

Abstract
Antiviral culinary plants are potential bioresources for preventive nutraceuticals and/or antiviral drugs in COVID-19. Structure-based virtual screening was undertaken to screen 173 compounds previously reported from Vernonia amygdalina and Occinum gratissimum for direct interaction with the active site of the 3-Chymotrypsin-Like Protease (3CLpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Based on docking scores and comparison with reference inhibitors, a hit-list of 10 top phytocompounds was defined, which also had strong interactions with the catalytic centre of 3CLpro from three related strains of coronavirus (SARS-CoV, MERS-CoV, HKU4). Among these, six compounds (neoandrographolide, vernolide, isorhamnetin, chicoric acid, luteolin, and myricetin) exhibited the highest binding tendencies to the equilibrated conformers of SARS-CoV-2 3CLpro in an in-depth docking analysis to 5 different representative conformations from the cluster analysis of the molecular dynamics simulation (MDS) trajectories of the protein. In silico drug-likeness analyses revealed two drug-like terpenoids viz: neoandrographolide and vernolide as promising inhibitors of SARS-CoV-2 3CLpro. These structures were accommodated within the substrate-binding pocket; and interacted with the catalytic dyad (Cys145 and His41), the oxyanion loop (residues 138–145), and the S1/S2 sub-sites of the enzyme active site through the formation of an array of hydrogen bonds and hydrophobic interactions. Molecular dynamics simulation and binding free energy calculation revealed that the terpenoid-enzyme complexes exhibit strong interactions and structural stability. Therefore, these compounds may stabilize the conformation of the flexible oxyanion loop; and thereby interfere with the tetrahedral oxyanion intermediate formation during the proteolytic activity of the enzyme.
Keywords: Food herbs, Coronavirus, COVID-19, Phytochemicals, Terpenoids, Molecular docking, Molecular dynamics simulations
Graphical abstract

1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the current global health crisis called coronavirus disease 2019 (COVID-19). Coronavirus infections have been life-threatening and difficult to treat due to their rapid outbreak, ease of adaptation, emergence of new and resistant viral strains and the inapplicability of antibiotics [1]. SARS-CoV-2 was shown to share a close genome sequence with previously reported strains such as SARS-CoV, MERS-CoV, and HKU4, mostly in the open reading frame a (ORF1a) [2]. Hence, SARS-CoV-2 has been clustered with beta-coronavirus genera, including SARS and SARS-like coronaviruses. The genome of SARS-CoV-2 contains a positive-sense, single-stranded RNA of about 30 kb size [3]. It is made up of a number of ORFs, with the first ORF being the largest, representing about 66 % of the genome [4]. During viral infection of host cell, the genome of the virus is emptied into host cell. This is followed by translation of the genomic content using the host's ribosomes to generate the viral structural and non-structural proteins [5]. The first ORF results in about 16 non-structural proteins (nsps), including the non-structural protein 5 (nsp5), also called 3-Chymotrypsin-like protease (3CLpro). The protease 3CLpro consists of 306 amino acid residues, ranging from amino acid 3264 to amino acid 3569 of the polyprotein 1 ab.
The 3-Chymotrypsin-like protease (3CLpro) is a cysteine protease that facilitates the proteolytic processing of the viral polyproteins to yield functional proteins essential for the packaging of new virions [6]. It is one of the most important components of viral replication as it cleaves the replicase polyprotein after its translation at 11 different sites releasing most of the functional protein components of the replicase-transcriptase complex, hence it is also known as main protease (Mpro) of coronaviruses [7]. Amino acid sequence alignments of this protease revealed that, SARS-CoV-2 3CLpro had ~96 % sequence identity with the previous SARS-CoV 3CLpro, and ~50 % sequence identity with MERS-CoV 3CLpro [8]. Its substrate specificity is primarily defined by the residues at the P1, P1’ and P2 positions of the peptide substrate. These positions are highly conserved in all coronaviruses in particular the presence of glutamine at the P1 position (N-terminus of the scissile bond) of the substrate is strictly required for 3CLpro binding across all coronaviruses [7]. Inhibition of 3CLpro by compounds is not expected to interfere with human proteases since the protease has no homologue in human [6]. In addition, since 3CLpro, plays critical role in the survival, replication and infectivity of SARS-CoV-2, it is an attractive drug target [9]. Inhibitors of this protease such as lopinavir and ritonavir, used for the treatment of HIV are reported for their potential use against COVID-19 [10].
While drug repurposing towards targeting important proteins in SARS-CoV-2 are still under development, considerable volume of scientific evidence suggest that novel natural compounds with potential antiviral activities can be deployed against SARS-CoV-2 [[11], [12], [13]]. Studies have revealed that such compounds from indigenous herbs and medicinal plants may inhibit replication of coronaviruses, especially SARS-CoV-2 [[14], [15], [16]]. African tea leaf (Vernonia amygdalina Del.), is a small shrub growing predominantly in tropical Africa widely used for culinary purposes [17,18]. This plant has been used earlier in Western Africa against several viral diseases [19,20]. African basil (Occinum gratissimum), a culinary herb with strong spicy flavour widely consumed in West Africa, is known to exhibit a wide range of biological roles including antiviral activities [21]. In vitro studies showed that its leaf extract inhibited HIV-1 and HIV-2 replication with antiviral indices of 110 [22]. These culinary herbs alongside others with well documented antiviral activities such as Aframomum melagueta and Piper guineense have been suggested as potential bioresources for preventive nutraceuticals and antiviral drugs against COVID-19 [23,24].
Structure-based virtual screening (SBVS) has been widely employed to search chemical compound libraries towards bioprospecting novel bioactive molecules against viral drug targets in the on-going campaign against coronavirus pandemic [25,26]. It is a fast, environmentally sound, and cost effective approach used in early-stage of drug discovery process [27]. In this technique, a dataset of compounds is docked into the binding site of the three-dimensional (3D) structure of the biological target obtained from X-ray, NMR, or computational modelling, in order to select a subset of these compounds based on the predicted binding scores for further biological evaluation. Rapid identification and documentation of antiviral structures from widely consumed African antiviral culinary herbs and spices such as Vernonia amygdalina and Occinum gratissimum may help to support the current drive towards developing safe, accessible and economically feasible antiviral preparations to be used as home-grown preventive nutraceuticals, food supplements, and antiviral drugs against the pandemic. Therefore, this study was carried out to screen an in-house library of 173 compounds from Vernonia amygdalina and Occinum gratissimum for druggable phytochemicals with direct interactions with the active site of SARS-CoV-2 3CLpro in silico.
2. Materials and methods
2.1. Retrieval and preparation of protein structure for molecular docking
The recently published three-dimensional structure of 3CLpro of SARS-CoV-2 (PDB ID: 6Y84), and those of SARS-CoV (PDB ID: 2DUC), MERS-CoV (PDB ID: 4YLU) and HKU4 (PDB ID: 2YNA), were retrieved from the Protein Data Bank (http://www.rcsb.org). All the crystal structures were prepared by removing existing ligands and water molecules, while missing hydrogen atoms were added using Autodock version 4.2 programs (Scripps Research Institute, La Jolla, CA).
2.2. Ligand preparation for molecular docking
Structure Data Format of the 173 bioactive phytocompounds derived from Vernonia amygdalina and Occinum gratissimum were retrieved from the PubChem database (www.pubchem.ncbi.nlm.nih.gov) alongside the reference inhibitors viz: Lopinavir, Ritonavir and N-{4-[(1H-benzotriazol-1-ylacetyl) (thiophen-3-ylmethyl)amino]phenyl}propanamide (R30) of 3CLpro. They were converted to mol2 chemical format using Open babel [28], while compounds that were not available on the database were drawn with ChemDraw version 19, and converted to mol2 chemical format. Polar hydrogen charges of the Gasteiger-type were assigned to atoms, while the non-polar hydrogen molecules were merged with the carbons. Ligand molecules were further converted to the dockable PDBQT format using AutoDock Tools.
2.3. Molecular docking study
An active site targeted molecular docking of the 173 phytochemicals and reference inhibitors against SARS-CoV-2 3CLpro was initially performed using AutoDock Vina in PyRx 0.8 [29]. Based on the docking scores, binding poses and interaction in the catalytic site, a hit-list of 21 phytochemicals was defined. The top docked compounds were further docked into the active sites of 3CLpro of SARS-CoV, MERS-CoV and HKU4. Before docking analyses, all ligands were imported and energy minimization was performed with OpenBabel [28] incorporated into PyRx 0.8. The Universal Force Field (UFF) was used as the energy minimization parameter and conjugate gradient descent as the optimization algorithm. The grid boxes used for docking studies were obtained by selecting the amino acid residues that define the active site of enzyme and drawing the grid box to enclose them (Table 1 ). All the other parameters were kept as default. The molecular interactions were viewed with Discovery Studio Visualizer version 16.
Table 1.
Binding site coordinates of 3-Chymotrypsin-like protease of Coronaviruses.
| Dimensions | SARS-CoV-2 (Å) | SARS-CoV (Å) | MERS-CoV (Å) | HKU4 (Å) |
|---|---|---|---|---|
| center_x | 11.06 | 44.40 | 35.38 | 29.28 |
| center_y | 4.06 | 14.71 | 21.37 | 44.91 |
| center_z | 14.93 | 11.43 | 38.10 | 16.60 |
| Size x | 18.50 | 23.16 | 20.37 | 20.89 |
| Size y | 23.02 | 18.95 | 15.65 | 18.37 |
| Size z | 23.02 | 21.66 | 20.38 | 20.47 |
2.4. Molecular dynamics simulation
The structure of SARS-CoV-2 3CLpro was downloaded from the Protein Data Bank with code 6Y84 [30,31]. The unliganded was subjected to a 100 ns production run at the NVT ensemble (normal volume and temperature with a constant number of atoms) molecular dynamics simulation (MDS). Before the production run, the system was subjected to minimization for 10,000 steps using a conjugate gradient algorithm. CHARMM 36 force field was used in the MDS using the Nanoscale Molecular Dynamics (NAMD 2.13) software [32,33]. Visualizing Molecular Dynamics (VMD 1.9.3) software was used to prepare the input files and analyze the output trajectories [34]. A water box was added to the protein system after adding the missing Hydrogen atoms and removing any ligands. TIP3P water model was used to resemble the added water box, with 10 Å padding, for the periodic boundary condition to be applied [35]. Nose-Hoover Langevin piston was used to control the pressure at 1.01325 bar. In contrast, Langevin dynamics controlled the system's temperature at the physiological value. The temperature, pH, and salt concentration were set at the physiological values (310 K, 7.0, and 0.154 M NaCl, respectively) during the simulation period. The time step was set at its default two fs with SHAKE approximation in action. Subsequently, cluster analysis of the trajectories was performed using the UCSF Chimera software using its default values [36]. A representative conformation from each cluster was used in the in-depth docking experiment as discussed below.
After a series of docking experiments, the backbone protein (3CLpro) and the best two complexes (3CLpro-Neoandrographolide and 3CLpro-vernolide complexes) were chosen for Molecular Dynamic Simulation (MDS) using NAMD 2.13. The necessary MDS files were generated using CHARMM-GUI [[37], [38], [39]] while setting the salt concentration and temperature to 0.154 NaCl and 310 K, respectively, to mimic the physiological conditions. Before running the production of 50 ns, the system was minimized for 10,000 steps in a constant number of atoms, constant volume, and constant temperature (NVT) ensemble using a conjugate gradient algorithm, then equilibrated in a constant number of atoms, constant pressure, and constant temperature (NPT) ensemble for one ns. The pressure was controlled by the Nose-Hoover Langevin piston set to atmospheric pressure (1.01325 bar), while the temperature was controlled by Langevin dynamics. The force field used was the CHARMM36 force field.
Binding affinity was calculated using Molecular Mechanics Generalized Born Surface Area (MM-GBSA) utilizing MMPBSA.py script implemented in Amber tools 17 [40,41]. All frames (500 frames with time interval of 100 ps between frames) were used in the calculation with salt concentration set to 0.154 Mol, while the rest of the settings were left as default.
2.4.1. Molecular docking to different clusters from molecular dynamics trajectories
Five different coordinates of 3CLpro after cluster analysis of the MDS trajectories were used to dock the best ten compounds (vernolide, vernomygdin, 11, 13-dihydrovernodalin, neoandrographolide, vernomenin, myricetin, chicoric acid, luteolin, rosmarinic acid, and isorhamnetin) along with the reference inhibitors (ritonavir and lopinavir) using AutoDock Vina software [29,42]. The 3D structures of these ten were generated using the Avogadro software [43], while the Universal Force Field (UFF) was employed to optimize it using the steepest descent algorithm with energies (806, 1046, 2015, 1740, 394, 741, 428, 388, 272, 241, 213, and 875 kJ/mol, respectively) [[43], [44], [45]]. The Protein-Ligand Interaction Profiler (PLIP) web server and PyMOL 2.4 software were utilized to analyze the docking complexes [46].
2.5. In silico physicochemical properties and ADMET study
The top ranked compounds based on their binding affinity and docked poses with the 5 different representative structures were subjected to various drug-likeness and ADMET filtering analysis. The drug-likeness analysis which includes Lipinski, Veber, Ghose, Egan and Muegge were performed on the SwissADME (http://www.swissadme.ch/index.php) webserver. [47], while the predicted Absorption, Distribution, Metabolism, Excretion and toxicity (ADME/tox) study was analysed using the SuperPred webserver (http://lmmd.ecust.edu.cn/admetsar1/predict/) [48]. The SDF file and canonical SMILES of the compounds were downloaded from PubChem Database or copied from ChemDraw to calculate ADMET properties using default parameters.
3. Results and discussion
3.1. Screening of phytochemicals against the active site of SARS-CoV-2 3CLpro
Structure-based virtual screening has been used widely to identify potential inhibitors of SARS-CoV-2 replication [16,49]. Common techniques such as molecular docking simulations use scoring functions to estimate the force of non-covalent interactions between a ligand and molecular target in order to predict the best mode of interaction between two molecules to form a stable complex. The preliminary results of molecular docking of the phytochemicals from Vernonia amygdalina and Occinum gratissimum against the 3CLpro of the novel SARS-CoV-2 alongside with the reference inhibitors (lopinavir and ritonavir) are represented in Table S1 (supplementary material). From the results, a hit list of 21 phytochemicals (Table S2) were selected based on their orientation at the catalytic site, the interacting residues and binding affinities comparable to those of reference inhibitors, lopinavir (ΔG = −7.2 kcal/mol) and ritonavir (ΔG = −7.2 kcal/mol).
Further binding docking of the topmost 10 compounds (Table 2 ) against the active regions of the target protein in SARS-CoV, MERS-CoV and HKU4 (Table 1), revealed that, these chemical structures (Table 2) had considerable docking scores (Table 3 ) and interactions with the coronavirus strains. Early homology models of SARS-CoV-2 3CLpro indicated close structural relation to those of other coronaviruses. Superimposition of the X-ray crystal structures of the 3CLpro of SARS-CoV-2 and other coronavirus strains indicates a considerable degree of structural similarity and conservation of the active site [8]. This is currently exploited for the development of SARS-CoV-2 3CLpro inhibitors that were based on previous compounds targeting the 3CLpro of these related coronaviruses [8].
Table 2.
Structures of reference inhibitors and top docked phytochemicals with the active sites of 3CLpro of Coronaviruses.
| S/N | Compounds | Class of compounds | Chemical Structure | Source Plants |
|---|---|---|---|---|
| S1 | Lopinavir | 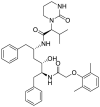 |
||
| S2 | Ritonavir | 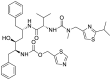 |
||
| S3 | R30 | 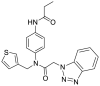 |
||
| 1 | Vernolide | Sesquiterpene lactones | 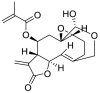 |
Vernonia amygdalina |
| 2 | Vernomygdin | Sesquiterpene lactones | 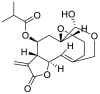 |
Vernonia amygdalina |
| 3 | 11, 13-dihydrovernodalin | Sesquiterpene lactones | 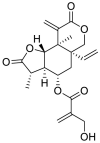 |
Vernonia amygdalina |
| 4 | Chicoric acid | Phenolic acids | 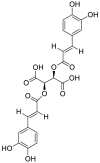 |
Occinum gratissimum |
| 5 | Rosmarinic acid | Phenolic acids | 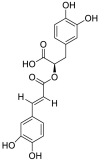 |
Occinum gratissimum |
| 6 | Luteolin | Flavonoids | 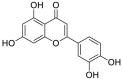 |
Occinum gratissimum |
| 7 | Neoandrographolide | Diterpenoid lactone | 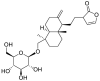 |
Vernonia amygdalina |
| 8 | Vernomenin | Sesquiterpene | 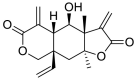 |
Vernonia amygdalina |
| 9 | myricetin | Flavonoids | 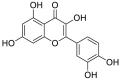 |
Occinum gratissimum |
| 10 | Isorhamnetin | Flavonoids | 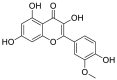 |
Vernonia amygdalina |
Table 3.
Binding energies of top ten ranked phytochemicals docked in the active sites of 3-Chymotrypsin-like proteases of coronaviruses.
| S/N | Compounds | Binding energies (Kcal/mol) |
||||
|---|---|---|---|---|---|---|
| PubChem ID | SARS-CoV-2 | SARS-CoV | MERS-CoV | HKU4 | ||
| S1 | Lopinavir | 92,727 | −7.2 | −8.3 | −7.5 | |
| S2 | Ritonavir | 392,622 | −7.2 | −7.2 | −7.1 | |
| S3 | R30 | −7.5 | ||||
| 1 | Vernolide | 5,281,508 | −8.0 | −7.9 | −7.7 | −7.7 |
| 2 | Vernomygdin | – | −7.9 | −7.7 | −7.6 | −7.2 |
| 3 | 11, 13-dihydrovernodalin | 23,786,372 | −7.8 | −7.8 | −7.4 | −7.2 |
| 4 | Chicoric acid | 5,281,764 | −7.7 | −7.4 | −8.3 | −8.9 |
| 5 | Rosmarinic acid | 5,281,792 | −7.7 | −7.2 | −8.0 | −8.7 |
| 7 | Neoandrographolide | 9,848,024 | −7.7 | −8.3 | −7.8 | −8.1 |
| 6 | Luteolin | 5,280,445 | −7.7 | −7.7 | −7.7 | −8.3 |
| 8 | Vernomenin | 442,324 | −7.7 | −6.9 | −6.4 | −6.7 |
| 9 | Myricetin | 5,281,672 | 7.7 | −7.5 | −7.9 | −8.0 |
| 10 | Isorhamnetin | 5,281,654 | −7.6 | −8.0 | −7.7 | −8.4 |
While the top three ranked phytochemicals SARS-CoV-2 3CLpro were found to be vernolide, vernomygdin and 11, 13-dihydrovernodalin (−8.0, −7.9 and −7.8 kcal/mol respectively); neoandrographolide, isorhamnetin and vernolide (−8.3, −8.0 and −7.8 kcal/mol respectively) were topmost against SARS-CoV 3CLpro; chicoric acid, rosmarinic acid and myricetin (−8.3, −8.0 and −7.9 kcal/mol respectively) against MERS-CoV and chicoric acid, rosmarinic acid and isorhamnetin (−8.9, −8.7 and −8.4 kcal/mol respectively) against HKU4 (Table 3). It was observed that the top three ranked phytochemicals for SARS-CoV-2 and SAR-CoV 3CLpros were isolated from Vernonia amygdalina while those for HKU4 3CLpro were from Occinum gratissimum.
3.2. Molecular interactions between the top docking phytochemicals and the active sites of 3CLpro of coronaviruses
A monomer of 3CLpro is made up of three domains: domain I (residues 8–101), domain II (residues 102–184), and domain III (residues 201–303) and a long loop (residues 185–200) connects domains II and III. Domains I and II comprise six-stranded antiparallel β-barrels with the substrate binding site at the intersection of the two domains. The enzymatic activity of 3CLpro resides in the catalytic dyad of Cys145 and His41 [50]. The substrate-binding pocket lies in the cleft between domains I and II, and features the catalytic dyad residues Cys145 and His41. The substrate-binding pocket is divided into a series of subsites (including S1, S2, S4 and S1’), each accommodating a single but consecutive amino acid residue in the substrate. Ser1 in each one monomer interacts with Phe140 and Glu166 of the other monomer to stabilize the S1 subsite, a structural feature that is essential for catalysis [51]. The current study revealed that, the reference drugs and the top-docking phytocompounds form complexes with SARS-CoV-2 3CLpro that are stabilized by numerous non-covalent interactions in the active regions of the target protein of the coronaviruses as shown in Table 4 .
Table 4.
Interacting amino acid residues of the 3CLpro of Coronaviruses with the top phytochemicals of Vernonia amygdalina and Occinum gratissimum.
|
Compounds |
Coronavirus | Residues involved in hydrogen bonding (bond distance, Å) | Residues involved in hydrophobic interactions | Residues involved in others interactions |
|---|---|---|---|---|
| Lopinavir (S1) | SARS-Cov-2 | GLU166(2.97) ASN142(2.97) PRO168(2.97) SER144(2.97) | MET49 HIS41 LEU27 | CYS145 |
| Ritonavir (S2) | SER46(2.46) THR26(3.24) | MET49 MET165 | GLU166 | |
| Vernolide | GLY143(2.00) MET165(3.63) HIS41(2.25) | CYS145 | ||
| Vernomygdin | GLU166(2.97) HIS163(2.97) ASN142(2.97) GLY143(2.97) MET165(2.97) | CYS145 LEU27 MET4 | ||
| 11, 13-dihydrovernodalin | CYS145(2.74) ASN142(2.25) | MET165 HIS41 LEU27 | ||
| Lopinavir (S1) | SARS-CoV | CYS145(2.49) THR25(2.74) GLU166(2.10, 2.08) | MET49 | HIS41 |
| Ritonavir (S2) | THR24(2.31) HR25(2.47) THR26(2.92) ASN142(3.12) | CYS44 CYS145 MET49 | ||
| Neoandrographolide | ASP48 (2.92) GLU166(3.13, 3.37) GLU47 (2.47) | HIS41 CYS145 CYS44 MET165 MET49 | HIS163 | |
| Isorhamnetin | HIS41 (2.47) CYS145 (2.47) MET165 (2.47) THR25(2.74) MET49 (2.47) THR24(2.74) | MET49 | GLU47 | |
| Vernolide | ALA46 (2.47) THR26(2.74) GLY26(2.74) | HIS41 CYS145 | ||
| Lopinavir | MERS-CoV | GLN169(2.81) GLY167(2.66) | HIS41 CYS145 CYS44 MET25 LEU49 ALA46 | |
| R30 | GLU169 (3.42) GLN167 (3.09) | CYS145 MET168 GLN192 LEU170 HIS41 LEU49 | ||
| Chicoric acid | MET168 (2.88) HIS41 (3.40) LYS191 (2.45) LEU170 (2.52) GLN195 (2.42) HIS166 (2.31) PHE143(2.41) | CYS145 CYS148 | ||
| Rosmarinic acid | GLN167 (2.16) GLU169 (2.20) GLN192 (2.68) THR26 (2.97) | HIS41 LEU49 | ||
| Myricetin | PHE143 (2.40) HIS166 (2.82) GLU169 (2.10, 2.14) | CYS148 LEU49 HIS41 | ||
| Ritonavir | HKU4 | CYS145 (3.31) CYS148 (3.67, 2.75) GLY167 (2.92, 3.03) GLY192 (2.06) ASN122 (2.34) | LEU49 ALA46 GLN169 | HIS41 |
| Chicoric acid | CYS145 (3.68) GLY146 (2.36) HIS166 (2.90, 1.98) SER147 (2.48) LEU144 (2.32) THR193 (1.92) LYS191 (1.99) GLU169 (2.70) | LEU49 | ||
| Rosmarinic acid | HIS41 (3.04) LEU49 (2.92) TRY54 (2.68) THR193 (2.72) | GLY192 MET168 | ||
| Isorhamnetin | GLU169 (2.70) HIS41 (3.06) LEU144 (2.70) | MET168 CYS145 |
Lopinavir and ritonavir the antiretroviral protease inhibitors which were originally developed for use against HIV and later recommended for the treatment of SARS and MERS infections [52], were used as reference drugs. The interactions of lopinavir were majorly through hydrogen bonds and hydrophobic interactions, with few electrostatic interactions. While the 4-hydroxyl and acetyl group of lopinavir interacted via hydrogen bond with GLU166 and SER144 of the domains I and II of 3CLpro of SARS-CoV-2, its 3-methyl and 1-phenyl moieties interacted via a hydrogen bonds. The 1-phenyl and the methyl moieties of the 2,6-dimethylphenoxy interacted via hydrophobic interactions with the catalytic dyad (Cys145 and His41) residues of 3CLpro of SARS-CoV-2 (Fig. 1 ). For 3CLpro of SARS-CoV, the 1-amino group of 2-oxo-1,3-diazinan-1-yl, 4-hydroxyl and acetyl groups of lopinavir interacted via H-bond with GLU166, THR25 and CYS145 in the same domain as SARS-CoV-2 while the 3-methyl and 1-phenyl groups formed an alkyl and pi-sulfur interaction with MET49 and CYS145 respectively (Figure S1). In the case of HKU4, two hydrogen bonds were observed between GLU169 and GLN167 and the carbonyl group and amino group of the butanamide moiety of lopinavir respectively (Figure S2), while hydrophobic interactions were formed by the phenyl rings. In the same vein, ritonavir having the same binding affinities as lopinavir interacted in a different manner with 3CLpro of the coronaviruses. The 15-hydroxy, 7-oxatetracyclo moiety and the carbonyl group of methylprop-2-enoate of vernolide interacted via H-bond with HIS41, GLY143 and MET165 of 3CLpro of SARS-CoV-2, while the heptadec-9-en-3-yl ring formed an alkyl interaction with CYS145 (Table 4). The hydrogen bonds observed between vernomygdin and HIS163, GLU166, and GLY143 of 3CLpro of SARS-CoV-2 were contributed by dihydrofuran-2 (3H)-one and the carbonyl group of methylpropanoate. The heptadec-9-en-3-yl ring and the alkyl group of methylpropanoate moiety were responsible for the alkyl interactions with amino acids of 3CLpro of SARS-CoV-2. The hydroxyl group of hydroxymethyl-prop-2-enoate of 11, 13-dihydrovernodalin contributed the only hydrogen bonds with CYS145 of 3CLpro of SARS-CoV-2. Several alkyl and pi-alkyl interactions were formed by the rings and methyl group of the furan ring of 11, 13-dihydrovernodalin and 3CLpro of SARS-CoV-2.
Fig. 1.

Amino acid interactions of top binding phytochemicals in the active site of 3-Chymotrypsin-like protease of SARS-CoV-2 3CLpro. (S) Surface view (a–e) interactive view. Ligands in stick representation are presented in different colours: (a) green: lopinavir (b) red: ritonavir (c) blue: vernolide (d) orange: vernomygdin (e) purple: 11,13-dihydrovernodalin. Types of interactions are represented by: Green-dotted line-hydrogen bonding; light purple-dotted line-hydrophobic interaction (pi-alkyl, alkyl and pi-stacking); purple-dotted line-pi-pi T-Shaped interaction; light purple-dotted line - pi-stacking interaction yellow-dotted line-pi-sulfur interaction and 3-letter abbreviation of amino acids are in red colour.
Vernolide, vernomygdin and 11, 13-dihydrovernodalin, the best docked phytochemicals in the SARS-CoV-2 3CLpro were observed to interact with the S1 subsite residues such as HIS41, ASN142, GLY143, SER144 and the GLU166 of β11. Interactions with the S1 and β11 residues have been reported for some other inhibitors of SARS-CoV-2 replication [6,51], suggesting that these three phytochemicals may effectively inhibit the proliferation of the virus. Interactions of the compounds at the S2 subsite were predominantly hydrophobic except for vernomygdin that formed a hydrogen bond with HIS163 and important residue in the hydrophobic pack that have been implicated in its catalytic activity [6] (Table 4). The binding of the top three ranked compounds docked in 3CLpro of the coronaviruses revealed that isorhamnetin and all the phytochemicals of V. amygdalina interacted with both amino acids of the catalytic dyad, indicating that they may be more effective inhibitors of the enzyme. The stability of the complexes formed stemmed from the vast number of interactions with some important active site residues HIS41, MET49, MET165, THR25, LEU27, ASP48, LEU50, LEU141, CYS145, HIS164, LEU167, PRO168, AEP187, and ALA191 which have been reported to be significant for the binding of the inhibitors with 3CLpro [53]. SARS-CoV 3CLpro had the highest binding affinity for neoandrographolide, a diterpene lactone obtained from V. amygdalina. The 2H-Furan-5-one ring formed two hydrogen bonds to ASP48 and GLU47. An alkyl interaction was formed by the methyl group at the oxan-2-yl-oxymethyl junction with CYS145 while the several pi-alkyl interactions were majorly formed by the 1H-naphthalen-1-yl and 2H-Furan-5-one ring (Figure S1). Isorhamnetin, an O-methylated flavonol obtained from Vernonia amygdalina interacted via conventional H-bonds with GLU166, GLY143 and THR45. A carbon hydrogen interaction was observed with CYS145 and THR24, while pi-cation, pi-sulfur and pi-alkyl were observed between the rings and HIS45, MET49 and CYS145 respectively. The carbonyl group of methylprop-2-enoate moiety and 15-hydroxyl group of vernolide formed a conventional hydrogen bond with GLY143 and THR25 of SARS-CoV 3CLpro. Pi-alkyl and alkyl interactions of the heptadec-9-en-3-yl with HIS41 and CYS145 were also observed. Phytocompounds from the plants had comparable interactions with MERS-CoV as the co-crystalized compound N-{4-[(1H-benzotriazol-1-ylacetyl) (thiophen-3-ylmethyl)amino]phenyl}propanamide (Tables 3 and 4). Chicoric acid and rosmarinic acid, from Occinum gratissimum, were the top docked phytochemicals to MERS-CoV and HKU4 3CLpro. The 3,4-hydroxyl group on the two phenyl moiety of chicoric acid were major donors of hydrogen atoms for the H-bonds, while the first phenyl ring made Pi-sulfur and Pi-alkyl contacts to the active site cysteine (CYS148 and CYS145) of MERS-CoV. The hydroxyl and the carbonyl group of the prop-2-enoyl moiety of rosmarinic acid formed hydrogen bonds to GLN167 and GLU169 respectively. The phenyl ring linked to the prop-2-enoyl group formed Pi-Pi T-shaped and Pi-Alkyl contact to HIS41 and LEU49 of MERS-CoV respectively (Figure S2). The 3 hydroxyl unit attached to ring B of myricetin formed 2 hydrogen bonds with PHE143 and GLU169, while the 4-hydroxyl formed a hydrogen bond with HIS166. The A and C rings of myricetin made Pi-Pi-stacked contacts with HIS41, while the B ring made Pi-Alkyl and Pi-sulfur contacts with LEU49 and CYS148 (Figure S2). In the case of HKU4, the hydroxyl and carbonyl groups on prop-2-enoyl [oxy]butanedioic moiety of chicoric acid interacted via several H-bonds with the residues at the active site. The hydroxyl and carbonyl groups on prop-2-enoyl [oxy]propanoic moiety of rosmarinic acid contributed the 3 hydrogen bonds to TYR54, LEU49 and HIS41 (Figure S3), while the first 3,4-dihydroxyphenyl moiety formed the hydrophobic interactions. HIS41 formed both hydrogen bond and pi-pi T-shaped interaction with the carbonyl group on the chromen-4-one moiety of isorhamnetin. The 4-hydroxy-3-methoxyphenyl moiety of isorhamnetin formed carbon hydrogen and pi-alkyl interactions with CYS145 (Figure S3).
3.3. Optimization of docking interactions of phytocompounds with SARS-CoV-2 3CLPro conformations
An in-depth docking simulation of the phytocompounds and reference inhibitors was performed to optimize the docking experiment and interactions with the target protein using previously reported protocols [54,55]. Fig. 2 shows the average binding affinities of the best ten phytocompounds along with the reference inhibitors (positive controls) against the five different representative conformations gotten from the clustering analysis of the SARS-CoV-2 3CLpro MDS trajectories (see Figure S3). The means and the standard errors of the mean of the 5 binding energies for each representative conformation of SARS-CoV-2 3CLpro were calculated for each phytochemicals and reference inhibitors. As reflected from the binding energy values, the ten phytochemicals are able to bind effectively to the SARS-CoV-2 3CLpro different conformations, just like the positive controls. The binding energy values ranged from −6.1 Kal/mol (rosmarinic acid) down to −8.1 kcal/mol (neoandrographolide and chicoric acid). As reflected from Fig. 2, vernolide, neoandrographolide, myricetin, chicoric acid, luteolin, and Isorhamnetin (green columns) are the compounds with best the binding affinities to SARS-CoV-2 3CLpro. The interactions of the best docked phytocompounds with SARS-CoV-2 3CLpro were further analysed using the PLIP webserver.
Fig. 2.

The average binding energy values of the reference compounds (Ritonavir and Lopinavir) and the best ten natural compounds calculated with AutoDock Vina software.
From the docking results, five complexes for each phytochemical were generated. The best representative complex for each phytochemical was selected based on the binding affinity for further analysis using the PLIP webserver. The details of the interactions established upon docking of the reference inhibitors and the best ten phytochemicals against SARS-CoV-2 3CLpro are presented in Table 5 . The most reported types of interactions are hydrogen bonding and few hydrophobic contacts in some complexes. At least three hydrogen bond, and up to seven were reported in the docking complexes between the compounds and SARS-CoV-2 3CLpro. The most-reported residues from the 3CLpro that interacted with the ligands (represented in bold in Table 5) are ASN142, GLY143, SER144, CYS145, and GLU166, and these formed 6, 9, 14, 7, and 5 interactions with the ligands, respectively. CYS145 is one of the 3CLpro active site dyads (HIS41 and CYS145), and it was reported in all the ligands except myricetin and luteolin. So far, two terpenoid structures viz: vernolide and neoandrographolide with strong interactions with the active region of SARS-CoV-2 3CLpro have been identified (Fig. 2, Table 5 and Fig. 3 ). The surface views of these structures in the substrate binding pocket of SARS-CoV-2 3CLPro are shown in Fig. 4 .
Table 5.
The interactions of the top 10 ranked phytochemicals of Vernonia amygdalina and Occinum gratissimum and positive control (Ritonavir and Lopinavir) for the best representative conformation from the cluster analysis of SARS-CoV-2 3CLpro molecular dynamics simulation (MDS) trajectories.
| Compound | Binding energies (kcal/mol) |
H-bonding |
Hydrophobic interactions |
||
|---|---|---|---|---|---|
| Number | Residues from SARS-CoV-2 3CLpro | Number | Residues from SARS-CoV-2 3CLpro | ||
| Ritonavir | −6.4 | 6 | ASN142(2), GLY143, SER144, CYS145, and GLU166 | 1 | MET165 |
| Lopinavir | −6.3 | 5 | ASN142, GLY143, ASP178(2), and GLN189 | 1 | THR25 |
| Vernolide | −7.5 | 3 | GLY143, SER144, and CYS145 | 1 | MET165 |
| Vernomygdin | −6.9 | 5 | ASN142, GLY143, SER144, CYS145, and GLN189 | 2 | MET165, and GLU166 |
| 11, 13-dihydrovernodalin | −6.6 | 6 | ASN28(2), GLY143, SER144, CYS145, and GLU166 | 3 | LEU27 (2), and MET165 |
| Neoandrographolide | −7.7 | 7 | THR45, SER46, LEU50, ASN142, GLY143, SER144, and CYS145 | 1 | THR25 |
| Vernomenin | −6.4 | 3 | GLY143, SER144, and CYS145 | 2 | THR25, and LEU27 |
| Myricetin | −7.1 | 7 | LEU141, ASN142, GLY143, SER144(3), and GLU166 | 0 | |
| Chicoric acid | −7.3 | 6 | LEU141, ASN142, GLY143, SER144(2), and CYS145 | 1 | GLN189 |
| Luteolin | −7.2 | 4 | SER144, GLU166(2), and GLN189 | 0 | |
| Rosmarinic acid | −6.8 | 7 | THR26 (2), PHE140, LEU141, GLY143, SER144, and CYS145 | 0 | |
| Isorhamnetin | −7.4 | 6 | ASN142, GLY143, SER144(3), and CYS145 | 0 | |
Residues in bold represent the most reported residues that interacted with the compounds.
Fig. 3.

The interaction pattern of the best six phytochemical structures with the active site of the best representative conformation from the cluster analysis of SARS-CoV-2 3CLpro MDS trajectories. The residues of the 3CLpro are shown in blue sticks labelled by its one-letter code. The ligands are represented in yellow sticks with cyan aromatic rings. H-bonds are shown in blue lines while hydrophobic contacts in dashed-gray lines.
Fig. 4.

Surface representation of (a) vernolide and (b) neoandrographolide in the substrate-binding pocket of SAR-CoV-2 3CLpro.
Binding interactions of neoandrographolide at the enzyme catalytic site is stabilized by several H bonds between its 2H-Furan-5-one ring and key residues (ASN142, GLY143, SER144, CYS145) of catalytic pocket of the enzyme, which led this ring to be sandwiched between CYS145 and ASN142 (Fig. 3). Furthermore, neoandrographolide structure inserts into the bulky hydrophobic S1/S2 subsites (composed of the side chains of HIS41, MET49, HIS41, ASN142, GLY143, SER144, and MET165) (Figs. 3 and 4b). Consequently, neoandrographolide was accommodated in the substrate-binding pocket and interacted with the catalytic residues, the oxyanion loop (residues 138–145), and the S1/S2 subsites, which are the key elements for the recognition of substrates. Interactions with the S1 have been reported for some other inhibitors of SARS-CoV-2 replication [6,51] suggesting that this structure may effectively inhibit the proliferation of the virus. With the aid of an array of direct and indirect hydrogen bonds with ASN142/GLY143/SER144/CYS145, neoandrographolide may fix the conformation of the flexible oxyanion loop, which served to stabilize the tetrahedral transition state of the proteolytic reaction. This binding mode of neoandrographolide is similar in many respect to that of baicalein, the first natural noncovalent, nonpeptidomimetic inhibitor of SARS-CoV-2 3CLpro derived from Shuanghuanglian [56]. Vernolide, another terpenoid structure (sesquiterpene lactone) isolated from Vernonia amygdalina is a potential non-covalent inhibitor of SARS-CoV-2 3CLPro inhibitor. Its interactions with the active site of this enzyme mimic the non-covalent interactions of carmofur, a potent covalent inhibitor of this enzyme which also establishes non-covalent interactions with its target [57]. The carbonyl group of methylprop-2-enoate moiety of vernolide occupies the oxyanion hole and forms hydrogen bonds with the backbone amides of Gly143, and Cys145 (Figs. 3 and 4a), mimicking the tetrahedral oxyanion intermediate formed during protease cleavage. A side chain of vernolide inserts into the bulky hydrophobic S2 subsite (composed of the side chains of HIS41 and MET165) (Figs. 3 and 4a). Therefore, these terpenoid structures alongside other phytocompounds from the source plants may be suggested as inhibitors of SARS-CoV-2 3CLpro.
3.4. In silico drug-likeness and pharmacokinetic properties of topmost phytocompounds
The top 6 phytocompounds (Neoandrographolide, vernolide, isorhamnetin, chicoric acid, luteolin, and Myricetin) from the docking analysis to the representative conformation gotten from the clustered MDS trajectories were subjected to the predictive pharmacokinetics drug-likeness and ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) filtering analyses. The result of the analyses for the six-top phytocompounds is represented in Table 6 . Several pharmacokinetic and ADMET molecular descriptors were used for the assessment (Table 6). From these six, four phytocompounds (vernolide, neoandrographolide, isorhamnetin and luteolin), fulfilled the requirement for at least four from the five physicochemical analyses (Lipinski, Veber, Ghose, Egan and Muegge). Five of the compounds except chicoric acid having 2 violations of greater than 5 H-bond donors and 10 H-bond acceptors atoms from Lipinski filter and a higher number of rotatable bonds above 10 and TPSA greater than 140 for the Veber filter. The five compounds are predicted to have good absorption or permeation from Lipinski filters [58] and good oral bioavailability from Veber filters [59]. The Ghose's filter that is based on computed physicochemical property profiles such as log P, molar refractivity, molecular weight, number of atoms as well of functional groups [60] screened out neoandrographolide and myricetin with two and one violations respectively. The Egan's and Muegge's filters screened out chicoric acid and myricetin. The Egan's filter is based on the physical processes involved in membrane permeability [61] while the Muegge's filter is based on the underfunctionalized properties of nondrug compounds [59]. Vernolide, neoandrographolide, isorhamnetin and luteolin also presented good Abbot Bioavailability Score [62] that is based on their predominant charges at biological pH. The molecular properties of the four phytochemicals based on the severally computed partition coefficient (log P) showed that the drugs had relatively good lipophilicity with logP values were less than 5 [63].
Table 6.
In silico Physicochemical and ADMETa parameters of the top-binding phytochemicals of Vernonia amygdalina and Occinum gratissimum with 3CLpro of SARS-CoV-2.
| a) Physicochemical properties | Vernolide | Neoandrographolide | Isorhamnetin | Chicoric acid | Luteolin | Myricetin |
|---|---|---|---|---|---|---|
| Molecular weight (g/mol) | 362.37 | 480.59 | 316.26 | 474.37 | 286.23 | 318.24 |
| Num. heavy atoms | 26 | 34 | 23 | 34 | 21 | 23 |
| Num. arom. Heavy atoms | 0 | 0 | 16 | 12 | 16 | 16 |
| Num. rotatable bonds | 3 | 7 | 2 | 11 | 1 | 1 |
| Num. H-bond acceptors | 7 | 8 | 7 | 12 | 6 | 8 |
| Hydrogen bond donor | 1 | 4 | 4 | 6 | 4 | 6 |
| iLogP | 2.45 | 3.27 | 2.35 | 1.00 | 1.86 | 1.08 |
| XLogP | 0.93 | 2.63 | 1,87 | 2.01 | 2.53 | 1.18 |
| WLogP | 1.17 | 1.83 | 2.29 | 1.01 | 2.28 | 1.69 |
| MLogP | 1.18 | 1.26 | −0.31 | 0.14 | −0.03 | −1.08 |
| Molar Refractivity | 89.51 | 125.27 | 82.50 | 114.00 | 76.01 | 80.06 |
| TPSA (Å2) | 94.59 | 125.68 | 120.36 | 208.12 | 111.13 | 151.59 |
| Drug-likeness | ||||||
| Lipinski | Yes | Yes | Yes | No (2 violations: NorO>10, NHorOH>5) | Yes | Yes |
| Veber | Yes | Yes | Yes | No (2 violations: Rotors>10, TPSA>140) | Yes | Yes |
| Ghose | Yes | No (2 violations: MW>480, No. of atoms>70) | Yes | Yes | Yes | No (1 violation: TPSA>140) |
| Egan | Yes | Yes | Yes | No(1 violation: TPSA>131.6) | Yes | No (1 violation: TPSA>131.6) |
| Muegge | Yes | Yes | Yes | No (3 violations: TPSA>150, H-acc>10, H-don>5) | Yes | No (2 violations: TPSA>150, H-don>5) |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.11 | 0.55 | 0.55 |
| Absorption (Probability) | ||||||
| (b) Admet SAR | ||||||
| HIA | HIA+ (0.58) | HIA- (0.127) | HIA- (0.498) | HIA+ (0.883) | HIA+ (0.9650) | HIA- (0.437) |
| Caco-2 Permeability Cm/s | Caco2+ (−5.096) | Caco2+ (−5.84) | Caco2+ (−5.217) | Caco2+ (−6.709) | Caco2+ (−5.12) | Caco2+ (−6.63) |
| P-glycoprotein Substrate | Neg. (0.484) | Pos. (0.778) | Neg. (0.015) | Neg. (0.051) | Neg. (0.038) | Neg. (0.208) |
| P-glycoprotein Inhibitor | Neg. (0.027) | Neg. (0.007) | Pos. (0.538) | Neg. (0.193) | Neg. (0.366) | Neg. (0.064) |
| Distribution (Probability) | ||||||
| Blood-Brain Barrier | BBB+ (0.4.39) | BBB- (0.476) | BBB- (0.34) | BBB+ (0.552) | BBB-(0.464) | BBB- (0.4.27) |
| PPB % | 65.501 | 72.039 | 90.707 | 76.782 | 91.796 | 76.595 |
| VD L/kg | −0.147 | −0.452 | −0.932 | −1.406 | −1.406 | −1.39 |
| Metabolism (Probability) | ||||||
| CYP450 1A2 Inhibitor | Neg. (0.069) | Neg. (0.028) | Pos. (0.941) | Neg. (0.239) | Neg. (0.069) | Neg. (0.133) |
| CYP450 1A2 Substrate | Neg. (0.33) | Neg. (0.258) | Neg. (0.456) | Neg. (0.262) | Pos. (0.968) | Pos. (0.968) |
| CYP450 3A4 Inhibitor | Neg. (0.149) | Neg. (0.262) | Pos. (0.768) | Neg. (0.087) | Neg. (0.412) | Neg. (0.376) |
| CYP450 3A4 Substrate | Neg. (0.562) | Neg. (0.523) | Neg. (0.428) | Neg. (0.15) | Pos. (0.867) | Neg. (0.459) |
| CYP4502C9 Inhibitor | Neg. (0.116) | Neg. (0.144) | Neg. (0.183) | Neg. (0.071) | Neg. (0.328) | Pos. (0.656) |
| CYP450 2C9 Substrate | Neg. (0.313) | Neg. (0.408) | Pos. (0.772) | Pos. (0.504) | Neg. (0.0496) | Pos. (0.557) |
| CYP4502C19 Inhibitor | Neg. (0.093) | Neg. (0.103) | Neg. (0.24) | Neg. (0.157) | Neg. (0.124) | Neg. (0.068) |
| CYP450 2C19 Substrate | Neg. (0.474) | Neg. (0.462) | Pos. (0.54) | Neg. (0.334) | Pos. (0.542) | Neg. (0.345) |
| CYP4502D6 Inhibitor | Neg. (0.296) | Neg. (0.329) | Neg. (0.468) | Neg. (0.248) | Neg. (0.463) | Neg. (0.318) |
| CYP450 2D6 Substrate | Neg. (0.267) | Neg. (0.274) | Neg. (0.41) | Neg. (0.415) | Neg. (0.401) | Neg. (0.18) |
| Elimination | ||||||
| T 1/2 (Half Life Time) | 0.883 h | 1.53 h | 0.658 h | 1.79 h | 0.745 h | 1.915 h |
| CL (Clearance Rate) mL/min/kg | 1.914 | 1.032 | 1.951 | 0.823 | 1.919 | 1.709 |
| Toxicity | ||||||
| hERG Blockers | Ng. (0.256) | Neg. (0.474) | Neg. (0.301) | Neg. (0.578) | Neg. (0.436 | Neg. (0.353) |
| H-HT | Neg. (0.444) | Pos. (0.584) | Pos. (0.654) | Neg. (0.348) | Pos. (0.592) | Neg. (0.332) |
| AMES | Neg. (0.411) | Neg. (0.224) | Neg. (0.044) | Neg. (0.224) | Pos (0.74) | Neg. (0.074) |
| SkinSen | Neg (0.340 | Neg (0.256) | Neg (0.186) | Neg (0.414) | Neg (0.278) | Neg. (0.278) |
| LD50 (LD50 of acute toxicity) | 3.211 -log mol/kg (222.927 mg/kg) | 3.448-log mol/kg (171.31 mg/kg) | 2.71-logmol/kg (604.02mg/kg) | 2.38-logmol/kg (1945.92mg/kg) | 2.58 -log mol/kg (737.444 mg/kg) | 2.69 -log mol/kg (648.262 mg/kg) |
| DILI | Neg. 0.424 | Neg. (0.196) | Pos. 0.904 | Pos. 0.84 | Pos. 0.9 | Pos. 0.9 |
| Pharmacokinetics | ||||||
| GI absorption | High | High | High | High | High | High |
| Log Kp (skin permeation) cm/s | −7.85 | −7.36 | −6.93 | −7.77 | −6.25 | −7.40 |
ADMET: Absorption, distribution, metabolism, elimination, and toxicity; GI: Gastro-intestinal; BBB: Blood Brain Barrier; P-gp: permeability glycoprotein; CYP: cytochrome P450; hERG: human Ether-à-go-go-Related Gene; HIA: Human Intestinal Absorption; H-HT: Human Hepatotoxicity AMES: Ames Mutagenicity; DILI: Drug Induced Liver Injury; VD: Volume Distribution; PPB: Plasma Protein Binding.
The Caco-2 permeability and intestinal absorption (HIA) descriptors determine the ultimate bioavailability of the drug. Compounds with low Caco-2 permeability potential (<8 × 10−6 cm/s) could be absorbed through the human intestinal wall [64]. The permeability glycoprotein (P-gp) is expressed in the intestinal epithelium, kidney cells liver cells, blood-brain barrier and blood-testis barrier capillary endothelial cells, where it functions by pumping xenobiotics back into the intestinal lumen, urine-conducting ducts bile ducts, and capillaries [65]. Vernolide and luteolin, expressed positive and high probability of human intestinal absorption and non-substrate to the permeability-glycoprotein (P-gp), while the all the six phytochemicals presented positive Caco-2 permeability. It is thereby suggested that vernolide will be absorbed into the blood stream subverting the capability of P-gp to pumps them back into the intestinal lumen, bile ducts, urine-conducting ducts and capillaries [65]. Blood brain barrier (BBB) penetration, predicts the blood brain barrier penetration of a molecule. Vernolide displayed properties that indicated their ability to cross the BBB. SARS-CoV-2 has been reported to infect the brain, thus indicating its ability to cross the blood brain barrier (BBB) [66], compounds that can cross the BBB will be beneficail in the overal all viral clearance. compounds that can cross the BBB will be beneficail in the overall viral clearance. The estimated half-life time (less than 2 h) and clearance ratefall within the moderate range. Vernolide, neoandrographolide, isorhamnetin presented a tolerable LD50 between (51–500 mg/kg), Among the descriptors for the in silico toxicities analysis, hERG channel plays a vital role in the repolarization and termination stages of action potential in cardiac cells [67]. Compounds that block the hERG channel have the potential to cause cardiotoxicity [68]. All the six phytocompounds did not exhibit the potential of being hERG channel blockers, suggesting that they may not cause hERG channel-related cardiotoxicity [68]. The three compounds did not exhibited mutagenicity in silico, thereby they may not cause genetic mutations, which do initiate the pathophysiology of other diseases, such as cancer [69]. The impact of the compounds on phase I drug metabolism in the liver was also analysed using the various cytochrome P450 descriptors. Vernolide, neoandrographolide did not display inhibitory potential for the various cytochrome P450, thus may not adversely affect phase I drug metabolism in the liver. Hence, vernolide, neoandrographolide seem to demonstrate high probability of absorption, subcellular distribution, and low toxicity.
3.5. Molecular dynamic simulations and binding free energy calculation for the best two complexes and the reference complex
MDS for the best two complexes in addition to the 3CLpro-ritonavir were performed for 50 ns using NAMD software, and then the MM-GBSA was done using Amber tools. In Table 7 , the residual contribution for the binding of 3CLpro against the best two compounds (Neoandrographolide and Vernolide) and the reference compound (ritonavir) are listed with the bold residues for the highest contributed residues in the binding (bold). The active site dyads (H41 and C145) are shown underlined in the table as well. For the 3CLpro- Neoandrographolide complex, C44 is the main contributor for the binding (−0.60 kcal/mol), while for the 3CLpro-Vernolide complex, H41, C145, and M165 are the main contributors (−1.16, −1.00, and −1.22 kcal/mol, respectively). The contribution of the active site dyads (H41 and C145) of the 3CLpro in the binding of Vernolide to the protein is evident from Table 7 (−2.16 kcal/mol). In comparison, a lower contribution of these two residues was reported in the case of the 3CLpro- Neoandrographolide and 3CLpro -Ritonavir complexes (−0.13 and −0.04 kcal/mol, respectively). The D187 and E166 (red coloured) have negative contribution to the binding of the 3CLpro to Neoandrographolide and Vernolide, respectively (+0.22 and + 0.27 kcal/mol). The total binding energy for the Vernolide is the lowest (−8.61 kcal/mol) compared to Neoandrographolide (−4.23 kcal/mol) and Ritonavir (−6.47 kcal/mol) hence it is the suggested compound that bind to 3CLpro.
Table 7.
The MM-GBSA calculations for the best two complexes after 50 ns MDS. Red coloured residues represent the residue have negative contribution on the binding (positive binding energies). The average binding free energies and its individual terms are shown at the bottom for each complex with its standard deviations.

The results shown in Fig. 5 supports previous observations. The three complexes were equilibrated for 50 ns as reflected from the flattened Root Mean Square Deviation (RMSD) and the Radius of Gyration (RoG) curves in Fig. 5A. The Root Mean Square Fluctuations (RMSF) in Å was plotted for the apo-3CLpro (red line) and the three complexes (3CLpro-Ritofenavir (blue line), 3CLpro- Neoandrographolide (orange line), and 3CLpro-Vernolide (gray line) (Fig. 5B).
Fig. 5.

A) The Root Mean Square Deviation (RMSD) and the Radius of Gyration (RoG) versus the simulation time in nanoseconds for the 3CLpro-Ritofenavir (blue line), 3CLpro- Neoandrographolide (orange line), and 3CLpro- Vernolide (gray line). B) The per-residue RMSF calculated for the apo-protein (blue), 3CLpro-Neoandrographolide (red), and 3CLpro-Vernolide (green). The structure of the protein is represented in a green cartoon with some residues in coloured sticks. C) An enlarged panel of the RMSF curves at the S46–P52 region for the three complexes and the apo form of 3CLpro.
Two regions of the RMSF plots that have higher fluctuations (greater than 2 Å) in addition to the N and C termini, are the S46–P52 region (red cartoon) and the T190-A193 region (yellow cartoon). As shown in the RMSF at the S46–P52 region, the apo-3CLpro shows higher fluctuations than the 3CLpro-Ritonavir (blue line), 3CLpro-Neoandrographolide (orange line) and 3CLpro-Vernolide (gray line) complexes. This region forms a loop that is important in substrate recognition since its presence near the protein's active site (blue sticks). The stabilization effect of the ligand binding to S46–P52 region of the protein is due to C44 (magenta sticks) in the case of 3CLpro-Neoandrographolide, which has the most contribution in the protein-ligand binding (−0.60 kcal/mol). In comparison, H41 and C145 (blue sticks) are the most contributed residues in the binding in 3CLpro-Vernolide (−2.16 kcal/mol). Additionally, the residues D48 and M49 (see Fig. 5C) in the case of 3CLpro-Neoandrographolide complex (orange line) show lower RMSF value (2.1 Å) compared to other complexes (3.2 Å) and the Apo form (4.2 Å). So, the stabilization of the S46–P52 loop is more observed in the case of Neoandrographolide than other compounds which in turn is more stable than the Apo protein.
4. Conclusion
Structure-based virtual screening of our in-house library of Vernonia amygdalina- and Occinum gratissimum-derived compounds against 3CLpro revealed two drug-like terpenoid structures viz: neoandrographolide and vernolide, alongside other phytochemicals as promising non-covalent inhibitors of SARS-CoV-2 3CLpro. These terpenoid structures were found accommodated within the substrate-binding pocket, and interacted with the catalytic dyad, the oxyanion loop (residues 138–145), and the S1/S2 subsites of the enzyme active site. With the aid of an array of hydrogen bonds and hydrophobic interactions with residues 142–145, these phytocompounds may stabilize the conformation of the flexible oxyanion loop; and thereby interfere with the tetrahedral oxyanion intermediate formation during proteolytic cleavage. Binding affinity calculation using Molecular Mechanics Generalized Born Surface Area (MM-GBSA) and Root Mean Square Fluctuations (RMSF) analyses through Molecular Dynamics Simulations (MDS) further revealed that the terpenoid-enzyme complexes exhibit strong interactions and structural stability, which could be adapted for experimental models towards development of preventive nutraceuticals, food supplement, and antiviral drugs in coronavirus diseases.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of competing interest
No potential conflict of interest is reported by the authors.
Acknowledgements
The authors acknowledge with thanks resources offered by the PhytoBioNet platform. MDS and MM-GBSA calculations are done on the Bibliotheca Alexandrina HPC facility, Alexandria, Egypt.
Biographies
Dr. Gideon A. Gyebi is currently a lecturer at the Department of Biochemistry, Faculty of Science and Technology Bingham University, Karu, Nasarawa, Nigeria. He obtained his Ph.D in Biochemistry in cancer chemoprevention and toxicology from the Department of Biochemistry, University of Ilorin, Kwara state, Nigeria. He was a former faculty member of the College of Natural and Applied Sciences, Salem University, Nigeria. He was a visiting researcher to the Department of Chemistry and Biomolecular Sciences, Faculty of Engineering, and the Anaerobic Research Unit of Gifu University, (1-1 Yanagido Gifu) Japan. His research interests are natural product Chemistry, Bioinformatics malaria and cancer chemotherapy. He is a pioneer member of the PhytoBioNet: Phytochemistry and Bioinformatic Network.
Dr. Abdo A. Elfiky I'm working in the Biophysics Department, Faculty of Sciences, Cairo University, Giza, Egypt as a Lecturer starting from the year 2013 then Associate Professor 2019. My interest in Molecular Biophysics pushes me to work in Structural Bioinformatics and Drug Design to find potent inhibitors against viral and pathogenic fungal proteins utilizing in silico techniques. During the past 6 years, I focused my research on polymerase protein due to its vital role in the life cycle of the pathogenic organisms. I studied also other proteins like protease, estrogen receptors and kinases. Currently, I'm working on fungal CotH kinase family proteins crucial for Mucormycosis virulence and the unfolded protein response master chaperone protein, GRP78.
Oludare M. Ogunyemi is an advanced PhD candidate in the Department of Biochemistry, College of Medicine, University of Ibadan and currently a lecturer in the Department of Biochemistry, Salem University, Nigeria. He is a recipient of several awards which include the African-German Network of Excellence in Science Mobility grant, 2016. He was a visiting pre-doctoral researcher to the Food Security and Safety Research Niche, North-West University, Mafikeng, South Africa. He is a reviewer of manuscripts for reputable journals. He is a pioneer member of the Phytochemistry and Bioinformatic Network (PhytoBioNet). His hopes to conduct research in the area of Human nutraceuticals and bioinformatics.
Ibrahim Mohamed Ibrahim is a master degree candidate in Department of Biophysics, Cairo University, Egypt. He is a researcher in Dr. Abdo A. Elfiky research group. He has published several papers related to structural bioinformatics.
Dr. Adegbenro P. Adegunloye is a Clinical Biochemist with keen interest in malaria chemotherapy and biochemical toxicology. He obtained his Ph.D. in Biochemistry from University of Ilorin, Nigeria. He was a former lecturer at Wesley University of Science and Technology, Ondo, Nigeria.
Professor Joseph O. Adebayo is currently a professor of clinical biochemistry and biochemical toxicology at the Department of Biochemistry, Faculty of Life Sciences, University of Ilorin, Nigeria. He obtained his Ph.D in Biochemistry (Clinical option) from the University of Ilorin, Kwara state, Nigeria. He is a former postdoctoral fellow at the Instituto Ren'e Rachou, FIOCRUZ, Belo Horizonte MG, Brasil. His research interests are malaria chemotherapy and diagnosis. He has presented various academic as well as research-based papers at several national and international conferences including Society of Toxicology USA. He has supervised several Ph.D students, one of such is Dr. Gideon A. Gyebi.
Professor C.O.O Olaiya is a professor and the Director of Nutritional and Industrial Biochemistry Research Unit, Department of Biochemistry, College of Medicine, University of Ibadan, Nigeria. He obtained his Ph.D. in Biochemistry from the University of Ibadan, Nigeria. He is a recipient of several awards such as the TWAS Postdoctoral Research Fellowship Award, 2009 among others. He was a former postdoctoral fellow at the International Center for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan and Visiting Professor at Kampala International University, Uganda. His current research interests include: Plant metabolites and biofortification of food crops, modulation of plant responses to abiotic stress, fermentation technology, and nutraceuticals. He has presented various academic as well as research-based papers at many national and international conferences. He has trained and supervised quite a number of Ph.D students to date.
Joshua O. Ocheje received his M.Sc. Analytical Chemistry from the University of Ibadan and currently works as a Lecturer in the Department of Pure and Industrial Chemistry, Nnamdi Azikiwe University, Nigeria. He is a recipient of the National Youth Service Corps (NYSC) Presidential Honors Award 2019. His research interests include the use of biochemistry and X-ray crystallography to study the structure and function of enzymes and other important proteins as drug targets.
Modupe Mercy Fabusiwa recently completed her B.Sc. degree in Biochemistry from Salem University, Nigeria. She has published papers related to food and nutritional biochemistry.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2021.104671.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/%2Facscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang H., Li X., Li T., Zhang S., Wang L., Wu X., Liu J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020;39:1629–1635. doi: 10.1007/s10096-020-03899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fani M., Teimoori A., Ghafari S. Comparison of the COVID-2019 (SARS-CoV-2) pathogenesis with SARS-CoV and MERS-CoV infections. Future Virol. 2020 doi: 10.2217/fvl-2020-0050. [DOI] [Google Scholar]
- 4.Pal M., Berhanu G., Desalegn C., Kandi V. Severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12 doi: 10.7759/%2Fcureus.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.-H., Nitsche A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.) 2020;368:409–412. doi: 10.1126/%2Fscience.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shahid M., Shahzad-Ul-Hussan S. Structural insights of key enzymes into therapeutic intervention against SARS-CoV-2. J. Struct. Biol. 2020;213 doi: 10.1016/%2Fj.jsb.2020.107690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30:127377. doi: 10.1016/j.bmcl.2020.127377. 127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gyebi G.A., Adegunloye A.P., Ibrahim I.M., Ogunyemi O.M., Afolabi S.O., Ogunro O.B. Prevention of SARS-CoV-2 cell entry: insight from in silico interaction of drug-like alkaloids with spike glycoprotein, human ACE2, and TMPRSS2. J. Biomol. Struct. Dyn. 2020:1–25. doi: 10.1080/07391102.2020.1835726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/nejmoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panyod S., Ho C.-T., Sheen L.-Y. Dietary therapy and herbal medicine for COVID-19 prevention: a review and perspective. J Tradit Complement Med. 2020;10:420–427. doi: 10.1016/j.jtcme.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta S., Singh V., Varadwaj P.K., Chakravartty N., Katta A.V.S.K.M., Lekkala S.P., Thomas G., Narasimhan S., Reddy A.R., Reddy Lachagari V.B. Secondary metabolites from spice and herbs as potential multitarget inhibitors of SARS-CoV-2 proteins. J. Biomol. Struct. Dynam. 2020:1–20. doi: 10.1080/07391102.2020.1837679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akindele A.J., Agunbiade F.O., Sofidiya M.O., Awodele O., Sowemimo A., Ade-Ademilua O., Akinleye M.O., Ishola I.O., Orabueze I. COVID-19 pandemic: a case for phytomedicines. Natural Product Communications. 2020;15 doi: 10.1177/1934578X20945086. 1934578X20945086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choy K.T., Wong A.Y., Kaewpreedee P., Sia S.F., Chen D., Hui K.P.Y., Chu D.K.W., Chan M.C.W., Cheung P.P., Huang X., Peiris M., Yen H.L. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pandey A., Khan M.K., Hamurcu M., Gezgin S. Natural plant products: a less focused aspect for the COVID-19 viral outbreak. Front. Plant Sci. 2020;11 doi: 10.3389/fpls.2020.568890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gyebi G.A., Ogunro O.B., Adegunloye A.P., Ogunyemi O.M., Afolabi S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CL(pro)): an in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dynam. 2021;39:3396–3408. doi: 10.1080/%2F07391102.2020.1764868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igile G.O., Oleszek W., Jurzysta M., Burda S., Fafunso M., Fasanmade A.A. Flavonoids from Vernonia amygdalina and their antioxidant activities. J. Agric. Food Chem. 1994;42:2445–2448. doi: 10.1021/jf00047a015. [DOI] [Google Scholar]
- 18.Oyeyemi I.T., Akinlabi A.A., Adewumi A., Aleshinloye A.O., Oyeyemi O.T. Vernonia amygdalina: a folkloric herb with anthelminthic properties. Beni-Suef University Journal of Basic and Applied Sciences. 2018;7:43–49. doi: 10.1016/j.bjbas.2017.07.007. [DOI] [Google Scholar]
- 19.Nittya D., Suresh K. A review on ethno-medicinal uses and pharmacology of Vernonia cinerea Less. Nat. Prod. Res. 2014;29:1102–1117. doi: 10.1080/14786419.2014.981814. [DOI] [PubMed] [Google Scholar]
- 20.Oladunmoye M.K., Afolami O., Oladejo B., Amoo I.A., Osho B. Derivatized extracts from aframomium melegueta K. Schum. And Vernonia amygdalina delile contain organic compounds that showed antiviral effects against atypical fowl pox virus (FPV kabete) J. Antivir. Antiretrovir. 2020;11 [Google Scholar]
- 21.Oso B.J., Ogunyemi O.M. Assessment of in vitrobiological properties of aqueous extracts of Murraya koenigii(L.) Spreng, Thymus vulgaris L.,and Ocimum gratissimum L. leaves. Croat. J. Food Sci. Technol. 2020;12 doi: 10.17508/CJFST.2020.12.2.13. [DOI] [Google Scholar]
- 22.Pandey S., Singh S., Kumar N., Botany R. ANTIVIRAL, antiprotozoal, antimalarial and insecticidal activities OF ocimum gratissimum L. Asian J. Pharmaceut. Res. Dev. 2017;5:1–9. [Google Scholar]
- 23.Bongo G.N., Matondo A., Mbadiko Mutunda C., Inkoto C., Lengbiye E., Kabengele C., Tshibangu D., Tshilandaa D., Ngbolua K.-T.-N., Mpiana P.T. Ocimum species as potential bioresources against COVID-19: a review of their Phytochemistry and antiviral activity. International Journal of Pathogen Research. 2020;5:42–54. [Google Scholar]
- 24.Esimone C.O., Omobuwajo O., Amadi C., Adiku M.U., Edrada-Ebel R., Chaidir C. Antiviral potentials of Nigerian aframomum melagueta roscoe and piper guineense schum. And thonn. Nigerian Journal of Natural Product and Medicine. 2006;10:51–54. [Google Scholar]
- 25.Gyebi G.A., Adegunloye A.P., Ibrahim I.M., Ogunyemi O.M., Afolabi S.O., Ogunro O.B. Prevention of SARS-CoV-2 cell entry: insight from in silico interaction of drug-like alkaloids with spike glycoprotein, human ACE2, and TMPRSS2. J. Biomol. Struct. Dynam. 2020:1–25. doi: 10.1080/07391102.2020.1835726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Borquaye L.S., Gasu E.N., Ampomah G.B., Kyei L.K., Amarh M.A., Mensah C.N., Nartey D., Commodore M., Adomako A.K., Acheampong P., Mensah J.O., Mormor D.B., Aboagye C.I. Alkaloids from cryptolepis sanguinolenta as potential inhibitors of SARS-CoV-2 viral proteins: an in silico study. BioMed Res. Int. 2020;2020 doi: 10.1155/2020/5324560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maia E.H.B., Assis L.C., de Oliveira T.A., da Silva A.M., Taranto A.G. Structure-based virtual screening: from classical to artificial intelligence. Frontiers in Chemistry. 2020;8 doi: 10.3389/fchem.2020.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berman H., Henrick K., Nakamura H. Announcing the worldwide protein Data Bank. Nat. Struct. Biol. 2003;10:980. doi: 10.1038/nsb1203-980. [DOI] [PubMed] [Google Scholar]
- 31.COVID-19 main protease with unliganded active site. 2020. http://www.rcsb.org/structure/6Y842020 accessed 13 June 2020, 3.
- 32.Jo S., Kim T., Iyer V.G., Im W. CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 33.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(33–38):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 35.Mark P., Nilsson L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. 2001;105:9954–9960. [Google Scholar]
- 36.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 37.Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. PMID: 18351591. [DOI] [PubMed] [Google Scholar]
- 38.Brooks B.R., Brooks C.L., III, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S.J., et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J., Cheng X., Swails J.M., Yeom M.S., Eastman P.K., Lemkul J.A., Wei S., Buckner J., Jeong J.C., Qi Y.J., et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016;12:405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller B.R., III, McGee T.D., Jr., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- 41.Case D.S.C.D.A., Cheatham T.E., III, Darden T.A., Duke R.E., Giese T.J., Gohlke H., Goetz A.W., D, Greene N.H., Izadi S., Kovalenko A., Lee T.S., LeGrand S., Li P., Lin C., Liu J., Luchko T., Luo R., Mermelstein K.M.M.D., Monard G., Nguyen H., Omelyan I., Onufriev A., Pan F., Qi R., Roe D.R., A, Roitberg C.S., Simmerling C.L., Botello-Smith W.M., Swails J., Walker R.C., Wang J., Wolf R.M., X, Wu L.X., York D.M., Kollman P.A. University of California; San Francisco: 2017. AMBER 2017. [Google Scholar]
- 42.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fliege J., Svaiter B.F. Steepest descent methods for multicriteria optimization. Math. Methods Oper. Res. 2000;51:479–494. [Google Scholar]
- 45.Rappe A.K., Casewit C.J., Colwell K.S., Goddard W.A., Skiff W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992;114:10024–10035. [Google Scholar]
- 46.Salentin S., Schreiber S., Haupt V., Adasme M., Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng F., Li W., Zhou Y., Shen J., Wu Z., Liu G., Lee P.W., Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 49.Trezza A., Iovinelli D., Santucci A., Prischi F., Spiga O. An integrated drug repurposing strategy for the rapid identification of potential SARS-CoV-2 viral inhibitors. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 52.Li J.Y., You Z., Wang Q., Zhou Z.J., Qiu Y., Luo R., Ge X.Y. The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microb. Infect. 2020;22:80–85. doi: 10.1016/j.micinf.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020 doi: 10.26434/%2Fchemrxiv.11875446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ogunyemi O.M., Gyebi G.A., Elfiky A.A., Afolabi S.O., Ogunro O.B., Adegunloye A.P., Ibrahim I.M. Alkaloids and flavonoids from African phytochemicals as potential inhibitors of SARS-Cov-2 RNA-dependent RNA polymerase: an in silico perspective. Antiviral Chem. Chemother. 2020;28 doi: 10.1177/2040206620984076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Paris R., Quevedo C.V., Ruiz D.D., Norberto de Souza O., Barros R.C. Computational intelligence and neuroscience; 2015. Clustering Molecular Dynamics Trajectories for Optimizing Docking Experiments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su H.-x., Yao S., Zhao W.-f., Li M.-j., Liu J., Shang W.-j., Xie H., Ke C.-Q., Hu H.-c., Gao M.-n., Liu H., Shen J.-s., Tang W., Zhang L., Xiao G.-f., Ni L., Wang D.-w., Zuo J.-P., Xu Y.-c. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jin Z., Zhao Y., Sun Y. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020;27:529–532. doi: 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- 58.Lipinski C., a, Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 59.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 60.Ghose A.K., Viswanadhan V.N., Wendoloski J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Combin. Chem. 1999;1:55–68. doi: 10.1021/cc9800071. [DOI] [PubMed] [Google Scholar]
- 61.Egan W.J., Merz K.M., Baldwin J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000;43:3867–3877. doi: 10.1021/jm000292e. [DOI] [PubMed] [Google Scholar]
- 62.Martin Y.C. A bioavailability score. J. Med. Chem. 2005;48:3164–3170. doi: 10.1021/jm0492002. [DOI] [PubMed] [Google Scholar]
- 63.Hughes J.D., Blagg J., Price D.A., Bailey S., DeCrescenzo G.A., Devraj R.V., Ellsworth E., Fobian Y.M., Gibbs M.E., Gilles R.W. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008;18:4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- 64.Larregieu C.A., Benet L.Z. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J. 2013;15:483–497. doi: 10.1208/s12248-013-9456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin J.H., Yamazaki M. Role of P-glycoprotein in pharmacokinetics. Clin. Pharmacokinet. 2003;42:59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 66.Zanin L., Saraceno G., Panciani P.P., Renisi G., Signorini L., Migliorati K., Fontanella M.M. SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir. 2020:1–4. doi: 10.1007/%2Fs00701-020-04374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raschi E., Vasina V., Poluzzi E., De Ponti F. The hERG K+ channel: target and antitarget strategies in drug development. Pharmacol. Res. 2008;57:181–195. doi: 10.1016/j.phrs.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 68.Kratz J.M., Grienke U., Scheel O., Mann S.A., Rollinger J.M. Natural products modulating the hERG channel: heartaches and hope. Nat. Prod. Rep. 2017;34:957–980. doi: 10.1039/c7np00014f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shen J., Cheng F., Xu Y., Li W., Tang Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010;50:1034–1041. doi: 10.1021/ci100104j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.