

Abstract
DNA/RNA motif mining is the foundation of gene function research. The DNA/RNA motif mining plays an extremely important role in identifying the DNA- or RNA-protein binding site, which helps to understand the mechanism of gene regulation and management. For the past few decades, researchers have been working on designing new efficient and accurate algorithms for mining motif. These algorithms can be roughly divided into two categories: the enumeration approach and the probabilistic method. In recent years, machine learning methods had made great progress, especially the algorithm represented by deep learning had achieved good performance. Existing deep learning methods in motif mining can be roughly divided into three types of models: convolutional neural network (CNN) based models, recurrent neural network (RNN) based models, and hybrid CNN–RNN based models. We introduce the application of deep learning in the field of motif mining in terms of data preprocessing, features of existing deep learning architectures and comparing the differences between the basic deep learning models. Through the analysis and comparison of existing deep learning methods, we found that the more complex models tend to perform better than simple ones when data are sufficient, and the current methods are relatively simple compared with other fields such as computer vision, language processing (NLP), computer games, etc. Therefore, it is necessary to conduct a summary in motif mining by deep learning, which can help researchers understand this field.
Keywords: motif mining, deep learning, protein binding site, recurrent neural networks, convolutional neural network
Introduction
Motif plays a key role in the gene-expression regulating both transcriptional and posttranscriptional levels. DNA/RNA motifs involve many biological processes, including alternative splicing, transcription and translation [1–4]. From the late 1990s to the early 21st century, researchers through biological experiments gradually identified a large number of proteins with binding functions and their corresponding binding sites on the genome sequences, the binding sites of the same protein are certain conservative short sequences regarded as motifs, people initially used conservative sequences to describe protein binding sites [5–8]. With the deepening of researchers’ understanding of motif research, various motif mining algorithms emerge [9]. Early motif mining methods are mainly divided into two principal types: enumeration methods and probabilistic methods: enumeration approach and probabilistic method [10].
The first class is based on simple word enumeration. Yeast Motif Finder (YMF) algorithm used consensus representation to detect short motifs with a small number of degenerate positions in the yeast genome developed by Sinha et. al [11]. YMF is mainly divided into two steps: the first step enumerates all motifs of search spaces and the second step calculates the z-score of all motifs to find the greatest one. Bailey proposed discriminative regular expression motif elicitation algorithm that calculated the significance of motifs using Fisher’s Exact test [12].
To accelerate the running speed of word enumeration-based motif mining methods, some special methods were used, like suffix trees, parallel processing [13]. Besides, motif mining algorithms, such as LMMO [14], DirectFS [9], ABC [15], DiscMLA [16], CisFinder [12], Weeder [17], Fmotif [18] and MCES [19] all used this idea in the model.
In probabilistic-based motif mining methods, a probabilistic model that needs a few parameters will be constructed [20]. These methods provided a base distribution of bases for each site in the binding region to distinguish the motif is exist or not [21]. These methods usually built distribution by the position-specific scoring matrix (PSSM/PWM) or motif matrix [22]. PWM was an m by n size matrix (m represents the length of a specific protein binding site, and n represents the type of nucleotide base), which was used to indicate the degree of preference of a specific protein binding motif at each position [23]. Just as Figure 1 shows, PWM can intuitively express the binding preference of a specific protein with fewer parameters, so if a set of specific protein binding site data is given, the parameters of PWM can be learned from these binding site data. Some methods are based on PWM approaches such as MEME [11], STEME [24], EXTREME [25], AlignACE [26] and BioProspector [27].
Figure 1 .
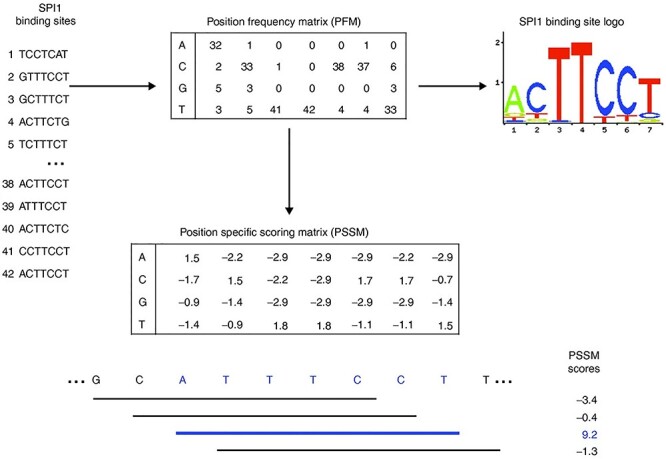
The process of generating PSSM, position frequency matrix (PFM) and logo of SPI1 [104]. The process of as follows generating PSSM, PFM and logo of SPI1. First, generate a PFM based on the number of times each type of nucleotide appears in each position of the alignment. Then, convert the PFM into a logarithmic scale PSSM/PWM. By adding the corresponding nucleotide values of PSSM, the score of any DNA sequence window with the same length as the matrix can be calculated and drawn into a logo map.
ChIP-seq and high-throughput sequencing have tremendously increased the amount of data available in vivo [28], which makes it possible to study the motif mining by deep learning [29]. In bioinformatics, although deep learning methods are not many at present, it is now on the rise [30]. Known applications include DNA methylation [31, 32], protein classification [33–35], splicing regulation and gene expression [36–38] and biological image analysis tasks [39–42]. Of particular relevance to our work is the development of applications for motif mining, such as DNA-/RNA-protein binding sites [43], chromatin accessibility [36, 44–46], enhancer [47–49], DNA-shape [50, 51].
DeepBind [43] is the first study to apply deep learning in motif mining. Just as Figure 2 shows, DeepBind attempted to describe the method by CNN and predicts DNA-protein/RNA-protein binding sites in a way that machine learning or genomics researchers can easily understand. It treated a genome sequence window as a picture. Unlike an image composed of pixels with three color channels (R, G, B), it treated the genomic sequence as a fixed-length sequence window composed of four channels (A, C, G, T) or (A, C, G, U). Therefore, the problem of DNA protein binding site prediction is similar to the problem of binary classification of pictures.
Figure 2 .
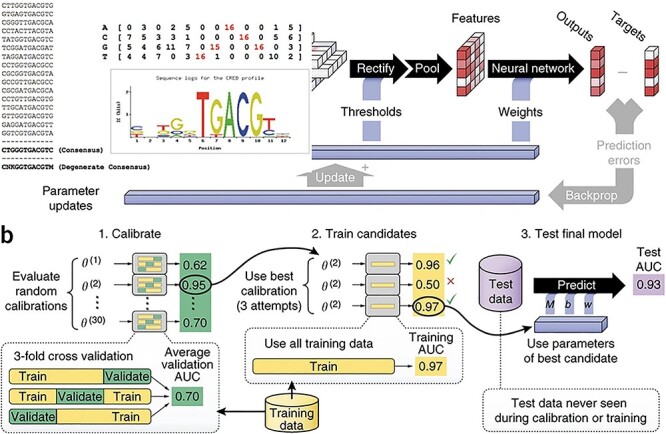
The parallel training process of Deepbind [43]. (A) The DeepBind model processes five independent sequences in parallel. The data first passes through the convolutional layer to extract features, then passes through the pooling layer to optimize the features. Finally, features go through the activation function to output the prediction result and compare with the target to calculate the loss and update weight to improve the prediction accuracy. (B) It is shown in detail that the dataset is divided into validate set, train set and test set, which are used to calculate validate AUC (area under the curve), training AUC and test AUC, respectively, to select the optimal parameters.
After this, a series of research on deep learning in motifs mining appeared. Some researchers focused on the impact of various parameters in deep learning, such as the number of layers, on motif mining [52]. Some researchers have made more attempts for deep learning frameworks, adding a long short-term memory (LSTM) layer to DeepBind, and obtained a new model combining CNN and RNN for motif mining [53]. Besides, there are methods such as iDeepS that combine CNN and RNN to target specific RNA binding proteins (RBP) [54]. The advantage of the combined model of RNN and CNN is that the newly added RNN layer can capture the long-term dependency between sequence features by learning the features extracted by the CNN layer to improve the accuracy of prediction. Other researchers used a pure RNN-based method: the KEGRU method [55] created an internal state of the network by using a k-mer representation and embedding layer, and it captures long-term dependencies by combining with a layer of bidirectional gated recurrent units (bi-GRUs). Besides, many researchers have done a lot of works based on three basic models, for example, Xiaoyong Pan [56], Qinhu Zhang [51, 57], Wenxuan Xu [58], Dailun Wang [59] and Wenbo Yu [60].
Although, there are currently many deep learning methods in motif mining. Those methods compared to the deep learning methods in the field of computer vision and NLP, such as image field [61, 62], video field [63] and question answering field [64], are also relatively primitive and simple. Therefore, it is necessary to summarize the motif mining through deep learning to help researchers to better understand the field. In this paper, we introduce the basic biological background knowledge about motif mining and provide insights into the differences between the basic models of deep learning CNN and RNN, and discuss some new trends in the development of deep learning. This article hopes to help researchers who do not have basic deep learning or basic biology Background knowledge to quickly understand topic mining.
The remainder of this paper is organized as follows: The second section describes the basic biological background knowledge, several common databases and the basic knowledge of motif. Then, the third section describes different models of deep learning algorithms for DNA/RNA motif mining. Finally, we further discuss some new developments and challenges in motif mining deep learning and possible future directions in the fourth section.
Basic Knowledge of Motif
In this section, we introduce the some basic knowledge of motif mining. Motif mining (or motif discovery) in biological sequences can be defined as the problem of finding a set of short, similar, conserved sequence elements (‘motifs’) that are often short and similar in nucleotide sequence with common biological functions [65]. Motif mining has been one of the widely studied problems in bioinformatics, such as transcription factor binding site (TFBS) because its biological significance and bioinformatics significance is highly significant [66, 67].
As shown in Figure 3, it shows how multiple sequences recognize the same transcription factor (CREB). Their ‘consensus’ means that each position has its own more friendly nucleic acid by the transcription factor. Since transcription factor binding can tolerate approximate values, all oligos that differ from the consensus sequence to the maximum number of nucleotide substitutions can be considered as valid instances of the same TFBS.
Figure 3 .
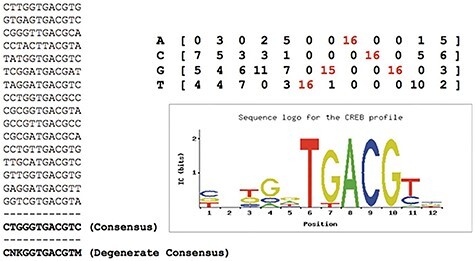
A set of binding sites recognized by the same TF (CREB) [65]. It shows how multiple sequences recognize the same transcription factor (CREB). First, Zambelli built their ‘consensus’ (bottom left) by counting the frequency of each nucleic acid in the sequence [65]. And ‘consensus’ (bottom left) with the highest frequency of nucleotides at each position to indicate the motifs they form a ‘degenerate’ consensus, which includes nucleotides that have no obvious preference position (K = G or T; M = A or C; N = any nucleotide; according to IUPAC codes [105]). Besides, motifs can be converted into an alignment matrix of the nucleotide frequency (top right) by dividing each column by the number of sites used, as well as a ‘sequence logo’ (bottom left) [106] showing nucleotide conservation and corresponding information.
After understanding the basic concept of motif, we introduce common databases and data preprocessing methods. The commonly used motif mining database is as follows: TCGA database [68], NCBI database [69] and ENCODE database [70]. Generally speaking, two data preprocessing methods are the following methods as shown in Figure 4, bottom left.
Figure 4 .
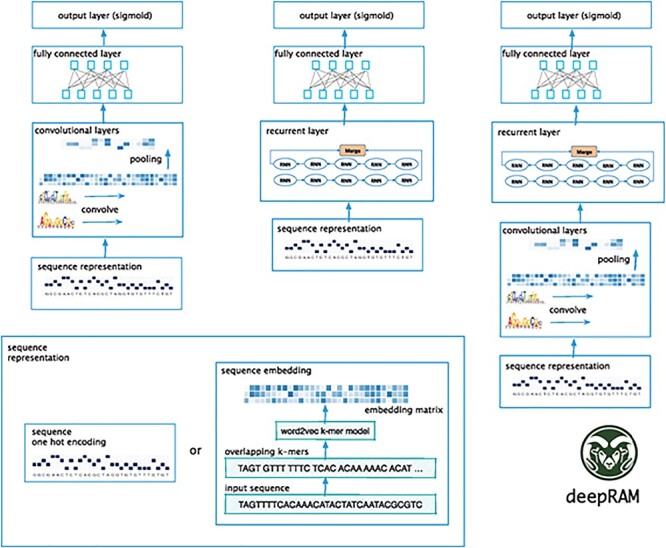
Sequence representation of motif mining [78]. It shows two data preprocessing methods(bottom left) and three architectures include CNN-only (left), RNN-only (center) and hybrid CNN–RNN models (right).
The simple method is to use the one-hot encoding. One-hot is often used for indicating the state of a state machine [71]. For example, using one-hot codes to encode DNA sequences as binary vectors: A = (1,0,0,0), G = (0,1,0,0), C = (0,0,1,0) and T = (0,0,0,1). RNA sequences can also be encoded similarly by simply changing T to U. It is easy to design and modify, and easy to detect illegal states. However, it is easily sparse and context-free.
Another method is to label with k-mers and vectorize by embedding [44]. For example, we can tokenize the DNA sequence ‘ATCGCGTACGATCCG’ as different k-mers, as shown in Table 1. Different k-mers can be vectorized using the embedding method widely used in the NLP field [72], such as word2vec [73]. RNA sequences can be represented similarly.
Table 1.
Different parameters for k-mers
| Length | Window | Tokenized | Vectorization |
|---|---|---|---|
| 3 | 3 | ATC GCG TAC GAT CCG | 0321 3412 4532 4214 |
| 4 | 4 | ATCG CGTA CGAT | 0123 3412 4532 |
| 5 | 5 | ATCGC GTACG ATCCG | 4124 5124 2134 |
| 4 | 2 | ATCG CGCG CGTA TACG CGAT ATCC | 2563 3124 4236 3578 2145 |
| 4 | 3 | ATCG GCGT TACG GATC | 4252 5134 2136 3451 2411 |
It shows DNA sequence ‘ATCGCGTACGATCCG’ is cut into multiple different k-mers and his vector when the length is (3,4,5,4,4) and the window is (3,4,5,2,3).
Deep Learning in Motif Mining
In recent years, deep learning has achieved great success in various application scenarios, which makes researchers try to apply it to DNA or RNA motif mining. Next, we introduce these models in detail. There are three main types of deep learning frameworks in motif mining: CNN-based models (Figure 4, left), RNN-based models (Figure 4, center), hybrid CNN–RNN-based models (Figure 4, right). We summarize several classic deep learning methods in motif mining, as shown in Table 2.
Table 2.
Deep learning algorithm in DNA motif mining
| Model | DeepBind | DeepSNR | DeepSEA | Dilated | DanQ | BiRen | KEGRU | iDeeps |
|---|---|---|---|---|---|---|---|---|
| Architecture | CNN | CNN | CNN | CNN | CNN + RNN | CNN + RNN | RNN | CNN + RNN |
| Embedding | NO | NO | NO | NO | NO | NO | YES | NO |
| Input | One-hot | One-hot | One-hot | One-hot | One-hot | k-mer | k-mer | One-hot |
It shows the architecture, embedding and input of eight classic deep learning models in motif mining.
DeepBind [43] is the first attempt to use CNN to predict DNA or RNA motifs from original DNA or RNA sequences. DeepBind used a single CNN layer, which consists of one convolutional layer, followed by rectification and pooling operation, and one fully connected network (FCN) augmented at the end to transform feature vectors into a scalar binding score. It also opened up a precedent for deep learning in motif mining and provides a basic framework for other deep learning methods. It corresponded to each base to four channels similar to the RGB channel in color and used one-hot encoding to complete vectorization. Many subsequent methods use this to build their models.
DeepSEA [38] was a deep learning method based on CNN, which used three convolution layers with 320, 480 and 960 kernels, respectively. Higher-level convolutional layers receive input from a larger spatial range, and lower-level convolutional network layers can represent more complex features. DeepSEA added an FCN layer on top of the third convolutional layer, in which all neurons receive input from all outputs of the previous layer so that the information of the entire sequence data can be completely obtained. The convolution step of the DeepSEA model consisted of three convolutional layers and two maximum merge layers, and the motif was learned in alternating order.
DeepSNR [74] was a deep learning method based on CNN. The convolution part of the DeepSNR model had the same structure as the DeepBind network. But DeepSNR added that the deconvolution network is a mirrored version of the convolution network, which can reduce the size of the activation and enlarges the activations through combinations of unpooling and deconvolution operations.
Dilated [75] was a deep learning method based on dilated multilayer CNN. This method learns the mapping from the DNA region of the nucleotide sequence to the position of the regulatory marker in this region. The dilated convolution can capture a hierarchical representation of the input space that is larger than the standard convolution so that they can be scaled to larger before and after sequences.
DanQ [53] used a single layer CNN followed by a bidirectional LSTM (BLSTM). The first layer of the DanQ model aimed to scan the position of the motif in the sequence through convolution filtering. The convolution step of the DanQ model was much simpler than DeepSEA. It contained a convolutional layer and a maximum merge layer to learn the motif. After the largest pooling layer was the BLSTM layer. Motifs can follow the adjustment grammar determined by physical constraints, which determine the spatial arrangement and frequency of the pattern combination in vivo, which is a feature related to tissue-specific functional elements (such as enhancers). So the LSTM layer is after the maximum pooling layer. The last two layers of the DanQ model were dense layers of rectified linear units and multitask sigmod output, similar to the DeepSEA model. The advantage of the combined model of RNN and CNN was that the newly added RNN layer can capture the long-term dependency between sequence features by learning the features extracted by the CNN layer to improve the accuracy of prediction.
BiRen [49] developed a hybrid architecture based on deep learning, which combines the sequence encoding and representation capabilities of CNN and bidirectional recurrent neural network of processing long sequences of DNA excellent ability. BiRen had undergone limited experimental verification of enhancer element training, which comes from the VISTA enhancer browser [76], and has enhanced gene activity, as evaluated in transgenic mice. BiRen could learn regulatory codes directly from genomic sequences, and demonstrate excellent recognition accuracy, overcoming the robustness of noisy data, and two new methods for other species based on sequence features for other species General k-mer for enhancer prediction. BiRen enabled researchers to have a deeper understanding of the regulatory codes of enhancer sequences.
KEGRU [55], which usesd a layer of GRU and k-mer embedding, was a pure RNN layer model without CNN layer. KEGRU mainly used the k-mer and embedding layer to achieve the purpose of CNN feature extraction tasks in other models. Such a structure made it perform better in sequence relationships and achieves a good structure in RNA motif mining.
iDeeps [54] which used convolutional neural networks (CNNs) and a BLSTM network to simultaneously identify the binding sequence and structure motifs from RNA sequences. The CNN module embedded in iDeep can also automatically capture the interpretable binding motif of RBP. The BLSTM network made the iDeep framework to not only achieve better performance on binding sequence but also easily capture structure motifs.
Model selection may be the most challenging step in deep learning because the performance of deep learning algorithms is very sensitive to different parameters [77]. The deepRAM [78] provides implementations of several existing architectures and their variants: DeepBind (single layer CNN), DeepBind* (multilayer CNN), DeepBind-E* (multilayer CNN, k-mer embedding), DanQ (single layer CNN, bidirectional LSTM), DanQ* (multilayers CNN, bidirectional LSTM), Dilated (multilayer dilated CNN), KEGRU (k-mer embedding, single layer GRU), ECLSTM (k-mer embedding, single-layer CNN and LSTM) and ECBLSTM (k-mer embedding, single-layer CNN and bidirectional LSTM). They conducted a lot of experimental comparisons, which gave researchers a deeper understanding of these methods.
Before introducing the experimental results of deepRAM [78], we introduce two sets of datasets used in the experiment. The first group is the DNA datasets include 83 ChIP-seq data from the ENCODE project [70]. The second group is the RNA datasets include 31 CLIP-seq data for 19 proteins [79–81].
The deepRAM [78] has conducted a large number of experiments on these two datasets of experimental data and conducted an in-depth comparison and description of the above deep learning models. The experimental results of the model on these datasets are shown in Figure 5.
Figure 5 .
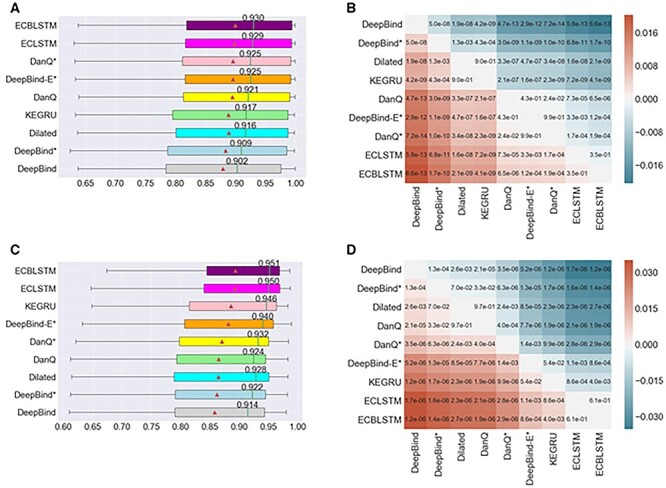
Comparison results of nine deep learning models [78]. It compares the performance of these models in predicting DNA and RNA motif mining tasks. (A) The AUC distribution of nine models in 83 ChIP-seq datasets. (B) P-value annotated heat maps using paired models of nine models in 83 ChIP-seq datasets. (C) The AUC distribution of nine models in 31 CLIP-seq datasets. (D) P-value annotated heat maps using paired models of nine models in 31 datasets.
Among all models, the ECBLSTM model performed best, whether it was a median AUC of 0.930 on ChIP-seq data or a median AUC of 0.951 on CLIP-seq data, and the simplest DeepBind of all models is here. The median AUC on the two datasets was 0.902 and 0.914, respectively. DeepBind is the simplest model considered here: it uses a single hot sequence encoding and a single convolutional layer. By comparing the performance of ECBLSTM with the model of DeepBindE*, it can be seen that adding an LSTM layer can further improve performance. Because LSTM layers are better at capturing long-term dependencies than CNN layers. Compared with the original DeepBind, both DeepBind* or DeepBind-E* can provide improved performance. By comparing the performance of DanQ and DanQ*, it is further found that the performance of models deeper than single-layer CNN tends to perform better. Experiment results demonstrate the performance advantages of deeper and more complex networks. Zhang [17] found that the simpler model performs best in this task, and the conclusions found through deepRAM’s experiment are just the opposite. Based on the experimental results and theoretical analysis, it is found that the complexity of the model should be related to the task and data. Too many parameters can easily cause over-fitting [82]. Generally, the parameters of our task model should not exceed the data sample too much.
Discussion
From the traditional method of motif to the latest development process of deep learning, we can find great progress with the development of sequencing technology and new algorithms. We analyzed the existing models, and their variants found that the more complex models tend to perform better when data are sufficient in the third section. The recent research trends can be found that the model is usually more and more complex. For example, researchers try to combine existing models with new models, such as combining attention units [83, 84], capsule network [85], multiscale convolutional gated recurrent unit networks [86], weakly supervised CNN [87] and multiple-instance learning [88]. However, the existing deep learning models in motif mining are too simple, no more than three layers, compared to the model in the image field usually over 10 layers. Therefore, there is still much room for improvement.
Recently, since the adversarial training of neural networks can lead to regularization to provide higher performance, this field has developed rapidly, including involving adversarial generative networks [89] and a series of related research such as Wasserstein GAN [90], MolGAN [91] and NetGAN [92]. In motif mining, GAN may be used to automatically generate negative examples instead of simple random generation or shuffling the positive sequence. Besides, pretraining models [93] that have achieved significant results in the NLP field, from word2vec [73, 94] to now Bert [95] and GPT [96]. In motif mining, pretraining can be used to enhance the robustness and generalization ability of the model. The great success of AlphaGo [97] has set off an unprecedented change in the Go world, and it has made deep reinforcement learning familiar to the public. In particular, AlphaGo Zero does not require any history of human chess, and only uses deep reinforcement learning [98]. The achievement of training from 0 to 3 days has far exceeded the knowledge of Go that humans have accumulated for thousands of years. In motif mining, reinforcement learning may enable people to learn more motifs beyond human knowledge.
As we enter the era of big data, whether it is in academic or industrial, deep learning is already a very important development direction. In bioinformatics, which has made great progress in traditional machine learning, deep learning is expected to produce encouraging results [99]. In this review, we conducted a comprehensive review of the application of deep learning in the field of motif mining. We desire that this review will provide help researchers understand this field and promote the application of motif mining in research.
Of course, we also need to recognize the limitations of deep learning methods and the promising direction of future research. Although deep learning is promising, it is not a panacea.
In many applications of motif mining, there are still many potential challenges, including unbalanced or limited data, interpretation of deep learning results [71] and the choice of appropriate architecture and hyperparameters. For unbalanced or limited data, the common methods are enhanced datasets [48] or few-shot learning [100]. For interpretation of deep learning results, common methods are the interpretability of the model itself [101] or the interpretation after the prediction [71]. For the choice of appropriate architecture and hyperparameters, frameworks such as Spearmint [102], Hyperopt [103] and DeepRAM [78] allow to automatically explore the hyper-parameter space. Besides, how to make full use of the ability of deep learning to accelerate the training process of deep learning also needs further research. Therefore, we hope that the issues discussed in this article will be helpful to the success of future deep learning methods in motif mining.
Key Points
Motif mining (or motif discovery) in biological sequences can be defined as the problem of finding a set of short, similar, conserved sequence elements (‘motifs’) that are often short and similar in nucleotide sequence with common biological functions. Motif plays a key role in the gene-expression regulating both transcriptional and posttranscriptional levels.
In recent years, deep learning has achieved great success in various application scenarios, which makes researchers try to apply it to DNA or RNA motif mining. There are three main types of deep learning frameworks in motif mining: CNN-based models, RNN-based models and hybrid CNN–RNN-based models.
Briefly, we also introduce the application of deep learning in the field of motif mining in terms of data preprocessing, features of existing deep learning architectures and comparing the differences between the basic deep learning models.
Acknowledgement
This work was supported by the grant of National Key R&D Program of China (Nos. 2018AAA0100100 & 2018YFA0902600) and partly supported by National Natural Science Foundation of China (Grant nos. 61861146002, 61520106006, 61732012, 61932008, 61772370, 61672382, 61702371, 61532008, 61772357, and 61672203) and China Postdoctoral Science Foundation (Grant no. 2017M611619) and supported by ``BAGUI Scholar'' Program and the Scientific & Technological Base and Talent Special Program, GuiKe AD18126015 of the Guangxi Zhuang Autonomous Region of China and supported by Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), LCNBI and ZJLab.
Ying He is pursuing a Ph.D. degree in computer science and technology at Tongji University, China. His research interests include bioinformatics, machine learning and deep learning.
Zhen Shen is pursuing a Ph.D. degree in computer science and technology at Tongji University, China. His research interests include bioinformatics, machine learning and deep learning.
Qinhu Zhang received a Ph.D. degree in computer science and technology at Tongji University, China, in 2019. He is currently working at Tongji University as a post-doctor. His research interests include bioinformatics, machine learning and deep learning.
Siguo Wang is working toward the Ph.D. degree in computer science and technology, Tongji University, China. Her research interests include bioinformatics, machine learning and deep learning.
De-Shuang Huang is a chaired professor at Tongji University. At present, he is the Director of the Institute of Machines Learning and Systems Biology, Tongji University. Dr. Huang is currently IAPR Fellow and a senior member of the IEEE. His current research interest includes bioinformatics, pattern recognition and machine learning.
Contributor Information
Ying He, computer science and technology at Tongji University, China.
Zhen Shen, computer science and technology at Tongji University, China.
Qinhu Zhang, computer science and technology at Tongji University, China.
Siguo Wang, computer science and technology at Tongji University, China.
De-Shuang Huang, Institute of Machines Learning and Systems Biology, Tongji University.
References
- 1. Ferre F, Colantoni A, Helmer-Citterich M. Revealing protein–lncRNA interaction. Brief Bioinform 2016;17:106–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet 2014;15:829–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajyaguru P, She M, Parker R. Scd6 targets eIF4G to repress translation: RGG motif proteins as a class of eIF4G-binding proteins. Mol Cell 2012;45:244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo W-L, Huang D-S. An efficient method to transcription factor binding sites imputation via simultaneous completion of multiple matrices with positional consistency. Mol Biosyst 2017;13:1827–37. [DOI] [PubMed] [Google Scholar]
- 5. Stormo GD, Hartzell GW. Identifying protein-binding sites from unaligned DNA fragments. Proc Natl Acad Sci 1989;86:1183–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Welch W, Ruppert J, Jain AN. Hammerhead: fast, fully automated docking of flexible ligands to protein binding sites. Chem Biol 1996;3:449–62. [DOI] [PubMed] [Google Scholar]
- 7. Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein–protein binding sites. J Mol Biol 2004;338:181–99. [DOI] [PubMed] [Google Scholar]
- 8. Bradford JR, Westhead DR. Improved prediction of protein–protein binding sites using a support vector machines approach. Bioinformatics 2005;21:1487–94. [DOI] [PubMed] [Google Scholar]
- 9. Zhu L, Li N, Bao W, et al. Learning regulatory motifs by direct optimization of Fisher Exact Test Score. In: 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2016, pp. 86-91.
- 10. Hashim FA, Mabrouk MS, Al-Atabany W. Review of different sequence motif finding algorithms. Avicenna J Med Biotechnol 2019;11:130. [PMC free article] [PubMed] [Google Scholar]
- 11. Sinha S, Tompa M. YMF: a program for discovery of novel transcription factor binding sites by statistical overrepresentation. Nucleic Acids Res 2003;31:3586–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011;27:1653–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pavesi G, Mereghetti P, Mauri G, et al. Weeder web: discovery of transcription factor binding sites in a set of sequences from co-regulated genes. Nucleic Acids Res 2004;32:W199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu L, Zhang H-B, Huang D-S. LMMO: a large margin approach for refining regulatory motifs, IEEE/ACM Transactions on Computational Biology and Bioinformatics 2017;15:913-25. [DOI] [PubMed] [Google Scholar]
- 15. Karaboga D, Aslan S. A discrete artificial bee colony algorithm for detecting transcription factor binding sites in DNA sequences. Genet Mol Res 2016;15:1–11. [DOI] [PubMed] [Google Scholar]
- 16. Zhang H, Zhu L, Huang D. DiscMLA: AUC-based discriminative motif learning. In: 2015 IEEE International Conference on Bioinformatics and Biomedicine. 2015, pp. 250-5.
- 17. Zhang Y, Wang P, Yan M. An entropy-based position projection algorithm for motif discovery. Biomed Res Int 2016;2016:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sharov AA, Ko MS. Exhaustive search for over-represented DNA sequence motifs with CisFinder. DNA Res 2009;16:261–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jia C, Carson MB, Wang Y, et al. A new exhaustive method and strategy for finding motifs in ChIP-enriched regions. PLoS One 2014;9:e86044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sinha S. On counting position weight matrix matches in a sequence, with application to discriminative motif finding. Bioinformatics 2006;22:e454–63. [DOI] [PubMed] [Google Scholar]
- 21. Yu Q, Huo H, Chen X, et al. An efficient algorithm for discovering motifs in large DNA data sets. IEEE Trans Nanobioscience 2015;14:535–44. [DOI] [PubMed] [Google Scholar]
- 22. Stormo GD. DNA binding sites: representation and discovery. Bioinformatics 2000;16:16–23. [DOI] [PubMed] [Google Scholar]
- 23. Xia X. Position weight matrix, gibbs sampler, and the associated significance tests in motif characterization and prediction. Forensic Sci 2012;2012:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Helden J, André B, Collado-Vides J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J Mol Biol 1998;281:827–42. [DOI] [PubMed] [Google Scholar]
- 25. Thomas-Chollier M, Herrmann C, Defrance M, et al. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res 2012;40:e31–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma X, Kulkarni A, Zhang Z, et al. A highly efficient and effective motif discovery method for ChIP-seq/ChIP-chip data using positional information. Nucleic Acids Res 2012;40:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pavesi G, Mauri G, Pesole G. An algorithm for finding signals of unknown length in DNA sequences. Bioinformatics 2001;17:S207–14. [DOI] [PubMed] [Google Scholar]
- 28. Myllykangas S, Buenrostro J, Ji HP. Overview of sequencing technology platforms. In: Bioinformatics for High Throughput Sequencing. Berlin: Springer, 2012, 11–25. [Google Scholar]
- 29. Zhu L, Guo W-L, Huang D-S, et al. Imputation of ChIP-seq datasets via Low Rank Convex Co-Embedding. In: 2015 IEEE International Conference on Bioinformatics and Biomedicine. 2015, pp. 141-4.
- 30. Angermueller C, Pärnamaa T, Parts L, et al. Deep learning for computational biology. Mol Syst Biol 2016;12:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vidaki A, Ballard D, Aliferi A, et al. DNA methylation-based forensic age prediction using artificial neural networks and next generation sequencing. Forensic Sci Int Genet 2017;28:225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angermueller C, Lee HJ, Reik W, et al. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 2017;18:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Asgari E, Mofrad MR. Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One 2015;10:e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pärnamaa T, Parts L. Accurate classification of protein subcellular localization from high-throughput microscopy images using deep learning. G3: Genes, Genomes, Genet 2017;7:1385–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Almagro Armenteros JJ, Sønderby CK, Sønderby SK, et al. DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 2017;33:3387–95. [DOI] [PubMed] [Google Scholar]
- 36. Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res 2016;26:990–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leung MK, Xiong HY, Lee LJ, et al. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014;30:i121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bar Y, Diamant I, Wolf L, et al. Deep learning with non-medical training used for chest pathology identification. In: Medical Imaging 2015: Computer-Aided Diagnosis. Bellingham, WA: International Society for Optics and Photonics, 2015, 94140V. [Google Scholar]
- 40. Tron R, Zhou X, Daniilidis K. A survey on rotation optimization in structure from motion. Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition Workshops 2016, 77–85.
- 41. Mahmud M, Kaiser MS, Hussain A, et al. Applications of deep learning and reinforcement learning to biological data. IEEE Trans Neural Netw Learn Syst 2018;29:2063–79. [DOI] [PubMed] [Google Scholar]
- 42. Affonso C, Rossi ALD, Vieira FHA, et al. Deep learning for biological image classification. Expert Syst Appl 2017;85:114–22. [Google Scholar]
- 43. Alipanahi B, Delong A, Weirauch MT, et al. Predicting the sequence specificities of DNA-and RNA-binding proteins by deep learning. Nat Biotechnol 2015;33:831–8. [DOI] [PubMed] [Google Scholar]
- 44. Min X, Zeng W, Chen N, et al. Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics 2017;33:i92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nair S, Kim DS, Perricone J, et al. Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. Bioinformatics 2019;35:i108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Q, Xia F, Yin Q, et al. Chromatin accessibility prediction via a hybrid deep convolutional neural network. Bioinformatics 2018;34:732–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kleftogiannis D, Kalnis P, Bajic VB. DEEP: a general computational framework for predicting enhancers. Nucleic Acids Res 2015;43:e6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohn D, Zuk O, Kaplan T. Enhancer identification using transfer and adversarial deep learning of DNA sequences. BioRxiv 2018; 264200. [Google Scholar]
- 49. Yang B, Liu F, Ren C, et al. BiRen: predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 2017;33:1930–6. [DOI] [PubMed] [Google Scholar]
- 50. Yang J, Ma A, Hoppe AD, et al. Prediction of regulatory motifs from human Chip-sequencing data using a deep learning framework. Nucleic Acids Res 2019;47:7809–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang Q, Shen Z, Huang D-S. Predicting in-vitro transcription factor binding sites using DNA sequence+ shape. IEEE/ACM Trans Comput Biol Bioinform 2019. [DOI] [PubMed] [Google Scholar]
- 52. Zhang S, Zhou J, Hu H, et al. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res 2016;44:e32–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quang D, Xie X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res 2016;44:e107–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pan X, Rijnbeek P, Yan J, et al. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics 2018;19:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen Z, Bao W, Huang D-S. Recurrent neural network for predicting transcription factor binding sites. Sci Rep 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pan X, Shen H-B. Predicting RNA–protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics 2018;34:3427–36. [DOI] [PubMed] [Google Scholar]
- 57. Zhang Q, Zhu L, Huang D-S. High-order convolutional neural network architecture for predicting DNA-protein binding sites. IEEE/ACM Trans Comput Biol Bioinform 2018;16:1184–92. [DOI] [PubMed] [Google Scholar]
- 58. Xu W, Zhu L, Huang D-S. DCDE: an efficient deep convolutional divergence encoding method for human promoter recognition. IEEE Trans Nanobioscience 2019;18:136–45. [DOI] [PubMed] [Google Scholar]
- 59. Wang D, Zhang Q, Yuan C-A, et al. Motif discovery via convolutional networks with K-mer embedding. In: International Conference on Intelligent Computing. Berlin: Springer, 2019, 374–82. [Google Scholar]
- 60. Yu W, Yuan C-A, Qin X, et al. Hierarchical attention network for predicting DNA-protein binding sites. In: International Conference on Intelligent Computing. Berlin: Springer, 2019, 366–73. [Google Scholar]
- 61. Xu K, Ba J, Kiros R, et al. Show, attend and tell: neural image caption generation with visual attention. International Conference on Machine Learning, 2015, 2048–57.
- 62. Tang P, Wang H, Kwong S. G-MS2F: GoogLeNet based multi-stage feature fusion of deep CNN for scene recognition. Neurocomputing 2017;225:188–97. [Google Scholar]
- 63. Yao L, Torabi A, Cho K, et al. Describing videos by exploiting temporal structure. In: Proceedings of the IEEE International Conference on Computer Vision. 2015, pp. 4507-15.
- 64. Noh H, Hongsuck Seo P, Han B. Image question answering using convolutional neural network with dynamic parameter prediction. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition. 2016, pp. 30-8.
- 65. Zambelli F, Pesole G, Pavesi G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief Bioinform 2013;14:225–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pavesi G, Mauri G, Pesole G. In silico representation and discovery of transcription factor binding sites. Brief Bioinform 2004;5:217–36. [DOI] [PubMed] [Google Scholar]
- 67. Sandve GK, Drabløs F. A survey of motif discovery methods in an integrated framework. Biol Direct 2006;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tomczak K, Czerwińska P, Wiznerowicz M. The cancer genome atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncol 2015;19:A68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sherry ST, Ward M-H, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Consortium EP. The ENCODE (ENCyclopedia of DNA elements) project. Science 2004;306:636–40. [DOI] [PubMed] [Google Scholar]
- 71. Lanchantin J, Singh R, Wang B, et al. Deep motif dashboard: visualizing and understanding genomic sequences using deep neural networks. In: Pacific Symposium on Biocomputing, Vol. 2017. Singapore: World Scientific, 2017, 254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goldberg Y, Levy O. word2vec explained: deriving Mikolov et al.'s negative-sampling word-embedding method. arXiv:1402.3722. 2014.
- 74. Salekin S, Zhang JM, Huang Y. A deep learning model for predicting transcription factor binding location at single nucleotide resolution. In: 2017 IEEE EMBS International Conference on Biomedical & Health Informatics. 2017, pp. 57-60.
- 75. Gupta A, Rush AM. Dilated convolutions for modeling long-distance genomic dependencies. arXiv:1710.01278. 2017.
- 76. Visel A, Minovitsky S, Dubchak I, et al. VISTA enhancer browser—a database of tissue-specific human enhancers. Nucleic Acids Res 2007;35:D88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lipton ZC, Steinhardt J. Troubling trends in machine learning scholarship. arXiv:1807.03341. 2018.
- 78. Trabelsi A, Chaabane M, Ben-Hur A. Comprehensive evaluation of deep learning architectures for prediction of DNA/RNA sequence binding specificities. Bioinformatics 2019;35:i269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Blin K, Dieterich C, Wurmus R, et al. DoRiNA 2.0—upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res 2015;43:D160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. iCount . iCount. http://icount.biolab.si/.
- 81. Stražar M, Žitnik M, Zupan B, et al. Orthogonal matrix factorization enables integrative analysis of multiple RNA binding proteins. Bioinformatics 2016;32:1527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cawley GC, Talbot NL. On over-fitting in model selection and subsequent selection bias in performance evaluation. J Mach Learn Res 2010;11:2079–107. [Google Scholar]
- 83. Hong Z, Zeng X, Wei L, et al. Identifying enhancer–promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 2020;36:1037–43. [DOI] [PubMed] [Google Scholar]
- 84. Shen Z, Zhang Q, Kyungsook H, et al. A deep learning model for RNA-protein binding preference prediction based on hierarchical LSTM and attention network. IEEE/ACM Trans Comput Biol Bioinform 2020. [DOI] [PubMed] [Google Scholar]
- 85. Shen Z, Deng S-P, D-S H. Capsule network for predicting RNA-protein binding preferences using hybrid feature. IEEE/ACM Trans Comput Biol Bioinform 2019. [DOI] [PubMed] [Google Scholar]
- 86. Shen Z, Deng S-P, Huang D-S. RNA-protein binding sites prediction via multi scale convolutional gated recurrent unit networks. IEEE/ACM Trans Comput Biol Bioinform 2019. [DOI] [PubMed] [Google Scholar]
- 87. Zhang Q, Zhu L, Bao W, et al. Weakly-supervised convolutional neural network architecture for predicting protein-DNA binding. IEEE/ACM Trans Comput Biol Bioinform 2018, 2672–80. [DOI] [PubMed] [Google Scholar]
- 88. Zhang Q, Shen Z, Huang D-S. Modeling in-vivo protein-DNA binding by combining multiple-instance learning with a hybrid deep neural network. Sci Rep 2019;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goodfellow I, Pouget-Abadie J, Mirza M, et al. Generative adversarial nets. In: Advances in Neural Information Processing Systems, 2014, 2672–80.
- 90. Arjovsky M, Chintala S, Bottou L. Wasserstein GAN. arXiv:1701.07875. 2017.
- 91. De Cao N, Kipf T. MolGAN: an implicit generative model for small molecular graphs. arXiv:1805.11973. 2018.
- 92. Bojchevski A, Shchur O, Zügner D, et al. Netgan: generating graphs via random walks. arXiv:1803.00816. 2018.
- 93. Mikolov T, Grave E, Bojanowski P, et al. Advances in pre-training distributed word representations. arXiv:1712.09405. 2017.
- 94. Rong X. word2vec parameter learning explained. arXiv:1411.2738. 2014.
- 95. Devlin J, Chang M-W, Lee K, et al. Bert: pre-training of deep bidirectional transformers for language understanding. arXiv:1810.04805. 2018.
- 96. Radford A, Narasimhan K, Salimans T, et al. Improving language understanding by generative pre-training. 2018.
- 97. Silver D, Hassabis D. Alphago: mastering the ancient game of go with machine learning. Res Blog 2016;9. https://ai.googleblog.com/2016/01/alphago-mastering-ancient-game-of-go.html. [Google Scholar]
- 98. Silver D, Schrittwieser J, Simonyan K, et al. Mastering the game of go without human knowledge. Nature 2017;550:354–9. [DOI] [PubMed] [Google Scholar]
- 99. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2017;18:851–69. [DOI] [PubMed] [Google Scholar]
- 100. Snell J, Swersky K, Zemel R. Prototypical networks for few-shot learning. In: Advances in Neural Information Processing Systems, Long Beach, CA, USA: NIPS Foundation, 2017, 4077–87. [Google Scholar]
- 101. Hu H-J, Wang H, Harrison R, et al. Understanding the prediction of transmembrane proteins by support vector machine using association rule mining. In: 2007 IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology. 2007, pp. 418-25.
- 102. Snoek J, Larochelle H. Spearmint. https://github.com/JasperSnoek/spearmint2012.
- 103. Bergstra J, Yamins D, Cox DD. Hyperopt: a python library for optimizing the hyperparameters of machine learning algorithms. In: Proceedings of the 12th Python in Science Conference. 2013, p. 20.
- 104. Worsley-Hunt R, Bernard V, Wasserman WW. Identification of cis-regulatory sequence variations in individual genome sequences. Genome Med 2011;3:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cornish-Bowden A. Nomenclature for incompletely specified bases in nucleic acid sequences: recommendations 1984. Nucleic Acids Res 1985;13:3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Crooks GE, Hon G, Chandonia J-M, et al. WebLogo: a sequence logo generator. Genome Res 2004;14:1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]