

Abstract
Rationale:
While thrombin is the key protease in thrombus formation, other coagulation proteases, such as fXa or activated protein C (aPC), independently modulate intracellular signaling via partially distinct receptors.
Objectives:
To study the differential effects of fXa or fIIa inhibition on gene expression and inflammation in myocardial ischemia-reperfusion injury (IRI).
Methods and results:
Mice were treated with a direct fIIa inhibitor (fIIai) or direct fXa inhibitor (fXai) at doses that induced comparable anticoagulant effects ex vivo and in vivo (tail bleeding assay and FeCl3-induced thrombosis). Myocardial IRI was induced via LAD ligation. We determined infarct size and in vivo aPC generation, analyzed gene expression by RNAseq, and performed immunoblotting and ELISA. The signaling-only 3K3A-aPC variant and inhibitory antibodies that blocked all or only the anticoagulant function of aPC were used to determine the role of aPC. Doses of fIIai and fXai that induced comparable anticoagulant effects resulted in a comparable reduction in infarct size. However, unbiased gene expression analyses revealed marked differences, including pathways related to sterile inflammation and inflammasome regulation. fXai but not fIIai inhibited sterile inflammation by reducing the expression of proinflammatory cytokines (IL-1β, IL-6, and TNFα) as well as NF-κB and inflammasome activation. This anti-inflammatory effect was associated with reduced myocardial fibrosis 28 days post myocardial IRI. Mechanistically, in vivo aPC generation was higher with fXai than with fIIai. Inhibition of the anticoagulant and signaling properties of aPC abolished the anti-inflammatory effect associated with fXai, while inhibiting only the anticoagulant function of aPC had no effect. Combining 3K3A-aPC with fIIai reduced the inflammatory response, mimicking the fXai-associated effect.
Conclusion:
We showed that specific inhibition of coagulation via DOACs had differential effects on gene expression and inflammation, despite comparable anticoagulant effects and infarct sizes. Targeting individual coagulation proteases induces specific cellular responses unrelated to their anticoagulant effect.
Keywords: Animal Models of Human Disease, Basic Science Research, Inflammation, Ischemia, Myocardial Biology, Coagulation, thrombosis, fXa, DOACs, activated protein C, myocardial infarction, inflammasome, ischemia/reperfusion injury, thrombin
Graphical Abstract
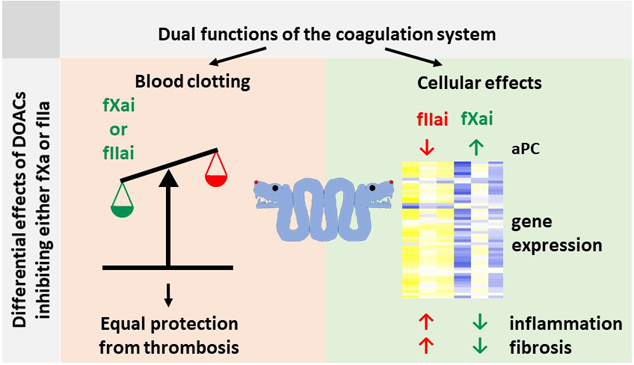
Myocardial infarction and related thrombosis are a major cause of morbidity and mortality worldwide. Direct Oral Anti-Coagulants (DOACs) have changed the way we treat and prevent thrombosis and thromboembolism, but rely on a new mechanism - the specific inhibition of a single clotting protease. We show that the direct inhibition of the coagulation factors fIIa and fXa gives different anti-inflammatory effects despite the same anticoagulant effectiveness. These effects depend at least in part on the differential activation of protein C, a signal-competent and cytoprotective protease. These results show that the efficacy of DOACs depends not only on their antithrombotic effects, but also on cellular effects such as regulation of gene expression, sterile inflammation and fibrosis. These studies show that not all DOACs are equal, even if they convey comparable antithrombotic effects, as they affect cellular responses differently. Future preclinical and clinical studies must consider additional endpoints such as inflammation and fibrosis when evaluating the safety and effectiveness of anticoagulants.
INTRODUCTION
Long-term anticoagulation is an established therapy in patients who have increased risk of thromboembolic disease. Direct oral anticoagulants (DOACs) are emerging as the therapy of choice and are increasingly replacing vitamin K antagonists (VKAs). DOACs have overall been proven safe and efficient in comparison to VKAs.1 DOACs differ conceptually from most anticoagulant therapies used in the past and are particularly different from the standard long-term outpatient treatment with VKAs. Unlike VKAs, DOACs do not inhibit multiple coagulation regulators, which dampens the entire coagulation system. DOACs inhibit specific coagulation proteases while retaining all other coagulation proteases. Based on the established “waterfall” or “cascade” model of the coagulation system, in which thrombin (fIIa) is the key protease, inhibiting specific coagulation proteases may be as efficient and safe as VKAs. However, it is now well established that individual coagulation proteases convey effects that are independent of hemostasis, in part through receptor-dependent mechanisms.2 Thus, while fIIa is the key protease in thrombus formation, the same is not true in regard to protease-dependent signaling. Individual proteases, including fIIa, fXa, and activated protein C (aPC) or complexes of proteases and receptors, such as TF/fXa or TF/fXa/fVIIa, can elicit specific and differential cellular effects through protease-activated receptors (PARs) and coreceptors.3–6
The signaling-dependent function of coagulation proteases is emerging as an important regulator of cellular homeostasis independent of hemostasis (defined as the regulation of platelet and fibrinogen activation).4 Functions of coagulation proteases beyond the regulation of hemostasis are well established and modulate inflammatory responses or cell death, for example.7 Assuming that balanced signaling in the coagulation system is required to modulate inflammatory responses or cell survival in physiological or pathophysiological settings, the consequences of specifically targeting individual coagulation proteases with DOACs remain unknown. Thus, it is currently uncertain whether the homeostatic function of the coagulation system, which depends on signaling via specific coagulation proteases, is disturbed by DOACs, despite a comparable effect in regard to blood clotting.
Several reports support the concept that DOACs differ in some respects.8, 9 In particular, a small and, in some reports, significant increase of acute coronary syndromes has been observed with various low molecular weight direct thrombin inhibitors (e.g., ximelagatran and dabigatran)10–13, while direct fXa inhibitors appear to reduce the incidence of myocardial infarction.14–17 These observations triggered clinical follow-up studies and preclinical studies, providing evidence that the direct fIIa inhibitor dabigatran and direct fXa inhibitor rivaroxaban have different effects on platelet activation.18–21 However, studies directly comparing fXa and fIIa inhibitors in the setting of cardiovascular disease are lacking.
Mice do not spontaneously develop myocardial infarction, precluding a direct comparison of fIIa and fXa inhibitors in mice in the context of spontaneous incidence of myocardial infarction. However, both protective and detrimental effects of coagulation factors and their receptors in myocardial ischemia-reperfusion injury have been described.22, 23 For example, thrombin and thrombin-derived peptides convey both detrimental and protective effects.24, 25 In addition, protease activated receptor 1 (PAR1) and PAR4 negatively affect ventricular remodeling and myocardial fibrosis after myocardial IRI22, 23, while the PAR1-derived peptide that is released upon activation by thrombin (parstatin) protects against myocardial IRI.26 The anticoagulant and cytoprotective coagulation protease activated protein C (aPC) reduces myocardial IRI through various mechanisms, including inflammasome inhibition through a receptor-dependent mechanism.27, 28 aPC is generated by the thrombin and thrombomodulin (TM) complex and is therefore thrombin dependent.29 Of note, direct thrombin inhibitors have been proposed to prevent aPC generation, resulting in an apparently paradoxical increase in thrombin generation.29–31
Based on these data, we hypothesized that exposure of mice to a direct fIIa inhibitor (fIIai) or a direct fXa inhibitor (fXai) may induce different responses to myocardial IRI. To this end, we pretreated mice with fIIai and fXai at doses that induced comparable anticoagulant effects ex vivo and in vivo and then induced myocardial IRI in these mice. In an unbiased approach, we aimed to identify potential differentially regulated responses in these mice despite a comparable anticoagulant effect and comparable size of the myocardial infarct volume.
METHODS
Data Availability.
See online supplemental material for additional information. The data that support the findings of this study are available from the corresponding authors upon reasonable request.
RNAseq data access.
RNA-Seq data have been deposited in the Gene Expression Omnibus (GEO) database (accession number GSE161325).
Mice.
Wild-type mice (C57BL/6, age 8–9 weeks) were obtained from Janvier Lab (S.A.S., St. Berthevin Cedex, France). Only male mice were used throughout this study in accordance with the approved procedures. All animal experiments were conducted according to standards and procedures approved by the local Animal Care and Use Committee (Landesverwaltungsamt Halle and Landesverwaltungsamt Leipzig, Germany).
In vivo intervention.
Mice (age 9–10 weeks) were treated with anticoagulants for 7 days prior to the interventions. In some experiments (tail bleeding time; FeCl3 in vivo thrombosis model, see below), increasing doses of a direct thrombin inhibitor (fIIai; dabigatran®, range 5–30 mg/kg, mixed in chow diet) or direct fXa inhibitor (fXai; rivaroxaban®, range 1–20 mg/kg, mixed in chow diet) were used. In the myocardial IRI model (see below), mice received fXai (3 mg/kg) or fIIai (10 mg/kg) for 7 days prior to surgery, and treatment was reinitiated 2 h post-surgery and continued for 24 h (short-term model) or 28 days (long-term model). A subgroup of fXai-treated mice was concomitantly injected with the monoclonal antibody MAPC1591 (10 mg/kg, i.p., every 2nd day), which blocks aPC anti-coagulant activity, or with the monoclonal antibody MPC1609 (10 mg/kg, i.p., every 2nd day), which blocks the anticoagulant and signaling activity of aPC.32, 33 A subgroup of fIIai-treated mice was concomitantly injected with wild-type aPC (1 mg/kg, i.p.) or a recombinant aPC variant specifically lacking its anticoagulant function (3K3A-aPC, 1 mg/kg, i.p.) 30 min prior to myocardial IRI.27, 34, 35
Statistical Analysis.
The data are summarized as the mean ± SEM (standard error of the mean) or as boxplots. Statistical analyses are mentioned in the supplementary section for analyses of RNAseq data and in the corresponding figure legends. Post hoc comparisons of ANOVA were corrected with the Tukey method. Statistical significance was accepted at values of p<0.05. The Kolmogorov-Smirnov test or the D’Agostino-Pearson normality test was used to determine whether the data were consistent with a Gaussian distribution. Statistics XL (www.statistixl.com) and Prism 5 (www.graphpad.com) software were used for statistical analyses.
RESULTS
Defining the dosing regiments of fIIai and fXai with comparable anticoagulant effects.
We conducted dose-escalation studies in mice to identify dosing regimens that resulted in comparable anticoagulant effects of direct fIIai and fXai. First, tail bleeding time was determined in mice that received increasing doses of a direct thrombin inhibitor (fIIai; dabigatran, 5–30 mg/kg, mixed in chow diet) or a direct fXa inhibitor (fXai; rivaroxaban, 1–20 mg/kg, mixed in chow diet). Tail bleeding time was determined 1 week after starting anticoagulant treatment. The average tail-bleeding time in untreated control mice (Cont) was 4 ± 1 min. As expected, both fIIai and fXai dose-dependently prolonged the tail-bleeding time. The D50 in mice, which is defined as the dose at which the tail-bleeding time was 50% of the maximal observed tail-bleeding time, was ~10 mg/kg for fIIai and ~3 mg/kg for fXai (Fig. 1a, b).
Fig. 1: Comparative analyses of fIIai and fXai in tail-bleeding and in vivo thrombosis assays.
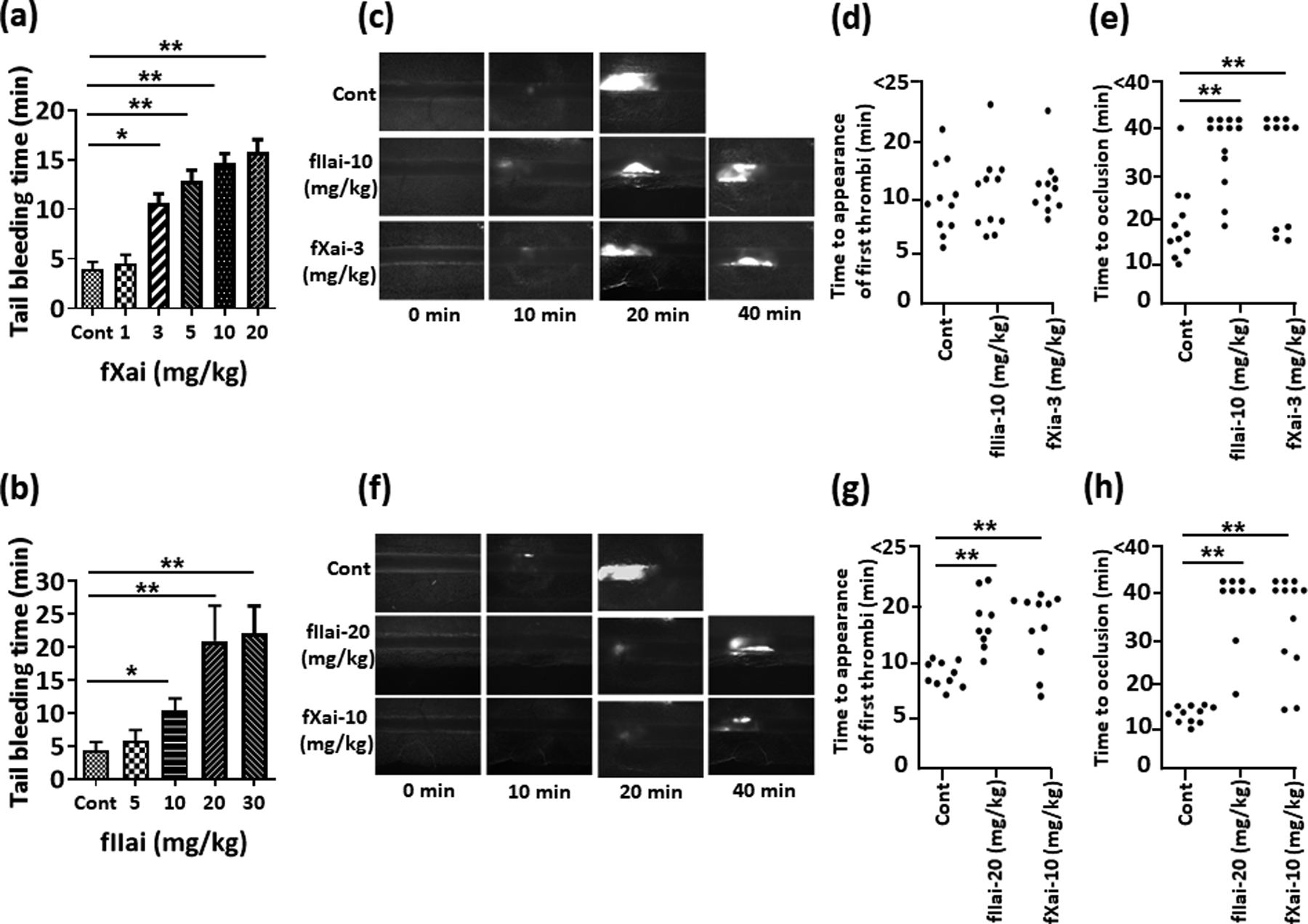
a-b: Dose dependent effects of fIIai and fXai in the tail-bleeding assay. The D50 (50 % effective dose) is 3 mg/mg for fXai (a) and 10 mg/kg for fIIai (b), while the D100 (100 % effective dose) is 10 mg/kg for fXai and 20 mg/kg for fIIai. Bar graph summarizing data.
c-h: Dose dependent effects of fXai and fIIai on FeCl3-induced thrombus in mice. Thrombus formation was visualized over 40 min using fluorescence microscopy. Time to appearance of first thrombi (d,g) and time to first occlusion (e,h) were recorded using a low (D50, c-e) and high (D100, f-h) dose. Representative fluorescent images of thrombosis formation 10, 20, 30 and 40 min after induction of injury are shown and dot-plots summarizing data for time till appearance of first thrombi and time to occlusion.
Mice without (Cont) or with fXai or with fIIai pretreatment. Data shown in a and b represent mean ± SEM; a, b: n=9 for each group; d, e, g, h: each dot represents one mouse; *P<0.05,**P<0.01 (a, b, d, e, g, h: ANOVA).
Next, we used an in vivo thrombosis assay. We induced thrombosis in mesenteric arterioles by local application of FeCl3. Mice were pretreated for 1 week with either the determined D50 (10 mg/kg for fIIai, 3 mg/kg for fXai) or the lowest dose that resulted in the maximal prolongation of bleeding time (D100; 20 mg/kg for fIIai, 10 mg/kg for fXai). To avoid excessive hemorrhage during the surgical procedure, anticoagulants were withheld 12 h prior to the intervention. At the lower dose (D50), the time until the appearance of the first thrombi was not different in treated mice compared to control mice, while the time to occlusion was comparably prolonged for both anticoagulants (Fig. 1c–e). At the higher dose (D100), both anticoagulants comparably prolonged the time until the appearance of the first thrombi, and both anticoagulants had a comparable effect on the time to occlusion (Fig. 1f–h).
Using the ED50 dosing scheme we observed plasma levels of anti-fIIa and anti-fXai in the range of plasma concentrations observed in patients (Dabigatran: ~22 ng/ml after 16 h; Rivaroxaban: ~46 ng/ml after 4 h) (Online Fig. Ia, b).36, 37 Additionally, using the ED50 dosing scheme comparable effects on prothrombin time (PT) and plasma levels of thrombin anti-thrombin complex (TAT) and platelet factor 4 (PF4) were observed (Online Fig. Ic–e), corroborating that the chosen dosing resulted in similar anti-coagulation effects. We observed no spontaneous bleeding in any of the treated mice. Taken together, we identified dosing regimens for fXai and fIIai with comparable anticoagulant effects both in vivo (tail-bleeding time, in vivo thrombus formation) and ex vivo.
fXai and fIIai differentially regulate myocardial IRI-associated inflammation.
We next determined the impact of fIIai and fXai in a myocardial IRI model using dosing regimens with comparable anticoagulant effects (ED50; 10 mg/kg for fIIai, 3 mg/kg for fXai; LAD ligation for 90 min followed by 24 h of reperfusion, Fig. 2a). Mice were pretreated for 1 week with either fIIai or fXai. To avoid excessive bleeding during surgically induced myocardial IRI, both anticoagulants were withheld 12 h prior to IRI. Infarct size was determined after 24 h and was markedly and comparably reduced by both anticoagulants (Fig. 2b, c). The comparable reduction in infarct size in mice treated with either direct anticoagulant was in agreement with the comparable anticoagulant effects observed using the ED50 in the tail-bleeding time analysis and the mesenteric thrombosis model.
Fig. 2: Comparable reduction of infarct size, but disjunct effect on gene expression by fIIai and fXai following myocardial IRI.
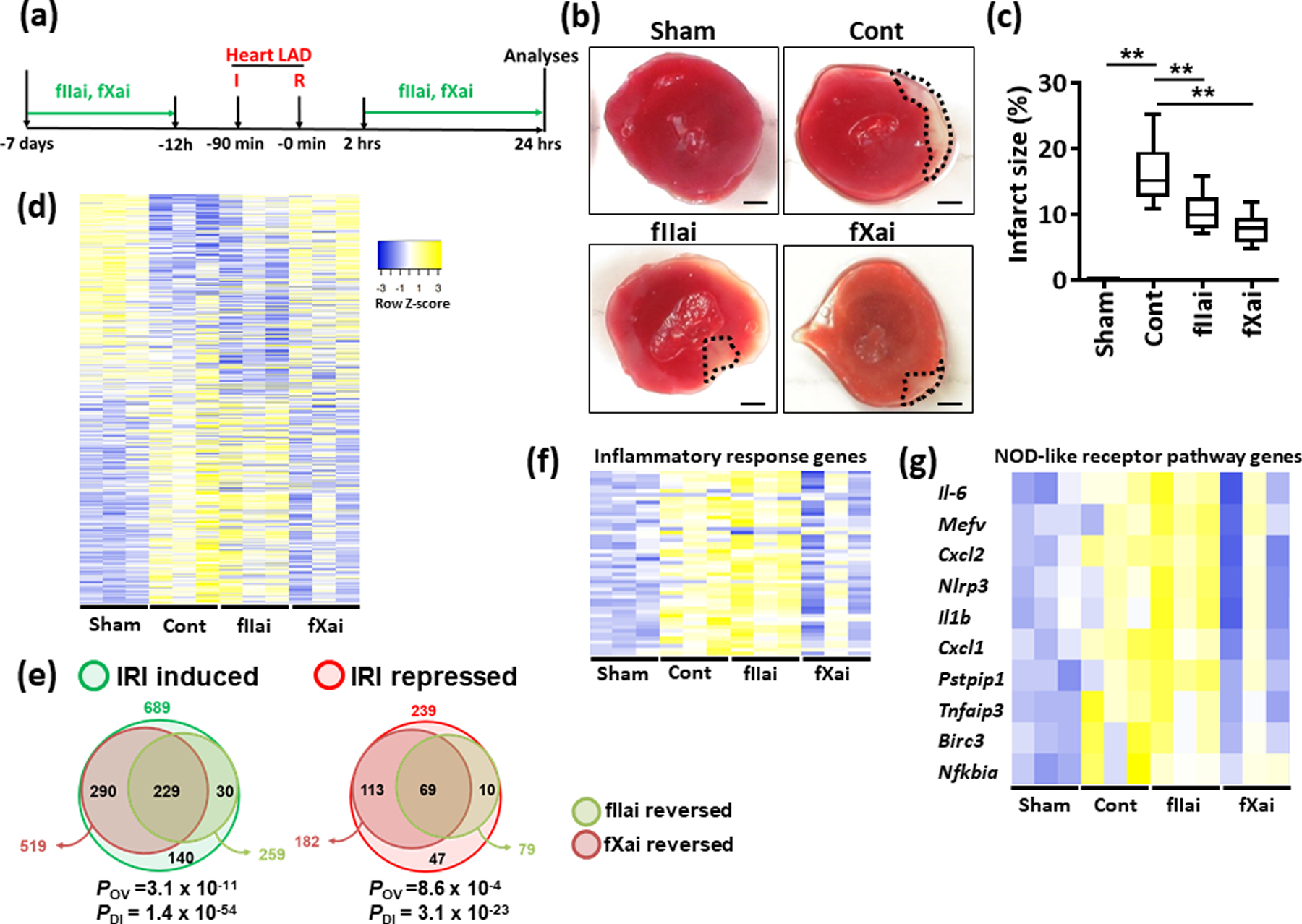
a: Schematic experimental plan.
b,c: Inhibition of fIIa- and fXa reduces infarct size. Representative heart sections showing infarcted area detected by TTC staining (b, black doted encircled area, size bar: 20 μm) and box-plot summarizing data (c).
d-g: Heat maps (d, f, g) and Venn diagram (e) summarizing differential gene-expression identified by RNA-seq. Gene count values larger than the average are represented in yellow, while lower counts than the average are represented in blue. Whenever transcript values are close to the average value, samples are colored in white (d, f, g). Venn diagram (e) showing overlap of genes significantly changed (induction or repression) in fIIai- or fXai-treated mice in relation to gene-expression in control-IRI mice. The overlap of genes regulated together by fIIai or fXai (POV, hypergeometric testing), but also the disparity of genes exclusively regulated by either fIIai or fXai were significant (PDI, exact binomial test). Heat maps of genes involved in Inflammatory response (f) and NOD-like receptor pathway (g) from panel d.
Mice without (Cont) or with fXai or with fIIai pretreatment. c: n=8; d, f, g: n=3 for each group;**P<0.01 (c: ANOVA).
Since directly targeting coagulation proteases with DOACs may differentially affect coagulation-protease-dependent signaling despite a similar anticoagulant effect, we next conducted unbiased expression analyses (RNAseq). In infarcted heart tissue of mice undergoing myocardial IRI without anticoagulant treatment (control group), the expression of 689 genes was induced and that of 239 genes was reduced compared to those of the hearts of sham-operated mice (Fig. 2d, e). Of the 689 induced genes and the 239 repressed genes in IRI, fXai treatment resulted in at least 1.5-fold reversal of 519 genes (75.3%) and 182 genes (76.15%), respectively (Fig. 2d, e). Gene expression in the hearts of fIIai-treated IRI mice was strikingly different from that of fXai-treated IRI mice and more closely followed the changes observed in control IRI hearts (Fig. 2d, e). Of the 689 induced genes and 239 repressed genes in IRI, fIIai treatment resulted in at least 1.5-fold reversal of 259 genes (37.5%) and 79 genes (33.05%), respectively (Fig. 2d, e). While genes regulated by fXai and fIIai overlapped significantly (229 upregulated and 69 repressed, Fig. 2e), the number of genes specifically reversed by fXai were larger than the number of genes specifically reversed by fIIai (fXai: 290 upregulated, 113 repressed and fIIai: 30 upregulated, 10 repressed, respectively, Fig. 2e). Hence, fXai treatment prevented changes in gene-expression upon myocardial IRI to a larger extent than fIIai treatment.
We performed functional annotation to study the pathways that contributed to these differential effects. Genes that were induced due to myocardial IRI were mainly involved in cytokine-cytokine receptor interaction, TNF signaling, NOD-like receptor signaling, Jak-STAT signaling, but also chemokine-signaling, phosphatidylinositol signaling, or NF-κB signaling (Online Fig. II). Of these IRI-induced and pathway-related genes, fXai treatment prevented the induction of genes related to the cytokine-cytokine receptor interaction, TNF signaling, NOD-like receptor signaling, and NF-κB signaling (Online Fig. III). Genes that were suppressed by fIIai treatment were related to pathways involved in aldosterone synthesis and secretion, TGF-β signaling, calcium signaling, and phosphatidylinositol signaling (Online Fig. IV).
We further performed gene ontology analysis on genes that were induced by myocardial IRI and repressed upon fIIai or fXai treatment. Gene ontology analysis of differentially expressed genes revealed that amongst genes upregulated due to IRI, the most relevant biological process was involved in inflammatory response, including 49 differential genes assessed (Fig. 2f). While genes that were regulated by fXai treatment were predominately involved in biological processes related to inflammatory responses (Fig. 2f, g), those that were regulated by fIIai treatment were not. Taken together, although fIIai and fXai resulted in comparable infarct sizes, unbiased gene expression analyses revealed marked differences, particularly in pathways related to inflammation and NOD-like receptor signaling (inflammasome) associated genes (Fig. 2f, g).
To validate the data obtained from the unbiased analyses, we determined the expression levels of sterile inflammation and inflammasome regulators in injured tissue by qRT-PCR. mRNA expression of IL-6, IL-1β, TNF-α, MIF, and MCP-1 was increased in injured tissue in comparison to sham-operated mice (Fig. 3a). The expression of IL-6, IL-1β, TNF-α, and MCP-1 was increased to the same or an even greater extent in fIIai-treated mice, while the expression of mRNA for these cytokines was reduced in fXai-treated mice compared to that of control mice (Fig. 3a). Thus, fXai specifically reduced mRNA expression of inflammation associated genes. Alternative gene expression patterns as identified by RNAseq were likewise confirmed by qRT-PCR, including genes not reduced by either anticoagulant (Pik3r5, Tlr2), regulated by both anticoagulants (Prkcg), or specifically regulated by fIIai (Ucp3) (Online Fig. V). Expression of the proinflammatory cytokines IL-1β, IL-6, TNF-α, and IL-6 is in part controlled by NF-κB.38 Congruently, phosphorylation of IκBα (NF-κB inhibitor α) and expression of p65 NF-κB were increased in mice with myocardial IRI compared to those of sham-operated mice (Fig. 3b, c). In fXai-treated but not fIIai-treated mice, IκBα phosphorylation and p65 NF-κB levels were markedly reduced (Fig. 3b, c). Concurrently, monocyte/macrophage frequency was increased to the same extent in control and fIIai-treated mice, while monocyte/macrophage frequency was markedly lower in fXai-treated mice (Online Fig. VI). The blunted inflammatory response in fXai-treated mice was associated with improved VE-cadherin staining (linear staining pattern and higher staining intensity) and less signs of intramural edema upon electron microscopical analyses as compared to fIIai-treated or control mice (Online Fig. VII), reflecting improved vascular barrier function post myocardial IRI in fXai-treated mice. These results establish that fIIai less potently inhibits the inflammatory response associated with myocardial IRI than fXai, despite a comparable anticoagulant effect and a comparable reduction in infarct size.
Fig. 3: fXai, but not fIIai, restricts inflammation and NF-κβ activation following myocardial IRI.
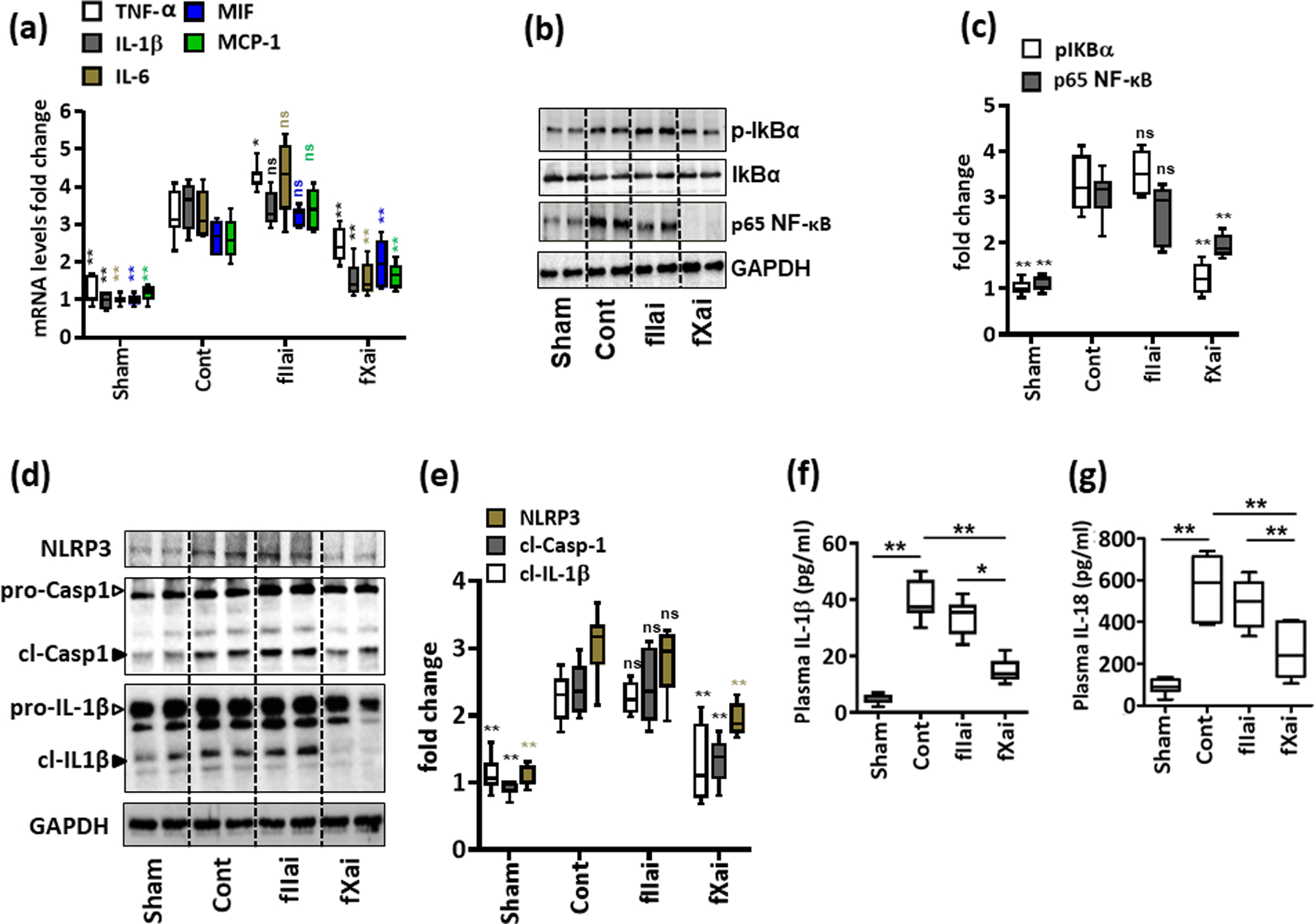
a: fIIai- and fXai differentially regulate inflammation in infarcted myocardial tissue. mRNA expressions (quantitative RT-PCR) of pro-inflammatory cytokines interleukin-6 (IL-6), IL-1β, tumor necrosis factor-α (TNF-α), macrophage migration inhibitory factor (MIF) and monocyte chemoattractant protein-1 (MCP-1) were induced following myocardial IRI. fXai but not fIIai inhibits mRNA expression of IL-6, IL-1β, TNF-α, MIF and MCP-1. Box-plots summarizing data of qRT-PCR. GAPDH was used for normalization.
b,c: fXai reduces NF-κB pathway activation following myocardial IRI. Representative immunoblots (b, GAPDH as loading control) and box-plot summarizing data for phosphorylated levels of IκBα and p65 NF-κB (c).
d-g: Treatment of mice with fXai restricts markers of inflammasome activation following myocardial IRI. Representative immunoblots showing cardiac NLRP3 expression and cleaved caspase- 1 (cl-Casp1) and cleaved IL-1β (cl-IL-1β), loading control: GAPDH (d). Arrowheads indicate inactive (white arrowheads) and active (black arrowheads) form of caspase-1 or IL-1β. The active form was quantified. Box-plots summarizing results (e). Box-plots summarizing plasma levels of IL-1β (f) and IL-18 (g).
Mice without (Cont) or with fXai or with fIIai pretreatment. a, c, e: n=8; f, g: n=10 for each group; *P<0.05,**P<0.01, ns: non-significant (compared to Cont), a, c, e-g: ANOVA comparing control to other groups.
fXai but not fIIai ameliorates inflammasome activation following myocardial IRI.
Activation of the NF-κB pathway drives NLRP3 inflammasome activation, thus promoting myocardial IRI-associated sterile inflammation.39, 40 Accordingly, both NF-κB and the NLRP3 inflammasome are linked with the severity of myocardial IRI.27, 41 Hence, we next determined whether fIIai and fXai differentially modulate myocardial IRI-associated NLRP3 inflammasome activation. The expression of NLRP3 and cleaved caspase-1 (cl-Casp1) and IL-1β (cl-IL-1β) were increased following myocardial IRI compared to that of sham-operated mice (Fig. 3d, e). fXai but not fIIai markedly reduced NLRP3 expression and cleavage of procaspase-1 and pro-IL-1β (Fig. 3d, e). Concurrently, plasma levels of the inflammasome-associated cytokines IL-1β and IL-18 were reduced in fXai-treated but not fIIai-treated mice (Fig. 3f, g, and Online Fig. VIII). Thus, fXai but not fIIai efficiently restricts inflammasome activation associated with myocardial IRI.
The pronounced difference in regard to gene-expression and inflammasome activation upon myocardial IRI in mice pretreated with fIIai or fXai raises the question as to whether anticoagulant treatment “preconditions” the heart and affects the NLRP3 inflammasome or gene expression already before myocardial IRI and whether the observed changes may be linked with altered epigenetic regulation. Analyses of mice after 1 week of treatment with fIIai or fXai did not result in different expression of selected cytokines and in particular had no impact on the NLRP3 inflammasome (Fig. 4a, b). Apparently IRI-dependent inflammasome activation is required to uncover differences in inflammasome regulation. However, unbiased expression analyses (RNAseq) revealed altered gene expression (Fig. 4c, d). Thus, compared to controls, 69 genes were induced by both fIIai and fXai (significant overlap) and a comparable number of genes were specifically induced by fIIai or fXai (23 and 20, respectively, Fig. 4d). Additionally, 32 genes were suppressed by both fIIai and fXai (significant overlap), while a low, but significantly different number of genes were specifically suppressed by fIIai or fXai (5 and 20 genes, respectively, Fig. 4d). Thus, the total number of genes regulated by both anticoagulants or specifically by either anticoagulant was lower in the resting state compared to changes observed post myocardial IRI. Accordingly, the total number of pathways identified was less as compared to the observations made post myocardial IRI (Online Fig. IX and X). Neither anticoagulant targeted pathways related to cytokine-cytokine receptor interaction or NOD-like receptors signaling, which is in agreement with the above results obtained by qRT-PCR and immunoblotting (Fig. 4a, b). In addition, we observed differential effects of fXai and fIIai on H3K9me3, an epigenetic histone H3 modification reflecting heterochromatin, and epigenetic regulators (DNMT1, SIRT1, Fig. 4e, f). Taken together, these results demonstrate that anticoagulation with fXai or fIIai “prime” or “precondition” the myocardium, but that additional factors are required for the differential inflammasome regulation during myocardial IRI.
Fig. 4: fXai and fIIai treatment does not induce myocardial inflammation but differentially regulate gene expression and epigenetic marks in healthy mice.
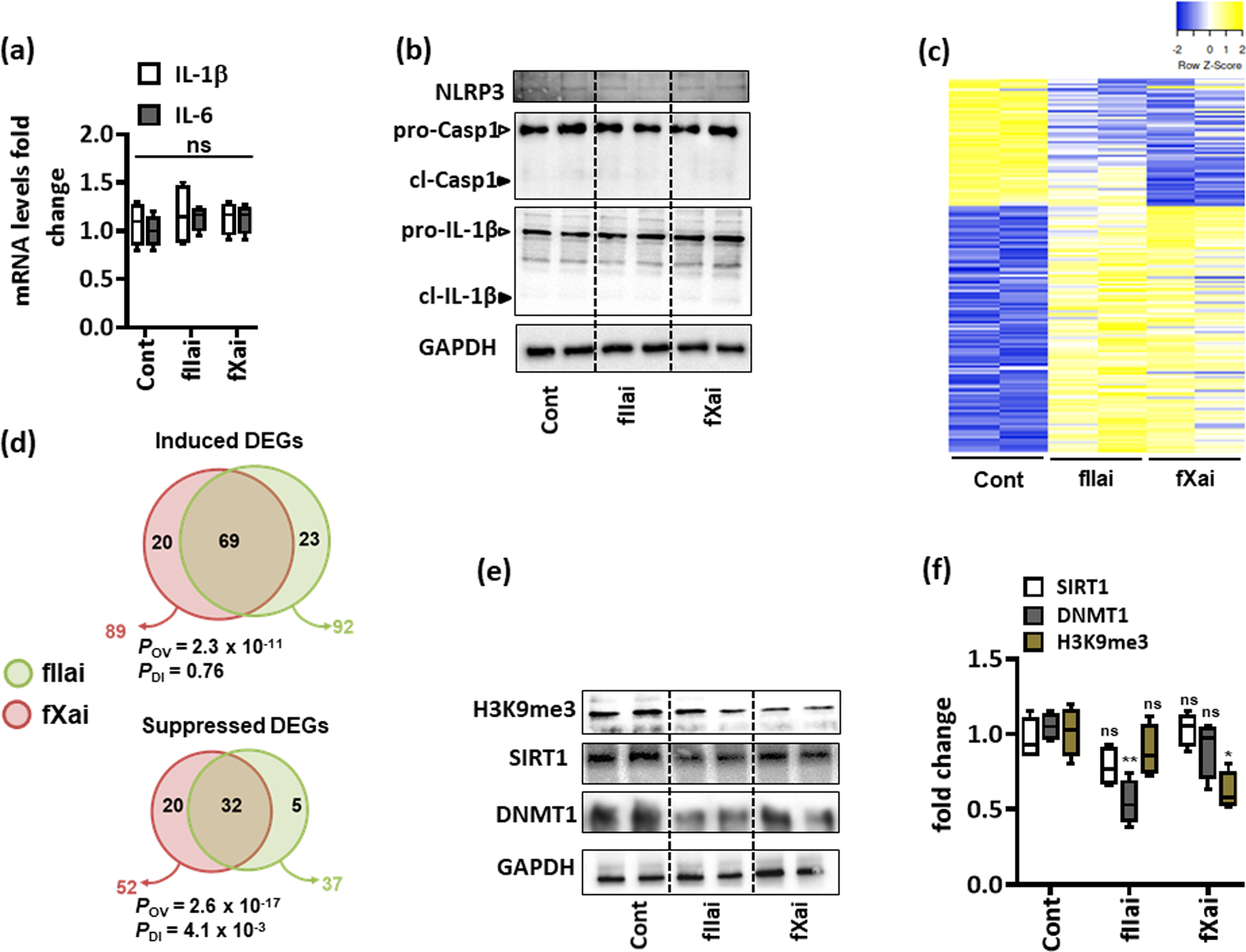
a: mRNA expression (qRT-PCR) of interleukin-6 (IL-6) and IL-1β was not altered in fIIai- and fXai-treated mice as compared to controls; box-plots summarizing data; GAPDH was used for normalization.
b: Representative immunoblots reflecting cardiac NLRP3 expression and total and cleaved forms of caspase-1 (pro-Casp1 and cl-Casp1) and IL-1β (pro-IL-1β and cl-IL-1β); loading control: GAPDH (d); arrowheads indicate inactive (white arrowheads) and active (black arrowheads) forms of caspase-1 or IL-1β.
c,d: Heat map (c) and Venn diagrams (d) summarizing differential gene-expression identified by RNA-seq. Gene count values larger than the average are represented in yellow, while lower counts than the average are represented in blue. Whenever transcript values are close to the average value, samples are colored in white (c). Venn diagram (d) showing overlap of genes significantly changed (induction, top or repression, bottom) in fIIai- or fXai-treated mice in relation to gene-expression in control (untreated) mice. The overlap of genes commonly regulated by fIIai or fXai was significant (POV, hypergeometric testing). The number of genes exclusively regulated by either fIIai or fXai was significant for downregulated genes (PDI, exact binomial test).
e,f: Representative immunoblots showing cardiac levels of H3K9me3, DNMT1, and SIRT1 after one-week treatment with fIIai or fXai compared to control; representative immunoblots (e) and box-blots summarizing results (f); GAPDH was used as loading control.
Mice without (Cont) or with fXai or with fIIai pretreatment. a: n=6; c: n=2; f: n=5 for each group; *P<0.05,**P<0.01, ns: non-significant (compared to Cont), a, f: ANOVA, comparing control to other groups).
fIIai but not fXai reduces protein C activation following myocardial IRI.
Considering potential mechanisms underlying the observed differences in sterile inflammation and the NLRP3 inflammasome upon treatment with fIIai and fXai, we evaluated the differential regulation of the serine protease activated protein C (aPC).30, 42 As activation of the zymogen protein C depends on the thrombin – thrombomodulin complex, direct fIIai and fXai may interfere with aPC generation to variable extents. Differential regulation of aPC appeared to be an attractive explanation, as the anti-inflammatory effects of aPC are well established and depend at least in part, particularly in the context of myocardial IRI, on NLRP3 inflammasome inhibition.27 To determine whether direct fIIai and fXai differentially interfere with protein C activation, we used an established in vivo protein C activation assay.43, 44 Myocardial IRI was induced in mice after 7 days of fIIai or fXai treatment, following the same dosing regimens as above (D50; 10 mg/kg for fIIai, 3 mg/kg for fXai), and in vivo aPC generation was determined after 24 h. In vivo aPC generation was markedly reduced in mice following myocardial IRI (Fig. 5a). In vivo aPC generation was slightly but significantly further reduced in fIIai-treated animals (Fig. 5a, P=0.028). Conversely, in vivo aPC generation was increased in fXai-treated mice compared to that of control IRI mice (Fig. 5a). These data indicate that direct fIIai interferes with aPC generation, while fXai does not impair and even appears to promote aPC generation in comparison to that of control myocardial IRI mice.
Fig. 5: fIIai-treatment is associated with lower aPC levels in myocardial IRI.
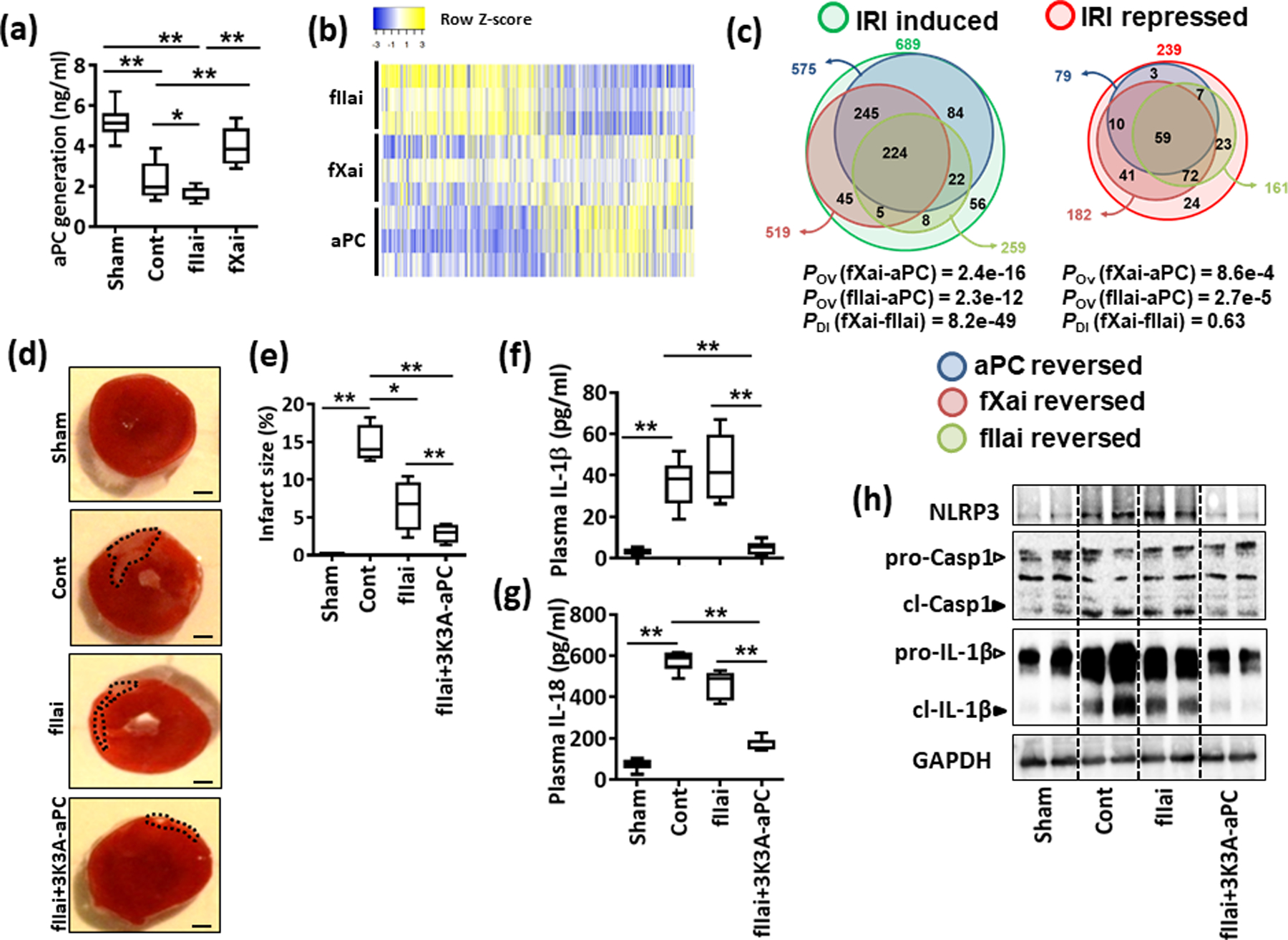
a: Plasma levels of aPC upon myocardial IRI in control and fIIai or fXai pretreated mice. fIIai but not fXai decreases aPC generation following myocardial IRI. Box-plot summarizing results.
b,c: Heat map (b) and Venn diagrams (c) summarizing differential gene-expression identified by RNA-seq. Gene count values larger than the average are represented in yellow, while lower counts than the average are represented in blue. Whenever transcript values are close to the average value, samples are colored in white (b). Venn diagrams (c) showing overlap of genes significantly changed (induction or repression) in fIIai, fXai, or aPC-treated mice, in relation to gene-expression in control-IRI mice. The overlap of genes regulated together by aPC and either fIIai or fXai was significant (POV, hypergeometric testing). The number of IRI-induced genes targeted by aPC and exclusively fXai was larger than that of genes regulated by aPC and exclusively fIIai (PDI, exact binomial test).
d-h: Concomitant treatment of fIIai and 3K3A-aPC reduces infarct size and the myocardial inflammatory response. Representative heart sections showing infarcted area detected by TTC staining (d, black doted encircled area, size bar: 20 μm) and box-plot summarizing data (e). Box-plots summarizing plasma levels of IL-1β (f) and IL-18 (g). Representative immunoblots showing cardiac NLRP3 expression and cleaved caspase- 1 (cl-Casp1) and cleaved IL-1β (cl-IL-1β); loading control: GAPDH (h). Arrowheads indicate inactive (white arrowheads) and active (black arrowheads) form of caspase-1 or IL-1β. Mice without (Cont) or with fIIai pretreatment alone (fIIai) or 3K3A-aPC on the top fIIai pretreatment (fIIai+3K3A-aPC). a: n=8; b: n=3; e: n=7; f, g: n=10 for each group; *P<0.05, **P<0.01 (a, e-g,: ANOVA).
aPC has been shown to induce a gene expression profile that is associated with cytoprotection.45–47 The increased aPC generation in fXai- and reduced aPC generation in fIIai-treated mice suggests that the observed differences in gene expression reflect aPC-dependent gene regulation. To directly determine whether the observed changes in gene expression reflect, at least in part, aPC-dependent effects, we compared gene expression in fXai- or fIIai-treated mice to that of aPC-treated mice.27 In support of the proposed aPC-dependent gene regulation in fXai-treated mice, changes in gene expression in aPC-treated mice were similar to those observed in fXai-treated mice but were strikingly different from those in fIIai-treated mice (Fig. 5b). Thus, of the 689 induced genes and 239 repressed genes upon myocardial IRI, aPC treatment resulted in an at least 1.5-fold reversal of 575 of the induced genes (83.4 %) and of 161 of the repressed genes (67.3 %). There was a large overlap of genes that were regulated by aPC or fXai treatment: 469 (245 + 224, 68.06%) of the 689 IRI-induced genes and 131 (72 + 59, 54.8 %) of the 239 IRI-repressed genes were similarly (1.5-fold) reversed by either intervention (Fig. 5c). Similar to fXai treatment, aPC treatment in the myocardial IRI mouse model repressed genes that are involved in cytokine-cytokine receptor interactions, NOD-like receptor signaling, pathways related to TNF signaling, NF-κB signaling, and biological processes related to inflammatory responses (Online Fig. III and XI).
In contrast, the overlap of genes regulated by fIIai with genes regulated by aPC was less pronounced: of 689 induced genes and 239 suppressed genes in the control group, 246 genes (224 + 22, 35.7 %) of the IRI-induced genes and 66 (59 + 7, 27.6 %) of the IRI-repressed genes were similarly (1.5 fold) reversed by both aPC and fIIai (Fig. 5c). The overlapping pathways regulated by aPC and fIIai treatment were related to HIF-1 signaling and phosphatidylinositol signaling (Online Fig. IV and XI). The number of IRI-induced genes normalized by aPC and fXai was larger than the number of genes regulated by aPC and fIIai (PDI (fXai-fIIai), Fig. 5c). Thus, in the context of myocardial IRI, fXai promotes aPC generation and alters gene expression profiles that mimics the effects of aPC and resulting in suppression of inflammatory pathways.
aPC signaling restores cardiac protection post myocardial IRI in fIIai-treated mice.
We next investigated whether restoring aPC signaling in mice treated with fIIai is sufficient to mimic the cytoprotective effect observed in response to fXai. To this end, mice that were treated only with the fIIai (following the same protocol as above) were compared to fIIai-treated mice that received a signaling-selective aPC mutant (3K3A-aPC, 1 mg/kg, i.p., 30 min before I/R; controls: PBS, equal volume, i.p.;). 3K3A-aPC is an aPC variant that is largely devoid of its anticoagulant function but retains the cytoprotective properties of aPC.27, 34, 35 3K3A-aPC on top of fIIai markedly reduced infarct size (Fig. 5d, e). The proinflammatory effect of fIIai on cytokine expression was lost upon concomitant 3K3A-aPC treatment (Online Fig. XIIa, b). Furthermore, 3K3A-aPC treatment in addition to fIIai restricted NLRP3 expression, cleavage of procaspase-1 and pro-IL-1β (Fig. 5h and Online Fig. XIIa, c) and plasma levels of NLRP3 inflammasome-associated cytokines (IL-1β and IL-18) following myocardial IRI (Fig. 5f, g). Hence, the unfavorable proinflammatory profile following myocardial IRI and pretreatment with fIIai can be compensated for by restoring aPC signaling.
The protective effect associated with fXai depends on aPC signaling.
To determine whether the beneficial anti-inflammatory effects of fXai depend on increased generation of endogenous aPC, we used inhibitor antibodies either to completely block all aPC activities using the mAb MPC1609 or selectively block the anticoagulant activity of aPC while retaining its cytoprotective activities using the mAb MAPC1591.32 Mice were treated for one week with fXai (3 mg/kg) and randomly assigned to injections with either MPC1609, MAPC1591 (each 10 mg/kg, i.p., every 2nd day), or PBS (control, equal volume, i.p.). Myocardial IRI was induced, and the mice were analyzed after 24 h as described above (Fig. 6a). Again, fXai markedly reduced infarct size (Fig. 6b, c) and mRNA expression of IL-6, TNF-α, MIF, and MCP-1 in comparison to those of control IRI mice (Fig. 6d–g). The protective effect of fXai on infarct size and cytokine expression was lost upon concomitant MPC1609 treatment, whereas MAPC1591 treatment did not impede the protective function of fXai (Fig. 6b–g). Furthermore, fXai-mediated inhibition of the NLRP3 inflammasome following myocardial IRI was lost upon MPC1609 but not MAPC1591 treatment, indicating that fXai-mediated inflammasome inhibition is independent of aPC-mediated coagulation inhibition but depends on aPC signaling (Fig. 7). Taken together, these data demonstrate that fXai does not interfere with aPC generation or with cytoprotective signaling by aPC following myocardial IRI, while fIIa inhibition by fIIai abolishes aPC-dependent protective effects.
Fig. 6: The protective effects associated with fXai depend on aPC signaling.
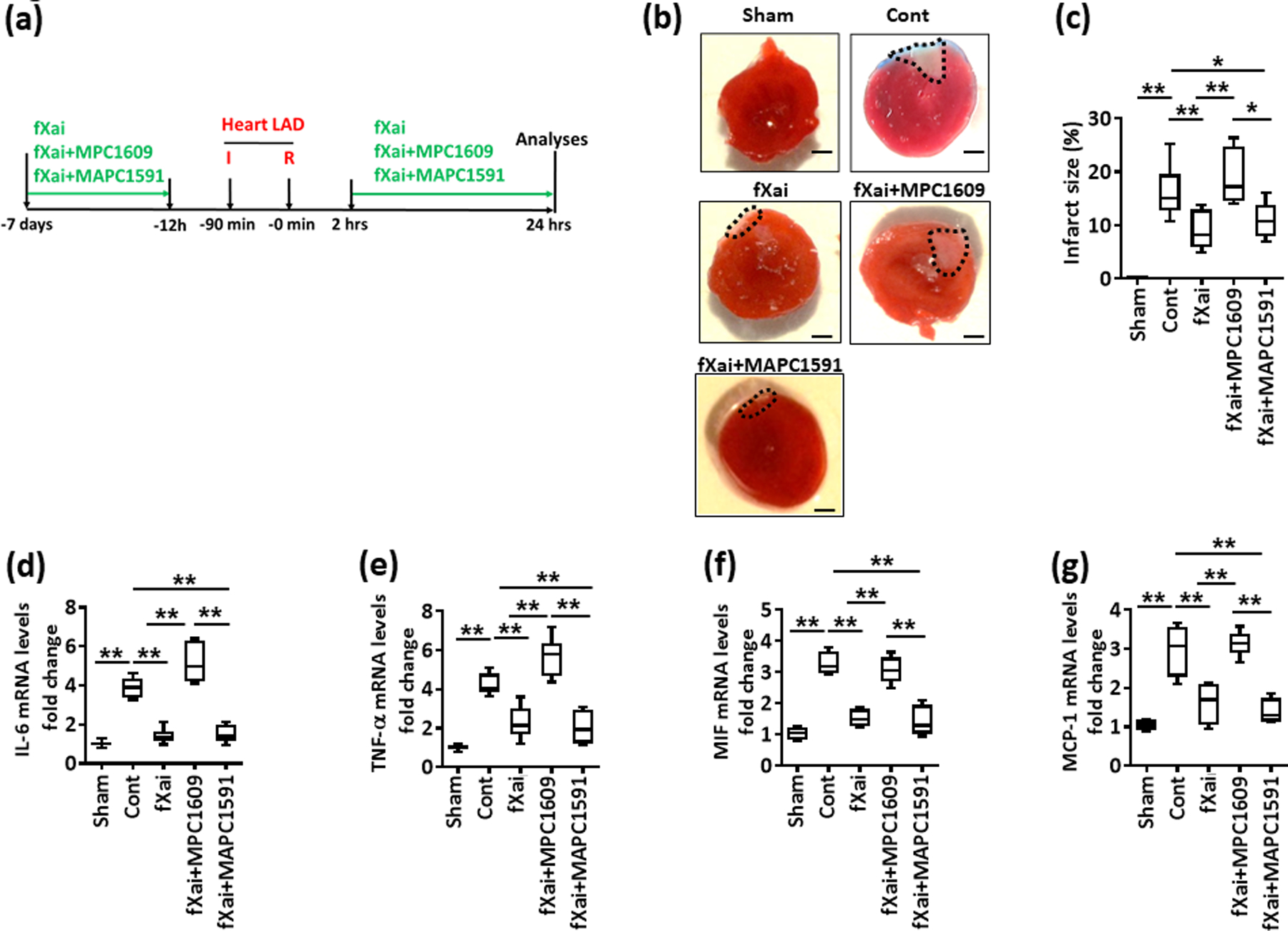
a: Schematic experimental plan.
b, c: The protective effect of fXai in regard to infarct size and cytokine expression is lost upon concomitant MPC1609 but not MAPC1591 treatment. Representative heart sections showing infarcted area detected by TTC staining (b, black doted encircled area; size bar: 20 μm) and box-plot summarizing data (c).
d-g: Box-plots summarizing data of qRT-PCR showing relative fold change expression for IL-6 (d), TNF-α (e), MIF (f) and MCP-1 (g); GAPDH was used for normalization.
Mice without (Cont) or with fXai (fXai), fXai plus MPC1609 (fXai+MPC1609) or fXai plus MAPC1591 (fXai+MAPC1591) pretreatment. c: n=8; d-g: n=6 for each group; *P<0.05, **P<0.01 (c-g: ANOVA).
Fig. 7: Inflammasome inhibition by fXai depends on aPC signaling.
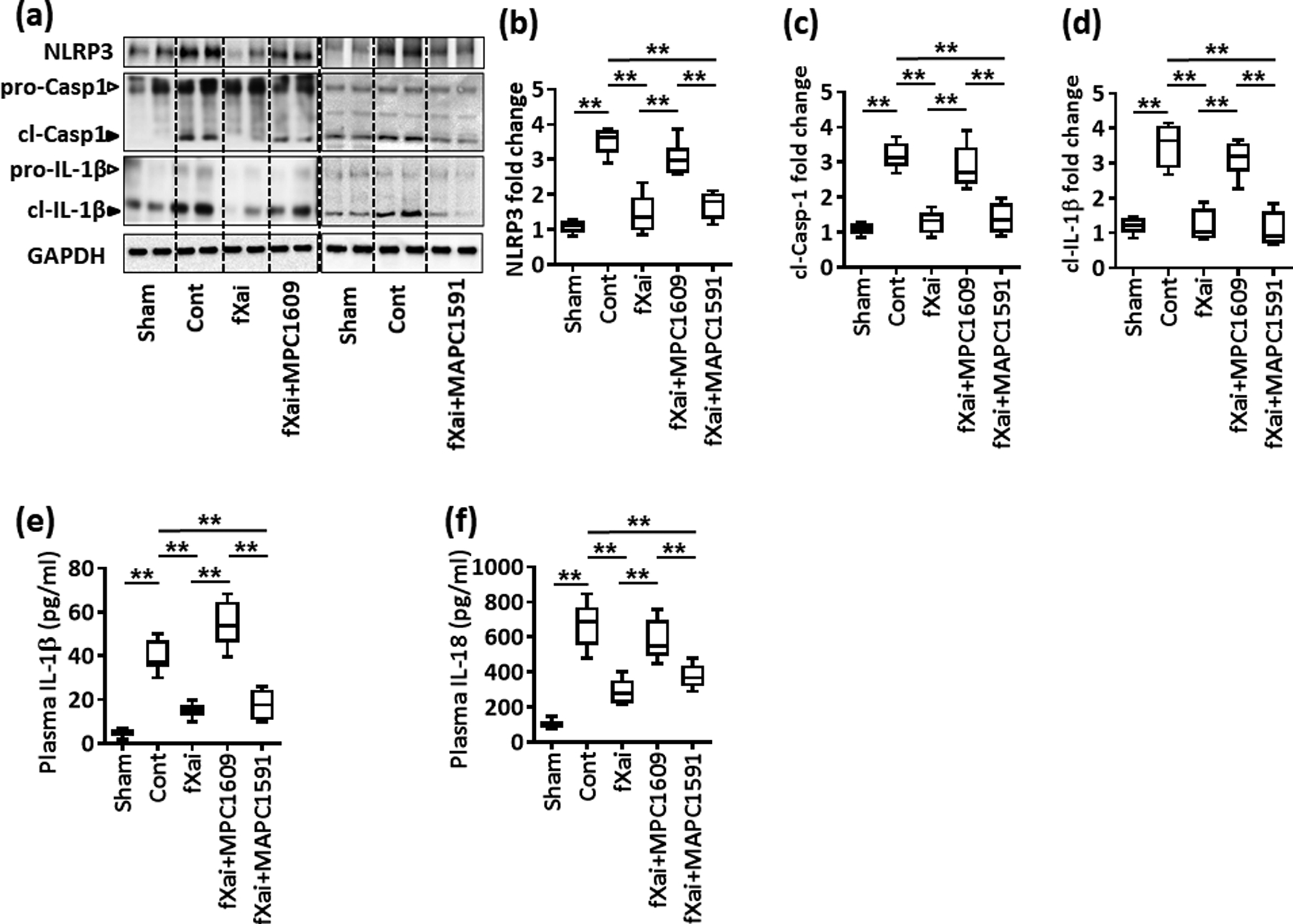
Reduced inflammasome activation following myocardial IRI by fXai is lost upon concomitant MPC1609, but not MAPC1591 treatment. Representative immunoblots (a) showing cardiac NLRP3 expression and cleaved caspase- 1 (cl-Casp1) and cleaved IL-1β (cl-IL-1β); loading control: GAPDH; arrowheads indicate inactive (white arrowheads) and active (black arrowheads) form of caspase-1 or IL-1β. The active form was quantified. Box-plots summarizing results of immunoblots (b-d). Box-plots summarizing plasma levels of IL-1β (e) and IL-18 (f).
Mice without (Cont) or with fXai (fXai), fXai plus MPC1609 (fXai+MPC1609) or fXai plus MAPC1591 (fXai+MAPC1591) pretreatment. b–d: n=8; e, f: n=12 for each group; *P<0.05, **P<0.01 (c-f: ANOVA).
Direct fXa inhibition ameliorates cardiac fibrosis after myocardial IRI.
Failure to resolve inflammation following myocardial IRI facilitates tissue destruction, ultimately leading to replacement of the myocardium with extracellular matrix.48 Considering the differential effect of fIIai and fXai on myocardial IRI-associated inflammation, we hypothesized that these anticoagulant strategies differentially affect cardiac fibrosis after myocardial infarction. We evaluated cardiac fibrosis 28 days post myocardial IRI in control mice and mice that were treated with fIIai or fXai (Fig. 8a and Online Fig. XIII). Masson trichrome staining revealed marked cardiac fibrosis post myocardial IRI in control IRI mice compared to sham-operated mice (Fig. 8b–c and Online Fig. XIV). Concomitantly, mRNA expression of fibrosis-related genes (collagen 1α1, collagen 3α1, α smooth muscle actin (α-SMA), and connective tissue growth factor (CTGF)) was markedly increased 28 days post myocardial IRI in control mice (Fig. 8d–g). Thus, despite comparable infarct sizes 24 h after myocardial IRI (Fig. 2), the extent of fibrosis 28 days after myocardial IRI, as reflected by Masson trichrome staining and mRNA expression of fibrosis-related genes, was markedly reduced in fXai-treated but not fIIai-treated mice (Fig. 8b–g).
Fig. 8: fXai, but not fIIai, ameliorates cardiac fibrosis following myocardial IRI.
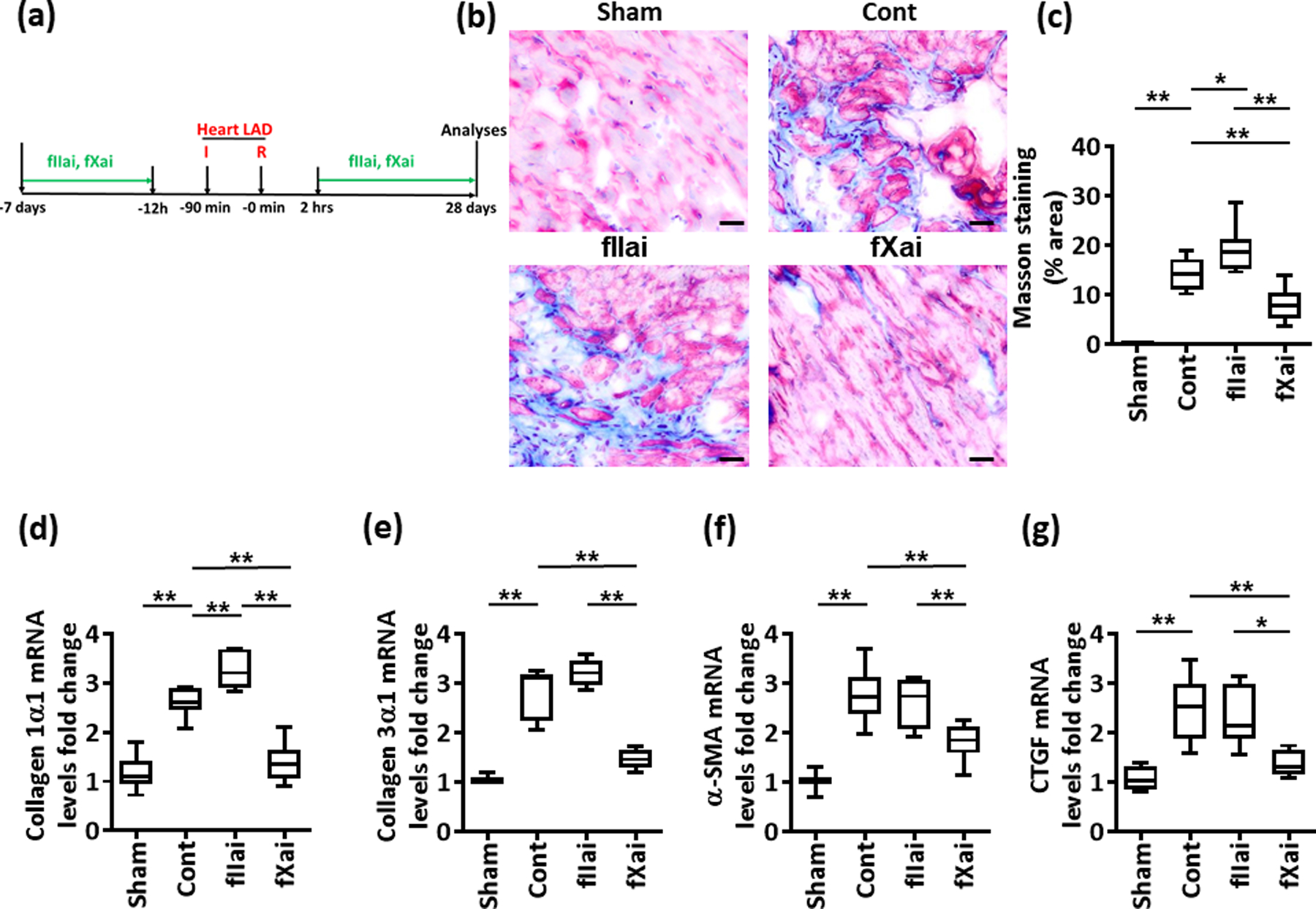
a: Schematic experimental plan.
b,c: fXai but not fIIai reduces cardiac fibrosis following myocardial IRI. Representative Masson’s trichrome stained mouse heart sections 4 weeks after myocardial IRI (b). The collagen-positive area is significantly lower in fXai, but not in fIIai treated mice; size bar: 20 μm. Bot-plot summarising results (c).
d-g: Box-plots summarizing data of qRT-PCR showing fold change expression for collagen 1α1 (d), collagen 3α1 (e), alpha smooth muscle actin (α-SMA, f), and connective tissue growth factor (CTGF, g); GAPDH was used for normalization.
Mice without (Cont) or with fIIai or fXai pretreatment. c: n=7; d-g: n=10 for each group; *P<0.05, **P<0.01 (c-g: ANOVA).
DISCUSSION
Direct oral anticoagulants have emerged as an alternative therapeutic strategy to prevent or treat thrombosis. Their safety profiles have been evaluated, showing noninferiority or even superiority in regard to hemostatic endpoints (incidence of thrombosis and/or hemorrhage).49, 50 It is, however, well established that coagulation proteases not only regulate hemostasis but also modulate inflammatory responses, cell death, and cellular responses, largely through receptor-dependent mechanisms.3–6, 51, 52 Modulation of inflammation and cellular function are typically not taken into account when evaluating the safety and efficacy of anticoagulants. Here, we show that specific inhibition of either fIIa or fXa differentially affects gene expression upon myocardial IRI, despite a similar impact on infarct size. The gene expression profile of mice treated with fIIai differed markedly from all other groups, including fXai-treated mice. Conversely, gene expression in fXai-treated mice was similar to that of sham-operated mice and showed a large concordance with the gene expression profile of aPC-treated mice. Because the dosing regimens were experimentally determined to provide comparable anticoagulant effects and the infarct sizes were comparable, the observed differences in gene expression appear to be independent of anticoagulant efficacy.
Among the pathways that were most prominently affected was the sterile inflammatory response, including the NLRP3 inflammasome. Differential regulation of the NLRP3 inflammasome was mechanistically linked to preserving aPC generation in the presence of fXai. The current findings are consistent with the recently demonstrated aPC-mediated reduction in inflammasome activation in myocardial IRI through receptor- and signaling-dependent mechanisms.27 Taken together, the current study provides experimental evidence that targeting individual coagulation proteases (as achieved with DOACs) has pronounced effects on nonhemostatic functions such as the inflammasome, despite comparable anticoagulant efficacies. These insights, if confirmed in humans, are of the uttermost clinical relevance, considering the increasing use of direct fIIa and direct Xa inhibitors in patients who are at risk of myocardial infarction. In a broader context, the current data suggest that in addition to hemostatic endpoints (thrombosis and hemorrhage), nonhemostatic endpoints (such as inflammasome activation reflected in inflammation biomarkers) need to be evaluated as endpoints in clinical studies evaluating anticoagulant strategies.
The mechanism(s) through which direct fIIai versus direct fXai differentially affect aPC generation remains to be fully established. A straightforward explanation is blockage of the thrombin active site, which is required for aPC generation. However, other mechanisms may also be involved. Thrombin activity and specificity are regulated by exosites 1 and 2.53 Thrombin is highly plastic, and binding to its exosites or active site induces allosteric changes.53–55 Intriguingly, the binding of dabigatran to active site of thrombin attenuates its binding to immobilized γA/γA-fibrin, which is exosite 1-dependent.56 Exosite 1 interacts with EGF domains four, five, and six of thrombomodulin, and this interaction is crucial for the cofactor activity of thrombomodulin.57 Reduced binding of thrombin exosite 1 to thrombomodulin diminishes aPC generation by 60 % to 80 %.53 Whether a reduction in the thrombin-thrombomodulin interaction by dabigatran, blocking the thrombin active site by dabigatran, or a combination of both factors contributes to the observed reduction in aPC generation remains to be shown. Of note, the allosteric changes induced by dabigatran upon thrombin binding may also impair other functions of thrombin, since exosite 1 is required for thrombin inhibition by heparin cofactor II.58
Preclinical studies support that direct fIIa inhibitors impede protein C activation by the thrombomodulin-thrombin complex.30, 31, 42 Thus, various direct antithrombin-independent fIIai (including dabigatran or melagatran) increase thrombin generation at low but not at high concentrations (“biphasic response”) in a thrombomodulin and protein C dependent fashion.30,31 The apparently paradoxical increase in thrombin generation observed with low concentration of fIIai is paralleled by reduced aPC-PCI (protein C inhibitor) complex formation, reflecting reduced aPC generation, supporting the notion that direct thrombin inhibitors (fIIai) impede protein C activation.30 The biphasic response of thrombin generation and reduced aPC-PCI formation by fIIai is in agreement with a model in which low fIIai concentrations primarily prevent protein C activation by the thrombomodulin-thrombin complex, while high fIIai concentrations are sufficient to inhibit thrombin regardless of thrombomodulin-thrombin dependent protein C activation.
The induction of thrombin generation upon fIIa inhibition is observed regardless of the method used (CAT method, fragment 1+2 generation).31 These direct fIIa inhibitors likewise promote coagulation activation in rodent models of tissue factor-induced coagulation activation.30, 31 In contrast to direct antithrombin-independent fIIa inhibitors, direct fXa inhibitors, such as rivaroxaban, edoxaban, or DX-9065a, do not inhibit protein C activation or increase thrombin generation.30, 59 Of note, those studies included direct fIIai and fXai that were different from those used in the current study, suggesting that the results observed are not specific to the pharmacological agent used but may reflect group-specific effects.
Relevance of the current finding that differential regulation of inflammasome activation by direct fIIa and direct fXa inhibitors for the clinical setting is supported by some clinical and preclinical studies. The direct fIIa inhibitor melagatran increased inflammatory markers, including the inflammasome-dependent cytokine IL-18, in patients with myocardial infarction in a retrospective analysis.60 In contrast, in a rodent stroke model, fXai pretreatment reduced inflammation, including a marked reduction in the inflammasome-related IL-1β gene.61 In an in vitro model, fXai dose-dependently suppressed proinflammatory gene expression in HUVECs exposed to recalcified human plasma, while dabigatran showed a biphasic response with enhanced proinflammatory gene expression at lower concentrations.62 The biphasic response in gene expression is congruent with the previously mentioned biphasic thrombin generation response observed with different concentrations of fIIa inhibitors.30, 31, 42
We cannot exclude other mechanisms underlying the differential effect of direct fIIa versus direct fXa inhibition. Thus, Chan et al. observed an induction of soluble thrombomodulin (sTM) plasma levels in humans treated with fXai for 24 weeks.63 In the atherosclerosis Risk in Communities (ARIC) study, levels of sTM were inversely correlated with coronary heart disease in a prospective analysis, suggesting that increased levels of sTM, which may reflect basal expression levels of thrombomodulin, are protective.63, 64 However, the study by Chan et al. (1) failed to evaluate whether increased sTM plasma levels observed in fXai-treated individuals reflected enhanced expression or shedding of endothelial thrombomodulin, (2) determine whether increased plasma sTM levels resulted in increased protein C activation, or (3) directly compare the fXai effect to that of a direct thrombin inhibitor.63
Another factor that contributes to our results may be the ability of rivaroxaban to reduce fXa-mediated platelet activation.19, 21, 65, 66 Since platelet-derived ATP and extracellular vesicles promote inflammasome activation67, 68, the observed reduction in inflammasome activation may reflect reduced platelet activation upon fXai treatment. In contrast, direct fIIai exacerbates platelet adhesion and aggregation, apparently by supporting the interaction of thrombin with GPIbα on platelets.20 Accordingly, fXai may provide cardioprotection in IRI via two complimentary mechanisms: (A) inhibition of platelet activation that reduces platelet-dependent inflammasome activation, and (B) enhanced aPC generation that conveys cytoprotective effects, including direct inhibition of inflammasome activation. Previous studies and the present work indicate that direct thrombin inhibitors do not employ either mechanism.12, 30, 42
An important aspect of the current study is that mice were pretreated with both anticoagulants, thus mimicking a clinical situation when patients receive anticoagulant treatment prior to myocardial infarction, rather than immediate therapeutic drug interventions in an acute setting. The differences in gene expression and of some epigenetic marks observed after 1 week of fIIai- or fXai-treatment in the absence of myocardial IRI support the concept of a homeostatic effect of coagulation proteases in addition to their well-established hemostatic effects.4 While no direct effect on the inflammasome was apparent after 1 week treatment, it is conceivable that the observed differences may “prime” differential responses in regard to inflammasome activation. Epigenetic gene regulation by coagulation proteases provide a rationale for the pronounced differences observed upon myocardial IRI despite the rather short half-life of the anticoagulant fXai itself (~7 h) and of aPC (~20 min in vivo) and are congruent with previous reports linking the cytoprotective effects of aPC with epigenetic mechanisms69, 70. Further studies are needed to define epigenetic mechanisms and associated homeostatic effects of coagulation proteases and inhibitors.
We believe that the current results provide important new insights for discussions regarding the risk of myocardial infarction in patients treated with DOACs. While most studies evaluated the impact of DOACs on the risk of myocardial infarction, we delineate here a mechanism through which DOACs may differentially influence the outcome of myocardial IRI. In addition, we provide evidence that direct anticoagulants differentially regulate inflammation-related endpoints that are not directly related to thrombus formation. If these findings are confirmed in humans, inflammation-related endpoints and yet to define homeostatic endpoints should be taken into account when evaluating DOACs anticoagulant strategies in the future.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Unlike most former anticoagulants, direct orally active anticoagulants (DOACs) target specific coagulation proteases.
DOACs provide efficient protection from venous thrombosis and thromboembolism in patients with atrial fibrillation.
Coagulation proteases regulate not only blood clot formation, but also cellular function via independent, largely receptor-dependent mechanisms.
What New Information Does This Article Contribute?
Despite equal antithrombotic effects, different DOACs differentially regulate gene expression, sterile inflammation, and fibrosis post myocardial ischemia reperfusion injury with a potential impact on outcome.
Differential gene-expression by different DOACs depends on divergent effects on protein C activation, a cytoprotective and signaling competent coagulation protease.
The efficacy of anticoagulant therapy is not only reflected by their antithrombotic effect, but also by their cellular effects.
ACKNOWLEDGEMENT
We thank Johannes Lauf, Kathrin Deneser, Julia Judin, and Rumiya Makarova for excellent technical support.
SOURCE OF FUNDING
This work was supported by grants from the ‘Deutsche Forschungsgemeinschaft’ (IS67/5–3, IS-67/8–1, IS-67/11–1, CRC 1118/B07 and CRC854/B26 to B.I., SH849/1–2 & SH849/4–1 to K.S., KO5736/1–1 to S. Kh., 361210922/GRK2408/P7&P9 to B.I., 361210922/GRK2408/P5 to K.S., and 361210922/GRK2408/P4 to R.B.D.), the Stiftung Pathobiochemie und Molekulare Diagnostik (SPMD, to K.S.), “Scientific project funding in the field of heart medicine” at the Medical Faculty-University Hospital Leipzig to K.S., B.I., C.B., and the National Institute of Health (TACTIC; UM1-HL120877 to C.T.E.; R01 HL142975 to J.H.G).
Nonstandard Abbreviations and Acronyms:
- LAD
- DOACs
- IL
- aPC
activated protein C
- fIIai
factor IIa inhibitor
- fXai
factor Xa inhibitor
- fVIIa
factor VIIa
- TNF-α
tumor necrosis factor-α
- VKAs
vitamin K antagonists
- TF
tissue factor
- NLRP3
NLR Family Pyrin Domain Containing 3
- RNAseq
RNA sequencing
- α-SMA
α smooth muscle actin
- CTGF
connective tissue growth factor
- sTM
soluble thrombomodulin
- ATP
adenosine triphosphate
- TAT
thrombin anti-thrombin
- PT
prothrombin time
- PF4
platelet factor 4
- DNMT1
DNA methyltransferase 1
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
The authors have nothing to disclose. J.H.G. is a Consultant for ZZ Biotech LLC.
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: A meta-analysis of randomised trials. Lancet. 2014;383:955–962 [DOI] [PubMed] [Google Scholar]
- 2.Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, Kaufmann R. Proteinase-activated receptors (pars) - focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun Signal. 2013;11:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin JH, Zlokovic BV, Mosnier LO. Activated protein c, protease activated receptor 1, and neuroprotection. Blood. 2018;132:159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Isermann B Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors. J Thromb Haemost. 2017;15:1273–1284 [DOI] [PubMed] [Google Scholar]
- 5.Shahzad K, Kohli S, Al-Dabet MM, Isermann B. Cell biology of activated protein c. Curr Opin Hematol. 2019;26:41–50 [DOI] [PubMed] [Google Scholar]
- 6.Zelaya H, Rothmeier AS, Ruf W. Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemost. 2018;16:1941–1952 [DOI] [PubMed] [Google Scholar]
- 7.Jackson SP, Darbousset R, Schoenwaelder SM. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood. 2019;133:906–918 [DOI] [PubMed] [Google Scholar]
- 8.Marin F, Anguita-Sanchez M, Sanmartin M. Direct oral anticoagulants and cardiovascular prevention in patients with nonvalvular atrial fibrillation. Expert Opin Pharmacother. 2017;18:67–77 [DOI] [PubMed] [Google Scholar]
- 9.Tornyos A, Kehl D, D’Ascenzo F, Komocsi A. Risk of myocardial infarction in patients with long-term non-vitamin k antagonist oral anticoagulant treatment. Prog Cardiovasc Dis. 2016;58:483–494 [DOI] [PubMed] [Google Scholar]
- 10.Eikelboom JW, Weitz JI. Anticoagulation therapy. Dabigatran and risk of myocardial infarction. Nat Rev Cardiol. 2012;9:260–262 [DOI] [PubMed] [Google Scholar]
- 11.Mak KH. Coronary and mortality risk of novel oral antithrombotic agents: A meta-analysis of large randomised trials. BMJ Open. 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polzin A, Dannenberg L, Wolff G, Helten C, Achilles A, Hohlfeld T, Zeus T, Kelm M, Massberg S, Petzold T. Non-vitamin k oral anticoagulants (noac) and the risk of myocardial infarction: Differences between factor iia and factor xa inhibition? Pharmacol Ther. 2019;195:1–4 [DOI] [PubMed] [Google Scholar]
- 13.Uchino K, Hernandez AV. Dabigatran association with higher risk of acute coronary events: Meta-analysis of noninferiority randomized controlled trials. Arch Intern Med. 2012;172:397–402 [DOI] [PubMed] [Google Scholar]
- 14.Chatterjee S, Sharma A, Uchino K, Biondi-Zoccai G, Lichstein E, Mukherjee D. Rivaroxaban and risk of myocardial infarction: Insights from a meta-analysis and trial sequential analysis of randomized clinical trials. Coron Artery Dis. 2013;24:628–635 [DOI] [PubMed] [Google Scholar]
- 15.Committee AS, Investigators, Alexander JH, et al. Apixaban, an oral, direct, selective factor xa inhibitor, in combination with antiplatelet therapy after acute coronary syndrome: Results of the apixaban for prevention of acute ischemic and safety events (appraise) trial. Circulation. 2009;119:2877–2885 [DOI] [PubMed] [Google Scholar]
- 16.Granger CB, Alexander JH, McMurray JJ, et al. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med. 2011;365:981–992 [DOI] [PubMed] [Google Scholar]
- 17.Lee CJ, Gerds TA, Carlson N, Bonde AN, Gislason GH, Lamberts M, Olesen JB, Pallisgaard JL, Hansen ML, Torp-Pedersen C. Risk of myocardial infarction in anticoagulated patients with atrial fibrillation. J Am Coll Cardiol. 2018;72:17–26 [DOI] [PubMed] [Google Scholar]
- 18.Nehaj F, Sokol J, Mokan M, Ivankova J, Mokan M. Thrombin receptor agonist peptide-induced platelet aggregation is reduced in patients receiving dabigatran. Clin Appl Thromb Hemost. 2018;24:268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perzborn E, Heitmeier S, Laux V. Effects of rivaroxaban on platelet activation and platelet-coagulation pathway interaction: In vitro and in vivo studies. J Cardiovasc Pharmacol Ther. 2015;20:554–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petzold T, Thienel M, Konrad I, et al. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci Transl Med. 2016;8:367ra168. [DOI] [PubMed] [Google Scholar]
- 21.Pignatelli P, Pastori D, Bartimoccia S, Menichelli D, Vicario T, Nocella C, Carnevale R, Violi F. Anti xa oral anticoagulants inhibit in vivo platelet activation by modulating glycoprotein vi shedding. Pharmacol Res. 2016;113:484–489 [DOI] [PubMed] [Google Scholar]
- 22.Kolpakov MA, Rafiq K, Guo X, et al. Protease-activated receptor 4 deficiency offers cardioprotection after acute ischemia reperfusion injury. J Mol Cell Cardiol. 2016;90:21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J, Strande JL. Protease-activated receptor 1 inhibition by sch79797 attenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts. J Cardiovasc Pharmacol Ther. 2013;18:460–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu LM, Osipov RM, Robich MP, Feng J, Sheller MR, Sellke FW. Effect of thrombin fragment (tp508) on myocardial ischemia reperfusion injury in a model of type 1 diabetes mellitus. Circulation. 2010;122:S162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirabet M, Garcia-Dorado D, Ruiz-Meana M, Barrabes JA, Soler-Soler J. Thrombin increases cardiomyocyte acute cell death after ischemia and reperfusion. J Mol Cell Cardiol. 2005;39:277–283 [DOI] [PubMed] [Google Scholar]
- 26.Routhu KV, Tsopanoglou NE, Strande JL. Parstatin(1–26): The putative signal peptide of protease-activated receptor 1 confers potent protection from myocardial ischemia-reperfusion injury. J Pharmacol Exp Ther. 2010;332:898–905 [DOI] [PubMed] [Google Scholar]
- 27.Nazir S, Gadi I, Al-Dabet MM, et al. Cytoprotective activated protein c averts nlrp3 inflammasome-induced ischemia-reperfusion injury via mtorc1 inhibition. Blood. 2017;130:2664–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren D, Giri H, Li J, Rezaie AR. The cardioprotective signaling activity of activated protein c in heart failure and ischemic heart diseases. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loghmani H, Conway EM. Exploring traditional and nontraditional roles for thrombomodulin. Blood. 2018;132:148–158 [DOI] [PubMed] [Google Scholar]
- 30.Kamisato C, Furugohri T, Morishima Y. A direct thrombin inhibitor suppresses protein c activation and factor va degradation in human plasma: Possible mechanisms of paradoxical enhancement of thrombin generation. Thromb Res. 2016;141:77–83 [DOI] [PubMed] [Google Scholar]
- 31.Perzborn E, Heitmeier S, Buetehorn U, Laux V. Direct thrombin inhibitors, but not the direct factor xa inhibitor rivaroxaban, increase tissue factor-induced hypercoagulability in vitro and in vivo. J Thromb Haemost. 2014;12:1054–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Sluis GL, Niers TM, Esmon CT, Tigchelaar W, Richel DJ, Buller HR, Van Noorden CJ, Spek CA. Endogenous activated protein c limits cancer cell extravasation through sphingosine-1-phosphate receptor 1-mediated vascular endothelial barrier enhancement. Blood. 2009;114:1968–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu J, Ji Y, Zhang X, Drake M, Esmon CT. Endogenous activated protein c signaling is critical to protection of mice from lipopolysaccaride-induced septic shock. J Thromb Haemost. 2009;7:851–856 [DOI] [PubMed] [Google Scholar]
- 34.Kerschen EJ, Fernandez JA, Cooley BC, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein c. J Exp Med. 2007;204:2439–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein c variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744 [DOI] [PubMed] [Google Scholar]
- 36.Kubitza D, Becka M, Voith B, Zuehlsdorf M, Wensing G. Safety, pharmacodynamics, and pharmacokinetics of single doses of bay 59–7939, an oral, direct factor xa inhibitor. Clinical pharmacology and therapeutics. 2005;78:412–421 [DOI] [PubMed] [Google Scholar]
- 37.van Ryn J, Stangier J, Haertter S, Liesenfeld KH, Wienen W, Feuring M, Clemens A. Dabigatran etexilate--a novel, reversible, oral direct thrombin inhibitor: Interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost. 2010;103:1116–1127 [DOI] [PubMed] [Google Scholar]
- 38.Liu T, Zhang L, Joo D, Sun SC. Nf-kappab signaling in inflammation. Signal Transduct Target Ther. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jo EK, Kim JK, Shin DM, Sasakawa C. Molecular mechanisms regulating nlrp3 inflammasome activation. Cell Mol Immunol. 2016;13:148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takahashi M Nlrp3 inflammasome as a novel player in myocardial infarction. Int Heart J. 2014;55:101–105 [DOI] [PubMed] [Google Scholar]
- 41.Kawano S, Kubota T, Monden Y, Tsutsumi T, Inoue T, Kawamura N, Tsutsui H, Sunagawa K. Blockade of nf-kappab improves cardiac function and survival after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;291:H1337–1344 [DOI] [PubMed] [Google Scholar]
- 42.Furugohri T, Morishima Y. Paradoxical enhancement of the intrinsic pathway-induced thrombin generation in human plasma by melagatran, a direct thrombin inhibitor, but not edoxaban, a direct factor xa inhibitor, or heparin. Thromb Res. 2015;136:658–662 [DOI] [PubMed] [Google Scholar]
- 43.Isermann B, Hendrickson SB, Zogg M, Wing M, Cummiskey M, Kisanuki YY, Yanagisawa M, Weiler H. Endothelium-specific loss of murine thrombomodulin disrupts the protein c anticoagulant pathway and causes juvenile-onset thrombosis. J Clin Invest. 2001;108:537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein c protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358 [DOI] [PubMed] [Google Scholar]
- 45.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203 [DOI] [PubMed] [Google Scholar]
- 46.Madhusudhan T, Wang H, Ghosh S, et al. Signal integration at the pi3k-p85-xbp1 hub endows coagulation protease activated protein c with insulin-like function. Blood. 2017;130:1445–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein c pathway. Science. 2002;296:1880–1882 [DOI] [PubMed] [Google Scholar]
- 48.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Investigators E, Bauersachs R, Berkowitz SD, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363:2499–2510 [DOI] [PubMed] [Google Scholar]
- 50.van Es N, Coppens M, Schulman S, Middeldorp S, Buller HR. Direct oral anticoagulants compared with vitamin k antagonists for acute venous thromboembolism: Evidence from phase 3 trials. Blood. 2014;124:1968–1975 [DOI] [PubMed] [Google Scholar]
- 51.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814 [DOI] [PubMed] [Google Scholar]
- 52.Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, Ramachandran R. Biased signalling and proteinase-activated receptors (pars): Targeting inflammatory disease. Br J Pharmacol. 2014;171:1180–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen K, Stafford AR, Wu C, Yeh CH, Kim PY, Fredenburgh JC, Weitz JI. Exosite 2-directed ligands attenuate protein c activation by the thrombin-thrombomodulin complex. Biochemistry. 2017;56:3119–3128 [DOI] [PubMed] [Google Scholar]
- 54.Treuheit NA, Beach MA, Komives EA. Thermodynamic compensation upon binding to exosite 1 and the active site of thrombin. Biochemistry. 2011;50:4590–4596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeh CH, Stafford AR, Leslie BA, Fredenburgh JC, Weitz JI. Dabigatran and argatroban diametrically modulate thrombin exosite function. PLoS One. 2016;11:e0157471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pospisil CH, Stafford AR, Fredenburgh JC, Weitz JI. Evidence that both exosites on thrombin participate in its high affinity interaction with fibrin. J Biol Chem. 2003;278:21584–21591 [DOI] [PubMed] [Google Scholar]
- 57.Stearns DJ, Kurosawa S, Esmon CT. Microthrombomodulin. Residues 310–486 from the epidermal growth factor precursor homology domain of thrombomodulin will accelerate protein c activation. J Biol Chem. 1989;264:3352–3356 [PubMed] [Google Scholar]
- 58.Boyle AJ, Roddick LA, Bhakta V, Lambourne MD, Junop MS, Liaw PC, Weitz JI, Sheffield WP. The complete n-terminal extension of heparin cofactor ii is required for maximal effectiveness as a thrombin exosite 1 ligand. BMC Biochem. 2013;14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furugohri T, Shiozaki Y, Muramatsu S, Honda Y, Matsumoto C, Isobe K, Sugiyama N. Different antithrombotic properties of factor xa inhibitor and thrombin inhibitor in rat thrombosis models. Eur J Pharmacol. 2005;514:35–42 [DOI] [PubMed] [Google Scholar]
- 60.Christersson C, Oldgren J, Wallentin L, Siegbahn A. Treatment with an oral direct thrombin inhibitor decreases platelet activity but increases markers of inflammation in patients with myocardial infarction. J Intern Med. 2011;270:215–223 [DOI] [PubMed] [Google Scholar]
- 61.Dittmeier M, Kraft P, Schuhmann MK, Fluri F, Kleinschnitz C. Pretreatment with rivaroxaban attenuates stroke severity in rats by a dual antithrombotic and anti-inflammatory mechanism. Thromb Haemost. 2016;115:835–843 [DOI] [PubMed] [Google Scholar]
- 62.Ellinghaus P, Perzborn E, Hauenschild P, Gerdes C, Heitmeier S, Visser M, Summer H, Laux V. Expression of pro-inflammatory genes in human endothelial cells: Comparison of rivaroxaban and dabigatran. Thromb Res. 2016;142:44–51 [DOI] [PubMed] [Google Scholar]
- 63.Chan MY, Lin M, Lucas J, et al. Plasma proteomics of patients with non-valvular atrial fibrillation on chronic anti-coagulation with warfarin or a direct factor xa inhibitor. Thromb Haemost. 2012;108:1180–1191 [DOI] [PubMed] [Google Scholar]
- 64.Salomaa V, Wu KK. Soluble thrombomodulin as predictor of incident coronary heart disease. Lancet. 1999;354:1646–1647 [DOI] [PubMed] [Google Scholar]
- 65.Petzold T, Thienel M, Dannenberg LK, et al. Rivaroxaban reduces arterial thrombosis by inhibition of fxa driven platelet activation via protease activated receptor-1. Circ Res. 2019 [DOI] [PubMed] [Google Scholar]
- 66.Sokol J, Nehaj F, Ivankova J, Mokan M, Mokan M. First evidence: Rivaroxaban and apixaban reduce thrombin-dependent platelet aggregation. J Thromb Thrombolysis. 2018;46:393–398 [DOI] [PubMed] [Google Scholar]
- 67.Kohli S, Ranjan S, Hoffmann J, et al. Maternal extracellular vesicles and platelets promote preeclampsia via inflammasome activation in trophoblasts. Blood. 2016;128:2153–2164 [DOI] [PubMed] [Google Scholar]
- 68.Vats R, Brzoska T, Bennewitz MF, et al. Platelet extracellular vesicles drive inflammasome-il-1beta-dependent lung injury in sickle cell disease. Am J Respir Crit Care Med. 2020;201:33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bock F, Shahzad K, Wang H, et al. Activated protein c ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66shc. Proc Natl Acad Sci U S A. 2013;110:648–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shahzad K, Gadi I, Nazir S, et al. Activated protein c reverses epigenetically sustained p66(shc) expression in plaque-associated macrophages in diabetes. Commun Biol. 2018;1:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merienne N, Meunier C, Schneider A, et al. Cell-type-specific gene expression profiling in adult mouse brain reveals normal and disease-state signatures. Cell reports. 2019;26:2477–2493 e2479 [DOI] [PubMed] [Google Scholar]
- 73.Saddic LA, Sigurdsson MI, Chang TW, et al. The long noncoding rna landscape of the ischemic human left ventricle. Circulation. Cardiovascular genetics. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang C, Gong B, Bushel PR, et al. The concordance between rna-seq and microarray data depends on chemical treatment and transcript abundance. Nature biotechnology. 2014;32:926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oliveros JC (2007. –2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
See online supplemental material for additional information. The data that support the findings of this study are available from the corresponding authors upon reasonable request.