

Abstract
DNA damage can take many forms such as double-strand breaks and/or the formation of abasic (apurinic/apyrimidinic; AP) sites. The presence of AP sites can be used to determine therapeutic efficacy of many drugs, such as doxorubicin. While there are different assays to search for DNA damage, they are fraught with limitations, such as the need for large amounts of DNA secured from millions of cells. This is challenging due to the growing importance of using liquid biopsies as a source of biomarkers for many in vitro diagnostic assays. To accommodate the mass limits imposed by the use of liquid biopsies, we report a single-molecule DNA damage assay that uses plastic nanofluidic chips to stretch DNA to near its full contour length when the channel dimensions (width and depth) are near the persistence length (~50 nm) of double-stranded (ds) DNA. The nanofluidic chip consisted of input funnels for high loading efficiency of single DNA molecules, entropic traps to store the DNA and simultaneous load a series of nanochannels for high throughput processing, and an array of stretching nanochannels to read the AP sites. Single dsDNA molecules, which were labeled with an intercalating dye and a biotinylated aldehyde reactive probe (bARP), could be parked in the stretching nanochannels, where the AP sites were read directly using a dual-color fluorescence microscope equipped with an EMCCD camera. One color of the microscope was used to read the DNA length and the second color detected the AP sites. The nanofluidic chip was made from thermoplastics via nanoimprint lithography, which obviated the need for direct writing the devices in glass or quartz using focused ion beam milling. We show that we can read the frequency of AP sites in single dsDNA molecules with the frequency of AP sites determined by associating fluorescently-labeled streptavidin with bARP through a biotin/streptavidin complex.
Keywords: Nanofluidics, single-molecule detection, DNA Abasic Sites
Graphical Abstract
Nanofluidic circuit for detecting damage (abasic sites) in single DNA molecules.
Introduction
Cellular DNA is constantly subjected to various exogenous and endogenous reactive species that result in DNA damage. DNA damage can generate a loss of a nucleotide base, usually from spontaneous hydrolysis of N-glycosylic bonds leading to the formation of apurinic/apyrimidinic (AP) sites, commonly referred to as abasic sites.1 Exogenous factors such as ultraviolet radiation, X-rays, tobacco smoke, and endogenous factors such as metabolic processes that produce reactive oxygen species (ROS) can generate AP sites in DNA.2 These factors can alter the bases in several ways including methylation or oxidation of guanine bases producing N7-methylguanine (N7-meG) or 8-oxo-7,8-dihydroguanine (8-oxoG), respectively.1
These damaged bases can be repaired through a variety of in vivo mechanisms but the predominant repair mechanism is the base excision repair (BER) pathway. BER repairs single bases with modifications3 by removal of the damaged bases via DNA glycosylases resulting in AP sites that are intermediates of the BER pathway.1
Many therapies for cancer depend on DNA damaging agents that cause cytotoxicity and cell death, which impeded tumor growth and spreading. Thus, increase in the number of AP sites often serve as biomarkers of a patient’s response to therapy.4 Most AP sites in healthy individuals are repaired, while the excess number of AP sites produced in cancer patients receiving chemotherapy are higher in abundance than those not receiving DNA damaging chemotherapeutic agents. An example of a damaging chemotherapeutic agent is Doxorubicin (DOX), an anthracycline antibiotic that is used for treating breast cancer patients,5 and is a potent exogenous agent that causes extensive production of ROS.6 DOX works by interfering with topoisomerase II enzyme action in addition to the formation of several ROS and H2O2.
Several methods to quantify AP sites have been reported. One method uses capillary electrophoresis with laser-induced fluorescence (CE-LIF), where the DNA possessing AP sites are labelled with a fluorescent aldehyde reactive probe (FARP).7 Robert et al. developed a chromatographic/tandem mass spectrometry method for the detection of AP sites pre-labelled with O-4-nitrobenzylhydroxylamine.8 Wang et al. used Positron Emission Tomography, where a positron emitting carbon-11 was integrated into methoxyamine that identified AP sites by forming covalent bonds with them. Some of the above-mentioned methods are expensive and/or labor intensive and as such, are clinically incompatible.4, 9
The use of a biotin containing aldehyde reactive probe (bARP) to quantify AP sites10 was initially developed by Kubo et al.11 In this method, the aldehyde group in the deoxyribose sugar of the open ring form of the AP site reacts with the alkoxyamine in bARP forming an oxime. The bARP binds to dye-labelled streptavidin to allow securing an optical signature indicative of an AP site.11–14
Commercially available benchtop techniques to detect AP sites include a microtiter method, where bARP binds specifically to the aldehyde groups in the AP sites followed by ELISA, where alkaline phosphatase reacts with bARP and absorbance is measured at 650 nm.13 However, because this is a colorimetric assay, it can only determine the number of AP sites, but cannot deduce the position of AP sites within the DNA molecule. In addition, the concentration of DNA has to be above a certain limit for the benchtop method to detect the AP sites. Unfortunately, this technique requires large quantities of DNA, which is not achievable when analyzing DNA secured from liquid biopsy markers. For example, circulating tumor cells (CTCs) recovered from liquid biopsies typically yield only 2–200 cells per mL with the number recovered depending on the cancer type and stage.15, 16 This small number of cells corresponds to a DNA mass in the range of picograms, which is well below the lower limit of sample required for the colorimetric method (see SI for more information).
Another method to detect AP sites is based on PCR. But the issue with this method is when amplifying the DNA containing AP sites, the DNA polymerase results in mis-incorporation of nucleotides leading to mutations or stalling of the PCR at the 3’ hydroxyl end of the missing nucleobase. Sikorsky et al. suggested that the presence of AP sites leads to the DNA polymerase during PCR to preferentially incorporate dATP and inducing n-1 deletions opposite the AP sites.17 As such, researchers have been investigating methods to quantify AP sites using techniques that require smaller amounts of input DNA.
Single-molecule imaging can process small amounts of DNA. Detection of AP sites on a 250 bp DNA with 2 AP sites was performed using atomic force microscopy.18 A method to directly visualize AP sites on λ-DNA stretched on a glass slide19 used scanning near-field optical microscopy.20 Chastain and coworkers mapped the frequency of AP sites in DNA by optically interrogating single DNA molecules spread on a glass slide.21 These methods suffered from several shortcomings, such as time consuming optical scanning, and the need for complicated sample preparation workflows.22
The ability to detect smaller masses of DNA, even single molecules, has led to the development of nanofluidics, which includes 1D structures like nano-slits, 2D structures like nanochannels, and 3D structures like nanopores.23 Nano-confined environments, for example nanochannels, overcome the static and dynamic motions of nucleic acids by confining and stretching them in a space smaller than their persistence length, which is approximately 50 nm for double stranded (ds) DNA. Nanometer channels also reduce the sample volume requirements and analysis time.24 One approach for visualizing single molecule in a high throughput format is DNA curtain technology developed by Greene et al., where DNA is anchored to one end of a lipid bilayer membrane and is pushed using hydrodynamic forces to the other end. The DNA molecules were aligned and stretched simultaneously along these barriers. But, the bottleneck of this approach is the requirement of a lipid barrier in the absence of which the DNA would not align.25 In addition, the technique uses electron beam lithography (EBL) of fused silica glass, which makes the fabrication process both complicated and expensive.26 Recently, a DNA-skybridge (3D-structure) was reported, which used the same principles for stretching DNA molecules and imaging them in a high throughput format using light sheet microscopy.27As with the aforementioned techniques, hydrodynamic forces are required to stretch the DNA and the DNA must be functionalized with a biotin molecule on both ends to anchor the DNA to a surface.
Here we present a high throughput, novel sensing platform for the direct reading of AP sites with optical quantification by the controlled stretching of single dsDNA molecules to near their full contour length using a nanochannel array imprinted into a thermoplastic. Our nanostructures were fabricated in poly(methyl methacrylate), PMMA, using nanoimprint lithography (NIL), where the structures from a Si master were transferred into the PMMA polymer using a three-step process. The first step consisted of the transfer of nanoscale features into a UV resin. The second step used the UV resin stamp to transfer the structures into another polymer (PMMA) under certain pressure and temperature depending on the glass transition temperature (Tg) of the polymer. Finally, the nano-features were covered with a cyclic olefin copolymer (COC) plate using thermal fusion bonding according to a previously published method from our group.28–30
Imprinted channels were ≤100 nm (width and depth) to allow stretching of single dsDNA molecules close to their contour length. The device consisted of an array of nanochannels with each channel possessing a funnel entrance capable of efficient loading of single DNA molecules and connected to an entropic nano-trap for storing the DNA that enters the nanofluidic circuit randomly. The stored DNA could then be loaded simultaneously into an array of stretching nanochannels, where they were parked for direct reading of the AP sites using single-molecule imaging. AP sites were labeled with bARP, which associated to a fluorescently-labeled streptavidin molecule (red fluorescent label). The dsDNA molecule was also labeled with an intercalating dye (green fluorescence intercalating dye) to read DNA length. After loading into the nanochannels, fluorescently-labeled AP sites were optically visualized using wide-field epi-illumination microscopy with dual color laser excitation and direct quantification of AP sites by analyzing optical signatures of the stretched DNA.
Experimental Methods
Materials and reagents.
Silicon (Si) <100> wafers were secured from University Wafers (Boston, MA). PMMA (Tg = 105°C) sheets were purchased from Good Fellow (Berwyn, PA) and cyclic olefin copolymer (COC 8007, Tg = 78°C) cover plates were purchased from TOPAS Advanced Polymers (Florence, KY). The anti-adhesion coating material, (tridecafluoro–1,1,2,2-tetrahydrooctyl) trichlorosilane (T-silane), was secured from Gelest, Inc. (Morrisville, PA). Tripropylene glycol diacrylate (TPGDA), trimethylolpropane triacrylate (TMPTA), Irgacure 651 (photo-initiator), sodium citrate buffer (pH = 5.0), and β-mercaptoethanol were obtained from Sigma-Aldrich (St Louis, MO). λ-DNA, 48.5 Kbp (New England BioLabs, Ipswich MA) and T4 DNA, 166 Kbp (Wako Chemicals USA, Inc. Richmond VA) were used as received. The DNA damage quantification kit was purchased from Dojindo Molecular Technologies, Inc. Rockville MD. Aldehyde Reactive Probe with biotin (bARP), streptavidin/Alexa Fluor® 594 conjugate, streptavidin/Alexa Fluor® 647 conjugate, and YOYO-1 were all purchased from Life Technologies (Carlsbad, NY). Chroma spin TE-1000 columns were obtained from Clontech Laboratories, Inc. (San Francisco CA). Aqueous citrate buffer and Tris-Borate-EDTA (TBE, 89 mM Tris, 89 mM Borate, 2 mM EDTA) buffer were secured from Fisher Scientific® (Philadelphia, PA). All dilutions of reagents and model samples were made using 18 MΩ/cm milliQ water (Millipore). Solutions were filtered through 0.2 μm filters (Thermo Scientific® Nalgene syringe filters) prior to use.
Fabrication of the multi-structured nanofluidic devices.
Nanofluidic devices were fabricated using our published procedures (see FIGURE S1).31, 32 Briefly, microstructures were fabricated into a Si substrate using photolithography and wet-etching. Photolithography was done using AZ1518 photoresist, spin coated on oxide coated wafers at 3000 rpm for 60 s. The wafers were baked at 100°C for 2 mins and exposed to UV light for 4 s and the features were developed using MIF-300 developer. Subsequently, they are wet-etched using 40% KOH until the Si is etched for a depth of 6–7 μm. Nanochannels were milled into the Si substrate by FIB milling (FEI Helios 600 Nanolab Dual Beam System). A multi-structure device with an array of nanochannels (100 nm x 80 nm) possessing a 3D tapered funnel with/without a nano-trap (400 nm and 600 nm diameter) were fabricated by FIB milling. The Si master was first replicated into a UV-resin consisting of 70% TPGDA, 28% TMPTA, and 2% Irgacure 651 via UV-NIL to produce a UV-resin stamp. The patterns in the UV-resin stamp were then transferred into a PMMA substrate by thermal NIL at 130°C for 120 s under 1910 kN/m2 (HEX03, JenOptik). The structures were enclosed with a low Tg COC 8007 cover plate by thermal fusion bonding at 70°C under a pressure of 680 kN/m2 for 900 s.
Finite element analysis (COMSOL).
The nano-trap structure was simulated using a 2D model in COMSOL to explore the electric field drop across nano-traps of different sizes (400 nm and 600 nm) for λ-DNA (48.5 Kbp). The electric field was simulated with a driving potential of 0.1 V at a temperature of 293 K. A line plot across the nano-trap was created and the drop in electric field strength across the corresponding nano-trap was determined from the simulation.
Current-voltage measurements.
The current across the multi-structured nanofluidic device with no nano-trap, 400 nm X 400 nm (dia X depth), and 600 nm X 600 nm (dia X depth) nano-traps were measured using an Axopatch 200B. Devices were initially filled with 50% methanol (v/v) to facilitate easy filling with aqueous buffers, followed by filling with a 1 M KCl solution in TBE buffer. The Pt electrodes were placed at each end of the fluidic circuit equipped with large reservoirs to make ohmic contact and the device was placed inside a Faraday cage. Then, different voltages and the corresponding currents were recorded. Each data point represented the mean ±standard deviation from five different measurements.
Dual color laser system (see Figure S2).
The Gaussian beam from a 488 nm laser (λex = 488 nm; P = 2000 mW; 2 mm beam diameter) was passed through a short pass dichroic filter (DF1) and expanded 10 times with a Galilean beam expander (focal lengths were −25 mm and 250 mm for L3 and L4 plano-concave and plano-convex lenses, respectively). The 641 nm laser beam (λex = 641 nm; P = 140 mW; 0.7 mm beam diameter) was expanded 5 times with a Galilean beam expander (focal lengths were −25 mm and 125 mm for L1 and L2 plano-concave and plano-convex lenses, respectively). Then, it was passed through an iris used to knock off the wings of the beam and was reflected by DF1. Both laser beams were directed into the microscope to ensure uniform laser intensity in the field-of-view and complete back-filling of the microscope objective (OBJ). The beam was passed through an iris into the back of a 100x oil immersion objective lens (1.25 NA) using lens L5 after passing through a laser line filter (F1) and reflected by a dichroic filter (DF2). A collimated laser beam impinged upon the nanofluidic device. Fluorescence emission was collected by the objective and passed through DF2 and spectrally selected using a long pass filter (F2). A mirror was used to steer the fluorescence onto an EMCCD camera. The videos and data were collected at ~150 fps using a Hamamatsu EMCCD C9100–13 digital camera with an EM gain and subsequently captured using Metamorph software. Faster frame rates (up to 405 fps) could be achieved using a smaller ROI and a binning option of the EMCCD camera.
Labelling λ-DNA using a bis-intercalating dye.
λ-DNA (NEB) was stained with a bis-intercalating dye, YOYO-1 (Molecular Probes, Eugene, OR) in a 5:1 base-pair/dye ratio in 1X TBE (89 mM Tris, 89 mM Borate, 2 mM EDTA) buffer solution. The TBE solution also contained 4% v/v β-mercaptoethanol as an anti-photobleaching agent to reduce photo-induced damage by acting as a radical scavenger.
ARP-streptavidin assay.
Genomic DNA in biological cells can be damaged due to reactive oxygen species (ROS) present in the cell. ROS inside the cell can be generated by radiation, chlorinated compounds, ultraviolet light, metal ions, peroxide compounds, or chemotherapeutic drugs.21 Generated AP sites in a DNA reside in equilibrium with a ring opened and a ring closed structure (see Figure 1).
Figure 1.

ROS inducing a loss of a nucleotide base (AP site) in dsDNA followed by ring opening, which generates an aldehyde group that is available for conjugation to an aldehyde reactive probe (ARP). ARP reacts with an active aldehyde group in the ring-opened form of the AP site. The biotin group in the ARP (bARP) has a strong affinity toward streptavidin, which allows for the generation of fluorescence at each AP site due to streptavidin being labeled with Alexa Fluor 647.33
It has been reported that under native conditions, 5% of AP sites are in the ring-opened form containing an active aldehyde group.13 Through the use of molar excess of reactive species or a probe with the aldehyde group, it is possible to convert all of the closed AP sites into the open-ring form.13 A biotinylated hydroxylamine, called “aldehyde reactive probe” (bARP), readily conjugates to the active aldehyde group in the ring-opened form and attaches strongly through an imine bond (Figure 1). After labeling with bARP, it is possible to produce an association complex with streptavidin. Horseradish peroxidase assays13 and Alexa Fluor labeled streptavidin assays19, 20 can then be used to produce biotin-streptavidin complexes.
Colorimetric assay for AP site quantification.
A DNA damage quantification kit was purchased from Dojindo Molecular Technologies, Inc. Standard AP DNA with a known number of AP sites was labeled with ARP. It included calf-thymus DNA (0.5 μg/mL) with 0, 2.5, 5, 10, 20, and 40 ARP-labeled AP sites per molecule, DNA binding solution, substrate solution, HRP-streptavidin, TE buffer, and washing buffer. Calibration curves were obtained using the standard protocol provided by the manufacturer.
To prepare the colorimetric assay using DNA standards in a 96 well plate, 60 μL of each standard ARP-DNA was added in 3 wells for each sample. Then, 100 μL of DNA binding solution was mixed by pipetting into each well followed by overnight incubation at room temperature. The next day, 1/4000 diluted HRP-streptavidin solution was freshly prepared by mixing 10 μL of HRP-streptavidin in 40 mL of washing buffer (due to its instability, this solution was freshly prepared every time). Binding solutions were discarded, and the wells were washed with washing buffer 5 times. After washing, 150 μL of diluted HRP-streptavidin was added to each well and incubated at 37°C for 1 h. After incubation, the washing step was repeated. Then, 100 μL of substrate solution was added to each well and incubated at 37°C for another 1 h. The optical density (OD) was recorded using a 96-well plate reader (SpectraMax M5 Multimode Plate Reader, Molecular Devices, LLC).
Fluorescence dyes for AP site labeling.
Suitable dyes for direct imaging of the AP sites were selected prior to the experiment and the feasibility of imaging with YOYO-1 was evaluated (YOYO-1 is an intercalating dye and is used to measure the DNA length to normalize the AP site frequency with respect to DNA length). We selected Alexa Fluor 647 for scoring the presence of AP sites (Alexa Fluor 647 was covalently attached to streptavidin and carried from 1–3 dyes per molecule) due to its deep red fluorescence, which showed minimal amounts of spectral overlap with the YOYO-1 intercalating dye. In addition, both YOYO-1 and Alexa Fluor 647 could be excited with both lasers found in our imaging system.
Labeling bARP AP DNA.
bARP AP DNA was tagged with streptavidin covalently labeled with Alexa Fluor 647. The corresponding streptavidin molecule reacts with the biotin group present in bARP to produce Alexa Fluor labeled AP sites in the dsDNA. Figure 1 shows the amine group of the bARP probe reacting with an aldehyde group of the open-ringed form to produce an imine bond. Alexa Fluor labeled streptavidin solution was diluted by volume of 1 to 5,000. Ten μL of the diluted streptavidin solution was mixed with 10 μL of bARP AP DNA and the mixture was incubated at 37°C for 1 h. Following this step, gel filtration was performed to remove the excess Alexa Fluor labeled streptavidin from the solution. Finally, the DNA backbone was labeled with YOYO-1 at 5:1 bp/dye ratio in 1X TBE including 4% v/v β-mercaptoethanol as an anti-photobleaching agent.
DNA trapping and extension in nanostructures.
Labeled DNA samples were directed towards the multi-structured nanochannel by an applied electric potential. Single DNA molecules entered through the funnel entrance and were injected towards the nano-trap where it was stored. The electric field was then adjusted to eject the single DNA molecules into the array of nanochannels, where they were elongated to near their full contour length. When the DNA entered the stretching nanochannels, the electric field was turned off such that the DNA would be confined and extended inside the nanochannels so that they could be imaged to detect the AP sites. DNA molecules were allowed to relax before imaging. Fluorescence images were acquired using the dual color microscope system described previously. Image acquisition was performed using a Hamamatsu EMCCD camera (digital mode, 1024 ×1024 pixels, high-resolution) with Metamorph software.
Results and Discussion
Multi-structured nanofluidic device.
We have discussed in our previous publications the use of thermoplastics as substrates for nanofluidic devices.28–30, 34 These materials, which can be linear or branched polymers, have different physiochemical properties such as glass transition temperatures (Tg), hardness, and chemical composition, which allows for the selection of a material best suited for a particular application. In addition, the surface properties of thermoplastics can be modified using techniques such as UV/O3 irradiation or O2 plasma treatment.29 For example, PMMA has a water contact angle of 69° and following exposure to UV/O3 light, the water contact angle is reduced to 36° making the nanochannel more receptive to wetting reducing bubble formation, which can cause device failure when requiring aqueous buffers. COC is actually a copolymer consisting of ethylene and norbornene monomers with the mole fraction of each monomer in the COC grade affecting its physiochemical properties such as the Tg and has excellent optical clarity with low background autofluorescence. The PMMA substrate with COC cover plate was selected here due to the high optical clarity of the COC cover plate to allow for single-molecule detection of the AP sites, the fact that PMMA can be used with NIL to provide high replication fidelity for nanometer structures, and finally the lower Tg of COC compared to PMMA allowed for the ability to thermally fusion bond the COC cover plate to the PMMA substrate to minimize nanostructure deformation in the PMMA substrate.31
The deformability of thermoplastics due to their relatively low Tg and melting temperature makes them useful for the fabrication of micro- and nanofluidic channels via hot embossing, injection molding, compression molding, thermal forming, and casting techniques. A method for the fabrication of nanochannels in thermoplastics is NIL, which is a variant of hot embossing. Since its first report in the 1990s by Steven Chou and co-workers,35–37 NIL has become an extensively used tool for the fabrication of nanostructures even with sub-10 nm sizes. The main advantage of NIL is the ability to build multi-scale patterns in a single imprinting step with high reproducibility. Thus, higher rates of production of nanofluidic devices at lower cost can be realized using NIL compared to direct writing of devices using electron beam or focused ion beam milling.38 In this paper, our mixed-scale nanofluidic devices were made via NIL. However, we note that nanofluidic devices can also be made using injection compression molding at even higher rates of production and lower device cost compared to NIL.39
The detailed procedure of nanofluidic device fabrication has been described elsewhere, but a schematic of this process can be found in Figure S1.28–30 Figure 2A shows SEM images of the PMMA nanofluidic circuit investigated herein, which consisted of 3 main components: (i) A tapered 3D funnel that facilitates the entry of dsDNA into the nanofluidic circuit; (ii) a nano-trap (600 μm) that stores the dsDNA entropically during the loading stage of operation. DNA molecules randomly enter the funnel and placing a trap in the fluidic circuit stores them until they are simultaneously ejected into the stretching nanochannel array simultaneously so they can be stretched and parked for analysis in a high throughput format; and (iii) a stretching nanochannel that stretches the molecule to assist in visualizing the AP sites within a single dsDNA molecule.40
Figure 2.

(A) SEM images of the multi-structured nanofluidic circuit comprised of a 3D funnel, nano-trap, and a stretching nanochannel. Its equivalent circuit diagram with resistors including an adjustable resistor for the nano-trap (variable resistance is predicated on changing the trap size) is also shown. (B) Array of multi-structured nanofluidic circuits for high throughput processing of single DNA molecules to search for AP sites. (C) Entrance funnel used to extend the electric field into the adjoining microchannel for assisting in loading single DNA molecules into the nanofluidic circuit from access microchannels. (D) SEM image of a 400 nm nano-trap (diameter and depth). The device shown was made from PMMA using thermal NIL from a resin stamp produced via UV-NIL from a Si master.
The DNA molecule entering into the nanofluidic circuit must overcome the free energy barrier created as a result of the reduction in the conformational entropy associated with the confined molecule when resident within a nanochannel.41–43 The effect of inlet structures connecting a microchannel to nanochannel influences the rate of DNA entry into a nanofluidic network.44 DNA molecules can be efficiently introduced into nanochannels by using a 3D funnel structure. This funnel is shaped to increase electrostatic and hydrodynamic forces so that they are greater than the repelling entropic force. As the funnel tapers towards the nanochannel, there is a gradual increase in confinement of the DNA molecule.44 The funnel entrance also increases the probability of dsDNA molecules entering into the nanochannel by increasing the effective electric field penetration depth into the adjoining microchannel. As can be seen from Figure S3A–B, we found that the injection efficiency of single λ-DNA molecules into the nanofluidic circuit increased nearly 6-fold compared to a blunt interface between the microfluidic network and nanofluidic circuit. We also tested a groove-type input and found that it was not as efficient as the 3D funnel (Figure S3B).
Following the 3D funnel, there was placed a nano-trap (Figure 2B) in the circuit that served to “store” the dsDNA molecules entering into the device. Craighead et al. used entropic traps, which consisted of expansion chambers, to separate DNA molecules based on entropic considerations the effects of which were dependent on the DNA length (i.e., longer DNA molecules moved out of the expansion chamber faster than shorter DNAs and thus, had a shorter trapping time).45–48 In this paper, we used a similar structure, but as a storage unit during loading so that dsDNA molecules could be uniformly introduced into a parallel array of stretching nanochannels so that multiple molecules could be parked and imaging in stretching nanochannels. Two different nano-traps sizes were evaluated, a 400 (diameter and depth) and 600 nm (diameter and depth) trap. The region following the nano-trap leads to the stretching nanochannel, which was 100 × 80 nm (width x depth; the length was 80 μm to allow the accommodation of dsDNA molecules with relatively large contour lengths; Figure 2C) to allow for detection of the AP sites. Figure 2B shows the array of nanofluidic circuits, which facilitated high throughput processing of DNA molecules. Figure 2C and 2D show high resolution SEMs of the 3D funnel and the nano-trap, respectively. Furthermore, COMSOL simulations were performed to evaluate the electric field strength distributions across the nano-traps of 400 nm and 600 nm. This assisted in determining the approximate driving voltage required to trap the DNA molecules and then, subsequently inject them into the stretching nanochannel array (Figure S4).
Translocation of dsDNA through the nanofluidic circuit.
λ-DNA stained with YOYO-1 in 1X TBE buffer was introduced into the nanofluidic circuit under the influence of an applied potential of 0.1 V. The electrodes (Pt) made electrical contact to the fluidic circuit as they were placed at each end of the circuit in large reservoirs to accommodate large amounts of buffer so as to not shift the pH during the measurements. The dsDNA was loaded into the nanofluidic circuit with a negative voltage applied to the funnel side of the circuit and the opposite size was kept at earth ground. Therefore, the electrophoretic mobility of the stained dsDNA was higher than the electroosmotic flow of the device and drove the translocation process through the circuit.28–30
Fluorescence measurements were collected using the dual-color, single-molecule microscope (see Figure S2). Representative images are shown in Figure 3 as a single λ-DNA molecule translocated through the circuit. These experiments were performed for both trap sizes and DNA entered through the funnel and moved to the trap, where it coiled inside the nano-trap (trapping) and was then subsequently ejected from the trap and into the stretching nanochannel (translocation time). Complete trapping was observed in Figure 3, panel 2, image (viii) where the molecule was observed to form a blob. In addition, we could manipulate the applied voltage to control the storage/trapping time. At voltages <0.1 V, the molecule was found to remain in the trap until a higher voltage was applied (data not shown). We should note that at the voltage (0.1 V) used in these experiments, the λ-DNA molecules were found to transiently remain in the nano-trap. We note as well that using the 600 nm trap and T4 DNA, which has a length of 160 Kbp, we were unable to even intermittently trap this DNA molecule using the 600 nm trap (data not shown).
Figure 3.

(A) Representative frames showing the translocation of stained λ-DNA through the multi-structured nanofluidic circuit. The frames were divided into 4 sections; (i) ‘Enter the funnel,’ where the DNA enters the funnel from the microfluidic channel and into the nanofluidic circuit. (ii) ‘From the funnel to trap,’ where the DNA leaves the funnel, stretches briefly in the short nanochannel section and enters into the nano-trap. (iii) ‘Escape from trap,’ where the DNA resides in the trap for a period of time and subsequently ejected from the nano-trap and inserted into the stretching nanochannel. (iv) “Stretching of dsDNA” in the stretching nanochannel to near its full contour length and is parked so as to detect the AP sites. For these experiments, the driving voltage was 0.1 V DC and the buffer used was 1× TBE (pH = 8.0). The scale bar (5 μm) is shown in Section 4, image (viii) at the top right corner of the image. Box plots showing (B) trapping and (C) translocation times of λ-DNA electrokinetically transported through the multi-structured nanofluidic circuit under a 0.1 V driving voltage having a 400 nm or 600 nm trap. The average trapping time for the 400 nm trap was ~138 ms, while for the 600 nm trap it was 225 ms. The average translocation time through the nanofluidic circuit with a 400 nm trap was 660 ms, while for the 600 nm trap it was 1847 ms. All measurements were performed using 1X TBE buffer (pH = 7.9). Events were captured at 10 ms exposure times resulting in a frame rate of 90 fps and 1 × 1 binning for the EMCCD. The p value calculated between the 400 and 600 nm traps for the trapping and translocation times (Wilcoxon signed rank test) were statistically different at the 95% confidence interval (p<0.05).
The effect of different trap sizes were evaluated. We considered these sets of experiments to be important as we are using a high throughput approach to increase the number of DNA strands that can be effectively analyzed simultaneously. When multiple channels are used, the stochastic nature of DNA entering into the circuit could lead to some molecules exiting the channels while other molecules are imaged. We should note as well that the dsDNA molecules must be parked in the stretching nanochannels to provide sufficient time to image the single molecule. Figure 3B shows the trapping time of λ-DNA in the nano-traps under the influence of 0.1 V DC. The average trapping time of λ-DNA (contour length ≈16 μm) in the 400 nm trap was ~138 ms, which was shorter than the trapping time in the 600 nm trap (225 ms). The experimental results agree to the observations from our simulations (Figure S4A–B). Figure 4B shows boxplots of the translocation times of λ-DNA through the nanofluidic circuit. The translocation time for a circuit with the 400 nm trap ranged from 400 to 800 ms, while for a 600 nm trap, the translocation times ranged from 700 to 1900 ms. We should note that in the present case, we were using λ-DNA as the model, which has a fixed length defined by the base composition of this DNA molecule, which is 48.5 Kbp in length.
Figure 4.

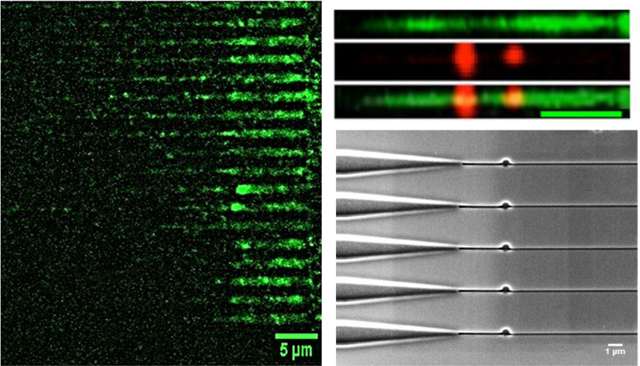
Images of λ-DNA confined in 100 nm × 80 nm stretching nanochannels. (A) Raw images of λ-DNAs’ backbones labeled with YOYO-1 intercalating dye (green) in the multi-structured device having 20 stretching nanochannels. (B) Examples of unprocessed raw images, where the AP sites were labeled with Alexa Fluor 647 streptavidin (red) with AP sites identified within the dsDNA molecules. (C) Processed composite image showing the AP sites co-localized in the DNA strand (yellow). Three specific strands are shown, 1 AP site was identified in 8.9 μm strand, 2 AP sites were identified in a 14 μm length strand and 3 AP sites were identified in a 16.2 μm length DNA strand. The images were processed in Fiji and images of DNA and the AP sites were imaged separately and merged. A scale bar of 5 μm is represented in the images unless otherwise indicated.
DNA stretching and detection of AP sites using the nanofluidic circuit.
Before we embarked upon the detection of AP sites using single streptavidin molecules labeled with Alex Fluor 647 through association with the bARP (see Figure 1), we investigated the ability to detect single dye-labeled streptavidin molecules by dispensing a dilute solution on a COC cover slip (12 pM; 2 μL volume deposited on a glass slide; see Figure S5A–E). We were able to detect single streptavidin molecules with at an average SNR of 15. The fluorescence intensity of single dye-labeled streptavidin molecules was calculated using Image J as a function of gray scale value. The average fluorescence intensity of a single streptavidin molecule was found to be 2,000 – 4,000 arbitrary units. A few streptavidin molecules had an intensity of ~14,000 and this could have been due to those streptavidin molecules being multiply labeled. To assure that we were indeed detecting single streptavidin molecules, the fluorescence image was tracked over time with the intensity of a single spot measured (see Figure S5E). As can be seen from this intensity plot, there was an abrupt cessation of fluorescence observed at that spot indicative of a single bleaching event.
Single dsDNA molecules have been stretched on a glass slide via electrostatic interactions between the DNA and the glass slide with AP sites analyzed using fluorescence.21 The challenge with this approach was the non-controlled stretching of dsDNA, which made it difficult to determine quantitatively the frequency of AP sites (reported as number of AP sites per 105 nucleotides). As we have shown from our previous work, stretching of dsDNA molecules in thermoplastic nanochannels is highly reproducible with the degree of stretching dependent on the nanochannel cross-sectional area.31
For analyzing the AP sites herein, calf thymus DNA with a standard number of AP sites per unit length was used. On application of the appropriate DC voltage (>0.1 V), the DNA was ejected from the nano-trap and moved into the stretching nanochannel where it could be parked (i.e., removal of electric field) for reading AP sites as well as the length of DNA strands by staining with YOYO-1.
The dimension of the stretching nanochannel (100 × 80 nm) falls under the regime between the classic Odijk (50 nm) and extended de Gennes (100 nm) regimes. For a stretching nanochannel that was 100 nm x 80 nm, Dave = 89, and from our previous work using a PMMA/COC nanofluidic devices, the DNA extension here was determined to be 60%.31 Figure 4A shows single DNA molecules parked in the stretching nanochannels. Towards the left of the image is where the trap and funnel would be located There were 20 stretching nanochannels in the device and the image shows single DNA molecules stretched in 18 of them. The purpose of using multiple channels was to increase the throughput of the AP site determinations.49 The DNA molecules shown here are of different lengths as the mixture contains calf-thymus DNA, which has variable lengths (see Figure S6). As the DNA molecules entered the stretching nanochannels from the trap, the electric field was turned off to park the DNA. The 488 nm laser line was used to image the DNA backbone labelled with YOYO-1 first. The AP sites labelled with streptavidin molecules were then imaged using the 647 nm laser. Once both images were acquired, they were merged for colocalization of the DNA strand (YOYO-1 image) and the AP sites (Alexa Fluor 647 image), which also assisted to discriminate between true AP sites and speckles generated from the background in the fluorescence image (see Figure S5C).
Figure 4B shows two unprocessed images with three rows for each image (only one stretching nanochannel is shown). The first row in the image is the DNA molecule imaged with the 488 nm laser stained with YOYO-1 that allowed determining the DNA molecule’s length. For imaging AP sites (second row), the filter cube on the fluorescence microscope was changed to the 647 nm laser and red fluorescence was correspondingly imaged. The DNA with their AP sites was merged (third row) with the dsDNA back bone to find the number of AP sites/105 nucleotides. While the calorimetric assay determined the number of AP sites based on absorbance (see Figure S7 in the SI),50 we determined the number and position of AP sites at the single-molecule level. The distance between nucleotides is 0.34 nm, so the corresponding length of 105 nucleotides is 34 μm. The standard DNAs from calf thymus had 0, 2.5, 5, 10, 20, 40 AP sites in a 34 μm length of dsDNA. As can be seen from Figure S7, we could produce calibration plots for AP sites spectrophotometrically, but this required high concentrations of DNA standards (0.5 ng/μL).
DNA lengths were measured from the fluorescence images using Image J based on 8.44 pixels equivalent to 1 μm. Figure 4C shows three representative images where the lengths of the single dsDNA molecules were measured to be 8.9, 14 and 16.2 μm having 1, 2 and 3 AP sites (red spots colocalized with the DNA green strand), respectively. The AP sites were quantified using the intensity profile observed when imaging single streptavidin molecules. The intensity observed over the integrated intensity of 1,000 arbitrary units were counted as a single AP site and for values that exceeded 6,000 arbitrary units, two AP sites were scored. Twenty calf thymus DNA strands were analyzed and the average AP site frequency was found to be 6.0/105 ±1.8/105, which was close to the 5.0/105 nucleotide AP site standard used here. We should note that for these AP site standards, the length of DNA fragments was highly variable and ranged from 250 to ~48,500 bp based on gel electrophoresis data; see Figure S5 (average Mw of calf-thymus DNA is 8,580,000).51 The negative control for these experiments were measured using λ-DNA molecules that did not contain AP sites, and no streptavidin molecules were found to be colocalized with the YOYO-1 stained DNA strand.
Conclusion
We report a nanofluidic device made from thermoplastics via NIL for measuring AP sites in single DNA molecules. The use of NIL provided reasonable production rates at modest costs to allow implementing the device for in vitro diagnostics that requires one-time use to minimize false positive results from possible sample carryover artifacts. Following NIL of the fluidic substrate, the device could be sealed with a cover plate using thermal fusion bonding of a plastic with a lower Tg than that of the substrate to minimize deformation of the underlying structures, especially the nanostructures.31 The fluidic device possessed three components: (i) Input tapered funnel to increase loading rates of DNA into the nanofluidic circuit; (ii) entropic nano-trap for DNA storage to allow simultaneous loading of DNAs into nanochannel arrays; and (iii) stretching nanochannels for extending the dsDNA to near its contour length to read AP sites with high spatial resolution. In microscale channels, the DNA would be randomly coiled and thus, hard to read individual AP sites nonetheless determine DNA length to arrive at AP site frequency.
The ability to analyze the position of AP sites within a DNA strand are important as studies have shown preferential formation of AP sites in areas undergoing replication.21 The ability of detecting AP sites in DNA will have important applications to cancer chemotherapy, where the frequency of AP sites can be used to track their response to therapy. The nanofluidic assay reported herein will allow for the quantification of the number of AP sites even from single molecules. The nanofluidic circuit was able to efficiently capture single DNA molecules, store them in nano-traps, and subsequently eject them into stretching nanochannels to read the AP sites directly.
One of the advantages of the AP site detection method employed herein is its ability to detect AP sites in single molecules as shown in Figure 4. Spectrophotometric kits require large amounts of DNA to get viable results. For example, the spectrophotometric kit for detecting AP sites required ≥0.5 ng/μL of DNA to provide quantitative data (see Figure S7 in SI). This will become extremely important when considering sourcing the DNA for AP frequency determinations from circulating tumor cells, CTCs. For example, in a single CTC 6 pg of DNA are found and even 100 CTCs (600 pg) may not provide sufficient amounts of DNA for the spectrophotometric assay. Also, amplification of the DNA via PCR to accommodate the mass limits associated with the commercial kit is not feasible, because polymerization through the AP site can incorporate any nucleotide and thus, create errors in the analysis. Our assay can work directly on non-amplified DNA.
Supplementary Material
Acknowledgements
The authors would like to thank the NIH for funding of this work (NIBIB: P41 EB020594; NCI: P30 CA168524; P20 GM130423). The authors also thank the Kansas University Nanofabrication Facility and the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for assistance in making the nanofluidic devices used in this work.
References
- 1.Boiteux S. and Guillet M, DNA Repair, 2004, 3, 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya A, Chattopadhyay R, Mitra S. and Crowe SE, Physiological reviews, 2014, 94, 329–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.COOKE MS, EVANS MD, DIZDAROGLU M. and LUNEC J, The FASEB Journal, 2003, 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Liu L, Wu C, Bulgar A, Somoza E, Zhu W. and Gerson SL, Nuclear medicine and biology, 2009, 36, 975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozcelikkale A, Shin K, Noe-Kim V, Elzey BD, Dong Z, Zhang J-T, Kim K, Kwon IC, Park K. and Han B, Journal of controlled release : official journal of the Controlled Release Society, 2017, 266, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asensio-López MC, Soler F, Pascual-Figal D, Fernández-Belda F. and Lax A, PloS one, 2017, 12, e0172803-e0172803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fundador E. and Rusling J, Anal Bioanal Chem, 2007, 387, 1883–1890. [DOI] [PubMed] [Google Scholar]
- 8.Roberts KP, Sobrino JA, Payton J, Mason LB and Turesky RJ, Chemical Research in Toxicology, 2006, 19, 300–309. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Liberman RG, Skipper PL, Margolin Y, Tannenbaum SR and Dedon PC, Analytical Biochemistry, 2005, 343, 84–92. [DOI] [PubMed] [Google Scholar]
- 10.Atamna H, Cheung I. and Ames BN, Proceedings of the National Academy of Sciences of the United States of America, 2000, 97, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubo K, Ide H, Wallace SS and Kow YW, Biochemistry, 1992, 31, 3703–3708. [DOI] [PubMed] [Google Scholar]
- 12.Ide H, Akamatsu K, Kimura Y, Michiue K, Makino K, Asaeda A, Takamori Y. and Kubo K, Biochemistry, 1993, 32, 8276–8283. [DOI] [PubMed] [Google Scholar]
- 13.Kow YW and Dare A, Methods, 2000, 22, 164–169. [DOI] [PubMed] [Google Scholar]
- 14.Shalhout S, Haddad D, Sosin A, Holland TC, Al-Katib A, Martin A. and Bhagwat AS, Molecular and cellular biology, 2014, 34, 4019–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ and Uhr JW, 9. [DOI] [PubMed] [Google Scholar]
- 16.He W, Kularatne SA, Kalli KR, Prendergast FG, Amato RJ, Klee GG, Hartmann LC and Low PS, International journal of cancer, 2008, 123, 1968–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sikorsky JA, Primerano DA, Fenger TW and Denvir J, Biochem Biophys Res Commun, 2007, 355, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun HB, Qian L. and Yokota H, Analytical Chemistry, 2001, 73, 2229–2232. [DOI] [PubMed] [Google Scholar]
- 19.Hirose T, Ohtani T, Muramatsu H. and Tanaka A, Photochemistry and Photobiology, 2002, 76, 123–126. [DOI] [PubMed] [Google Scholar]
- 20.Kim J, Muramatsu H, Lee H. and Kawai T, FEBS letters, 2003, 555, 611–615. [DOI] [PubMed] [Google Scholar]
- 21.Chastain PD, Nakamura J, Rao S, Chu H, Ibrahim JG, Swenberg JA and Kaufman DG, The FASEB Journal, 2010, 24, 3674–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan T-F, Ha C, Phong A, Cai D, Wan E, Leung L, Kwok P-Y and Xiao M, Nucleic acids research, 2006, 34, e113-e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C, Qu Y, Luo Y. and Fang N, Electrophoresis, 2011, 32, 3308–3318. [DOI] [PubMed] [Google Scholar]
- 24.Fijen C, Fontana M, Lemay SG, Mathwig K. and Hohlbein J, bioRxiv, 2017, DOI: 10.1101/201079,201079. [DOI] [PubMed] [Google Scholar]
- 25.Gibb B, Silverstein TD, Finkelstein IJ and Greene EC, Analytical Chemistry, 2012, 84, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins BE, Ye LF, Duzdevich D. and Greene EC, in Methods in Cell Biology, eds. Waters JC and Wittman T, Academic Press, 2014, vol. 123, pp. 217–234. [DOI] [PubMed] [Google Scholar]
- 27.Kim D, Rashid F, Cho Y, Zaher MS, IIHwanCho S. Hamdan r., Jeong C. and Lee J-B, Nucleic Acids Research, 2019, 47, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uba FI, Hu B, Weerakoon-Ratnayake K, Oliver-Calixte N. and Soper SA, Lab on a Chip, 2015, 15, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uba FI, Pullagurla SR, Sirasunthorn N, Wu J, Park S, Chantiwas R, Cho YK, Shin H. and Soper SA, Analyst, 2015, 140, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weerakoon-Ratnayake KM, O’Neil CE, Uba FI and Soper SA, Lab Chip, 2017, 17, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uba FI, Hu B, Weerakoon-Ratnayake K, Oliver-Calixte N. and Soper SA, Lab Chip, 2015, 15, 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uba FI, Pullagurla S, Sirasunthorn N, Wu J, Park S, Chantiwas R, Cho Y-K, Shin H. and Soper SA, Analyst, 2014, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weerakoon Ratnayake K, LSU Doctoral Dissertations, 2015. [Google Scholar]
- 34.O’Neil C, Amarasekara CA, Weerakoon-Ratnayake KM, Gross B, Jia Z, Singh V, Park S. and Soper SA, Analytica Chimica Acta, 2018, 1027, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou SY, Krauss PR and Renstrom PJ, Applied Physics Letters, 1995, 67, 3114–3116. [Google Scholar]
- 36.Chou SY, Krauss PR and Renstrom PJ, Journal of Vacuum Science & Technology B, 1996, 14, 4129–4133. [Google Scholar]
- 37.Chou SY and Krauss PR, Microelectronic Engineering, 1997, 35, 237–240. [Google Scholar]
- 38.Duan C, Wang W. and Xie Q, Biomicrofluidics, 2013, 7, 026501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utko P, Persson F, Kristensen A. and Larsen NB, Lab on a Chip, 2011, 11, 303–308. [DOI] [PubMed] [Google Scholar]
- 40.Lin J, Qi R, Aston C, Jing J, Anantharaman TS, Mishra B, White O, Daly MJ, Minton KW, Venter JC and Schwartz DC, Science, 1999, 285, 1558–1562. [DOI] [PubMed] [Google Scholar]
- 41.Hsieh S-F and Wei H-H, Phys. Rev. E, 2009, 79, 021901. [DOI] [PubMed] [Google Scholar]
- 42.Klotz AR, 92. [Google Scholar]
- 43.Austin R, Nature Materials, 2003, 2, 567–568. [DOI] [PubMed] [Google Scholar]
- 44.Zhou J, Wang Y, Menard LD, Panyukov S, Rubinstein M. and Ramsey JM, Nature Communications, 2017, 8, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner SWP, Cabodi M. and Craighead HG, Physical Review Letters, 2002, 88, 128103. [DOI] [PubMed] [Google Scholar]
- 46.Han J, Turner SW and Craighead HG, Physical Review Letters, 1999, 83, 1688–1691. [Google Scholar]
- 47.Hana J. and Craighead HG, J. Vac. Sci. Technol, 1999, 17, 2142–2147. [Google Scholar]
- 48.Han J. and Craighead HG, Science, 2000, 288, 1026–1029. [DOI] [PubMed] [Google Scholar]
- 49.Weerakoon-Ratnayake KM, Vaidyanathan S, Amarasekara CA, Johnson CK and Soper SA, in Spectroscopy and Dynamics of Single Molecules, ed. Johnson CK, Elsevier, 2019, pp. 335–377. [Google Scholar]
- 50.Masyuk AI, Masyuk TV and Larusso NF, J Hepatol, 2013, 59, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porsch B, Laga R, Horsky J, Konak C. and Ulbrihc K, Biomacromolecules, 2009, 10, 3148–3150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.