

Abstract
Altered gut microbiomes may play a role in rapid evolution to anthropogenic change but remain poorly understood. Atlantic killifish (Fundulus heteroclitus) in the Elizabeth River, VA have evolved resistance to polycyclic aromatic hydrocarbons (PAHs) and provide a unique opportunity to examine the links between shifts in the commensal microbiome and organismal physiology associated with evolved resistance. Here, 16S rRNA sequence libraries derived from fish guts and sediments sampled from a highly PAH contaminated site revealed significant differences collected at similar samples from an uncontaminated site. Phylogenetic groups enriched in the libraries derived from PAH-resistant fish were dissimilar to their associated sediment libraries, suggesting the specific environment within the PAH-resistant fish intestine influence the gut microbiome composition. Gut metabolite analysis revealed shifts between PAH-resistant and non-resistant subpopulations. Notably, PAH-resistant fish exhibited reduced levels of tryptophan and increased levels of sphingolipids. Exposure to PAHs appears to impact several bacterial in the gut microbiome, particularly sphingolipid containing bacteria. Bacterial phylotypes known to include species containing sphingolipids were generally lower in the intestines of fish subpopulations exposed to high concentrations of PAHs, inferring a complex host-microbiome relationship. Overall, killifish microbial community shifts appear to be related to a suppression of overall metabolite level, indicating a potential role of the gut in organismal response to anthropogenic environmental change. These results on microbial and metabolomics shifts are potentially linked to altered bioenergetic phenotype observed in the same PAH-resistant killifish populations in other studies.
Keywords: gut microbes, gut metabolome, Atlantic killifish, adaptive resistance, polycyclic aromatic hydrocarbons
1. Introduction
The Elizabeth River system is in the southern region of the Chesapeake Bay estuary near the cities of Chesapeake, Norfolk, Virginia Beach, and Portsmouth, Virginia, USA. Sediments in parts of the river, including those near the former site of Republic Creosoting, Inc. (RCI; a wood-preservation company), are heavily contaminated with creosote, a complex chemical mixture that includes unsubstituted, heterocyclic, and phenolic polycyclic aromatic hydrocarbons (PAHs) (Jayasundara et al., 2017; Osterberg et al., 2018; Reid et al., 2016). PAHs are highly toxic, with deleterious effects reported in several organisms, including in fish (Delistraty, 1997; Hodson, 2017; Mu et al., 2017). Exposure to PAHs result in a range of adverse outcomes in eukaryotes including developmental deformities (Corrales et al., 2014), nuclear and mitochondrial DNA damage (Jung et al., 2011), and cancer (Wang and Xue, 2015).
Atlantic killifish (Fundulus heteroclitus, commonly known as mummichogs), are found throughout the Elizabeth River system, including the RCI site where a subpopulation of these fish has evolved resistance to high concentrations of creosote contaminated sediment. Some adaptations to PAH exposure in Elizabeth River killifish subpopulations have already been described, including modifications in the aryl hydrocarbon receptor (AHR) pathway and upregulation of antioxidant defense systems (Di Giulio and Clark, 2015). Further explanation of the mechanisms underlying this evolved resistance, are described in Di Giulio and Clark (2015).
In addition, these fish subpopulations have limited home range, the PAH-resistant phenotype is highly site-specific, and population genomic studies show minimum mixing between subpopulations (Osterberg et al., 2018). Therefore, these exposed fish represent an opportunity to examine evolved chemical resistance and have been extensively studied in that context in the past (Clark et al., 2013; Di Giulio and Clark, 2015; Reid et al., 2016).
Commensal microbiomes of fish are thought to play a role in defining the physiology of host organisms and are known to produce a variety of endogenous biomolecules (Davis et al., 2016; Kelly and Salinas, 2017; Sheng et al., 2018). The microbiome, especially the intestinal microbial community, can respond to or change in response to environmental conditions such as diet or exposure to xenobiotics (Dai et al., 2011; Macfarlane et al., 1986). These changes in the intestinal microbial community may potentially affect the physiology and metabolism of the eukaryotic host in a variety of ways, including the synthesis of amino acids and their metabolites (Dai et al., 2011). Such metabolites may be synthesized by either the host or the commensal microbes. Gut microbial communities in particular may be critical in determining aspects of overall organismal fitness, including metabolic plasticity and metabolic disorders (Li et al., 2008; Parekh et al., 2014). Intestinal microflora can also potentially directly affect the chemical transformation, bioavailability, and bioactivity of ingested harmful chemicals (Gómez and Balcázar, 2008; Ni et al., 2014). Importantly, symbiotic gut bacteria show trends of co-evolution with their fish hosts (Sullam et al., 2012).
While the genomic changes underlying PAH-resistance in these killifish subpopulations have been studied extensively for several decades, this is the first study that has explored the shifts in the gut microbiomes and metabolome. PAH-adapted RCI subpopulations have altered energy demands and metabolic capacity (Lindberg et al., 2017). Genomic data (Osterberg et al., 2018) explain some of the bioenergetic shifts, but the connection between observed altered metabolic phenotype and the fish gut microbiome have not been investigated.
Here, we hypothesized that the intestinal microbial communities and metabolomes of PAH-adapted subpopulations of killifish will be significantly different from that of non-exposed killifish from a similar, but uncontaminated habitat. Furthermore, to explore potential mechanistic underpinnings, we examined shifts in the gut metabolome and attempted to correlate them with the observed differences in fish microbial community.
2. Materials and Methods
2.1. Sample Collection.
This study was run in parallel with an embryonic toxicology study (Volkoff et al., 2019). at Duke University (Durham, NC). Adult killifish were collected from King’s Creek (“KC”; 37°18′16.2″ N, 76°24′58.9″ W), a site oftentimes used as a reference for PAH contaminated sites in the Elizabeth River, and the RCI site in the Elizabeth River (36°47′39.65″ N, 76°17′31.94″ W). The KC and RCI sites averaged 953 and 295,903 ng/g total selected PAHs/dry sediment, respectively. Polychlorinated bisphenols (PCBs) were below detection in KC sediments and averaged 26 ng/g at RCI. The amount of organic matter in the sediments was measured using the loss on ignition (LOI) test. LOI values (% of sample by dry weight) were 9.00% and 10.44% for KC and RCI, respectively. Chemical contaminant data are provided in Table 1. Details regarding analytical chemistry methods are available in Volkoff et al., 2019.
Table 1.
Sediment chemical and metal concentrations in sediments collected from King’s Creek (KC) and Republic Creosoting Industries (RCI).
| Site | Lead | Chromium | Nickel | Copper | Zinc | Arsenic | Cadmium | Mercury | PAHs (total) |
PCB |
|---|---|---|---|---|---|---|---|---|---|---|
| μg/g | ng/g | |||||||||
| KC | 18.9±3.6 | 21.5±3.5 | 10.2 ± 1.7 | 12.7 ± 2.3 | 62.4 ± 12.3 | 3.6 ± 0.6 | 0.3 ± 0.1 | 0.09 ± 0.01 | 953.3 | ND |
| RCI | 48.2±22.9 | 22.8±6.8 | 13.1 ± 2.2 | 41.3±2.8 | 185.7±28.4 | 6.5 ± 0.5 | 0.5 ± 0.03 | 0.24±0.01 | 295,902.9 | 26.1 |
Fish were collected at both sites within 4 hours of each other (2014 June) and sorted based on sex and size in the field. RCI fish collected during this time period (2013-2015) and since then show significant resistance to PAH toxicity (Lindberg et al., 2017; Riley et al., 2016). Ten similarly sized male fish from each site were anesthetized on ice and field dissected using ethanol-sterilized lancets and forceps. The mid- to hind-portion of the intestines from the duodenum to the anus were removed and immediately placed in liquid nitrogen. For sediment analyses, four 50-mL grab samples of river sediment were also collected at each site and mechanically homogenized. Sediment samples were transported on ice. Tissue and sediment samples were thereafter stored at −80°C prior to DNA isolation or metabolite analysis. For all samples the time between collection/storage on ice to storage in the −80°C did not exceed 18 hours.
2.2. DNA Extraction.
Four fish intestinal samples from each of the KC and RCI sites were thawed to room temperature, 400 μL of 1X phosphate buffered saline (PBS; pH 7.4) was added, and the contents vortexed for 5 sec at maximum speed. Samples were then placed in a Bransonic® 1510 Ultrasonic Cleaner (Branson Ultrasonics Corporation, Danbury, CT) on ice for 15 min to separate microbes from the intestinal lining. The supernatant was decanted into a new tube. A second volume of 400 μL PBS was added to the remaining tissue and sonicated for an additional 15 min. The supernatants were combined (~800 μL) and centrifuged for 10 min at 8,000 × g. DNA was isolated from the resulting pellet using the PowerLyzer® PowerSoil® DNA Isolation Kit as instructed by the manufacturer (Mo Bio Laboratories, Carlsbad, CA), except that cells were physically lysed by horizontal agitation for 20 min (rather than 10 min) using a VWR VX-2500 Multitube Vortexer (VWR, Radnor, PA). Sediment DNA was extracted from quadruplicate 300 mg samples of sediment from KC and RCI sites using the PowerLyzer® PowerSoil® DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA). DNA was extracted as instructed by the manufacturer, except that the tubes were vortexed horizontally for 20 minutes on the VWR VX-2500 Multitube Vortexer (VWR,Radnor, PA). All DNA samples were quantified in triplicate using a Qubit® 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA) with dsDNA BR reagents according to manufacturer’s instructions and stored at −20°C until further use.
2.3. LC-MS Targeted Metabolomics.
Targeted metabolomics was conducted on 10 μL homogenized triplicate fish intestinal samples using electrospray ionization liquid chromatography-mass spectrometry (ESI-LC-MS/MS) and MS/MS measurements using the AbsoluteIDQ™ p180 kit (Biocrates Life Sciences AG, Innsbruck, Austria). This kit simultaneously quantifies 163 metabolites, including acyl carnitines (Cx:y), amino acids (AA – proteinogenic amino acids and ornithine), hexose (sum of hexoses-about 90–95% glucose), glycerophospholipids (lysophosphatidylcholines (lysoPC) and phosphatidylcholines (PC diacyl (aa) and acyl-alkyl (ae)), and sphingolipids (SMx:y). The assay procedures of the AbsoluteIDQ™ p150 kit as well as the metabolite nomenclature have been described in detail previously (Kit, 2006). Absolute metabolite values were quantified using internal standards. Statistical significance was determined using the Holm-Sidak method and an alpha value of 5%. Each metabolite was analyzed individually without assuming a consistent standard deviation. The Biocrates data were analyzed using MetaboAnalyst version 2.0 (Xia et al., 2015, 2012; Xia and Wishart, 2011a, 2011b). Variable Importance in Projection (VIP) scores, a weighted sum of squares of the partial least squared (PLS) loadings taking into account the amount of explained Y-variation, were calculated for each metabolite. The metabolites with VIP scores over 1.25 were further used to calculate pathway impact scores (0.1) and to determine the impact of pathways using the MetaboAnalyst pathway analysis tool (Xia and Wishart, 2011b).
2.4. Microbial Community Analyses.
Primers targeting the V3 and V4 regions of prokaryotic 16S rRNA genes (Pro341F: 5′-CCT ACG GGN BGC ASC AG-3′; Pro805R 5′-GAC TAC NVG GGT ATC TAA TCC-3′) were used to simultaneously determine the bacterial and archaeal communities of fish intestine and sediment samples (Lefèvre et al., 2018; Takahashi et al., 2014). DNA was PCR amplified using 5Prime HotMasterMix® (Qiagen). PCR conditions consisted of 94°C for 3 min, 27 cycles of 94°C for 30 sec, 53°C for 40 sec and 72°C for 1 min with a final elongation step at 72°C for 5 min. Each DNA sample was PCR amplified in triplicate for four samples from each of the guts and three samples from each of the sediments. The amplicon products from different samples were then mixed in equal concentrations and purified using Agencourt® AMPure XP beads (Agencourt Bioscience Corporation, Beverly, MA, USA). Sequences were dual-indexed using 20 (12 forward, 8 reverse) distinct indices, purified once again using Agencourt® AMPure XP beads, and the DNA quantified as previously described. Samples were pooled to construct the sequencing library, and 2×300 pair-ended reads were obtained using an Illumina MiSeq at the Duke IGSP Genome Sequencing and Analysis Core Facility (Durham, NC).
2.5. Illumina MiSeq Sequencing Data Analysis.
Raw data files were demultiplexed using the Illumina BaseSpace App (http://basepace.illumina.com) and deposited into the Sequence Read Archive (SRA) under the bioproject accession number PRJNA385860. Two libraries representing a fourth sampling of killifish guts from the two sites (KG4 and RG3, biosamples SRR5528680 and SRR5228689) possessed few sequences and were not included in analyses. The remaining demultiplexed mate-paired fastq files representing triplicate libraries of RCI and KC sediments and fish intestines were joined, screened, and analyzed using mothur v.1.39.5 (v. 1.39.4 was used for the “sub.sample” and “dist.shared” commands due to crashes of those programs in the Windows 1.39.5 release) (Kozich et al., 2013). The analysis workflow generally followed the online mothur MiSeq standard operating procedure (SOP) protocol (https://www.mothur.org/wiki/MiSeq_SOP; last accessed February 2018) using an OTU-based analysis method with a dissimilarity of 0.03. Bacterial and archaeal sequences were analyzed simultaneously. The targeted V3-V4 16S rRNA gene region resulted in non-overlapping regions at contig termini with less certain accuracy and a correspondingly greater number of OTUs than might be expected with other gene regions. To account for this, phylogenetic analyses were combined with OTU-based analyses in some instances, especially when statistical analyses (e.g., “metastats”) indicated multiple OTUs with identical taxonomic classification were responsible for significant differences between treatments. Diversity analyses and library comparisons were performed using libraries sub-sampled to the smallest library size, but all sequences were used when determining relative abundance of OTUs/phylotypes in each library. Taxonomic classification was determined within mothur using the RDP training set v.16 (cutoff = 80) (Wang et al., 2007). The closest relatives of OTUs were determined by blastn searches (Altschul et al., 1990) of the NCBI ‘nr’ database using representative sequences obtained using the “get.oturep” command of mothur. Figures were otherwise generated or visualized using Grapher v.9 and Inkscape v.0.92.2.
3. Results and Discussion
3.1. Metabolomic Analyses.
Broader metabolite level comparisons highlighted key differences in overall bioenergetic phenotype between RCI and KC populations. Examination of 163 metabolites also revealed key differences in metabolite levels of sphingolipids, glycerophospholipids, acylcamitines and gut-associated amino acids between KC and RCI fish intestines, while biogenic amines showed no difference between the populations. Thirty four of the 81 custom metabolic indicators measured using the AbsoluteIDQ® p150 Kit showed disruption in the RCI samples (p < 0.015) suggesting impacts on the metabolism in killifish at that location. In particular, C2+C3/Co, an indicator ratio for fatty acid oxidation related to short-term metabolic control, was twice as high in KC samples as RCI samples (p = 0.003) suggesting marked differences in metabolic phenotype. RCI samples also displayed significantly lower spermidine/putrescine and spermine/spermidine ratios (p < 0.001), and lower hexose concentrations (p < 10−7) compared to KC samples, which are indicative of differences in metabolic capabilities of the fish subpopulations. Spermine and spermidine are known radical oxygen species scavengers and may be associated with preventing oxidative stress in the vertebrate central nervous system. Elevated levels of reactive oxidative species are an established outcome of PAH exposure and as discussed in Di Giulio and Clark (2015), PAH-adapted killifish in the Elizabeth River differed in their response to oxidative stressors when compared to reference site fish. An intriguing future research direction is to explore the role of spermine and spermidine concentrations in mediating oxidative stressor response in PAH-resistant fish.
Both acylcarnitines and amino acid metabolite levels were higher in KC fish compared to RCI fish. Of the 21 amino acids analyzed, 19 were found to be significantly higher in total concentration in KC compared to RCI samples (p < 0.01); only the low abundance amino acids citrulline (Cit) and ornithine (Orn) displayed no significant differences (Figure 1). Similarly, 14 of the 40 acylcarnitines (~35%) were significantly higher in KC samples (p-values ranging from 4.15×10−4 to 2.78×10−2) while only one (C5; p=3.34×10−2) was higher in the RCI gut samples.
Figure 1.
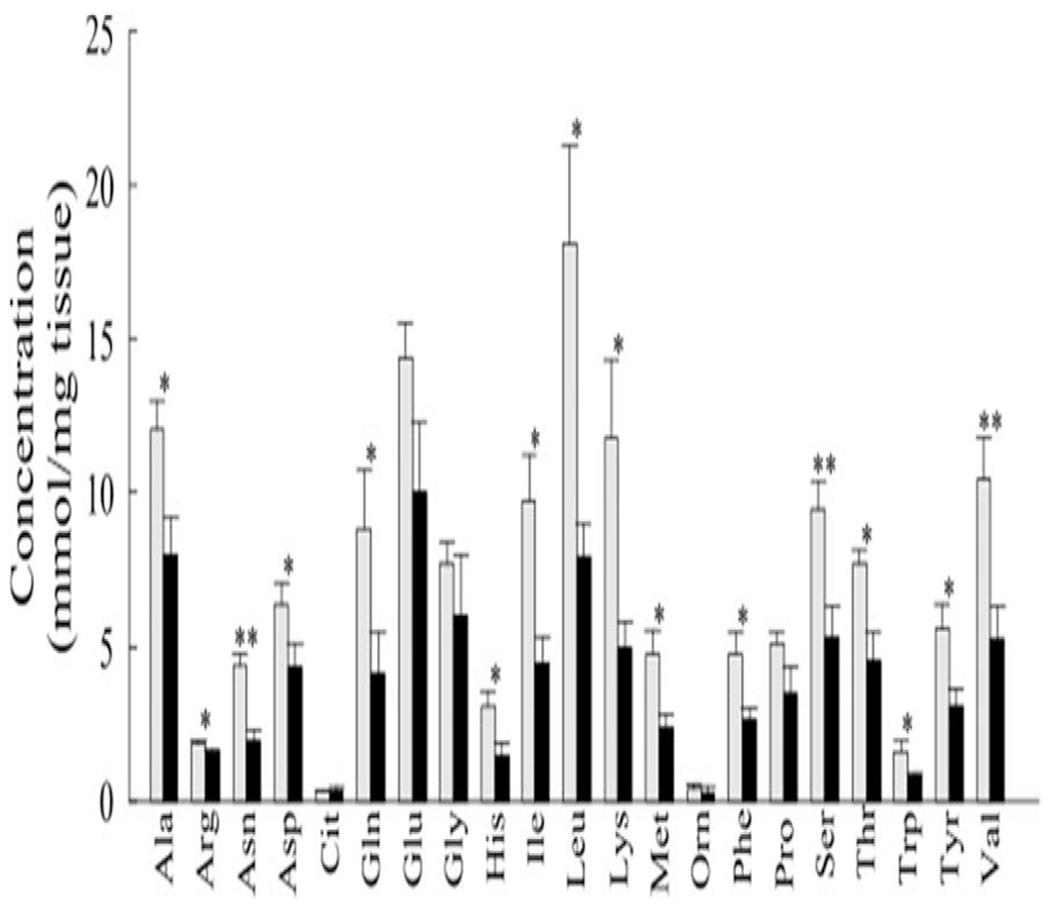
Concentrations of amino acids from fish guts of King’s Creek (KC; grey bars) and Republic Creosote, Inc. (RCI; black bars) samples. Triplicate measurements from each of three samples were averaged and the bars and error bars represent means and standard deviation of those samples (n=3). Single and double asterisks (*) indicate significant difference at p<0.05 and p<0.01, respectively. Abbreviations follow standard 3-letter nomenclature for amino acids.
In contrast to amino acids and acylcamitines, a number of sphingolipid and glycerophospholipid metabolites showed an overall trend of being significantly higher in RCI samples. Of the 90 quantified glycerophospholipids, 19 (21%) were significantly higher in KC gut samples while 30 (33%) were found to be higher in RCI gut samples. Of the 15 sphingolipids analyzed, 5 were significantly higher (p < 0.05) in one of the sample types: SM (OH) C22:1, SM (OH) C24:1, and SM C24:1 were higher in RCI samples while SM 08:0 and SM C26:1 were elevated in KC fish guts (Figure 2). Overall concentrations of sphingolipids were 107.9 and 121.0 μ;mol/mg tissue on average for KC and RCI, respectively (p=0.043, Wilcoxon matched-pairs signed rank test).
Figure 2.
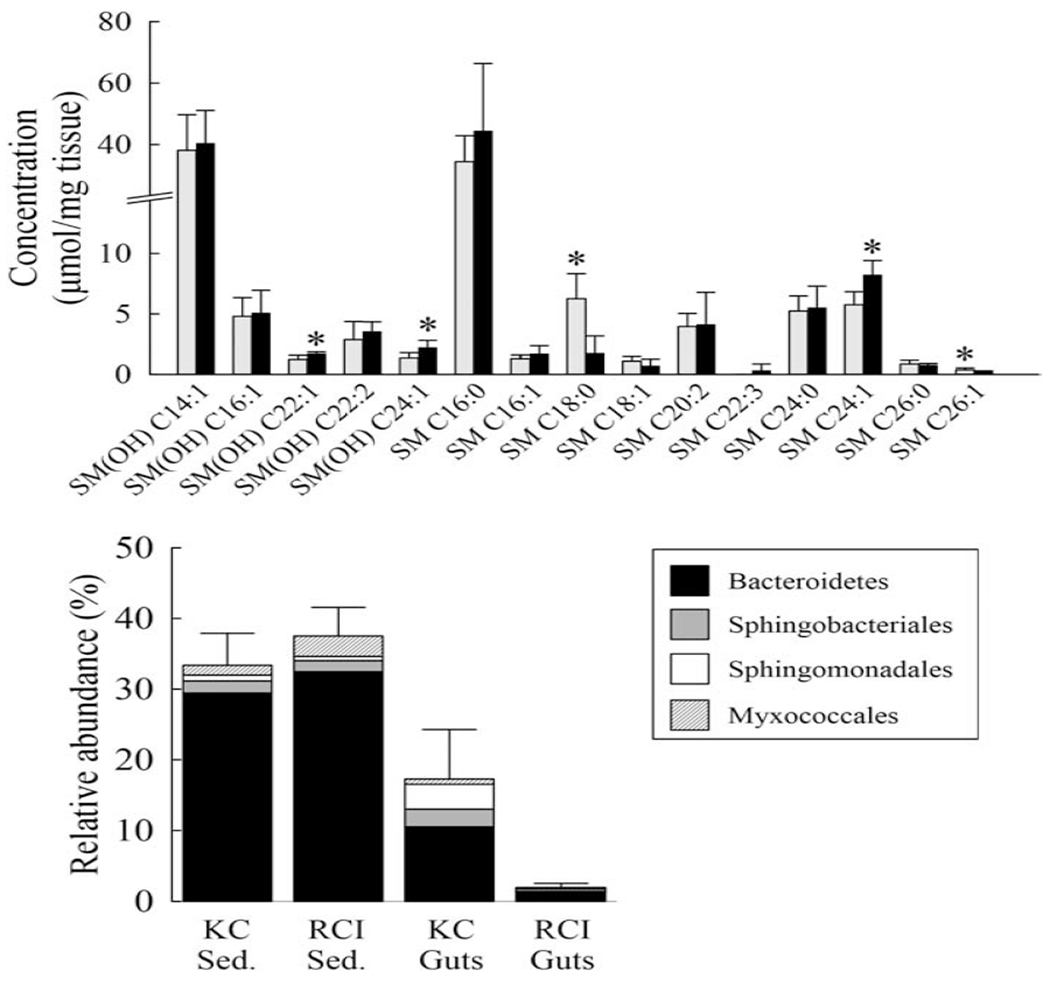
Sphingolipids and relative abundance of some bacterial groups associated with sphingolipids. (Top) Bar graph showing levels of quantified sphingolipids in fish guts from KC (grey) and RCI (black) samples. Values shown are averages of triplicate samples with error bars representing standard deviations. Asterisks (*) above bars represent samples with significantly greater values (p < 0.05). (Bottom) Stacked bar graph showing average relative abundance of taxa potentially containing sphingolipid-producing bacteria. KC, sediment sample does not include the outlier library. Error bars account for propagation of error of either range (KC, sed.) or standard deviation (all other samples) for all four taxa.
Collectively, metabolomic analysis revealed fundamental bioenergetic phenotype differences in fish resistant to PAHs compared to fish living in clean sites that are consistent with previous analyses at the genomic, cellular, and organismal level (Jayasundara et al., 2017; Lindberg et al., 2017; Osterberg et al., 2018; Reid et al., 2016). Considering that amino acids are major dietary components and the products of protein anabolism, and also regulate energy and protein homeostasis in eukaryotic hosts, it is possible that overall protein metabolism is suppressed and signaling is altered in RCI fish compare to KC fish. Acylcamitines play a critical role in the transfer of fatty acids into mitochondria for β-oxidation (Houten et al., 2016; Houten and Wanders, 2010). Further studies are warranted to examine if reduced acylcarnitine levels, coupled with altered mitochondrial function (Lindberg et al., 2017) in RCI fish is linked to observed changes in sphingolipid and glycerophospholipid levels (Rico et al., 2018). For example, glycerophospholipids (a.k.a, phosphoglycerides) are phospholipids with a glycerol-base that have been shown to interact with important pathways such as cholesterol regulation and neurological function (Farooqui et al., 2000; Schmid et al., 1990), and further suggest an altered lipid metabolic phenotype in RCI fish. Notably, sphingolipids, also known as glycosylceramides, play structural roles in eukaryotic cellular membranes (Bartke and Hannun, 2009) including as defense structures against xenobiotics, and are also involved in numerous signaling cascades (Spiegel and Milstien, 2002). It is possible that the changes in sphingolipid levels are a result of altered metabolic processes in RCI fish as well as a consequence of increased need for protection against xenobiotics in the PAH contaminated site. Additionally, these changes in metabolite levels could be linked to changes in the intestinal microbial communities. For example, sphingolipids are of particular interest in this analysis due to their presence in the membranes of some anaerobic intestinal bacteria (Kato et al., 1995) and members of the order Sphingomonadales (Olsen and Jantzen, 2001), the latter of which are commonly found in PAH-contaminated environments (Lafortune et al., 2009; Sohn et al., 2004; Zhou et al., 2006). As such, the observed changes in metabolite levels are possibly linked to shifts in the gut microbial community in RCI fish that are discussed next.
3.2. Microbial Community Analyses.
The prokaryotic microbial community of KC and RCI fish gut and river sediment populations were analyzed by high-throughput sequencing of archaeal and bacterial 16S rRNA genes. Triplicate libraries of mixed prokaryotic genes obtained for each of the four treatments comprised ~2.5M contigs. After eliminating sequences of incorrect length, containing ambiguous bases in the overlapping region, containing homopolymer runs > 10, classifying outside of the bacteria or archaea, or representing potential chimeras, approximately 220k sequences were considered for community comparisons (Table 2). Sequence reads and OTUs were within and above the range expected for fish gut samples (Ghanbari et al., 2015). Sequencing data were uploaded to NCBI (STUDY: PRJNA385860).
Table 2.
Statistics concerning 16S rRNA gene libraries derived from King’s Creek (KC) and Republic Creosoting, Inc (RCI) sediments and fish gut samples.
| Sample | No. Sequencesa |
No. OTUsa |
H′b | Inv. Simpsonb |
Chao1 | Coverage |
|---|---|---|---|---|---|---|
| KC, sediment |
KS1: 16175 KS2: 22044 KS3: 15024 |
1815 (782) |
6.48 (1.00) |
256 (177) |
4698 (2356) |
0.82 (0.09) |
| RCI, sediment |
RSI: 18761 RS2: 22961 RS3: 21495 |
2766 (120) |
7.43 (0.08) |
475 (14) |
8178 (568) |
0.70 (0.02) |
| KC, fish guts |
KG1: 6547 KG2: 10985 KG3: 8661 |
1050 (32) |
6.21 (0.22) |
184 (111) |
1259 (97) |
0.96 (0.01) |
| RCI, fish guts |
RG1: 15407 RG2: 9658 RG4: 15790 |
1107 (158) |
5.48 (0.14) |
45 (6) |
2457 (651) |
0.90 (0.02) |
With the exception of number of sequences per library, all values are presented as averages with standard deviations in parentheses (n = 3) based on a randomly subsampled library of 6547 sequences. No. – number.
H′ – non-parametric Shannon diversity index; Inv. Simpson – inverse of the Simpson diversity index.
Microbial diversity and richness were lower in fish gut samples compared to sediment samples. The lowest richness was observed in fish gut samples from the contaminated RCI site. Principal coordinate analyses (PcoA) indicated that most sediment-derived samples clustered together irrespective of the contaminant level, while distinct and separate clusters were observed for microbial community from the KC and RCI fish gut samples (Figure 3) indicating PAH-contaminant level dependent striking differences in the gut microbes.
Figure 3.
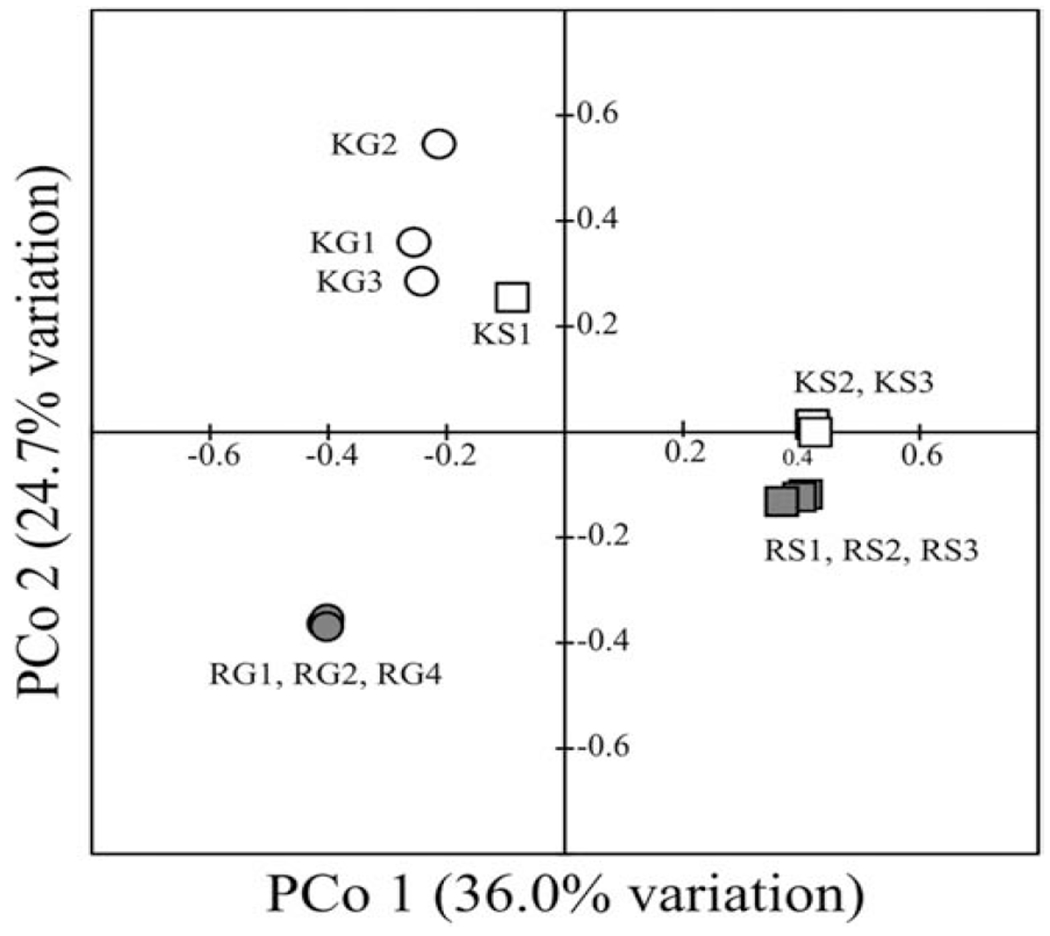
Principal coordinate analysis (PCoA) of libraries constructed from King’s Creek (K) and Republic Creosoting, Inc. (R) associated sediments (S) and fish guts (G). Triplicates of each library are shown (numbered 1-4), with some symbols partially obscuring others representing libraries in the same treatment.
The partial 16S rRNA gene sequence libraries of fish guts from the KC and RCI river regions were examined for organisms more highly represented in the PAH-exposed subpopulation. Using the metastats implementation of mothur, a total of 67 phylogenetic groups representing 184 OTUs were significantly greater in RCI gut libraries than in the corresponding KC libraries (p < 0.05). A summary of the most abundant of these groups (summed contribution of ≥ 1% relative abundance) are presented in Table 3. Note that within each indicated phylotype, each of the summed OTUs were individually significantly enriched in RCI gut libraries, but don’t necessarily represent the total diversity of the indicated phylotype present in those samples. Each of these phylotypes was not only more abundant in RCI fish gut samples compared to KC fish guts but were also better represented in those libraries than either of the sediment treatments. No archaeal phylotypes were significantly enriched in comparisons of the RCI and KC fish gut libraries. While increased relative abundance in sequence libraries are likely indicative of increased representation of those organisms in gut microbiomes, they may not necessarily be directly correlated with adaptations in response to increased contamination. However, such increases are indicators that these groups may be important for such adaptations and provide direction for further investigations.
Table 3.
Relative abundance of 16S rRNA genes from bacterial taxa in sediments and fish gut samples significantly increased in fish guts from highly contaminated sites.
| Phylotypea | No. OTUsa |
KC Sed.a, b | RCI Sed. | KC Fish | RCI Fish |
|---|---|---|---|---|---|
| Uncl. Rhodobacteraceae | 24 | 0.45 (0.05) | 0.58 (0.11) | 1.02 (0.08) | 11.9 (0.50) |
| Halochromatium | 2 | 0.17 (0.16) | 0.74 (0.02) | 0.89 (0.75) | 11.1 (1.46) |
| Uncl. Actinomycetales | 9 | 0.16 (0.13) | 0.84 (0.13) | 1.48 (1.20) | 7.53 (0.64) |
| Ilumatobacter | 2 | 0.26 (0.06) | 1.56 (0.24) | 0.79 (0.68) | 7.19 (1.03) |
| Anderseniella | 1 | 0.01 (0.01) | 0.11 (0.09) | 0.45 (0.43) | 2.96 (0.25) |
| Uncl. Acidobacteria Gp16 | 2 | 0.04 (0.04) | 0.27 (0.02) | 0.76 (0.55) | 2.30 (0.22) |
| Methyloceanibacter | 1 | 0.05 (0.04) | 0.14 (0.09) | 0.64 (0.46) | 1.88 (0.59) |
| Uncl. Actinobacteria | 10 | 0.16 (0.18) | 1.16 (0.05) | 0.33 (0.24) | 1.70 (0.19) |
| Litorilinea | 12 | 0.06 (0.07) | 0.39 (0.03) | 0.23 (0.21) | 1.67 (0.09) |
| Uncl. Desulfobulbaceae | 5 | 0.02 (0.02) | 0.34 (0.08) | 0.54 (0.41) | 1.59 (0.25) |
| Uncl. Chromatiaceae | 9 | 0.04 (0.04) | 0.60 (0.15) | 0.15 (0.16) | 1.55 (0.04) |
| Tetrasphaera | 1 | 0.04 (0.07) | 0.04 (0.00) | 0.31 (0.16) | 1.52 (0.06) |
| Uncl. cand. Saccharibacteria |
6 | 0.03 (0.03) | 0.15 (0.02) | 0.21 (0.14) | 1.49 (0.34) |
| Sulfurovum | 4 | 0.34 (0.31) | 0.85 (0.09) | 0.02 (0.04) | 1.46 (0.66) |
| Oceanicola | 1 | 0.13 (0.11) | 0.03 (0.02) | 0.17 (0.10) | 1.42 (0.06) |
| Uncl. Desulfobacteraceae | 2 | 0.43 (0.39) | 0.88 (0.09) | 0.16 (0.08) | 1.04 (0.42) |
Abbreviations: Uncl. – unclassified, cand. – candidate, No. – number., Sed. – sediment.
Values reported are the mean and standard deviation (in parentheses) of the relative abundance of sequences in those libraries classified to those phylotypes, considered all available sequences in the libraries. Values greater than 1% are highlighted in bold. Phylotype refers to the highest taxonomic level the OTU could be assigned with confidence and does not necessarily include all OTUs in the analysis representing that taxa, only those showing increased representation in RCI fish libraries. For phylotypes containing multiple OTUs, each OTU comprising the group individually displayed signficantly greater representation in RCI fish versus KC fish samples prior to clustering.
Collectively, the largest phylogenetic group in RCI gut samples could only be classified as members of the family Rhodobacteraceae but was represented by 24 different OTUs. Within this clade, the representative sequences of abundant individual OTUs were most similar to a variety of organisms including Celeribacter indicus (2.0% of RCI gut samples; 99% 16S rRNA gene similarity), Oceanicella actignis (1.7%; 97%), Oceanicola granulosus (1.3%, 98%), and Seohaeicola sp. (1.0%; 100%). Interestingly, Celeribacter indicus was previously isolated from deep-sea sediments and can degrade a variety of low-molecular-weight PAHs; it is possible that similar organisms may play the same role in RCI fish populations adapted to PAH contamination (Lai et al., 2014).
Two OTUs were classified as members of the Halochromatium genus with the more abundant OTU representing 10.7% of all RCI gut sample sequences. Of described organisms in this genus, the representative sequence of this OTU was most similar to H. roseum and H. glycolicum (96%). Halochromatium species are generally photolithotrophic marine purple-sulfur bacteria that utilize sulfur compounds as electron donors for carbon fixation. However, some species of purple-sulfur bacteria (including H. glycolicum) are capable of chemolithotrophic growth with sulfide and thiosulfate, or chemoorganotrophic growth with glycolate (Caumette et al., 1997; de Wit and van Gemerden, 1987).
Consistent with the above observation, other phylotypes with potential impacts on sulfur cycling were also present in significantly elevated levels in the RCI fish gut samples (p < 0.05). Sequences taxonomically classified to the families Desulfobulbaceae (1.6% of RCI gut sequences) and Desulfobacteraceae (1.0%), and genus Sulfurovum (1.5%) were significantly more abundant in intestinal samples of PAH-exposed fish subpopulations. Members of the Desulfobacteraceae and Desulfobulbaceae are strict anaerobes that reduce sulfates to sulfides, while characterized Sulfurovum strains are sulfur-oxidizing aerobes. The dominant Desulfobulbaceae OTU representative sequence (representing 1.0% of all RCI gut sequences) possessed only 94% sequence similarity to any cultivated organism (Desulfobacterium catecholicum) but possessed 99% identity to sequences recovered from Sapelo Island marsh sediment. Interestingly, the recovered Sulfurovum sequences from this study were equally similar to described Sulfurovum species as to environmental sequences representing endosymbionts of tubeworms (Forget and Kim Juniper, 2013) and clams (Rodrigues et al., 2010), suggesting these uncultivated organisms are successful at colonizing a variety of aquatic fauna.
OTUs representing the Ilumatobacter genus Actinobacteria, found in marine and freshwater sediments and waters (Fang et al., 2015; Gugliandolo et al., 2016), were also found in high abundance in RCI fish gut libraries (7.2% of all sequences). Ilumatobacter sequences have been found in abundance on the gills of Chinese mitten crabs (Eriocheir sinensis), although not in the intestines of those animals (Zhang et al., 2016). Their prevalence in the RCI fish samples in this study suggests they are potentially a part of a microbiome adaption to the contaminated environment, but the description of closely related isolates provide few clues as to what role they may play (Matsumoto et al., 2013, 2009). However, it is worth noting that Ilumatobacter sequences also comprised a larger percentage of the RCI sediment libraries than the lesser-contaminated KC sediment libraries, suggesting that the presence of PAHs may have influenced their abundance in both sample types. Unclassified members of the order Actinomycetales comprised 7.5% of the RCI fish gut libraries with the two most abundant OTUs representing 2.8% and 2.0% of all sequences in those libraries. These OTUs were most similar to various environmental sequences from water and sediments. A group of uncharacterized Actinobacteria sequences displayed increased abundance in both RCI gut libraries (1.7%) compared to KC gut libraries, and also comprised > 1% of the highly contaminated RCI sediment libraries.
The sole OTU representing Anderseniella in RCI fish libraries displayed 97% sequence similarity to the only described member of that genus (A. baltica) (Brettar et al., 2007), but 99% similarity to environmental sequences from sediments (Stauffert et al., 2013) and the intestines of marine shrimp (Rungrassamee et al., 2016). Anderseniella sequences were also detected as core members of the microbiome of one oyster species (García Bernal et al., 2017). Unclassified members of the Acidobacteria Gp16, were also well-represented in RCI gut samples (2.3%), and shared high similarly (98-100%) to various environmentally-derived marine sediment sequences, as well as those derived from the guts of fish (Miyake et al., 2016).
For the most part, phylogenetic groups enriched in PAH-exposed fish subpopulation-derived libraries were not highly abundant in their associated sediment libraries. This suggests that for the majority of these groups, it was the specific environment of the PAH-exposed fish intestine, and not the sediment from which that community may have been derived, that accounted for their increased abundance. It is also possible that altered physiological processes associated with PAH adaption in RCI fish may have contributed to the observed shifts in their gut microbial communities.
A total of 124 OTUs were implicated as containing significantly greater numbers of sequences (p < 0.05) in KC fish gut samples compared to RCI fish gut samples. However, contrary to what was observed in the RCI intestinal libraries in which several highly abundant OTUs represented a significant portion of the libraries’ total sequences, most of the significant OTUs in KC libraries represented only a small percentage of their total libraries (data not shown). In fact, only four OTUs averaged at least 1% representation; Methanobacterium (1.62%), Bacillus (1.44%), unclassified Chloroflexi (1.41%), and unclassified Synerigistaceae (1.33%) sequences. The lower abundance of these (and other) OTUs in RCI libraries is likely at least partially attributable to dilution by the vastly increased representation of the aforementioned dominant groups found in RCI fish gut samples derived from PAH-exposed fish subpopulations.
The single outlier sediment sample that did not cluster with any of the other five sediment libraries (Figure 3, KS1) possessed 17 OTUs with a combination of a relative abundance >1% and a low or non-existent presence in any of the other libraries (fish or sediment; data not shown). Among the most abundant were sequences from the family Rhodocyclaceae (9.0% of all sequences from that library), unclassified Betaproteobacteria (5.4%), Novosphingobium spp. (3.9%), and Cloacibacterium spp. (3.0%). Both the Rhodocyclaceae and unclassified Betaproteobacteria sequences were most similar (99% sequence similarity) to environmental sequences derived from freshwater samples (Watanabe et al., 2012). Cloacibacterium strains have been isolated from human wastewater treatment systems (Allen et al., 2006), and were found among the most abundant organisms in petroleum hydrocarbon-exposed freshwater systems (Jurelevicius et al., 2013). Novosphingobium organisms are aerobes from various environments and are commonly associated with hydrocarbon degradation (Kertesz and Kawasaki, 2010). Combined with a lack of archaeal sequences present in abundance in all other sediment samples from this study (data not shown), this dramatic microbiome shift suggests that this sample may have been uniquely affected by higher oxygen concentrations.
3.3. Sphingolipid-containing Phylotypes.
Sphingolipids are found in a variety of bacteria, many associated with animal intestines. These include not only members of the order Sphingomonadales (including the PAH-degrading genera Sphingomonas, Sphingobium, and Novosphingobium), but also the genera Bacteroides and Sphingobacteria (Olsen and Jantzen, 2001), and organisms within the Myxococcales (e.g., the genera Myxococcus, Sorangium) (Keck et al., 2011; Lorenzen et al., 2014). As sphingolipids in fish guts may be derived from bacterial, fungal, or animal sources, we examined the relative abundance of sequences affiliated with those bacterial phylogenetic groups as a surrogate for measuring the potential contribution of prokaryotic sphingolipids to fish guts, and to determine whether PAH-exposure may have influenced those populations (Figure 2, Table S1). Sequences associated with the order Sphingomonadales were generally most abundant in fish gut libraries from the KC river region and found in lower abundance or absent in the PAH-exposed fish subpopulations samples. Sequences of the order Bacteroidales and unclassified members of the Myxococcales displayed similarly low abundance in PAH-impacted fish sample libraries but were otherwise present in significant numbers in sediment libraries. The majority of KC fish-associated Sphingobacteriales sequences were from the genus Terrimonas (which are not known to possess sphingolipids) and unclassified members of the family Chitinophagaceae (data not shown). These two taxa were also present among the most well-represented Sphingobacteriales-associated OTUs in RCI fish libraries, although the relative abundances of Sphingobacteriales OTUs were generally much smaller in RCI fish compared to KC fish samples (Figure 2b). Based on these data, it seems likely that any prokaryotic contribution to the sphingolipid pool (and their associated effects on eukaryotic systems) might be minimal and reduced particularly in RCI sub-populations, though the exact microbial contributions are difficult to determine from these sequence data alone.
The lower relative abundance of sequences from putative sphingolipid-containing bacterial taxa may have implications for the fitness of animals adapted to higher levels of PAH contamination. Some bacterial sphingolipids, particularly those in the Bacteroides genus, have structural similarities to animalian sphingomyelins (Heaver et al., 2018) which are hypothesized to affect lipid metabolism—altered lipid metabolism is a likely consequence of PAH-adaption. Bacterial-derived sphingolipids have also been found to impact immune responses of fish (Sepahi et al., 2016). The lower representation of bacterial taxonomic groups putatively containing sphingolipids in guts of PAH-exposed fish therefore may have implications for the adaptability and success of those populations in changing environments, and may explain in part the observed phenomenon of higher susceptibility to environmental stressors (Jayasundara et al., 2017).
3.4. Metabolite and Microbial Community Relationships.
Adult fish from the Elizabeth River have previously been shown to display metabolic disparities, especially in their mitochondrial function between exposed and non-exposed subpopulations (Jayasundara et al., 2017; Meyer and Di Giulio, 2003), and the shifts in microbe and metabolite concentrations between RCI and KC guts may be due to this altered metabolic phenotype. Metabolite level changes in tryptophan and sphingolipids are two notable examples of observed metabolic changes of fish in PAH contaminated sites. The altered sphingolipid producing bacterial community in RCI fish is potentially coupled to both altered sphingolipid metabolism. Additionally, tryptophan is generated by the gut microbiome and has been implicated in at least five AHR activation pathways, including modulation of central nervous system inflammation, brain cell activity, and the differentiation of regulatory T cells (Quintana and Sherr, 2013; Rothhammer et al., 2016; Sridharan et al., 2014). PAH-adapted killifish also show a recalcitrance to induction of cytochrome P450 (CYP1) and other xenobiotic metabolic enzymes, along with elevated antioxidant response (Clark et al., 2014). Elizabeth River killifish exposed to PAH contamination have previously been observed to exhibit changes in CYP1A and CYP1B1 gene expression (Meyer et al., 2002; Wills et al., 2010). As tryptophan ligands activate AHR which in turn upregulate the transcription of CYP1A and CYP1B1 genes, reduced tryptophan levels could potentially contribute to decreased CYP1 expression (Quintana and Sherr, 2013).
4. Conclusions.
This study provides the first evidence to suggest a potential link between the gut microbial community, the metabolome, and the PAH-resistant phenotype of killifish living in a highly contaminated site. There exist several limitations in studying environmental samples, rather than lab-controlled samples, since sediment and gut systems are in constant flux. While these limitations do not allow us to establish direct connections to metabolic and bioenergic differences, we were still able to quantify major shifts in microbes and some metabolites, such as amino acids. Further, these shifts were observed in a complex and realistic environmental landscape, providing valuable data for future lab-controlled and field studies. For instance, we identified striking differences between amino acids in the two fish subpopulations, a potential relationship between sphingolipids and sphingobacteria, and a tentative connection between spermine/spermidine and oxidative stress. The dynamics of these relationships should be further explored; for instance, it is unclear if the sphingolipid regulation is due to the bacterial community, the fish host, or a combination of both. These associations provide important future areas of research in the context of killifish, as well as when examining adaptation to anthropogenic pollution in aquatic habitats.
Overall, this study revealed that exposure to PAH-contamination alters the structure of fish gut prokaryotic communities as well as the gut-associated metabolome. We postulate that shifts in the microbiome and the metabolome may be related to other physiological changes, although the mechanisms of such outcomes are unclear. In defining influences on the gut microbial community and metabolome, there is an opportunity to predict the role of the microbiome in organismal response to environmental change, monitor ecological change, and potentially forecast changes in disease susceptibility or other adaptations (Ley et al., 2006).
Supplementary Material
Highlights.
When compared to fish in a clean location, fish inhabiting a highly PAH contaminated site with evolved resistance to PAHs have shifts in their gut prokaryotic communities and metabolome.
The gut metabolome of PAH-resistant fish shows comparatively reduced levels of tryptophan and sphingolipids.
A potential relationship exists between altered sphingolipid metabolism and gut-associated sphingobacteria in PAH-resistant fish.
5. Acknowledgment
The authors would like to thank Mike Unger for his help with fish collection. CKG would like to thank RTI International for providing partial financial support for this work in the context of the RTI University Scholar’s Program. This work was funded by the NIEHS supported Duke University Superfund Research Center (P42ES010356). The metabolomics portion of this study was performed under NIH Common Fund award through NIDDK, Project Number 1U24DK097193-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. Supporting Information Available
Relative abundance of sequences from 16S rRNA gene libraries associated with bacterial phyla known to harbor members containing sphingolipids. This material is available free of charge via the Internet at http://pubs.acs.org.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
7. References
- Allen TD, Lawson PA, Collins MD, Falsen E, Tanner RS, 2006. Cloacibacterium normanense gen. nov., sp. nov., a novel bacterium in the family Flavobacteriaceae isolated from municipal wastewater. International journal of systematic and evolutionary microbiology 56,1311–1316. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, 1990. Basic local alignment search tool. Journal of molecular biology 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bartke N, Hannun YA, 2009. Bioactive sphingolipids: metabolism and function. Journal of lipid research 50, S91–S96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieri RH, Hein C, Huggett RJ, Shou P, Slone H, Smith C, Su C-W, 1986. Polycyclic aromatic hydrocarbons in surface sedicments from the Elizabeth River subesturary. International Journal of Environmental Analytical Chemistry 26, 97–113. [Google Scholar]
- Brettar I, Christen R, Bötel J., Lünsdorf H, Höfle MG., 2007. Anderseniella baltica gen. nov., sp. nov., a novel marine bacterium of the Alphaproteobacteria isolated from sediment in the central Baltic Sea. International journal of systematic and evolutionary microbiology 57, 2399–2405. [DOI] [PubMed] [Google Scholar]
- Caumette P, Imhoff JF, Süling J, Matheron R, 1997. Chromatium glycolicum sp. nov., a moderately halophilic purple sulfur bacterium that uses glycolate as substrate. Archives of microbiology 167,11–18. [DOI] [PubMed] [Google Scholar]
- Clark BW, Bone AJ, Di Giulio RT, 2014. Resistance to teratogenesis by F1 and F2 embryos of PAH-adapted Fundulus heteroclitus is strongly inherited despite reduced recalcitrance of the AHR pathway. Environmental Science and Pollution Research 21,13898–13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BW, Cooper EM, Stapleton HM, Di Giulio RT, 2013. Compound-and mixture-specific differences in resistance to PAHs and PCB-126 among Fundulus heteroclitus subpopulations throughout the Elizabeth River estuary (Virginia, USA). Environmental science & technology 47,10556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales J, Thornton C, White M, Willett KL, 2014. Multigenerational effects of benzo [a] pyrene exposure on survival and developmental deformities in zebrafish larvae. Aquatic toxicology 148,16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z-L, Wu G, Zhu W-Y, 2011. Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Frontiers in bioscience 16,1768–86. [DOI] [PubMed] [Google Scholar]
- Davis DJ, Bryda EC, Gillespie CH, Ericsson AC, 2016. Microbial modulation of behavior and stress responses in zebrafish larvae. Behavioural brain research 311, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit R, van Gemerden H, 1987. Chemolithotrophic growth of the phototrophic sulfur bacterium Thiocapsa roseopersicina. FEMS microbiology ecology 3,117–126. [Google Scholar]
- Delistraty D, 1997. Toxic equivalency factor approach for risk assessment of polycyclic aromatic hydrocarbons. Toxicological & Environmental Chemistry 64, 81–108. [Google Scholar]
- Di Giulio RT, Clark BW, 2015. The Elizabeth River story: a case study in evolutionary toxicology. Journal of toxicology and environmental Health, Part B 18, 259–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Chen L, Liu Y, Tao W, Zhang Z, Liu H, Tang Y, 2015. Planktonic and sedimentary bacterial diversity of Lake Sayram in summer. MicrobiologyOpen 4, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA, Farooqui T, 2000. Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chemistry and physics of lipids 106, 1–29. [DOI] [PubMed] [Google Scholar]
- Forget NL, Kim Juniper S, 2013. Free-living bacterial communities associated with tubeworm (R idgeia piscesae) aggregations in contrasting diffuse flow hydrothermal vent habitats at the Main Endeavour Field, Juan de Fuca Ridge. MicrobiologyOpen 2, 259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García Bernal M, Trabal Fernández N, Saucedo Lastra PE, Medina Marrero R, Mazón-Suástegui JM, 2017. Streptomyces effect on the bacterial microbiota associated to Crassostrea sikamea oyster. Journal of applied microbiology 122, 601–614. [DOI] [PubMed] [Google Scholar]
- Ghanbari M, Kneifel W, Domig KJ, 2015. A new view of the fish gut microbiome: advances from next-generation sequencing. Aquaculture 448, 464–475. [Google Scholar]
- Gómez GD, Balcázar JL, 2008. A review on the interactions between gut microbiota and innate immunity of fish. FEMS immunology & medical microbiology 52,145–154. [DOI] [PubMed] [Google Scholar]
- Gugliandolo C, Michaud L, Giudice AL, Lentini V, Rochera C, Camacho A, Maugeri TL, 2016. Prokaryotic community in lacustrine sediments of byers peninsula (Livingston Island, Maritime Antarctica). Microbial ecology 71, 387–400. [DOI] [PubMed] [Google Scholar]
- Heaver SL, Johnson EL, Ley RE, 2018. Sphingolipids in host-microbial interactions. Current opinion in microbiology 43, 92–99. [DOI] [PubMed] [Google Scholar]
- Hodson PV, 2017. The toxicity to fish embryos of PAH in crude and refined oils. Archives of environmental contamination and toxicology 73,12–18. [DOI] [PubMed] [Google Scholar]
- Houten SM, Violante S, Ventura FV, Wanders RJ, 2016. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annual review of physiology 78, 23–44. [DOI] [PubMed] [Google Scholar]
- Houten SM, Wanders RJ, 2010. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. Journal of inherited metabolic disease 33, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasundara N, Fernando PW, Osterberg JS, Cammen KM, Schultz TF, Di Giulio RT, 2017. Cost of tolerance: physiological consequences of evolved resistance to inhabit a polluted environment in teleost fish Fundulus heteroclitus. Environmental science & technology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Matson CW, Collins LB, Laban G, Stapleton HM, Bickham JW, Swenberg JA, Di Giulio RT, 2011. Genotoxicity in Atlantic killifish (Fundulus heteroclitus) from a PAH-contaminated Superfund site on the Elizabeth River, Virginia. Ecotoxicology 20, 1890–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurelevicius D, Alvarez VM, Marques JM, de Sousa Lima LRF, de Almeida Dias F, Seldin L, 2013. Bacterial community response to petroleum hydrocarbon amendments in freshwater, marine, and hypersaline water-containing microcosms. Applied and environmental microbiology 79, 5927–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Muto Y, Tanaka-Bandoh K, Watanabe K, Ueno K, 1995. Sphingolipid composition in Bacteroides species. Anaerobe 1,135–139. [DOI] [PubMed] [Google Scholar]
- Keck M, Gisch N, Moll H, Vorhölter F-J, Gerth K, Kahmann U, Lissel M, Lindner B, Niehaus K, Holst O, 2011. Unusual outer membrane lipid composition of the gram-negative, lipopolysaccharide-lacking myxobacterium Sorangium cellulosum So ce56. Journal of biological chemistry 286, 12850–12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C, Salinas I, 2017. Under pressure: interactions between commensal microbiota and the teleost immune system. Frontiers in immunology 8, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz MA, Kawasaki A, 2010. Hydrocarbon-degrading sphingomonads: Sphingomonas, sphingobium, novosphingobium, and sphingopyxis, in: Handbook of Hydrocarbon and Lipid Microbiology. [Google Scholar]
- Kit A, 2006. Analytical Specifications p150. BIOCRATES Life Sciences, Innsbruck, Austria. [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD, 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and environmental microbiology 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafortune I, Juteau P, Déziel E, Lépine F, Beaudet R, Villemur R, 2009. Bacterial diversity of a consortium degrading high-molecular-weight polycyclic aromatic hydrocarbons in a two-liquid phase biosystem. Microbial ecology 57, 455–468. [DOI] [PubMed] [Google Scholar]
- Lai Q, Cao J, Yuan J, Li F, Shao Z, 2014. Celeribacter indicus sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium from deep-sea sediment and reclassification of Huaishuia halophila as Celeribacter halophilus comb. nov. International journal of systematic and evolutionary microbiology 64, 4160–4167. [DOI] [PubMed] [Google Scholar]
- Lefèvre E, Bossa N, Gardner CM, Gehrke GE, Cooper EM, Stapleton HM, Hsu-Kim H, Gunsch CK, 2018. Biochar and activated carbon act as promising amendments for promoting the microbial debromination of tetrabromobisphenol A. Water research 128,102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, Gordon JI, 2006. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. [DOI] [PubMed] [Google Scholar]
- Li M, Wang B, Zhang Menghui, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X., Zhang Meiling, 2008. Symbiotic gut microbes modulate human metabolic phenotypes. Proceedings of the National Academy of Sciences 105, 2117–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg CD, Jayasundara N, Kozal JS, Leuthner TC, Di Giulio RT, 2017. Resistance to polycyclic aromatic hydrocarbon toxicity and associated bioenergetic consequences in a population of Fundulus heteroclitus. Ecotoxicology 26, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen W, Bozhüyük KA, Cortina NS, Bode HB, 2014. A comprehensive insight into the lipid composition of Myxococcus xanthus by UPLC-ESI-MS. Journal of lipid research 55, 2620–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane GT, Cummings JH, Allison C, 1986. Protein degradation by human intestinal bacteria. Microbiology 132,1647–1656. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Kasai H, Matsuo Y, Ōmura S, Shizuri Y, Takahashi Y, 2009. Ilumatobacter fluminis gen. nov., sp. nov., a novel actinobacterium isolated from the sediment of an estuary. The Journal of general and applied microbiology 55, 201–205. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Kasai H, Matsuo Y, Shizuri Y, Ichikawa N, Fujita N, Ōmura S, Takahashi Y, 2013. Ilumatobacternonamiense sp. nov. and Ilumatobactercoccineum sp. nov., isolated from seashore sand. International journal of systematic and evolutionary microbiology 63, 3404–3408. [DOI] [PubMed] [Google Scholar]
- Meyer JN, Di Giulio RT, 2003. Heritable adaptation and fitness costs in killifish (Fundulus heteroclitus) inhabiting a polluted estuary. Ecological applications 13, 490–503. [Google Scholar]
- Meyer JN, Nacci DE, Di Giulio RT, 2002. Cytochrome P4501A (CYP1A) in killifish (Fundulus heteroclitus): heritability of altered expression and relationship to survival in contaminated sediments. Toxicological sciences 68, 69–81. [DOI] [PubMed] [Google Scholar]
- Miyake S, Ngugi DK, Stingl U, 2016. Phylogenetic diversity, distribution, and cophylogeny of giant bacteria (Epulopiscium) with their surgeonfish hosts in the Red Sea. Frontiers in microbiology 7, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Chernick M, Dong W, Di Giulio RT, Hinton DE, 2017. Early life co-exposures to a real-world PAH mixture and hypoxia result in later life and next generation consequences in medaka (Oryzias latipes). Aquatic Toxicology 190,162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Yan Q, Yu Y, Zhang T, 2014. Factors influencing the grass carp gut microbiome and its effect on metabolism. FEMS microbiology ecology 87, 704–714. [DOI] [PubMed] [Google Scholar]
- Olsen I, Jantzen E, 2001. Sphingolipids in bacteria and fungi. Anaerobe 7,103–112. [Google Scholar]
- Osterberg JS, Cammen KM, Schultz TF, Clark BW, Di Giulio RT, 2018. Genome-wide scan reveals signatures of selection related to pollution adaptation in non-model estuarine Atlantic killifish (Fundulus heteroclitus). Aquatic Toxicology 200, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh PJ, Arusi E, Vinik A, Johnson DA, 2014. The role and influence of gut microbiota in pathogenesis and management of obesity and metabolic syndrome. Frontiers in endocrinology 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Sherr DH, 2013. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacological reviews 65,1148–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid NM, Proestou DA, Clark BW, Warren WC, Colbourne JK, Shaw JR, Karchner SI, Hahn ME, Nacci D, Oleksiak MF, 2016. The genomic landscape of rapid repeated evolutionary adaptation to toxic pollution in wild fish. Science 354,1305–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico JE, Zang Y, Haughey NJ, Rius AG, McFadden JW, 2018. Circulating fatty acylcarnitines are elevated in overweight periparturient dairy cows in association with sphingolipid biomarkers of insulin resistance. Journal of dairy science 101, 812–819. [DOI] [PubMed] [Google Scholar]
- Riley AK, Chernick M, Brown DR, Hinton DE, Di Giulio RT, 2016. Hepatic responses of juvenile Fundulus heteroclitus from pollution-adapted and nonadapted populations exposed to Elizabeth River sediment extract. Toxicologic pathology 44, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues CF, Webster G, Cunha MR, Duperron S, Weightman AJ, 2010. Chemosynthetic bacteria found in bivalve species from mud volcanoes of the Gulf of Cadiz. FEMS microbiology ecology 73, 486–499. [DOI] [PubMed] [Google Scholar]
- Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao C-C, Patel B, Yan R, Blain M, 2016. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aiyl hydrocarbon receptor. Nature medicine 22, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungrassamee W, Klanchui A, Maibunkaew S, Karoonuthaisiri N, 2016. Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. Journal of invertebrate pathology 133,12–19. [DOI] [PubMed] [Google Scholar]
- Schmid HH, Schmid PC, Natarajan V, 1990. N-acylated glycerophospholipids and their derivatives. Progress in lipid research 29,1–43. [DOI] [PubMed] [Google Scholar]
- Sepahi A, Cordero H, Goldfine H, Esteban MÁ, Salinas I, 2016. Symbiont-derived sphingolipids modulate mucosal homeostasis and B cells in teleost fish. Scientific reports 6,1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y, Ren H, Limbu SM, Sun Y, Qiao F, Zhai W, Du Z-Y, Zhang M, 2018. The presence or absence of intestinal microbiota affects lipid deposition and related genes expression in zebrafish (Danio rerio). Frontiers in Microbiology 9,1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JH, Kwon KK, Kang J-H, Jung H-B, Kim S-J, 2004. Novosphingobium pentaromativorans sp. nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. International journal of systematic and evolutionary microbiology 54,1483–1487. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S, 2002. Sphingosine 1-phosphate, a key cell signaling molecule. Journal of biological chemistry 277, 25851–25854. [DOI] [PubMed] [Google Scholar]
- Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, 2014. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nature communications 5,1–13. [DOI] [PubMed] [Google Scholar]
- Stauffert M, Cravo-Laureau C, Jezequel R, Barantal S, Cuny P, Gilbert F, Cagnon C, Militon C, Amouroux D, Mahdaoui F, 2013. Impact of oil on bacterial community structure in bioturbated sediments. PLoS One 8, e65347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullam KE, Essinger SD, Lozupone CA, O’CONNOR MP, Rosen GL, Knight ROB, Kilham SS, Russell JA, 2012. Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Molecular ecology 21, 3363–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M, 2014. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PloS one 9, el05592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkoff SJ, Osterberg JS, Jayasundara N, Cooper E, Hsu-Kim H, Rogers L, Gehrke GE, Jayaraman S, Di Giulio RT, 2019. Embryonic Fundulus heteroclitus responses to sediment extracts from differentially contaminated sites in the Elizabeth River, VA. Ecotoxicology 28,1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR, 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and environmental microbiology 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xue Y, 2015. Characterization of solid tumors induced by polycyclic aromatic hydrocarbons in mice. Medical science monitor basic research 21, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Komatsu N, Kitamura T, Ishii Y, Park H-D, Miyata R, Noda N, Sekiguchi Y, Satou T, Watanabe M, 2012. Ecological niche separation in the Polynucleobacter subclusters linked to quality of dissolved organic matter: a demonstration using a high sensitivity cultivation-based approach. Environmental microbiology 14, 2511–2525. [DOI] [PubMed] [Google Scholar]
- Wills LP, Matson CW, Landon CD, Di Giulio RT, 2010. Characterization of the recalcitrant CYP1 phenotype found in Atlantic killifish (Fundulus heteroclitus) inhabiting a Superfund site on the Elizabeth River, VA. Aquatic toxicology 99, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Mandal R, Sinelnikov IV, Broadhurst D, Wishart DS, 2012. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic acids research 40, W127–W133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Sinelnikov IV, Han B, Wishart DS, 2015. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic acids research 43, W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Wishart DS, 2011a. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nature protocols 6, 743–760. [DOI] [PubMed] [Google Scholar]
- Xia J, Wishart DS, 2011b. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Current protocols in bioinformatics 34,14–10. [DOI] [PubMed] [Google Scholar]
- Zhang M, Sun Y, Chen L, Cai C, Qiao F, Du Z, Li E, 2016. Symbiotic bacteria in gills and guts of Chinese mitten crab (Eriocheir sinensis) differ from the free-living bacteria in water. PloS one 11, e0148135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HW, Guo CL, Wong YS, Tam NFY, 2006. Genetic diversity of dioxygenase genes in polycyclic aromatic hydrocarbon-degrading bacteria isolated from mangrove sediments. FEMS microbiology letters 262,148–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.