

Abstract
Advances in high-throughput sequencing technologies have resulted in an exponential growth of publicly accessible biological datasets. In the ‘big data’ driven ‘post-genomic’ context, much work is being done to explore human protein–protein interactions (PPIs) for a systems level based analysis to uncover useful signals and gain more insights to advance current knowledge and answer specific biological and health questions. These PPIs are experimentally or computationally predicted, stored in different online databases and some of PPI resources are updated regularly. As with many biological datasets, such regular updates continuously render older PPI datasets potentially outdated. Moreover, while many of these interactions are shared between these online resources, each resource includes its own identified PPIs and none of these databases exhaustively contains all existing human PPI maps. In this context, it is essential to enable the integration of or combining interaction datasets from different resources, to generate a PPI map with increased coverage and confidence. To allow researchers to produce an integrated human PPI datasets in real-time, we introduce the integrated human protein–protein interaction network generator (IHP-PING) tool. IHP-PING is a flexible python package which generates a human PPI network from freely available online resources. This tool extracts and integrates heterogeneous PPI datasets to generate a unified PPI network, which is stored locally for further applications.
Keywords: high-throughput technology, protein–protein interaction, network analysis, post-genomic analysis, human proteome
Introduction
Advances in high-throughput sequencing technologies coupled with analytical innovations are yielding novel computational tools. This has enabled access to entire genomic contents of individuals and research methodologies have shifted accordingly. Millions of genetic variants have been uncovered and documented in the public domain, some of which are associated with diseases or affect response to therapeutics. Effects of these variants revealed that some disease outcomes, in particular complex diseases, such as cancer and tuberculosis, or a response to therapeutics are influenced by multiple genes. Considering that many genes may contribute to disease, it is understandable that modern attempts to associate phenotypes with genetic variations do so on a genomic scale [1], using the interactome, i.e. the complete set of physical protein–protein interactions (PPIs) within a cell [2–4]. This context has been described as the post-genomic era and involves the exploration of available sources of information at the systems level [5]. This provides new opportunities for genomic analysis to correlate phenotypes to interactions between candidate or target genes [5]. This functional information can be inferred using PPI networks [6, 7], highlighting how genes/proteins interact and influence each other in the same sub-network (motifs or module), in which case, reconstructing the complete interactome map is essential. This is in stark contrast to the older geno-centric view [8], which sought to describe simple links connecting individual genes to particular phenotypes [9].
Currently, one approach for exploring the interactome is through the generation and analysis of PPI networks [10–13]. These networks display proteins and the interactions between protein pairs in the form of mathematical graphs, which consist of edges and nodes [1, 14]. PPI networks are currently being used for several biological applications, including protein function prediction; candidate gene or target prediction [6] and prioritization; post genome-wide association analyses [15]; prediction of disease phenotype trends and identification of disease-related genetic patterns or properties [1]. Interactions between protein pairs can be predicted experimentally by high-throughput yeast two-hybrid screens and/or mass spectrometry [16]. Alternatively, interactions can be inferred from literature [17, 18], or predicted based on sequence data [19]. These PPI datasets are curated and stored in several online resources [7], including STRING [20], IntAct [21], MINT [22], BioGRID [23], DIP [24], HPRD [25] and MPPI-MIPS [26]. Some of these resources update their datasets regularly using manual curation guidelines from the international molecular exchange (IMEx) initiative [27], e.g. IntAct, MINT, DIP or automated curation schemes, e.g. STRING. Thus, new versions are often released, rendering older networks potentially outdated and existing human PPI networks are still incomplete [28, 29]. Furthermore, while many of these existing PPIs are shared [30] between these resources, none of them covers all reported PPIs [28, 31]. This suggests that the generation of an aggregate network that combines interaction datasets from different sources, increasing the network coverage and confidence [14, 28, 31, 32], in real-time would aid researchers in producing outputs that are continually based on current information.
Existing PPI generally list pairs of proteins, usually accompanied by references to the literature that documents these interactions with confidence scores. There are some differences between the data stored within these resources in terms of the protein identifier (ID) system used as well as the structure of the dataset files. So, the IMEx consortium [27] was set to harmonize curation efforts in standardizing public interaction datasets through common computational query interfaces (PSICQUIC) [33, 34] using Human proteome organisation proteomics standards-initiative molecular interaction (HUPO PSI-MI) to produce a non-redundant set of PPIs following the minimum information about a molecular interaction experiment (MIMIx) guidelines [35]. Each resource contains the interaction data that can be extracted to perform analyses of human PPI networks. Integrating datasets from existing PPI resources to produce a unified PPI network is still challenging due to the lack of tools which easily generate a unified human PPI network on demand and in real-time for use in the generation and testing of hypotheses. Existing attempts to overcome this challenge have focused on pre-designing databases only accessible via web platforms [36, 37]. These web platforms offer some advantages, e.g. ease of access and being more user-friendly than terminal application interfaces. However, depending on server hosts, a web service may become unavailable at any time. Furthermore, users are required to trust the database developer updates. Here, we introduce an integrated human protein–protein interaction network generator, IHP-PING, a user-friendly and accessible tool, easing integration of PPI datasets from multiple sources into a unified PPI network on-the-fly, which is stored locally for further user applications.
Implementation of the IHP-PING Package
IHP-PING is a portable and expandable package implemented in Python version ≥2.7 [38] and tested on a Linux operating system. It runs using a single command-line terminal on any computer or any operating system running Python and satisfying the IHP-PING requirements (see Supplementary File, Appendix A1: Section 2.1 and Section 2.2). It is freely available and accessible at http://web.cbio.uct.ac.za/ITGOM/post-analysis-tools/ihp-ping-dev/ and https://github.com/gkm-software-dev/post-analysis-tools under the GNU General Public License (GPL:https://www.gnu.org/licenses/gpl-3.0.en.html).
Overview of different online PPI databases
There exist several online databases (see http://www.pathguide.org/) storing different PPI datasets. We classify these databases into three main categories depending on types of PPIs stored: experimentally inferred and/or computationally predicted. These categories are: source experimentally inferred, type computationally predicted and integrated metadatabase. The source experimentally inferred category consists of primary databases capturing experimentally verified PPIs from literatures, including published high-throughput experiments. Some illustrations of source experimentally inferred databases are IntAct, MINT, DIP and BioGRID. The type computationally predicted databases are those storing only computationally derived PPIs. Type computationally predicted databases include human PIPs [39] and Prediction of Interactome (POINT) [40]. The integrated metadatabase category is composed of databases that merge PPIs from source experimentally inferred or computationally predicted PPIs. Generally, databases from this category retrieve experimentally PPIs from IntAct, MINT, DIP, BioGRID, MIPS-MPPI and HPRD (see Table 1 for description). Currently, there exist several metadatabases, e.g. Protein InteraCtion KnowLedgebase (PICKLE 2.0) [36], Molecular Interaction Search Tool (MIST) [37], High-quality INTeractomes (HINT) [41], the Human Integrated Protein–Protein Interaction Reference (HIPPIE) [42], Integrated Interactions Database (IID) [43], Agile Protein Interactomes DataServer (APID) [44], Protein Interaction Network Analysis (PINA) platform [45], the Integrated Interactome System (IIS) [46], the Unified Human Interactome database (UniHI) [47] and STRING, which also includes computationally predicted PPIs. STRING is the largest integrated metadatabase containing computationally predicted PPIs using all known computational models, namely conserved genomic context (neighbourhood, gene fusion events, phylogenetic profile or gene co-occurrence across multiple genomes and sequence homology), interolog, gene co-expression and text mining models. Considering the large number of existing online PPI database, it would be impossible to explore each of them individually, so we refer the interested user to the interoperable link (http://www.pathguide.org/) [48] containing links to most of these online databases, enabling users to retrieve information needed about each database.
Table 1.
The resources used by IHP-PING to retrieve PPI datasets for building an integrated human PPI network. In column 2, Arg stands for argument for each tool as used in the IHP-PING package when running the tool (refer to Appendix A1). Note the number of proteins and that of PPIs are only those contained in the PPI network generated
| Scheme | Arg | Resource | Description | Curation guidelines/Last release date | Number of proteins extracted | Number of PPIs extracted | URL | References |
|---|---|---|---|---|---|---|---|---|
| STRING | stringdb | Search tool for retrieval of interacting genes/proteins | A database of known and predicted protein interactions, with interactions extracted from literature, large-scale experiments, other databases, genomic context, and co-expression. | Automated (01/2019) | 18 558 | 5545 701 | https://string-db.org | [20] |
| BioGRID | biogrid | Biological general repository for interaction datasets | A source of literature curated human protein interactions, with high throughput and literature curated protein interactions for other species such as S. cerevisiae. | Manual 08/2020 | 16 356 | 323 522 | https://thebiogrid.org | [23] |
| DIP | dip | Database of interacting proteins | A collection of protein interactions obtained from literature curation of experimental data. | IMEx and Automated | 2950 | 4661 | https://dip.mbi.ucla.edu/dip | [24] |
| HPRD | hprd | Human protein reference database | A resource specifically focused on manual literature curation of interactions within the human proteome. | Manual (2009) | 5091 | 13 450 | http://www.hprd.org | [25] |
| IntAct | intact | Open source molecular interaction database | A database of protein interactions extracted largely from large-scale experiments, with some interactions obtained through literature curation in collaboration with Swiss-Prot. | IMEx | 16 904 | 251 532 | http://www.ebi.ac.uk/intact | [21] |
| MINT | mint | Molecular interaction database | A database of protein interactions manually curated from literature, with the majority of interactions extracted from large-scale experiments. | IMEx | 7485 | 22 953 | https://mint.bio.uniroma2.it/ | [22] |
| MPPI-MIPS | mips | The Munich information centre for protein sequences mammalian protein–protein interaction database | A database of protein interactions manually curated from literature, with a specific focus on interactions within mammals. | Manual (2010) | 313 | 239 | http://mips.helmholtz-muenchen.de/proj/ppi | [54] |
| UniProt | sequence | Universal Protein knowledge-base | Centralized resource for protein sequences and functional information: A collection of manually and automatically curated protein sequences and annotations, which includes a list of human proteins reviewed by Swiss-Prot and mapping information to convert protein IDs in other databases to the IDs used by UniProt. | Manual and Automated (08/2020) | 18 068 | 2586 676 | http://www.uniprot.org | [31] |
Table 2.
Different data sources and combined or unified scores of each interaction in Figure 9
| Prot-ID | Q92820 | Q9UI17 | P42898 | Q86XF0 | Q6UB35 | P04818 | P31939 | P00374 | P11586 | O95954 | P22102 | P34897 | P34896 | Q05932 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q8NFQ8 | - | - | - | - | - | - | - | - | - | - | - | - | - | Intact stringdb biogrid 0.86688 |
| P35914 | - | - | - | - | - | - | - | - | - | - | - | - | - | Stringdb biogrid 0.72520 |
| Q5T1C6 | - | - | - | - | - | - | - | - | - | - | - | - | - | Intact stringdb biogrid 0.87024 |
| P13473 | - | - | - | - | - | - | - | - | - | - | - | - | - | Intact biogrid 0.84000 |
| P49257 | - | - | - | - | - | - | - | - | - | - | - | - | - | Stringdb 0.80000 |
| Q8IXS6 | - | - | - | - | - | - | - | - | - | - | - | - | - | Intact stringdb 0.69560 |
| Q86X76 | - | - | - | - | - | - | - | - | - | - | - | Intact stringdb 0.69200 | - | Intact stringdb biogrid 0.88352 |
| P30793 | - | - | - | - | - | - | - | Stringdb 0.83000 | - | - | Stringdb 0.76200 | - | - | Stringdb 0.84000 |
| P49914 | - | - | - | - | - | - | - | - | Stringdb 0.97100 | Stringdb 0.94400 | Stringdb 0.96200 | - | Stringdb 0.72300 | Stringdb 0.73500 |
| P41440 | Stringdb 0.90200 | - | Stringdb 0.79100 | - | - | Stringdb 0.71100 | - | - | Stringdb 0.73100 | - | - | - | Stringdb 0.70600 | Stringdb 0.74400 |
| Q9UL12 | - | Sequence stringdb 0.99165 | - | - | - | - | Stringdb 0.72500 | - | - | Stringdb 0.82200 | - | Stringdb 0.97900 | Stringdb 0.97700 | Stringdb 0.76800 |
| Q92820 | - | - | Stringdb 0.71400 | Stringdb 0.90300 | - | Stringdb 0.76800 | - | Stringdb 0.96700 | - | - | - | - | - | Stringdb 0.98600 |
| Q9UI17 | - | - | - | - | - | - | Stringdb 0.71500 | - | - | Stringdb 0.81000 | - | Stringdb 0.81500 | Stringdb 0.74800 | Stringdb 0.75900 |
| P42898 | - | - | - | - | Stringdb 0.82000 | Stringdb 0.99200 | - | Stringdb 0.85000 | Stringdb 0.98900 | Stringdb 0.72900 | Stringdb 0.87000 | Stringdb 0.98100 | Stringdb 0.98800 | Stringdb 0.86800 |
| Q86XF0 | - | - | - | - | Stringdb 0.92900 | Stringdb 0.98700 | Stringdb 0.90400 | Sequence stringdb 0.98658 | Stringdb 0.92900 | Stringdb 0.92000 | Stringdb 0.94400 | Stringdb 0.95400 | Stringdb 0.94600 | Stringdb 0.97100 |
| Q6UB35 | - | - | - | - | - | Stringdb 0.71900 | Stringdb 0.95900 | Stringdb 0.96800 | Sequence stringdb 0.99425 | Stringdb 0.96100 | Stringdb 0.99600 | Stringdb 0.98600 | Stringdb 0.98300 | Stringdb 0.97100 |
| P04818 | - | - | - | - | - | - | Stringdb biogrid 0.94440 | Stringdb 0.99900 | Stringdb 0.98400 | - | Stringdb 0.93300 | Stringdb 0.98500 | Stringd 0.98800 | Stringdb 0.98600 |
| P31939 | - | - | - | - | - | - | - | Stringdb 0.96000 | Stringdb 0.98700 | Stringdb 0.96300 | Stringdb biogrid 0.99960 | Stringdb 0.99300 | Stringdb biogrid 0.99800 | Stringdb 0.74000 |
| P00374 | - | - | - | - | - | - | - | - | Stringdb 0.97900 | Stringdb 0.94400 | Stringdb 0.98800 | Stringdb 0.98100 | Stringdb 0.98300 | Stringdb 0.99000 |
| P11586 | - | - | - | - | - | - | - | - | - | Stringdb 0.96200 | Stringdb 0.99600 | Stringdb 0.98600 | Stringdb 0.99100 | Stringdb 0.97800 |
| O95954 | - | - | - | - | - | - | - | - | - | - | Stringdb 0.95600 | Stringdb 0.97700 | Stringdb 0.95900 | Stringdb 0.79200 |
| P22102 | - | - | - | - | - | - | - | - | - | - | - | Stringdb 0.99700 | Stringdb 0.99700 | Stringdb 0.85900 |
| P34897 | - | - | - | - | - | - | - | - | - | - | - | - | Sequence intact stringdb biogrid 0.99816 | Stringdb 0.79400 |
| P34896 | - | - | - | - | - | - | - | - | - | - | - | - | - | Stringdb 0.85000 |
Heterogeneous PPI dataset sources of the unified human PPI network
IHP-PING retrieves PPI datasets from eight different online resources shown in Figure 1 with complete descriptions in Table 1. It is worth noting that there exist several online PPI databases as indicated in the previous subsection, however, the sample selected by IHP-PING is representative as it exhaustively considers source experimentally inferred PPI databases, used by most of integrated metadatabases, and STRING, the largest integrated metadatabase implementing different models for retrieving computationally predicted PPIs. In addition, we have also included protein sequence information, which consists of protein sequences and InterPro domains [49] retrieved from the UniProt database [50] and used to predict further interactions with scores computed using an information theory-based scheme described in [19]. Each PPI is integrated with its score from its source or estimated for sources with no PPI scores, depending on the source. These interaction scores provide an indication about the confidence of predicted interactions. This is important due to relatively high noise related to high-throughput data or experiments from which interactions are inferred. So, the PPI network produced may contain incorrectly classified interactions, i.e. may fail to detect interactions (false negatives) or wrongly identify some other interactions (false positives), which is technology-dependent. The likelihood of incorrectly classifying an interaction may be minimized computationally by (1) using a data integration model, combining information from multiple interacting data sources into one unified network, and (2) applying a strict interaction reliability or confidence score cut-off. These techniques are expected to significantly reduce the false negative and positive rate of the network produced, leading to a PPI network of high confidence interactions with an increased coverage [14, 28, 31, 32]. With the advances in computational models and big data analytics, these computational methods enable the prediction of relatively high accurate PPIs and the subsequent validation of experimental results [51].
Figure 1 .
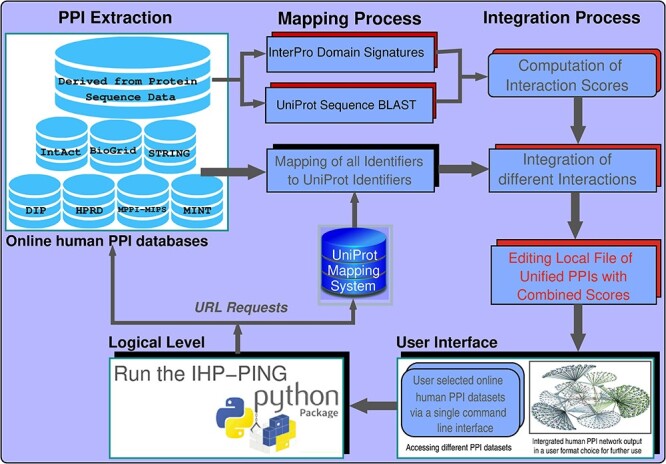
Overall workflow of the IHP-PING tool. The scheme goes through three main steps from user input to a generated human PPI network: Input is parsed via a simple single command-line terminal, then the selected human PPI datasets are retrieved and network generated in tsv, csv or csv2 format.
PPI confidence score estimation
PPI datasets are retrieved sequentially, stored in memory with each interaction being extracted from the downloaded files, cleaning the memory space for each source, once PPI extraction process is done. IHP-PING stores these interactions in the output file alongside a score for each interaction, which is calculated differently depending on the dataset from which the interaction was obtained. In the case of MINT and STRING, there is an interaction score within the dataset which is extracted by IHP-PING and entered into the output directly. The HPRD dataset does not contain interaction scores but, for each interaction, it lists the publications and evidence sources that support the interaction, which are then used to estimate the reliability or confidence score. Given proteins p and q, this score is calculated as follows:
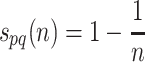 |
where n is the total number of confidence sources and publications. It is worth noting that, if a PPI has been identified by one source, a random score of 0.5 is assigned. This means that if an interaction is confirmed only by one source, this interaction may be true or false, in which case, a reliability score is simply the probability of this interaction being true. From the formula, it is clear that this confidence score increases as the number of sources increases, as expected.
BioGRID, DIP, IntAct and MPPI-MIPS datasets do not contain interaction scores and thus a default reliability scores at the middle range (medium confidence): 0.7 for DIP and 0.6 for others is assigned by IHP-PING, which the authors set based on the confidence level or the trustworthiness of the dataset under consideration provided relatively high noise related to high-throughput data or experiments which has shown to produce both high false positive and false negative PPIs [51]. The slight difference of the DIP dataset score was decided considering the internal curation strategy as DIP also considers the use of computational or automated curation approaches on top of the manual curation (https://dip.mbi.ucla.edu/dip/page?id=about). Users should be aware that the estimation of these scores is based on a relative perception and only reflects the IHP-PING developers’ subjective beliefs. So, in case a user feels that the reasoning fundamentally misjudged or underestimated these scores, he can easily adjust them from the output file as it clearly separates different PPI sources and related scores per column (see the IHP-PING parameter inputs and result outputs section below).
The interactions predicted from protein sequences receive a score according to sequence similarity and shared protein signatures computed using an information theory-based scheme described in [19], calculating the cumulative standard normal distribution function, 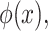 as:
as:
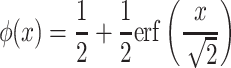 |
with 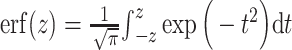 the Gauss error function implemented in the Python math library. This speeds up sequence-predicted interaction score computation and avoids the use of any other specific Python libraries.
the Gauss error function implemented in the Python math library. This speeds up sequence-predicted interaction score computation and avoids the use of any other specific Python libraries.
PPI combined score, harmonizing and integrating different PPI datasets
After calculating the reliability or confidence score for each functional association protein pair, the combined confidence score 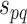 for interacting proteins p and q, integrating confidence scores in a unified PPI network needs to be computed. Of note, this reliability or confidence score of an interaction between proteins
for interacting proteins p and q, integrating confidence scores in a unified PPI network needs to be computed. Of note, this reliability or confidence score of an interaction between proteins 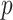 and
and 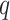 measures our confidence level in this interaction, which is the probability or likelihood that this interaction occurred. So, let us assume that
measures our confidence level in this interaction, which is the probability or likelihood that this interaction occurred. So, let us assume that 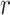 different sources were used to retrieve this interaction and let
different sources were used to retrieve this interaction and let 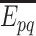 be an event that interaction between proteins
be an event that interaction between proteins 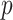 and
and 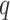 could not be retrieved from any of these
could not be retrieved from any of these 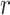 sources, that is:
sources, that is:
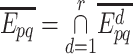 |
where 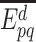 is the event that this interaction could not be retrieved from the source
is the event that this interaction could not be retrieved from the source 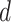 . As these sources are assumed to be independent, the probability,
. As these sources are assumed to be independent, the probability, 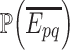 , of the event
, of the event 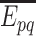 is then given by
is then given by
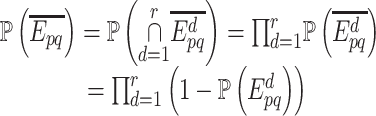 |
with 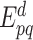 the contrary event of
the contrary event of 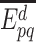 , i.e. the event that the interaction between
, i.e. the event that the interaction between 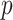 and
and 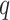 is retrieved from the source
is retrieved from the source 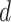 and thus
and thus 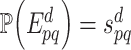 with
with 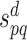 the confidence or reliability score of an interaction between
the confidence or reliability score of an interaction between 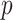 and
and 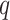 retrieved from the source
retrieved from the source 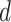 . Thus, the combined confidence score
. Thus, the combined confidence score 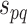 for interacting proteins
for interacting proteins 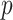 and
and 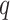 , which is the probability of the event,
, which is the probability of the event, 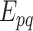 , indicating that the interaction between
, indicating that the interaction between 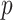 and
and 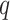 is retrieved from at least one of the sources, contrary to
is retrieved from at least one of the sources, contrary to 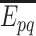 , is given by
, is given by
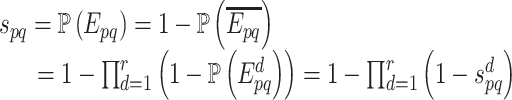 |
Therefore, the combined confidence score 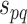 for interacting proteins p and q is given by the following formula [1, 10]:
for interacting proteins p and q is given by the following formula [1, 10]:
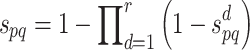 |
noting that r is the total number of PPI data sources and 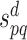 is the confidence score of an interaction between p and q retrieved from the PPI data source d. Thus, for minimizing the likelihood of false positive interactions, a reliability cut-off can be applied, which may lead to a highly reliable PPI network. Note that IHP-PING includes computationally predicted interactions (e.g. from protein shared domains and sequences) with interaction reliability scores computed on-the-fly, i.e. when PPIs are being inferred, in contrast to the STRING scheme, which also integrates computationally predicted PPIs by pre-computing PPI reliability scores.
is the confidence score of an interaction between p and q retrieved from the PPI data source d. Thus, for minimizing the likelihood of false positive interactions, a reliability cut-off can be applied, which may lead to a highly reliable PPI network. Note that IHP-PING includes computationally predicted interactions (e.g. from protein shared domains and sequences) with interaction reliability scores computed on-the-fly, i.e. when PPIs are being inferred, in contrast to the STRING scheme, which also integrates computationally predicted PPIs by pre-computing PPI reliability scores.
Naturally, there are some issues when integrating interactions from multiple sources as datasets often use different ID systems. For example, STRING [20] uses a unique ID system while DIP [24] includes its own protein ID alongside the corresponding UniProt protein ID [50]. Different protein identifiers are mapped to reviewed proteins only from Swiss-Prot under the non-redundant UniProt identifier system for harmonization before integration. Once all IDs have been mapped successfully, the interactions from each database are integrated into a single data frame. The final output of IHP-PING is a local file stored in the desired directory which contains all PPI information extracted from the datasets, namely the two protein IDs and the score from each source with a combined score in the last column for each interaction. Under the IHP-PING package, the interacting proteins in the output file may be in UniProt or gene name ID systems, depending on the user choice (see Supplementary File, Appendix A1: Section 2.4). This file may then be used to perform downstream PPI network analysis.
IHP-PING parameter inputs and result outputs
In order to use IHP-PING, the user invokes a specific module through Python alongside command arguments (see Supplementary File for details, Appendix A1: Section 2.5 and Section 2.6 for details). The main argument to be provided is the list of datasets (see Table 1) to be incorporated into the final unified network, with other parameters changing the operation of the program according to user preferences, such as the format of the output file, which can be tab, comma or column separated value (tsv, csv or csv2 format) as described in Figure 2. Each requested database is downloaded from specified Uniform Resource Locations (URLs) and there are different types of resources which may be integrated by IHP-PING, including PPI databases and PPIs predicted from protein sequences and domain signatures.
Figure 2 .
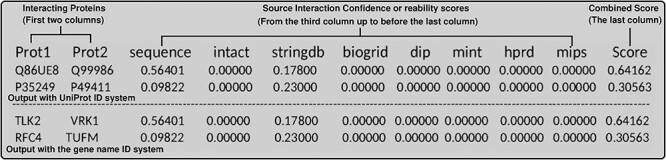
The IHP-PING output structure. The first two columns are the two interacting proteins, following columns are interaction source confidence or reliability scores and the last column is the interaction combined score. Space between columns can be tabs, commas or semicolons depending on whether the user chooses tsv, csv or csv2.
PPI datasets are retrieved sequentially, stored in memory with each interaction being extracted from the downloaded files, cleaning the memory space for each source, once the PPI extraction process is done. IHP-PING stores these interactions in the output file alongside a score for each interaction, which is calculated differently depending on the dataset from which the interaction was obtained, as described in previous sections. Upon running the IHP-PING package, the output file named after the requested databases and containing all PPIs retrieved is created within the folder of the package by default, if no specific folder which should contain the output file has been provided. This output file format depends on the user specifications, as indicated above, with tsv being the default format.
Building a unified human PPI network
A unified human PPI network has been built within the IHP-PING package without any parameter for the protein UniProt identifier (ID) system within a tsv output file format (see Supplementary, Appendix A1: Section 2.5 for details File for details). This network is generated in three steps highlighted in Figure 1, synchronizing different protein IDs to reviewed proteins from Swiss-Prot under the non-redundant UniProt ID system for harmonization before integration. Each PPI is integrated with its score from its source or estimated for sources with no PPI scores, as described previously. IHP-PING retrieved PPIs from all the sources and generated an output file in a tabular (tsv) format with 11 columns with each row representing a unique PPI (see Figure 2). The first two fields contain the IDs of the two proteins involved in the interaction, with columns 3 through 10 showing the scores for the interaction from each source. The last value in the row contains the combined score of the interaction. The total number of interactions obtained from each source is shown in Figure 3, distributed in low, medium and high confidence levels, with score less than 0.3, ranging between 0.3 and 0.7, and greater than 0.7, respectively, in Figure 4. These thresholds are shown in Figure 5 and have been analytically and statistically inferred and are lower and upper tail inflection points of the kernel density distribution of an interaction score sample. In fact, Figures 3 and 4 are contextual and time specific as most of PPI databases are regularly updated, i.e. these figures evolve with any new release from IHP-PING PPI data sources.
Figure 3 .
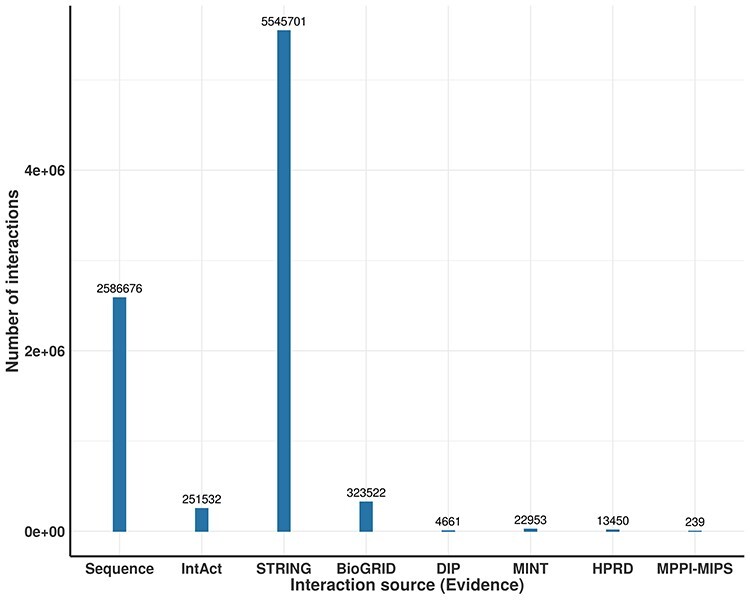
Distribution of interactions obtained from different resources contributing to a unified human PPI network—All interactions per source.
Figure 4 .
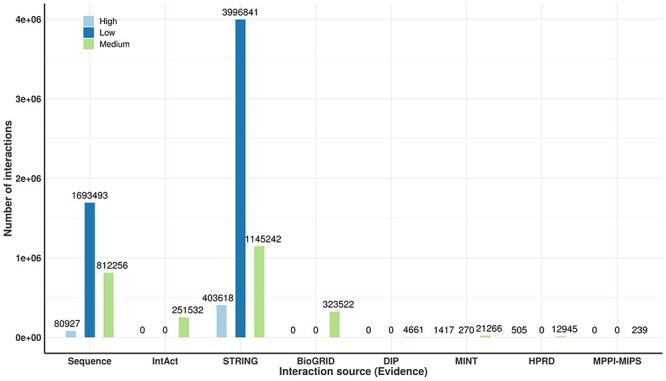
Distribution of interactions obtained from different resources contributing to a unified human PPI network—All interactions per source in high-low-medium confidence level interaction frequencies.
Figure 5 .
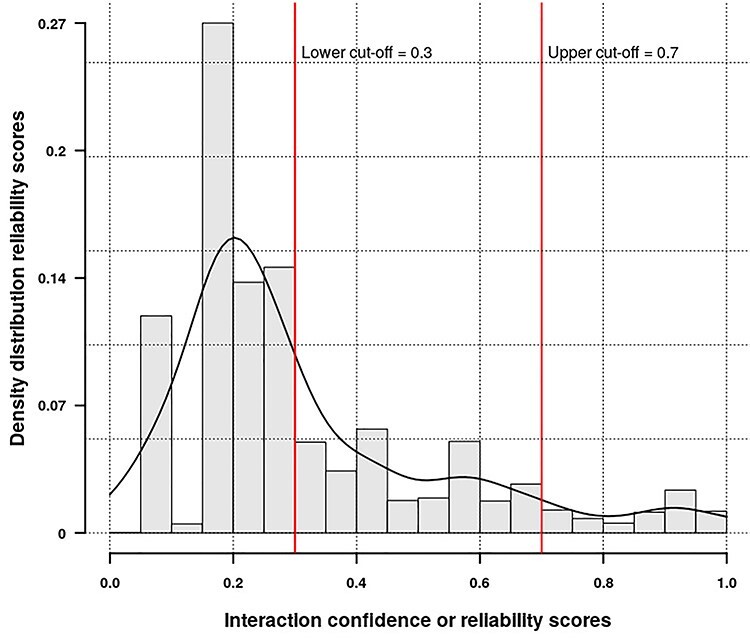
Highlighting lower and upper score thresholds for low-medium-high confidence levels.
The human PPI network generated in this instance contained 8017 087 interactions connecting 19 957 proteins out of 20 366 reviewed human proteins. The number of interacting proteins being less than that in the reviewed human proteome suggests that these PPI datasets are still incomplete despite the high number of PPIs predicted by computational approaches from STRING and protein sequence data (see Figure 3), likely at the cost of more noise. Predicted PPIs contain a total of 5276 025 interactions with low confidence, 2045 319 with medium confidence and 695 743 with high confidence level. In analysis of these interactions, 51 466 interactions with low confidence (interaction score less than 0.3) were predicted by at least two different datasets. Distributions of PPIs shared between different types of PPIs, experimentally inferred (BioGRID, IntAct, MINT, DIP, HPRD and MPPI-MIPS) and computationally inferred (STRING and those predicted from protein sequence datasets), are shown in the Venn diagrams in Figure 6. These Venn diagram results suggest that there is a relatively large number of PPIs shared between experimentally and computationally inferred PPIs, for example, PPIs from MINT are shared by all IHP-PING dataset sources. As pointed out previously, biases may exist in the PPI network generated due to relatively high noise related to high-throughput data or experiments from which interactions are inferred. In the absence of gold standard PPIs, the data integration model and the application of a strict interaction reliability or confidence score cut-off are computationally explored to reduce the impact of these biases, leading to a PPI network of high confidence interactions with an increased coverage [14, 28, 31, 32].
Figure 6 .
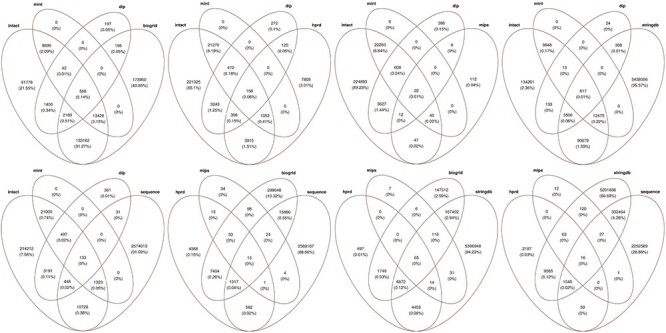
Venn diagrams showing the general distributions of shared PPIs between experimentally derived PPIs from online databases following IMEx curation guidelines (IntAct, MINT and DIP), as well as BioGRID, HPRD and MPPI-MIPS, and computationally predicted PPIs from STRING and Sequence.
Here, we used a high confidence human PPI network, extracted from the unified network generated, considering only interactions with score >0.7 or predicted by two different sources, to check general topological properties of the biological networks, namely power-law and small-world properties. This network consisted of 960 514 interactions linking 19 345 proteins. The distribution of degree plotted is shown in Figure 7 and distribution of shortest path lengths within the network in Figure 8. The power exponent, 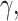 was estimated to 1.38942 with P-value <0.0001, implying that the network obtained fits perfectly the power-law property [14, 52]. Furthermore, analysing in terms of the distribution of path lengths within the network shows that with average path length of 2.92607 ≈ 3. These results indicate that the human PPI network conforms to the properties of biological networks [53, 54]. Finally, for illustration we used igraph and tcltk R software libraries to plot Folylpolyglutamate synthase, mitochondrial (Q05932) protein in human predicted to be targeted by the Mycobacterium tuberculosis pathogen [1] and the output is shown in Figure 9.
was estimated to 1.38942 with P-value <0.0001, implying that the network obtained fits perfectly the power-law property [14, 52]. Furthermore, analysing in terms of the distribution of path lengths within the network shows that with average path length of 2.92607 ≈ 3. These results indicate that the human PPI network conforms to the properties of biological networks [53, 54]. Finally, for illustration we used igraph and tcltk R software libraries to plot Folylpolyglutamate synthase, mitochondrial (Q05932) protein in human predicted to be targeted by the Mycobacterium tuberculosis pathogen [1] and the output is shown in Figure 9.
Figure 7 .
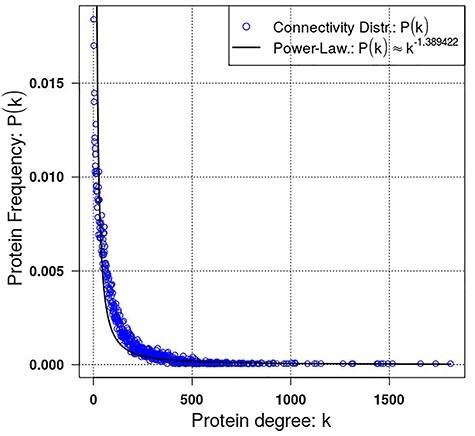
The unified human PPI network topological property. Power-law property visualizing protein degree against connections frequency in the network.
Figure 8 .
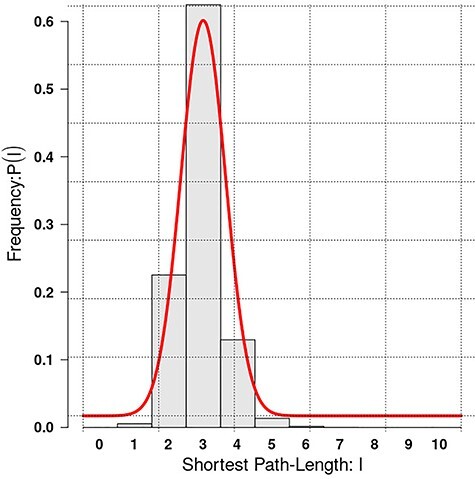
The unified human PPI network topological property. Small-world property—the distribution of shortest path lengths within the interaction network.
Figure 9 .
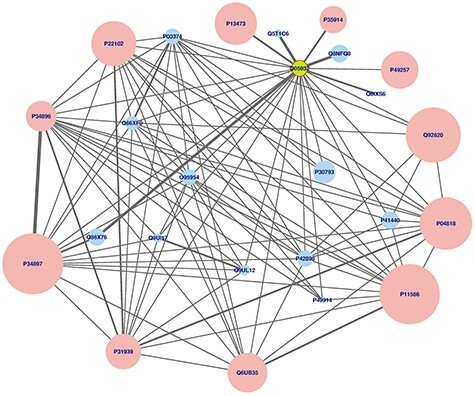
Using igraph and tcltk R software libraries to plot the Folylpolyglutamate synthase, mitochondrial (Q05932) protein for illustration. Node or protein size is now proportional to the its degree in the protein–protein interaction network and each link is proportional to the number of its data sources provided in Table 2, together with final or unified confidence scores.
Discussion
Other existing integrated human PPI network builders
Existing integrated human PPI network builders have attempted to generate new databases which integrate PPI information from some or all existing online PPI sources highlighted in Table 1. Such attempts include PINA, IIS, UniHI, PICKLE 2.0, HINT, HIPPIE, APID and MIST. As for IHP-PING, these tools retrieve experimentally inferred PPI datasets from commonly used databases, including IntAct, MINT, BioGRID, DIP, HPRD and MPPI-MIPS. It is worth noting that some of these databases keep up with updating their datasets, which are manually curated following IMEx guidelines as full members of the consortium [27], e.g. IntAct, MINT, DIP. BioGRID, an observer member of the IMEx consortium, also regularly updates its datasets, however, some databases, such as HPRD [25] and MPPI-MIPS [55], have not updated their datasets for almost 10 years now (see Table 1). This should be taken into account when retrieving PPI datasets from these sources.
The common thread linking most of these human PPI builders to integrated PPI network generation and analysis is firstly, the access via a web interface and, secondly, the existence of prebuilt databases which are reportedly updated regularly. Though these approaches have some advantages, they also present some limitations to the users of these services. Web platforms are considered to be easily accessible and more user-friendly when compared to terminal interfaces or application software that requires an initial installation. Web platforms allow for users to access the platform or service at any device with an internet connection. The limitations of a web platform include the requirement of a stable and constant internet connection and the resource is dependent on the server host. At any time, the web resource may become unavailable to the user through the actions of the web hosting service.
Issues related to existing builders and the IHP-PING solution
With regards to pre-constructed databases, the benefit of their use is that queries are fast as no dataset integration steps need to be processed; the data are immediately available for use. The cost to this increased speed is the loss of user customization as the user is not able to specify which datasets they would like to be included in the database, though UniHI [47] attempts to mitigate this by informing users of the original resource from which an interaction was extracted. By using pre-constructed databases, the user is also required to trust the curation process of the database architects.
As an attempt to avoid pre-constructed databases, and the issues resultant from PPI datasets being regularly updated, we present the IHP-PING tool. IHP-PING is a software tool which generates a human PPI network from freely available online resources in real-time. This tool is user-friendly and accessible to biologists so that it may be used without extensive training in software applications. This tool downloads and integrates the PPI data of multiple sources to generate a PPI network which is stored locally for the user. While an internet is required for the download of the datasets specified by the user, any downstream analysis of the network produced can be run locally without an internet connection.
Firstly, while IHP-PING requires a stable internet connection at runtime, it is not implemented as a web platform. Web-based tools do have certain advantages such as ease of access but, in the case of large-scale protein interaction networks, a network stored locally can be analysed on local hardware, reducing the user’s reliance on server hardware of the platform and allowing the user to make use of any available cluster resources. The network produced by IHP-PING is stored locally and can integrate different datasets into the output network according to the user’s preference. Secondly, the modular structure of the code allows for the extension of IHP-PING to support additional datasets. Beyond supporting new PPI databases as they become hosted online, any novel method that predicts protein interactions could theoretically be incorporated into the software tool presented here given that a protein pair with an interaction score can be obtained. In the opposite fashion, datasets that become deprecated can be easily excluded by the user from the network.
IHP-PING perspective, evolution and improvements
Looking at the network output shown in Figure 3, it is clear that the majority of interactions extracted by IHP-PING from PPI datasets come from STRING and are predicted from sequence data. This is due to the additional computational approaches used to predict some of PPIs, which is not the case for other datasets. We believe that the number of interactions that are curated from literature will increase over time as more experiments are performed and published. IHP-PING is uniquely equipped to retrieve the most recent data from its supported resources, thus once a supported PPI database is updated, subsequent runs of IHP-PING will generate a new network based on the updated information. Therefore, the number of interactions obtained by the software is likely to increase over time.
Finally, advances in high-throughput technology have enabled the generation of tissue-specific gene expression information and the inclusion of this information may improve the coverage of the network produced and reduce false negative PPIs. In its current form, IHP-PING does not directly include gene expression information, even though STRING supports gene expression information and retrieving PPIs from STRING implies that this information is implicitly included. There is a need for IHP-PING to support explicitly gene expression information as it is the case for sequence data—this is an area of potential future expansion. In addition, there is a need for a visualization technique, like the Cytoscape software [56] or igraph and tcltk R software libraries, to support drawing of a graph or network from edge and node data, and which is compatible with the IHP-PING output, supporting the flexibility of the software. This is also an area of future work, where we will assess the dynamic python-networkx and python-matplotlib libraries to implement a graphical user interface (GUI) to support a systematic network visualization.
Conclusion
Considering the rapid expansion of the bioinformatics field with highly dynamic datasets, tools used to manipulate these data should be flexible and extensible to adequately manage the regular updates. IHP-PING, a Python adaptable and easy-to-use application, presents such a tool for generating integrated PPI networks, with clear benefits when compared to similar solutions. IHP-PING enables the generation of an aggregate human PPI network in real-time that is continually based on current information. These PPIs are experimentally and computationally inferred and scored based on the nature of the datasets and technology used to derive these datasets. Unlike the existing web platform based application, in which case a stable and constant internet connection is required, IHP-PING requires the internet connection only for downloading the datasets specified by the user to generate a PPI network which is stored locally for the user. Any downstream analysis of the network produced can be run locally without an internet connection. Moreover, the IHP-PING package may be easily adapted to support new PPI databases as they become hosted online as well as any novel method that predicts protein interactions and estimates source interaction scores. Additionally, datasets that become deprecated are automatically removed from the network, producing a unified PPI network that is up to date.
Key Points
Harmonizing and integrating human protein–protein interactions (PPIs) experimentally and computationally inferred from multiple heterogeneous sources into a unified human PPI network.
Providing an easy-to-use, portable and flexible Python package, IHP-PING, which enables the generation of human PPI network in real-time with available online datasets.
Enabling estimating confidence scores to produce a unified human PPI network with high confidence and coverage, and producing a network output that is continually based on current information.
Enabling the user to choose the output format and protein identifier system to ease the use of the PPI network generated in some potential further applications.
Supplementary Material
Gaston K. Mazandu is an honorary senior member of the Computational Biology Division at UCT and an associate researcher at the African Institute for Mathematical Sciences (AIMS), and holds a senior lecturer position in the Division of Human Genetics, Department of Pathology, University of Cape Town (UCT).
Christopher Hooper is an MBChB student, UCT.
Kenneth Opap is a PhD candidate in Bioinformatics, the Computational Biology Division, UCT.
Funmilayo Makinde is a PhD candidate in Bioinformatics, Computational Biology Division at UCT in collaboration with AIMS.
Victoria Nembaware is a senior staff member, the Division of Human Genetics, Department of Pathology and the coordinator of mobile genomics in Africa (mGenAfrica) project at UCT.
Nicholas E. Thomford is a researcher, Division of Human Genetics, Department of Pathology, UCT and lecturer at the University of Cape Coast, School of Medical Science.
Emile R. Chimusa is a professor in Bioinformatics, Division of Human Genetics, Department of Pathology, UCT.
Ambroise Wonkam is a professor in Medical Genetics, Division of Human Genetics, Department of Pathology and senior specialist medical geneticist, HPCSA MP0686980, UCT.
Nicola J. Mulder is a professor and Head of the Computational Biology Division, UCT.
Contributor Information
Gaston K Mazandu, Computational Biology Division, Department of Integrative Biomedical Sciences, IDM, CIDRI-Africa WT Centre, University of Cape Town, Health Sciences Campus. Anzio Rd, Observatory, 7925, South Africa; African Institute for Mathematical Sciences, 5-7 Melrose Road, Muizenberg, 7945, Cape Town, South Africa; Division of Human Genetics, Department of Pathology, University of Cape Town, Health Sciences Campus, Anzio Rd, Observatory, 7925, South Africa.
Christopher Hooper, Computational Biology Division, Department of Integrative Biomedical Sciences, IDM, CIDRI-Africa WT Centre, University of Cape Town, Health Sciences Campus. Anzio Rd, Observatory, 7925, South Africa.
Kenneth Opap, Computational Biology Division, Department of Integrative Biomedical Sciences, IDM, CIDRI-Africa WT Centre, University of Cape Town, Health Sciences Campus. Anzio Rd, Observatory, 7925, South Africa.
Funmilayo Makinde, Computational Biology Division, Department of Integrative Biomedical Sciences, IDM, CIDRI-Africa WT Centre, University of Cape Town, Health Sciences Campus. Anzio Rd, Observatory, 7925, South Africa; African Institute for Mathematical Sciences, 5-7 Melrose Road, Muizenberg, 7945, Cape Town, South Africa.
Victoria Nembaware, Division of Human Genetics, Department of Pathology, University of Cape Town, Health Sciences Campus, Anzio Rd, Observatory, 7925, South Africa.
Nicholas E Thomford, Division of Human Genetics, Department of Pathology, University of Cape Town, Health Sciences Campus, Anzio Rd, Observatory, 7925, South Africa; School of Medical Sciences, University of Cape Coast, PMB, Cape Coast, Ghana.
Emile R Chimusa, Division of Human Genetics, Department of Pathology, University of Cape Town, Health Sciences Campus, Anzio Rd, Observatory, 7925, South Africa.
Ambroise Wonkam, Division of Human Genetics, Department of Pathology, University of Cape Town, Health Sciences Campus, Anzio Rd, Observatory, 7925, South Africa.
Nicola J Mulder, Computational Biology Division, Department of Integrative Biomedical Sciences, IDM, CIDRI-Africa WT Centre, University of Cape Town, Health Sciences Campus. Anzio Rd, Observatory, 7925, South Africa.
Acknowledgements
We would like to acknowledge all authors for their contributions in writing the paper, and developing the Python package. We extend our acknowledgements to everyone involved with free software, from the core developers to those who contributed to the documentation as well as those providing computational facilities, specifically the Centre for high performance computing (CHPC), South Africa (https://www.chpc.ac.za).
Funding
Some of the authors are supported partially by the South African Medical Research Council (SAMRC) through its Division of Research Capacity Development under the National Medical Students Research Training Programme. This study is supported by the National Institutes of Health (NIH), USA, Common Fund under SADaCC (1U01HG007459-1) and H3ABioNet (U24HG006941). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
References
- 1. Mazandu GK, Chimusa ER, Rutherford K, et al. Large-scale data-driven integrative framework for extracting essential targets and processes from disease-associated gene data sets. Brief Bioinform 2018;19(6):1141–52. [DOI] [PubMed] [Google Scholar]
- 2. Cusick ME, Klitgord N, Vidal M, et al. Interactome: gateway into systems biology. Hum Mol Genet 2005;14(2):R171–81. [DOI] [PubMed] [Google Scholar]
- 3. Mazandu GK, Mulder NJ. Using the underlying biological organization of the mycobacterium tuberculosis functional network for protein function prediction. Infect Genet Evol 2011;12(5):922–32. [DOI] [PubMed] [Google Scholar]
- 4. Mazandu GK, Opap K, Mulder NJ. Contribution of microarray data to the advancement of knowledge on the mycobacterium tuberculosis interactome: use of the random partial least squares approach. Infect Genet Evol 2011;11(4):725–33. [DOI] [PubMed] [Google Scholar]
- 5. Mazandu GK, Kyomugisha I, Geza E, et al. Designing data-driven learning algorithms: A necessity to ensure effective post-genomic medicine and biomedical research. In: Artificial Intelligence - Applications in Medicine and Biology. 5 Princes Gate Court, London, UK: IntechOpen Publisher, 2019, 3–18. [Google Scholar]
- 6. Li X, Li W, Zeng M, et al. Network-based methods for predicting essential genes or proteins: a survey. Brief Bioinform 2020;21(2):566–83. [DOI] [PubMed] [Google Scholar]
- 7. Wu Z, Liao Q, Liu B. A comprehensive review and evaluation of computational methods for identifying protein complexes from protein-protein interaction networks. Brief Bioinform 2020;21(5):1531–48. [DOI] [PubMed] [Google Scholar]
- 8. Perbal L. The case of the gene: Postgenomics between modernity and postmodernity. EMBO Rep 2015;16:777–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Proc Natl Acad Sci 1941;27:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Akinola RO, Mazandu GK, Mulder NJ. A quantitative approach to analyzing genome reductive evolution using protein–protein interaction networks: a case study of mycobacterium leprae. Front Genet 2016;7:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mulder NJ, Akinola RO, Mazandu GK, et al. Using biological networks to improve our understanding of infectious diseases. Comput Struct Biotechnol J 2014;11(18):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rapanoel HA, Mazandu GK, Mulder NJ. Predicting and analyzing interactions between mycobacterium tuberculosis and its human host. PLoS One 2013;8(7):e67472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mazandu GK, Mulder NJ. Function prediction and analysis of mycobacterium tuberculosis hypothetical proteins. Int J Mol Sci 2012;13(6):7283–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mazandu GK, Mulder NJ. Generation and analysis of large-scale data-driven mycobacterium tuberculosis functional networks for drug target identification. Advances in Bioinformatics 2011;2011:801478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chimusa ER, Dalvie S, Dandara C, et al. Post genome-wide association analysis: dissecting computational pathway/network-based approaches. Brief Bioinform 2019;20(2):690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stelzl U, Worm U, Lalowski M, et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 2005;122:957–68. [DOI] [PubMed] [Google Scholar]
- 17. Cusick ME, Yu H, Smolyar A, et al. Literature-curated protein interaction datasets. Nat Methods 2009;6(1):39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He M, Wang Y, Li W. PPI finder: a mining tool for human protein-protein interactions. PLoS One 2009;4(2):e4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mazandu GK, Mulder NJ. Scoring protein relationships in functional interaction networks predicted from sequence data. PLoS One 2011;6(4):e18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Orchard S, Ammari M, Aranda B, et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 2014;42(D1):D358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Licata L, Briganti L, Peluso D, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 2012;40:D857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oughtred R, Stark C, Breitkreutz B, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res 2019;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salwinski L, Miller CS, Smith AJ, et al. The database of interacting proteins: 2004 update. Nucleic Acids Res 2004;32:D449–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keshava PTS, Goel R, Kandasamy K, et al. Human protein reference database–2009 update. Nucleic Acids Res 2009;37:D767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mewes HW, Ruepp A, Theis F, et al. MIPS: curated databases and comprehensive secondary data resources in 2010. Nucleic Acids Res 2006;39:D220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orchard S, Kerrien S, Abbani S, et al. Protein interaction data curation: the international molecular exchange (IMEx) consortium. Nat Methods 2012;9(4):345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stojmirović A, Yu Y-K. ppiTrim: constructing non-redundant and up-to-date interactomes. Database 2011;2011:bar036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skinnider MA, Stacey RG, Foster LJ. Genomic data integration systematically biases interactome mapping. PLoS Comput Biol 2018;14(10):e1006474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Las Rivas J, Fontanillo C. Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput Biol 2010;6(6):e1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li T, Wernersson R, Hansen RB, et al. A scored human protein-protein interaction network to catalyze genomic interpretation. Nat Methods 2017;14(1):61–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Safari-Alighiarloo N, Taghizadeh M, Rezaei-Tavirani M. Protein-protein interaction databases: an overall view on interactome organization. International journal of analytical, pharmaceutical and biomedical. Sciences 2015;4(1):15–23. [Google Scholar]
- 33. Aranda B, Blankenburg H, Kerrien S, et al. PSICQUIC and PSISCORE: accessing and scoring molecular interactions. Nat Methods 2011;8:528–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. del-Toro N, Dumousseau M, Orchard S, et al. New reference implementation of the PSICQUIC web service. Nucleic Acids Res 2013;41(Web Server issue):W601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Orchard S, Salwinski L, Kerrien S, et al. The minimum information required for reporting a molecular interaction experiment (MIMIx). Nat Biotechnol 2007;25:894–8. [DOI] [PubMed] [Google Scholar]
- 36. Gioutlakis A, Klapa MI, Moschonas NK. PICKLE 2.0: a human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS One 2017;12(10):e0186039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu Y, Vinayagam A, Nand A, et al. Molecular interaction search tool (MIST): an integrated resource for mining gene and protein interaction data. Nucleic Acids Res 2018;46(D1):D567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Python Software Foundation . Python Language Reference, version 2.7. Available athttp://www.python.org.
- 39. McDowall MD, Scott MS, Barton GJ. PIPs: human protein-protein interactions prediction database. Nucleic Acids Res 2009;37:D651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang TW, Tien AC, Huang WS, et al. POINT: a database for the prediction of protein-protein interactions based on the orthologous interactome. Bioinformatics 2004;20(17):3273–6. [DOI] [PubMed] [Google Scholar]
- 41. Das J, Yu H. HINT: high-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol 2012;6(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alanis-Lobato G, Andrade-Navarro MA, Schaefer MH. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res 2016;45:D408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kotlyar M, Pastrello C, Sheahan N, et al. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res 2016;44:D536–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Alonso-López D, Campos-Laborie FJ, Gutiérrez MA, et al. APID database: redefining protein–protein interaction experimental evidences and binary interactomes. Database 2019;2019:baz005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cowley MJ, Pinese M, Kassahn, et al. PINA v2.0: mining interactome modules. Nucleic Acids Res 2012;40:D862–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carazzolle MF, Carvalho LM, Slepicka HH, et al. IIS – integrated interactome system: a web-based platform for the annotation, analysis and visualization of protein-metabolite-gene-drug interactions by integrating a variety of data sources and tools. PLoS One 2014;9(6):e100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kalathur RK, Pinto JP, Hernández-Prieto MA, et al. UniHI 7: an enhanced database for retrieval and interactive analysis of human molecular interaction networks. Nucleic Acids Res 2014;42:D408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bader GD, Cary MP, Sander C. Pathguide: a pathway resource list. Nucleic Acids Res 2006;34(suppl 1):D504–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mitchell AL, Attwood TK, Babbitt PC, et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res 2019;47:D351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. UniProt Consortium . UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zahiri J, Bozorgmehr JH, Masoudi-Nejad A. Computational prediction of protein-protein interaction networks: algorithms and resources. Curr Genomics 2013;14:397–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Almaas E, Barabási A-L. Power laws in biological networks. In: Koonin EV, Wolf YI, Karev GP (eds). Power Laws, Scale-Free Networks and Genome Biology (Molecular Biology Intelligence Unit). Austin, Texas, USA: Springer, 2006, 1–11. [Google Scholar]
- 53. Jeong H, Mason SP, Barabási A-L, et al. Lethality and centrality in protein networks. Nature 2001;411:41–2. [DOI] [PubMed] [Google Scholar]
- 54. Jeong H, Tombor B, Albert R, et al. The large-scale organization of metabolic networks. Nature 2000;407:651–4. [DOI] [PubMed] [Google Scholar]
- 55. Mewes HW, Ruepp A, Theis F, et al. MIPS: curated databases and comprehensive secondary data resources in 2010. Nucleic Acids Res 2011;39(Database issue):D220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Otasek D, Morris JH, Bouças J, et al. Cytoscape automation: empowering workflow-based network analysis. Genome Biol 2019;20:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.