

Summary
The ever-increasing size and scale of biological information have popularized network-based approaches as a means to interpret these data. We develop a network propagation method that integrates kinase-inhibitor-focused functional screens with known protein-protein interactions (PPIs). This method, dubbed KiRNet, uses an a priori edge-weighting strategy based on node degree to establish a pipeline from a kinase inhibitor screen to the generation of a predictive PPI subnetwork. We apply KiRNet to uncover molecular regulators of mesenchymal cancer cells driven by overexpression of Frizzled 2 (FZD2). KiRNet produces a network model consisting of 166 high-value proteins. These proteins exhibit FZD2-dependent differential phosphorylation, and genetic knockdown studies validate their role in maintaining a mesenchymal cell state. Finally, analysis of clinical data shows that mesenchymal tumors exhibit significantly higher average expression of the 166 corresponding genes than epithelial tumors for nine different cancer types.
Keywords: kinase, network, propagation, computational, modeling, protein, interaction, hepatocellular, carcinoma
Graphical abstract
Highlights
-
•
Network propagation is a powerful way to integrate and interpret biological data
-
•
KiRNet produces protein-protein interaction models from kinase inhibitor screens
-
•
We identify a network of molecular regulators of mesenchymal cancer cell state
-
•
KiRNet provides a systems-level insight from pharmacological screen results
Motivation
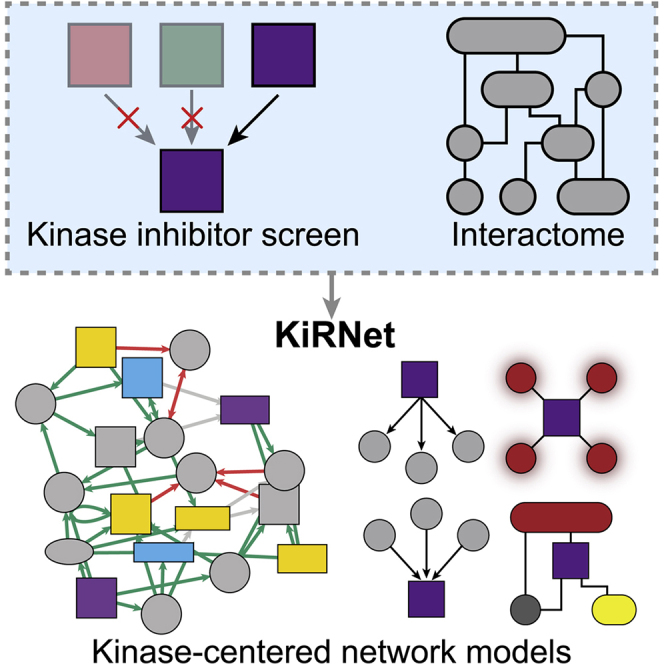
We have previously developed a powerful ensemble modeling approach called Kinome Regularization (KiR) that uses a functional kinase inhibitor screen to predict key kinases contributing to a phenotype, such as cell migration. However, kinase-mediated protein-protein interactions (PPIs) might also contribute to the phenotype. We sought to extend the method by integrating hits with known PPIs to accurately model relationships among them and robustly identify additional non-kinase targets. By adapting existing network propagation principles and methods and optimizing them for KiR's kinase predictions, we develop a complete, robust pipeline to go from a drug screen to kinase-centered, functional network modules.
Network propagation approaches use known protein-protein interactions to amplify biological results into new predictions. Bello et al. introduce KiRNet to produce kinase-centered network models from kinase inhibitor functional screen results. They apply KiRNet to predict and validate a functional network model of the molecular regulators of FZD2-driven mesenchymal cancer cells.
Introduction
In the past few decades, biosciences have seen an incredible rise in high-throughput technologies and large-scale databases (D'Argenio, 2018; Moutsatsos and Parker, 2016; Türei et al., 2016). Researchers are increasingly leveraging network-based approaches to integrate and interpret these data into actionable, functionally relevant output (Cowen et al., 2017). These techniques model biological systems as collections of nodes (genes, proteins, compounds, etc.) and edges (interactions between two nodes, e.g., phosphorylation, repression, etc.). Networks are often generated from large databases of protein-protein interactions (PPIs), such as STRING, HPRD, the Kyoto Encyclopedia of Genes and Genomes (KEGG), and OmniPath (Lehne and Schlitt, 2009). Network-based approaches yield powerful insights by connecting context-specific information to prior knowledge of biological interactions and enabling the use of topological and mathematical tools to interrogate the resulting model.
Our previously established KiR approach uses elastic net regularization to regress the results of a quantitative drug screen using a specific set of kinase inhibitors (KIs) against the previously quantified effects of those inhibitors on nearly 300 human protein kinases (Anastassiadis et al., 2011; Gujral et al., 2014a; Rata et al., 2020). The result is a list of hits (in this case, kinases) that are predicted to act as key mediators of the given cellular function. We have applied this approach to study phenotypes ranging from cell migration to malarial liver-stage infection (Arang et al., 2017; Gujral et al., 2014a). Here, we developed KiRNet, a method that applies a local network propagation approach to expand on the functional insight provided by KiR. KiRNet maps the results of KiR onto a previously generated PPI network to create kinase-centered, functional network models. These models bring three key innovations to KiR by: (1) extending KiR to non-kinase proteins; (2) integrating predictions with prior knowledge, including context-specific data, such as mRNA levels (although these data are not required to run KiRNet), and broader knowledge, such as PPIs; and (3) creating a cohesive, network-level understanding of the specific proteins and interactions that result in an observed phenotype.
We validated KiRNet performance by using an epithelial hepatocellular carcinoma (HCC) cell line, Huh7 (Huh7 wild-type [WT]), engineered to overexpress FZD2 (Huh7-Fzd2), a receptor for WNT5A/B that drives epithelial-mesenchymal transition (EMT) and cancer metastasis in HCC (Golkowski et al., 2020a; Gujral et al., 2014b). KiRNet produced a kinase-centered network model consisting of 166 proteins predicted to mediate this FZD2-driven mesenchymal cancer cell state. Mass spectrometry data confirmed that proteins in this network displayed strong differential phosphorylation in a FZD2-dependent manner, and perturbing this network significantly affected multiple EMT-associated phenotypes, including cell migration and E-cadherin expression. Furthermore, the expression of the network members was found to be elevated in mesenchymal tumors across nine cancer types in The Cancer Genome Atlas (TCGA), including HCC. These results demonstrate the power of KiRNet to turn functional screen results into predictive, functional network models of complex disease phenotypes, such as Fzd2-driven HCC.
Results
Transforming drug screen results into a context-specific PPI network
The first input step in KiRNet is our previously established KiR method to identify kinases predicted to be functionally important for HCC growth (Figure 1A). We screened a total of 42 KIs in both Huh7 WT and Huh7-Fzd2 cell lines via live-cell imaging, using endpoint confluence (percent of image area occupied by cells) as a quantitative measure of growth and proliferation (Figure S1A, Data S1; see STAR Methods). The quantitative effects of each of these 42 KIs on nearly 300 human protein kinases have been previously characterized (Anastassiadis et al., 2011; Rata et al., 2020). We used these quantitative inhibition profiles and each drug's responses as the explanatory and response variables, respectively, to generate elastic net-regularized multiple linear regression KiR models, as described previously (Gujral et al., 2014a) (Figures S1B and S1C; see STAR Methods for details and code). There were 17 kinases with positive coefficients in both the Huh7 WT and Huh7-Fzd2 models, designating these kinases as most explanatory in predicting cell confluence in response to a KI (Figure S1D, Data S1). Only two kinases, TAOK1 and TEK, were predicted in both models, giving us confidence that these predictions represented cell-specific dependencies rather than broadly essential genes. The 17 kinases predicted by the Huh7-Fzd2 KiR model are heretofore referred to as the key kinases or key functional nodes inputted into KiRNet.
Figure 1.

KiRNet expands lists of hits from functional screens into network-level hypotheses
(A) Schematic overview of the KiRNet method.
(B) Kinase enrichment and differential phosphorylation enrichment at different cutoffs of distance from key functional nodes in the network. Enrichment of both reaches a peak at a distance of 11 (arbitrary units), establishing the cutoff for the refined subnetwork. ∗∗∗p < 0.001, Fisher's exact test.
(C) Closeness for the 429 nodes in the optimally refined subnetwork, normalized to a minimum of 0 and mean of 1. Red bars indicate that the node exhibits significantly higher phosphorylation in Huh7-Fzd2 cells compared with Huh7 WT cells. Selected differentially phosphorylated nodes are labeled. An empirically determined closeness cutoff of 1.1149274 was enforced to identify the high-value subnetwork of 166 proteins (p < 0.0001, Fisher's exact test).
(D) Subnetworks produced by KiRNet for Huh7-Fzd2. High-value subnetwork (left) contains 166 proteins chosen via an empirical cutoff for closeness (as calculated in the refined subnetwork). The top 25 proteins (right) show the subnetwork of the 25 proteins with the highest closeness in the refined subnetwork. In both networks, sibling nodes (nodes with identical edges) have been collapsed into single nodes, thus each node might represent more than one protein.
To extend these KiR predictions into a network-based modeling approach, we next compiled a list of PPIs. In general, PPIs might be sourced from many public databases, such as STRING, HPRD, OmniPath, BioGrid, or others (Lehne and Schlitt, 2009; Szklarczyk et al., 2017; Türei et al., 2016). We chose to use the KEGG despite it containing fewer interactions than many others as it is more curated and well-annotated, potentially making our models more robust (Kanehisa, 1996). The interactions in KEGG were collated and simplified, resulting in a general PPI network of 6,021 nodes (genes or proteins) and 59,142 interactions (see STAR Methods).
The third (optional) input for KiRNet is any additional data annotating the nodes (genes/proteins), such as mRNA levels, mutational status, or (phospho)peptide levels. Although none of these data are needed to run KiRNet, incorporating them gives the user better metrics to optimize and assess the models (demonstrated below). We collected previously published data on genetic mutations and RNA expression levels for Huh7 WT and Huh7-Fzd2 cell lines (Cerami et al., 2012; Gao et al., 2013; Xue et al., 2020). Kinase protein and phosphopeptide expression for Huh7 WT and Huh7-Fzd2 cell lines were determined by using our kinobead and liquid chromatography-mass spectrometry (LC-MS) kinome profiling workflow as described previously (see STAR Methods) (Golkowski et al., 2017, 2020a). Based on these data, we excluded any nodes with an mRNA expression log2(CPM+1) level less than 1 to remove the lowest expressed ∼6% of genes (372 out of 6,021) mapped to the PPI network. We removed a further ∼6% (348) of nodes that were disconnected from the rest of the network in small (>33 nodes), isolated clusters, leaving a context-specific PPI network of 5,301 nodes and 48,425 interactions predicted to be present and occurring in Huh7-Fzd2 cells.
A priori edge weighting based on node degree
Mapping the KiR predictions to a contextualized PPI network allows us to leverage topological and computational techniques to predict subnetworks that are functionally important for Huh7-Fzd2 growth. Because we were particularly interested in kinome signaling, we chose to pursue a local network propagation approach rather than a global network or diffusion-based approach, as both methods have been shown to generate unique and valuable predictions (Navlakha and Kingsford, 2010). However, for both biological and technical reasons, PPI networks exhibit a “scale-free” structure; that is, the distribution of node degree (the number of edges connected to a node) follows a power-law distribution, with a small number of “hub” nodes exhibiting disproportionately large numbers of interactions (Albert, 2005; Pržulj et al., 2004). This poses two distinct challenges: (1) network propagation methods, especially local methods, are heavily biased toward these hub nodes (Erten et al., 2011); and (2) distance-based metrics (i.e., number of “steps” between two nodes in a network) are skewed by connections through these hubs, making them less reliable than global or diffusion-based metrics (Cao et al., 2013; Erten et al., 2011). To address these challenges, we developed an a priori strategy to assign weights, or distances, to each edge in the network, with higher weights signifying a less likely or more difficult interaction. We computed the edge weight between two nodes as the natural logarithm of the product of their degrees, such that edges between nodes with high degrees will have larger weights (see “edge weighting” in STAR Methods). This effectively penalizes paths through hub nodes and provides a much finer-grained distance metric to use in downstream analyses. By correcting for the primary sources of bias for distance-based metrics, we are able to use local network propagation with confidence (Cao et al., 2013; Erten et al., 2011; Navlakha and Kingsford, 2010). Notably, computing these edge weights requires no additional data beyond the PPI network. By quantitatively differentiating edges this way, we created a context-specific, PPI network ready to be topologically interrogated.
Identifying high-value nodes using network propagation and topological analysis
Armed with a contextualized PPI network and a priori edge weights to improve local network methods, we sought to identify a small, testable subnetwork that captures the differential signaling in mesenchymal HCC (Huh7-Fzd2) cells. To focus on kinase-centered subnetworks, we assigned every node a “functional distance” equal to the weighted, undirected path distance to the nearest key kinase. We then created series of hypothetical subnetworks by enforcing a functional distance cutoff at integer values between 1 and 30 (arbitrary units) and calculated the enrichment of kinases in each subnetwork (see STAR Methods). This enrichment peaked at a cutoff of 11 (p < 0.001; Figure 1B). For mesenchymal cancer cells (Huh7-Fzd2), we exploited quantitative phosphopeptide data that provided additional insight into which proteins are likely to be functionally important for these cells' growth. The enrichment of differentially phosphorylated nodes (compared with Huh7 WT) also peaked at a distance cutoff of 11 (p < 0.001), reinforcing this as a meaningful cutoff (Figure 1B). Although these two metrics are not independent, as the phosphopeptide data are biased toward the kinome- and kinase-interacting proteins, this validates the use of kinase enrichment as an a priori means of identifying a meaningful kinome-centered subnetwork and provides further support for the flexibility of KiRNet.
The resulting subnetwork, referred to as the “optimally refined” subnetwork, serves as our hypothesis-generating platform for determining the functionally important nodes and edges in regulating and mediating the phenotype of interest. For our mesenchymal HCC model, this subnetwork contained 429 nodes. We wanted to predict which of these nodes are “high-value” nodes that can be targeted to alter the cellular phenotype. To this end, we tested many established measures of node centrality, including closeness, betweenness, degree, and combinations thereof (Freeman, 1978). The previously defined edge weights ensure that even these local, distance-based centralities are not dominated by hub nodes. Comparing these measures with our quantitative phosphopeptide data, we found closeness, defined as the inverse of the average distance between a node and all other nodes in the network, was the most effective at predicting differentially phosphorylated proteins (Figures 1C and S2A). Thus, when there are no additional data present, this “refined closeness” can be used as a topological predictor of a node's differential regulation and, consequently, its functional importance for the kinase-mediated phenotype. We created a high-value subnetwork of 166 proteins by using an empirical closeness cutoff to maximize the enrichment of differentially phosphorylated nodes, while also minimizing the size of the subnetwork (Figures 1D and S2B; see STAR Methods). This subnetwork serves as the final KiRNet model for our experimental system: a focused subnetwork, centered around the predicted key kinases, that predicts the proteins and relationships most critical for the phenotype of interest.
Validating the KiRNet model predictions
Our KiRNet model predicts the kinase-centered subnetwork of proteins and interactions that regulate the mesenchymal cancer cell state observed in FZD2-expressing Huh7 cells. We compared differential kinase phosphorylation between Huh7-Fzd2 and Huh7 WT cells with differential kinase phosphorylation in FOCUS cells as a preliminary validation. The FOCUS HCC cell line exhibits endogenously high levels of FZD2 and is commonly used in Wnt signaling studies (Gujral et al., 2014b). We have previously generated a stable cell line with a short-hairpin knockdown of FZD2 that we refer to as FOCUS-shFZD2 (Gujral et al., 2014b), and we collected previously published quantitative kinase phosphopeptide expression data determined by kinobead/LC-MS kinome profiling (data available in Golkowski et al., 2020a). Given the central role of FZD2 in our mesenchymal HCC system, we hypothesized that the changes in phosphorylation of the Huh7-Fzd2 high-value proteins would mirror those in FOCUS-shFzd2; that is, high-value proteins with increased phosphorylation in Huh7-Fzd2 (compared with Huh7 WT) would exhibit decreased phosphorylation in FOCUS-shFzd2 (compared with FOCUS WT), and vice versa. We observed that 45 of the 96 (46.9%) measured phosphosites on high-value nodes showed this mirrored responses in Huh7-Fzd2 and FOCUS-shFzd2 (Figure 2A, left), exhibiting a modest enrichment in the high-value network compared with all phosphosites measured (p < 0.1, Fisher's exact test). These mirrored responses support the hypothesis that these high-value nodes, chosen by their topological closeness, are functionally important for the FZD2-mediated phenotype in both cell lines.
Figure 2.

KiRNet model identifies a critical subnetwork regulating a FZD2-driven, mesenchymal cancer cell state
(A) Heatmap of phosphorylation changes in Huh7-Fzd2 and FOCUS cells. The left panel shows all 96 detected phosphosites on all of the proteins from the high-value model; the right panel shows selected phosphosites from proteins chosen for further validation. Data are presented as the log fold change in phosphopeptide level between the modified cell line (Huh7-Fzd2 or FOCUS shFZD2) and the parental cell line (Huh7 WT or FOCUS WT, respectively). The top group contains sites decreased in Huh7-Fzd2 and increased in FOCUS shFZD2; the second group contains sites increased in Huh7-Fzd2 and decreased in FOCUS shFZD2; the third group contains sites decreased in both; the bottom group contains sites increased in both.
(B) Quantitative real-time PCR results for E-cadherin (CDH1) expression in Huh7-Fzd2 cells transfected with transient siRNA knockdowns targeting various high-value genes. Presented as the log2 fold change compared with a non-targeting siRNA. Data are presented as the mean of three technical replicates.
(C) Quantitative changes in cell migration as assessed by a wound-healing assay in Huh7-Fzd2 transfected with transient siRNA targeting various high-value genes. siFZD2 and siTGFBR1 are included as positive controls, and siMERTK is included as a negative control. Data are presented as mean ± SEM of at least three biological replicates. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, one-way ANOVA with two-tailed Holm-Sidak multiple comparisons test.
(D) Expression of the high-value nodes from the Huh7-Fzd2 KiRNet model in TCGA cohorts. Samples were stratified into epithelial or mesenchymal sample types based on gene expression signatures. LogCPM signature of the high-value genes was significantly higher in mesenchymal samples in 9 out of 17 tested cancer types, including HCC. Signature expression was significantly decreased in mesenchymal ovarian cancer samples, and not significantly different in the other cancer types (non-significant results not shown; see STAR Methods for a full list of cancer types assessed). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, Wilcoxon rank-sum test. Abbreviations are as follows: COAD, colon adenocarcinoma; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; LIHC, liver HCC; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma.
Next, we selected a panel of specific nodes from the mesenchymal cell KiRNet model and further validated their role in the mesenchymal phenotype. We chose nodes from the following categories: a kinase predicted by our original KiR model (PRKCQ); kinases not predicted by our functional screen (EPHA2, MAP2K2/6, MAP3K11, MAPK8/9/10, PAK1/2/4, LIMK1, MAP2K3/4, and MAP3K1/14) and non-kinases (ARHGEF12, AZI2, IRS1, RAC1, RHOA, TAB1, TAB2, and TBKBP1). The phosphosites on these chosen proteins were representative of the high-value network, with approximately half of the sites changing in ways that correspond to the FZD2 status of both Huh7 and FOCUS cells (Figure 2A, right).
To quantify these proteins' effects on the mesenchymal-like state driven by FZD2, we depleted their expression levels in Huh7-Fzd2 cells via RNAi (Figure S2D). Expression levels of E-cadherin, a common epithelial cell state marker, exhibited an over 3-fold increase for approximately half of the genes tested, including RAC1, MAPK8/9/10, PAK2/4, RHOA, TAB1/2, TBKBP1, and MAP3K1, reinforcing the role these proteins play in maintaining the mesenchymal state of these cells (Figure 2B). We also performed a wound-healing assay as an aggregate measure of the migratory and proliferative phenotype exhibited by FZD2-driven cells. We observed appreciable decreases in wound closure for the majority of tested small interfering RNA (siRNA) targets (Figure 2C). Depletions of MAPK10, PAK1/2, RHOA, TAB1/2, TBKBP1, and MAP3K1 each resulted in more than a 35% decrease in wound closure compared with scrambled siRNA control (p < 0.05; Figures 2C and S2E). Together, these data identify kinases and non-kinases that play a role in the mesenchymal cancer cell phenotype.
Finally, we asked whether the KiRNet identified nodes in the mesenchymal HCC cells are also expressed in a broader set of mesenchymal-like tumors. We analyzed mRNA expression of the high-value genes across 17 different cancer types in TCGA database (The Cancer Genome Atlas Research Network, 2017). Samples in TCGA were stratified into epithelial- or mesenchymal-like cell states based on the expression of key marker genes, as defined previously (Xue et al., 2020) (see STAR Methods). A high-value signature was computed for each sample from expression levels of the 166 high-value genes. Nine of the tested cancer types, including HCC, showed significantly higher expression of this high-value signature in mesenchymal-like cancers, whereas only ovarian cancer showed significantly decreased expression (Figure 2D). This provides further support that the high-value network predicted by using KiRNet mediates a mesenchymal-like phenotype, found in both FZD2-driven cells and mesenchymal-like cancers.
Discussion
As the cost and time needed for many functional screens decreases, there will be an ever-increasing need to follow up the resulting leads. Traditional approaches of manual literature curation and one-by-one experiments can take years to interpret and contextualize these results. KiRNet meets this need and capitalizes on the functional insights provided by KiR and similar screening methods by integrating them with contextualized PPI networks to provide a cohesive understanding of the biological system.
We demonstrated the power of KiRNet by identifying a network of proteins that are functionally important for mesenchymal cancer cells. We validated that these proteins are indeed critical for maintaining the migration and proliferation that accompanies the mesenchymal cell state. Some proteins, such as RAC1, RHOA, and SRC, have already been established as key players in this area (Jansen et al., 2018; Patel et al., 2016; Xia et al., 2019). Others, such as TBKBP1, PAK1/2/4, and MAPK8/9/10, expand on the initial functional screen hits and allow us to consider new proteins and interactions that otherwise would have been missed. KiRNet is especially powerful when handling limited amounts of data, as high-value predictions can be made based merely on a list of functional hits.
KiRNet draws heavily on existing network analysis principles and methods. At its core, KiRNet refines a global network into a local network propagation approach. Although many existing methods rely on global network approaches, focused network models have been shown to make valid predictions that are distinct and complementary to global methods (Navlakha and Kingsford, 2010). For example, RIDDLE developers demonstrated that the addition of a local extension step detected additional functionally associated genes when coupled to a global diffusion-based approach (Wang et al., 2012). Similarly, KiRNet's local network approach yields valuable insights that can augment and complement other global network approaches. A key innovation of KiRNet, the a priori edge weighting based on degree, seeks to address the same challenges many as other network approaches. These include the scale-free-like structure of biological networks, which biases distance-based analyses toward large hub nodes, and the “small world” nature of these networks, which causes local propagation methods to quickly balloon to intractable network sizes (Cao et al., 2013). Previously, DADA developers showed that both random walk with restarts and network propagation approaches are biased toward nodes with a high degree (Erten et al., 2011). Although this could be due to these proteins' more central role in mediating phenotypes (a variation of the “centrality-lethality hypothesis”), the authors demonstrate that, by including statistical adjustments that effectively penalize high-degree nodes, DADA lowered the false-positive rate in identifying disease-associated genes. Our edge-weighting strategy in KiRNet provides a similar correction to the bias toward hub nodes. Similarly, Cao et al. (2013) developed a new distance metric termed diffusion state distance (DSD) to address that pairs of nodes in PPI networks are separated by an average of only two to three edges, causing even small propagations to generate enormous networks. DSD transforms whole-number distances into a more continuous distribution of path lengths by using a global diffusion approach; KiRNet's a priori edge weighting accomplishes the same function using a local approach (Cao et al., 2013).
KiRNet's distinguishing features are its intrinsic relationship to our KiR method and the ability to generate predictive models without requiring any large-scale datasets. By directly extending KiR, KiRNet creates a complete pipeline from a functional drug screen to prioritized network modules. Although we validated the method in an in vitro cell line system, it can be applied to any model system in which the quantitative responses to ∼30 KIs can be assessed, from in vivo animal models to individual slices of a single tumor (Nishida-Aoki and Gujral, 2019). Because the resulting network models are kinase centered, they are extremely conducive to validation and perturbation via small-molecule KIs (Roskoski, 2019; Wu et al., 2016). The well-documented role of kinase signaling in many malignancies positions KiRNet at a critical intersection between computational modeling and translational medicine (Fleuren et al., 2016; Knapp, 2018; Knight et al., 2010). Finally, by removing the requirement for large-scale data, such as a differential expression or genetic sequencing while still providing a framework for incorporating these data, KiRNet has the potential to provide insights into systems ranging from fully characterized cell lines to primary patient samples.
Limitations of the study
The methods described in this paper continue to be an active area of development. In its current form, KiRNet does not utilize mRNA abundance, protein levels, or phosphopeptide levels in computing the edge weights of the network, potentially limiting valuable information regarding the relative frequency of these interactions. In the future, these data could be used to compute a posterior probability of interaction between two nodes, updating the a priori probabilities currently used via Bayes' theorem. In addition, KiRNet has been tailored to KiR kinase predictions as its seed nodes and relied on additional experimental data to validate the findings. Thus, KiRNet models are biased toward kinome- or kinase-associated nodes and make it most suitable for studying kinase-mediated phenotypes. As more complete chemical biology tools become available, KiRNet might serve as a blueprint for investigating other protein families, such as epigenetic regulators (Fuhrmann et al., 2015; Wu et al., 2019).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Kinase inhibitors used for initial KiR model screening | National Center for Advancing Translational Science, NIH | See Data S1 |
| Deposited data | ||
| Huh7 WT and Huh7-Fzd2 RNA expression data | Xue et al., 2020 | GEO: GSE138380 |
| Huh7 WT, FOCUS, and FOCUS-shFzd2 protein and phosphopeptide data | Golkowski et al., 2020a | MassIVE: MSV000083236 |
| Huh7-Fzd2 phosphopeptide data | This paper | MassIVE: MSV000086446 |
| Mutation data | cBioPortal; Cerami et al., 2012; Gao et al., 2013 | HuH7 Cancer Cell Line Encyclopedia (Novartis/Broad, Nature 2012) https://www.cbioportal.org/patient?studyId=cellline_ccle_broad&caseId=HuH-7 |
| Kinase family and annotations | KinHub Beta | http://www.kinhub.org/kinases.html |
| KEGG Pathway interactions | Kanehisa, 1996 | https://www.genome.jp/kegg/pathway.html |
| TCGA Cancer sample data | National Cancer Institute Genomic Data Commons data portal | https://portal.gdc.cancer.gov/ |
| Experimental models: cell lines | ||
| Huh7 | Gujral et al., 2014b | N/A |
| Huh7-Fzd2 | Gujral et al., 2014b | N/A |
| FOCUS | Gujral et al., 2014b | N/A |
| FOCUS shFzd2 | Gujral et al., 2014b | N/A |
| Oligonucleotides | ||
| qPCR primers |
RealtimePrimers.com BioRad |
See Data S2 |
| siRNA sequences | Horizon ON-TARGETplus siRNA-SMARTpool | See Data S2 |
| Software and algorithms | ||
| KiRNet R script | This paper | https://github.com/FredHutch/KiRNet-Public |
| R version 3.4.1 -- "Single Candle" | R Core Team, 2016 | https://www.r-project.org/ |
| R Studio Version 1.2.1335 | RStudio Team, 2018 | http://www.rstudio.com/ |
| Igraph | Csardi and Nepusz, 2006 | http://igraph.org |
| Tidyr | Wickham et al., 2019 | https://www.tidyverse.org/ |
| Prism 7 for Windows version 7.03 | Graphpad Software, Inc. | https://www.graphpad.com/ |
| KinMap | Eid et al., 2017 | http://www.kinhub.org/kinmap |
| Incuctye ZOOM 2016B | Sartorius | https://www.essenbioscience.com/en/products/software/ |
| Colorspace | Zeileis et al., 2019 | http://colorspace.r-forge.r-project.org/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Taranjit S. Gujral (tgujral@fredhutch.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The custom R script that implements both KiR KiRNet generated in this study is available on GitHub at https://github.com/FredHutch/KiRNet-Public. This repository also includes all associated files needed to execute the script and produce a sample model. The Huh7-Fzd2 mass spectrometry dataset generated in this study are available online in the MassIVE repository (https://massive.ucsd.edu/) under the dataset ID MSV000086446.
Experimental model and subject details
Cell culture and media
Hepatocellular Huh7 cells were obtained from American Type Culture Collection. FOCUS cells were obtained from J. Wands (Brown University). Both cell lines were grown at 37°C under 5% CO2, 95% ambient atmosphere and maintained in Dulbecco’s minimum essential medium (DMEM) supplemented with 10% FBS (Sigma) and 1% Penn Strep. Stable cell lines, FOCUS cells with depleted levels of FZD2 and Huh7 cells expressing FZD2, were generated and cultured as described previously (Gujral et al., 2014b; Xue et al., 2020).
Method details
Kinase inhibitor screening
Kinase inhibitor screening was performed as described previously (Gujral et al., 2014a). Huh7 WT and Huh7-Fzd2 cells were plated in 96 well plates and treated with a panel of 42 kinase inhibitors at 6–8 different doses each. Images of the cells were taken every 2 hours, and cell confluence (the proportion of total area occupied by cells) was quantified via Incucyte ZOOM™ software. Endpoint confluence at each dose was plotted and fitted with a dose-response curve using GraphPad Prism. These curves were interpolated at 500 nM (the dose profiled in the drug-target matrix), and the interpolated response was used as the response variable in building the KiR models.
Elastic Net regularized models
KiR models and the list of predicted key kinases or key functional nodes were generated as previously described (Gujral et al., 2014a). Briefly, a panel of 427 kinase inhibitors previously had their pairwise effects on 298 human kinases profiled (Anastassiadis et al., 2011; Rata et al., 2020). The result is a quantitative drug-target matrix, where each entry is a percentage between 0 and 100 that represents that kinases residual activity (as a percent of control, uninhibited activity) in the presence of that inhibitor. A small panel of these inhibitors were tested on both Huh7 WT and Huh7-Fzd2 cells as described above, with the end result being a single interpolated response for each drug that represents the cell growth (as % control) at the profiled dose of the inhibitor (usually 500 nM). The kinase inhibition profiles of each inhibitor and the quantitative responses to those inhibitors were used as the explanatory and response variables, respectively, for elastic net regularized multiple linear regression models (Zou and Hastie, 2005). Custom R scripts (available at https://github.com/FredHutch/KiRNet-Public) employing the glmnet package were used to generate the final models (Friedman et al., 2010). Leave-one-out cross validation (LOOCV) was used to select the optimal value for the penalty scaling factor λ (Figure S1B). Models were computed for 11 evenly-spaced values of α (the relative weighting between LASSO and Ridge regularization) ranging from 0 to 1.0 inclusive. Kinases with positive coefficients in at least one of these models (with the exception of , which always has non-zero coefficients for every kinase) were considered hits (Figure S1D). Model accuracy was assessed via the LOOCV error as well as the root-mean-squared error of the predictions for the tested inhibitors (Figure S1B–C).
Initial protein-protein interaction network generation
The initial protein-protein interaction network used in this paper was based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway database (Kanehisa, 1996). Custom R scripts (available at https://github.com/FredHutch/KiRNet-Public) were used to download the KEGG Markup Language (KGML) file for each pathway. All pathways that had human (hsa) identifiers were collated to form a single master list of interactions. Because many interactions are present in multiple pathways, this master list was further simplified by aggregating interactions with the same starting node, ending node, and interaction type, and concatenating their subtype annotations. This simplified list was used as the input to igraph to generate a single network consisting of all KEGG Pathway human interactions (Csardi and Nepusz, 2006). The final result contains 6,021 unique proteins and 59,142 unique interactions. Fifty-five non-silent genetic mutations present in Huh7 were retrieved from cBioPortal (https://www.cbioportal.org/patient?studyId=cellline_ccle_broad&caseId=HuH-7), and the corresponding nodes were annotated with a simple true/false (Cerami et al., 2012; Gao et al., 2013). This network was filtered to remove indirect interactions (often present in KEGG pathways to visually condense long signaling cascades into a single step). Nodes with mRNA expression log2(counts per million + 1) < 1 were removed to eliminate the lowest ∼8% of genes expressed in Huh7-Fzd2 cells and not include nodes and edges that are not expressed in our experimental system. The network was then examined to determine how many weakly connected “components” it contains. A “component” is defined as a subnetwork wherein every node is reachable by every other node in the subnetwork; a weakly connected component does not consider the direction of interactions. The largest weakly connected component in the initial Huh7-Fzd2 network contained 94% of all nodes (5,301 nodes out of 5,649 total), while the second largest component contained only 32 nodes. We therefore chose to consider only the largest weakly connected component.
Edge weighting
Once the initial network was generated, a priori weights (or “distances”) were assigned to each edge based on the degree of each node, so as to penalize edges connected to large hub nodes with many interactions. One strategy for assigning edge weights to represent distances (i.e. higher numbers mean less likely interactions) is to take the negative logarithm of the probability of the interaction (Costa et al., 2017):
| (Equation 1) |
In many cases, the probabilities of interactions are derived from the underlying evidence for that interaction. Because KEGG Pathway is manually curated and consists primarily of very well-studied pathways, there are no probabilities or confidence values associated with interactions, as they are presumed to be correct in at least some contexts. Therefore, we chose an a priori strategy that bases the probability of any single interaction occurring dependent only on the degree of the starting and ending nodes. This strategy assumes all interactions to or from a node are equally likely, but each starting node must select a single edge to which to transmit information or signals, and each ending node must similarly select a single edge from which to receive information or signals. Assuming that these selections are independent, the resulting probability of a specific interaction , starting at node and ending at node , is the product of the inverses of the degrees:
| (Equation 2) |
where is the out-degree of node A (the number of edges coming from node A) and is the in-degree of node B (the number of edges ending at node B). Substituting into Equation 1 yields:
| (Equation 3) |
All edges in the initial network were weighted using this strategy. These original edge weights were preserved throughout the modeling process, even when analyzing smaller subnetworks where some interactions have been removed.
Calculating optimally refined network via kinase enrichment
After edge weight assignments in the initial network, the following steps were undertaken to produce the optimally refined network. Each node in the network was assigned a key functional distance by calculating the (weighted) length of the shortest path between that node and the nearest key functional node (i.e. KiR predicted kinase) using the distances function from the igraph R package. Thirty distinct, hypothetical subnetworks were created by removing nodes from the network with a key functional distance greater than a cutoff value. We tested cutoffs at integer values between 1 and 30. For each of these networks, the enrichment of kinases and differentially phosphorylated nodes were computed as fold enrichments of the number of each in the subnetwork versus the expected number based on the size of the subnetwork and the composition of the entire initial network. p values were calculated using Fisher’s exact test for overrepresentation using the fisher.test function in R.
Identifying refined closeness cutoff for high-value subnetwork
The “refined closeness” of each node was computed using the “closeness” function in the igraph package. These values were shifted and scaled to a minimum of 0 and a mean of 1. In order to choose the best cutoff for refined closeness, we plotted the enrichment of differentially phosphorylated nodes that would be included in the high-value set versus the total number of nodes included for each value of the refined closeness. Because the enrichment was a (mostly) decreasing function of subnetwork size, we fit this relationship with a polynomial model and selected the point with the maximum residual; that is, the subnetwork that, relative to the fitted relationship, had the largest enrichment for its size (Figure S2B). The empirically derived cutoff value for (normalized) closeness was 1.1149, resulting in a final network size of 166 nodes and an enrichment of 1.76 (p < 0.0001, Fisher’s exact test).
KinMap
The KinMap in Figure S2C was generated using the online KinMap tool available at http://www.kinhub.org/kinmap (Eid et al., 2017).
Small interfering RNA transfection
All small interfering RNA (siRNA) were obtained from Dharmacon (Thermo. Specific sequences and identifiers are provided in Data. siRNA transfections in both 12-well plate for expression profiling and in 96-well plates for cell migration were carried out using Lipofectamine RNAiMax (Invitrogen) according to manufacturer instructions.
RNA extraction and quantitative real-time PCR
mRNA expression changes in CDH1 was determined using quantitative real-time PCR (qPCR). Total cellular RNA was isolated using an RNeasy Mini Kit (QIAGEN). Briefly, 1 μg of total RNA was reverse transcribed into first-strand cDNA using an RT2 First Strand Kit (QIAGEN). The resultant cDNA was subjected to qPCR using human gene-specific primers. Sequences and identifiers for all primers used are given in Data S2. The qPCR reaction was performed with an initial denaturation step of 10 min at 95°C, followed by 15 s at 95 °C and 60 s at 58°C for 40 cycles using Biorad CFX384 thermocycler (Biorad). The mRNA levels of each gene were normalized relative to the mean levels of the housekeeping gene GAPDH and compared using the 2−ΔΔCt method as described previously c(Gujral et al, 2014a, 2014b).
Migration analysis
To study the effect of KiRNet predicted nodes on migration of mesenchymal cancer cells, a wound-healing assay was employed as described previously (Gujral et al, 2014a, 2014b). Briefly, siRNAs targeting various proteins and scrambled control were transfected in Huh7-Fzd2 cells using Lipofectamine RNAiMax (Invitrogen) according to manufacturer instructions. Cells were plated on 96-well plates (Essen Image Lock, Essen Instruments) and 48 hours post transfections, a wound was scratched with wound scratcher (Essen Instruments). Wound confluence was monitored with Incucyte Live-Cell Imaging System and software (Essen Instruments). Wound closure was observed every 2 hours for 24–72 hours by comparing the mean relative wound density of at least three biological replicates.
TCGA analysis
Patient data and clinical manifests were downloaded from selected TCGA (The Cancer Genome Atlas) projects through the National Cancer Institute's Genomic Data Commons data portal (https://portal.gdc.cancer.gov/) using the GenomicDataCommons Bioconductor package in R. 17 TCGA patient cohorts, containing 7881 patients in total, were selected, representing both high incidence and highly aggressive cancer subtypes. Data was processed as described previously (Xue et al., 2020). To assess co-expression between KiRNet predicted nodes and EMT, we used previously reported epithelial and mesenchymal marker genes to rank patients in each TCGA cohort by calculating the mean-rank of their epithelial marker expression and mesenchymal marker expression, giving an E-score and M-score respectively. Patients with above median E-score and below median M-score were labelled as the epithelial-like group, while patients with above median M-score and below median E-score were labelled as mesenchymal-like. LogCPM of the high-value signature was calculated by taking the average of the log(CPM+1) values for all available genes from the signature within each sample. p values were calculated using a Wilcoxon rank-sum test. The comprehensive list of cancer types analyzed is as follows: breast invasive carcinoma, cervical squamous cell carcinoma and endocervical adenocarcinoma, colon adenocarcinoma, glioblastoma multiforme, head and neck squamous cell carcinoma, kidney renal clear cell carcinoma, liver hepatocellular carcinoma, lung adenocarcinoma, lung squamous cell carcinoma, mesothelioma, ovarian serous cystadenocarcinoma, pancreatic adenocarcinoma, prostate adenocarcinoma, rectum adenocarcinoma, sarcoma, skin cutaneous melanoma, and stomach adenocarcinoma.
Kinobead/LC-MS kinome profiling
To quantify kinase protein and phosphopeptide expression changes between Huh7-Fzd2 and Huh7 WT cells, we used kinobead/LC-MS kinome profiling, an approach based on kinase affinity purification coupled to phosphopeptide enrichment followed by LC-MS analysis (Golkowski et al., 2017, 2020b).
Kinase affinity enrichment and on-bead digestion
Kinase affinity enrichment and on-bead digestion was performed as previously described (Golkowski et al, 2020a, 2020b). Briefly, three micro tubes containing 35 μl of a 50% slurry of the in-house-made, optimized kinobead mixture in 20% aq. ethanol were prepared for each pulldown experiment. The beads were washed twice with 300 μl modified RIPA buffer (50 mM Tris, 150 mM NaCl, 0.25% Na-deoxycholate, 1% NP-40, 1 mM EDTA and 10 mM NaF, pH 7.8). 1 mg of protein extract in mod. RIPA buffer containing HALT protease inhibitor cocktail (100x, Thermo Fisher Scientific, Waltham, MA) and phosphatase inhibitor cocktail II and III (100x, Sigma-Aldrich, St Louis, MO) were added to the first tube. The mixture was incubated on a tube rotator for 1h at 4°C and then the beads were spun down rapidly at 2000 rpm on a benchtop centrifuge (5s). The supernatant was pipetted into the next tube with kinobeads for the second round of affinity enrichment. The procedure was repeated once more for a total of three rounds of affinity enrichment. After removal of the supernatant, the beads were rapidly washed twice with 300 μl of ice-cold mod. RIPA buffer and three times with 300 μl ice-cold tris-buffered saline (TBS, 50 mM tris, 150 mM NaCl, pH 7.8) to remove detergents. 100 μl of the denaturing buffer (20% trifluoroethanol (TFE) (Wang et al., 2005), 25 mM Tris containing 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP∗HCl) and 10 mM chloroacetamide (CAM), pH 7.8), were added and the slurry vortexed at low speed briefly. At this stage, kinobeads from the three tubes are combined and heated at 95°C for 5 min. The mixture was diluted 2-fold with 25 mM triethylamine bicarbonate (TEAB), the pH adjusted to 8-9 by addition 1 N aq. NaOH; 5 μg LysC were added and the mixture agitated on a thermomixer at 700 rpm at 37°C for 2 hrh. Then 5 μg MS-grade trypsin (Thermo Fisher Scientific, Waltham, MA) were added, and the mixture agitated on a thermomixer at 700 rpm at 37°C overnight. 600 μl of 1% formic acid was added and the mixture acidified by addition of another 6 μl of formic acid to yield 1.2 ml peptide solution in total. An aliquot of 120 μl (10%) of the peptide solution was desalted using StageTips and analyzed in single nanoLC-MS/MS runs for protein quantification. The remaining peptide solution (90%) was dried under vacuum at RT on a SpeedVac. 300 μl of 70% aq. ACN + 0.1 % TFA was added to each tube, the mixture vortexed, and sonicated in a bath sonicator until dried peptide residue dissolved. In case the dried residue could not be fully resuspended, additional 0.1% aq. TFA can be added in 10 μl increments until dissolved. The solution was subjected to IMAC phosphopeptide enrichment protocol and desalted using StageTips (see ‘IMAC phosphopeptide enrichment’ and ‘Peptide and phosphopeptide desalting with StageTips’ below).
IMAC phosphopeptide enrichment
IMAC phosphopeptide enrichment was performed according to the published protocol (in-tube batch version) with the following minor modifications (Villén and Gygi, 2008). 20 μl of a 50% IMAC bead slurry composed of 1/3 commercial PHOS-select iron affinity gel (Sigma Aldrich, St Louis, MO), 1/3 in-house made Fe3+-NTA Superflow agarose and 1/3 in-house made Ga3+-NTA Superflow agarose was used for phosphopeptide enrichment (Ficarro et al., 2009). The IMAC slurry was washed three times with 10 bed volumes of 80% aq. ACN containing 0.1% TFA and phosphopeptide enrichment was performed in the same buffer.
Peptide and phosphopeptide desalting with StageTips
Peptides and phosphopeptides were desalted using C18 StageTips according to the published protocol with the following minor modifications for phosphopeptides (Rappsilber et al., 2007). After activation with 50 μl methanol and 50 μl 80% aq. ACN containing 0.1% TFA the StageTips were equilibrated with 50 μl 1% aq. formic acid. Then the peptides that were reconstituted in 50 μl 1% aq. formic acid were loaded and washed with 50 μl 1% aq. formic acid. The use of 1% formic acid instead of 5% aq. ACN containing 0.1% TFA reduces the loss of highly hydrophilic phosphopeptides.
nanoLC-MS/MS analyses
The LC-MS/MS analyses were performed as described previously with the following minor modifications (Golkowski et al., 2017, 2020a). Peptide samples were separated on a Thermo-Dionex RSLCNano UHPLC instrument (Sunnyvale, CA) using 20 cm long fused silica capillary columns (100 μm ID) packed with 3 μm 120 Å reversed phase C18 beads (Dr. Maisch, Ammerbuch, DE). For whole peptide samples the LC gradient was 120 min long with 10−35% B at 300 nL/min. For phosphopeptide samples the LC gradient was 120 min long with 3%−30% B at 300 nL/min. LC solvent A was 0.1% aq. acetic acid and LC solvent B was 0.1% acetic acid, 99.9% acetonitrile. MS data was collected with a Thermo Fisher Scientific Orbitrap Elite (kinobead-MS experiments, global phosphoproteomics analyses) or Orbitrap Fusion Lumos Tribrid (global proteome analyses) spectrometer. Data-dependent analysis was applied using Top15 selection with CID fragmentation.
Computation of MS raw files
Data.raw files were analyzed by MaxQuant/Andromeda (Cox et al., 2011) version 1.5.2.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. MS/MS spectra were searched against the UniProt human database (updated July 22nd, 2015). MaxQuant search parameters: Variable modifications included Oxidation (M) and Phospho (S/T/Y). Carbamidomethyl (C) was a fixed modification. Max. missed cleavages was 2, enzyme was Trypsin/P and max. charge was 7. The MaxQuant “match between runs” feature was enabled. The initial search tolerance for FTMS scans was 20 ppm and 0.5 Da for ITMS MS/MS scans.
MaxQuant output data processing
MaxQuant output files were processed, statistically analyzed and clustered using the Perseus software package v1.5.6.0 (Tyanova et al., 2016). Human gene ontology (GO) terms (GOBP, GOCC and GOMF) were loaded from the ‘Perseus Annotations’ file downloaded on 01.08.2017. Expression columns (protein and phosphopeptide intensities) were log2 transformed and normalized by subtracting the median log2 expression value from each expression value of the corresponding data column. Potential contaminants, reverse hits and proteins only identified by site were removed. Reproducibility between LC-MS/MS experiments were analyzed by column correlation (Pearson’s r) and replicates with a variation of r > 0.25 compared to the mean r values of all replicates of the same experiment (cell line or knockdown experiment) were considered outliers and excluded from the analyses. Data imputation was performed using a modeled distribution of MS intensity values downshifted by 1.8 and having a width of 0.2. For statistical testing of significant differences in expression, a two-sample Student’s T-test with Benjamini-Hochberg correction for multiple hypothesis testing was applied (FDR = 0.05).
Quantification and statistical analysis
Enrichment tests for kinases and differentially phosphorylated nodes were performed using custom R scripts employing the fisher.test function to implement a Fisher’s Exact test for overrepresentation.
Significant differences in expression of peptides and phosphosites were assessed using a two-sample Student’s T-test with Benjamini-Hochberg correction for multiple hypothesis testing (FDR = 0.05).
Cell migration in the wound healing assay was quantified using the Incucyte ZOOM™ software, which computes the area percentage of the of the initial wound that is occupied by cells. Significance was assessed in GraphPad Prism using a one-way ANOVA with the Holm-Sidak multiple comparisons test.
TCGA significance was calculated using custom R scripts implementing a Wilcoxon rank sum test.
Acknowledgments
T.B. is a recipient of the Fred Hutch Interdisciplinary Training Grant in Cancer Research (T32CA080416). This work was supported by grants from the National Science Foundation (2047289), the American Cancer Society (133870-RSG-19-197-01-CDD), the Anderson and Heidner Foundations, the NIH (R01GM129090), and Scientific Computing Infrastructure at Fred Hutch funded by Office of Research Infrastructure Program (ORIP) grant S10OD028685. This work used an EASY-nLC1200 UHPLC and Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer purchased with funding from an NIH Shared Instrumentation Grant (SIG) grant (S10OD021502). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The results published here are in part based upon data generated by TCGA Research Network: https://www.cancer.gov/tcga. We thank Dr. Milka Kostic for her helpful comments and suggestions for the manuscript.
Author contributions
T.B. and T.S.G. conceived the study. T.B., M. Chan, M. Ceribelli, C.T., and T.S.G. performed the cellular experiments. M.G. and S.-E.O. performed the mass spectrometry and proteomic experiments and analyses. T.B. wrote the KiRNet script. A.G.X. and N.K. performed TCGA analysis. T.B. and T.S.G. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 1, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2021.100007.
Supplemental information
References
- Albert R. Scale-free networks in cell biology. J. Cell Sci. 2005;118:4947–4957. doi: 10.1242/jcs.02714. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T., Deacon S.W., Devarajan K., Ma H., Peterson J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arang N., Kain H.S., Glennon E.K., Bello T., Dudgeon D.R., Walter E.N.F., Gujral T.S., Kaushansky A. Identifying host regulators and inhibitors of liver stage malaria infection using kinase activity profiles. Nat. Commun. 2017;8:1232. doi: 10.1038/s41467-017-01345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M., Zhang H., Park J., Daniels N.M., Crovella M.E., Cowen L.J., Hescott B. Going the distance for protein function prediction: a new distance metric for protein interaction networks. PLoS One. 2013;8:76339. doi: 10.1371/journal.pone.0076339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A., Petrenko A.A., Guizien K., Doglioli A.M. On the calculation of betweenness centrality in marine connectivity studies using transfer probabilities. PLoS One. 2017;12 doi: 10.1371/journal.pone.0189021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L., Ideker T., Raphael B.J., Sharan R. Network propagation: a universal amplifier of genetic associations. Nat. Rev. Genet. 2017;18:551–562. doi: 10.1038/nrg.2017.38. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Csardi G., Nepusz T. The igraph software package for complex network research. InterJ. Compl. Syst. 2006:1695. [Google Scholar]
- D’Argenio V. The high-throughput analyses era: are we ready for the data struggle? High-Throughput. 2018;7:8. doi: 10.3390/ht7010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid S., Turk S., Volkamer A., Rippmann F., Fulle S. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics. 2017;18:16. doi: 10.1186/s12859-016-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erten S., Bebek G., Ewing R.M., Koyutürk M. DADA: degree-aware algorithms for network-based disease gene prioritization. BioData Min. 2011;4:19. doi: 10.1186/1756-0381-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro S.B., Adelmant G., Tomar M.N., Zhang Y., Cheng V.J., Marto J.A. Magnetic bead processor for rapid evaluation and optimization of parameters for phosphopeptide enrichment. Anal. Chem. 2009;81:4566–4575. doi: 10.1021/ac9004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleuren E.D.G., Zhang L., Wu J., Daly R.J. The kinome “at large” in cancer. Nat. Rev. Cancer. 2016;16:83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- Freeman L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978;1:215–239. [Google Scholar]
- Friedman J., Hastie T., Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J., Clancy K.W., Thompson P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015;115:5413–5461. doi: 10.1021/acs.chemrev.5b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M., Vidadala R.S.R., Lombard C.K., Suh H.W., Maly D.J., Ong S.E. Kinobead and single-shot LC-MS profiling identifies selective PKD inhibitors. J. Proteome Res. 2017;16:1216–1227. doi: 10.1021/acs.jproteome.6b00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M., Lau H.-T., Chan M., Kenerson H., Vidadala V.N., Shoemaker A., Maly D.J., Yeung R.S., Gujral T.S., Ong S.-E. Pharmacoproteomics identifies kinase pathways that drive the epithelial-mesenchymal transition and drug resistance in hepatocellular carcinoma. Cell Syst. 2020;11:196–207.e7. doi: 10.1016/j.cels.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golkowski M., Vidadala V.N., Lau H.T., Shoemaker A., Shimizu-Albergine M., Beavo J., Maly D.J., Ong S.E. Kinobead/LC-MS phosphokinome profiling enables rapid analyses of kinase-dependent cell signaling networks. J. Proteome Res. 2020;19:1235–1247. doi: 10.1021/acs.jproteome.9b00742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral T.S., Peshkin L., Kirschner M.W. Exploiting polypharmacology for drug target deconvolution. Proc. Natl. Acad. Sci. U S A. 2014;111:5048–5053. doi: 10.1073/pnas.1403080111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral T.S., Chan M., Peshkin L., Sorger P.K., Kirschner M.W., Macbeath G. A noncanonical frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell. 2014;159:844–856. doi: 10.1016/j.cell.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen S., Gosens R., Wieland T., Schmidt M. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol. Ther. 2018;183:1–21. doi: 10.1016/j.pharmthera.2017.09.002. [DOI] [PubMed] [Google Scholar]
- Kanehisa M. KEGG) toward pathway engineering: a new database of genetic and molecular pathways. Sci. Technol. Jpn. 1996;59:34–38. [Google Scholar]
- Knapp S. New opportunities for kinase drug repurposing and target discovery. Br. J. Cancer. 2018;118:936–937. doi: 10.1038/s41416-018-0045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z.A., Lin H., Shokat K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer. 2010;10:130–137. doi: 10.1038/nrc2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehne B., Schlitt T. Protein-protein interaction databases: keeping up with growing interactomes. Hum. Genomics. 2009;3:291–297. doi: 10.1186/1479-7364-3-3-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutsatsos I.K., Parker C.N. Recent advances in quantitative high throughput and high content data analysis. Expert Opin. Drug Discov. 2016;11:415–423. doi: 10.1517/17460441.2016.1154036. [DOI] [PubMed] [Google Scholar]
- Navlakha S., Kingsford C. The power of protein interaction networks for associating genes with diseases. Bioinformatics. 2010;26:1057–1063. doi: 10.1093/bioinformatics/btq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida-Aoki N., Gujral T.S. Emerging approaches to study cell–cell interactions in tumor microenvironment. Oncotarget. 2019;10:785–797. doi: 10.18632/oncotarget.26585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Sabbineni H., Clarke A., Somanath P.R. Novel roles of Src in cancer cell epithelial-to-mesenchymal transition, vascular permeability, microinvasion and metastasis. Life Sci. 2016;157:52–61. doi: 10.1016/j.lfs.2016.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pržulj N., Wigle D.A., Jurisica I. Functional topology in a network of protein interactions. Bioinformatics. 2004;20:340–348. doi: 10.1093/bioinformatics/btg415. [DOI] [PubMed] [Google Scholar]
- Rappsilber J., Mann M., Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- Rata S., Scott Gruver J., Trikoz N., Lukyanov A., Vultaggio J., Ceribelli M., Thomas C., Singh Gujral T., Kirschner M.W., Peshkin L. An optimal set of inhibitors for reverse engineering via kinase regularization. bioRxiv. 2020 doi: 10.1101/2020.09.26.312348. [DOI] [Google Scholar]
- Roskoski R. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019;144:19–50. doi: 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Türei D., Korcsmáros T., Saez-Rodriguez J. OmniPath: Guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods. 2016;13:966–967. doi: 10.1038/nmeth.4077. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Villén J., Gygi S.P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 2008;3:1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Qian W.J., Mottaz H.M., Clauss T.R.W., Anderson D.J., Moore R.J., Camp D.G., Khan A.H., Sforza D.M., Pallavicini M., et al. Development and evaluation of a micro- and nanoscale proteomic sample preparation method. J. Proteome Res. 2005;4:2397–2403. doi: 10.1021/pr050160f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.I., Hwang S., Kincaid R.P., Sullivan C.S., Lee I., Marcotte E.M. RIDDLE: reflective diffusion and local extension reveal functional associations for unannotated gene sets via proximity in a gene network. Genome Biol. 2012;13:R125. doi: 10.1186/gb-2012-13-12-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P., Nielsen T.E., Clausen M.H. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov. Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4:1686. [Google Scholar]
- Wu Q., Heidenreich D., Zhou S., Ackloo S., Krämer A., Nakka K., Lima-Fernandes E., Deblois G., Duan S., Vellanki R.N., et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019;10:1–14. doi: 10.1038/s41467-019-09672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L., Lin J., Su J., Oyang L., Wang H., Tan S., Tang Y., Chen X., Liu W., Luo X., et al. Diallyl disulfide inhibits colon cancer metastasis by suppressing Rac1-mediated epithelial-mesenchymal transition. Onco. Targets Ther. 2019;12:5713–5728. doi: 10.2147/OTT.S208738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue A.G., Chan M., Gujral T.S. Pan-cancer analysis of the developmental pathways reveals non-canonical wnt signaling as a driver of mesenchymal-type tumors. Transl. Res. 2020;224:1–15. doi: 10.1016/j.trsl.2020.06.003. [DOI] [PubMed] [Google Scholar]
- Zeileis A., Fisher J.C., Hornik K., Ihaka R., McWhite C.D., Murrell P., Stauffer R., Wilke C.O. colorspace: A Toolbox for Manipulating and Assessing Colors and Palettes. ArXiv. 2019 [Google Scholar]
- Zou H., Hastie T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005;67:301–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom R script that implements both KiR KiRNet generated in this study is available on GitHub at https://github.com/FredHutch/KiRNet-Public. This repository also includes all associated files needed to execute the script and produce a sample model. The Huh7-Fzd2 mass spectrometry dataset generated in this study are available online in the MassIVE repository (https://massive.ucsd.edu/) under the dataset ID MSV000086446.