

Abstract
Stimulator of interferon genes (STING) activation by intratumoral STING agonist treatment has been recently shown to eradicate tumors in preclinical models of cancer immunotherapy, generating intense research interest and leading to multiple clinical trials. However, there are many challenges associated with STING agonist-based cancer immunotherapy, including low cellular uptake of STING agonists. Here, biodegradable mesoporous silica nanoparticles (bMSN) with an average size of 80 nm are developed for efficient cellular delivery of STING agonists. STING agonists delivered via bMSN potently activate innate and adaptive immune cells, leading to strong antitumor efficacy and prolonged animal survival in murine models of melanoma. Delivery of immunotherapeutic agents via biodegradable bMSN is a promising approach for improving cancer immunotherapy.
Keywords: biodegradable nanoparticles, silica nanoparticles, cancer immunotherapies, STING agonists
Stimulator of interferon genes (STING) molecule is one of the pattern recognition receptor proteins that detect intracellular DNA. It serves to protect the host against intracellular invasions by pathogens, such as virus and bacteria. Foreign DNA is detected by cyclic GMP-AMP (cGAMP) synthase (cGAS), which produces cGAMP from adenosine triphosphate (ATP) and guanosine triphosphate (GTP), leading to subsequent activation of STING. The cGAS-STING pathway activates a cascade of immune pathways, including IRF3 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-rB), resulting in potent type I interferon (IFN) response.[1] Importantly, a number of recent studies revealed that STING plays a crucial role in sensing spontaneously arising tumors and initiating innate immune responses.[2] Numerous studies have reported promising anticancer effects of STING agonists,[3,4] including preclinical studies that have shown STING agonist-mediated tumor regression and durable antitumor immunity.[5,6] Thus, STING has sparked intense research interest in the field of cancer immunotherapy[7,8] and there are multiple clinical trials underway to evaluate the therapeutic efficacy of STING agonists.
However, there are major challenges associated with STING activators for cancer immunotherapy. As STING agonists are based on cyclic dinucleotides with negative charges, their cellular permeability is minimal. Due to their small molecular weight, STING agonists diffuse rapidly into systemic circulation upon injection, limiting drug exposure in tumor tissues and potentially causing off-target toxicities. In addition, the amount of STING agonist injected to tumor has been shown to dictate CD8+ T cell response,[9] underscoring the importance of regulating the dose and pharmacokinetics of STING agonist to achieve robust antitumor immunity. To address these issues, various nanoparticles (NPs) carrying STING agonists and polymers with STING-activating properties have been developed with varying degrees of success reported in preclinical models.[10–14] Here, we sought to promote cellular delivery of STING agonists using a well-established NP system with a strong track record of biocompatibility, safety, and manufacturability. In particular, mesoporous silica nanoparticle (MSN) is a widely used inorganic drug delivery nanocarrier with tunable size, low immunogenicity, controlled release ofcargo materials, and facile and low-cost preparation process.[15,16] However, while synthetic MSNs composed of amorphous silica are generally considered to be biocompatible, their limitations include relatively small pore sizes for drug loading, slow biodegradation, and long-term tissue retention.[17–21]
To address these issues, we have developed biodegradable mesoporous silica nanoparticles (bMSN) for cytosolic delivery of STING agonists (Figure 1). We have recently shown that bMSN with a less-dense Si—O—Si matrix undergoes faster biodegradation process than conventional MSN and that bMSN’s large pore size ranging 5–10 nm allows for efficient loading and delivery of biomacromolecules for combination cancer treatments.[22] Here, we report that bMSN surface-modified with amine serves as a promising platform for cellular delivery of STING agonists and immune activation. Using two murine models of melanoma, we demonstrate the therapeutic efficacy of bMSN carrying STING agonists, highlighting the potential of bMSN for applications in cancer immunotherapy.
Figure 1.
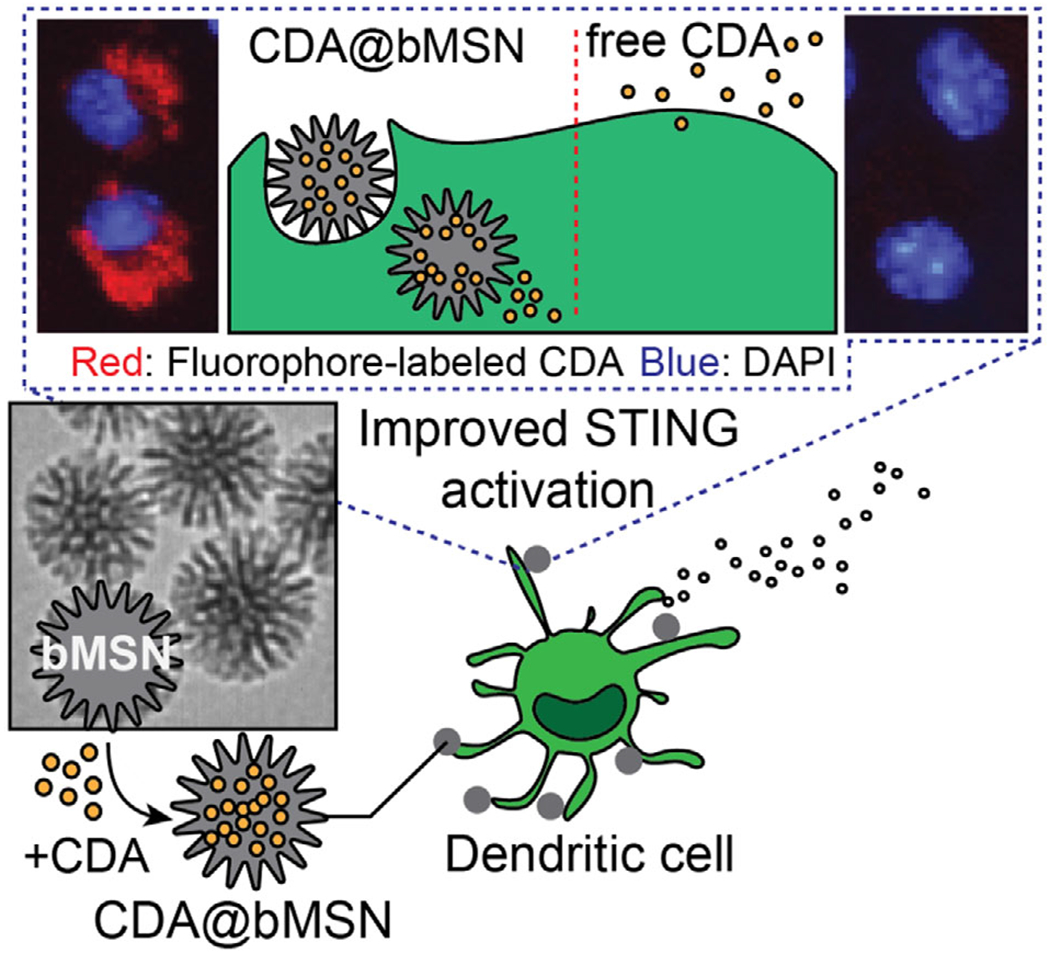
Schematic illustration of bMSN for delivery of STING agonists.
We synthesized bMSN as we previously reported with some modifications for cytosolic delivery of STING agonists.[22] The resulting bMSN had an approximate average diameter of 80 nm, as visualized by transmission electron microscopy (TEM) (Figure 2a). Compared with conventional mesoporous silica nanoparticles, bMSN had a larger pore size (5–10 nm) and a thinner Si—O—Si matrix that allowed for rapid degradation within 120 h in a physiological condition (Figure 2b). We loaded bMSN with a model STING agonist, bis-(3’−5’)-cydic dimeric adenosine monophosphate (CDA). Since CDA is a dicyclic nucleotide with anionic charges, the silica surface of bMSN was modified with amines (−NH2) to facilitate charge-mediated drug loading. The amine-modification changed the zeta potential of bMSN from −27.6 to 9.3 mV but did not affect the size of the nanoparticles (Figure 2c,d). The resulting bMSN-NH2 (henceforth referred to as bMSN) was incubated at various concentrations with a fixed amount of CDA (8 μg). After CDA was simply mixed and incubated with preformed bMSN for 1 h, we observed >90% loading of 8 pg CDA into 25 pg of bMSN (Figure 2e), indicating drug loading of ≈290 μg mg−1 of bMSN (CDA@bMSN). CDA was released from CDA@bMSN within 1 h at pH 7.0 (Figure 2f). However, when incubated at a slightly acidic condition of pH 6.0 mimicking the conditions within the tumor microenvironment (TME),[23] we observed slower release of CDA (Figure 2f).
Figure 2.
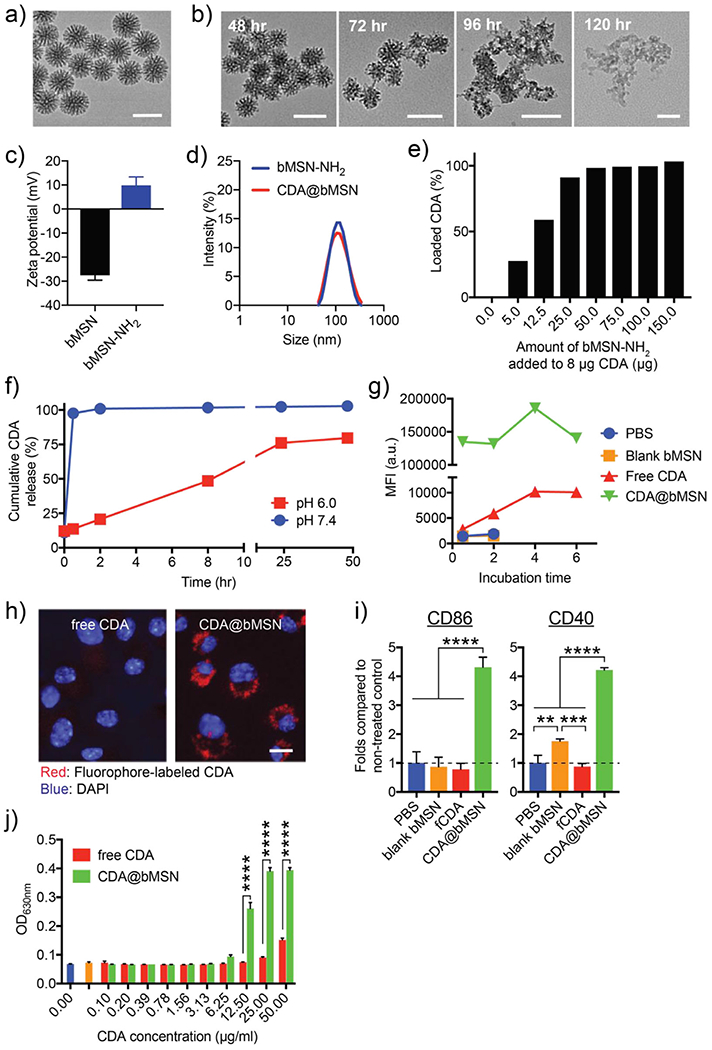
Cellular delivery of STING agonists using bMSN. a) TEM image of bMSN. b) Degradation of bMSN in a physiological condition (Krebs-Henseleit solution at 37 °C). c) Surface charge and d) hydrodynamic size of bMSN measured before and after amine-modification using dynamic light scattering (DLS). Particles were transferred to water for measurements. e) CDA-loading capacity of bMSN. f) CDA Release profiles in different pH conditions. g,h) Uptake of CDA by BMDCs assessed in vitro with g) flow cytometry and h) confocal microscopy. i) Activation of BMDCs measured by flow cytometry after 4 h of incubation. j) STING activation of human monocyte-derived THP1-Blue ISG cells measured after overnight incubation. All data are presented as mean ± SEM, showing representative results from two independent studies with n = 3, with an exception of g) with n = 1. Scale bars in a,b) = 100 nm and h) = 5 μm. **P < 0.01, *** P < 0.001, ****P < 0.0001 analyzed by one-way or two-way ANOVA with Tukey’s honestly significant difference (HSD) multiple comparison post hoc test.
Next, we examined cellular uptake of CDA@bMSN using mouse bone marrow-derived dendritic cells (BMDCs), as a widely used surrogate for APCs. BMDCs incubated with free fluorophore-tagged CDA for 6 h showed a minimal increase in the fluorescence signal (Figure 2g), whereas BMDCs incubated with CDA@bMSN exhibited markedly enhanced CDA signal (Figure 2g). Similarly, confocal microscopy revealed that BMDCs and B16F10 melanoma cells incubated with fluorophore-tagged CDA@bMSN exhibited much stronger cytosolic fluorescence signal, compared with minimal signal in cells treated with free CDA (Figure 2h and Figure S1, Supporting Information), demonstrating efficient bMSN-mediated cytosolic delivery of STING agonist.
In addition, CDA@bMSN treatment led to robust BMDC activation, as evidenced by significantly increased expression of CD40 (P < 0.0001) and CD86 (P < 0.001), compared with free CDA treatment (Figure 2i), likely due to the increased cellular uptake of CDA@bMSN. Blank bMSN slightly increased the expression level of CD40, but not CD86, indicating minimal immune activation by the blank bMSN itself. Next, we used THP1-Blue interferon-stimulated genes (ISG) (human monocyte-derived cells expressing a reporter gene for STING activation) to study the effects of CDA@bMSN on STING activation. Compared with free CDA, CDA@bMSN induced significantly stronger STING activation even at 12.5 μg mL−1 dose of CDA (P < 0.0001, Figure 2j), indicating amplification STING activation by bMSN-mediated delivery of CDA. Notably, both THP1-Blue ISG cells and primary CD8+ T cells incubated with either free CDA or CDA@bMSN exhibited similar levels of viability (Figures S2 and S3, Supporting Information), showing biocompatibility of CDA@bMSN. We have also previously reported biological safety of the bMSN platform.[22] Taken together, compared with free soluble CDA, CDA@bMSN significantly improved cellular uptake of CDA and amplified STING activation, without negatively affecting cytotoxicity.
We examined secretion of cytokines and chemokines from BMDCs treated with CDA formulations. In line with the enhanced uptake of CDA and activation of BMDCs (Figure 2g–j), CDA@bMSN treatment significantly increased the release of interleukin-6 (IL-6), IL-12p40, IFN-β, CXCL10, CCL2, CCL3, and CCL5 from BMDCs, compared with free CDA (P < 0.0001, Figure 3a). Although tumor-infiltrating immune cells are known as the major cell types that are activated by STING agonists and initiate antitumor immune response, tumor cells also have been shown to respond to STING agonists.[24,25] We examined whether CDA treatment can promote cytokine and chemokine production from melanoma cell lines, B16F10 and B16F10OVA. Tumor cells incubated with free CDA did not release any detectable levels of cytokines or chemokines; however, CDA@bMSN treatment led to significantly amplified secretion of CXCL10 and CCL5 from B16F10 cells and CCL2 and CCL5 from B16F10OVA cells (P < 0.01 for CCL2 and P < 0.0001 for the others, Figure 3b). In order to confirm STING-dependent activation, we pretreated BMDCs with an STING inhibitor, C-178, followed by incubation with CDA formulations. Pretreatment with C-178 significantly decreased the secretion of cytokines and chemokines induced by both free CDA and CDA@bMSN (Figure 3c), showing that CDA-mediated immune activation is indeed dependent on the STING pathway.
Figure 3.
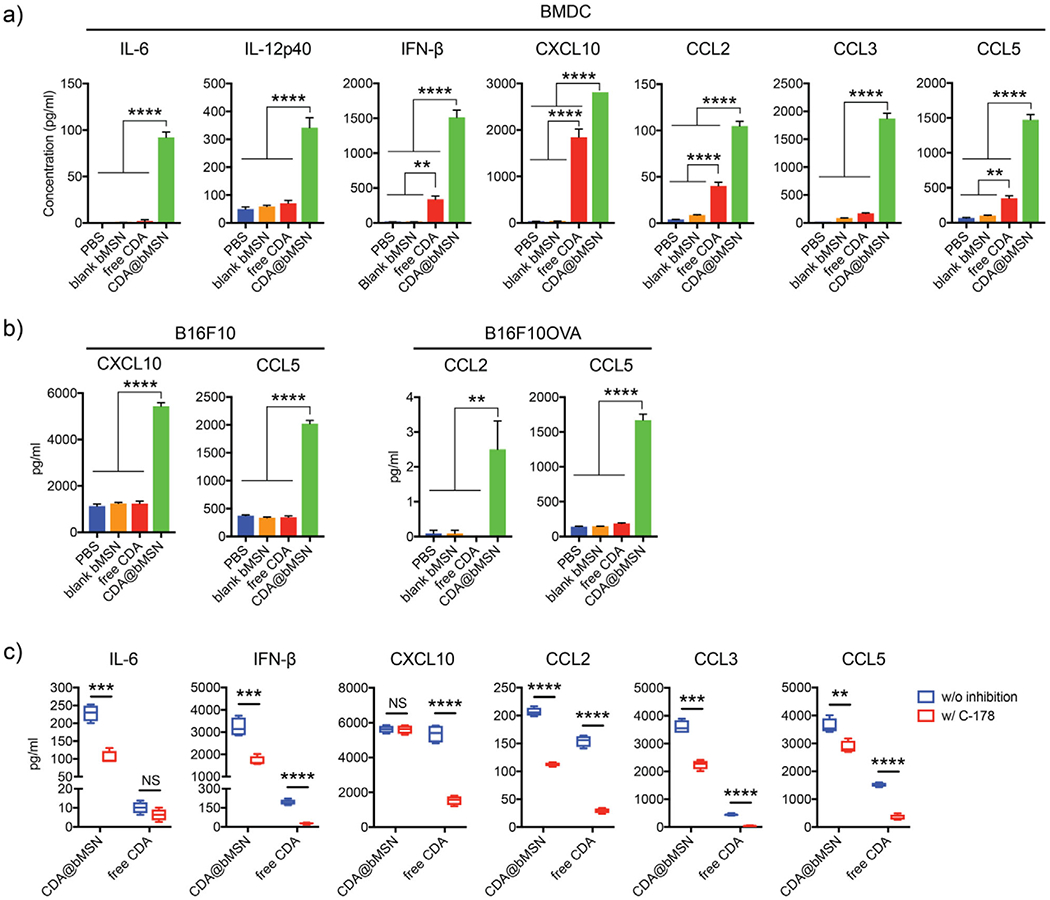
CDA@bMSN promotes cytokine and chemokine release from BMDCs and tumor cells. a) Mouse BMDCs and b) mouse melanoma cell lines, B16F10 and B16F10OVA, were treated with 10 μg mL−1 of CDA for 6 h in vitro. Supernatants were assessed by enzyme-linked immunosorbent assay (ELISA) for cytokines and chemokines. c) BMDCs pretreated for 1 h with 0.5 × 10−6 m of an STING inhibitor, C-178, followed by treatment with 10 μg mL−1 of CDA for 6 h. Data are presented as mean ± SEM, showing representative results from two independent studies with n = 3–4. **P < 0.01, *** P < 0.001, ****P < 0.0001 analyzed by one-way or two-way ANOVA with Tukey’s HSD multiple comparison post hoc test.
Next, antitumor effects of CDA@bMSN was investigated in a murine melanoma model of B16F10OVA. C57BL/6 mice were injected subcutaneously with 3 × 105 B16F10OVA cells on the right-side flank. When tumors reached >100 mm3 on day 6 after tumor inoculation, we performed a single intratumoral administration of 2 μg CDA in either bMSN or free form (Figure 4a). Interestingly, both free CDA and CDA@bMSN treatments were able to induce regression of established tumors with minimal tumor volume by day 14 (Figure 4b). However, 50% of mice in the free CDA-treated group quickly relapsed and had to be euthanized by day 30. In stark contrast, 100% mice in the CDA@bMSN treatment group remained tumor free for the duration of the study (Figure 4c). To understand the differences between the free CDA and CDA@bMSN treatment groups, we analyzed the levels of cytokines and chemokines as well as various immune cell subsets. In line with our in vitro results (Figure 3), after 3 h of intratumoral administration, CDA@bMSN induced strong release of IFN-β, CXCL10, CCL2, and CCL5 in TME and serum (Figure 4d,e). Notably, even after 24 h of administration, higher levels of CXCL10 and CCL2 were detected within the TME for the CDA@bMSN group, compared with free CDA group (P < 0.05, Figure 4d), suggesting sustained immune activation mediated by bMSN.
Figure 4.
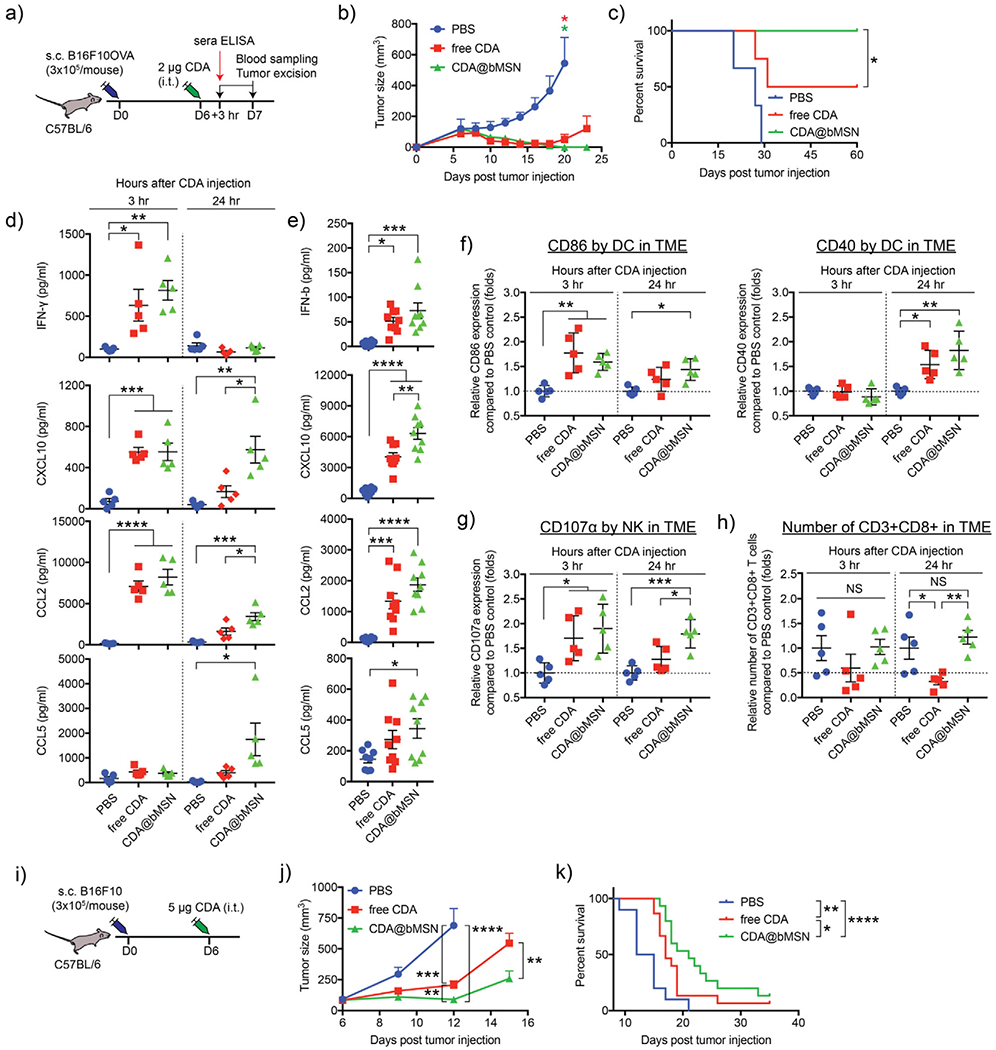
A single intratumoral treatment with CDA@bMSN exerts potent antitumor efficacy. a) C57BL/6 mice were subcutaneously injected with 3 × 105 B16F10OVA cells on the right-side flank. After 6 d, each mouse received intratumoral injection of 2 μg CDA as a soluble or bMSN formulations. After 3 or 24 h, blood sampling and tumor excision were performed. b) Tumor growth curves and c) animal survival are shown. d) Cytokine levels within tumor tissues or e) sera were measured by ELISA after 3 and 24 h or 3 h of CDA injection, respectively. Flow cytometric analyses were performed to examine f) CD86 and CD40 expression on DCs, g) CD107α expression on NK cells, and h) the number of CD8+ T cells within the B16F10OVA TME. i) C57BL/6 mice were subcutaneously injected with 3 × 105 B16F10 cells on the right-side flank. After 6 d, each mouse received intratumoral injection of 5 μg free CDA or CDA@bMSN. j) Tumor growth curves and k) animal survival curves are shown. Data are presented as mean ± SEM, showing representative results from two independent studies with n = 3–4 for (b) and (c) and n = 5 for (d)-(f). n = 15 for (j) and (k) pooled from two independent studies. *P < 0.05, **P < 0.01, *** P < 0.001, ****P < 0.0001 analyzed by one-way or two-way ANOVA with Tukey’s HSD multiple comparison post hoc test. Animal survival curves were analyzed by the log-rank (Mantel–Cox) test.
We also investigated the effects of CDA treatment on the innate and adaptive immune responses. Intratumoral administration of CDA@bMSN led to robust activation of DCs within the TME, as evidenced by upregulation of CD86 within 3 h and CD40 within 24 h post-treatment (Figure 4f). There was a trend for higher expression levels of CD86 and CD40 on DCs after 24 h of treatment with CDA@bMSN, compared with free CDA (Figure 4f) although their differences were not statistically significant. While free CDA treatment induced transient upregulation and down-regulation of CD107α, a degranulation marker, on intratumoral natural killer (NK) cells, CDA@bMSN treatment led to sustained expression of CD107α on intratumoral NK cells for up to 24 h (P < 0.5, Figure 4g). By 24 h of CDA@bMSN treatment, we also observed activation of NK cells in the circulation, as shown by increased levels of CD107α and NKG2D[26,27] (Figure S4, Supporting Information).
Intratumoral injection of free CDA significantly decreased the number of CD8+ T cells within the tumor by 24 h post-CDA injection, compared with no treatment group (P < 0.05, Figure 4h). This is in line with the literature reporting decreased tumor-infiltration of lymphocytes after intratumoral administration of free STING agonist.[9,24] In contrast, intratumoral CDA@bMSN treatment resulted in significantly higher number of intratumoral CD8+ T cells at 24 h time point, compared with free CDA (P < 0.01, Figure 4h), suggesting that bMSN-mediated CDA delivery reversed the decrease in intratumoral infiltration of CD8+ T cells associated with free CDA.[9,24] CDA@bMSN treatment also significantly increased the expression of a degranulation marker, CD107α, on intratumoral CD8+ T cells by 24 h, compared with untreated control (P < 0.05, Figure S5, Supporting Information). Overall, these results suggest that bMSN-mediated delivery of CDA amplifies the magnitude and duration of cytokine and chemokine release within TME and potently activates intratumoral DCs, NK cells, and CD8+ T cells, thus leading to regression of established tumors.
Having observed strong antitumor efficacy of CDA@bMSN, we evaluated CDA@bMSN in the setting of established B16F10 melanoma. As B16F10 is a poorly immunogenic, highly aggressive tumor model, we increased the dose of CDA@bMSN to 5 μg. C57BL/6 mice were inoculated subcutaneously (s.c.) with 3 × 105 B16F10 cells. When tumors were >50 mm3 on day 6, we performed a single intratumoral administration of CDA either in a soluble or bMSN forms (Figure 4i). Whereas mice in the untreated control group quickly succumbed to B16F10 tumor with a median survival of 12 d, free CDA treatment slowed the B16F10 tumor growth and extended the median survival to 18 d (P < 0.01, Figure 4j,k). Compared with free CDA group, CDA@bMSN treatment further inhibited B16F10 tumor growth (P < 0.01, Figure 4j) and extended the median survival to 24 d (P < 0.05, Figure 4k), thus highlighting the therapeutic potential of CDA@bMSN.
In summary, we have developed bMSN for efficient cytosolic delivery of STING agonists. While previous studies have reported various STING agonist-loaded NP systems,[10,11,28–30] including liposomes and polymeric NPs, their fabrication and drug loading procedures are often complicated with multiple synthesis and separation steps, and many of these NP platforms have not been clinically tested. On the other hand, silica-based NPs offer a promising platform with biocompatibility, facile manufacturing process, and clinical safety.[15,16,31] Notably, we have achieved >90% loading of CDA into bMSN simply by admixing CDA with preformed bMSN for 1 h. We show that bMSN carrying STING agonists improves STING activation by DCs and tumor cells and elicits potent innate and adaptive immune responses in vivo, leading to strong antitumor efficacy and prolonged animal survival in murine models of melanoma. While the mechanisms underlying bMSN-mediated STING activation and subsequent cascades of innate and adaptive immune responses remain to be elucidated, our results suggest that bMSN is a biodegradable and biocompatible carrier for efficient delivery of STING agonists. It is also notable that the typical dose of STING agonists reported in the literature ranges from 10 to 240 μg, often used in combination with chemotherapeutic or immunotherapeutic agents. [5,9,32–36] In contrast, we report that a single injection of CDA@bMSN at the dose of 5 μg or less exerted potent antitumor efficacy, thus highlighting the dose-sparing effect of the bMSN platform. Recent advances in cancer immunotherapy have generated intense research interest in drug delivery vehicles for improving immune activation,[37–40] and our bMSN system may offer a promising platform for delivery of immunomodulatory agents for cancer immunotherapy. Nevertheless, as our current studies have mainly focused on the acute responses mounted by innate immune cells after intratumoral CDA injection, our future studies will address the effects ofbMSN-based CDA delivery on adaptive immune responses and examine systemic delivery of CDA@bMSN. Further research is warranted to optimize the bMSN platform for the delivery of STING agonists in the context of combination cancer immunotherapy.
Experimental Section
Synthesis of bMSN and CDA Loading:
bMSN was synthesized by an oil-water biphase reaction approach.[22,41] Twenty-four milliliters of (25 wt%) cetyltrimethylammonium chloride (CTAC) solution and 0.18 g of triethylamine (TEA) were added to 36 mL of water and stirred gently at 60 °C for 1 h. Twenty milliliters of tetraethyl orthosilicate (TEOS) in cyclohexane (10% v/v) was carefully added to 60 mL of the water-CTAC-TEA solution (0.3 m CTAC and 20 × 10−3 m TEA) and kept at 60 °C. The reaction was kept at a constant temperature with continuous stirring for 18 h to obtain nanoparticles. They were washed with ethanol for three times and water for two times with centrifugation at 15 500 × g for 15 min. Surfactant was removed by incubating the nanoparticles in 10% NH4NO3/ethanol v/v at 50 °C overnight, followed by washing. The resulting nanoparticles were freeze dried and stored at 4 °C until use. To load CDA, 40 μg of CDA was mixed with 225 μg of bMSN in 5 × 10−3 m histidine buffer, followed by 1 min of bath sonication. The mixture was incubated in 37 °C for 1 h under constant shaking, then centrifuged at 10 000 × g for 5 min. After removing the supernatant, the pellet was dispersed into phosphate buffered saline (PBS). The resulting CDA@bMSN showed 96.3% of CDA loading efficiency.
In Vivo Studies:
Animals were cared for following the federal, state, and local guidelines. The University of Michigan, Ann Arbor is an Assessment and Accreditation of Laboratory Animal Care (AAALAC) international accredited institution and all work conducted on animals was in accordance with and approved by the Institutional Animal Care and Use Committee (IACUC) with the protocol # PRO00008587. Female C57BL/6 mice, 5–6 weeks in age (the Jackson Laboratory) were inoculated subcutaneously with 3 × 105 mouse melanoma cells (either B16F10 or B16F10OVA) on the right-side flank. After 6 d, mice received CDA formulations (PBS as control) via intratumoral injection. Blood sampling and tumor excision were performed on the indicated time points. Blood samples were collected from the facial vein using a lancet. In a separate study, tumor sizes were measured every 2–3 d for monitoring tumor growth.
Statistical Analysis:
Data are presented as mean ± standard error of the mean (SEM). Data were approximately normally distributed, and variance was similar between the groups. Experiments were repeated multiple times as independent experiments with the sample size indicated in the figure captions. Data were analyzed using either one-way analysis of variance (ANOVA) or two-way ANOVA, followed by Tukey’s multiple comparison test for comparison of multiple groups using Prism 7.0e (Graph-Pad Software). Animal survival was analyzed by the log-rank (Mantel–Cox) test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgements
This work was supported in part by National Institutes of Health (NIH) (R01EB022563, R01AI127070, R01CA210273, R01CA223804, R01DK125087, and U01CA210152), The Michigan Translational Research and Commercialization for Life Sciences Hub, and Emerald Foundation. J. J.M. was supported by The Department of Defense/Congressionally Directed Medical Research Programs Peer Reviewed Cancer Research Program (W81XWH-16-1-0369) and National Science Foundation (NSF) Faculty Early Career Development Program (CAREER) Award (1553831). K. S.P. acknowledges financial support from the University of Michigan Tissue Engineering and Regeneration Training Program (DE007057 from National Institute for Dental and Craniofacial Research). C.L. was supported by a predoctoral fellowship from the Cellular Biotechnology Training Program (T32GM008353) and a Graduate Assistance in Areas of National Need Fellowship awarded to the University of Michigan. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. The authors acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for provision of major histocompatibility complex class-I tetramers.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adtp.202000130
Contributor Information
Kyung Soo Park, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA, Biointerfaces Institute, University of Michigan, Ann Arbor MI 48109, USA.
Cheng Xu, Biointerfaces Institute, University of Michigan, Ann Arbor MI 48109, USA, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor MI 48109, USA.
Xiaoqi Sun, Biointerfaces Institute, University of Michigan, Ann Arbor MI 48109, USA, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor MI 48109, USA.
Cameron Louttit, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA, Biointerfaces Institute, University of Michigan, Ann Arbor MI 48109, USA.
James J. Moon, Department of Biomedical Engineering, University of Michigan, Ann Arbor, MI 48109, USA, Biointerfaces Institute, University of Michigan, Ann Arbor MI 48109, USA, Department of Pharmaceutical Sciences, University of Michigan, Ann Arbor MI 48109, USA
References
- [1].Motwani M, Pesiridis S, Fitzgerald KA, Nat. Rev. Genet 2019, 20, 657. [DOI] [PubMed] [Google Scholar]
- [2].Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF, Immunity 2014, 41, 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li A, Yi M, Qin S, Song Y, Chu Q, Wu K, J. Hematol. Oncol 2019, 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Woo SR, Corrales L, Gajewski TF, Trends Immunol. 2015, 36, 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW, Gajewski TF, Cell Rep. 2015, 11, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gadkaree SK, Fu J, Sen R, Korrer MJ, Allen C, Kim YJ, Head Neck 2017, 39, 1086. [DOI] [PubMed] [Google Scholar]
- [7].Barber GN, Nat. Rev. Immunol 2015, 15, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Su T, Zhang Y, Valerie K, Wang XY, Lin S, Zhu G, Theranostics 2019, 9, 7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, Hudson TE, Vu UT, Francica BJ, Banda T, Katibah GE, Kanne DB, Leong JJ, Metchette K, Bruml JR, Ndubaku CO, McKenna JM, Feng Y, Zheng L, Bender SL, Cho CY, Leong ML, van Elsas A, Dubensky TW, McWhirter SM, Cell Rep. 2018, 25, 3074. [DOI] [PubMed] [Google Scholar]
- [10].Shae D, Becker KW, Christov P, Yun DS, Lytton-Jean AKR, Sevimli S, Ascano M, Kelley M, Johnson DB, Balko JM, Wilson JT, Nat. Nanotechnol 2019, 14, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ,J. Clin. Invest 2015, 125, 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y, Du M, Huang G, Wang C, Chen X, Porembka MR, Lea J, Frankel AE, Fu YX, Chen ZJ, Gao J, Nat. Nanotechnol 2017, 12, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Miao L, Li L, Huang Y, Delcassian D, Chahal J, Han J, Shi Y, Sadtler K, Gao W, Lin J, Doloff JC, Langer R, Anderson DG, Nat. Biotechnol 2019, 37, 1174. [DOI] [PubMed] [Google Scholar]
- [14].Louttit C, Park KS, Moon JJ, Biomaterials 2019, 217, 119287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI, Chem. Soc. Rev 2012, 41, 2590. [DOI] [PubMed] [Google Scholar]
- [16].Slowing II, Vivero-Escoto JL, Wu CW, Lin VS, Adv. Drug Delivery Rev 2008, 60, 1278. [DOI] [PubMed] [Google Scholar]
- [17].Shen D, Yang J, Li X, Zhou L, Zhang R, Li W, Chen L, Wang R, Zhang F, Zhao D, Nano Lett. 2014, 14, 923. [DOI] [PubMed] [Google Scholar]
- [18].Cauda V, Schlossbauer A, Bein T, Microporous Mesoporous Mater. 2010, 132, 60. [Google Scholar]
- [19].Li Z, Barnes JC, Bosoy A, Stoddart JF, Zink JI, Chem. Soc. Rev 2012, 41, 2590. [DOI] [PubMed] [Google Scholar]
- [20].Benne N, van Duijn J, Kuiper J, Jiskoot W, Slütter B, Controlled Release J 2016, 234, 124. [DOI] [PubMed] [Google Scholar]
- [21].Kwon D, Cha BG, Cho Y, Min J, Park EB, Kang SJ, Kim J, Nano Lett. 2017, 17,2747. [DOI] [PubMed] [Google Scholar]
- [22].Xu C, Nam J, Hong H, Xu Y, Moon JJ, ACS Nano 2019, 13, 12148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ, Gao J, Nat. Mater 2014, 13, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Francica BJ, Ghasemzadeh A, Desbien AL, Theodros D, Sivick KE, Reiner GL, Hix Glickman L, Marciscano AE, Sharabi AB, Leong ML, McWhirter SM, Dubensky TW, Pardoll DM, Drake CG, Cancer Immunol. Res 2018, 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sundararaman SK, Barbie DA, Immunity 2018, 49, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alter G, Malenfant JM, Altfeld M, Immunol J. Methods 2004, 294, 15. [DOI] [PubMed] [Google Scholar]
- [27].Aktas E, Kucuksezer UC, Bilgic S, Erten G, Deniz G, Cell Immunol. 2009, 254, 149. [DOI] [PubMed] [Google Scholar]
- [28].Lin LC, Huang CY, Yao BY, Lin JC, Agrawal A, Algaissi A, Peng BH, Liu YH, Huang PH, Juang RH, Chang YC, Tseng CT, Chen HW, Hu CJ, Adv. Funct. Mater 2019, 29, 1807616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Atukorale PU, Raghunathan SP, Raguveer V, Moon TJ, Zheng C, Bielecki PA, Wiese ML, Goldberg AL, Covarrubias G, Hoimes CJ, Karathanasis E, Cancer Res. 2019, 79, 5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wilson DR, Sen R, Sunshine JC, Pardoll DM, Green JJ, Kim YJ, Nanomedicine 2018, 14, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Anselmo AC, Mitragotri S, AAPSJ. 2015, 17, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, Sivick KE, Zeng Q, Soares KC, Zheng L, Portnoy DA, Woodward JJ, Pardoll DM, Dubensky TW Jr., Kim Y, Sci. Transl. Med 2015, 7, 283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M, Proc. Natl. Acad. Sci. USA 2015, 112, 15408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sallets A, Robinson S, Kardosh A, Levy R, Blood Adv. 2018, 2, 2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ghaffari A, Peterson N, Khalaj K, Vitkin N, Robinson A, Francis JA, Koti M, Br. J. Cancer 2018, 119, 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Foote JB, Kok M, Leatherman JM, Armstrong TD, Marcinkowski BC, Ojalvo LS, Kanne DB, Jaffee EM, Dubensky TW Jr., Emens LA, Cancer Immunol. Res 2017, 5, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Milling L, Zhang Y, Irvine DJ, Adv. Drug Delivery Rev 2017, 114, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Scheetz L, Park KS, Li Q, Lowenstein PR, Castro MG, Schwendeman A, Moon JJ, Nat. Biomed. Eng 2019, 3, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Nam J, Son S, Park KS, Zou W, Shea LD, Moon JJ, Nat. Rev. Mater 2019, 4, 398. [Google Scholar]
- [40].Riley RS, June CH, Langer R, Mitchell MJ, Nat. Rev. Drug Discovery 2019, 18, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Shen D, Yang J, Li X, Zhou L, Zhang R, Li W, Chen L, Wang R, Zhang F, Zhao D, Nano Lett. 2014, 14, 923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.