons, Inc: 2014; pp 105–135. [Google Scholar]
- 8.Zeglis BM; Mohindra P; Weissmann GI; Divilov V; Hilderbrand SA; Weissleder R; Lewis JS Modular Strategy for the Construction of Radiometalated Antibodies for Positron Emission Tomography Based on Inverse Electron Demand Diels-Alder Click Chemistry. Bioconjugate Chem. 2011, 22 (10), 2048–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holland JP; Divilov V; Bander NH; Smith-Jones PM; Larson SM; Lewis JS 89Zr-DFO-J591 for ImmunoPET of Prostate-Specific Membrane Antigen Expression In Vivo. J. Nucl. Med 2010, 51 (8), 1293–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dijkers EC; Oude Munnink TH; Kosterink JG; Brouwers AH; Jager PL; de Jong JR; van Dongen GA; Schroder CP; Lub-de Hooge MN; de Vries EG Biodistribution of 89Zr-trastuzumab and PET Imaging of HER2-Positive Lesions in Patients With Metastatic Breast Cancer. Clin. Pharmacol. Ther 2010, 87 (5), 586–592. [DOI] [PubMed] [Google Scholar]
- 11.Dijkers ECF; Kosterink JGW; Rademaker AP; Perk LR; van Dongen GAMS; Bart J; de Jong JR; de Vries EGE; Lub-de Hooge MN Development and Characterization of Clinical-Grade 89Zr-Trastuzumab for HER2/neu ImmunoPET Imaging. J. Nucl. Med 2009, 50 (6), 974–981. [DOI] [PubMed] [Google Scholar]
- 12.Perk L; Visser O; Stigter-van Walsum M; Vosjan M; Visser G; Zijlstra J; Huijgens P; van Dongen G Preparation and evaluation of 89Zr-Zevalin for monitoring of 90Y-Zevalin biodistribution with positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2006, 33 (11), 1337–1345. [DOI] [PubMed] [Google Scholar]
- 13.Börjesson PKE; Jauw YWS; Boellaard R; de Bree R; Comans EFI; Roos JC; Castelijns JA; Vosjan MJWD; Kummer JA; Leemans CR; Lammertsma AA; van Dongen GAMS Performance of Immuno–Positron Emission Tomography with Zirconium-89-Labeled Chimeric Monoclonal Antibody U36 in the Detection of Lymph Node Metastases in Head and Neck Cancer Patients. Clin. Cancer Res 2006, 12 (7), 2133–2140. [DOI] [PubMed] [Google Scholar]
- 14.Perk LR; Visser OJ; Stigter-van Walsum M; Vosjan MJWD; Visser GWM; Zijlstra JM; Huijgens PC; Dongen GAMS Preparation and evaluation of 89Zr-Zevalin for monitoring of 90Y-Zevalin biodistribution with positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2006, 33 (11), 1337–1345. [DOI] [PubMed] [Google Scholar]
- 15.Perk LR; Visser GWM; Vosjan MJWD; Stigter-van Walsum M; Tijink BM; Leemans CR; van Dongen GAMS 89Zr as a PET Surrogate Radioisotope for Scouting Biodistribution of the Therapeutic Radiometals 90Y and 177Lu in Tumor-Bearing Nude Mice After Coupling to the Internalizing Antibody Cetuximab. J. Nucl. Med 2005, 46 (11), 1898–1906. [PubMed] [Google Scholar]
- 16.Verel I; Visser GWM; Boellaard R; Stigter-van Walsum M; Snow GB; van Dongen GAMS 89Zr Immuno-PET: Comprehensive Procedures for the Production of 89Zr-Labeled Monoclonal Antibodies. J. Nucl. Med 2003, 44 (8), 1271–1281. [PubMed] [Google Scholar]
- 17.Poreddy AR; Schall OF; Osiek TA; Wheatley JR; Beusen DD; Marshall GR; Slomczynska U Hydroxamate-Based Iron Chelators: Combinatorial Syntheses of Desferrioxamine B Analogues and Evaluation of Binding Affinities. J. Comb. Chem 2004, 6 (2), 239–254. [DOI] [PubMed] [Google Scholar]
- 18.Price EW; Orvig C Matching Chelators to Radiometals for Radiopharmaceuticals. Chem. Soc. Rev 2014, 43 (1), 260–290. [DOI] [PubMed] [Google Scholar]
- 19.Meijs WE; Haisma HJ; Klok RP; van Gog FB; Kievit E; Pinedo HM; Herscheid JDM Zirconium-Labeled Monoclonal Antibodies and Their Distribution in Tumor-Bearing Nude Mice. J. Nucl. Med 1997, 38 (1), 112–118. [PubMed] [Google Scholar]
- 20.Meijs WE; Haisma HJ; Van Der Schors R; Wijbrandts R; Van Den Oever K; Klok RP; Pinedo HM; Herscheid JDM A facile method for the labeling of proteins with zirconium isotopes. Nucl. Med. Biol 1996, 23 (4), 439–448. [DOI] [PubMed] [Google Scholar]
- 21.Meijs WE; Herscheid JDM; Haisma HJ; Pinedo HM Evaluation of desferal as a bifunctional chelating agent for labeling antibodies with Zr-89. Int. J. Rad. Appl. Instrum. [A] 1992, 43 (12), 1443–1447. [DOI] [PubMed] [Google Scholar]
- 22.Perk L; Vosjan M; Visser G; Budde M; Jurek P; Kiefer G; van Dongen G p-Isothiocyanatobenzyl-desferrioxamine: a new bifunctional chelate for facile radiolabeling of monoclonal antibodies with zirconium-89 for immuno-PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2010, 37 (2), 250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vosjan MJWD; Perk LR; Visser GWM; Budde M; Jurek P; Kiefer GE; van Dongen GAMS Conjugation and radiolabeling of monoclonal antibodies with zirconium-89 for PET imaging using the bifunctional chelate p-isothiocyanatobenzyl-desferrioxamine. Nat. Protocols 2010, 5 (4), 739–743. [DOI] [PubMed] [Google Scholar]
- 24.Price EW; Zeglis BM; Lewis JS; Adam MJ; Orvig C H6phospa-Trastuzumab: Bifunctional Methylenephosphonate-based Chelator with 89Zr, 111In and 177Lu. Dalton Trans. 2014, 43, 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jauw YWS; Menke-van der Houven van Oordt CW; Hoekstra OS; Hendrikse NH; Vugts DJ; Zijlstra JM; Huisman MC; van Dongen GAMS Immuno-Positron Emission Tomography with Zirconium-89-Labeled Monoclonal Antibodies in Oncology: What Can We Learn from Initial Clinical Trials? Frontiers in pharmacology 2016, 7, 131–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandit-Taskar N; O’Donoghue JA; Ruan S; Lyashchenko SK; Carrasquillo JA; Heller G; Martinez DF; Cheal SM; Lewis JS; Fleisher M; Keppler JS; Reiter RE; Wu AM; Weber WA; Scher HI; Larson SM; Morris MJ First-in-Human Imaging with 89Zr-Df-IAB2M Anti-PSMA Minibody in Patients with Metastatic Prostate Cancer: Pharmacokinetics, Biodistribution, Dosimetry, and Lesion Uptake. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 2016, 57 (12), 1858–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandit-Taskar N; O’Donoghue JA; Durack JC; Lyashchenko SK; Cheal SM; Beylergil V; Lefkowitz RA; Carrasquillo JA; Martinez DF; Fung AM; Solomon SB; Gönen M; Heller G; Loda M; Nanus DM; Tagawa ST; Feldman JL; Osborne JR; Lewis JS; Reuter VE; Weber WA; Bander NH; Scher HI; Larson SM; Morris MJ A Phase I/II Study for Analytic Validation of 89Zr-J591 ImmunoPET as a Molecular Imaging Agent for Metastatic Prostate Cancer. Clin. Cancer Res 2015, 21 (23), 5277–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousseau J; Zhang Z; Wang X; Zhang C; Lau J; Rousseau E; Čolović M; Hundal-Jabal N; Bénard F; Lin K-S Synthesis and evaluation of bifunctional tetrahydroxamate chelators for labeling antibodies with 89Zr for imaging with positron emission tomography. Bioorg. Med. Chem. Lett 2018, 28 (5), 899–905. [DOI] [PubMed] [Google Scholar]
- 29.Briand M; Aulsebrook ML; Mindt TL; Gasser G A solid phase-assisted approach for the facile synthesis of a highly water-soluble zirconium-89 chelator for radiopharmaceutical development. Dalton Trans. 2017, 46 (47), 16387–16389. [DOI] [PubMed] [Google Scholar]
- 30.Allott L; Da Pieve C; Meyers J; Spinks T; Ciobota DM; Kramer-Marek G; Smith G Evaluation of DFO-HOPO as an octadentate chelator for zirconium-89. Chem. Commun 2017, 53 (61), 8529–8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandya DN; Bhatt N; Yuan H; Day CS; Ehrmann BM; Wright M; Bierbach U; Wadas TJ Zirconium tetraazamacrocycle complexes display extraordinary stability and provide a new strategy for zirconium-89-based radiopharmaceutical development. Chemical Science 2017, 8 (3), 2309–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deri MA; Ponnala S; Zeglis BM; Pohl G; Dannenberg JJ; Lewis JS; Francesconi LC Alternative Chelator for 89Zr Radiopharmaceuticals: Radiolabeling and Evaluation of 3,4,3-(LI-1,2-HOPO). J. Med. Chem 2014, 57 (11), 4849–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patra M; Bauman A; Mari C; Fischer CA; Blacque O; Haussinger D; Gasser G; Mindt TL An octadentate bifunctional chelating agent for the development of stable zirconium-89 based molecular imaging probes. Chem. Commun 2014, 50 (78), 11523–11525. [DOI] [PubMed] [Google Scholar]
- 34.Adams CJ; Wilson JJ; Boros E Multifunctional Desferrichrome Analogues as Versatile 89Zr(IV) Chelators for ImmunoPET Probe Development. Mol. Pharm 2017, 14 (8), 2831–2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhai C; Summer D; Rangger C; Franssen GM; Laverman P; Haas H; Petrik M; Haubner R; Decristoforo C Novel Bifunctional Cyclic Chelator for 89Zr Labeling–Radiolabeling and Targeting Properties of RGD Conjugates. Mol. Pharm 2015, 12 (6), 2142–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma MT; Meszaros LK; Paterson BM; Berry DJ; Cooper MS; Ma Y; Hider RC; Blower PJ Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with 89Zr4+: comparison with desferrioxamine-B. Dalton Trans. 2015, 44 (11), 4884–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandya DN; Pailloux S; Tatum D; Magda D; Wadas TJ Di-macrocyclic terephthalamide ligands as chelators for the PET radionuclide zirconium-89. Chem. Commun 2015, 51 (12), 2301–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seibold U; Wängler B; Wängler C Rational Design, Development, and Stability Assessment of a Macrocyclic Four-Hydroxamate-Bearing Bifunctional Chelating Agent for 89Zr. ChemMedChem 2017, 12 (18), 1555–1571. [DOI] [PubMed] [Google Scholar]
- 39.Boros E; Holland JP; Kenton N; Rotile N; Caravan P Macrocycle-Based Hydroxamate Ligands for Complexation and Immunoconjugation of (89)Zirconium for Positron Emission Tomography (PET) Imaging. ChemPlusChem 2016, 81 (3), 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tinianow JN; Pandya DN; Pailloux SL; Ogasawara A; Vanderbilt AN; Gill HS; Williams S-P; Wadas TJ; Magda D; Marik J Evaluation of a 3-hydroxypyridin-2-one (2, 3-HOPO) based macrocyclic chelator for 89Zr4+ and its use for immunoPET imaging of HER2 positive model of ovarian carcinoma in mice. Theranostics 2016, 6 (4), 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tieu W; Lifa T; Katsifis A; Codd R Octadentate Zirconium(IV)-Loaded Macrocycles with Varied Stoichiometry Assembled From Hydroxamic Acid Monomers using Metal-Templated Synthesis. Inorg. Chem 2017, 56 (6), 3719–3728. [DOI] [PubMed] [Google Scholar]
- 42.Rudd SE; Roselt P; Cullinane C; Hicks RJ; Donnelly PS A desferrioxamine B squaramide ester for the incorporation of zirconium-89 into antibodies. Chem. Commun 2016, 52 (80), 11889–11892. [DOI] [PubMed] [Google Scholar]
- 43.Price EW; Zeglis BM; Cawthray JF; Lewis JS; Adam MJ; Orvig C What a Difference a Carbon Makes: H4octapa vs. H4C3octapa, Ligands for In-111 and Lu-177 Radiochemistry. Inorg. Chem 2014, 53 (19), 10412–10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guérard F; Lee Y-S; Brechbiel MW Rational Design, Synthesis, and Evaluation of Tetrahydroxamic Acid Chelators for Stable Complexation of Zirconium(IV). Chemistry – A European Journal 2014, 20 (19), 5584–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guérard F; Lee Y-S; Tripier R; Szajek LP; Deschamps JR; Brechbiel MW Investigation of Zr(IV) and 89Zr(IV) complexation with hydroxamates: progress towards designing a better chelator than desferrioxamine B for immuno-PET imaging. Chemical communications (Cambridge, England) 2013, 49 (10), 1002–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris WR; Yang B; Abdollahi S; Hamada Y Steric restrictions on the binding of large metal ions to serum transferrin. J. Inorg. Biochem 1999, 76 (3–4), 231–242. [DOI] [PubMed] [Google Scholar]
- 47.Sun H; Li H; Sadler PJ Transferrin as a Metal Ion Mediator. Chem. Rev 1999, 99 (9), 2817–2842. [DOI] [PubMed] [Google Scholar]
- 48.Harris W In Less Common Metals in Proteins and Nucleic Acid Probes; Clarke MJ, Ed.; Springer-Verlag, Berlin Heidelberg: 1998; Vol. 92, pp 121–162. [Google Scholar]
- 49.Tsung SH; Rosenthal WA; Milewski KA Immunological Measurement of Transferrin Compared with Chemical Measurement of Total Iron-Binding Capacity. Clin Chem 1975, 21 (8), 1063–1066. [PubMed] [Google Scholar]
- 50.Hider RC; Bickar D; Morrison IEG; Silver J Siderophore iron-release mechanisms. J. Am. Chem. Soc 1984, 106 (23), 6983–6987. [Google Scholar]
- 51.Raymond KN; Müller G; Matzanke B In Top. Curr. Chem; Boschke FL, Ed.; Springer-Verlag, Berlin, Heidelberg: 1984; Vol. 123, pp 49–102. [Google Scholar]
- 52.Delley B An all-electron numerical method for solving the local density functional for polyatomic molecules. J Chem Phys 1990, 92 (1), 508–517. [Google Scholar]
- 53.Delley B From molecules to solids with the DMol3 approach. J Chem Phys 2000, 113 (18), 7756–7764. [Google Scholar]
- 54.Perdew JP; Burke K; Ernzerhof M Generalized Gradient Approximation Made Simple. Physical Review Letter 1996, 77 (18), 3865–3868. [DOI] [PubMed] [Google Scholar]
- 55.Peverati R; Truhlar DG M11-L: A Local Density Functional That Provides Improved Accuracy for Electronic Structure Calculations in Chemistry and Physics. The Journal of Physical Chemistry Letters 2011, 3 (1), 117–124. [Google Scholar]
- 56.Klamt A; Schuurmann G COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J Chem Soc Perkin Trans 1993, 2, 799–805. [Google Scholar]
- 57.Andzelm J; Kölmel C; Klamt A Incorporation of solvent effects into density functional calculations of molecular energies and geometries. J Chem Phys 1995, 103 (21), 9312–9320. [Google Scholar]
- 58.Pandya DN; Bhatt N; An GI; Ha YS; Soni N; Lee H; Lee YJ; Kim JY; Lee W; Ahn H; Yoo J Propylene Cross-Bridged Macrocyclic Bifunctional Chelator: A New Design for Facile Bioconjugation and Robust 64Cu Complex Stability. J. Med. Chem 2014, 57 (17), 7234–7243. [DOI] [PubMed] [Google Scholar]
- 59.Holland JP; Caldas-Lopes E; Divilov V; Longo VA; Taldone T; Zatorska D; Chiosis G; Lewis JS Measuring the Pharmacodynamic Effects of a Novel Hsp90 Inhibitor on HER2/neu Expression in Mice Using 89Zr-DFO-Trastuzumab. PLoS One 2010, 5 (1), e8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.